
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Global optimization of extraframework ensembles in zeolites: structural analysis of extraframework aluminum species in MOR and MFI zeolites†
Elena V.
Khramenkova‡
 ,
Harshini
Venkatraman‡
,
Victor
Soethout
and
Evgeny A.
Pidko
*
,
Harshini
Venkatraman‡
,
Victor
Soethout
and
Evgeny A.
Pidko
*
Inorganic Systems Engineering group, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: E.A.Pidko@tudelft.nl
First published on 11th October 2022
Abstract
Metal-modified zeolites are versatile catalytic materials with a wide range of industrial applications. Their catalytic behaviour is determined by the nature of externally introduced cationic species, i.e., its geometry, chemical composition, and location within the zeolite pores. Superior catalyst designs can be unlocked by understanding the confinement effect and spatial limitations of the zeolite framework and its influence on the geometry and location of such cationic active sites. In this study, we employ the genetic algorithm (GA) global optimization method to investigate extraframework aluminum species and their structural variations in different zeolite matrices. We focus on extraframework aluminum (EFAl) as a model system because it greatly influences the product selectivity and catalytic stability in several zeolite catalyzed processes. Specifically, the GA was used to investigate the configurational possibilities of EFAl within the mordenite (MOR) and ZSM-5 frameworks. The xTB semi-empirical method within the GA was employed for an automated sampling of the EFAl–zeolite space. Furthermore, geometry refinement at the density functional theory (DFT) level of theory allowed us to improve the most stable configurations obtained from the GA and elaborate on the limitations of the xTB method. A subsequent ab initio thermodynamics analysis (aiTA) was chosen to predict the most favourable EFAl structure(s) under the catalytically relevant operando conditions.
1. Introduction
The development of advanced predictive models in heterogeneous catalysis requires a deep insight into the molecular-level structure of the active sites, their catalytic mechanism and evolution under operating conditions.1,2 The structural complexity and heterogeneity of common solid catalysts are major challenges in constructing representative active site models, which are conventionally performed using a combination of various indirect characterization data and chemical intuition of researchers.3 Global optimization techniques provide a practical approach to reducing the expert bias in addressing the active site structure in heterogeneous catalysts.4One of the most popular global optimization (GO) techniques is the genetic algorithm (GA) which allows scanning of the potential energy surface (PES) of a given system and prediction of the most stable structures via an evolutionary algorithm.3 The underlying principles of the algorithm are based on learning the “good” features of the possible structural moieties and improving them throughout the alterations of continuous structures. In this manner, a pool of advanced solutions can be identified which is otherwise challenging to guess based on chemical intuition and spectroscopic data only.5,6 GO algorithms have been successfully used to address the structural problem of the active sites in heterogeneous catalysis including oxides,7,8 supported metal nanoparticles,9 electrocatalytic interfaces,10 and, since quite recently, also zeolite-based catalysts.11,12
Zeolites are porous, crystalline aluminosilicates, whose reactivity and catalytic behaviour can be widely adjusted by modifying them with extraframework (EF) metal-containing species.13,14 The structural and catalytic properties of such EF species depend on the chemical composition (lattice Si/Al ratio), the presence of heteroatoms, and the topological properties of the confining zeolite matrix.15 As the spectrum of plausible variations in features is large, it is important to understand the role of zeolite topology and the nature of the extraframework aluminum (EFAl) species, which largely influence the activity of zeolites.
The extraframework aluminum species situated in zeolites hold an important position in zeolite chemistry and catalysis. The formation of Lewis acidic EFAl in the zeolite pores during the synthesis or post-synthetic activation modulates the acidity, catalytic reactivity, and long-term stability of zeolite-based catalysts.16,17 The formation of EFAl often results in releasing aluminum from the framework in procedures of high-temperature steaming, calcination or acid/base leaching.18 The structures of the EFAl are not completely known; however, some authors quote the condensed and uncondensed species. The former are the phases of Al2O3, which are formed at the external surfaces of zeolites, while the latter are aluminum and oxoaluminum cations such as AlO+, AlOH2+, and Al3+.19 The formation of these mononuclear species has been proposed based on the results of NMR spectroscopy and DFT calculations, which, until recently, have been mostly considering small mononuclear EF aluminum.16,20 It has also been demonstrated that the strength of the BAS can be enhanced in the presence of AlOH2+. However, DFT calculations combined with ab initio thermodynamic analysis (aiTA) have shown that, under the conditions of zeolite activation, the formation of the multinuclear Al3O4H34+ species inside FAU can be observed.16 A driving force for the formation of the EFAl complexes with high nuclearity was stated to be the high basicity of terminal O-containing groups and the unsaturation of Al centers in these complexes. The experimental observation of the multinuclear species was demonstrated by Zheng et al. using the combined 31P solid-state NMR and 1,2-bis(dimethylphosphine) ethane probe molecule method.21
EFAl species are speculated to reduce the effective pore size within the zeolitic framework leading to better stabilization of intermediates and transition states during the catalytic process.22 Another proposal states that EFAl species polarize the BAS in their vicinity which in turn increases the reactivity of the BAS.23,24 Although several insights were gained through earlier spectroscopic and theoretical studies of EFAl moieties, their exact geometry still remains elusive. The spectroscopic characterization of EFAl yields a general understanding of the coordination environment but provides limited information on the structural changes induced by the reactive conditions which are essential to discern the catalytic role of these species.3
Herein, we present a computational study aimed at addressing the EFAl structures in zeolites (Scheme 1(a)). The computational workflow implemented to achieve this is schematically illustrated in Scheme 1(b). For a given selection of EFAl stoichiometries encapsulated in MOR or ZSM-5 zeolites, an exhaustive scanning of the PES was carried out using the GA based on xTB semi-empirical calculations. The structures produced at the end of the GA configurational search were further refined by fully periodic DFT calculations. Subsequently, the stabilities of the structurally refined stoichiometries under catalytically relevant conditions were evaluated in the framework of aiTA. The workflow implemented in this study provides a bias-free approach for the structural analysis of extraframework species in zeolites and allows configurations to be found that could not be previously anticipated.
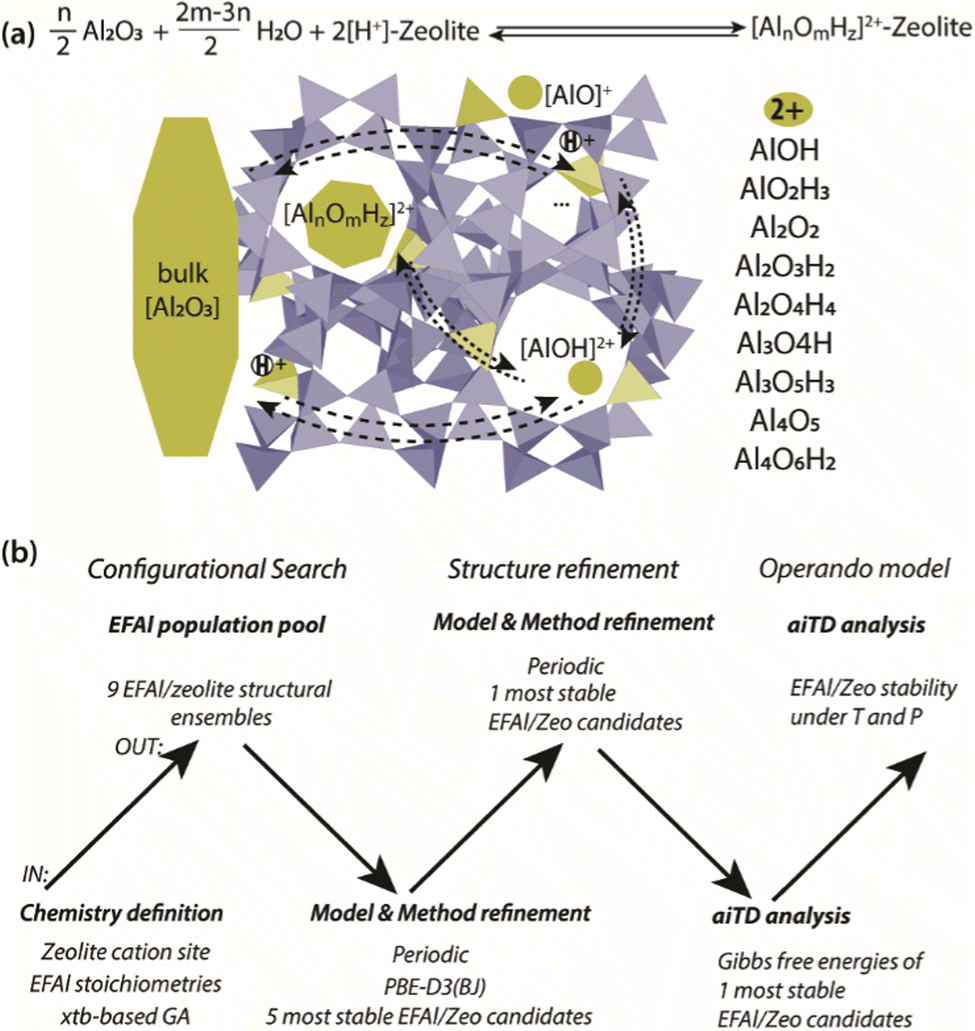 | ||
| Scheme 1 (a) The model definition for the interconversion of EFAl species in a zeolite matrix with a list of stoichiometries included in the current stability analysis of molecular intrazeolite EFAl with a reference to separate phases of bulk Al2O3 and the acidic zeolite matrix (2[H+]–zeolite). The structure prediction and stability evaluation of the EFAl–zeolites followed a hierarchical approach (b) involving the initial configurational search using a semi-empirical (xTB)-based configurational search followed by structural refinement using a full periodic DFT modelling and subsequent aiTA to evaluate the stability of the obtained extraframework ensembles under the catalytically relevant conditions. | ||
2. Computational methods
Configurational search
A genetic algorithm (GA) was used to exhaustively explore the configurational space formed by the isomeric structures of 9 different EFAl stoichiometries, individually. Fig. 1 presents the cluster models of the zeolites utilized in the GA search, which are highlighted as a part of their respective periodic structures. Such structural models were used earlier as they resemble the model catalysts of EFAl-containing zeolites prepared by impregnation and ion exchange processes. By selecting a side pocket of MOR and γ-site of ZSM-5 as the cation sites accommodating EFAl, a uniform confinement effect was created across EFAl in respective frameworks.25,26 The models assumed the defect-free zeolite lattice and uniform confinement environment to facilitate analysis and structural exploration. The detailed information on the pore size and zeolitic topologies is present in Section S.1. of the ESI.† The choice of the EFAl stoichiometries considered in this study (Scheme 1(a)) was inspired by evidence from previous findings on the formation of mononuclear species such as AlOH2+ and AlO2H32+, binuclear species such as Al2O22+, Al2O3H22+, and Al2O4H42+, and the spontaneous formation of multinuclear EF aluminum such as Al3O4H2+, Al3O5H32+, Al4O52+ and Al4O6H22+.20,27–30 Note that these stoichiometries correspond to complete ensembles. These ensembles within zeolites can be represented as a single species with a +2 overall charge and by a combination of species having the same total charge (e.g. a +1 EFAl cation and a zeolite BAS or a neural EFAl and two BAS). The cationic charge of the EF ensemble was compensated by two negatively charged AlO2− lattice units.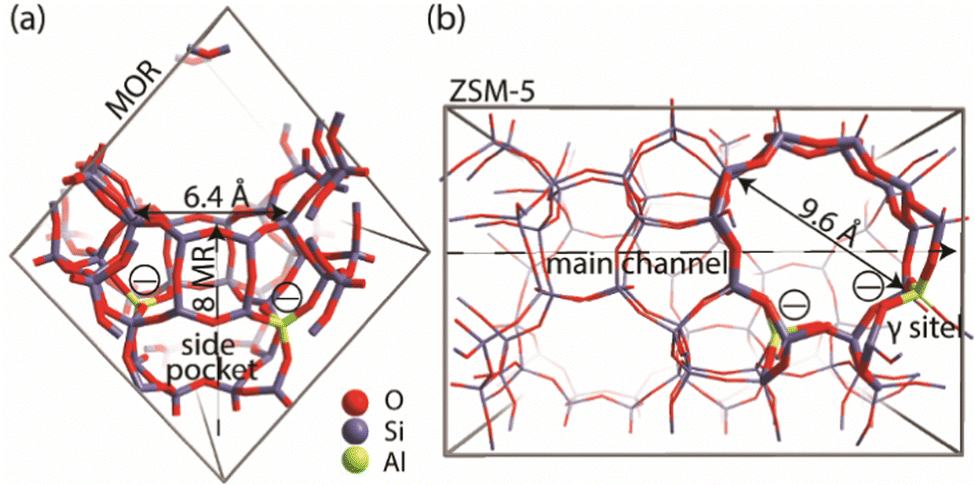 | ||
| Fig. 1 (a) Location of the cluster model utilized in the genetic algorithm for the global minima search of cationic sites incorporated in the periodic model of the mordenite. (b) Location of the cluster model utilized in the genetic algorithm for the global minima search of cationic sites incorporated in the periodic model of the ZSM. | ||
The configuration exploration was carried out using the version of the GA developed by Vilhelmsen and Hammer.31 The operations of the fitness assignment, crossover, mutation, and selection were executed in the GA using the atomic simulation environment (ASE) with the semi-empirical tight-binding calculator GFN1 – xTB.31–34 The mutation probability of the GA was chosen as 30%, and the population size at the end of each generation was set to consist of 20 structures. During the GA search, only the positions of the extraframework species were allowed to change, while the coordinates of the cluster model atoms were kept frozen. The parameters of the maximum energy difference, the maximum difference in the interatomic difference and the maximum interatomic distances were set to 0.02 eV, 0.7 Å, and 0.015 Å, respectively. The convergence was assumed to be reached when the last 5 consecutive generations of the population pool were identical.
Structure refinement
Since the GFN-xTB methods proved to be reliable for the geometries and frequencies calculations, but not the energies, GFN1-xTB optimized configurations were further refined at the DFT level using more realistic periodic zeolite models. The structural refinement was carried out at the PBE-D3(BJ) level of theory.35 The final pool of each GA run was examined and five lowest-energy configurations were extracted for the structural refinement. The compromise choice of 5 lowest lying candidates is motivated by our preliminary cluster calculations on mononuclear EFAl species in MOR (see Section S.2. in the ESI†), showing that such a choice allows the minimum-energy considerations to be found, whilst providing a good compromise between the accuracy and computational costs. For the structure refinement using periodic DFT (pDFT) calculations, the configurations obtained with the xTB GA on cluster models were directly transferred into the respective positions of the fully periodic zeolite models.pDFT calculations were carried out using the Vienna Ab Initio Simulation Package (VASP)36 with the generalized gradient approximation PBE functional and Grimme's semiempirical dispersion correction method D3 (BJ).37–39 A plane wave (PW) basis set with a cut-off energy of 400 eV was used in combination with the projected augmented wave (PAW)40 method. The Brillouin zone sampling was restricted to the Γ point. The convergence was assumed to be reached when the forces on the atoms were below 0.05 eV Å−1. The ZSM-5 unit cell was optimized, and the following lattice parameters were used: a = 20.24, b = 20.01 and c = 13.44 Å. A supercell of MOR was constructed by a doubling monoclinic primitive cell along the c axis and with the lattice parameters of a = b = 13.65, c = 15.02 Å and α = β = 90.0° and γ = 97.2°. Bulk α-Al2O3 with optimized unit cell parameters as a = b = 7.72, c = 12.96 and α = β = 90.0° and γ = 120.0° was modelled to be used as the reference state for the EFAl aiTA calculations. The Brillouin zone sampling was performed using a 3 × 3 × 1 mesh. For comparison, the thermodynamic analysis involving a less stable bulk Al(OH)3 phase as a reference was carried out with the results summarized in Section S.3. of the ESI.†
Operando model
The relative stabilities of the EFAl species in zeolite pores under the catalytically relevant conditions were next assessed in the framework of the aiTA method.41–43 The following equilibrium was considered for the stability assessment: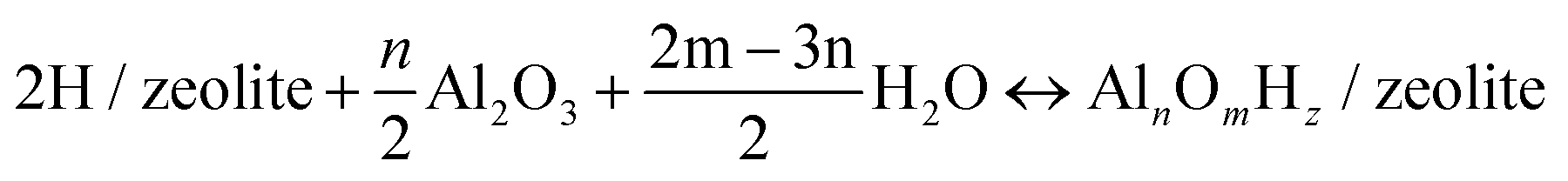 | (E1) |
The reaction free energy is defined as
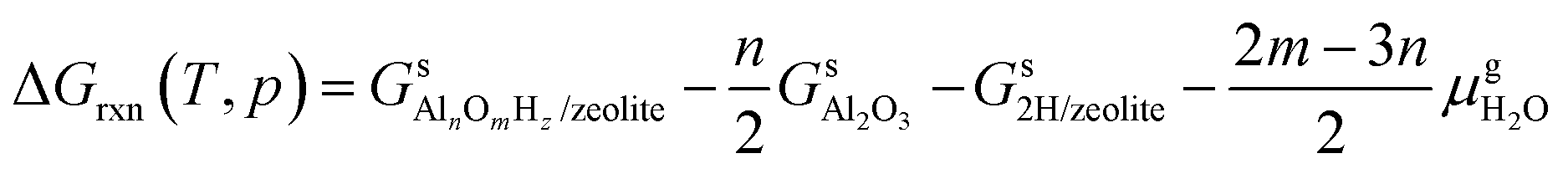 | (E2) |
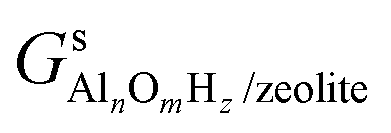 ,
, 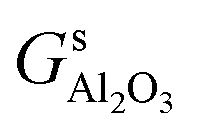 and Gs2H/zeolite are the Gibbs free energies of the EFAl-containing zeolite model, bulk Al2O3 and parent EFAl-free acidic zeolite matrix (2H/zeolite). The bulk, 2H/zeolite and AlnOmHz/zeolite are the DFT-approximated energies of the aluminum oxide, the respective zeolite framework with two hydrogens and the frameworks with situated EFAls. The PV-contribution entropy of the solids could be neglected and the expression for the Gibbs free energy can be written as follows:
and Gs2H/zeolite are the Gibbs free energies of the EFAl-containing zeolite model, bulk Al2O3 and parent EFAl-free acidic zeolite matrix (2H/zeolite). The bulk, 2H/zeolite and AlnOmHz/zeolite are the DFT-approximated energies of the aluminum oxide, the respective zeolite framework with two hydrogens and the frameworks with situated EFAls. The PV-contribution entropy of the solids could be neglected and the expression for the Gibbs free energy can be written as follows: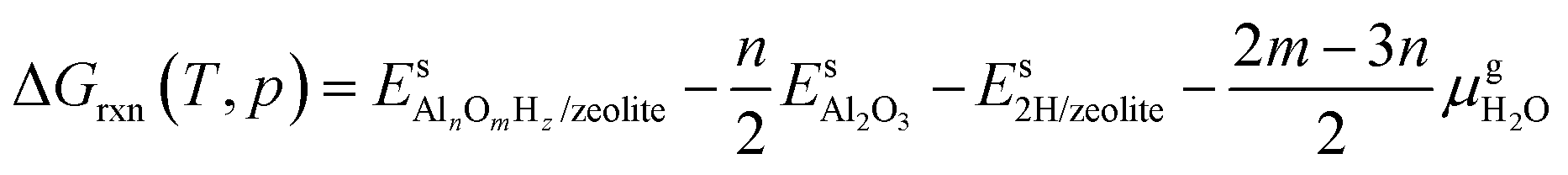 | (E3) |
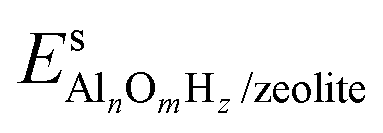 and Es2H/zeolite are the pDFT-energies of the given EFAl–zeolite and the parent H–zeolite structures, respectively. The condition dependencies of the reaction free energy are explicitly accounted for with
and Es2H/zeolite are the pDFT-energies of the given EFAl–zeolite and the parent H–zeolite structures, respectively. The condition dependencies of the reaction free energy are explicitly accounted for with 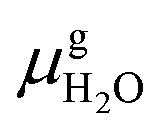 defined as
defined as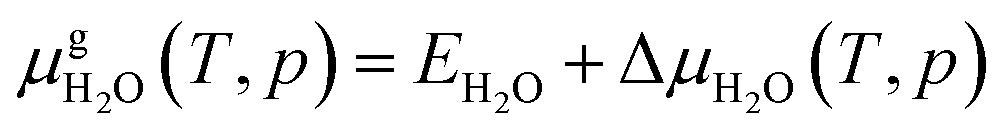 | (E4) |
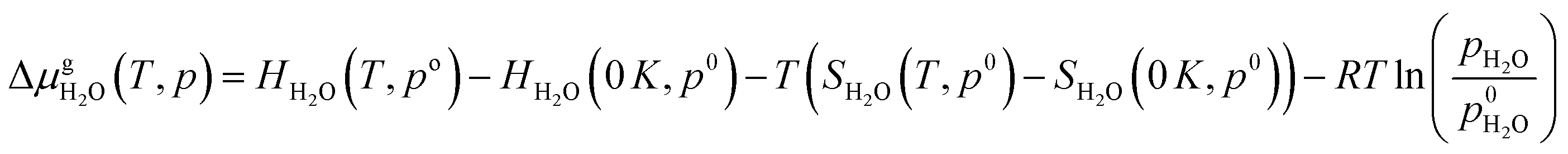 | (E5) |
Thermodynamic tables were used to calculate the values of entropy and enthalpy at a standard pressure (1 bar) and different temperatures.44 Incorporating eqn (E4) and (E5) in eqn (E3) gives the change in the Gibbs free energy of the formation of the EFAl stoichiometries. This methodology has been successfully employed earlier on a wide range of solid systems including zeolite catalysts.12,41,45
3. Results and discussion
Fig. 2 summarizes the most stable EFAl geometries among the five DFT-optimized configurations (Fig. S5 and S6, ESI†) that were initially extracted from the bias-free xTB-based GA configurational search (Fig. S3 and S4, ESI†). The pDFT refinement of the xTB semi-empirically optimized structural isomers results in their substantial geometrical alterations. The presence of the artefacts in xTB predictions has been previously shown by Vicent-Luna in a form of undesirable structural distortions of geometries with lower symmetry after the optimization.46 This could be attributed to the key approximation within GFN1 representing the noncovalent interactions via the atom pair-wise electrostatic interactions described as spherically symmetric and monopole-types.47 Nevertheless, we found that the xTB-driven GA search provides a robust and efficient way to explore the configurational space of the intrazeolite extraframework species to obtain a good structural guess for the subsequent pDFT refinement. In the following, we predominantly discuss the pDFT refined data, while the xTB-derived predecessor structures will only briefly be discussed where appropriate.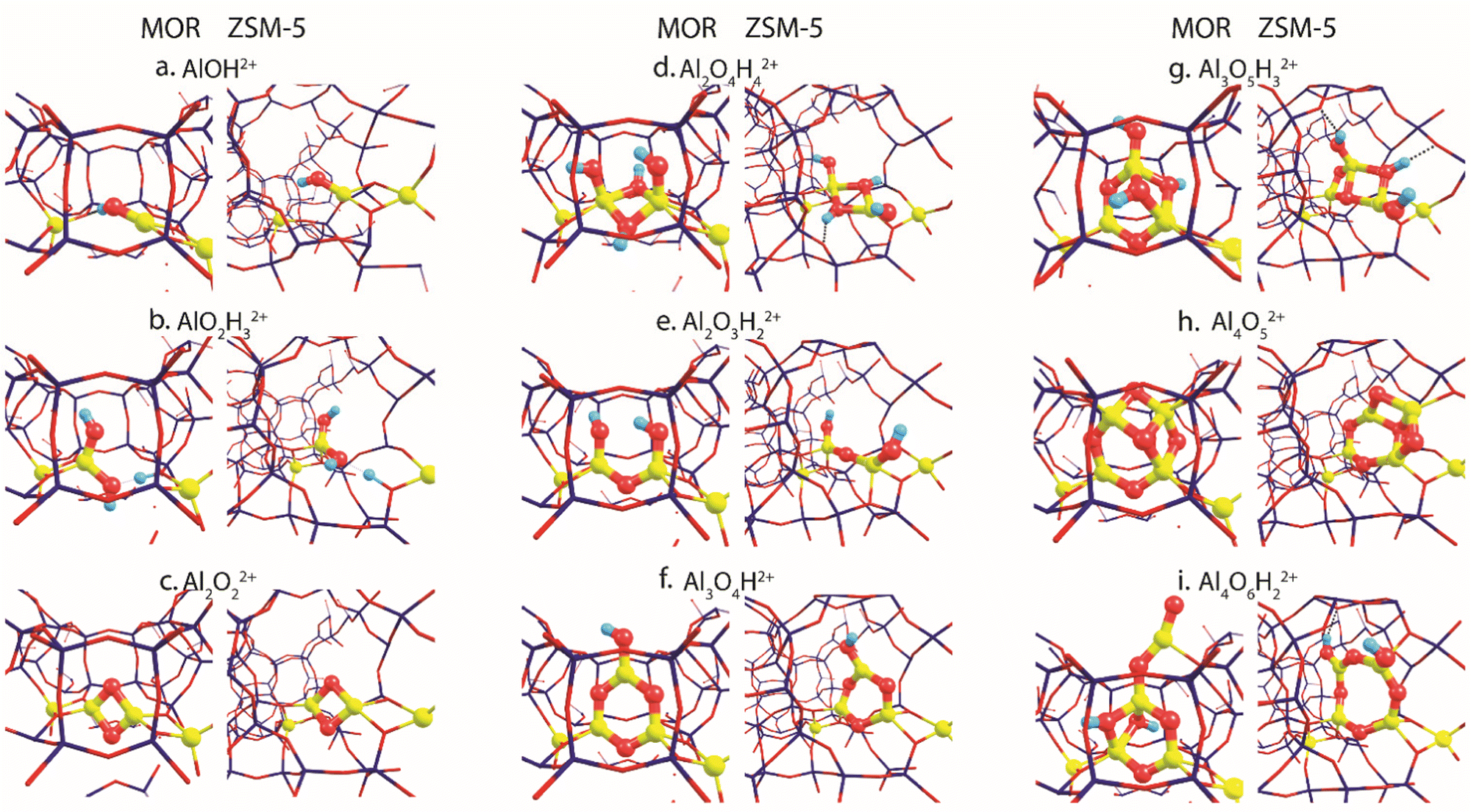 | ||
| Fig. 2 The global minima structures of various EFAl stoichiometries produced by GA runs and subsequently optimized in the MOR and ZSM-5 zeolite frameworks. | ||
The global minima geometries summarized in Fig. 2 indicate that, apart from the little distortions in the coordinational environment caused by the formation of the hydrogen bonds, similar EFAl geometries are found for particular stoichiometries in MOR and ZSM-5 zeolites. This stands for a majority of stoichiometries, except for the multinuclear Al3O5H3 and Al4O6H2 complexes, where structurally distinct features are promoted by the different confinement spaces.
For the smallest AlOH stoichiometry, a single stable EFAl type is revealed by pDFT represented by an AlOH2+ cation coordinated directly to the lattice basic oxygen ions of one of the framework AlO2− anions with the second one providing the indirect charge-compensation of the cationic EFAl (Fig. 2a). The GA structural exploration has also revealed alternative configurations for the mordenite-confined AlOH represented by Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O+ and an adjacent Brønsted acid site (Fig. S3c, ESI†), additionally stabilized within the narrower confinement space of the MOR side-pocket. Such configurations have been discussed in the previous EFAl literature.19,48 However, they were found unstable or underwent restructuring into the charge-alternative states during the pDFT refinement. Similarly, a single stable configuration is found for another mononuclear stoichiometry – AlO2H3, represented by a tetrahedral Al(OH)2+ EFAl cation coordinated to one lattice AlO2− anion with the second one charge-compensated by the H+ BAS site (Fig. 2b).
O+ and an adjacent Brønsted acid site (Fig. S3c, ESI†), additionally stabilized within the narrower confinement space of the MOR side-pocket. Such configurations have been discussed in the previous EFAl literature.19,48 However, they were found unstable or underwent restructuring into the charge-alternative states during the pDFT refinement. Similarly, a single stable configuration is found for another mononuclear stoichiometry – AlO2H3, represented by a tetrahedral Al(OH)2+ EFAl cation coordinated to one lattice AlO2− anion with the second one charge-compensated by the H+ BAS site (Fig. 2b).
Binuclear stoichiometries include Al2O2 and more hydrated/hydroxylated complexes such as Al2O4H4 and Al2O3H2. For both zeolites, the former species adopts a symmetrical diamond-shape binuclear Al(μ-O)2Al2+ configuration (Fig. 2c). This geometry is described as the most stable among all DFT-optimized structures, whereas some of their xTB-optimized predecessors reveal the presence of a linear isomer OAl(μ-O)Al which has one bridging oxygen and one terminal oxygen (Fig. S4a, ESI†), similar to the structure proposed earlier for the EF–Ga species.49 In Fig. 2d and e, Al2O4H4 and Al2O3H2 geometries respectively form symmetrical complexes that have minor distinctions in the hydrogen bond arrangements and proximity to the Al framework sites, affecting their effectivity of compensating the negative charge of the framework.
More complex trinuclear species such as Al3O4H and Al3O5H3 display more possibilities of structural arrangements. In Fig. 2f, Al3O4H stoichiometries exhibit the formation of a 6-membered ring with a coordinated OH-group, which is slightly distorted in ZSM-5 due to the formation of the hydrogen bonds between the OH group and the framework. The optimization of the A3O5H3 complexes gives a ring-shaped Al3O3H core moiety in ZSM-5 and an envelope-like Al3O3H core structure in MOR, which also coordinates two additional OH-groups of the structure. (Fig. 2g).
Fig. 2h and i show the EFAl moieties with the highest number of aluminum atoms considered in this study. A more hydrated Al4O6H2 (2i) species in mordenite accommodates a ring-shaped Al3O3H moiety with the bound OH-group at the same time expelling AlO2 species out of the pocket. Its ZSM-5 counterpart, on the other hand, could accommodate an 8-membered EFAl ring with two coordinated OH-groups. Another stable configuration of Al4O6H2 is a 6-membered EFAl with the OH-group and expelled AlO2H species as shown in Fig. S6i of the ESI.† For the Al4O5 stoichiometry in Fig. 2h, the most stable isomers adapt envelope-like Al3O3 geometries linked to an AlO2 moiety species in both zeolite frameworks. However, a more structurally diverse picture could be seen in a detailed 5-structure analysis. In Fig. S5h (ESI†), the Al4O5 geometry, when hosted in mordenite, tends to adapt in some cases coordination resembling the one from Al4O6H2 with an AlO2 species dislodged from the side pocket. The observed accommodation of the smaller species in the side pocket of MOR than that in ZSM-5 can be explained by a stronger confinement and limited pore size in mordenite.
With the results of DFT optimization being sufficiently elaborated, the respective stabilities of the structures under operando conditions could be compared. In Fig. 3, the ab initio thermodynamics analysis shows the Gibbs free energies of the formation of different stoichiometries as a function of water chemical potential (ΔμH2O). The values of the Gibbs free energies are computed using eqn (E3) and the ΔμH2O is derived from E5. The logarithmic pressure scales above the diagram represent the pressures required for the high (700 K and 500 K) and moderate (300 K) temperature processes.
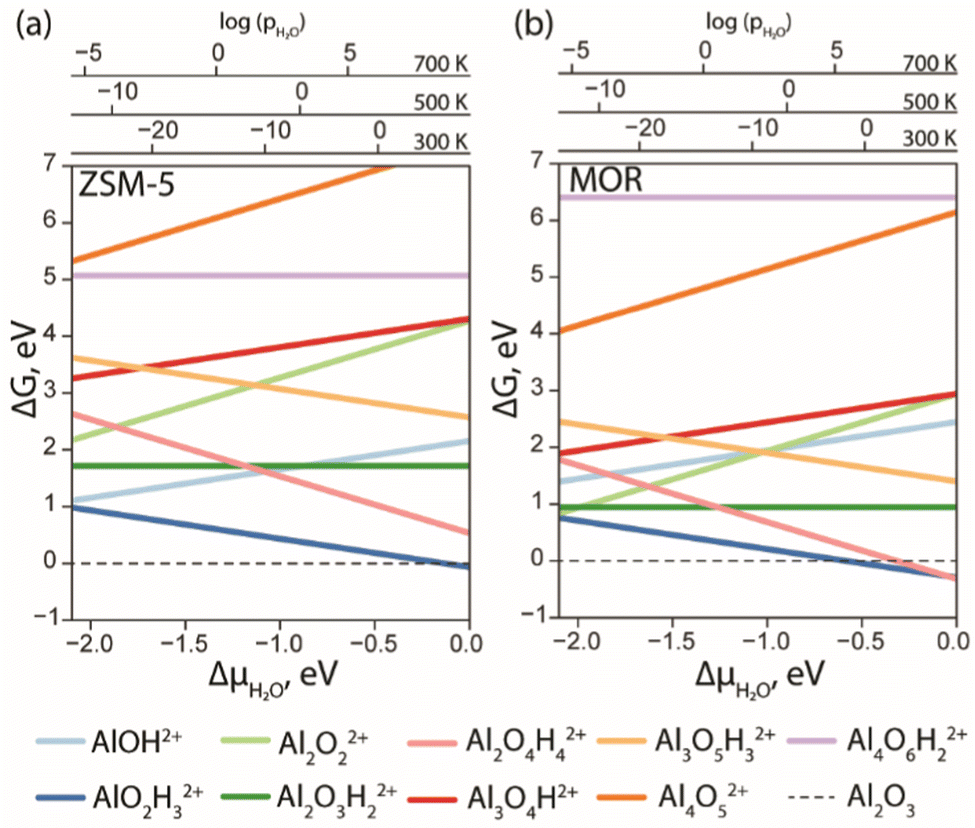 | ||
| Fig. 3 The Gibbs free energy diagram of the EFAl species confined in ZSM-5 (a) and MOR (b) was calculated with respect to the α-Al2O3 phase as a function of water chemical potential. | ||
The computed results reveal a general trend of the thermodynamic preference for the formation of mononuclear and binuclear cationic EFAl species in both zeolite topologies at high water chemical potentials, which is in line with the experimental findings.16,19,28 At low water chemical potential, the formation of a separate bulk aluminum oxide and zeolite BAS is predicted for the current highly stable α-Al2O3 reference structure. The cationic EFAl species are better stabilized when confined within the mordenite side pocket, as is evident from generally lower relative free energies predicted for all EFAl stoichiometries compared to those in the MFI. The largest cation Al4O6H22+ deviates from this trend, which because of its size cannot be accommodated within the narrow MOR side pocket resulting in the expulsion of quite an unstable O–AlO moiety into the main channel.
The thermodynamics of EFAl speciation is highly condition-dependent. At low chemical water potentials (μH2O < −1.0 eV), the formation of bulk α-Al2O3 oxide is preferred over the molecular cationic EFAl species indicating the limited possibility of the redispersion of Al upon the calcination zeolite activation procedures. Nevertheless, in the MOR zeolite, AlO2H3, Al2O3H2, Al2O2 and Al2O4H4 EFAl species can be found within 1 eV from the reference value, whereas they are an additional 1 eV less stable in ZSM-5. The mononuclear AlO2H3 and binuclear Al2O4H4 species are thermodynamically favoured at high μH2O, indicating that such isolated cationic sites can potentially be formed upon e.g. steaming or ion exchange and further stabilized kinetically in the activated material. The formation of EFAl species with a higher degree of nuclearity (three- and four-nuclear) is strongly thermodynamically unfavourable under all conditions, which is in sharp contrast to the situation previously discussed for low-silica faujasite, where trinuclear multiple-charged EFAl confined in small sodalite cages were among the most stable species.16
The predicted EFAl stabilities depend on the specific choice of the bulk Al reference phase. We have also considered a fully hydrated Al(OH)3 as a bulk reference state in aiTA (see Fig. S7 in Section S.3. of the ESI†). Because of the hydrated nature of the reference structure, the less hydrated multinuclear aggregates appear more stable at low μH2O in this case. Nevertheless, the preferential formation of mono- and binuclear cationic species at higher μH2O values relevant to the conditions of steam-calcination remains unchanged by the choice of the bulk Al reference.
Our results point to a preference for the increased hydration of EFAl at higher μH2O; however, we should also emphasize that the higher degrees of hydration could destabilize the EFAl/zeolite systems as water holds the potential to cleave Al–O–(H)–bonds,50 which could potentially manifest itself for stoichiometries with formal H2O contents above those considered in this study. The outcome of the GA search and the following aiTA procedure vividly indicated that a smaller pore size in mordenite offers a tighter confinement effect and better stabilization of the cationic species up to a certain size. The relative stability of the EFAl species when compared to the α-Al2O3 reference phase indicated a more pronounced stabilization of the mononuclear and binuclear species and the destabilization of the larger agglomerates due to the localized and disperse nature of the lattice negative charge and unfavourable confinement in MOR and ZSM-5. This explains the differences in EFAl speciation predicted for low-silica cage-type faujasite zeolites and suggests a different promoting effect of these species in the different zeolite topologies.
Conclusions
In this study, we applied the semi-empirical-based GA to investigate the structural uncertainty surrounding the EFAl species confined within zeolites. The catalytic importance of these extraframework species motivated us to research the configurations of different EFAl nuclearities in two zeolitic frameworks such as MOR and ZSM-5. Varying the stoichiometry of the EFAl accounts for the different concentrations of aluminum. For this purpose, the number of aluminum atoms ranging from 1 to 4 was considered in our study. Different zeolite topologies impose a distinct confinement effect on the anchored extraframework species. Here, we have shown that, within a chosen level of theory, the xTB-based GA method is effective for the structural exploration of complex EFAl species in zeolites by considering 9 different stoichiometries in MOR and ZSM-5. However, the structural and energy refinement at the DFT level is necessary to form a basis for the accurate and reliable structural evaluation and stability analysis.The low-nuclearity EFAl structures are coherent with the findings reported by a previous study, thereby validating the proposed computational workflow. In addition, more complex multinuclear EFAl configurations were investigated and included in the stability analysis. To analyse the EFAl stability under operando catalytically relevant conditions, the relative energetics of the pDFT-refined low-lying structures was estimated by embedding them in the ab initio thermodynamics analysis. It was shown that both ZSM-5 and MOR topologies favour the formation of the cationic mononuclear EFAl in the presence of water/water vapors. Specifically, the vicinal Brønsted acid site and mononuclear Al(OH)2+ cation representing the AlO2H3 EFAl stoichiometry were found to be the most thermodynamically stable at high μH2O in both zeolite frameworks. However, at low water chemical potential values, the formation of bulk Al2O3 is more thermodynamically favourable. This can be attributed to the restricted confinement space in MOR and ZSM-5 channels. Our calculations suggest a generally better stabilization of a majority of the EFAl species in the MOR zeolite.
Thus, we show that GA-based global optimization in combination with pDFT structural refinement and ab initio thermodynamics analysis provides an efficient computational workflow for the expert-bias-free structural prediction on intrazeolite extraframework ensembles and evaluating their stabilities under catalytically relevant conditions. By using the catalytically important EFAl species embedded in MOR and ZSM-5 frameworks, we demonstrate that our proposed approach can be a powerful tool to address the structural conundrum of extraframework speciation in zeolite-based catalysts.
Author contributions
The manuscript was written via contributions from all the authors. All the authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the European Research Council (ERC) under European Union's Horizon 2020 research and innovation programme (grant agreement no. 725686). We acknowledge the Netherlands Organization for Scientific Research (NWO) for sponsoring the supercomputer facilities.References
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Cu-ZSM-5 zeolites for the formation of methanol from methane and oxygen: Probing the active sites and spectator species, Catal. Lett., 2010, 138, 14–22 CrossRef CAS
.
- C. Vogt and B. M. Weckhuysen, The concept of active site in heterogeneous catalysis, Nat. Rev. Chem., 2022, 6, 89–111 CrossRef
- L. Grajciar, C. J. Heard, A. A. Bondarenko, M. V. Polynski, J. Meeprasert, E. A. Pidko and P. Nachtigall, Towards operando computational modeling in heterogeneous catalysis, Chem. Soc. Rev., 2018, 47, 8307–8348 RSC
- A. A. Peterson, Global optimization of adsorbate-surface structures while preserving molecular identity, Top. Catal., 2014, 57, 40–53 CrossRef CAS
- N. Chakraborti, Genetic algorithms in materials design and processing, Int. Mater. Rev., 2004, 49, 246–260 CrossRef CAS
- R. Leardi, Genetic algorithms in chemistry, J. Chromatogr. A, 2007, 1158, 226–233 CrossRef CAS PubMed
- C. Roberts and R. L. Johnston, Investigation of the structures of MgO clusters using a genetic algorithm, Phys. Chem. Chem. Phys., 2001, 3, 5024–5034 RSC
- M. H. Lee, J. Xu and W. Xie, Exploring the Stability of Single-Atom Catalysts Using the Density Functional Theory-Based Global Optimization Method: H2Formation on VO x/γ-Al2O3(100), J. Phys. Chem. C, 2022, 126, 6973–6981 CrossRef CAS
- G. Sun, A. N. Alexandrova and P. Sautet, Pt8 cluster on alumina under a pressure of hydrogen: Support-dependent reconstruction from first-principles global optimization, J. Chem. Phys., 2019, 151, 194703 CrossRef PubMed
- Z. Zhang, Z. Wei, P. Sautet and A. N. Alexandrova, Hydrogen-induced restructuring of a Cu(100) electrode in electroreduction conditions, ChemRxiv, 2022, preprint DOI:10.26434/chemrxiv-2022-bwx53.
- D. Hou, L. Grajciar, P. Nachtigall and C. J. Heard, Origin of the Unusual Stability of Zeolite-Encapsulated Sub-Nanometer Platinum, ACS Catal., 2020, 10, 11057–11068 CrossRef CAS
- C. Liu, E. A. Uslamin, E. Khramenkova, E. Sireci, L. T. L. J. Ouwehand, S. Ganapathy, F. Kapteijn and E. A. Pidko, High Stability of Methanol to Aromatic Conversion over Bimetallic Ca,Ga-Modified ZSM-5, ACS Catal., 2022, 12, 3189–3200 CrossRef CAS PubMed
- G. Li and E. A. Pidko, The Nature and Catalytic Function of Cation Sites in Zeolites: a Computational Perspective, ChemCatChem, 2019, 11, 134–156 CrossRef CAS
- N. Kosinov, C. Liu, E. J. M. Hensen and E. A. Pidko, Engineering of Transition Metal Catalysts Confined in Zeolites, Chem. Mater., 2018, 30, 3177–3198 CrossRef CAS PubMed
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Mesopore-modified zeolites: Preparation, characterization, and applications, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, Nature and Catalytic Role of Extraframework Aluminum in Faujasite Zeolite: A Theoretical Perspective, ACS Catal., 2015, 5, 7024–7033 CrossRef CAS
- T. T. Le, A. Chawla and J. D. Rimer, Impact of acid site speciation and spatial gradients on zeolite catalysis, J. Catal., 2020, 391, 56–68 CrossRef CAS
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Towards a better understanding of Lewis acidic aluminium in zeolites, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed
- D. L. Bhering, A. Ramírez-Solís and C. J. A. Mota, A density functional theory based approach to extraframework aluminum species in zeolites, J. Phys. Chem. B, 2003, 107, 4342–4347 CrossRef CAS
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S. Bin Liu and A. Zheng, Origin and Structural Characteristics of Tri-coordinated Extra-framework
Aluminum Species in Dealuminated Zeolites, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS PubMed
- A. Zheng, Y. Xiao, Z. Liu, G. Li and W. Chen, Observation of Multinuclear Extra-framework Aluminum Species in Dealuminated Zeolite Catalysts, Res. Sq., 2020 DOI:10.21203/rs.3.rs-50498/v1
- R. Gounder, A. J. Jones, R. T. Carr and E. Iglesia, Solvation and acid strength effects on catalysis by faujasite zeolites, J. Catal., 2012, 286, 214–223 CrossRef CAS
- J. Jiao, J. Kanellopoulos, W. Wang, S. S. Ray, H. Foerster, D. Freude and M. Hunger, Characterization of framework and extra-framework aluminum species in non-hydrated zeolites Y by 27Al spin-echo, high-speed MAS, and MQMAS NMR spectroscopy at B0 = 9.4 to 17.6 T, Phys. Chem. Chem. Phys., 2005, 7, 3221–3226 RSC
- J. Jiao, S. S. Ray, W. Wang, J. Weitkamp and M. Hunger, Effect of dehydration on the local structure of framework silicon atoms in zeolites Y investigated by solid-state NMR spectroscopy, Z. Anorg. Allg. Chem., 2005, 631, 484–490 CrossRef CAS
- H. Issa, J. Toufaily, T. Hamieh, A. Sachse and L. Pinard, Pyridine assisted desilication of mordenite, Appl. Catal., A, 2019, 583, 117139 CrossRef CAS
- J. Holzinger, P. Beato, L. F. Lundegaard and J. Skibsted, Distribution of Aluminum over the Tetrahedral Sites in ZSM-5 Zeolites and Their Evolution after Steam Treatment, J. Phys. Chem. C, 2018, 122, 15595–15613 CrossRef CAS
- C. Liu, I. Tranca, R. A. Van Santen, E. J. M. Hensen and E. A. Pidko, Scaling Relations for Acidity and Reactivity of Zeolites, J. Phys. Chem. C, 2017, 121, 23520–23530 CrossRef CAS PubMed
- S. Li, A. Zheng, Y. Su, H. Zhang, L. Chen, J. Yang, C. Ye and F. Deng, Brønsted/Lewis acid synergy in dealuminated HY zeolite: A combined solid-state NMR and theoretical calculation study, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS PubMed
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, Relationship between acidity and catalytic reactivity of faujasite zeolite: A periodic DFT study, J. Catal., 2016, 344, 570–577 CrossRef CAS
- A. Gola, B. Rebours, E. Milazzo, J. Lynch, E. Benazzi, S. Lacombe, L. Delevoye and C. Fernandez, Effect of leaching agent in the dealumination of stabilized Y zeolites, Microporous Mesoporous Mater., 2000, 40, 73–83 CrossRef CAS
- L. B. Vilhelmsen and B. Hammer, A genetic algorithm for first principles global structure optimization of supported nano structures, J. Chem. Phys., 2014, 141, 044711 CrossRef PubMed
- S. Grimme, C. Bannwarth and P. Shushkov, A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1-86), J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB - An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed
- P. Pracht, E. Caldeweyher, S. Ehlert and S. Grimme, A Robust Non-Self-Consistent Tight-Binding Quantum Chemistry Method for large Molecules, ChemRxiv, 2019 DOI:10.26434/chemrxiv.8326202.v1
- J. P. Menzel, M. Kloppenburg, J. Belić, H. J. M. de Groot, L. Visscher and F. Buda, Efficient workflow for the investigation of the catalytic cycle of water oxidation catalysts: Combining GFN-xTB and density functional theory, J. Comput. Chem., 2021, 42, 1885–1894 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized
gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol, J. Catal., 2016, 338, 305–312 CrossRef CAS
- H. Guhl, W. Miller and K. Reuter, Water adsorption and dissociation on SrTiO3 (001) revisited: A density functional theory study, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155455 CrossRef
- E. V. Khramenkova, M. G. Medvedev, G. Li and E. A. Pidko, Unraveling the Nature of Extraframework Catalytic Ensembles in Zeolites: Flexibility and Dynamics of the Copper-Oxo Trimers in Mordenite, J. Phys. Chem. Lett., 2021, 12, 10906–10913 CrossRef CAS PubMed
-
D. R. Stull and H. Prophet, JANAF Thermochemical Tables, US National Bureau of Standards, Washington, DC, 2nd edn, 1971 Search PubMed
- K. Reuter, Ab Initio Thermodynamics and First-Principles Microkinetics for Surface Catalysis, Catal. Lett., 2016, 146, 541–563 CrossRef CAS
- J. M. Vicent-Luna, S. Apergi and S. Tao, Efficient Computation of Structural and Electronic Properties of Halide Perovskites Using Density Functional Tight Binding: GFN1-xTB Method, J. Chem. Inf. Model., 2021, 61, 4415–4424 CrossRef CAS PubMed
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended tight-binding quantum chemistry methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS
- X. Pu, N. W. Liu and L. Shi, Acid properties and catalysis of USY zeolite with different extra-framework aluminum concentration, Microporous Mesoporous Mater., 2015, 201, 17–23 CrossRef CAS
- E. A. Pidko, R. A. Van Santen and E. J. M. Hensen, Multinuclear gallium-oxide cations in high-silica zeolites, Phys. Chem. Chem. Phys., 2009, 11, 2893–2902 RSC
- M. C. Silaghi, C. Chizallet, J. Sauer and P. Raybaud, Dealumination mechanisms of zeolites and extra-framework aluminum confinement, J. Catal., 2016, 339, 242–255 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2195171–2195188. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cp03603g. Data related to this publication is available via the 4TU database under the DOI: 10.4121/21400845. |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2022 |