
Visible-light and near-infrared fluorescence and surface-enhanced Raman scattering point-of-care sensing and bio-imaging: a review
Yingjie
Hang
 ,
Jennifer
Boryczka
and
Nianqiang
Wu
*
,
Jennifer
Boryczka
and
Nianqiang
Wu
*
Department of Chemical Engineering, University of Massachusetts Amherst, Amherst, MA 01003-9303, USA. E-mail: nianqiangwu@umass.edu; Tel: +1-413-545-6175
First published on 13th December 2021
Abstract
This review article deals with the concepts, principles and applications of visible-light and near-infrared (NIR) fluorescence and surface-enhanced Raman scattering (SERS) in in vitro point-of-care testing (POCT) and in vivo bio-imaging. It has discussed how to utilize the biological transparency windows to improve the penetration depth and signal-to-noise ratio, and how to use surface plasmon resonance (SPR) to amplify fluorescence and SERS signals. This article has highlighted some plasmonic fluorescence and SERS probes. It has also reviewed the design strategies of fluorescent and SERS sensors in the detection of metal ions, small molecules, proteins and nucleic acids. Particularly, it has provided perspectives on the integration of fluorescent and SERS sensors into microfluidic chips as lab-on-chips to realize point-of-care testing. It has also discussed the design of active microfluidic devices and non-paper- or paper-based lateral flow assays for in vitro diagnostics. In addition, this article has discussed the strategies to design in vivo NIR fluorescence and SERS bio-imaging platforms for monitoring physiological processes and disease progression in live cells and tissues. Moreover, it has highlighted the applications of POCT and bio-imaging in testing toxins, heavy metals, illicit drugs, cancers, traumatic brain injuries, and infectious diseases such as COVID-19, influenza, HIV and sepsis.
 Yingjie Hang | Yingjie Hang is a graduate student under the supervision of Prof. Wu at the University of Massachusettes Amherst. She received her BS degree from Soochow University in 2016. Her research interest lies in point-of-care testing, plasmon-enhanced fluorescence and surface-enhanced Raman scattering (SERS) for protein and nucleic acid detection. |
 Jennifer Boryczka | Jennifer Boryczka received her MS degree in materials science and engineering from West Virginia University, USA. Currently she is a graduate student under the supervision of Prof. Wu at the University of Massachusetts Amherst. She has co-authored about 5 journal articles. Her research interest lies in plasmon-enhanced fluorescent sensing, imaging and photodynamic therapy. |
 Nianqiang Wu | Dr Nianqiang (Nick) Wu received his PhD degree in materials science & engineering from Zhejiang University, China. Currently he is an Armstrong-Siadat Endowed Chair Professor in materials science at the University of Massachusetts Amherst. He is Fellow of the Electrochemical Society (FECS) and The Royal Society of Chemistry (FRSC). He received several honors and awards such as Highly Cited Researcher (Clarivate Analytics, Thomson Reuters) and ECS Sensor Division Outstanding Achievement Award. He has authored or co-authored about 200 journal articles, 3 book chapters and 1 book entitled “Biosensors Based on Nanomaterials and Nanodevices”. |
1. Introduction
Chemical compounds in human fluids and tissues reflect physiological or pathological processes, and can be used for diagnosis of diseases and assessment of medical intervention efficacy. It is essential to perform in vitro analysis of analytes such as metal ions, small molecules, proteins and nucleic acids in human fluids (blood, serum, plasma, urine, saliva and cerebrospinal spinal fluid).1–5 In most cases, in vitro analysis is performed using large-scale analytical techniques such as spectrophotometry, mass spectrometry, immunoassays, and electrophoresis. For example, enzyme-linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR) techniques are considered the gold standards for quantitation of soluble proteins and nucleic acids, respectively. Although ELISA and PCR are accurate methods, they are expensive, tedious, time-consuming, and require professionals to operate in a central laboratory. These shortcomings have motivated the development of point-of-care testing (POCT) tools to meet the needs of inexpensive, rapid, high-throughput, field-deployable, in vitro analyses by laypersons.6–8 The emergence of pandemics (such as swine flu (HIN1), severe acute respiratory syndrome (SARS), and COVID-19) and prevalence of epidemic diseases (such as malaria, dengue, chikungunya, yellow fever and Zika) have aroused an increasing demand for POCT tools to rapidly test for these infectious diseases at home, clinics, schools, employer sites, communities or hospitals.To enable portability, numerous sensors have been developed for in vitro analysis. According to the signal transduction mechanism, sensors can be categorized into different types such as electric, electrochemical, acoustic, magnetic, colorimetric, fluorescent, Raman devices, etc. Among these types of sensors, fluorescence and surface-enhanced Raman scattering (SERS) devices show great potential in in vitro analysis of human fluids due to their high sensitivity and high resistance to interference from sample matrices.9–11 For sensing applications, visible-light fluorescence and SERS probes are widely used because fluorescence dyes have large fluorescence quantum yields and Raman reporters have large scattering cross-sections, leading to strong signals in buffer sample matrices. Although most NIR-fluorescence dyes exhibit lower quantum yields than visible-light counterparts, and NIR Raman dyes have smaller scattering cross-sections, they are subject to less interference from the sample matrices of blood and plasma due to the reduced light absorption and less auto-fluorescence. On the other hand, it is worth noting that not all sensors are POCT tools because these sensors still require multi-step operations performed by well-trained personnel in a field or even in a laboratory. One of the effective means to enable POCT is the integration of sensors into microfluidic chips to create lab-on-chips, which are characteristic of militarization, field-deployment, automation, swiftness, minimal or non-invasive detection, and easy operation by laypersons.12,13 The availability of POCT tools will extend in vitro diagnosis (IVD) from hospitals to clinics and communities.
Besides in vitro testing, NIR fluorescence and SERS are finding increasing applications in in vivo imaging due to their minimal or non-invasive detection and relatively deep penetration into human tissues compared to other ultraviolet and visible-light analytical techniques. Current common bio-imaging technologies include magnetic resonance imaging (MRI), computed tomography (CT), ultrasound, X-ray radiography and positron-emission tomography scanning (PETS).14 However, these techniques are quite time-consuming and require minutes to hours to achieve imaging results. Besides, they are unable to obtain target-specific results and usually need professionals to distinguish the target area from the background. Also, long-time exposure to radiation under X-ray radiography, CT and PETS may cause harm to human health. Thus, it is difficult to obtain long-term and real-time imaging.15,16 In contrast to these imaging modalities, NIR fluorescence and SERS enable real-time monitoring of expressed genes, pathogens, metabolites, and drug compounds in living cells and tissues. Moreover, the target-specific results enhance diagnostic accuracy.
This article will first clarify the importance of biological transparency windows for selecting NIR fluorescence and SERS for POCT and bio-imaging, and then describe how to use plasmon to enhance the intensity of fluorescence emission and SERS. Next, it will give a summary of the design strategies of fluorescence and SERS sensors in the detection of metal ions, small molecules, proteins and nucleic acids, placing an emphasis on signal transduction and signal amplification. Subsequently, this article will describe a combination of sensors and sample handling modules in single microfluidic devices to create lab-on-chips toward POCT. Lastly, it will give an overview of bio-imaging based on NIR fluorescence and SERS, including instruments, probes and applications, and highlight how the principles of signal transduction and amplification are used in imaging living cells and tissues.
2. Optical properties of SERS and fluorescence
2.1 Biological transparency windows
For optical detection and imaging, an incident light beam is directed towards the sample to excite the light–matter interaction, and a detector is used to collect the light emitted or scattered from the sample. Biological samples may strongly absorb both the incident and the emitting light. Also, auto-fluorescence emission can occur under the excitation of the incident light, leading to interference. These effects may attenuate the incident light quickly with an increase in the penetration depth, reduce the intensity of emitting/scattering light, and increase the signal-to-noise ratio. Such side effects become deteriorated in the ultraviolet (UV) and visible-light spectral ranges, which make it difficult or even impossible for deep-tissue imaging and trace analyte detection. Fortunately, these side effects are minimized in three NIR spectral ranges, which are called “biological transparency windows”. The first biological window covers the wavelength from 650 nm to 950 nm (NIR-I), the second biological window ranges from 1000 nm to 1350 nm (NIR-II), and the third biological window spans from 1550 nm to 1870 nm (NIR-III).16–18 In short, this interprets why NIR fluorescence sensing or SERS imaging systems should be designed within the biological transparency windows. For example, traditional organic fluorescent dyes exhibit fluorescence in the visible-light or NIR-I window, such as indocyanine green (ICG) and methylene blue (MB) NIR-I probes.15 Recently, many newly developed organic dyes show the extended emission wavelengths at the NIR-II window.19 The fluorescence of quantum dots can be tuned from visible-light, NIR-I to NIR-II or NIR-III windows by tailoring the size or/and chemical composition. Semiconducting single-walled carbon nanotubes exhibit fluorescence emission in the NIR spectral range mainly between 900 and 1600 nm, corresponding to NIR-II or NIR-III windows.16Fig. 1 shows the light absorption spectra obtained from human skin, which guides the selection of biological transparency windows for in vivo optical imaging (IVI).16Fig. 2 reveals the molecules that cause strong light absorption and scattering in human tissues.20 The major light-extinction molecules in human tissues are, but not limited to, melanin, collagen, fat, water and blood (e.g., Ox-Hgb and D-Hgb). Light below 650 nm is strongly absorbed by oxy-hemoglobin (Ox-Hgb) and deoxy-hemoglobin (D-Hgb), while water is the main absorber in the spectral range over 1350 nm (Fig. 2).20 It can be seen from Fig. 1 that both absorption and scattering of light are reduced dramatically when the wavelength increases from the UV and visible-light regions to the NIR regions. As the wavelength falls into the biological transparency windows, light scattering decreases significantly, and light absorption is maintained at a low level in the spectral range of 500 nm to 1300 nm with a small increase in the NIR-III region. Therefore, optical loss in human tissues is minimal in the three biological transparency windows.
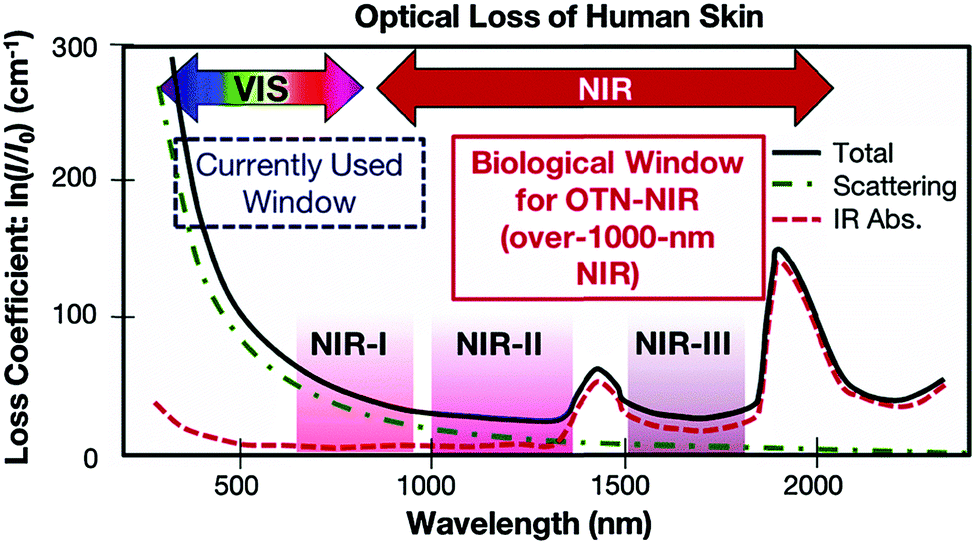 | ||
| Fig. 1 Optical loss of human skin. Reproduced from ref. 16, copyright 2016, The Royal Society of Chemistry. | ||
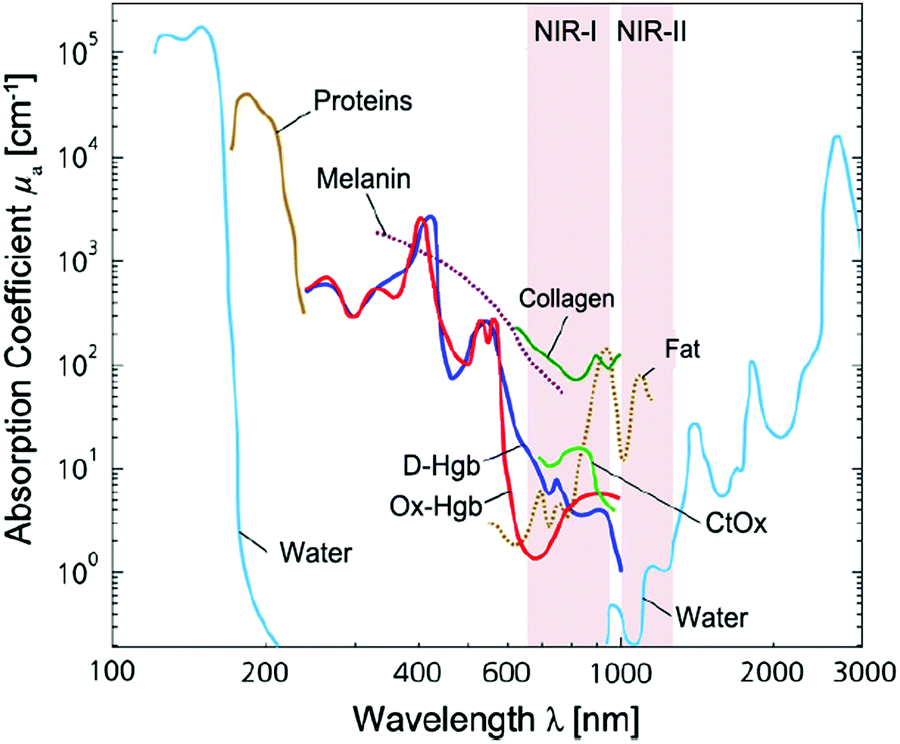 | ||
| Fig. 2 Light absorption spectra with X-axis on the natural logarithm base for different chromophores present in human tissues. Spectra for oxy-hemoglobin (Ox-Hgb) and deoxy-hemoglobin (D-Hgb), proteins, water, collagen, fat and cytochrome oxidase (CtOx) in the region from 100 nm to 3000 nm with respect to the specific concentration in mM are shown. This figure is further modified from ref. 20, copyright 2014, Elsevier. | ||
It is worth noting that light scattering is evident in a narrow spectral region (1350–1550 nm) between the NIR-II and NIR-III windows. Also, a narrow spectral region (950–1000 nm) between the NIR-I and NIR-II windows should be avoided for bio-imaging and detection although light scattering is negligible. Light is absorbed in this spectral region by water in biological samples, as shown in Fig. 3, and the absorbed light can heat biological samples.21 This is why NIR upconversion rare-earth nanoparticles such as NaYF4:Yb,Er, which are typically excited with a 980 nm laser, may encounter a problem when they are used for bio-imaging and detection.
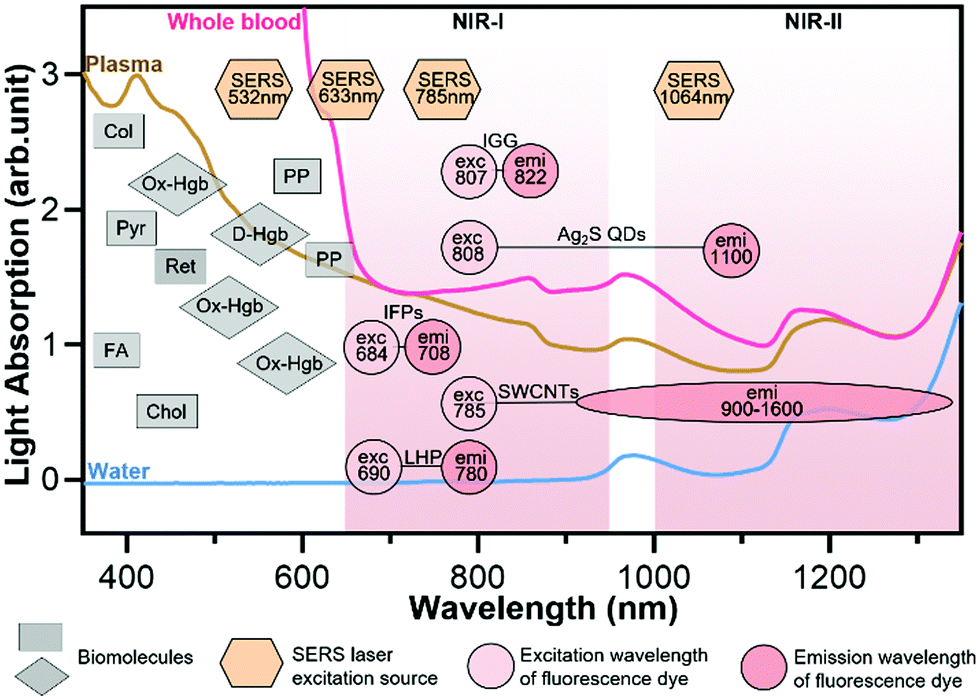 | ||
| Fig. 3 Light absorption and autofluorescence species in human blood as well as excitation and emission wavelengths of common fluorophores and SERS probes. Rectangles represent the auto-fluorescence spectral bands of plasma/blood proteins including pyridoxine (Pyr), retinol A (Ret), collagen (Col), cholecalciferol (Chol), folic acid (FA), and porphyrin (PP); diamonds represent the absorbent peaks of oxy-hemoglobin (OxHe) and deoxy-hemoglobin (D-He); hexagons represent the excitation wavelength of SERS probes; circles represent the excitation and emission of NIR fluorescence probes including indocyanine green (IGG), Ag2S quantum dots (Ag2S QDs), single-walled carbon nanotubes (SWCNTs), infrared fluorescent proteins (IFPs) and lead halide perovskites (LHPs). The light spectra in this figure were acquired in Wu's lab. Some data in this figure were taken from ref. 21 with permission from the Oxford University Press. | ||
In buffer sample matrices, a better limit of detection can be achieved using visible-light fluorescence probes because of their higher fluorescence quantum yields than their NIR counterparts. However, the visible-light fluorescence intensity can be reduced largely in blood or plasma matrices due to the strong light absorption and auto-fluorescence of the sample matrix. In contrast, the intensity of NIR fluorescence (or SERS) probes may be reduced slightly or even not changed when the sample matrix is switched from buffer to blood/plasma. Hence, NIR fluorescence and SERS probes are attractive for in vitro diagnosis (IVD), especially for blood or plasma samples. Fig. 3 shows the light absorption spectra obtained from human blood and plasma, which guides the selection of excitation light sources, dyes and fluorophores for in vitro optical detection in blood or plasma sample matrices. This figure reveals the major molecules in blood that cause strong light absorption and auto-fluorescence.21 The light-absorption molecules include oxy-hemoglobin (Ox-Hgb), deoxy-hemoglobin (D-Hgb) and bilirubin. D-Hgb exhibits the strongest light absorption at around 420 nm and the second light absorption peak at around 580 nm. Ox-Hgb displays its strongest light absorption at around 410 nm, and two secondary peaks at 550 nm and 600 nm. Overall, light absorption by Ox-Hgb and D-Hgb decreases with an increase in the wavelength, and becomes much weaker in the spectral range above 770 nm. Auto-fluorescence leads to a low signal-to-background ratio. The typical auto-fluorescence molecules in blood are amino acids and proteins, pyridoxine (Pyr), retinol A (Ret), collagen (Col), cholecalciferol (Chol), folic acid (FA), and porphyrin (PP).22–25 These molecules may exhibit strong auto-fluorescence in the visible-light regions, as shown in Fig. 3. Interestingly, auto-fluorescence becomes low in both the NIR-I and NIR-II windows in a blood sample matrix. The authors' group has compared the behavior of visible-light and NIR fluorescence probes in buffer and plasma, respectively. It has been found that the fluorescence intensity of the visible-light fluorescence dye (rhodamine 6G) was reduced by more than 50% when the sample matrix was switched from buffer to blood or plasma. In stark contrast, the NIR fluorescent dye (Cyanine 7) exhibited a negligible change in the fluorescence intensity.
The prevailing colorimetric paper-based lateral flow assays employ gold nanoparticles as colorimetric probes. These gold nanoparticles typically exhibit a light absorption maximum at around 520–560 nm, showing a red wine-like color on the test line of the assay, which is visible to human eyes. When these colorimetric paper-based lateral flow assays are applied to blood, serum and plasma samples, they suffer from severe interference of biomolecules in sample matrices, leading to poor sensitivity and repeatability. In contrast, NIR fluorophores and SERS probes that fall into the biological transparency windows (NIR-I and NIR-II) have advantages when testing plasma, serum and whole blood samples. Typically, the 785 nm or 1064 nm laser is used to excite SERS probes. And the typical examples of NIR fluorophores are organic dyes (e.g., indocyanine green, IGG), quantum dots (QDs, e.g., Ag2S), carbon nanomaterials (e.g., single-walled carbon nanotubes, SWCNTs), infrared fluorescent proteins (IFPs) and halide perovskite nanocrystals (e.g., lead halide perovskites, LHPs).14,19,26–29
2.2 Surface plasmon resonance
Although NIR fluorescence or SERS detection systems in the biological transparency windows are characteristic of the reduced light absorption and the minimized autofluorescence from biological samples, their signals are relatively low compared to their visible-light counterparts. NIR fluorophores typically exhibit lower quantum efficiency than visible-light counterparts. The quantum yield of visible-light fluorophores typically ranges from 20% to 80%, and is reduced to ∼10% for NIR-I fluorophores, and only ∼0.01–1.4% for NIR-II counterparts except for some nanomaterials such as PbS quantum dots.30 On the other hand, Raman scattering of a molecule is a dipole-like interaction with the excitation light, which shows that Raman scattering strength is proportional to the frequency of the excitation light to the fourth power. Consequently, the Raman scattering signal is greatly reduced when shifting from the visible-light to the NIR spectral regions. Therefore, surface plasmon resonance (SPR) is typically tuned into the NIR spectral region to enhance NIR fluorescence and SERS. Thus, we introduce the SPR concept and the plasmon-enhancement principles before we talk about NIR fluorescence and SERS sensing/imaging. Understanding the underlying mechanism will enable “devices-by-design” and “materials-by-design”.SPR is a phenomenon that numerous conduction electrons collectively oscillate on the surface of a metallic nanostructure upon the resonant excitation of incident light.31 It can generate an intense local electromagnetic (EM) field surrounding the nanostructure, which can modulate optical processes including photon absorption, Raman scattering and fluorescence.32 SPR has two typical forms, namely, propagating surface plasmon polariton (SPP) and localized SPR (LSPR). SPP refers to electron oscillation on the surface of a thin metal film when the frequency of incident light is smaller than the plasma frequency (Fig. 4a).32 The SPP frequency is determined by the plasma frequency of the bulk metal (ωp) and the dielectric constant of the surrounding medium (εdiel), which is expressed as33,34
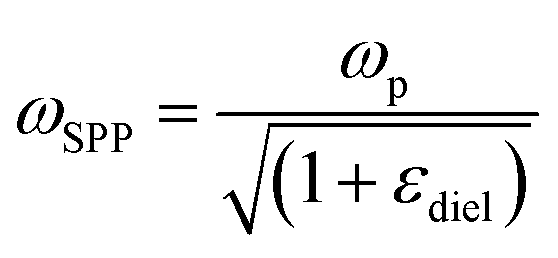 | (1) |
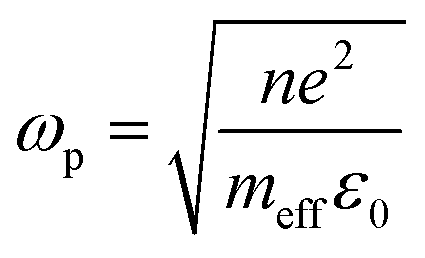 | (2) |
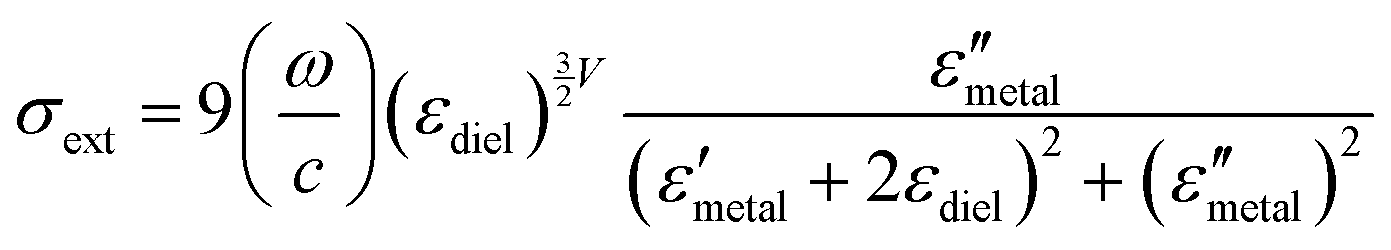 | (3) |
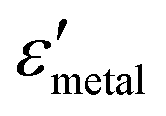 and
and 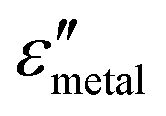 are the real and imaginary parts of the dielectric function, respectively.38 The denominator can vanish when
are the real and imaginary parts of the dielectric function, respectively.38 The denominator can vanish when 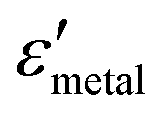 is negative and
is negative and 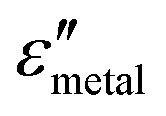 is small at the required wavelength range such as the near-infrared range. Strong plasmon resonance could be achieved at39,40
is small at the required wavelength range such as the near-infrared range. Strong plasmon resonance could be achieved at39,40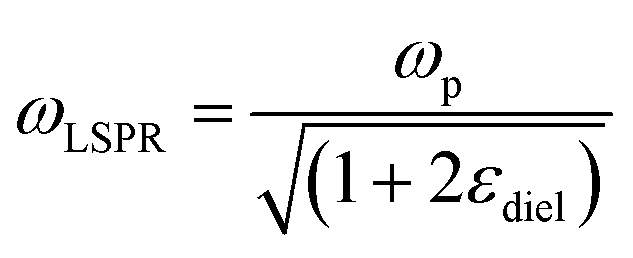 | (4) |
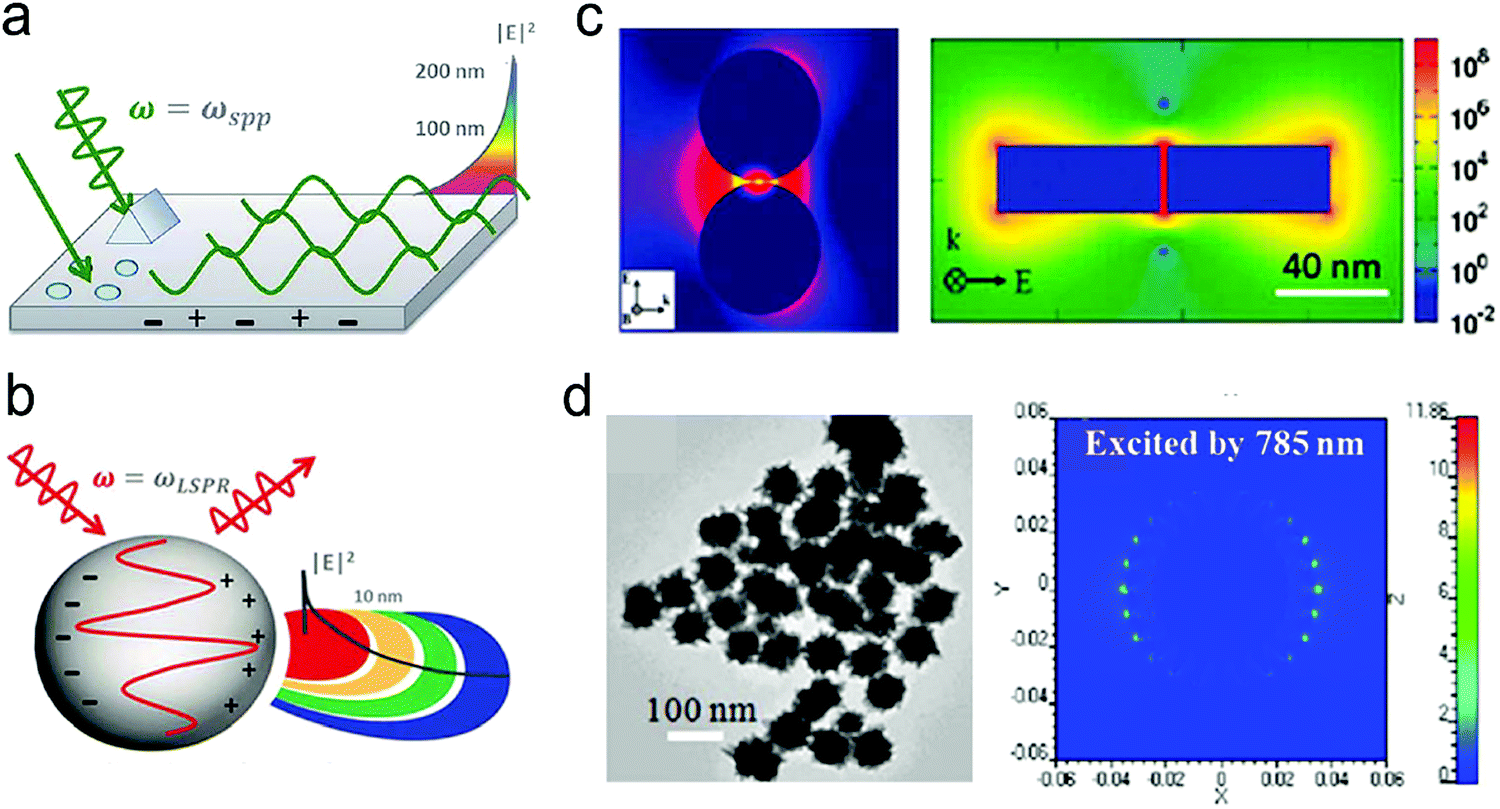 | ||
| Fig. 4 (a) The SPP-induced EM field and the decay. (b) The LSPR-induced EM field and the decay. (c) Finite-difference time-domain (FDTD) simulated EM field distributions for NPs with small gaps or sharp edges. (d) FDTD simulated EM field distributions for nanostars. Reproduced with permission from (a) ref. 32, copyright 2015, The Royal Society of Chemistry; (b) ref. 32, copyright 2015, The Royal Society of Chemistry; (c) ref. 44, copyright 2011, American Chemical Society; (d) ref. 45, copyright 2012, IOP Publishing Ltd. | ||
2.3 SERS-enhancement mechanisms and SERS probes
The EM enhancement mechanism is based on the enhanced localized EM field around a plasmonic metal nanostructure.11,56 In the case of the influence of both chemical enhancement and EM enhancement, the SERS signal intensity (PSERS) can be estimated by42
| PSERS ∝ chemical enhancement × EM enhancement | (5) |
| PSERS ∝ N·αRabs·|A(νe)|2·|A(νs)|2·I(νe) | (6) |
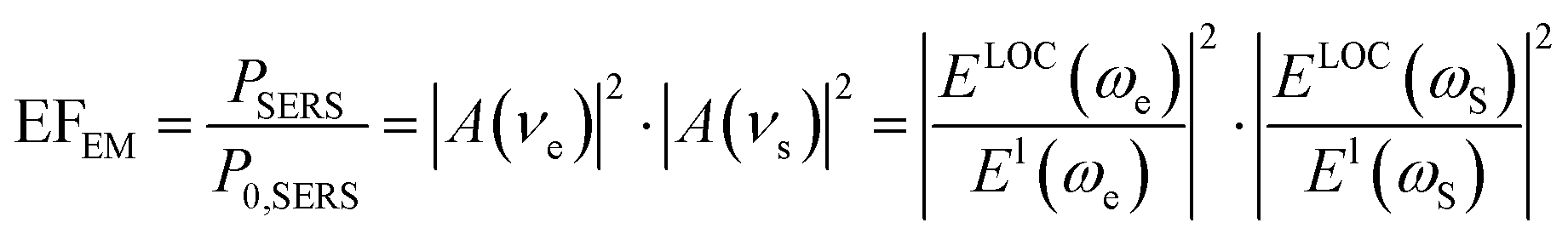 | (7) |
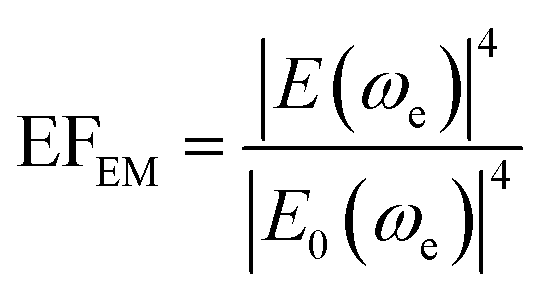 | (8) |
Apart from these two traditional SERS enhancement mechanisms, a new mechanism named “molecular hot spots” was proposed recently.58 It is found that the polarizability of aromatic molecules conjugated on a plasmonic metal nanoparticle interacts strongly with SPR, leading to a spatially extended enhanced EM field into the conjugated aromatic molecule assembly. This creates “molecular hot spots” surrounding the metal nanoparticle. The enhanced EM field is delocalized and spreads throughout the aromatic molecule layer on the surface of the metal nanoparticle. The “molecular hot spots” can be generated in aromatic molecules even on a small metal nanoparticle (<15 nm) with a weak LSPR. The “molecular hot spots” can result in a large enhancement of SERS signals of other molecules that are co-adsorbed with the aromatic molecules on the metal substrate. Even linear-chain molecules, which typically have extremely low polarizability, can exhibit strong SERS signals when they co-exist with aromatic molecules near the metal nanoparticle.
The second type is “metal@Raman reporter”, in which Raman reporter molecules are attached to a metal nanoparticle. Because the Raman scattering cross-section of Raman reporter molecules is quite small, only 10−28–10−30 cm2 per molecule, they alone cannot be used as SERS probes.49 Typically, plasmonic metal nanoparticles such as Au or Ag are utilized to generate LSPR to amplify Raman signals. Because LSPR-induced EM fields are decayed to a very low level beyond a distance of about 30 nm away from the particle surface, Raman reporters are required to stay in close proximity to the metal nanoparticles. Thus, metal@Raman reporters are formed through surface functionalization of plasmonic nanoparticles with Raman reporters.42 SH- or NH2-containing molecules with great affinity to the Au or Ag substrate are usually chosen as Raman reporters, such as malachite green isothiocyanate (MGITC), 4-aminothiophenol (4-ATP), 4-mercaptobenzoic acid (4-MBA), 4-mercaptopyridine. (4-MPY), and 5,5-dithiobis-2-nitrobenzoic acid (DTNB).64–66 To further enhance the SERS signal of Raman reporters, many researchers have engineered metal nanoparticle shapes from nanospheres to nanorods and nanostars. It was reported that more sharp tips in nanoparticles generate stronger EM fields, thus nanostars showed the highest SERS enhancement, nanorods took the second place, while nanospheres exhibited the lowest enhancement.44 The “metal@Raman reporter” particles may aggregate in an aqueous solution with high ionic strength or get detached from the metal particle surface during sensing/imaging, which leads to signal loss and poor repeatability.44,67 On the other hand, it is difficult to conjugate additional antibodies or DNA into the “metal@Raman reporter” particles due to the occupancy of Raman dyes on the metal surface.
The third type is sandwich-structured “metal core@Raman dye@shell” particles,44 in which the Raman reporter molecules are sandwiched between a metal core and an outer shell. This prevents from leaching out of Raman reporter molecules during detection. The plasmonic metal core is used to amplify SERS signals. The thin outer shell is typically made from silica (SiO2) or polymers such as polyethylene glycol (PEG).44,68–70 The silica shell not only provides a clean surface for bioconjugation with antibodies and DNA, but also renders excellent water-solidity, which make the sandwich-structured SERS probe stable in an aqueous solution with high ionic strength such as blood. In short, sandwich-structured SERS probes are characteristic of high sensitivity, robustness, water-solubility, and biocompatibility and repeatability.44
In general, both the second and the third types of SERS probes can be used in buffer sample matrices for sensing applications. Also, the sandwich-structured “metal core@Raman dye@shell” SERS probes are stable and robust in blood and plasma matrices. However, the “metal@Raman reporter” SERS probes may aggregate in blood or plasma due to the high ionic strength of sample matrices.
2.4 Plasmon-enhanced fluorescence mechanisms and NIR fluorophores
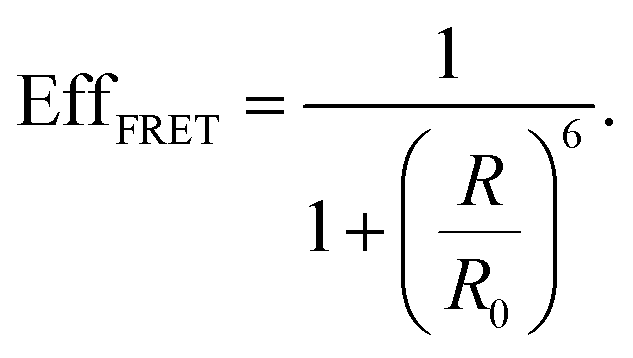 | (9) |
| ρLDOS(ω) ∼ |Eloc(ω)|2 | (10) |
Considering the spectral overlap of the plasmon and the fluorescence, and the gap, quenching or enhancement of fluorescence emission can be determined (Fig. 5a and b).32 First, when the plasmon band is overlapped with the fluorophore absorption band in Fig. 5a, fluorescence emission can be enhanced.32 It can be illustrated as follows. For metal nanoparticles smaller than ∼15 nm, the particles mainly absorb and generate an enhanced EM field, leading to excitation enhancement by FRET, followed by emission enhancement. It should be noticed that plasmon's radiation enhanced by the Purcell effect is negligible, thus excitation is only enhanced by FRET within a few nanometers of distance in this case. For metal nanoparticles larger than ∼15 nm, the particles mainly scatter, and both FRET at short distances and the Purcell effect at long distances can result in enhanced fluorescence emission.32,46,74 Second, when the plasmon band is overlapped with the fluorophore emission band in Fig. 5b, quenching or enhancement of fluorophore emission can be achieved.32 At a short distance (∼10 nm) between the plasmon and the fluorophore, fluorescence emission can be quenched through FRET. At a distance beyond FRET (>10 nm), strong Purcell enhancement is induced because of the large local density of states (LDOS) of the local plasmonic field, which increases the radiative rate of the fluorophore, and if the plasmon can scatter efficiently, strong emission enhancement can occur.32,46,74 Typically, the maximum enhancement takes place at an optimal gap of 10–30 nm between the plasmon and the fluorophore. Besides the overlap of the plasmon and absorption/emission band, it is important to choose a suitable laser line. For the overlap of the plasmon and absorption band, a signal laser line is enough to excite both the plasmon and fluorescence probe. When the plasmon is overlapped with the emission band, two laser lines are required. One laser line with a shorter wavelength is used to excite the fluorescence probe, and another with a longer wavelength can excite the plasmon.
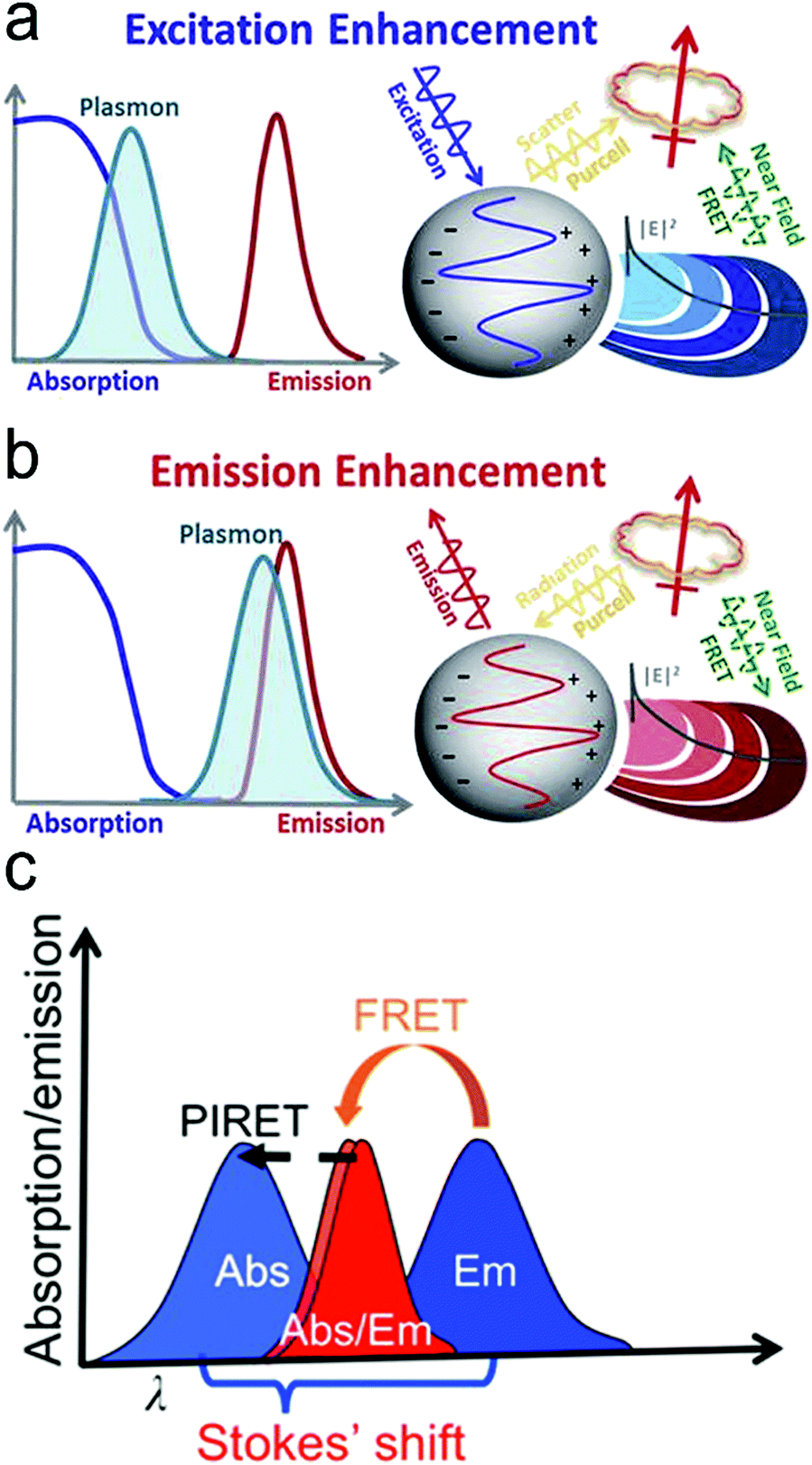 | ||
| Fig. 5 Mechanisms of plasmon-enhanced fluorescence. (a) When the plasmon overlaps with the fluorophore absorption spectrum, an enhanced fluorophore emission can be achieved. (b) When the plasmon overlaps with the fluorophore emission spectrum, quenching or enhancement of fluorophore emission can be achieved. (c) When the spectrum peak of the plasmon is longer than the absorption of the fluorophore, fluorescent intensity can be enhanced through the PIRET process if there is an overlap between the plasmon spectrum and fluorophore absorption. Reproduced with permission from (a) ref. 32, copyright 2015, The Royal Society of Chemistry; (b) ref. 32, copyright 2015, The Royal Society of Chemistry; (c) ref. 45, copyright 2016, American Chemical Society. | ||
Recently, a new mechanism, plasmon-induced resonance energy transfer (PIRET), is proposed for plasmon enhanced fluorescence.75 It is also a non-radiative energy transfer via the dipole–dipole interaction. Different from FRET which only occurs from the donor at a shorter wavelength to the acceptor at a longer wavelength, PIRET can realize the blue-shift coherent energy transfer from the longer-wavelength donor to the shorter-wavelength acceptor. For a plasmon–fluorophore pair, when the SPR band of the plasmon is overlapped with the absorption band of the fluorophore, the emission intensity of the fluorophore can be enhanced through PIRET. The schematic mode of energy transfer is shown in Fig. 5c.45 The plasmon spectrum often overlaps simultaneously with the absorption and emission spectrum of the fluorophore, such that FRET occurs to quench fluorescence and PIRET takes place to enhance fluorescence simultaneously. The net influence of FRET and PIRET is determined by the dephasing time (T2), which is expressed by45,75
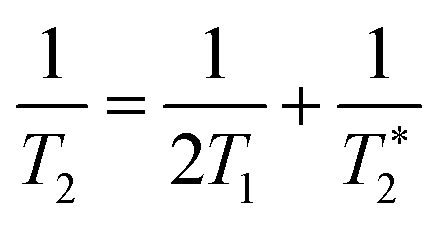 | (11) |
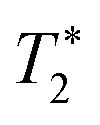 refers to the pure dephasing time of the plasmon (or fluorophore). Given
refers to the pure dephasing time of the plasmon (or fluorophore). Given 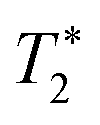 (around fs range) is much shorter than T1 (around ps range), and T2 is related to
(around fs range) is much shorter than T1 (around ps range), and T2 is related to 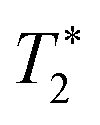 . If the fluorophore's dephasing time is faster than that of the plasmon, PIRET shows higher efficiency than FRET and the net influence is enhanced fluorescence. Conversely, FRET dominates in the whole process with the result of quenched fluorescence.
. If the fluorophore's dephasing time is faster than that of the plasmon, PIRET shows higher efficiency than FRET and the net influence is enhanced fluorescence. Conversely, FRET dominates in the whole process with the result of quenched fluorescence.
Plasmonic fluorescence probes based on PIRET show advantages in bio-sensing and bio-imaging.45,75,84,85 Because it is a blue-shifted energy transfer from the longer-wavelength end to the shorter-wavelength end, excitation can reduce photodamage to biological samples. The signal-to-noise ratio can also be improved. For example, Wang et al. developed Au nanorods covered by a merocyanine-doped poly(methyl methacrylate) film.45 It was observed that PIRET occurred when there was a significant overlap between the LSPR band and the fluorophore absorption band, which resulted in an average enhancement factor of up to 1854. As PIRET is a newly discovered mechanism for fluorescence enhancement, plasmonic fluorescence probes based on PIRET have just emerged.
In a special case, when Raman scattering and fluorescence spectra are overlapped for a fluorescent molecule at a given excitation wavelength, that is, both SERS and fluorescence are enhanced by the plasmonic electric field, the total cross-section (σ(total)) of the spectrum is theoretically the sum of the Raman scattering cross-section spectrum (σ(RS)) and the fluorescence cross-section spectrum (σ(FL))86
| σ(total) = σ(RS) + σ(FL) | (12) |
Owing to enhancement by the EM field, the total cross-section becomes
σ(enhanced![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) total) = σ(ERS) + σ(EFL) total) = σ(ERS) + σ(EFL) | (13) |
| σ(ERS) = σ(RS)·|A(νe)|2·|A(νs)|2. | (14) |
| σ(EFL) = q·σ(FL)·|A(νe)|2·|A(νs)|2 | (15) |
Combining eqn (13), (14) and (15),
| σ(enhancedtotal) = [σ(RS) + qσ(FL)]·|A(νe)|2·|A(νs)|2 | (16) |
3. Design of fluorescence and SERS sensors
In this section, we review the design of fluorescence and SERS sensors for the detection of typical analytes in healthcare and biomedical research. We discuss how to transduce and amplify sensing signals. In addition, we highlight the application examples of fluorescence and SERS sensors.Signals of both fluorescence and SERS can be amplified dramatically by surface plasmon resonance, achieving high sensitivity of detection, even reaching a single molecule level. It is worth noting that SERS signals increase with a decrease in the distance between the Raman reporter and the plasmonic nanostructure while the fluorescence signals of a fluorophore reach a maximum at an optimal distance between the fluorophore and the plasmonic nanostructure. On the other hand, both NIR fluorescence and SERS have strong resistance to interference from complex sample matrices (blood and plasma) and have deep penetration capability in bio-imaging. When designing sensors, keep in mind that few analyte molecules have signature or characteristic fluorescence peaks, while many analytes can generate fingerprinting or signature SERS spectra. Therefore, SERS can be used to conduct label-free detection of many trace analyte molecules, and is good at real-time monitoring metabolites from live cells. Low-cost fluorescence readers are well developed, and a lot of commercial products are available. In contrast, today Raman readers are more expensive than their fluorescence counterparts. But rapid technology progress will increase portability and reduce the costs of Raman readers in the future. Indeed, this trend was expedited in the last decade. Palm-sized Raman readers, which can be operated with cell phones via Bluetooth, are available commercially now. As for in vitro imaging, both modern fluorescence and Raman microscopes are able to real-time image live cells quickly. As for in vivo imaging, commercial fluorescent probes and imaging instruments are also well developed and available commercially. In vivo collection of Raman signals from deep tissues remains a challenge.
3.1 Protein sensors
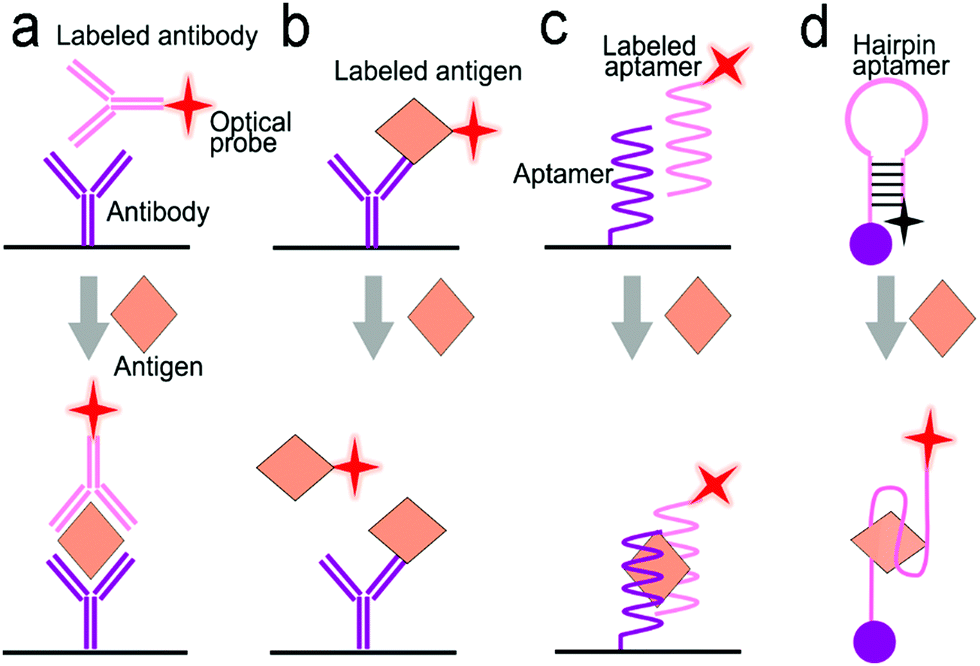 | ||
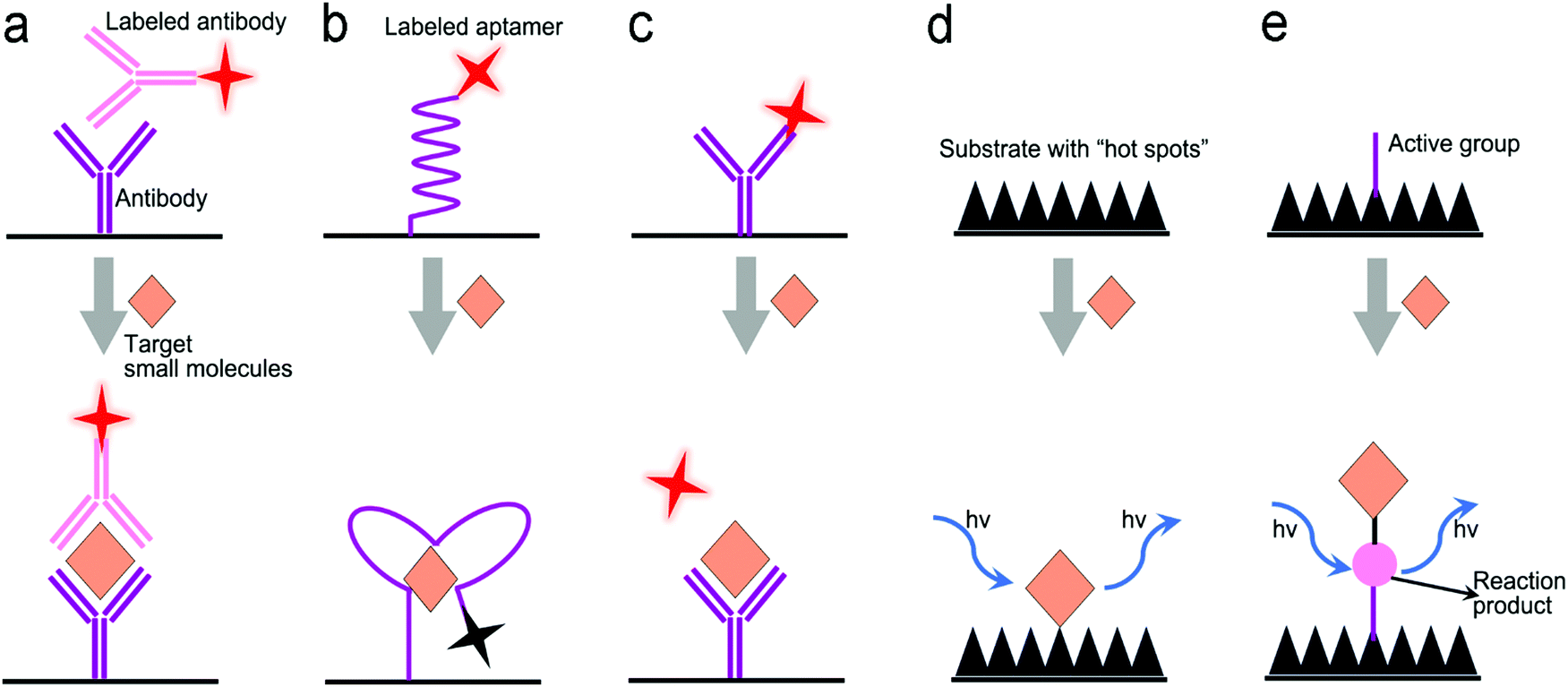
| Fig. 6 Signal transduction modes/principles for protein detection. (a) Sandwiched antibody–protein–antibody assay; (b) competitive assay; (c) sandwiched aptamer–protein–aptamer assay; (d) aptamer beacon that can change its conformation. | ||
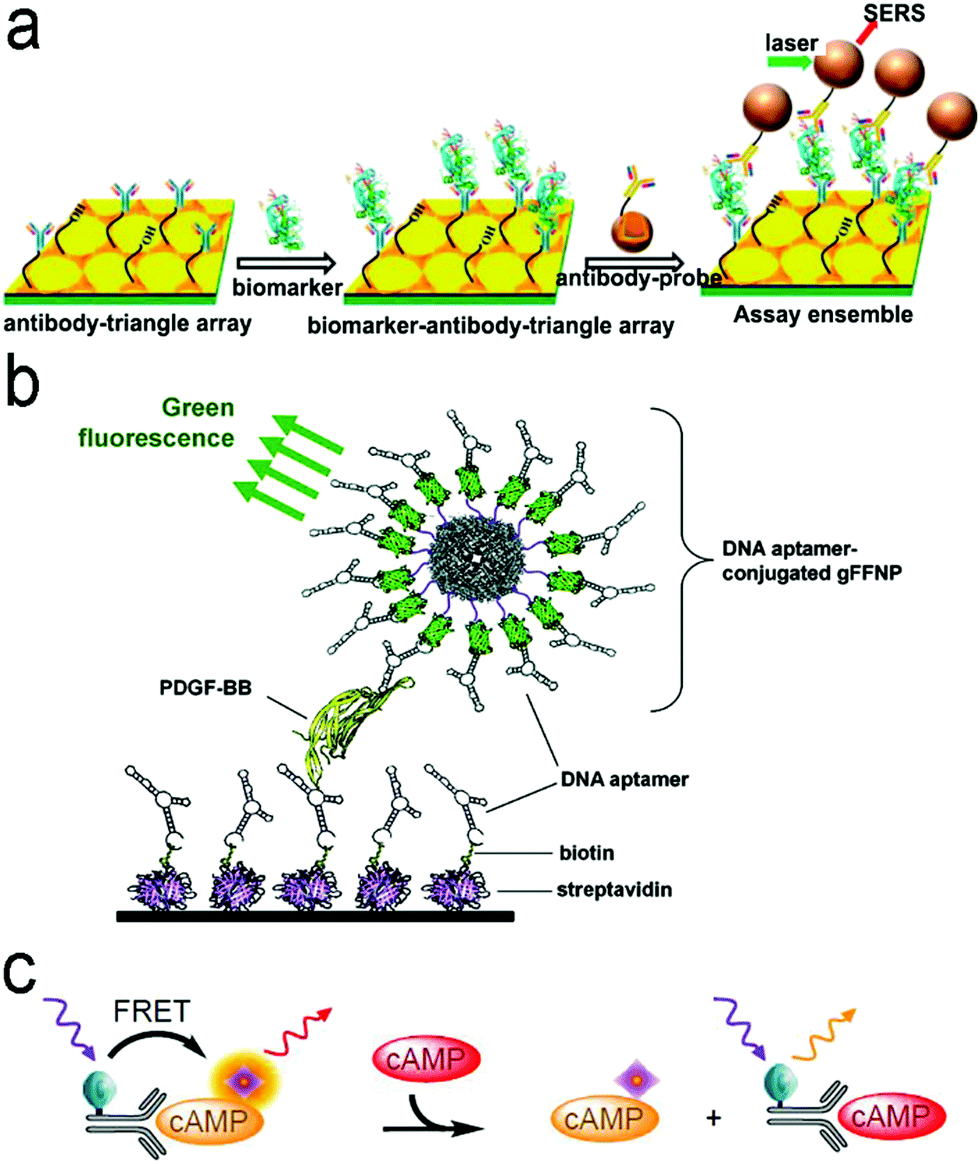 | ||
| Fig. 7 (a) ELISA-like SERS immunosensor using the antibody–antigen interaction for cancer biomarkers in blood plasma. (b) ELISA-like fluorescence immunosensor using the aptamer–antigen interaction. (c) Competitive assay based on FRET mechanism. Reproduced with permission from (a) ref. 87, copyright 2013, American Chemical Society; (b) ref. 100, copyright 2011, American Chemical Society; (c) ref. 102, copyright 2014, MDPI. | ||
Antibodies are expensive and produced by a complicated synthesis process, and require low temperature for storage. Hence, the substitution of antibodies with aptamers are receiving more attention because aptamers possess tailored specificity and affinity, relatively high thermostability, and can be produced massively to almost any epitope in a programable route.99Fig. 7b shows an aptamer-based sandwich assay for the detection of a cancer biomarker in buffer and diluted human serum.100 A pair of aptamers was immobilized on the surface of recombinant fluorescent ferritin nanoparticles as an optical probe, and the targeted cancer biomarker was sandwiched by the pair of aptamers. Such an aptamer sensor achieved a LOD down to 100 fM in a buffer.
Competitive immunoassays simplify the procedure and require the reduced steps of operation compared with sandwich-structured assays.91,101–103Fig. 7c reveals a FRET-based competitive sensor for the detection of the cyclic adenosine 3′,5′-monophosphate (cAMP) protein that was used for the evaluation of G protein-coupled receptor activation.102 Prior to detection, the fluorescence of the donor dye was quenched, but the fluorescence was emitted from the acceptor dye due to the FRET effect. When the analyte (cAMP) appeared in the assay, the FRET process was interrupted, and the donor emitted fluorescence. Competitive assays can also employ aptamers as molecular recognition elements.104–107 Xing et al. designed an aptamer-based competitive sensor for the determination of human epidermal growth factor receptor 2 (HER2).105 Initially, the HER2-specific aptamer linked with MnCuInS/ZnS QDs was hybridized with a complementary single-stranded DNA (ssDNA) linked to gold nanoparticles. The double-stranded DNA (dsDNA) was rigid and held the Au nanoparticles close to the QDs, and enabled FRET, leading to quenching of fluorescence of the QDs. During the detection process, the binding of the aptamer to HER2 led to the dissociation of dsDNA due to the higher affinity of the aptamer with HER2. As a result, the ssDNA became free-standing, and the gold nanoparticles were kept away from the QDs, which turned on the fluorescence of the QDs. This aptamer-based FRET sensor achieved a LOD of 1 ng mL−1 with a detection range from 2 to 100 ng mL−1, and successfully distinguished the healthy individuals (2–15 ng mL−1) from the individuals with the disease (15–75 ng mL−1).
3.2 Nucleic acid sensors
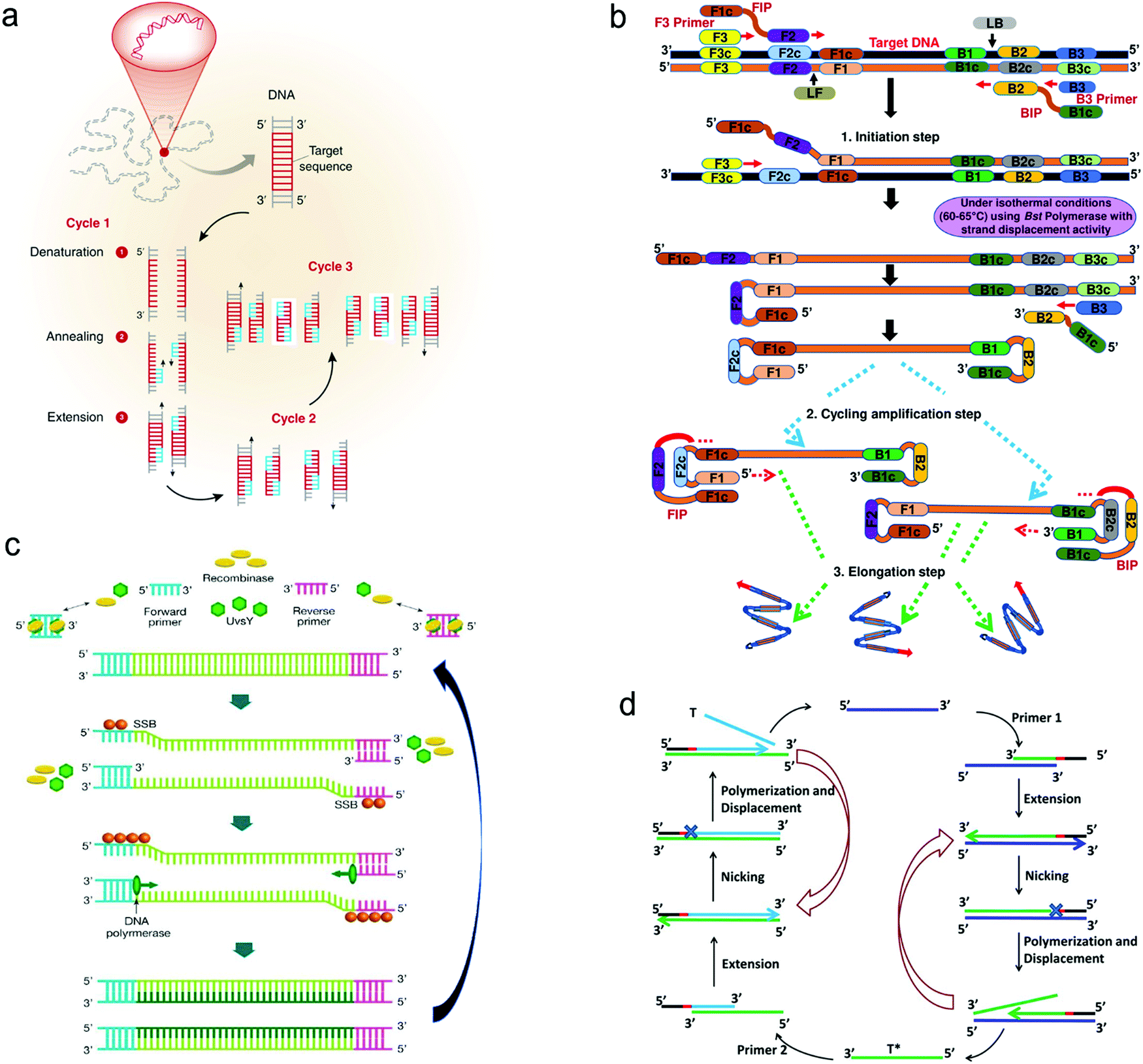 | ||
| Fig. 8 Schematic illustration of nucleic acid amplification. (a) Polymerase chain reaction (PCR). (b) Loop-mediated isothermal amplification (LAMP). (c) Recombinase polymerase amplification (RPA). (d) Strand displacement amplification (SDA). Reproduced with permission from (a) ref. 118, copyright 2013, Elsevier; (b) ref. 123, copyright 2019, PLOS; (c) ref. 125, copyright 2020, MDPI; (d) ref. 120, copyright 2014, American Chemical Society. | ||
(i) PCR is carried out by temperature cycling (Fig. 8a).118 At a high temperature of the denaturation step, two strands of duplex DNA are fully separated. The temperature is then reduced to allow primers to anneal to each ssDNA. In the presence of deoxynucleoside triphosphates (dNTPs) and heat-stable polymerase, primers are extended to form new double helical DNA at around 72 °C. Theoretically, the number of double-stranded DNA pieces is doubled in each cycle, achieving 2n copies of DNA after n cycles in the PCR process.
(ii) LAMP is performed isothermally at 60–65 °C for 45–60 min with the aid of the specific DNA polymerase, dNTPs and primers (Fig. 8b).123 The LAMP process starts when the F2 region of the forward inner primer (FIP) hybridizes to the F2c region of the target DNA and extends the strand, followed by the F3 primer which is complementary to the F3c region of the target elongating the strand. The FIP-linked strand is then displaced and forms a loop at the 5′-terminus, which also acts as a template for the backward inner primer (BIP). The B2 region of the BIP undergoes a similar strand synthesis as the F2 region and opens the 5′-terminus loop. The synthesized strand is then displaced by the B3 primer extended strand, forming a dumbbell-shaped DNA, which is an initiator of LAMP recycling. As a result, a mixture of stem-loop DNA structures with varied stem lengths and various cauliflower-shaped structures with multiple loops is obtained assisted by these primers.
(iii) RPA was developed by Niall Armes et al. in 2006 using three proteins (recombinase protein, single-stranded binding protein and a strand displacing DNA polymerase).124 A typical example is shown in Fig. 8c.125 The recombinase protein (T4 UvsX protein) binds to primers in the presence of a loading factor (T4 UvsY) and adenosine triphosphate (ATP) to form a recombinase-primer complex that seeks homologous sequences in duplex DNA and inserts the strand by primers at the cognate site. A single-stranded binding protein (SSB, T4 gp32 protein) is used to prevent the ejection of the invaded primer by branch migration. Subsequently, the recombinase protein disassembles from the complex and a strand displacing DNA polymerase (Bsu or Sau) binds to the 3′-end of the primer and initiates the elongation assisted by dNTPs, ultimately resulting in the exponential accumulation of the target double helical DNA.
(iv) SDA is also carried out under in vitro isothermal conditions. Fig. 8d gives an example of SDA for miRNA amplification.120 Primers used in SDA consist of a particular sequence (marked in red in Fig. 8d) as the recognition site of the nicking enzyme. SDA starts with the binding of the target miRNA to Primer 1 at the 3′-end, followed by the extension of Primer1 and miRNA in the presence of Klenow fragment polymerase. The nicking enzyme is added to cleave Primer 1 at the recognition site. Next, the elongation happens again at the nicking site, thus displacing the replicated strand that serves as a new template in the following step and the left duplex strand enters the nicking step to initiate the first cycle. The replicated strand bonds with Primer 2 to trigger the second cycle through extension, nicking, polymerization and displacement steps and ultimately achieves the new target miRNA, which could also enter the first cycle.
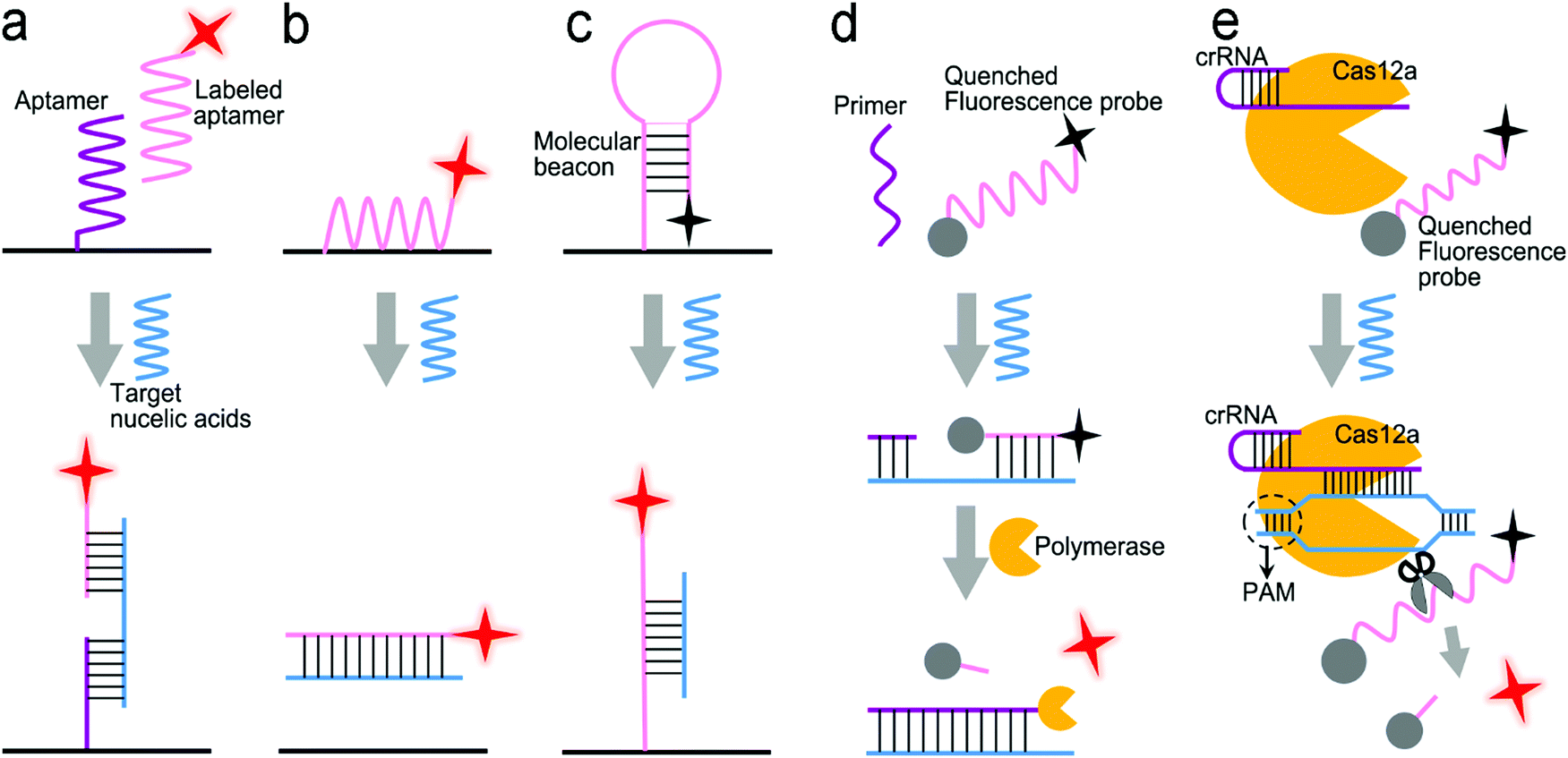 | ||
| Fig. 9 Signal transduction modes for nucleic acid detection. (a) “Three-strand” hybridization mode; (b) “two-strand” hybridization mode-based competitive assay; (c) “molecular beacon” mode; (d) enzyme-assisted assay using polymerase; (e) enzyme-assisted assay using CRISPR/Cas12a. | ||
000, achieving a LOD of 50 aM in a buffer solution with specific discrimination of a single-base mutant of DNA. It is desirable to combine nucleic acid amplification and detection in a single platform to perform the nucleic acid amplification test (NAAT).128–131 Guven et al. incorporated RCA with the “three-strand” detection scheme to detect the 35S promoter from cauliflower mosaic virus (CaMV35S) for tracking gene-modified organisms.129 After the analyte sequence was sandwiched by the primer and the capture strand on a gold substrate, the primer was catalyzed by DNA ligase and hybridized with an RCA template. With the assistance of Phi29 DNA polymerase and dNTPs, long nucleic acid products were generated via the RCA reaction, followed by hybridization with the detection strand-modified gold nanorods. This NAAT device reached a LOD of 6.3 fM with a wide detection range from 100 fM to100 nM.
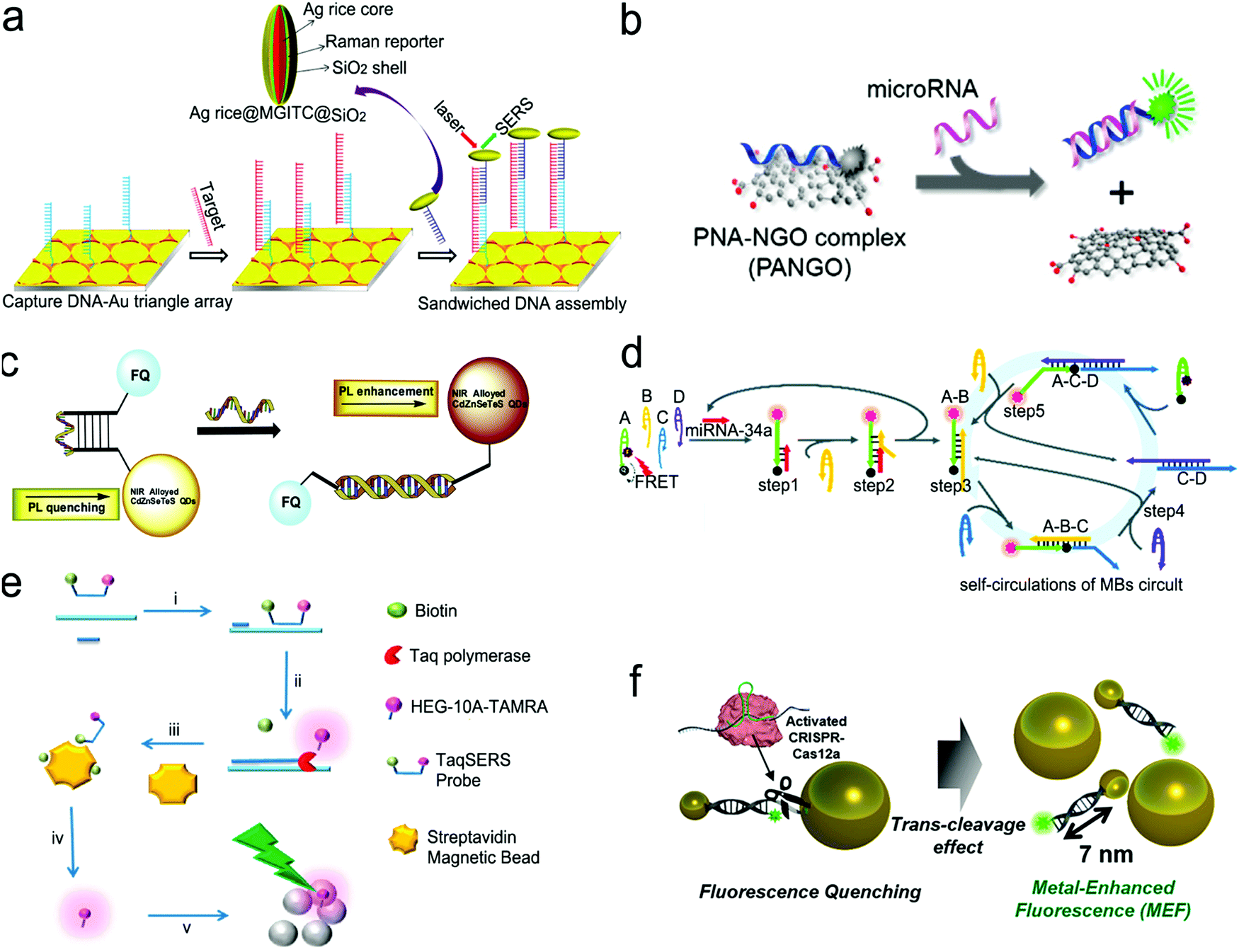 | ||
| Fig. 10 Nucleic acid detection with SERS or fluorescent sensors. (a) “Three-strand” hybridization assay using SERS probes for the detection of hepatitis B virus DNA. (b) “Two-strand” hybridization mode-based competitive interaction between single-stranded and double-stranded complex graphene oxide. (c) “Molecular beacon” mode for the detection of influenza virus RNA. (d) Nucleic acid amplification test (NAAT) using self-circulation of chain reaction for signal amplification and “molecule beacon” mode for detection. (e) Taq polymerase-assisted assay. (f) CRISPR/Cas12a-assisted assay with fluorescence probes determined by the size of metal nanoparticles and their distance. Reproduced with permission from (a) ref. 112, copyright 2013, American Chemical Society; (b) ref. 132, copyright 2013, American Chemical Society; (c) ref. 136, copyright 2016, Elsevier; (d) ref. 135, copyright 2019, The Royal Society of Chemistry; (e) ref. 137, copyright 2012, The Royal Society of Chemistry; (f) ref. 138, copyright 2020, American Chemical Society. | ||
Fig. 10b shows a sensor based on the “two-strand” mode as described in Fig. 9b.132 It has achieved a LOD of 1 pM toward miRNA in a buffer solution. The “two-strand” mode has also been used in NAAT.133–135 For instance, a biosensor was constructed via target cycling amplification to detect highly pathogenic avian influenza H7N9 virus.133 The number of analyte DNA after recycling was dependent on the released capture strands which were immobilized onto the surface of Ag nanorods. The detection strands labeled with the fluorescent dye Cyanine-5 (Cy5) were finally hybridized with the capture strands to record the analyte concentration. This NAAT assay exhibited a linear range from 1 fM to 100 pM at a LOD of 31 aM for H7 and 44 aM for N9 in buffer solutions, respectively.
Molecule beacons have been widely used in SERS and fluorescence sensors for nucleic acid detection.126,134–136,139Fig. 10c reveals a NIR fluorescent sensor base on a molecule beacon labeled with CdZnSeTeS QDs, showing a LOD of 2 copies per mL towards influenza virus H1N1 RNA in both buffer and human serum.136 In addition, the molecular beacon mode-based detection scheme has been incorporated with nucleic acid amplification methods. Fig. 10d presents a NAAT assay using self-circulation of chain reaction between the target miRNA and a molecular beacon (A) together with other three hairpin structures without probes (B, C, and D).135 In the presence of the analyte miRNA-34a, a breast cancer biomarker, the molecular beacon was unfolded via hybridization with miRNA-34a, forming a duplex with B (A–B) spontaneously. This released the target to activate the first cycle steps 1–2. The A–B duplex entered the next cycles by the formation of the C–D duplex, which catalyzed A and B to generate C–D in turn. This sensor detected 2.5 pM miRNA-34a in a buffer.
The enzymatic cleavage of phosphodiester linkages of DNA or RNA is also common in sensor design (Fig. 9d).140–142 Harper et al. utilized this method in a SERS sensor for the determination of mecA gene sequence of methicillin-resistant S. aureus (MRSA) bacterium (Fig. 10e).137 In this design, a sequence complementary to a specific region of mecA gene was modified with a 5′-biotin residue and a TAMRA dye-labeled 10 adenine bases at the 3′-end, respectively, which was termed as the TaqSERS probe. When the TaqSERS probe and a primer were hybridized to the target sequence, the Taq polymerase enzyme extended the primer with the addition of deoxynucleoside triphosphate (dNTP) and digested the TaqSERS probe simultaneously. The TAMRA dye-labeled DNA was then separated in the magnetic field after any free biotin and undigested TaqSERS probes were absorbed by streptavidin coated magnetic beads. The separated dye labeled DNA was aggregated by the negatively charged citrate surface of the Ag nanoparticles. In turn, these DNA sequences neutralized the charge of Ag nanoparticles, facilitating the formation of a nanoparticle cluster. Compared with the fluorescence intensity of the dye alone, the incorporation of the amplification method resulted in the LOD being an order of magnitude lower. Moreover, nucleic acid amplification by LAMP was combined with the enzyme-assisted detection scheme.141 Briefly, after amplification by LAMP, the products of the analyte sequence of DNA were complementary to a capture ssDNA strand labeled with Cy5 and Au nanoparticles. The Cy5 and Au nanoparticles were linked by oligonucleotides in close proximity (<2 nm), enabling remarkable enhancement of the SERS signal from Cy5. Upon adding the S1 nuclease, the SERS signal remained at high intensity in the presence of the target DNA strands. Otherwise, the S1 nuclease digested ssDNA into free dNTPs, and separated Cy5 away from the nanoparticles, leading to low SERS signals. Apart from the amplification feature, nuclease was used to digest any non-specific DNA or potential contamination to minimize the interference to SERS signals. This sensor was used to detect Salmonella enterica, a major foodborne pathogen, achieving a LOD of 66 CFU mL−1 in a buffer.
CRISPR/Cas systems are adaptive immunity found in bacteria and archaea to degrade intruding nucleic acids, which have been wildly developed as gene-editing tools because of their recognition and degradation ability toward the target nucleic acids induced by Cas protein under the direction of a guide RNA. CRISPR/Cas systems have been extended to sensor design for nucleic acid testing.110,127,138,143,144Fig. 10f shows the use of a CRISPR/Cas12a system to design a sensor for the detection of breast cancer gene-1 (BRCA-1).138 The sensor probe was designed as 60 nm sized Au nanoparticles (60-AuNP), and 20 nm sized Au nanoparticles (20-AuNP) were linked to a long fluorophore-labeled dsDNA (7 nm) and a short ssDNA (2 nm). Because the fluorescence probe was close to 60-AuNP, it was quenched by 60-AuNP at the initial state. When the analyte BRCA-1 gene was present, it was complementary to crRNA and formed a Cas12a–crRNA–BRCA-1 complex, activating the CRISPR/Cas system to cleave ssDNA in the surrounding environment. The ssDNA in the probe was then cut to dissociate the fluorophore from 60-AuNP. As a result, a fluorescence signal was emitted and enhanced by the 20-AuNP via FRET. The combination of CRISPR/Cas with FRET achieved a femtomolar level of LOD both in buffer and under human serum conditions within a short assay time (<30 min).
3.3 Small molecule sensors
Nutrients such as essential amino acids and vitamins cannot be synthesized to a sufficient amount in the body and must be obtained from food, and are indispensable for human health with limited amount.147,151 Specifically, in the family of essential amino acid, methionine is associated with greying of hair, depression and atherosclerosis; lysine plays a crucial role in proteinogenesis and epigenetic regulation; valine takes part in blood sugar regulation.152–157 In addition, adequate vitamin intake is important for the enzyme–substrate reaction, growth and development. The family of B complex vitamins forms coenzymes to facilitate catalysis processes. For instance, thiamine (B1) can function as thiamine pyrophosphate (TPP) in the body and this coenzyme is a vital player in carbohydrate metabolism.158 B9 participates in the synthesis of nucleic acids, metabolism of amino acids and cell proliferation.159 It is reported that long-term supplementation of B9 can lessen the risks of stroke, heart disease and prostate cancer.160 The deficiency of these amino acids and vitamins causes functional disorders, for example, insufficiency of vitamin E can lead to myopathies, spinocerebellar ataxia, peripheral neuropathy and retinopathy, etc.161,162
Illicit drugs, pesticides, herbicides, and food pollutants pose a threat to human health and the environment. Abuse of illicit drugs is a global problem with 271 million people (1 out of 18 people) from 15 to 64 years old.163 Drug abuse is one of the sources for hepatitis C and a quarter with human immunodeficiency virus (HIV).164 Therefore, drug testing is essential for drug control and prevention. Nitrite, the main species in nutrient pollution, is of concern to the ecological system.70 It is also widely present in meat products as food preservatives and excessive consumption causes diseases such as diabetes, cancer and neurodegenerative diseases.42,165 The United States Environmental Protection Agency (EPA) and the Legislation of the European Union have enacted the maximum contaminant level (MCL) of nitrite as 1.0 μg mL−1 in drinking water and the maximum allowable level as 50 mg kg−1 in meat products.70,165 Release of pesticides and herbicides into surface waters can result in pollution of the environment, accumulation in the food chain, uptake by human eventually, leading to adverse health effects, etc.166,167
 | ||
| Fig. 11 Signal transduction modes for small molecule detection. (a) Sandwiched antibody–molecule–antibody assay; (b) molecular beacon assay; (c) competitive assay; (d) direct detection using a substrate with “hot spots” to enhance Raman scattering; (e) target detection through monitoring their chemical reaction product using a SERS sensor. | ||
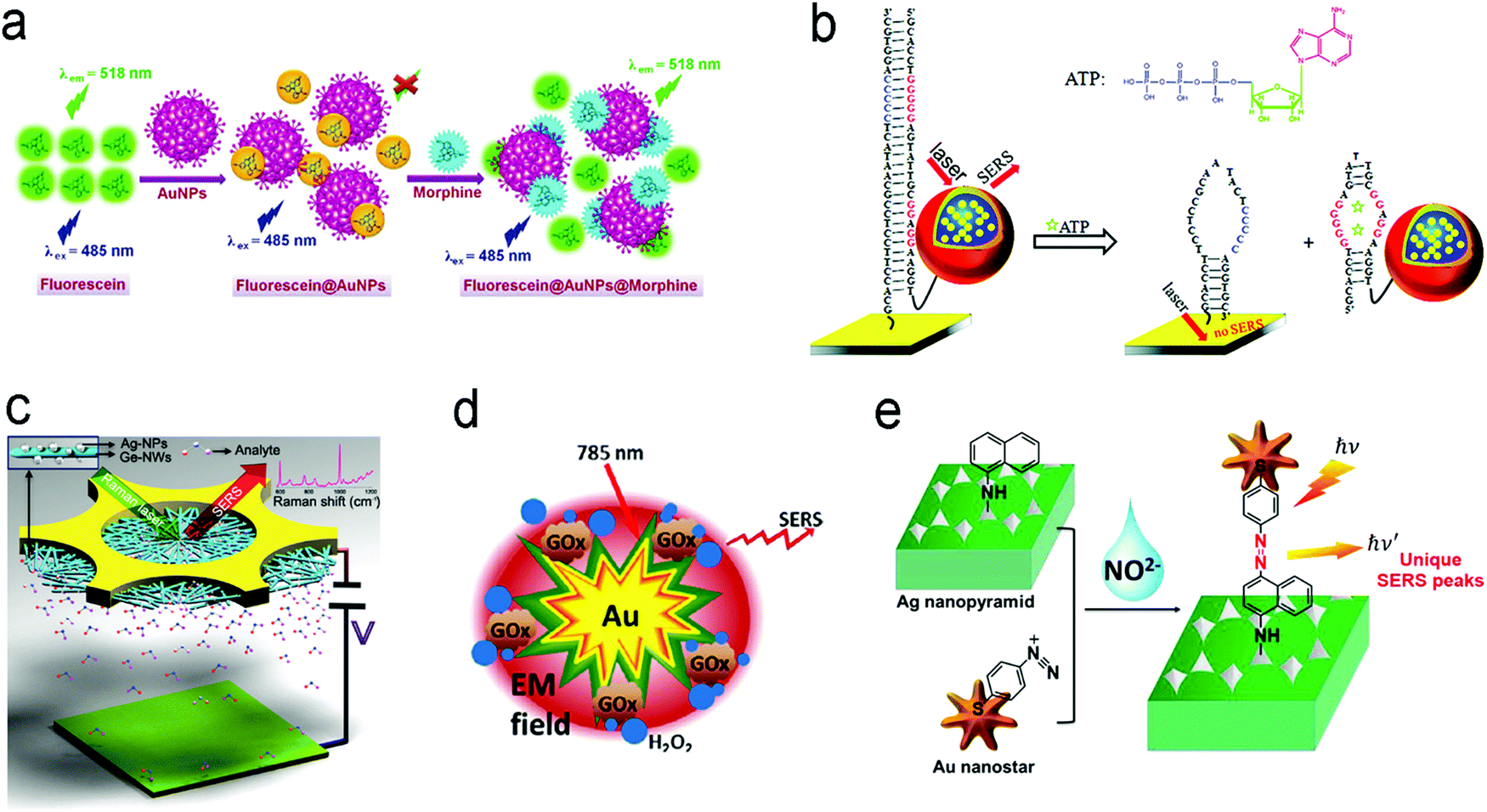 | ||
| Fig. 12 Detection of small molecules with fluorescence or SERS sensors. (a) A competitive assay based on the different affinities of fluorescein and morphine to Au nanoparticles. (b) Molecular beacon assay using SERS probe-linked aptamer for ATP detection. (c) SERS sensors with a “built-in” electrostatic preconcentration unit for the capture of the antibiotic analyte. (d) In situ generation and detection of SERS-active H2O2 for glucose detection. (e) In situ generation of SERS-active azo-moieties for nitrite detection. Reproduced with permission from (a) ref. 175, copyright 2018, Springer Nature; (b) ref. 64, copyright 2012, American Chemical Society; (c) ref. 166, copyright 2017, Wiley-VCH; (d) ref. 149, copyright 2014, Elsevier; (e) ref. 70, copyright 2018, Elsevier. | ||
Many small molecules contain benzene rings, which are usually SERS-active. Such molecules can be trapped on a plasmonic substrate to trigger SERS signals. Following this principle, various SERS sensors have been constructed.42,176,177 Huang et al. fabricated an Au@Ag nanopillar array with a 10 nm of gap between adjacent pillar tips.176 Due to its enhancement factor of up to 107, this SERS sensor was able to detect a 5 × 10−6 M 2,3,3′-trichloribiphenyl (PCB20) pesticide. To create stronger “hot spots”, Zhu et al. developed a hierarchically ordered array of Ag-nanorod bundles with about 2 nm of gaps between adjacent nanorods.178 This increased the SERS enhancement factor to 108. Furthermore, this sensor was also able to distinguish multiple analytes simultaneously, which benefited from the narrow band of each peak in SERS spectra. Using a Ag nanorod bundle array, methyl parathion had characteristic bands at 852, 1109, 1260, and 1328 cm−1 while 2,4-dichlorophenoxyacetic acid (2,4-D) exhibited specific peaks at 436, 915, 1298, 1373, and 1620 cm−1. The strong EM field enhancement (E/E0 ∼ 3 × 104) of the Ag nanorod bundle array not only achieved high sensitivity toward methyl parathion (a LOD of 21.5 nM) and 2,4-dichlorophenoxyacetic acid (2,4-D, a LOD of 61.9 nM) in water, but also discriminated two pollutants in a mixture. To trap and preconcentrate the charged or polar analytes, electrophoresis can be integrated into SERS sensors because charged or polar analytes are driven to the oppositely charged electrode under the electric field. This process was employed for the selective detection of polar molecules by altering potential polarity. Recently, a copper microgrid, which was modified with germanium nanowires and further decorated with Ag nanoparticles, served as both an electrode and a SERS substrate (Fig. 12c).166 After applying a voltage, the evenly distributed analytes in the solution were forced to move along the electric field to the SERS substrate, leading to a high concentration near the substrate. In this case, polar antibiotics, 2.4 nM 6-aminopenicillanic acid and 0.9 nM penicillin G, were detected, which were approximately 5.7 and 2.8 folds of improvement by electrostatic preconcentration.
In situ detection of chemical reaction products of an analyte can result in low interference and a high signal-to-noise ratio. Fig. 12d shows a SERS sensor for the detection of glucose in saliva.149 The Au nanostar@SiO2 core–shell nanoparticles were functionalized with glucose oxidase enzyme. The sensor catalyzed glucose to produce H2O2 that exhibited a finger-print SERS peak. The produced H2O2 molecules were close to the surface of core–shell nanoparticles, which were subject to a strong electromagnetic field generated by the “hot spot” near the Au nanostar tips, amplifying SERS signals remarkably. This sensor response was not affected by the co-existing ascorbic acid and uric acid, and it showed a dynamic range of concentration from 25 μM to 25 mM toward glucose detection in saliva. An additional intriguing example is that the product from the in situ chemical reaction served as a linker, which was covalently bound to two adjacent SERS substrates.70 In their design, the Ag nanopyramid array and the Au nanostars were modified with 1-naphthylamine (1-NA) and 4-aminothiophenol (4-ATP), respectively (Fig. 12e).70 After an analyte (nitrite) was present in the assay, it reacted with 1-NA and 4-ATP to form the azo group that was sandwiched between the Au nanostars and the Ag nanopyramid array. Owing to a small gap, the “hot spots” between the Ag nanopyramid and the Au nanostars significantly enhanced the SERS signals of the azo group, and showed a LOD of 0.6 pg mL−1 toward nitrite detection in water with a wide linear detection range from 1 pg mL−1 to 10 μg mL−1, which covered the MCL of 1.0 μg mL−1 of nitrite in drinking water regulated by the U.S. EPA.
3.4 Metal ion sensors
 | ||
| Fig. 13 Signal transduction modes/principles in SERS or fluorescence sensors for metal ion detection. (a) “One-to-one” model using single coordinate site between the optical probe and target ion; (b) aggregation of optical probes induced by multiple coordinate sites; (c) DNA hybridization assay; (d) molecular beacon-based competitive assay. | ||
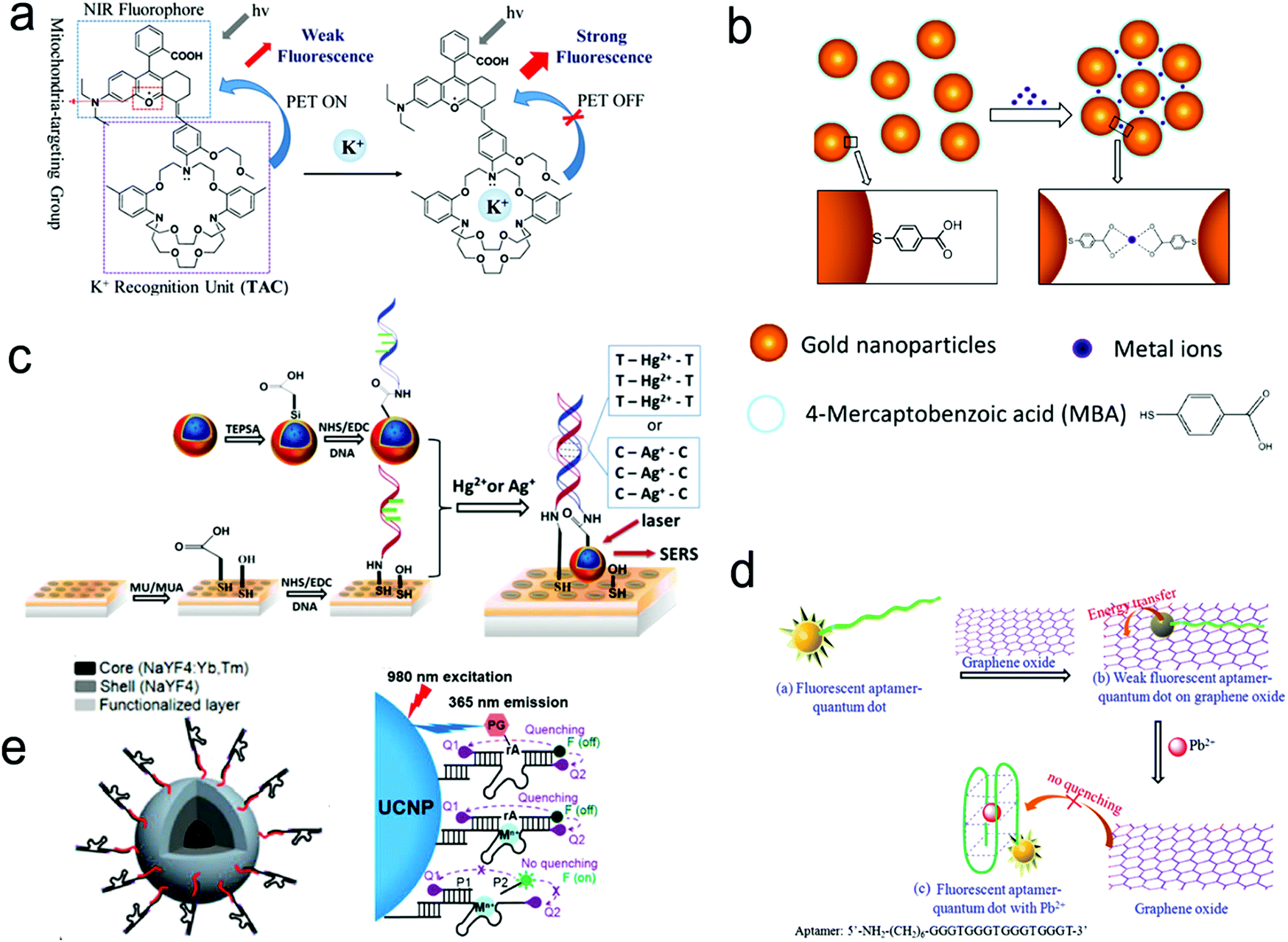 | ||
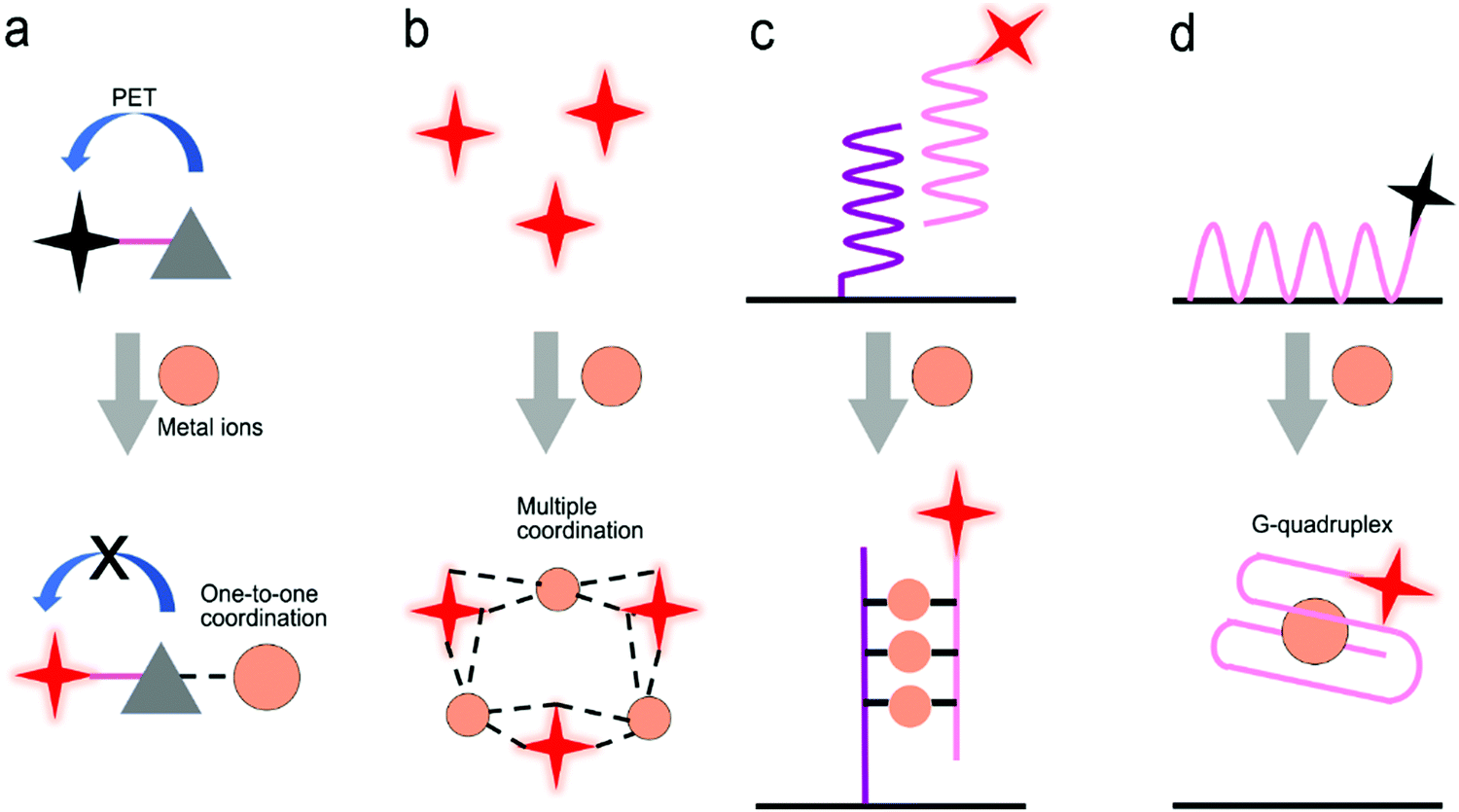
| Fig. 14 Detection of metal ions by the formation of metal ion–ligand complexes. (a) Coordination complex formed with one metal ion and one fluorescence probe, inhibiting photo-induced electron transfer (PET) to enhance the intensity of fluorescence. (b) Coordinate interaction induced aggregation of gold nanoparticles for SERS enhancement. (c) SERS probes coupled with an ordered Au nanohole array. (d) Competitive assay by the formation of a G-quadruplex/Pb2+ complex. (e) Catalytic nucleic acid (DNAzymes) activity activated by specific metal ions. Reproduced with permission from (a) ref. 181, copyright 2020, The Royal Society of Chemistry; (b) ref. 188, copyright 2017, IOP; (c) ref. 69, copyright 2015, The Royal Society of Chemistry; (d) ref. 189, copyright 2013, Elsevier; (e) ref. 190, copyright 2018, American Chemical Society. | ||
Fig. 14c shows a SERS sensor based on the metal ion-induced DNA hybridization (Fig. 13c).69 A single-stranded DNA (ssDNA) was initially immobilized on a gold nanohole array substrate, and another ssDNA labelled with a SERS probe (gold nanostar@Raman-reporter@silica nanoparticle) was suspended in the solution. Because Hg2+ and Ag+ showed strong affinity to thymine nucleobase (T) and cytosine nucleobase (C) to form the T–Hg2+–T and C–Ag+–C structures, respectively, the presence of analytes resulted in the formation of double-stranded DNA and moved the SERS probes close to the gold nanohole array substrate. The “hot spots” were formed between the gold nanostars and the Au nanohole array, amplifying the SERS signals. The sensor exhibited a LOD of 2.3 pM for Hg2+ and 0.17 nM for Ag+ in human saliva. This sensor can be used to monitor the release of metal ions from the silver–mercury amalgams in dental fillings.
Because aptamers can be programmed to selectively bind to different analytes, there is plenty of room for the application of aptamers in sensors.76,182,189 Wu's group used an aptamer to construct a fluorescent sensor for lead(II) ion detection (Fig. 14d).189 Initially, an aptamer conjugated with QDs was immobilized on the graphene oxide sheet through π–π stacking interactions. Upon binding to Pb2+ ions, the aptamer was transformed to a G-quadruplex/Pb2+ complex, turning on the fluorescence of the QDs. This sensor reached a LOD as low as 90 pM and was successfully applied to river water. Additionally, a special aptamer with catalytic nucleic acids (DNAzymes) activity, which can be activated by metal ions, provided an alternative method for detection.191,192 Lu's group demonstrated this method by conjugating Zn2+-specific DNAzyme with a quencher (Q2) at the end to lanthanide-doped upconversion nanoparticles, NaYF4:Yb,Tm@NaYF4 UCNP (Fig. 14e).190 The substrate strand of DNAzyme was linked with a fluorophore (F) and another quencher (Q1) at each end, respectively. The synergistic effect of Q1 and Q2 allowed complete quenching of the fluorophore in the initial state. The Zn2+ ions activated the catalytic ability of DNAzymes, leading to the cleavage of the scissile ribonucleotide adenosine (rA) and the dehybridization of the substrate strand. This triggered the emission of the fluorophore. In this design, the scissile ribonucleotide adenosine (rA) in the middle of the substrate strand was specifically protected by a 2′-nitrobenzyl photocage group (PG) that was photo-dissociated when the upconversion nanoparticles converted 980 nm NIR light into 365 nm emission. This showed a precise control of the location and time in detection by regulating NIR light. Similarly, Wang et al. introduced it into a SERS sensor where the gold-coated substrate was functionalized by DNAzyme. A cleavage substrate was used as a bridge with the bottom part hybridizing with the DNAzyme and the upper part hybridizing with the Au nanoparticles.193 Upon the addition of Pb2+, the SERS signal was reduced due to the detachment of Au nanoparticles from the gold surface.
4. Optofluidic devices toward point-of-care testing
Biosensors, which have been described in Section 3, have provided convenience for in vitro diagnosis. However, in many cases, these sensors have to be operated manually by professionals in a central laboratory, and thus cannot be used in POC settings. An effective solution is to incorporate SERS or fluorescent sensors into a microfluidic platform to form “all-in-one” optofluidic devices, which can pretreat samples, transport liquid, and detect analytes in a single chip. To enable POCT, optofluidic devices are desired to be miniaturized, portable, automotive, non- or minimally invasive, can be read out by a handheld or a portable reader, and operated by a layperson with minimized or no training. In this section, we highlight some important features and research progress in optofluidic devices that are relevant to POCT. In POC settings, devices are applied to various sample matrices such as water, buffer, saliva, urine and blood. Blood testing is most challenging among all these sample matrices in POC settings. So far, few commercial portable devices are available for direct and quantitative measurement of analytes in finger-prick whole blood samples in POC settings.4.1 Criteria and features of point-of-care-technologies
Many people call biosensors and microfluidic devices POC technologies, which is misleading. Not all biosensors and microfluidic devices can be identified as “point-of-care (POC)” technologies unless they meet the following criteria.194,1954.2 Active optofluidic devices
Pumps. For active microfluidic devices, traditional external pumping systems include syringe pumps, vacuum pumps, electro-osmotic pumps, electrowetting devices, etc.13,196–198 However, these pumps are large, expensive and some of them require training for operation. Thus, small and easy-operating pumps are desirable for POCT.199 For example, a commercially available latex balloon served as a pressure pump to drive liquid flow in microfluidic channels, and sodium polyacrylate with a high swelling ratio acted as a suction pump to accelerate flow by absorbing water.200,201 Additionally, some hand-operated systems including syringes and pipettes are used to inject liquid into microfluidic channels.199,202,203 Similarly, one can push a deformable elastomer chamber with a finger to change pressure to deliver fluid into the target zones.199
Valves. A valve is another important component that stops fluid flow, changes flow direction and sequentially delivers multiple preloaded reagents.13,199,204 Various types of valves have been developed to meet different needs in flow control. For instance, hydrophobic valves are introduced to stop or regulate liquid flow through generating a capillary barrier. Pneumatic valves operate flow based on a change in the pressure. Screwing, twisting, aligning holes or inducting magnetism can also realize fast switching and on-demand flow regulation.205–207 Compared with these manually operating valves, a series of automatic valves have been designed to better satisfy automation requirements in POCT. For example, a meltable valve made of wax can open in response to heating to separate assay steps.208 And an integrated pH-responsive hydrogel valve can sense the surrounding pH to display different extents of swelling or contracting to control flow.209 In addition, retention burst valves with predetermined heights and widths can encode capillary difference changes, enabling automatic sequential liquid delivery.210 Moreover, elastic reversible valves on a centrifugal microfluidic platform are used to release liquid on demand. Furthermore, a circular chip incorporated with peptide microarrays can automatically separate, collect and detect five different proteins from a single serum sample.211
Mixing modules. Typically, mixing is performed by confluence of fluids from different inlets to induce molecule diffusion. Multiple S-shaped channels are exploited to increase the diffusion efficiency.212 Apart from these passive micromixers with patterned channels, active stir mixing units can tune the mixing processes. For example, a stir mixing was powered by an electrical field using two aqueous electrolyte solutions,213 in which fluctuating waves were generated and propagated toward the downstream. Alternatively, flow instability conditions can be achieved by acoustic-driven micromixers, centrifugal mixers or magnetic mixers under magnetic fields.197,214,215
Purification/separation modules. In the case of handling complex sample matrices such as blood, urine and silva, separation may be required to extract desirable analytes, or to remove non-specific molecules prior to detection. Classical separation methods, such as nanoporous membrane filtration, cross-flow filtration, electrophoresis and isotachophoresis, rely on the size and/or charge difference of molecules in samples.13,199,216–219 For example, in a three-stage spiral channel device, multiplex cross-flow channels were patterned along the spiraling direction. As a result, tumor cells and blood cells were separated by optimizing the flow rate, achieving 1100-fold of analyte preconcentration.217 In order to increase specificity, specific ligand–analyte interaction, or additional surface modification with ovine serum albumin (BSA) is used for sample separation. For example, a porous hierarchical graphene foam served as the channel substrate, in which the electrospun titanium dioxide nanofibers were modified with the antibody of epidermal growth factor receptor 2.220 The surface-modified substrate in microchannels can capture the protein analyte and remove non-specific species by washing. Additional simpler purification methods include modulation of steric hindrance on the microchannel substrate and immobilization of the capture ligand on magnetic beads. The targets captured on magnetic beads can be isolated under the magnetic field.221,222
On-chip blood plasma separation unit. Testing blood in POC settings is much more challenging than saliva, urine and buffer. Whole blood consists of ∼55% plasma as well as ∼45% blood cells and platelets.223 Blood cells may cause severe interference during testing. Therefore, plasma is typically separated from whole blood prior to testing. In a central laboratory, plasma separation is processed by centrifugation and other lab-based equipment. However, such large-scale equipment is unavailable in POC settings, and is not a choice for laypersons. Hence, plasma separation is typically performed in situ by an on-chip unit in microfluidic devices using active or passive methods.196,224,225 Active separation methods include centrifugation, dielectrophoresis and acoustic waves (Fig. 15). Centrifugal separation, which utilizes difference in mass density between blood cells and plasma, was used in a compact-disk (CD) microfluidic device (Fig. 15a).226 By varying the disk rotation speed, the separated plasma was subsequently divided into smaller samples of equal volume to perform multiple testing simultaneously. Fig. 15b shows plasma separation by acoustic standing waves, in which blood cells were driven to the pressure nodes in the standing wave field. Acoustic microstreaming is usually observed, and air bubbles oscillate under the acoustic field. As shown in Fig. 15b, blood cells were trapped in the microstreaming vortex at a certain frequency while plasma flowed downstream.227Fig. 15c demonstrates a dielectrophoresis process, in which the polarization forces were formed in a non-uniform electric field to separate the suspended particles from fluid. Blood cells have dielectric properties different from proteins and small molecules, and can be forced to move in the microchannel. As shown in Fig. 15c, blood cells were trapped in the dead-end branches and left blood plasma in the middle of the channel.228
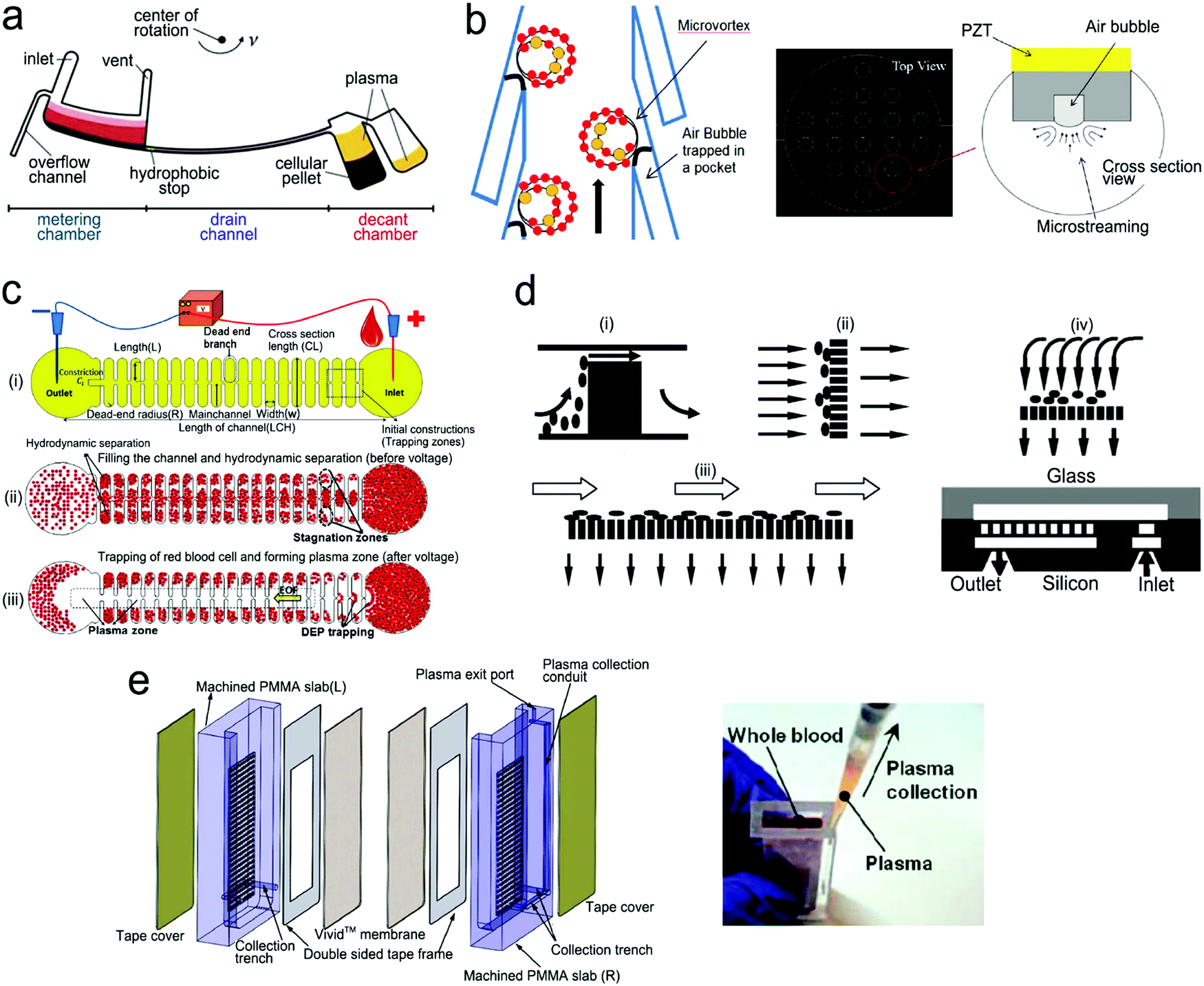 | ||
| Fig. 15 On-chip plasma separation methods including active (a–c) and passive (d and e) methods: (a) centrifugal separation in a CD-shaped disk. (b) Acoustic microstreaming vortex to trap blood cells. (c) Dielectrophoresis-induced plasma separation. (d) Silicon-based microfilter including (i) weir-type filter, (ii) pillar-type filter, (iii) cross-flow filter, and (iv) membrane filter. (e) Dual separation by combination of sedimentation and membrane filtration. Reproduced with permission from (a) ref. 226, copyright 2006, The Royal Society of Chemistry; (b) ref. 227, copyright 2021, Elsevier; (c) ref. 228, copyright 2015, Springer Nature; (d) ref. 229, copyright 2008, Springer Nature; (e) ref. 238, copyright 2013, American Chemical Society. | ||
Filtration is a common passive separation method, which is based on the size exclusion effect to impede cell movement. Pillar-type, grating-type, weir-type microchannels, packed-bead column, and filter membranes have been explored for plasma separation in microfluidic devices.225,229–231 Ji et al. compared four types of filtration structures, including weir-type, pillar-type, cross-flow and membrane filter (Fig. 15d).229 It was found that the cross-flow filter showed the highest efficiency in separating white blood cells from red blood cells in whole blood. However, blood cells could block the microchannels in these filters, limiting continuous operation. Hence, other passive separation methods such as hydrodynamic flow and capillary pumps have been exploited. Hydrodynamic flow is based on the Zweifach–Fung bifurcation law that cells prefer channels at a higher flow rate, leaving plasma in the channels at a lower flow rate.232,233 Both blood cells and plasma keep flowing in the microchannel, thus such devices can work for a longer time without blocking. For example, Yang et al. fabricated a “Y”-shaped channel with an asymmetric flow rate ratio to separate plasma.234 Heath's group designed a comb-liked channel to direct plasma skimming into high-resistance branches (Fig. 16b).235 By utilizing the Zweifach–Fung bifurcation effect, Heath's group further increased the efficiency of plasma separation with inertial forces.236 This method permitted direct separation in the same channel without skimming to other channels, leading to a larger proportion of plasma. Natural sedimentation is quite slow. The separation process will stop once the sediment gap is filled with cells. To accelerate this process, Maria et al. combined hydrodynamic effects and sedimentation to separate 2 μL of plasma from 10 μL of whole blood with 99.9% separation efficiency within 15 min.237 In Fig. 15e, Liu et al. combined sedimentation and membrane filtration into a plasma separator.238 It took less than 7 min to extract about 275 μL of plasma from 1.8 mL of whole blood. As a result, HIV virus was measured in the plasma matrix.
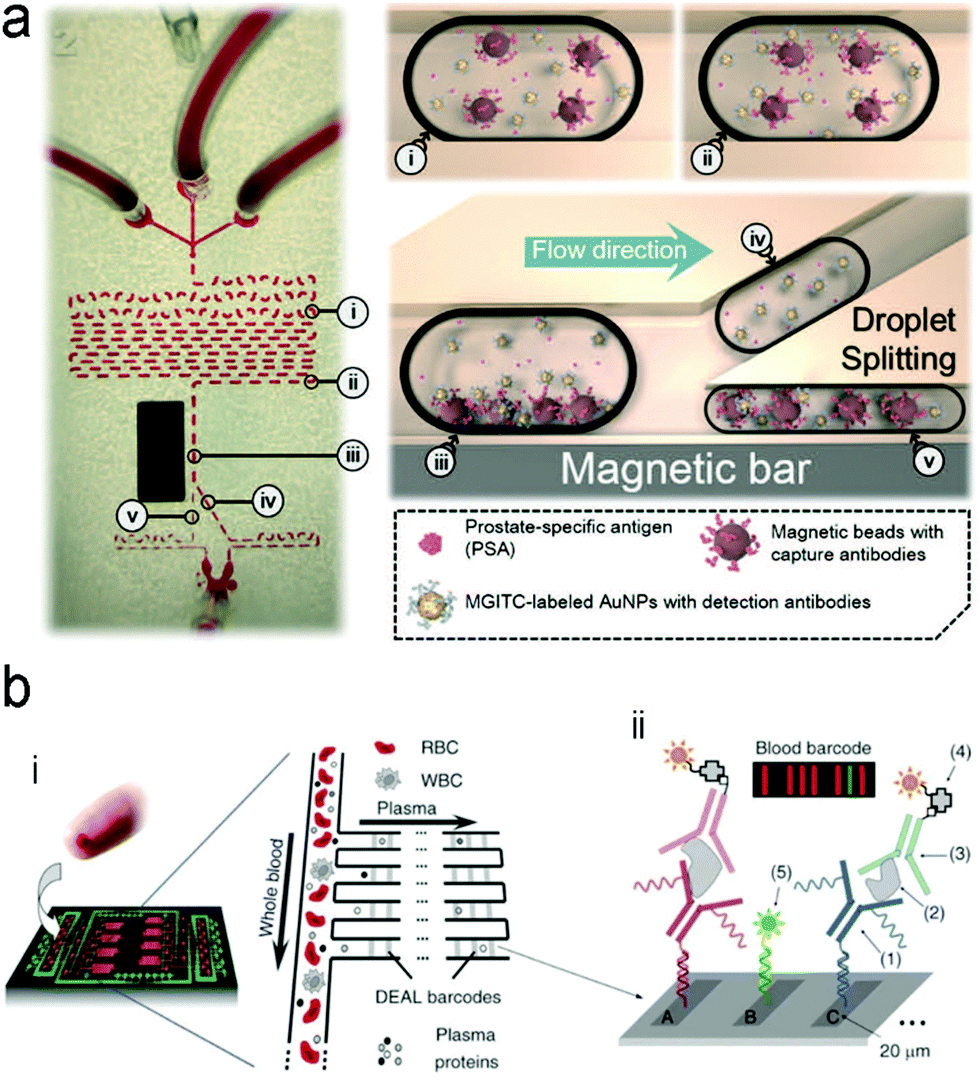 | ||
| Fig. 16 A typical structure of active optofluidic device for protein detection. (a) SERS-based microdroplet device for detection of prostate-specific antigen (PSA) cancer markers. It has 5 compartments with different functions: (i) generating microdroplet and mixing reagents; (ii) forming immunocomplexes on magnetic beads; (iii) separating immunocomplexes by a magnetic bar; (iv) moving the supernatant that contained unbound SERS probes; (v) separating magnetic immunocomplexes through droplet splitting. (b) In vitro fluorescence microfluidic device for multiplexed detection of proteins in finger-prick whole blood. Reproduced with permission from (a) ref. 218, copyright 2016, The Royal Society of Chemistry; (b) ref. 235, copyright 2008, Springer Nature. | ||
Digital microfluidic modules. Digital microfluidics (DMF) has been developed to precisely manipulate small quantities of liquid reagents in microfluidics.239 Following the electrowetting-on-dielectric principle, continuous liquid can form picoliter-to-microliter droplets due to the change in surface wettability (hydrophobic vs. hydrophilic) of discrete insulated electrodes laid out close to each other.240–242 Under the electric field, each droplet with different inclusions (targets, detection and capture compounds) can individually move, split, merge and dispense within a short period of time, realizing sample transport, mixing and separation.240,243 Digital microfluidics can also reduce sample consumption and avoid cross contamination besides their own merit of low power consumption.239 For instance, in a digital microfluidic device, the bottom of microfluidic channels was made of glass. An array of discrete ITO electrodes was deposited onto the glass substrate; the ITO electrodes were then completely covered with a dielectric material (SU-8/Al2O3). Subsequently, a hydrophobic layer was coated onto the surface of the dielectric material. The dielectric material served as a spacer between the top hydrophobic layer and the underlying ITO electrode. Without an applied electric field, the top surface layer was hydrophobic. When applying an electric field onto an ITO electrode, the top surface layer became polarized, showing a hydrophilic feature.244 Droplets were consequently created on the parallel substrates and transported to the target area to function as a fluorescence immunoassay. In this digital microfluidic device, the magnetic beads linked with the capture antibody were mixed with the sample droplets containing the protein analytes under electrowetting force, followed by incubation for capturing analytes on the magnetic beads. These magnetic beads can be extracted under the magnetic field and washed with buffer prior to fluorescence detection. Compared with conventional ELISA that requires at least 50 μL of a sample volume, this digital microfluidic device required only 520 nL, which made it possible to sensitively test a rare sample volume (5–10 μL) from fertilization in vitro. The total duration was shortened to less than 40 min for multiplexed detection of protein biomarkers.
Detection of proteins with optofluidic devices. Fig. 16a shows a typical example of optofluidic immunoassay where the liquid sample, the Raman dye-labelled Au nanoparticles with detection antibodies, and the magnetic beads with capture antibodies along with the carrier oil were externally injected into three microchannels, respectively.218 All liquids were fully mixed as the microdroplets passed through the winding channel, followed by the formation of sandwich magnetic immunocomplexes. These complexes were then separated and concentrated by a magnet. At the end of the device, the Au nanoparticles linked to magnetic beads were aggregated, leading to enhanced SERS signals. This device has achieved a LOD (0.1 ng mL−1) toward prostate-specific antigen (PSA) detection in serum. Like this device, aggregation of plasmonic nanoparticles in a microfluidic channel was commonly used for SERS transduction in optofluidic devices, which can be realized with other approaches such as micro–nano channel junctions and nanoporous silica microspheres besides the magnetic field.247 However, aggregation methods involve a lot of reagents in the device, which bears a burden on storage, shipping and operation. A different method is capture of analytes into a detection substrate on the bottom of the microfluidic chamber. In this way, the SERS or fluorescence probes are in situ immobilized onto the detection substrate.248–250 Heath's group has reported an integrated microfluidic system patterned with antibody barcode arrays as the multiplexed detection substrate (Fig. 16b).235 After a finger-prick blood sample was loaded onto the optofluidic device, plasma was quickly separated via the Zweifach–Fung effect. Different antigen biomarkers were then captured onto different barcode detection zones by forming the sandwiched configuration of the capture antibody–antigen–detection antibody labeled with a fluorescence probe. Such in situ immunoassays can detect multiple proteins simultaneously.
Detection of nucleic acids with optofluidic devices. When the level of nucleic acids is high, nucleic acids can be directly detected using optofluidic devices; the structure of devices is similar to that of optofluidic immunoassays. For example, Qi et al. reported an “in situ” measurement of nucleic acids using molecular beacons immobilized on a nanoporous gold disk inside the PDMS microchannel.248 When the targets flowed through the gold disk substrate, they were hybridized with the molecular beacons via base pairing, generating fluorescence signals on the detection zone. However, in most cases, the level of nucleic acid is very low in the samples. For example, the serum level of DNA in HIV patients within the first week of infection is as low as 50–1000 copies per mL. It is very difficult to directly measure such a level of nucleic acids. Hence, a nucleic acid amplification unit needs to be included into an optofluidic POCT device besides a detection unit.
A variety of nucleic acid amplification methods, such as polymerase chain reaction (PCR) and isothermal amplification, have been exploited in microfluidic devices to amplify signals.251 Particularly, digital PCR (dPCR) is a promising high-throughput approach.252 Amplicons are generated exclusively from a single DNA template in large-scale multiple diluted samples, converting fluorescent signals into linear and digital signals. Digital PCR is generally integrated into microfluidic systems in the form of droplets or integrated flow circuit chips. For example, a SlipChip was fabricated with two plates etched with wells and ducts (Fig. 17a).253 In the assembly of two plates, the elongated wells allowed for a continuous flow path to transport reagents into each reaction chamber. When the plates slipped, the misaligned channels and ridges broke off the flow path to obtain droplets. This chip with 1280 wells can achieve 1280 droplets and each of them contained ∼2.6 nL of the reaction solution, thus is able to perform single-copy PCR assay. Although PCR or digital PCR technology shows high sensitivity and accuracy in detection, 2–3 hours of thermal cycles cannot meet the POCT need. In contrast, isothermal amplification is relatively simpler and has shorter duration, which is suitable for microfluidic systems.254,255 For example, a centrifugal microfluidic chip was integrated with the recombinase (RAA)-aided amplification unit and the CRISPR/Cas12a detection unit.256 After centrifuging, the loaded sample was mixed with preloaded RAA–Cas12a reagents, followed with isothermal nucleic acid amplification at 37 °C.
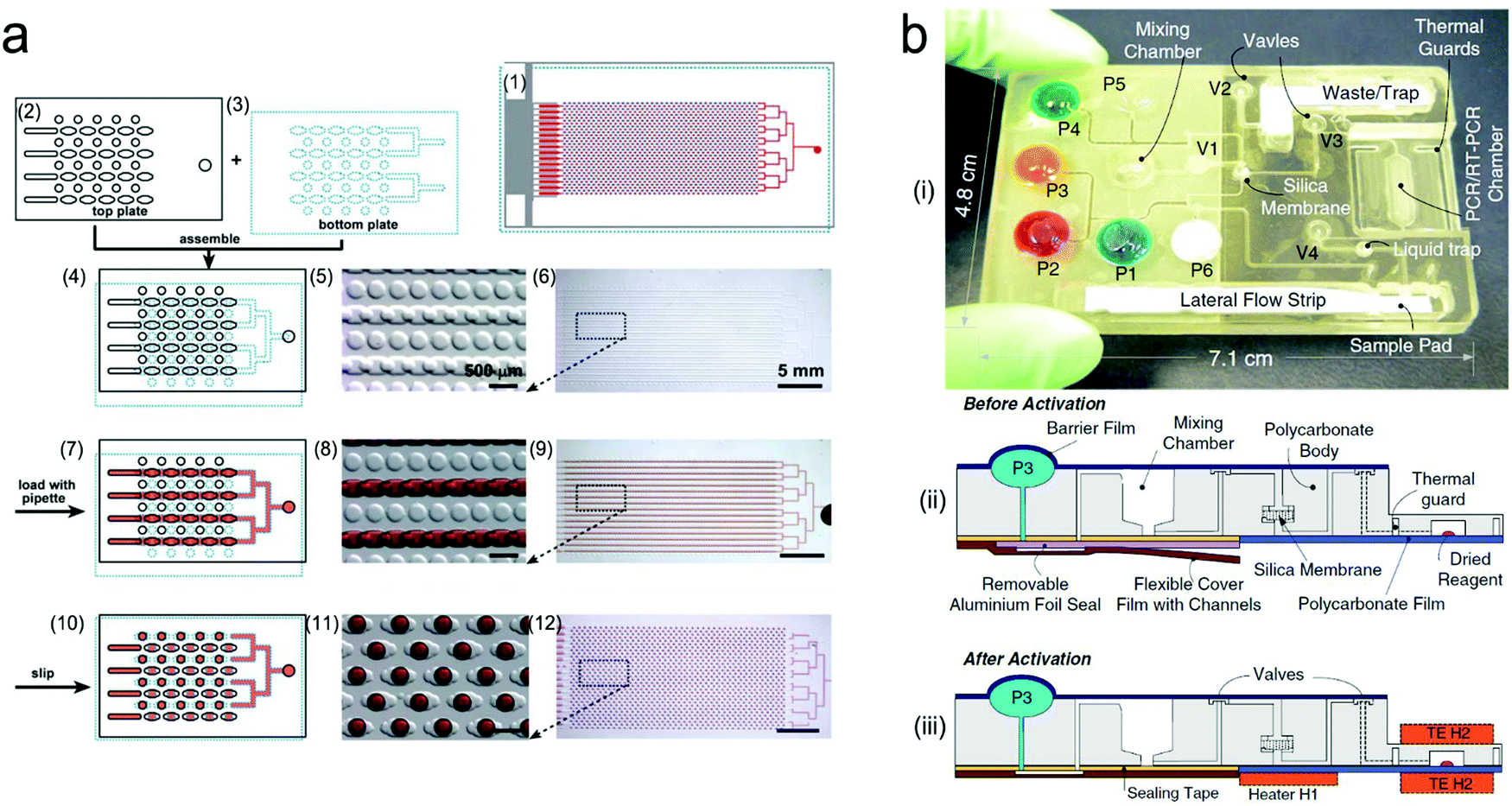 | ||
| Fig. 17 Integrated microfluidic devices for nucleic acid testing. (a) Schematic digital PCR process with a SlipChip: (1) assembled SlipChip after splitting; (2) and (3) the top plate and the bottom plate, respectively; (4–6) overlapping of the top and bottom plates to form a continuous fluidic path; (7–9) the aqueous reagent (red) filling the SlipChip through the connected elongated wells; (10–12) aqueous droplets formed in each compartment after splitting. (b) A self-contained microfluidic cassette integrated separation, amplification, and detection of nucleic acids. Reproduced with permission from (a) ref. 253, copyright 2010, The Royal Society of Chemistry; (b) ref. 267, copyright 2010, Springer Nature. | ||
Lysis and/or extraction of nucleic acids is sometimes required prior to detection. In this case, a lysis/extraction unit needs to be added into optofluidic devices so that nucleic acid tests can be done in POC settings. One of the common methods is chemical lysis, where lytic agents are mixed with the sample liquid in the microchannel to disrupt the cell membrane.257 For example, ammonium chloride is capable of red blood cell lysis; guanidinium thiocyanate and guanidinium chloride are used to disrupt membrane proteins; lysozyme works on the bacteria wall.257,258 Sethu et al. performed lysis by sandwiching whole blood with a lysis buffer on both sides.259 The mixture passed through a long serpentine channel to achieve full lysis. Besides chemical lysis, physical lysis is also common, including mechanical lysis, electroporation lysis, thermal lysis, laser lysis and sonication lysis.260,261 Kim et al. performed mechanical lysis with ZnO nanowires patterned in a microfluidics device.261 The ZnO nanowires were characteristic of high aspect ratios and sharp tips, which teared the plasma membrane and released nucleic acids but held cells. After lysis, nucleic acids may need to be extracted and separated for downstream amplification/detection. This can be done via micro-solid phase extraction, electrostatic interactions, nanoporous membrane filtration, or magnetic bead separation.262,263 Micro-solid phase extraction and magnetic bead separation technologies are commercially available for nucleic acid extraction.264 In microfluidic devices, silica micro-pillars or silica beads were built in the microchannel to perform micro-solid phase extraction.265 The negatively charged nucleic acids can bind to silica in a high ionic strength buffer, followed by washing with a non-polar solvent to remove other biological components. These nucleic acids were then eluted with a low ionic strength solution.266
“All-in-one” microfluidic devices are desirable for nucleic acid testing in POC settings, that is, combing lysis, extraction, amplification and detection modules in a single microfluidic chip.265,268Fig. 17b shows a typical example, where the self-contained microfluidic cassette can perform separation, amplification, and detection of nucleic acids. It employed the miniaturized linear motors to control fluid flow and valves.267 All the control units such as motors, heater controllers, micro-vacuum pump, and a liquid trap were packaged into a portable box as an analyzer. Before introducing the sample into the mixing chamber, the sealing layer was removed for the engagement of the connecting film. The cassette was then inserted into the analyzer to perform the preprogrammed process operation, including lysis with a prestored lysis buffer, extraction through the silica membrane, collection of pure nucleic acids with an elution buffer, amplification in the PCR chamber and detection in a lateral flow strip. This integrated system was successfully used for monitoring the DNA of bacteria (B. cereus) and the RNA of HIV, and it detected 1000 pathogen particles in the sample. In addition, centrifugal microfluidic CDs are well-known “all-in-one” devices for nucleic acid testing. Stumpf et al. designed a LabDisk with the pre-stored reagents.246 By controlling the rotational frequencies of the chip and using a magnet, a sample containing influenza H3N2 virus was sequentially transferred to each chamber and mixed with specific reagents. After the sample was mixed with the lysis buffer, nucleic acids were released and bound with the functionalized magnetic beads using a binding buffer. These magnetic beads were subsequently mixed with an elution buffer under the magnetic field to extract nucleic acids. Subsequently, nucleic acids were centrifuged into specific compartments to conduct reverse-transcript PCR, and to read fluorescence signals.
Moreover, the electric field gradients were used for preconcentration, purification, mixing, and acceleration of reactions among the sample and reagents.269,270 For example, the electric field gradients were formed using isotachophoresis (ITP), which utilized a low-mobility trailing electrolyte (TE) buffer and a high-mobility leading electrolyte (LE).271 Under an electric field, TE and LE ions can ensure that the target sample ions focus with the electro-migrating LE-to-TE interface. In this design, SARS-CoV-2 viral RNA and host DNA from the lysed samples of COVID-19 patients were extracted and preconcentrated in ITP, leaving impurities, proteins and other inhibitors. After loop-mediated isothermal amplification (LAMP), the electric field gradients in ITP were used to control and confocus the Cas12–gRNA enzymatic reaction. Such ITP-based nucleic acid device exhibited the test time down to 30 minutes.
4.3 Passive microfluidics
Active microfluidics usually uses external syringe pumps or pressure pumps, which provide great flexibility in designing the structure of microfluidic devices, selecting different liquids, and controlling liquid flow in a well-tuned manner. However, active microfluidics systems are relatively expensive and large, and require a lot of steps to operate. Therefore, there is an incentive to develop passive microfluidic devices, in which the “built-in” micropumps are used to drive liquid flow to realize self-powered fluidic systems, eliminating the use of external pumps and the associated electronic controllers. Although there are some constraints on regulating liquid flow and realizing sequential operation, passive microfluidic devices are attractive for POCT because they are relatively inexpensive, compact, and have a relatively simple structure. Lateral flow is the dominant form in passive microfluidics. Therefore, this section is focused on lateral flow devices that are divided into two different types: (i) plastic-/glass-based lateral flow devices and (ii) paper-based lateral flow assays.Basic structure of lateral flow devices. In lateral flow devices, fluids flow almost horizontally along the relatively long microchannels that are divided into different functional zones.275 It can consecutively or programmably perform sample handling and analyte detection as fluids flow to different functional zones. Fig. 18a shows the basic structure of a lateral flow device.272 It contains four main functional zones, including a pretreatment unit, a detection chamber, a fluid resistor and a “built-in” pump. A liquid sample is initially loaded into the pretreatment unit, and fills the pretreatment unit, the detection chamber and the fluid resistor sequentially, and finally reaches the “built-in” micropump. The resistor is used to adjust the flow rate and to optimize the incubation duration for fully mixing or reacting the reagents and analytes.221,273,276,277 In addition, a sample pretreatment unit may need to be placed on the upstream of the detection zone to perform separation or purification. To better regulate the separation and purification processes, an additional “built-in” micropump may be added into the pretreatment unit.
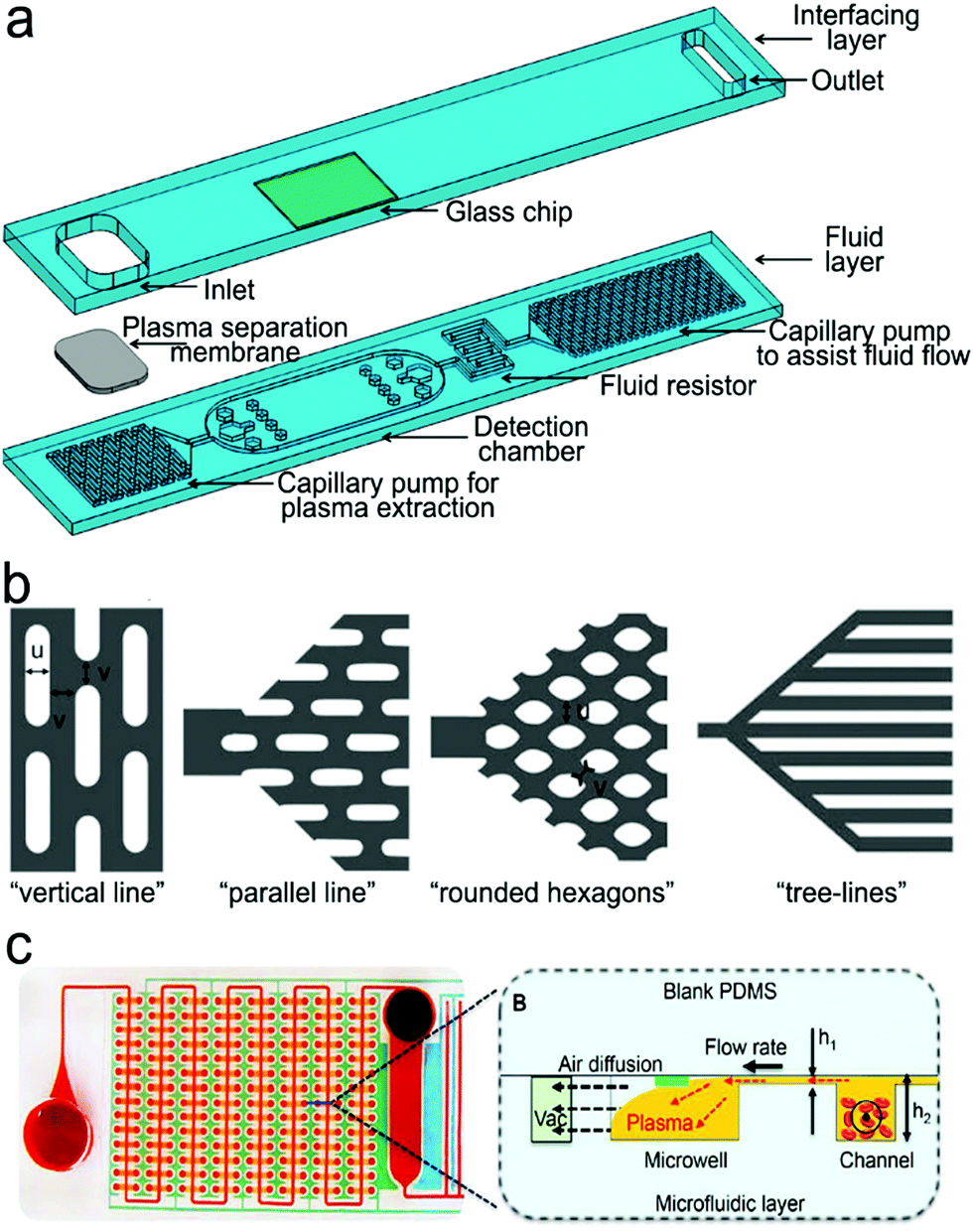 | ||
| Fig. 18 (a) Basic structure of plastic-/glass-based lateral flow device. It is composed of an interfacing layer (cover layer) and a fluid layer. The fluid layer contains four main functional zones, including a pretreatment unit (plasma separation and extraction), a detection chamber, a fluid resistor and a “built-in” micropump. (b) and (c) Examples of “built-in” micropumps: (b) capillary pumps such as “vertical lines”, “parallel lines”, “rounded hexagons” and “tree lines”. (c) A pneumatic pump fabricated by multiple vacuum pillars. Reproduced with permission from (a) ref. 272, copyright 2017, MDPI; (b) ref. 273, copyright 2007, The Royal Society of Chemistry; (c) ref. 274, copyright 2017, The Royal Society of Chemistry. | ||
“Built-in” micropumps. “Built-in” micropumps are used to drive liquid flow in passive microfluidic devices, and designed based on different mechanisms such as capillary forces, air transfer (solubility or permeability), diffusion and osmosis pressure, magnetic field, electrophoresis, electroosmosis, electrowetting, etc.273,278–280 The first two mechanisms are most popular. The capillary pumps drive flow via capillary forces, which are generated by interfacial tension. Once the device is dull of liquid, capillary pumping will stop. The flow rate can be regulated by the surface properties, size and shape of microstructures in a micropump, the distance between different pumps, and fluid viscosity.281,282Fig. 18b illustrates various microstructures in capillary pumps. Line-shaped microstructures are fabricated with a round periphery to reduce the occurrence of fluid pining.273 The fluids flow in both vertical and horizontal directions, and are modulated by the dimension of lines and the gap between neighboring lines. Zimmermann et al. fabricated a vertical line-shaped capillary pump, and found that 15 μm-sized lines exhibited the highest flow rate.273 However, such a microstructure showed a random filling front, which can be altered into a straight filling front by rotating the microstructure by 90°, namely, “parallel line”. In addition, this pump was designed with gradually increasing lateral dimension from the entrance of the pump, which can prevent the capillary pressure difference in the connection part to make the fluid efficiently flow to the capillary pump. Some capillary pumps exhibit an interlocked microstructure. For example, rounded hexagon-shaped pumps can achieve uniform flow rates due to the inhibited fluid pinning. Furthermore, a tree line-shaped capillary pump has been designed with branched filling paths separated from the main flow region (Fig. 18b).273 Such a structure can program sequential filling flow through generating different capillary pressures of the branched paths. This “tree-line” pump may suffer from a decrease in flow rate as fluids flow along each branch. Gradient surface wettability can solve this problem by moving spontaneously toward a more hydrophilic area owing to contact angle hysteresis.283
Pneumatic micropumps are also commonly used in passive microfluidic devices, in which pumping is realized through gas generation or storage of vacuum pillars inside microchannels. Zhu et al. fabricated a hydrogen generator using platinum-black to catalyze ammonia borane solution in a microchannel.280 The generated hydrogen bubbles pushed the liquid flow along the channel, and the velocity of the liquid was regulated by the gas removal rate at the gas outlet.279 Although this technology could achieve self-circulation by collecting hydrogen bubbles to add fresh reactants, it is quite difficult to ensure stable fluid flow, and the chemical reaction may be disturbed when loading the sample liquid or detection probes. Vacuum-driven microfluidics provides an alternative method utilizing gas solubility and permeability of PDMS.274,278,284 When it is degassed, the formed negative pressure forces the fluid to flow from the inlet to the dead-end microchannel. Compared with capillary pumps, surface treatment is typically not required in vacuum-driven microfluidics. However, it is challenging to maintain a constant flow rate due to a gradual decrease in the pressure difference with prolonging time. In this case, prolonging vacuuming time or changing channel patterns was proposed to mitigate this problem. Specifically, multiple vacuum pillars were introduced into the microchannel for continuous flow control (Fig. 18c).274
Purification/separation modules. Filtration, capillary pumping and hydrodynamic flow are commonly used for purification/separation in passive microfluidic devices. Hydrodynamic flow may be combined with filtration and capillary pumping to improve the separation efficacy. Maria's group designed a vertical cylinder with gradient hydrophilicity as a hydrophobic patch for blood plasma separation.237 The central portion of the vertical cylinder was hydrophobic while both sides of the well as well as their connected two parallel channels were hydrophilic. As blood flowed through the bottom hydrophilic microchannel and was elevated along the vertical cylinder by capillary action, the capillary force decreased due to the hydrophobic patch near the central part, slowing down the flow. The blood cells in the sample subsequently tended to be deposited because of their higher density while the plasma was lifted up at a higher velocity because of its lower viscosity. Ultimately, the separated plasma flowed into the upper channel for analyte detection. Similarly, a cyclic olefin copolymer was coated with a super-hydrophilic surface by the layer-by-layer assembly technique. Thus, asymmetric capillary forces were used for blood plasma separation.285 In addition, vacuum pillars were designed for automatic blood plasma separation without showing hemolysis. A slant capillary valve led the blood flow to the vacuum pillars. As a result, the blood cells were trapped while the plasma flowed into the separation channel.284
Integration of fluorescence or SERS units. Integration of SERS or NIR fluorescence sensing units into passive microfluidic devices is appealing for POCT. Gao et al. incorporated a SERS sensor into a polydimethylsiloxane (PDMS) lateral flow chip for monitoring prostate-specific antigen biomarkers (Fig. 19a).221 Driven by capillary forces, the liquid sample, the SERS probes conjugated detection antibodies and the magnetic beads linked with the capture antibodies were loaded from three different inlets and fully mixed in the winding-shaped channel due to the chaotic advection effect. The sandwiched structure was formed via the antigen–antibody interaction, and then purified by applying a magnetic field onto the magnetic beads. The SERS probes linked to the targeted antigen was related to the level of the analyte. The whole process was finished within 5 min. This integrated device achieved a LOD of 0.01 ng mL−1 in a human serum. Additionally, a blood plasma separation unit and a fluorescent hairpin molecular beacon were integrated into a vacuum-driven microfluidic chip for the detection of thrombin in whole blood (Fig. 19b).274 In this device, blood cells flowed into the vacuum microchambers while the plasma was driven to a plasma chamber, where the molecular beacons with the quenched fluorophores were stored. In the presence of thrombin, the molecular beacon opened the loop of the hairpin, pushing the quencher away from the fluorophore, and turning on the fluorescence.
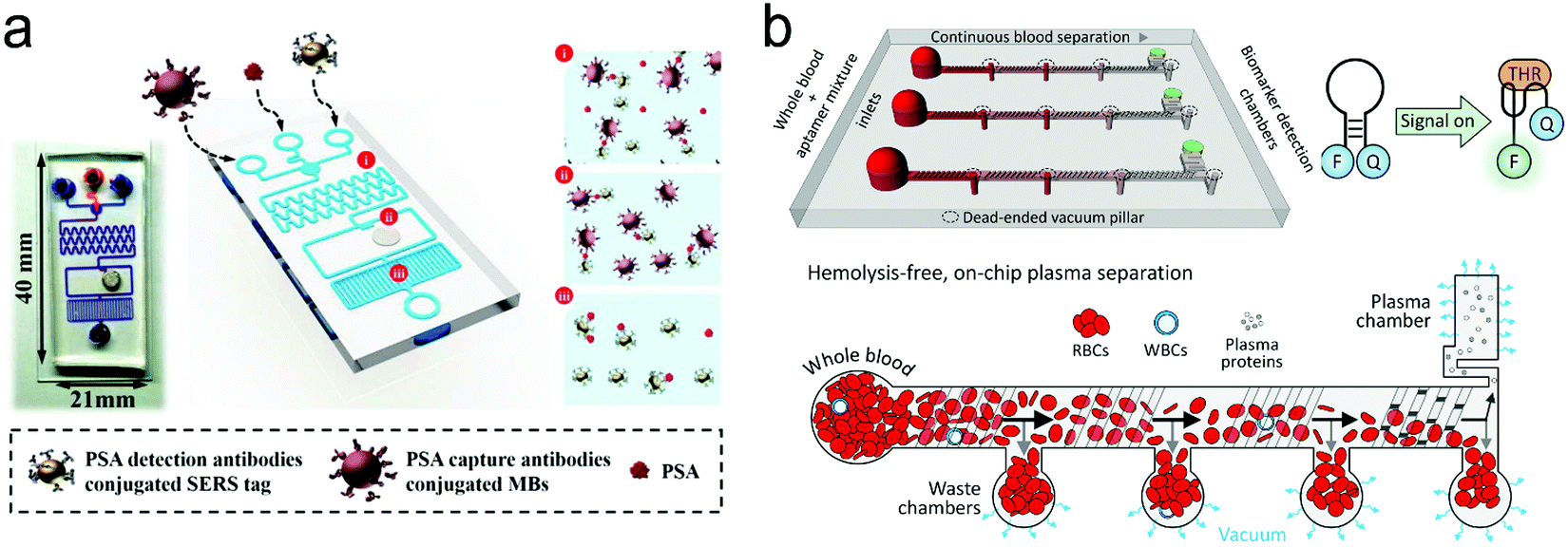 | ||
| Fig. 19 (a) A passive microfluidic device with an integrated capillary micropump and SERS probes, consisting of three compartments for (i) mixing the sample and reagents; (ii) preloading reagents and detecting; (iii) the capillary pump. (b) A passive microfluidic device with an integrated pneumatic micropump and fluorescence SERS probes. Reproduced with permission from (a) ref. 221, copyright 2019, American Chemical Society; (b) ref. 274, copyright 2017, Elsevier. | ||
Basic structure and principle. Fig. 20a shows the basic structure of a PLFS.67 It mainly consists of a sample pad, a conjugated pad, a nitrocellulose microfluidic membrane, an absorbent/wick pad, a control line and one to three test lines/zones. These pads/membranes are connected to each other with small overlaps to ensure continuous liquid flow from the sample pad all way to the absorbent pad. The sample pad is usually made of cellulose, woven meshes or filtration matrices to retain high tensile strength after wetting. The sample pad may be treated with a dry pH regulator to standardize the buffer condition of the sample liquid.286,288 The conjugate pad should prevent non-specific binding so that the detection probes can be released upon wetting by the liquid sample, and the analytes would not be held there. Hence, glass fibers, cellulose and polyester with high bed volume and low non-specific binding are suitable for the conjugate pad to store a large number of probes and release them quickly as liquid flows through.289 The nitrocellulose membrane is enriched by the amine group, which exhibits strong binding to antibodies or other types of bioreceptors, and is surface-modified with a surfactant or other reagents for prevention from non-specific binding. It also has tunable wicking properties to regulate capillary flow. This absorbent pad should have a sufficient volume so that it is not full even after the whole process is complete. Complete filling or overloading of the absorbent pad will stop the lateral flow of the liquid in the whole strip, and even cause the back flow or liquid accumulation on the nitrocellulose membrane, inducing the background noise on the test line.
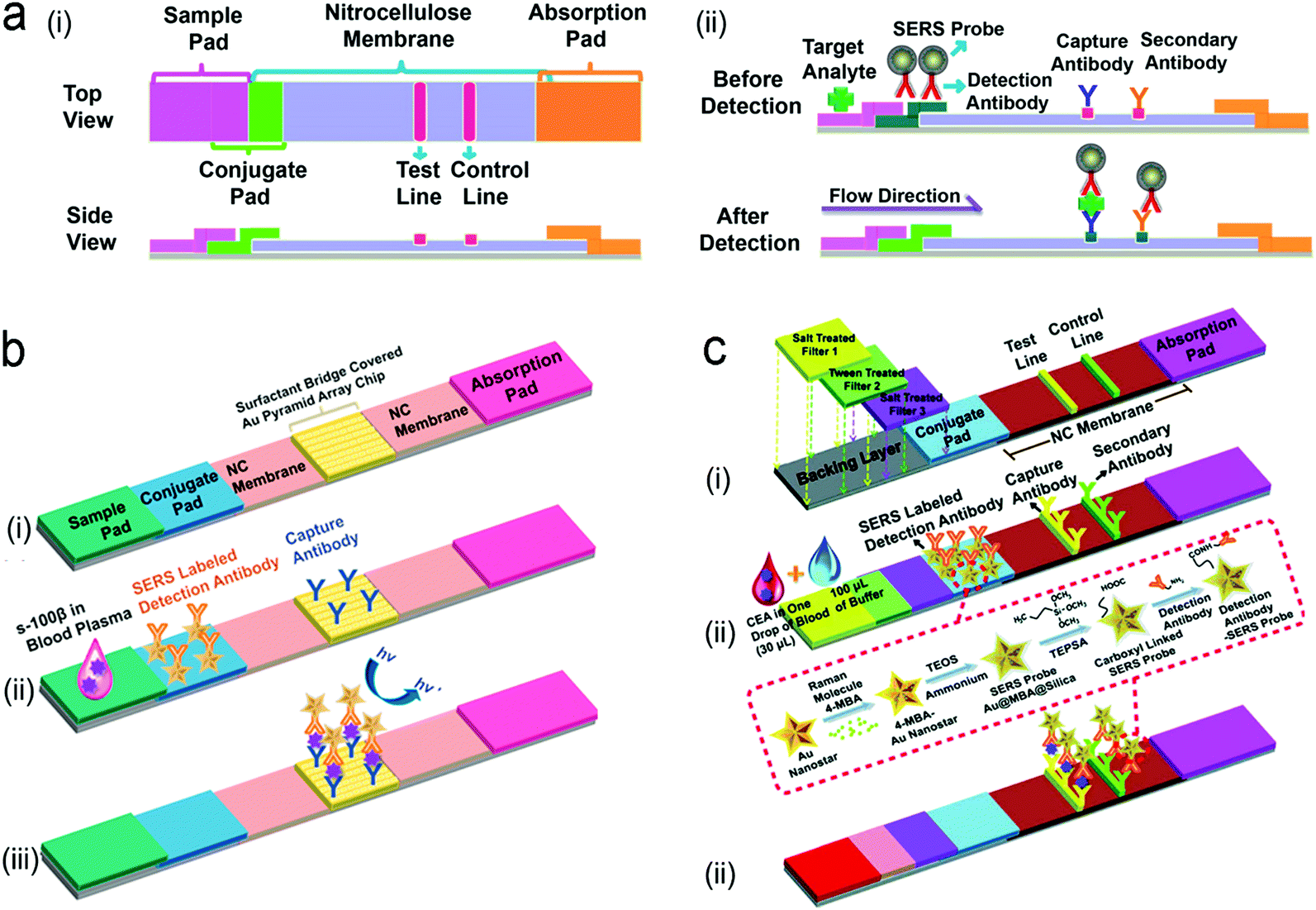 | ||
| Fig. 20 Structure of PLFS with integration SERS probes. (a) SERS-PLFS for the detection of traumatic brain injury (TBI) protein biomarker in diluted blood plasma samples. (i) Basic constructure of PLFS; (ii) side view of prestored reagents in the PLFS before detection; (iii) side view after detection. (b) Gold nanostar@Raman reporter@silica sandwiched nanoparticles coupled with a Au pyramid array for the detection of TBI biomarker S-100β in blood plasma: (i) structure; (ii) before detection; (iii) after detection. (c) SERS-PLFS with a built-in plasma separation unit for whole blood detection: (i) Structure; (ii) before detection; (iii) after detection. Reproduced with permission from (a) ref. 67, copyright 2017, American Chemical Society; (b) ref. 291, copyright 2021, Elsevier; (c) ref. 292, copyright 2020, American Chemical Society. | ||
Integration of SERS into PLFS. A traditional PLFS usually employs gold nanoparticles as colorimetric transducers and has achieved huge commercial success in home pregnancy testing in POC settings. A colorimetric PLFS can be read out by human eyes without the need for any instrument, and used for the qualitative measurement of a high level of analytes in water, buffer, saliva and urine. But they are unable to quantitatively measure a low level of analytes due to their low sensitivity. Particularly, when a PLFS is applied to plasma, serum or whole blood, it suffers from severe interference from sample matrices. One of the major reasons is that the strong red wine color of hemoglobin in such samples is overlapped with the color of gold nanoparticles on the test line. As described in Section 2, NIR fluorescence and SERS fall into the biological transparency windows, and are read out by portable readers. Thus, NIR fluorescence or SERS-based PLFS have great potential in the quantitative measurement of trace analytes even in complex sample matrices such as plasma, serum or whole blood.1,67,290
Many SERS probes are made by directly attaching Raman dye molecules to bare metal nanoparticles. Such SERS probes are unstable and easily aggregate in liquid with high ionic strength.44 Hence, Wu's group designed a sandwich-structured SERS probe, in which Raman dye molecules are sandwiched between a plasmonic metal core and a thin SiO2 outer shell, which exhibits excellent water solubility and stability in the blood sample matrix.44,64 His team also compared the SERS enhancement factors of Au nanospheres, nanorods and nanostars and found that Au nanostars contributed a lot of “hot spots”, achieving the highest SERS enhancement.44 The gold nanostar@Raman reporter@silica sandwiched SERS probes were incorporated into a PLFS to measure the neuron-specific enolase (NSE), a traumatic brain injury (TBI) biomarker, in clinical blood plasma, achieving a LOD of 0.86 ng/mL, which was three orders of magnitude lower than that of the colorimetric PLFS (Fig. 20a).67 In order to further enhance SERS signals, a gold nano-pyramid array chip was integrated into the detection zone of the PLFS (Fig. 20b).291 By covering the inorganic Au nano-array substrate with a surfactant bridge, the conjugate of SERS probes and analytes can flow smoothly throughout the chip. The “hot spots” formed between the gold nanostars in SERS probes and the gold nano-pyramid array created a hierarchical 3D plasmonic field, which greatly enhanced the SERS signals and achieved a LOD down to 5.0 pg mL−1 toward the TBI S-100β biomarker in blood plasma. This portable SERS-PLFS system showed a comparable LOD with ultrasensitive ELISA assays but in a much shorter assay time (∼30 min for PLFS vs. 2 h for ELISA). Furthermore, Wu's group integrated a blood plasma separation unit into a SERS-PLFS following the principle of salt-induced aggregation of blood cells (Fig. 20c).292 Such a SERS-PLFS has directly detected carcinoembryonic antigen (CEA), a cancer protein biomarker, in a drop of whole blood (30 μL) at a LOD of 1.0 ng mL−1. This portable “all-in-one” SERS-PLFS system can serve as a POCT tool for testing finger-prick blood samples, assisting in vitro diagnostics (IVD) in resource-limited settings. This SERS-PLFS can be adapted easily to detect other protein biomarkers in blood by replacing the antibodies.
Integration of fluorescence into PLFS. Fluorescent organic dyes and semiconducting quantum dots (QDs) have been used as fluorescent probes in the PLFS.288,293–295 Organic dyes usually suffer from low quantum yield, photobleaching and blinking/quenching. To avoid such problems, QDs have been explored in the PLFS. A PLFS integrated with dsDNA-functionalized QDs was developed to measure HIV-DNA in human serum.296 However, the small size of the QDs limits their conjugation with antibodies, especially for proteins over 100 kDa due to the steric effect. In addition, binding with large antibodies would possibly change the surface properties of the QDs and even cause instability in aqueous solutions. Macromolecule-coated nanobeads were thus introduced to not only stabilize the QDs during detection but also increase the fluorescence intensity in each entity. For instance, the dual-color CdSe/ZnS–COOH QDs were immobilized onto SiO2 nanoparticles via polyethyleneimine (PEI)-mediated electrostatic adsorption, and conjugated with antibodies.293 Such a fluorescent PLFS was used to quantify two sepsis biomarkers in human serum simultaneously, namely, procalcitonin (PCT) and C-reactive protein (CRP) (Fig. 21a).
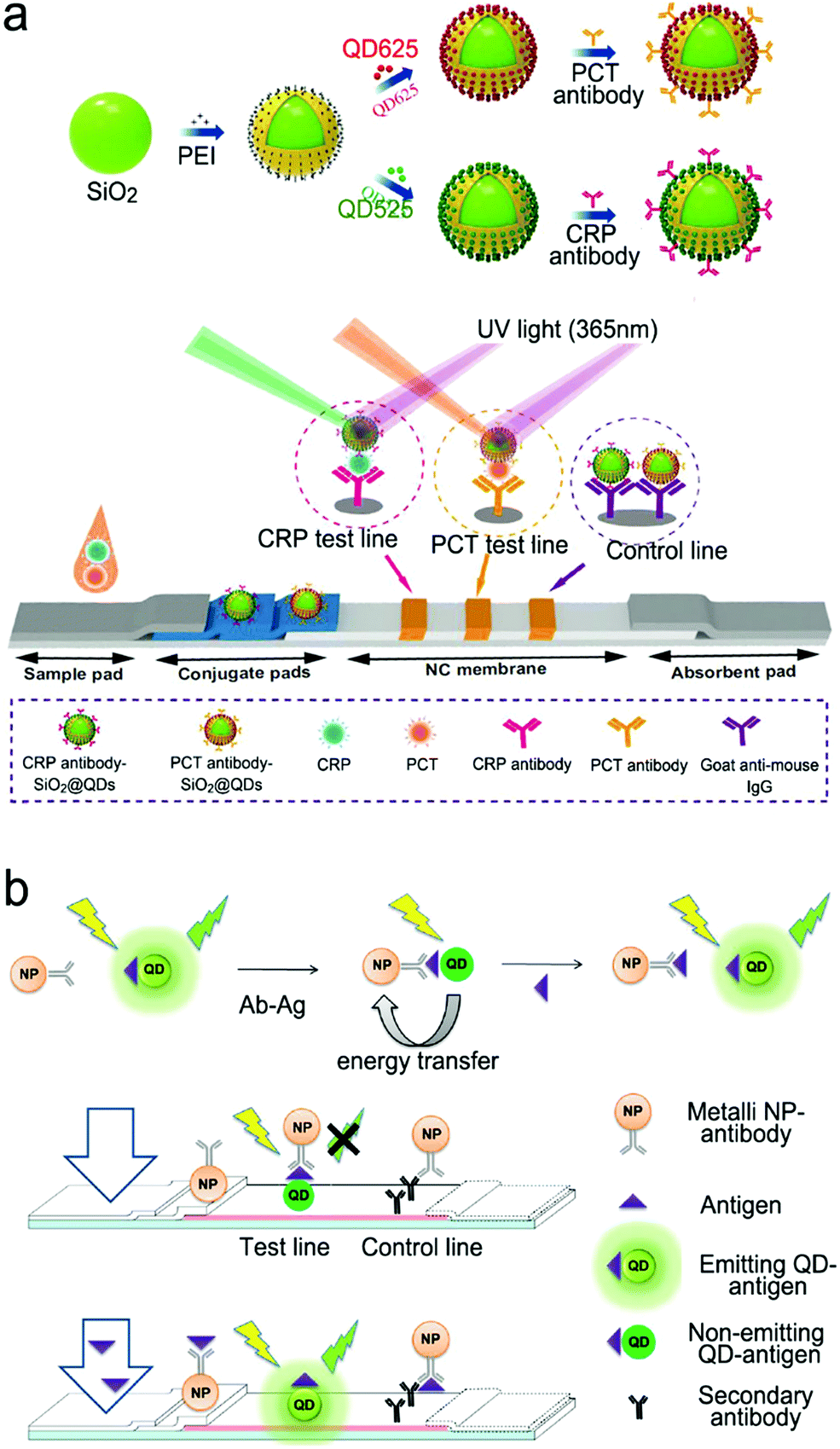 | ||
| Fig. 21 Fluorescent PLFS. (a) PLFS using dual QD nanobeads for the simultaneous detection of two sepsis biomarkers. (b) Competitive assay based on the “turn-on” fluorescence signal in PLFS. Reproduced with permission from (a) ref. 293, copyright 2020, Springer Nature; (b) ref. 295, copyright 2018, Springer Nature. | ||
FRET is of particular interest to PLFS because it can reduce the background signals. A quencher can either “turn-on” or “turn-off” fluorescence.295,297 For example, an immunoassay PLFS utilized FRET to detect carcino embryonic antigen where fluorescein isothiocyanate (FITC)-labeled antibodies were initially pre-immobilized onto the test zone of the PLFS.297 When analytes were present, the Au NPs functionalized with detection antibodies were brought in proximity to the fluorescent dye via the formation of a classic sandwich structure. Thus, the fluorescence of FITC was quenched by Au NPs via FRET. In addition, the “turn-on” FRET PLFS was developed for the measurement of fumonisin mycotoxins based on the competitive mechanism (Fig. 21b).295 In this design, the CdSe/ZnS QDs functionalized with antigen molecules were coated on the detection line of the PLFS where the metal NPs with antibodies stayed close to the QDs via the antigen–antibody interaction, keeping the quenching of QD fluorescence prior to detection. When analytes were present on the test line, they formed a complex with the metal NPs, which pushed the metal NPs from the QDs as the fluid flows, turning on the fluorescence of the QDs.
Engineering PLFS toward POCT – “on-strip” blood plasma separation. It is challenging to enable the testing of finger-prick blood samples with PLFS in POC settings. A promising route is to combine “on-strip” plasma separation and NIR fluorescence or SERS into a single paper-based microfluidic strip to create an “all-in-one” device. Filtration is a common approach for “on-strip” plasma separation in PLFS. Filter membranes with a pore size smaller than 2.5 μm have been placed in the sample pad to block blood cells.224,298,299 However, such small pores hinder the flow of plasma through the membrane. Hence, various agglutination reagents, such as agglutination antibody, salt and chitosan, have been used for the surface-modification of large pore membranes to induce aggregation of blood cells.291,300,301 The aggregated blood cells are too large to pass through the membrane while plasma can do.230 For example, an assembly of three-layer filtration membranes was treated with salts and then integrated into a SERS-PLFS.291 The first layer was functionalized with salts, which impaired the electric double-layer of the blood cell surface, causing deformation and aggregation of blood cells due to the osmotic pressure. The surfactant coated on the second layer accelerated the liquid flow to the third layer where salts held the residual blood cells. Such an “on-strip” plasma separation unit enabled the PLFS to directly measure whole blood samples.
Engineering PLFS toward POCT – tunning fluid. The flow rate is an important factor governing the performance of PLFS, which can be regulated by designing different path patterns and lengths.289,302 Computational simulation was combined with experimental analysis to investigate the transport processes in simple two-dimensional geometries and assisted optimization for sequential reagent delivery for sample pretreatment and analysis.303 In addition, valves were constructed for flow control and reagent loading.204,299,304,305 Paraffin wax, PDMS droplets or dissolvable reagents (e.g., sugar) were deposited on the paper surface as hydrophobic barriers to control the flow direction. Surfactant treatment can accelerate the flow.289,305 To slow down the flow, a cellulose pad was placed on the upstream of the test line as a shunt for time delay, tuning the flow rate by varying its size and number.305 Apart from these passive methods, active valves were built into PLFS by folding, stacking or cutting papers to initiate or stop the flow.304,306–309 For example, a paper-based device contained three parts and each part was loaded with different reagents. The loaded liquid can move to other parts by folding the specific part.310 Inspired by electrowetting on dielectrics, the “on-strip” electrical valves were fabricated by inkjet-printing and spraying technology on a paper-based device. The dielectric surface in the valve can be switched from hydrophobicity to hydrophilicity under an applied bias. Thus, the ionized sample solution can stop or flow by the external bias.
Engineering PLFS toward POCT – reshaping the overall structure. Besides the basic/classic structure shown in Fig. 20a, a lot of varieties have been developed to either improve the performance or realize new functions. An intriguing example is the origami paper-based devices that transfer and mix sample and reagents by folding papers.2,310,311 As shown in Fig. 22a,310 an origami device was composed of three layers where the target DNA, a short ssDNA with a quencher (Q1) and a long ssDNA with a fluorophore (F1) were loaded on each layer, respectively. Because Q1 was shorter than the target DNA, the target showed priority to hybridize with F1 compared with Q1, and the remaining F1 strands were complementary to Q1. The amount of the target DNA was proportional to the intensity of fluorescence.
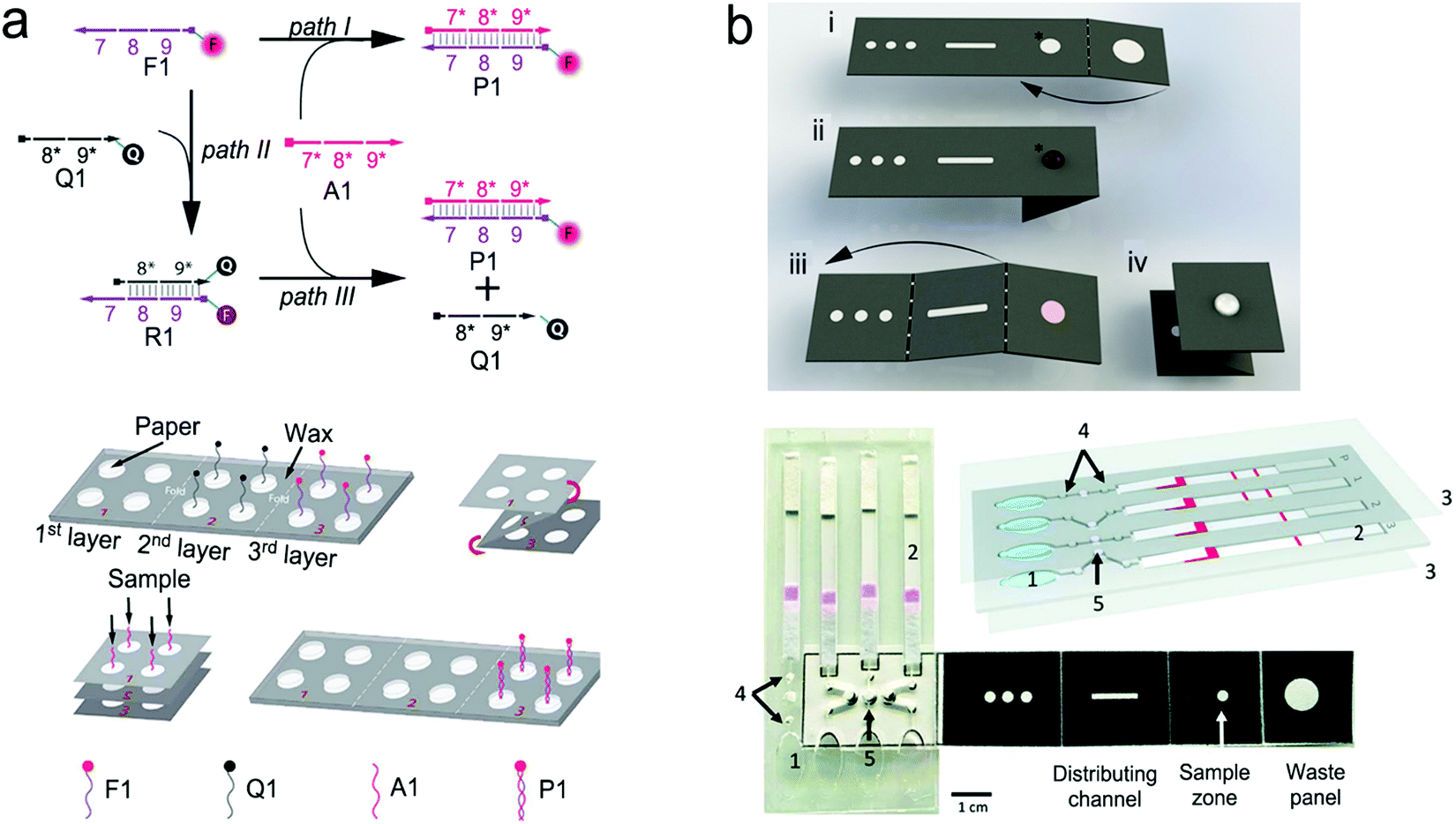 | ||
| Fig. 22 Advanced structures of PLFS. (a) An origami paper-based device for nucleic acid detection. (b) Hybrid device nucleic acid extraction, amplification and detection by the combination of an origami paper for the extraction of nucleic acid, a plastic microfluidic device for the LAMP reaction and a paper-based strip for the detection of malaria DNA. Reproduced with permission from (a) ref. 310, copyright 2013, American Chemical Society; (b) ref. 2, copyright 2019, National Academy of Sciences. | ||
Additionally, nucleic acid amplification sometimes needs to be conducted at a relative high temperature. For example, loop-mediated isothermal amplification (LAMP) of nucleic acids is typically performed at around 72 °C, which is difficult to be carried directly on the paper surface. Hence, a paper-based strip was combined with microfluidic PDMS to create hybrid structures, which not only reduced sample consumption but also enabled more functions.308,309,312–314 Phillips et al. modified the sample pad of PLFS to make a LAMP unit for the detection of HIV-1 virus in blood.315 The HIV-1 viral particles were separated by a size-selective filtration membrane, and migrated in a buffer solution to a polyether sulfone membrane preloaded with the LAMP reagents to perform LAMP at room temperature. The amplicons generated by fluorescence-labeled primers flowed along the downstream paper strip, finally captured on the test line for fluorescence recording. The whole process was finished within 90 minutes, achieving a LOD of 2.3 × 107 copies per mL in whole blood. Recently, Cooper's group developed a multiplex microfluidic diagnostic platform using hybrid devices.2 An additional example of a hybrid device is shown in Fig. 22b.2 The origami structure, plastic microfluidic modules and paper-based lateral flow strips were combined to create a hybrid device for the detection of malaria in blood. The liquid sample was pretreated with cell lysis. The released DNA was captured onto magnetic beads, and then loaded onto the sample zone of the origami paper part where excessive buffer and other residues were removed by the capillary force of the waste panel. The DNA bounded with the magnetic beads were distributed into three spots through distributing channels and transferred into different LAMP chambers. In each chamber, the LAMP reaction proceeded independently by adding different primers labeled with fluorophores. After that, finger pumps were used to push the liquid to flow to the paper test strips. As a result, it can detect two different types of malaria DNA together with the positive and the negative controls simultaneously.
5. NIR fluorescence and SERS imaging of live cells and tissues
NIR fluorescence and SERS take advantage of biological transparency windows, feature the extended penetration depth and the reduced interference from cells and tissues, and they are non-invasive techniques. Therefore, they find increasing applications in the real-time imaging of internal processes of living cells, tissues and organisms for diagnostic or biomedical research. This section gives an overview of the instruments, optical probes, imaging activation, and applications.5.1 Instruments
Developed from a basic fluorescence microscope, a confocal laser scanning microscope (CLSM) features controllable depth scanning, serial optical sectioning, spatial filtering, and adjustable magnification.323,324 The key to success is the application of pinholes located between the laser source and excitation filter (called the light source pinhole), and between the emission filter and eyepiece or detector (called the detector pinhole), respectively. With the addition of a light source pinhole, a point-liked laser is formed and reaches the specimen. Then, the emission fluorescence from the in-focus specimen passes through the detector pinhole to the detector. Otherwise, the fluorescence from the out-of-focus specimen is reflected. Thus, the use of pinhole not only realizes the high signal-to-noise ratio but also acquires multiple two-dimensional (2D) images at different depths in a sample, followed by reconstruction to form a three-dimensional (3D) image. Particularly, for uneven samples, depth scanning can collect and integrate serial optical sections into a single image, which help eliminate out-of-focus imaging. In addition, the confocal laser scanning microscope has a zoom function that adjusts magnification by altering the scanning rate and scanning area of the laser without changing the objectives. Given that a high zoom factor would lead to photobleaching, it is advisable to achieve a balance between the zoom factor and imaging. In terms of thick samples, such as embryos and brain slices, confocal microscopy with single-photon excitation is weak in serially scanning in deeper planes, thus multiphoton microscopy has emerged.325 A two-photon excitation fluorescence microscope depends on the simultaneous absorption of two photons, followed by non-radiative relaxation and fluorescence emission.326 It shows that the excitation wavelength is longer than the emission wavelength, which is opposite to the traditional fluorescence microscope. More importantly, NIR excitation light is commonly used in a two-photon microscope, leading to deep tissue penetration, low background signals and reduced photobleaching. Despite these advantages and commercial applications of a confocal microscope, the theoretical resolution limit is around 200 nm within the visible light spectrum because of the far-field diffraction limit.327 Fortunately, a series of super resolution techniques and microscopies has been introduced, such as 4Pi and I5M, stimulated emission depletion (STED), saturated structure illumination (SSIM), single-molecule localization and far-field microscopy.328–331 It is desirable that the nanometer scale can be resolved using a commercial fluorescence microscope in the near future.
In addition, an optical fiber-based imaging system is one of the in vivo imaging tools, which is referred to fluorescence microendoscopy (FME).334 The optical fiber in FME can be inserted into a tissue in a minimally invasive way, and flexibly maneuvered in the hollow tissue cavities. Various types of optical fibers are used in FME, such as fiber bundle, single-mode fiber, double-clad fiber, multi-mode fiber and graded index microlens.335 For example, a fiber bundle generally consists of ∼100000 individual step index fibers, which are closely packed with a total diameter, ranging from hundreds of micrometers to a few millimeters. Each core is of high-refractive index and serves as a pixel of an image. Because the arrangement of individual fibers in a fiber bundle is relatively maintained at both ends, the intensity of the image can be transmitted in a pixelated form. The fiber bundle can be introduced into basic fluorescence and confocal laser scanning as well as a two-photon excited fluorescence microscope. Specifically, each core of fiber bundles could also act as a pinhole in a confocal instrument. In a typical FME, the excitation light from the light source (e.g. lasers, LEDs and lamps) is transmitted to the distal end of the fiber bundle, illuminating the sample. The fluorescence emission light from the sample is then gathered by the fiber bundle, followed by sequentially passing through the objective, the dichroic mirror and the emission filter, and finally reaches a CCD camera.
Considering that the spatial resolution of a confocal Raman microscope is limited to the submicron scale, hyperspectral imaging is combined with Raman scattering into a single microscope to perform dual functions. Hyperspectral imaging can enhance the spatial resolution to tens of nanometers. In addition, Raman scattering is incorporated with a scanning near-field optical microscopy (SNOM) because SNOM can reach the nanometer scale resolution. In a single instrument, SNOM can be used to map the morphology of a sample and Raman scattering can be used to acquire chemical information.
Recent Raman microscopes are mainly used for in vitro imaging for cultured cells and biopsy tissues. The reason is that a sample under Raman microscopes needs to be focused within a specific length to the object. It is difficult to collect Raman scattering with a controlled distance under in vivo conditions. Also, to obtain a high-resolution image in a large area, a Raman microscope should scan the target area sequentially, which is a quite time-consuming process. These drawbacks impede its development in in vivo imaging. Different from Raman imaging, fluorescence emission from a sample is much easier to collect from a large-area or a large sized sample.
5.2 Materials for NIR fluorescence probes
The general design and materials of NIR fluorescence probes have been described in Section 2.4. Compared to SERS imaging, there are much more varieties of materials for NIR fluorescence bio-imaging. Hence, this sub-section is focused on materials for NIR fluorescence probes. For imaging of live cells and tissues, NIR fluorescence probes require a high molar extinction coefficient, high quantum yield, low- or even non-toxicity and good photostability in an aqueous environment. Particularly, in vivo imaging requires deep penetration and low background noise. In the past few decades, numerous NIR probes have been exploited, which can be classified into three types, namely, organic dyes, nanomaterials and proteins.Similarly, biocompatible and biodegradable carbon quantum dots (CQDs) are alternatives to traditional metal-based QDs. Their absorption and NIR emission spectra can also be tuned via size control, surface modification and heteroatom doping. Furthermore, carbon quantum dots display excitation-wavelength-dependent fluorescence due to the “giant red-edge effect”.352 Hence, they are able to present different fluorescence emission bands in imaging under different excitation wavelengths. Other carbon materials such as graphene oxides and carbon nanotubes (CNTs) have been explored for bio-imaging. Pristine graphene with solely sp2-carbon atoms do not show fluorescence. Graphene oxides and graphene quantum dots (GQDs) consist of a mixture of sp2 and sp3 carbon atoms, thus they have an opened band gap; their surface possess hydrophilic groups such as hydroxyl, carboxyl and carbonyl groups that make themselves water soluble. Like carbon quantum dots, free-standing graphene oxides and graphene quantum dots exhibit excitation-wavelength-dependent fluorescence, and they are photostable and biocompatible.
Recently halide perovskite nanocrystals have emerged for NIR fluorescence imaging.28,353 Among the vast perovskite families, lead-based halide perovskite (LHP) nanocrystals have attracted great interest due to their high quantum yield. It has been reported that lead-based halide perovskite nanocrystals achieved a photoluminescence quantum yield of up to 90% without any post-treatment.354 Lead-based halide perovskites have a very narrow fluorescence emission band that can be tuned by either their composition or/and size. However, halide perovskite nanocrystals typically contain toxic elements, and may exhibit poor stability in an aqueous solution.
5.3 Signal transduction
For fluorescence or SERS imaging of live cells and tissues, there are three common signal transduction modes/principles: (i) direct imaging with non-targeting optical probes, (ii) imaging with targeting optical probes, and (iii) activatable imaging with triggering modes. | ||
| Fig. 23 Direct imaging of tissues with stained NIR fluorescence or SERS probes. (a) In vivo fluorescence imaging of tumor tissues through the tail vein injection of PbS/CdS QDs into a mouse with xenograft MC38 tumors. (1) Schematic structure of PbS/CdS QDs probe. (2) TEM image of QDs. (3) Schematic illustration of non-invasive in vivo confocal imaging of the mouse through the skin; (4) in vivo wild-field fluorescence imaging of tumor tissues after the tail vein injection of QDs. Scale bar: 1 mm; (5) confocal fluorescence imaging of tumor vessels at a depth of ∼180 μm. Scale bar: 200 μm. (b) SERS imaging of characteristic biomolecules in cells. (1) SERS signals from absorbed molecules and changes with their distance to gold nanoparticles. (2) A photograph of a macrophage cell using a dark-field microscope and the gold nanoparticles shown as a small white spot (white arrow) under the dark-field microscope. (3) SERS spectra obtained from (a) the three characteristic peaks of phosphate at 997 cm−1, CH2 and CH3 at 1457 cm−1, and Amid II at 1541 cm−1. (4) Trajectory of gold nanoparticles under the dark-field microscope. (5) False-color mapping of the molecular distribution based on the trajectory of the nanoparticles (4). Reproduced with permission (a) ref. 360, copyright 2018, National Academy of Sciences; (b) ref. 361, copyright 2011, American Chemical Society. | ||
SERS probes without specific targeting moieties can also be used in tumor imaging by utilizing the permeability and retention effect. For example, PEGlyated sandwiched SERS nanostars with a LOD down to 1.5 fM were successfully used for the imaging of premalignant lesions of pancreatic and prostatic neoplasias.362 In addition, based on the “finger-printing” features of SERS spectra, SERS imaging can be directly used for the identification of cells and monitoring of molecular evolution in cells or tissues. After the Raman dye-free plasmonic metal nanoparticles entered the cells or tissues, the SERS-active molecules in the living body, such as proteins with chromophore groups (e.g. hemoglobin and myoglobin), nucleic acids, and metabolites (e.g. lactic acid and glucose), can be detected when they were in proximity with the plasmonic nanoparticles. In the meanwhile, a dark-field microscope was employed to map the position of plasmonic metal nanoparticles in living cells, tracking the SERS signals of characteristic biomolecules at different sites of cells, which can continuously monitor intracellular information (Fig. 23b).361 In order to mitigate the uncertainty and uncontrollability of the distribution of nanoparticles in living cells, researchers deposited metal nanoparticles on a solid substrate to form a SERS-active substrate.363 After seeding cells on such a SERS-active substrate, the biomolecules on the cell membrane get close to the substrate, thus obtaining the SERS signals of these biomolecules. The SERS images, which are obtained from the cells on the plasmonic substrate, can monitor the cell state because these biomolecules on the cell membrane play a vital role in regulating the signal pathway, cell behavior and in identifying the cell type and condition. The large amount of data generated in a dynamic system can be analyzed by statistical methods such as principal component analysis (PCA) and multivariate curve resolution (MCR).
 | ||
| Fig. 24 In vivo cancer targeting and SERS detection using the PEGlyated SERS probes conjugated with the single-chain variable fragment (ScFv) antibodies, which can recognize the tumor biomarker epidermal growth factor receptor (EGFR). (a) Schematic illustration of the structure of the PEGlyated SERS probes. (b) In vivo SERS spectra obtained from the targeting and non-targeting SERS probes. (c) Photographs illustrating a 785 nm laser beam focusing on the tumor site or on the anatomical location of the liver. Reproduced with permission ref. 368, copyright 2008, Springer Nature. | ||
In activable SERS imaging, the signal trigger is the environment-sensitive Raman reporter. The signal variation of Raman reporters reflects the physicochemical changes in living cells and tissues. For example, 4-mercaptobenzoic acid (MBA), 2-aminothiophenol and 4-mercaptopyridine are pH-sensitive Raman reporters, while indocyanine green (ICG) is a thermosensitive reporter.369–371
Fluorescence or SERS imaging can be triggered by various operating parameters (e.g. pH, viscosity, temperature) or biomarkers (e.g., small molecules, protein, nucleic acids) in cells and tissues. For example, the pH value plays a vital role in biological processes, such as ion transport, endocytosis and cell growth. Under normal physiological conditions, the pH value is around 7.2 and 7.4 in the cytosolic and extracellular environment, respectively; the pH value in lysosomes and mitochondria is approximately 5.0 and 8.0, respectively. Abnormal pH values may reflect pathological conditions, including cancer and Alzheimer's diseases. For instance, the pH value of lysosomes in the tumor cells becomes more acidic (pHly 3.8–4.7) than the normal one. Fig. 25a illustrates the effect of pH on the ET process resulting from protonation/deprotonation-induced electron distribution.372 Briefly, a pH-responsive probe was constructed using a heptamethine cyanine dye, which had good biocompatibility and a relatively high quantum yield. The NIR fluorophore (heptamethine cyanine derivatives IR783) served as the electron acceptor, and the amine moiety on one indole ring was the electron donor in neutral pH. When the probe was delivered into lysosomes through an organic anion transporting peptide, the acidic environment protonated the amine groups, triggering fluorescence emission. Visualization of a pH change in cancer cells can localize cancer cells specifically, and guide tumor ablation. In addition, Tang et al. developed a pH-triggered imaging method using the PET process, which strongly depends on a pH change from 6.70 to 7.90.373 It was used successfully for the real-time imaging of cellular pH in living HepG2 and HL-7702 cells. However, it is difficult to achieve a broad range of pH detection in these single-electron transfer modes, especially from acidity to alkalinity, to show the overall pH change in cells. A combinational electron transfer-energy transfer method was thus exploited to create an ICT-PET-FRET probe with dual pH-responsive sites.374 Therefore, the probe responded to a wide pH range from 4.0 to 8.0 with high sensitivity. It can be utilized not only in fluorescence imaging of lysosomal pH values with one of the pH sites targeting the lysosome place, but also in therapeutic effect evaluation based on the intracellular pH changes in cells after chloroquine treatment. In addition, taking advantage of the pH change from normal to tumor tissues, researchers have developed pH-sensitive imaging and drug delivery to realize imaging-guided surgery. For example, Song et al. labeled a hydrophilic polymer brush, a pH-sensitive hydrophobic brush and a Raman reporter on the surface of gold nanoparticles to form SERS-encoded amphiophilic nanoparticles (Fig. 25b).375 These nanoparticles can be further self-assembled into vesicles with the anticancer drugs embedded in the hollow cavity. After being tagged with the cancer-targeting ligands on the outer surface of SERS vesicles, these vesicles were successfully bound to the target cancer cells with strong SERS signals due to their assembly structure. Afterwards, the SERS vesicles were transferred into cytosol through endocytosis and the acidic intracellular compartments triggered the pH-responsive disassembly of SERS vesicles to form single gold particles. Compared with the SERS vesicles, these dispersed single gold nanoparticles showed much weaker signal intensity. Simultaneously, the embedded drugs were released with the disassembly of the SERS vesicles.
 | ||
| Fig. 25 Biomarker-triggered imaging. (a) pH activated electron transfer (ET) process in an organic NIR dye for cancer cell visualization. (b) Drug-loaded and targeted plasmonic vesicles for cancer cell targeting, imaging and therapy via pH-sensitive drug release. (c) Viscosity activated in vivo imaging of tumor tissues. Reproduced with permission (a) ref. 372, copyright 2016, The Royal Society of Chemistry; (b) ref. 375, copyright 2012, American Chemical Society; (c) ref. 376, copyright, American Chemical Society. | ||
Viscosity is one of the critical microenvironment parameters, which is associated with the transportation and reactions of biomolecules as well as chemical signals within cells. Viscosity perturbances are caused by diseases and pathologies, such as diabetes, atherosclerosis, cancers and Alzheimer's diseases. As it is an important indicator in disease diagnostics, a series of molecular rotors has been developed to map the cellular viscosity. For example, a molecular rotor, malononitrile moiety, was linked to 4H-chromene groups modified triphenylamine as the NIR fluorophore to form viscosity-sensitive probes. As shown in Fig. 25c, in low-viscosity media, after the fluorophore was excited by a proton, the excited electron was transferred into a twisted excited state due to the rotation of the malononitrile group, which reached a non-emissive decay.376 In contrast, in a high-viscosity media, molecular rotation was restricted, and the ET process was then inhibited, leading to the emissive decay of NIR fluorescence. This method was used to identify cancer, which showed higher viscosity than normal tissues, and to evaluate the efficacy of anticancer drugs.
Imaging can be triggered by the unique features of biomolecules or reactions, such as hydrophobic property of proteins, chain reactions of nucleic acids, or specific chemical reaction of small molecules. Fig. 26a shows the images of nucleic acids using FRET signal transduction and nucleic acid amplification to overcome the problems of intrinsic low-abundance analytes.377 First, the target microRNA-21 (miR-21), a crucial tumor-associated biomarker, was recycled via an Exo-III-assisted assay, and the exposed DNAzyme was further partially hybridized with a fluorophore/quencher pair. After experiencing biocatalysis by Mg2+ in cells, the fluorophore/quencher pair was cleaved, thus the fluorescence of the separated fluorophore was restored. In addition, the base-pairing complementarity can also be used in the intermolecular FRET process. For example, Hwang et al. developed a fluorophore-labeled nucleic acid as an imaging probe whose fluorescence was initially quenched by graphene oxide (GO) via FRET.378 After the probes were internalized into cancer cells through CD44 receptor-mediated endocytosis, the nucleic acid of the probe was specifically hybridized with the target when encountering the target miRNA, leading to the detachment of the probes from graphene oxide and recovery of fluorescence. Fig. 26b reveals an AIE imaging process activated by amyloid-β (Aβ) plaques, an important pathological hallmark for the early diagnosis of Alzheimer's disease.379 Initially, the AIE probe was under the “fluorescence-off” state due to the hydrophilicity of the sulfonate group of the probe. The fluorescence of the probe was turned on when binding to the Aβ aggregates via the N,N-dimethylamino group in the probe.
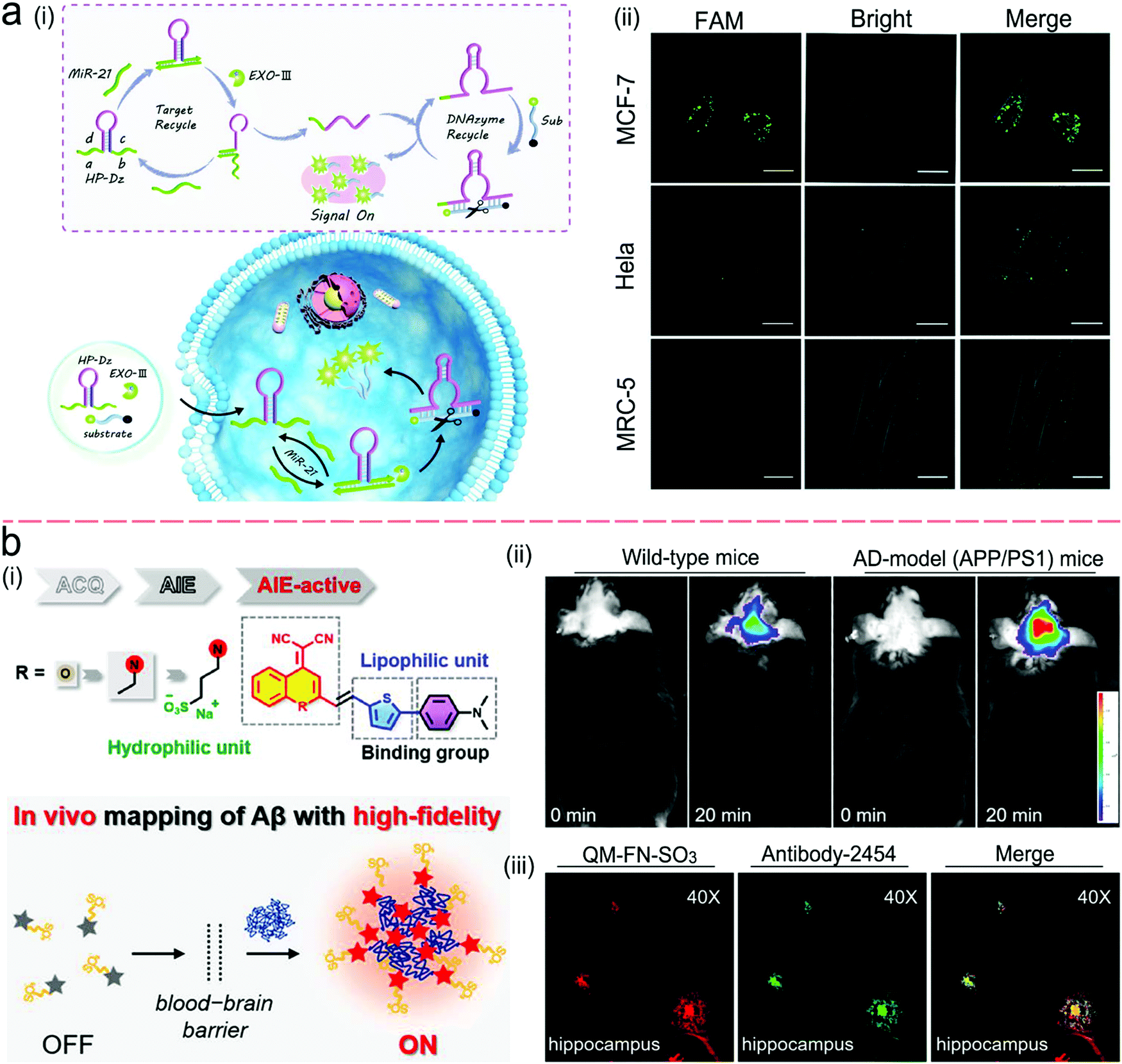 | ||
| Fig. 26 (a) MicroRNA-21 (miR-21)-activated imaging through a FRET process together with Exo-III-assisted nucleic acid amplification. (i) Schematic illustration of the imaging process; (ii) fluorescence imaging of miR-21 in breast cancer cells (MCF-7) with high miR-21 expression, HeLa cells with low miR-21expression and MRC-5 cells with no miR-21expression. The scale bar is 20 μm. (b) Amyloid-β (Aβ) plaque-activated AIE process for in situ imaging of Alzheimer's disease (AD). (i) Schematic illustration of synthesis of AIE probe and imaging process; (ii) in vivo imaging of Aβ deposition in the AD model (APP/PS1transgenic) mice in comparison to normal wild-type mice; (iii) ex vivo fluorescence imaging of brain slices from APP/PS1 mice after the injection of AIE probes (QM-FN-SO3) with the colocalization of Aβ plaques stained with anti-Aβ antibody-2454. Reproduced with permission from (a) ref. 377, copyright 2020, American Chemical Society; (b) ref. 379, copyright 2019, American Chemical Society. | ||
In cells, there exist a lot of chemically active molecules, such as reactive oxygen species (ROS), reactive nitrogen species (RNS), enzymes and metal ions. Thus, activable imaging can also be triggered by chemical reactions. Fig. 27 shows a NIR fluorescence imaging process activated by an alkaline phosphatase (ALP) reaction.380 Initially, fluorescence of merocyanine was quenched by the modified phosphate group via the electron transfer process. When it comes to ALP-positive tumor tissues, the membrane-bound ALP dephosphorylated the phosphate group, leading to the emission of NIR fluorescence. In this design, fluorescence imaging was combined with magnetic resonance imaging (MRI) through the modification of paramagnetic DOTA-Gd chelate to the probe with a hydrophobic dipeptide liner. After being dephosphorylated, the hydrophobic interaction of dipeptide promoted the molecular self-assembly into magnetic nanoparticles.
 | ||
| Fig. 27 In vivo bimodal imaging for tumor tissue visualization: alkaline phosphatase (ALP) reaction-triggered ET imaging combined with magnetic resonance imaging (MRI). (a) Chemical mechanism of the ALP reaction-triggered NIR fluorescence imaging and the self-assembly for further signal amplification; (b) schematic illustration of in vivo bimodal imaging of ALP-positive tumor cells; (c) ALP reaction-triggered NIR fluorescence imaging (left) and MRI imaging (right). Reproduced with permission from ref. 380, copyright 2019, American Chemical Society. | ||
6. Remarks and perspective
In summary, further efforts are needed to design and fabricate plasmonic nanostructures to achieve the effective coupling of plasmon with fluorescence or SERS to amplify signals. These efforts will include the commercialization of plasmonic NIR fluorescence and SERS probes. These probes should be inexpensive, water-soluble, “bright” (strong intensity), stable in storage, shipment and operation, and repeatable from batch to batch.44 Commercial availability of these plasmonic probes is the key step in boosting the application of NIR fluorescence and SERS in POCT and bio-imaging.The pregnancy test strip is the most successful example of a commercial POCT device. Like pregnancy test strips, most commercial lateral flow assays are based on colorimetric transduction. Such devices are inexpensive and can be used for rapidly and qualitatively detecting a relatively high level of analytes. However, they suffer from low sensitivity and severe interference from complex sample matrices such as blood. In contrast, NIR fluorescence and SERS microfluidic devices show great potential in sensitive and quantitative measurements of a low level of analytes in complex sample matrices. However, such POCT devices typically require the integration of a complex plasmonic nanostructure into a microfluidic chip in order to amplify NIR fluorescence or SERS signals. Although significant progress has been made in such a direction, there remain large technical barriers in fabricating and integrating 1D, 2D or 3D plasmonic nanostructures into microfluidic POCT devices.291
Today there are a lot of commercial POCT devices based on immunoassays with antibodies as molecular recognition elements. These immunoassays have been successfully used for detecting illicit drugs, HIV antibodies, biological warfare agents, pathogens and so on. They are typically used for the qualitative measurement of analytes in relatively simple sample matrices such as tap water, river water, saliva and urine. So far, few POCT devices are commercially available for the direct and quantitative measurement of analytes in finger-prick blood samples because whole blood is a much more challenging sample matrix as compared to buffer solutions, water, saliva and urine. Integration of a plasma separation unit and a NIR fluorescence or SERS detection platform into a single microfluidic or paper lateral flow strip is an effective solution to blood analysis in POC settings.292
There are few commercial POCT devices for nucleic acid testing. For example, no commercial POCT device is now available for reliable testing of SARS-COV-2 viral RNA in saliva or nasal swab samples in POC settings. One of the reasons is that the levels of nucleic acid biomarkers of interest, such as DNA, RNA and micro-RNA, are typically too low to be detected directly with a sensor. To enable nucleic acid testing in POC settings, both the amplification and detection of nucleic acid should be performed with a single lab-on-chip device. In some cases, an on-chip lysis and nucleic acid extraction module also need to be integrated into a POCT device. In short, nucleic acid POCT devices are complicated and quite challenging. Many researchers are tackling such challenges now. It is believed that more and more POCT devices will appear in the market for rapidly testing nucleic acids.
Besides test chips/strips/cartridges, readers are critical for POCT testing, and many inexpensive handheld fluorescence readers are commercially available for directly reading fluorescence signals from test chips/strips/cartridges. However, inexpensive handheld SERS readers are not commercially available to fit into test chips/strips/cartridges, which has hindered the commercialization of SERS-based POCT systems.
Smartphones show great potential in recording signals from fluorescence and SERS POCT test chips/strips/cartridges.381 One option is the integration of an optical detector into an existing smartphone, enabling the direct reading of signals with the smartphone. Another option is the connection of a smartphone with an existing reader via Bluetooth, enabling the wireless operation of the reader with the smartphone using an app, allowing for the wireless transmission of data to a doctor's office or a central station via a wireless 4G or 5G network. Such readers, which are wirelessly connected to smartphones, could be operated by someone at a remote central station.
Incorporation of numerous POCT devices, ambulances, clinics and hospitals into a wireless sensor network or a cloud-based network is an important direction in the future. Such a cloud-based network will play important roles in multi-facets of public health and security in smart-cities, -towns, or -countries. For example, it will allow for the real-time monitoring for the reoccurrence or of the treatment process of chronic diseases. And it can be used to track acute, infected, or asymptotic COVID-19 patients. Furthermore, it can be employed to monitor the occurrence and progression of pandemic or epidemic diseases.
Besides in vitro POCT, NIR fluorescence and SERS imaging can achieve fast screening and real-time tracking and monitoring of specific targets in living cells and tissues. They can greatly overcome the drawbacks of current large diagnostic instruments, such as high cost, time consumption and requirement of professional training. It is worth noting that longer-wavelength probes exhibit weaker signals than shorter-wavelength counterparts. However, the reduced light absorption and scattering by tissues and the depressed tissue autofluorescence not only enable deeper penetration but also allow for longer acquisition time with a greater signal-to-background ratio. On the other hand, in the case of rapid screening demand such as image-guided surgical applications, use of shorter NIR wavelengths and tolerance of higher background noise may be necessary for achieving stronger signals.
Some of the technical barriers for bio-imaging are the biocompatibility and biodegradability of NIR fluorescence and SERS probes. It is highly recommended to not only construct biocompatible and biodegradable probes, but also to reveal their mechanisms of action and metabolism, which can provide clear guidelines for choosing appropriate materials and structures to design probes. In addition, improvement in spatial resolution of imaging is an important research direction. This will benefit the site-specific detection or mapping of physiological or pathological processes in live cells, tissues and organs. Moreover, theranostics, which combines therapy with imaging functions into a single entity to form therapeutic agents, shows great potential. It will allow for time-solved and space-resolved tracking and adjustment of medical intervention.
It is estimated that the global POCT market will increase quickly from $29.5 billion in 2020 to $50.6 billion by 2025.382 The market will drive the advancement in POCT techniques. With the increasing need for sensitive and quantitative measurements of analytes in POC settings, the market share will shift from colorimetric and electrochemical devices to NIR fluorescence and SERS devices. To the best of our knowledge, we do not have quantitative data about the market value of NIR fluorescence and SERS bio-imaging. The technological development in NIR fluorescence and SERS bio-imaging will boost the growth of the market in these areas in the future.
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work is partially supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (1U01NS119647). Wu is partially supported by the Armstrong-Siadat Professorship Endowment. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- D. Wang, S. He, X. Wang, Y. Yan, J. Liu, S. Wu, S. Liu, Y. Lei, M. Chen, L. Li, J. Zhang, L. Zhang, X. Hu, X. Zheng, J. Bai, Y. Zhang, Y. Zhang, M. Song and Y. Tang, Nat. Biomed. Eng., 2020, 4, 1150–1158 CrossRef CAS PubMed.
- J. Reboud, G. Xu, A. Garrett, M. Adriko, Z. Yang, E. M. Tukahebwa, C. Rowell and J. M. Cooper, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4834–4842 CrossRef CAS PubMed.
- M. K. Yuzon, J.-H. Kim and S. Kim, BioChip J., 2019, 13, 277–287 CrossRef CAS.
- S. Chaiyo, E. Mehmeti, W. Siangproh, T. L. Hoang, H. P. Nguyen, O. Chailapakul and K. Kalcher, Biosens. Bioelectron., 2018, 102, 113–120 CrossRef CAS PubMed.
- L. Wu and X. Qu, Chem. Soc. Rev., 2015, 44, 2963–2997 RSC.
- B. Ning, T. Yu, S. Zhang, Z. Huang, D. Tian, Z. Lin, A. Niu, N. Golden, K. Hensley, B. Threeton, C. J. Lyon, X.-M. Yin, C. J. Roy, N. S. Saba, J. Rappaport, Q. Wei and T. Y. Hu, Sci. Adv., 2021, 7, eabe3703 CrossRef CAS PubMed.
- J. Meng, X. Tang, B. Zhou, Q. Xie and L. Yang, Talanta, 2017, 164, 693–699 CrossRef CAS PubMed.
- Y. Song, B. Lin, T. Tian, X. Xu, W. Wang, Q. Ruan, J. Guo, Z. Zhu and C. Yang, Anal. Chem., 2019, 91, 388–404 CrossRef CAS PubMed.
- X. Gao, H. Xu, M. Baloda, A. S. Gurung, L.-P. Xu, T. Wang, X. Zhang and G. Liu, Biosens. Bioelectron., 2014, 54, 578–584 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- M. Li, H. Gou, I. Al-Ogaidi and N. Wu, ACS Sustainable Chem. Eng., 2013, 1, 713–723 CrossRef CAS.
- Y. Song, Y. Y. Huang, X. Liu, X. Zhang, M. Ferrari and L. Qin, Trends Biotechnol., 2014, 32, 132–139 CrossRef CAS PubMed.
- S. F. Berlanda, M. Breitfeld, C. L. Dietsche and P. S. Dittrich, Anal. Chem., 2021, 93, 311–331 CrossRef CAS PubMed.
- C. H. Quek and K. W. Leong, Nanomaterials, 2012, 2, 92–112 CrossRef CAS PubMed.
- F. Yang, Q. Zhang, S. Huang and D. Ma, J. Mater. Chem. B, 2020, 8, 7856–7879 RSC.
- E. Hemmer, A. Benayas, F. Legare and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- L. Sordillo, Y. Pu, S. Pratavieira, Y. Budansky and R. Alfano, J. Biomed. Opt., 2014, 19, 056004 CrossRef PubMed.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, e1900321 CrossRef PubMed.
- F. Scholkmann, S. Kleiser, A. J. Metz, R. Zimmermann, J. Mata Pavia, U. Wolf and M. Wolf, NeuroImage, 2014, 85(part 1), 6–27 CrossRef PubMed.
- C. Swanson and A. D'Andrea, Clin. Chem., 2013, 59, 641–648 CrossRef CAS PubMed.
- Y. Haga, H. David Kay, M. A. Tempero and R. K. Zetterman, Clin. Biochem., 1992, 25, 277–283 CrossRef CAS PubMed.
- O. O. Abugo, R. Nair and J. R. Lakowicz, Anal. Biochem., 2000, 279, 142–150 CrossRef CAS PubMed.
- A. N. Semenov, B. P. Yakimov, A. A. Rubekina, D. A. Gorin, V. P. Drachev, M. P. Zarubin, A. N. Velikanov, J. Lademann, V. V. Fadeev, A. V. Priezzhev, M. E. Darvin and E. A. Shirshin, Molecules, 2020, 25, 1863 CrossRef CAS PubMed.
- H. Zeng, C. MacAulay, D. I. McLean and B. Palcic, J. Photochem. Photobiol., B, 1997, 38, 234–240 CrossRef CAS.
- R. A. Álvarez-Puebla, J. Phys. Chem. Lett., 2012, 3, 857–866 CrossRef PubMed.
- A. Miyawaki, Nat. Methods, 2016, 13, 729–730 CrossRef CAS PubMed.
- I. Lignos, V. Morad, Y. Shynkarenko, C. Bernasconi, R. M. Maceiczyk, L. Protesescu, F. Bertolotti, S. Kumar, S. T. Ochsenbein, N. Masciocchi, A. Guagliardi, C. J. Shih, M. I. Bodnarchuk, A. J. deMello and M. V. Kovalenko, ACS Nano, 2018, 12, 5504–5517 CrossRef CAS PubMed.
- X.-H. Chang, J. Zhang, L.-H. Wu, Y.-K. Peng, X.-Y. Yang, X.-L. Li, A.-J. Ma, J.-C. Ma and G.-Q. Chen, Micromachines, 2019, 10, 422 CrossRef PubMed.
- A. L. Antaris, H. Chen, S. Diao, Z. Ma, Z. Zhang, S. Zhu, J. Wang, A. X. Lozano, Q. Fan, L. Chew, M. Zhu, K. Cheng, X. Hong, H. Dai and Z. Cheng, Nat. Commun., 2017, 8, 15269 CrossRef CAS PubMed.
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS PubMed.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2015, 140, 386–406 RSC.
- J. Dostálek and W. Knoll, Biointerphases, 2008, 3, Fd12–Fd22 CrossRef PubMed.
- J. R. Lakowicz, Plasmonics, 2006, 1, 5–33 CrossRef CAS PubMed.
- N. W. Ashcroft and D. N. Mermin, Solid State Physics, Cengage Learning, 1976 Search PubMed.
- V. M. Shalaev and S. Kawata, Nanophotonics with Surface Plasmon, Elsevier, 2007 Search PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- A. M. Schwartzberg and J. Z. Zhang, J. Phys. Chem. C, 2008, 112, 10323–10337 CrossRef CAS.
- H. Tang, C. Zhu, G. Meng and N. Wu, J. Electrochem. Soc., 2018, 165, B3098–B3118 CrossRef CAS.
- J. M. McMahon, S. Li, L. K. Ausman and G. C. Schatz, J. Phys. Chem. C, 2011, 116, 1627–1637 CrossRef.
- M. Li, S. K. Cushing, J. Zhang, J. Lankford, Z. P. Aguilar, D. Ma and N. Wu, Nanotechnology, 2012, 23, 115501 CrossRef PubMed.
- M. Wang, B. B. Rajeeva, L. Scarabelli, E. P. Perillo, A. K. Dunn, L. M. Liz-Marzan and Y. Zheng, J. Phys. Chem. C, 2016, 120, 14820–14827 CrossRef CAS PubMed.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- I. Gryczynski, J. Malicka, Z. Gryczynski and J. R. Lakowicz, Anal. Biochem., 2004, 324, 170–182 CrossRef CAS PubMed.
- M. Käll, H. Xu and P. Johansson, J. Raman Spectrosc., 2005, 36, 510–514 CrossRef.
- S. D. Christesen, Appl. Spectrosc., 1988, 42, 318–321 CrossRef CAS.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- W. E. Doering and S. Nie, J. Phys. Chem. B, 2002, 106, 311–317 CrossRef CAS.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef CAS PubMed.
- J. Kim, Y. Jang, N.-J. Kim, H. Kim, G.-C. Yi, Y. Shin, M. H. Kim and S. Yoon, Front. Chem., 2019, 7, 582 CrossRef CAS PubMed.
- G. C. Schatz, M. A. Young and R. P. Van Duyne, Surface-Enhanced Raman Scattering: Physics and Applications, Springer, Berlin Heidelberg, 2006 DOI:10.1007/3-540-33567-6_2.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS.
- M. Li, S. K. Cushing, G. Zhou and N. Wu, Nanoscale, 2020, 12, 22036–22041 RSC.
- M. Schutz, C. I. Muller, M. Salehi, C. Lambert and S. Schlucker, J. Biophotonics, 2011, 4, 453–463 CrossRef PubMed.
- D. Graham, K. Faulds and W. E. Smith, Chem. Commun., 2006, 4363–4371 RSC.
- T. Keller, S. Brem, V. Tran, O. Sritharan, D. Schäfer and S. Schlücker, J. Biophotonics, 2020, 13, e201960126 CrossRef CAS PubMed.
- X. Mu, Y. Guo, Y. Li, Z. Wang, Y. Li and S. Xu, J. Raman Spectrosc., 2017, 48, 1196–1200 CrossRef CAS.
- L. Lin, X. Tian, S. Hong, P. Dai, Q. You, R. Wang, L. Feng, C. Xie, Z.-Q. Tian and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 7266–7271 CrossRef CAS PubMed.
- M. Li, J. Zhang, S. Suri, L. J. Sooter, D. Ma and N. Wu, Anal. Chem., 2012, 84, 2837–2842 CrossRef CAS PubMed.
- A. M. Gabudean, D. Biro and S. Astilean, J. Mol. Struct., 2011, 993, 420–424 CrossRef CAS.
- A. Michota and J. Bukowska, J. Raman Spectrosc., 2003, 34, 21–25 CrossRef CAS.
- X. Gao, P. Zheng, S. Kasani, S. Wu, F. Yang, S. Lewis, S. Nayeem, E. B. Engler-Chiurazzi, J. G. Wigginton, J. W. Simpkins and N. Wu, Anal. Chem., 2017, 89, 10104–10110 CrossRef CAS PubMed.
- X. Tan, Z. Wang, J. Yang, C. Song, R. Zhang and Y. Cui, Nanotechnology, 2009, 20, 445102 CrossRef PubMed.
- P. Zheng, M. Li, R. Jurevic, S. K. Cushing, Y. Liu and N. Wu, Nanoscale, 2015, 7, 11005–11012 RSC.
- P. Zheng, S. Kasani, X. Shi, A. E. Boryczka, F. Yang, H. Tang, M. Li, W. Zheng, D. E. Elswick and N. Wu, Anal. Chim. Acta, 2018, 1040, 158–165 CrossRef CAS PubMed.
- J. E. Park, J. Kim and J. M. Nam, Chem. Sci., 2017, 8, 4696–4704 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Academic, 2006 Search PubMed.
- T. Itoh, Y. S. Yamamoto and Y. Ozaki, Chem. Soc. Rev., 2017, 46, 3904–3921 RSC.
- J. B. Khurgin and G. Sun, J. Opt. Soc. Am. B, 2009, 26, B83–B95 CrossRef CAS.
- J. Li, S. K. Cushing, F. Meng, T. R. Senty, A. D. Bristow and N. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- M. Li, X. Zhou, W. Ding, S. Guo and N. Wu, Biosens. Bioelectron., 2013, 41, 889–893 CrossRef CAS PubMed.
- J. Yang, F. Zhang, Y. Chen, S. Qian, P. Hu, W. Li, Y. Deng, Y. Fang, L. Han, M. Luqman and D. Zhao, Chem. Commun., 2011, 47, 11618–11620 RSC.
- L. Trotsiuk, A. Muravitskaya, O. Kulakovich, D. Guzatov, A. Ramanenka, Y. Kelestemur, H. V. Demir and S. Gaponenko, Nanotechnology, 2020, 31, 105201 CrossRef CAS PubMed.
- F. Tang, F. He, H. Cheng and L. Li, Langmuir, 2010, 26, 11774–11778 CrossRef CAS PubMed.
- D. Gontero, A. V. Veglia, D. Boudreau and A. G. Bracamonte, J. Nanophotonics, 2017, 12, 012505 CrossRef.
- X. Wang, L. Zhang, A. Hao, Z. Shi, C. Dai, Y. Yang and H. Huang, ACS Appl. Nano. Mater., 2020, 3, 9796–9803 CrossRef CAS.
- Y. Gao, J. Wang, W. Wang, T. Zhao, Y. Cui, P. Liu, S. Xu and X. Luo, Anal. Chem., 2021, 93, 2480–2489 CrossRef CAS PubMed.
- I. G. Theodorou, Z. A. R. Jawad, Q. Jiang, E. O. Aboagye, A. E. Porter, M. P. Ryan and F. Xie, Chem. Mater., 2017, 29, 6916–6926 CrossRef CAS.
- H. Ren, J.-L. Yang, W.-M. Yang, H.-L. Zhong, J.-S. Lin, P. M. Radjenovic, L. Sun, H. Zhang, J. Xu, Z.-Q. Tian and J.-F. Li, ACS Mater. Lett., 2020, 3, 69–76 CrossRef.
- M. Yang, P. Moroz, Z. Jin, D. S. Budkina, N. Sundrani, D. Porotnikov, J. Cassidy, Y. Sugiyama, A. N. Tarnovsky, H. Mattoussi and M. Zamkov, J. Am. Chem. Soc., 2019, 141, 11286–11297 CrossRef CAS PubMed.
- T. Itoh, K.-i. Yoshida, H. Tamaru, V. Biju and M. Ishikawa, J. Photochem. Photobiol., A, 2011, 219, 167–179 CrossRef CAS.
- M. Li, S. K. Cushing, J. Zhang, S. Suri, R. Evans, W. P. Petros, L. F. Gibson, D. Ma, Y. Liu and N. Wu, ACS Nano, 2013, 7, 4967–4976 CrossRef CAS PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- X. Fu, L. Chen and J. Choo, Anal. Chem., 2017, 89, 124–137 CrossRef CAS PubMed.
- Z. Yang, H. Liu, Y. Tian, Y. Chen, Z. Niu, C. Zhou, F. Wang, C. Gu, S. Tang, T. Jiang and J. Zhou, J. Mater. Chem. C, 2020, 8, 2142–2154 RSC.
- T. Jiang, X. Wang, J. Zhou and H. Jin, Sens. Actuators, B, 2018, 258, 105–114 CrossRef CAS.
- M. Lee, H. Kim, E. Kim, S. Y. Yi, S. G. Hwang, S. Yang, E. K. Lim, B. Kim, J. Jung and T. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 37829–37834 CrossRef CAS PubMed.
- Z. Farka, M. J. Mickert, A. Hlavacek, P. Skladal and H. H. Gorris, Anal. Chem., 2017, 89, 11825–11830 CrossRef CAS PubMed.
- Z. Cheng, N. Choi, R. Wang, S. Lee, K. C. Moon, S. Y. Yoon, L. Chen and J. Choo, ACS Nano, 2017, 11, 4926–4933 CrossRef CAS PubMed.
- Z. Chen, S. M. Tabakman, A. P. Goodwin, M. G. Kattah, D. Daranciang, X. Wang, G. Zhang, X. Li, Z. Liu, P. J. Utz, K. Jiang, S. Fan and H. Dai, Nat. Biotechnol., 2008, 26, 1285–1292 CrossRef CAS PubMed.
- I. Al-Ogaidi, H. Gou, Z. P. Aguilar, S. Guo, A. K. Melconian, A. K. Al-Kazaz, F. Meng and N. Wu, Chem. Commun., 2014, 50, 1344–1346 RSC.
- Y. Luo, X. Wang, D. Du and Y. Lin, Biomater. Sci., 2015, 3, 1386–1394 RSC.
- M. Uchida, H. Kosuge, M. Terashima, D. A. Willits, L. O. Liepold, M. J. Young, M. V. McConnell and T. Douglas, ACS Nano, 2011, 5, 2493–2502 CrossRef CAS PubMed.
- H. Qian, Y. Huang, X. Duan, X. Wei, Y. Fan, D. Gan, S. Yue, W. Cheng and T. Chen, Biosens. Bioelectron., 2019, 140, 111350 CrossRef CAS PubMed.
- S.-E. Kim, K.-Y. Ahn, J.-S. Park, K. R. Kim, K. E. Lee, S.-S. Han and J. Lee, Anal. Chem., 2011, 83, 5834–5843 CrossRef CAS PubMed.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 22–32 CrossRef CAS PubMed.
- L. Norskov-Lauritsen, A. R. Thomsen and H. Brauner-Osborne, Int. J. Mol. Sci., 2014, 15, 2554–2572 CrossRef PubMed.
- S. Hepojoki, J. Rusanen, J. Hepojoki, V. Nurmi, A. Vaheri, A. Lundkvist, K. Hedman and O. Vapalahti, J. Clin. Microbiol., 2015, 53, 2292–2297 CrossRef CAS PubMed.
- K. Shao, L. Wang, Y. Wen, T. Wang, Y. Teng, Z. Shen and Z. Pan, Anal. Chim. Acta, 2019, 1068, 52–59 CrossRef CAS PubMed.
- H. Xing, T. Wei, X. Lin and Z. Dai, Anal. Chim. Acta, 2018, 1042, 71–78 CrossRef CAS PubMed.
- L. Zhao, M. Cheng, G. Liu, H. Lu, Y. Gao, X. Yan, F. Liu, P. Sun and G. Lu, Sens. Actuators, B, 2018, 273, 185–190 CrossRef CAS.
- J. Yang, Z. Zhang and G. Yan, Sens. Actuators, B, 2018, 255, 2339–2346 CrossRef CAS.
- E. Pyrak, J. Krajczewski, A. Kowalik, A. Kudelski and A. Jaworska, Molecules, 2019, 24, 4423 CrossRef CAS PubMed.
- B. C. Strachan, H. S. Sloane, J. C. Lee, D. C. Leslie and J. P. Landers, Analyst, 2015, 140, 2008–2015 RSC.
- L. Peng, J. Zhou, L. Yin, S. Man and L. Ma, Anal. Chim. Acta, 2020, 1125, 162–168 CrossRef CAS PubMed.
- X. Liu, F. Wang, R. Aizen, O. Yehezkeli and I. Willner, J. Am. Chem. Soc., 2013, 135, 11832–11839 CrossRef CAS PubMed.
- M. Li, S. K. Cushing, H. Liang, S. Suri, D. Ma and N. Wu, Anal. Chem., 2013, 85, 2072–2078 CrossRef CAS PubMed.
- S. Wang, M. A. Lifson, F. Inci, L. G. Liang, Y. F. Sheng and U. Demirci, Expert Rev. Mol. Diagn., 2016, 16, 449–459 CrossRef CAS PubMed.
- L. Huang, L. Ding, J. Zhou, S. Chen, F. Chen, C. Zhao, J. Xu, W. Hu, J. Ji, H. Xu and G. L. Liu, Biosens. Bioelectron., 2021, 171, 112685 CrossRef CAS PubMed.
- Y. Sun and T. Li, Anal. Chem., 2018, 90, 11614–11621 CrossRef CAS PubMed.
- Z. Liang, J. Zhou, L. Petti, L. Shao, T. Jiang, Y. Qing, S. Xie, G. Wu and P. Mormile, Analyst, 2019, 144, 1741–1750 RSC.
- Y. P. Wong, S. Othman, Y. L. Lau, S. Radu and H. Y. Chee, J. Appl. Microbiol., 2018, 124, 626–643 CrossRef CAS PubMed.
- L. Garibyan and N. Avashia, J. Invest. Dermatol. Symp. Proc., 2013, 133, 1–4 CrossRef PubMed.
- J. Li, J. Macdonald and F. von Stetten, Analyst, 2018, 144, 31–67 RSC.
- C. Shi, Q. Liu, C. Ma and W. Zhong, Anal. Chem., 2014, 86, 336–339 CrossRef CAS PubMed.
- Y. Zhou, B. Li, M. Wang, J. Wang, H. Yin and S. Ai, Mikrochim. Acta, 2017, 184, 4359–4365 CrossRef CAS.
- M. Fakruddin, K. S. Mannan, A. Chowdhury, R. M. Mazumdar, M. N. Hossain, S. Islam and M. A. Chowdhury, J. Pharm. BioAllied Sci., 2013, 5, 245–252 CrossRef PubMed.
- C. O. Nzelu, H. Kato and N. C. Peters, PLoS Neglected Trop. Dis., 2019, 13, e0007698 CrossRef PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, e204 CrossRef PubMed.
- A. S. James and J. I. Alawneh, Diagnostics, 2020, 10, 399 CrossRef CAS PubMed.
- N. Bidar, F. Oroojalian, B. Baradaran, S. Eyvazi, M. Amini, A. Jebelli, S. S. Hosseini, P. Pashazadeh-Panahi, A. Mokhtarzadeh and M. de la Guardia, TrAC, Trend Anal. Chem., 2020, 131, 116021 CrossRef CAS.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- I. Khalil, W. A. Yehye, N. M. Julkapli, S. Rahmati, A. A. Sina, W. J. Basirun and M. R. Johan, Biosens. Bioelectron., 2019, 131, 214–223 CrossRef CAS PubMed.
- B. Guven, I. H. Boyaci, U. Tamer, E. Acar-Soykut and U. Dogan, Talanta, 2015, 136, 68–74 CrossRef CAS PubMed.
- L. Ge, X. Sun, Q. Hong and F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 32089–32096 CrossRef CAS PubMed.
- J. Zhang, Y. Yang, X. Jiang, C. Dong, C. Song, C. Han and L. Wang, Biosens. Bioelectron., 2019, 141, 111402 CrossRef CAS PubMed.
- S.-R. Ryoo, J. Lee, J. Yeo, H.-K. Na, Y.-K. Kim, H. Jang, J. H. Lee, S. W. Han, Y. Lee, V. N. Kim and D.-H. Min, ACS Nano, 2013, 7, 5882–5891 CrossRef CAS PubMed.
- C. Song, Y. Liu, X. Jiang, J. Zhang, C. Dong, J. Li and L. Wang, Talanta, 2019, 205, 120137 CrossRef CAS PubMed.
- J. Huang, X. Su and Z. Li, Anal. Chem., 2012, 84, 5939–5943 CrossRef CAS PubMed.
- K. Guk, S. G. Hwang, J. Lim, H. Y. Son, Y. Choi, Y. M. Huh, T. Kang, J. Jung and E. K. Lim, Chem. Commun., 2019, 55, 3457–3460 RSC.
- O. Adegoke, T. Kato and E. Y. Park, Biosens. Bioelectron., 2016, 80, 483–490 CrossRef CAS PubMed.
- M. M. Harper, B. Robertson, A. Ricketts and K. Faulds, Chem. Commun., 2012, 48, 9412–9414 RSC.
- J. H. Choi, J. Lim, M. Shin, S. H. Paek and J. W. Choi, Nano Lett., 2021, 21, 693–699 CrossRef CAS PubMed.
- Y. Liu, A. Kannegulla, B. Wu and L.-J. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 18524–18531 CrossRef CAS PubMed.
- J. A. Dougan, D. MacRae, D. Graham and K. Faulds, Chem. Commun., 2011, 47, 4649 RSC.
- M. S. Draz and X. Lu, Theranostics, 2016, 6, 522–532 CrossRef CAS PubMed.
- G. A. Obande and K. K. Banga Singh, Infect. Drug Resist., 2020, 13, 455–483 CrossRef CAS PubMed.
- X. Zhao, S. Li, G. Liu, Z. Wang, Z. Yang, Q. Zhang, M. Liang, J. Liu, Z. Li, Y. Tong, G. Zhu, X. Wang, L. Jiang, W. Wang, G.-Y. Tan and L. Zhang, Sci. Bull., 2020, 66, 69–77 CrossRef.
- Y. Liu, S. Li, L. Zhang, Q. Zhao, N. Li and Y. Wu, RSC Adv., 2020, 10, 28037–28040 RSC.
- M. Yu, Y. Yao, B. Cui, C. Sun, X. Zhao, Y. Wang, G. Liu, H. Cui and Z. Zeng, ACS Appl. Nano Mater., 2018, 2, 48–57 CrossRef.
- W. Tan, L. Zhang, J. C. G. Doery and W. Shen, Lab Chip, 2020, 20, 394–404 RSC.
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52–67 RSC.
- R. Feng, Y. Xu, H. Zhao, X. Duan and S. Sun, Analyst, 2016, 141, 3219–3223 RSC.
- I. Al-Ogaidi, H. Gou, A. K. Al-Kazaz, Z. P. Aguilar, A. K. Melconian, P. Zheng and N. Wu, Anal. Chim. Acta, 2014, 811, 76–80 CrossRef CAS PubMed.
- W. Tan, L. Zhang, J. C. G. Doery and W. Shen, Sens. Actuators, B, 2020, 305, 127448 CrossRef CAS.
- J. Yin, Y. Kwon, D. Kim, D. Lee, G. Kim, Y. Hu, J. H. Ryu and J. Yoon, J. Am. Chem. Soc., 2014, 136, 5351–5358 CrossRef CAS PubMed.
- R. M. Wood, J. K. Rilling, A. G. Sanfey, Z. Bhagwagar and R. D. Rogers, Neuropsychopharmacology, 2006, 31, 1075–1084 CrossRef CAS PubMed.
- M. Rondanelli, A. Opizzi, N. Antoniello, F. Boschi, P. Iadarola, E. Pasini, R. Aquilani and F. S. Dioguardi, Clin. Nutr., 2011, 30, 571–577 CrossRef CAS PubMed.
- F. Xiao, J. Yu, Y. Guo, J. Deng, K. Li, Y. Du, S. Chen, J. Zhu, H. Sheng and F. Guo, Metabolism, 2014, 63, 841–850 CrossRef CAS PubMed.
- C. Martin and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2005, 6, 838–849 CrossRef CAS PubMed.
- J. M. Wood, H. Decker, H. Hartmann, B. Chavan, H. Rokos, J. D. Spencer, S. Hasse, M. J. Thornton, M. Shalbaf, R. Paus and K. U. Schallreuter, FASEB J., 2009, 23, 2065–2075 CrossRef CAS PubMed.
- I. C. G. Weaver, F. A. Champagne, S. E. Brown, S. Dymov, S. Sharma, M. J. Meaney and M. Szyf, J. Neurosci., 2005, 25, 11045–11054 CrossRef CAS PubMed.
- G. Mkrtchyan, V. Aleshin, Y. Parkhomenko, T. Kaehne, M. Luigi Di Salvo, A. Parroni, R. Contestabile, A. Vovk, L. Bettendorff and V. Bunik, Sci. Rep., 2015, 5, 12583 CrossRef CAS PubMed.
- N. Naderi and J. D. House, Advances in Food and Nutrition Research, Academic Press, 2018, vol. 83, pp. 195–213 Search PubMed.
- M. Visentin, N. Diop-Bove, R. Zhao and I. D. Goldman, Annu. Rev. Physiol., 2014, 76, 251–274 CrossRef CAS PubMed.
- Institute of Medicine, Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids, The National Academies Press, 2000 Search PubMed.
- R. Brigelius-Flohé and M. G. Traber, FASEB J., 1999, 13, 1145–1155 CrossRef.
- U. Nations, World Drug Report 2017, United Nations, 2017 DOI:10.18356/bdc264f4-en.
- E. De Rycke, C. Stove, P. Dubruel, S. De Saeger and N. Beloglazova, Biosens. Bioelectron., 2020, 169, 112579 CrossRef CAS PubMed.
- D. Li, Y. Ma, H. Duan, W. Deng and D. Li, Biosens. Bioelectron., 2018, 99, 389–398 CrossRef CAS PubMed.
- Q. Zhou, G. Meng, J. Liu, Z. Huang, F. Han, C. Zhu, D.-J. Kim, T. Kim and N. Wu, Adv. Mater. Technol., 2017, 2, 1700028 CrossRef.
- X. Hu, P. Zheng, G. Meng, Q. Huang, C. Zhu, F. Han, Z. Huang, Z. Li, Z. Wang and N. Wu, Nanotechnology, 2016, 27, 384001 CrossRef PubMed.
- W. Xue, X. Tan, M. K. Khaing Oo, G. Kulkarni, M. A. Ilgen and X. Fan, Analyst, 2020, 145, 1346–1354 RSC.
- W. Yu, C. Jiang, B. Xie, S. Wang, X. Yu, K. Wen, J. Lin, J. Wang, Z. Wang and J. Shen, Anal. Chim. Acta, 2020, 1102, 91–98 CrossRef CAS PubMed.
- X. Hua, Y. Ding, J. Yang, M. Ma, H. Shi and M. Wang, Sci. Total Environ., 2017, 583, 222–227 CrossRef CAS PubMed.
- M. Li, M. Ma, X. Hua, H. Shi, Q. Wang and M. Wang, RSC Adv., 2015, 5, 3039–3044 RSC.
- E. Guler, G. Bozokalfa, B. Demir, Z. P. Gumus, B. Guler, E. Aldemir, S. Timur and H. Coskunol, Drug Test. Anal., 2017, 9, 578–587 CrossRef CAS PubMed.
- S. R. Ahmed, R. Chand, S. Kumar, N. Mittal, S. Srinivasan and A. R. Rajabzadeh, TrAC, Trend Anal. Chem., 2020, 131, 116006 CrossRef CAS.
- L. Wu, G. Li, X. Xu, L. Zhu, R. Huang and X. Chen, TrAC, Trend Anal. Chem., 2019, 113, 140–156 CrossRef CAS.
- J. Nebu, J. S. Anjali Devi, R. S. Aparna, B. Aswathy, A. O. Aswathy and G. Sony, Mikrochim. Acta, 2018, 185, 532 CrossRef PubMed.
- Z. Huang, G. Meng, Q. Huang, Y. Yang, C. Zhu and C. Tang, Adv. Mater., 2010, 22, 4136–4139 CrossRef CAS PubMed.
- C. Qian, Q. Guo, M. Xu, Y. Yuan and J. Yao, RSC Adv., 2015, 5, 53306–53312 RSC.
- C. Zhu, G. Meng, P. Zheng, Q. Huang, Z. Li, X. Hu, X. Wang, Z. Huang, F. Li and N. Wu, Adv. Mater., 2016, 28, 4871–4876 CrossRef CAS PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- R. A. Alvarez-Puebla and L. M. Liz-Marzan, Angew. Chem., Int. Ed., 2012, 51, 11214–11223 CrossRef CAS PubMed.
- G. Song, D. Jiang, L. Wang, J. Ning, X. Sun, F. Su, M. Chen and Y. Tian, Chem. Commun., 2020, 56, 5405–5408 RSC.
- C. Li, D. Yao, X. Jiang, A. Liang and Z. Jiang, J. Mater. Chem. C, 2020, 8, 11088–11101 RSC.
- M. Li, Q. Wang, X. Shi, L. A. Hornak and N. Wu, Anal. Chem., 2011, 83, 7061–7065 CrossRef CAS PubMed.
- P. Chen, X. Jiang, K. Huang, P. Hu, X. Li, L. Wei, W. Liu, L. Wei, C. Tao, B. Ying, X. Wei and J. Geng, ACS Appl. Mater. Interfaces, 2019, 11, 36476–36484 CrossRef CAS PubMed.
- D. Li, Y. Ma, H. Duan, F. Jiang, W. Deng and X. Ren, Anal. Chim. Acta, 2018, 1038, 148–156 CrossRef CAS PubMed.
- X. Chen, Y. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120–2135 RSC.
- J. Cao, C. Zhao, X. Wang, Y. Zhang and W. Zhu, Chem. Commun., 2012, 48, 9897–9899 RSC.
- P. Zheng, X. Shi, K. Curtin, F. Yang and N. Wu, Mater. Res. Express, 2017, 4, 055017 CrossRef.
- M. Li, X. Zhou, S. Guo and N. Wu, Biosens. Bioelectron., 2013, 43, 69–74 CrossRef CAS PubMed.
- Z. Yang, K. Y. Loh, Y.-T. Chu, R. Feng, N. S. R. Satyavolu, M. Xiong, S. M. Nakamata Huynh, K. Hwang, L. Li, H. Xing, X. Zhang, Y. R. Chemla, M. Gruebele and Y. Lu, J. Am. Chem. Soc., 2018, 140, 17656–17665 CrossRef CAS PubMed.
- R. J. Lake, Z. Yang, J. Zhang and Y. Lu, Acc. Chem. Res., 2019, 52, 3275–3286 CrossRef CAS PubMed.
- Y. Lin, Z. Yang, R. J. Lake, C. Zheng and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 17061–17067 CrossRef CAS PubMed.
- Y. Wang and J. Irudayaraj, Chem. Commun., 2011, 47, 4394–4396 RSC.
- P. K. Drain, E. P. Hyle, F. Noubary, K. A. Freedberg, D. Wilson, W. R. Bishai, W. Rodriguez and I. V. Bassett, Lancet Infect. Dis., 2014, 14, 239–249 CrossRef PubMed.
- H. Kettler, K. White and S. Hawkes, Mapping the landscape of diagnostics for sexually transmitted infections, World Health Organization, Geneva, 2004 Search PubMed.
- F. Yang, Y. Zhang, X. Cui, Y. Fan, Y. Xue, H. Miao and G. Li, Biotechnol. J., 2019, 14, 1800181 CrossRef PubMed.
- R. Gorkin, J. Park, J. Siegrist, M. Amasia, B. S. Lee, J. M. Park, J. Kim, H. Kim, M. Madou and Y. K. Cho, Lab Chip, 2010, 10, 1758–1773 RSC.
- P. N. Nge, C. I. Rogers and A. T. Woolley, Chem. Rev., 2013, 113, 2550–2583 CrossRef CAS PubMed.
- J. Park, D. H. Han and J.-K. Park, Lab Chip, 2020, 20, 1191–1203 RSC.
- P. Thurgood, J. Y. Zhu, N. Nguyen, S. Nahavandi, A. R. Jex, E. Pirogova, S. Baratchi and K. Khoshmanesh, Lab Chip, 2018, 18, 2730–2740 RSC.
- J. Seo, C. Wang, S. Chang, J. Park and W. Kim, Lab Chip, 2019, 19, 1790–1796 RSC.
- B. Kim, S. Oh, D. You and S. Choi, Anal. Chem., 2017, 89, 1439–1444 CrossRef CAS PubMed.
- L. Gervais, N. de Rooij and E. Delamarche, Adv. Mater., 2011, 23, H151–H176 CrossRef CAS PubMed.
- J. Han, A. Qi, J. Zhou, G. Wang, B. Li and L. Chen, ACS Sens., 2018, 3, 1789–1794 CrossRef CAS PubMed.
- M. T. Guler, P. Beyazkilic and C. Elbuken, Sens. Actuators, A, 2017, 265, 224–230 CrossRef CAS.
- Y.-D. Ma, Y.-S. Chen and G.-B. Lee, Sens. Actuators, B, 2019, 296, 126647 CrossRef CAS.
- J. Park and J.-K. Park, Lab Chip, 2019, 19, 2973–2977 RSC.
- M. Díaz-González, G. Boix, C. Fernández-Sánchez and A. Baldi, SPIE, 2017.
- B. Jo, L. M. V. Lerberghe, K. M. Motsegood and D. J. Beebe, J. Microelectromech. Syst., 2000, 9, 76–81 CAS.
- A. O. Olanrewaju, A. Robillard, M. Dagher and D. Juncker, Lab Chip, 2016, 16, 3804–3814 RSC.
- M. M. Aeinehvand, L. Weber, M. Jiménez, A. Palermo, M. Bauer, F. F. Loeffler, F. Ibrahim, F. Breitling, J. Korvink, M. Madou, D. Mager and S. O. Martínez-Chapa, Lab Chip, 2019, 19, 1090–1100 RSC.
- A. Enders, I. G. Siller, K. Urmann, M. R. Hoffmann and J. Bahnemann, Small, 2019, 15, e1804326 Search PubMed.
- W. Luo, K. Yarn, M. Shih, Y. Wu, K. Chang and M. Weng, 3rd IEEE International Conference on NEMS, 2008, pp. 801–806.
- Q. Xiong, C. Y. Lim, J. Ren, J. Zhou, K. Pu, M. B. Chan-Park, H. Mao, Y. C. Lam and H. Duan, Nat. Commun., 2018, 9, 1743 CrossRef PubMed.
- M. R. Rasouli and M. Tabrizian, Lab Chip, 2019, 19, 3316–3325 RSC.
- A. T. Bender, M. D. Borysiak, A. M. Levenson, L. Lillis, D. S. Boyle and J. D. Posner, Anal. Chem., 2018, 90, 7221–7229 CrossRef CAS PubMed.
- N. Xiang, Q. Li and Z. Ni, Anal. Chem., 2020, 92, 6770–6776 CrossRef CAS PubMed.
- R. Gao, Z. Cheng, A. J. deMello and J. Choo, Lab Chip, 2016, 16, 1022–1029 RSC.
- C. Szydzik, K. Khoshmanesh, A. Mitchell and C. Karnutsch, Biomicrofluidics, 2015, 9, 064120 CrossRef PubMed.
- M. A. Ali, K. Mondal, Y. Jiao, S. Oren, Z. Xu, A. Sharma and L. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 20570–20582 CrossRef CAS PubMed.
- R. Gao, Z. Lv, Y. Mao, L. Yu, X. Bi, S. Xu, J. Cui and Y. Wu, ACS Sens., 2019, 4, 938–943 CrossRef CAS PubMed.
- M. Sharafeldin and J. J. Davis, Anal. Chem., 2021, 93, 184–197 CrossRef CAS PubMed.
- S. Tripathi, Y. V. B. Varun Kumar, A. Prabhakar, S. S. Joshi and A. Agrawal, J. Micromech. Microeng., 2015, 25, 084004 CrossRef.
- M. S. Maria, T. S. Chandra and A. K. Sen, Microfluid. Nanofluid., 2017, 21, 72 CrossRef.
- S. Tripathi, Y. V. B. Varun Kumar, A. Prabhakar, S. S. Joshi and A. Agrawal, J. Micromech. Microeng., 2015, 25, 083001 CrossRef.
- S. Haeberle, T. Brenner, R. Zengerle and J. Ducrée, Lab Chip, 2006, 6, 776–781 RSC.
- S. C. Liu, P. B. Yoo, N. Garg, A. P. Lee and S. Rasheed, Sens. Actuators, A, 2021, 317, 112482 CrossRef CAS.
- M. Mohammadi, H. Madadi, J. Casals-Terre and J. Sellares, Anal. Bioanal. Chem., 2015, 407, 4733–4744 CrossRef CAS PubMed.
- H. M. Ji, V. Samper, Y. Chen, C. K. Heng, T. M. Lim and L. Yobas, Biomed. Microdevices, 2008, 10, 251–257 CrossRef PubMed.
- X. Yang, O. Forouzan, T. P. Brown and S. S. Shevkoplyas, Lab Chip, 2012, 12, 274–280 RSC.
- H. Shimizu, M. Kumagai, E. Mori, K. Mawatari and T. Kitamori, Anal. Methods, 2016, 8, 7597–7602 RSC.
- K. Svanes and B. W. Zweifach, Microvasc. Res., 1968, 1, 210–220 CrossRef.
- Y.-C. Fung, Microvasc. Res., 1973, 5, 34–48 CrossRef CAS PubMed.
- S. Yang, A. Undar and J. D. Zahn, Lab Chip, 2006, 6, 871–880 RSC.
- R. Fan, O. Vermesh, A. Srivastava, B. K. Yen, L. Qin, H. Ahmad, G. A. Kwong, C. C. Liu, J. Gould, L. Hood and J. R. Heath, Nat. Biotechnol., 2008, 26, 1373–1378 CrossRef CAS PubMed.
- J. Wang, H. Ahmad, C. Ma, Q. Shi, O. Vermesh, U. Vermesh and J. Heath, Lab Chip, 2010, 10, 3157–3162 RSC.
- M. S. Maria, P. E. Rakesh, T. S. Chandra and A. K. Sen, Sci. Rep., 2017, 7, 43457 CrossRef PubMed.
- C. Liu, M. Mauk, R. Gross, F. D. Bushman, P. H. Edelstein, R. G. Collman and H. H. Bau, Anal. Chem., 2013, 85, 10463–10470 CrossRef CAS PubMed.
- G. Luka, A. Ahmadi, H. Najjaran, E. Alocilja, M. DeRosa, K. Wolthers, A. Malki, H. Aziz, A. Althani and M. Hoorfar, Sensors, 2015, 15, 30011–30031 CrossRef CAS PubMed.
- K. Choi, A. H. Ng, R. Fobel and A. R. Wheeler, Annu. Rev. Anal. Chem., 2012, 5, 413–440 CrossRef CAS PubMed.
- B. Coelho, B. Veigas, H. Águas, E. Fortunato, R. Martins, P. Baptista and R. Igreja, Sensors, 2017, 17, 2616 CrossRef PubMed.
- F. Mugele and J.-C. Baret, J. Phys.: Condens. Matter, 2005, 17, R705–R774 CrossRef CAS.
- A. M. Foudeh, D. Brassard, M. Tabrizian and T. Veres, Lab Chip, 2015, 15, 1609–1618 RSC.
- M. S. Lee, W. Hsu, H. Y. Huang, H. Y. Tseng, C. T. Lee, C. Y. Hsu, Y. C. Shieh, S. H. Wang, D. J. Yao and C. H. Liu, Biosens. Bioelectron., 2020, 150, 111851 CrossRef CAS PubMed.
- V. Srinivasan, V. K. Pamula and R. B. Fair, Lab Chip, 2004, 4, 310–315 RSC.
- F. Stumpf, F. Schwemmer, T. Hutzenlaub, D. Baumann, O. Strohmeier, G. Dingemanns, G. Simons, C. Sager, L. Plobner, F. von Stetten, R. Zengerle and D. Mark, Lab Chip, 2016, 16, 199–207 RSC.
- S. H. Yazdi, K. L. Giles and I. M. White, Anal. Chem., 2013, 85, 10605–10611 CrossRef CAS PubMed.
- J. Qi, J. Zeng, F. Zhao, S. H. Lin, B. Raja, U. Strych, R. C. Willson and W. C. Shih, Nanoscale, 2014, 6, 8521–8526 RSC.
- A. Kaminska, E. Witkowska, K. Winkler, I. Dziecielewski, J. L. Weyher and J. Waluk, Biosens. Bioelectron., 2015, 66, 461–467 CrossRef CAS PubMed.
- C. H. Sang, S. J. Chou, F. M. Pan and J. T. Sheu, Biosens. Bioelectron., 2016, 75, 285–292 CrossRef CAS PubMed.
- C. M. Chang, W. H. Chang, C. H. Wang, J. H. Wang, J. D. Mai and G. B. Lee, Lab Chip, 2013, 13, 1225–1242 RSC.
- P. J. Sykes, S. H. Neoh, M. J. Brisco, E. Hughes, J. Condon and A. A. Morley, Biotechniques, 1992, 13, 444–449 CAS.
- F. Shen, W. Du, J. E. Kreutz, A. Fok and R. F. Ismagilov, Lab Chip, 2010, 10, 2666–2672 RSC.
- M. Safavieh, M. K. Kanakasabapathy, F. Tarlan, M. U. Ahmed, M. Zourob, W. Asghar and H. Shafiee, ACS Biomater. Sci. Eng., 2016, 2, 278–294 CrossRef CAS PubMed.
- H. Yuan, Y. Chao and H. C. Shum, Small, 2020, 16, 1904469 CrossRef CAS PubMed.
- Y. Chen, Y. Mei, X. Zhao and X. Jiang, Anal. Chem., 2020, 92, 14846–14852 CrossRef CAS PubMed.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 CrossRef CAS PubMed.
- Z. Pang, A. Al-Mahrouki, M. Berezovski and S. N. Krylov, Electrophoresis, 2006, 27, 1489–1494 CrossRef CAS PubMed.
- P. Sethu, M. Anahtar, L. L. Moldawer, R. G. Tompkins and M. Toner, Anal. Chem., 2004, 76, 6247–6253 CrossRef CAS PubMed.
- L. Nan, Z. Jiang and X. Wei, Lab Chip, 2014, 14, 1060–1073 RSC.
- J. Kim, J. W. Hong, D. P. Kim, J. H. Shin and I. Park, Lab Chip, 2012, 12, 2914–2921 RSC.
- Y. Shin, A. P. Perera, C. C. Wong and M. K. Park, Lab Chip, 2014, 14, 359–368 RSC.
- Y. K. Cho, J. G. Lee, J. M. Park, B. S. Lee, Y. Lee and C. Ko, Lab Chip, 2007, 7, 565–573 RSC.
- W.-P. Hu, Y.-C. Chen and W.-Y. Chen, Sci. Rep., 2020, 10, 21132 CrossRef CAS PubMed.
- J. Yin, Y. Suo, Z. Zou, J. Sun, S. Zhang, B. Wang, Y. Xu, D. Darland, J. X. Zhao and Y. Mu, Lab Chip, 2019, 19, 2769–2785 RSC.
- S. Petralia, E. L. Sciuto and S. Conoci, Analyst, 2017, 142, 140–146 RSC.
- D. Chen, M. Mauk, X. Qiu, C. Liu, J. Kim, S. Ramprasad, S. Ongagna, W. R. Abrams, D. Malamud, P. L. Corstjens and H. H. Bau, Biomed. Microdevices, 2010, 12, 705–719 CrossRef CAS PubMed.
- C. Zhu, A. Hu, J. Cui, K. Yang, X. Zhu, Y. Liu, G. Deng and L. Zhu, Micromachines, 2019, 10, 537 CrossRef PubMed.
- M. Solsona, E. Y. Westerbeek, J. G. Bomer, W. Olthuis and A. van den Berg, Lab Chip, 2019, 19, 1054–1059 RSC.
- C. Liu, J. Sun, J. Li, C. Xiang, L. Che, Z. Wang and X. Zhou, Sci. Rep., 2017, 7, 7552 CrossRef PubMed.
- A. Ramachandran, D. A. Huyke, E. Sharma, M. K. Sahoo, C. Huang, N. Banaei, B. A. Pinsky and J. G. Santiago, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29518–29525 CrossRef CAS PubMed.
- V. Betancur, J. Sun, N. Wu and Y. Liu, Micromachines, 2017, 8, 376 CrossRef PubMed.
- M. Zimmermann, H. Schmid, P. Hunziker and E. Delamarche, Lab Chip, 2007, 7, 119–125 RSC.
- E.-C. Yeh, C.-C. Fu, L. Hu, R. Thakur, J. Feng and L. P. Lee, Sci. Adv., 2017, 3, e1501645 CrossRef PubMed.
- W. C. Mak, V. Beni and A. P. F. Turner, TrAC, Trends Anal. Chem., 2016, 79, 297–305 CrossRef CAS.
- L. Gervais and E. Delamarche, Lab Chip, 2009, 9, 3330 RSC.
- S. Ghosh and C. H. Ahn, Analyst, 2019, 144, 2109–2119 RSC.
- L. Xu, H. Lee, D. Jetta and K. W. Oh, Lab Chip, 2015, 15, 3962–3979 RSC.
- L. Qin, O. Vermesh, Q. Shi and J. R. Heath, Lab Chip, 2009, 9, 2016–2020 RSC.
- L. Zhu, N. Kroodsma, J. Yeom, J. L. Haan, M. A. Shannon and D. D. Meng, Microfluid. Nanofluid., 2011, 11, 569–578 CrossRef CAS.
- L. Gervais, M. Hitzbleck and E. Delamarche, Biosens. Bioelectron., 2011, 27, 64–70 CrossRef CAS PubMed.
- L. Xu, A. Wang, X. Li and K. W. Oh, Biomicrofluidics, 2020, 14, 031503 CrossRef CAS PubMed.
- J. M. Morrissette, P. S. Mahapatra, A. Ghosh, R. Ganguly and C. M. Megaridis, Sci. Rep., 2017, 7, 1800 CrossRef PubMed.
- S. Shin, B. Kim, Y. J. Kim and S. Choi, Biosens. Bioelectron., 2019, 133, 169–176 CrossRef CAS PubMed.
- K. K. Lee, M. O. Kim and S. Choi, J. Pharm. Biomed. Anal., 2019, 162, 28–33 CrossRef CAS PubMed.
- C. Parolo, A. Sena-Torralba, J. F. Bergua, E. Calucho, C. Fuentes-Chust, L. Hu, L. Rivas, R. Alvarez-Diduk, E. P. Nguyen, S. Cinti, D. Quesada-Gonzalez and A. Merkoci, Nat. Protoc., 2020, 15, 3788–3816 CrossRef CAS PubMed.
- J. Hu, S. Wang, L. Wang, F. Li, B. Pingguan-Murphy, T. J. Lu and F. Xu, Biosens. Bioelectron., 2014, 54, 585–597 CrossRef CAS PubMed.
- H. Wei, Y. Peng, Z. Bai, Z. Rong and S. Wang, Analyst, 2021, 146, 558–564 RSC.
- S. Kasetsirikul, M. J. A. Shiddiky and N.-T. Nguyen, Microfluid. Nanofluid., 2020, 24, 17 CrossRef.
- R. Wang, W. Zhang, P. Wang and X. Su, Mikrochim. Acta, 2018, 185, 191 CrossRef PubMed.
- X. Gao, J. Boryczka, P. Zheng, S. Kasani, F. Yang, E. B. Engler-Chiurazzi, J. W. Simpkins, J. G. Wigginton and N. Wu, Biosens. Bioelectron., 2021, 177, 112967 CrossRef CAS PubMed.
- X. Gao, J. Boryczka, S. Kasani and N. Wu, Anal. Chem., 2021, 93(3), 1326–1332 CrossRef CAS PubMed.
- X. Yang, X. Liu, B. Gu, H. Liu, R. Xiao, C. Wang and S. Wang, Mikrochim. Acta, 2020, 187, 570 CrossRef CAS PubMed.
- X. Weng and S. Neethirajan, Mikrochim. Acta, 2017, 184, 4545–4552 CrossRef CAS.
- L. Anfossi, F. Di Nardo, S. Cavalera, C. Giovannoli, G. Spano, E. S. Speranskaya, I. Y. Goryacheva and C. Baggiani, Mikrochim. Acta, 2018, 185, 94 CrossRef PubMed.
- X. Deng, C. Wang, Y. Gao, J. Li, W. Wen, X. Zhang and S. Wang, Biosens. Bioelectron., 2018, 105, 211–217 CrossRef PubMed.
- J. Wang, F. Cao, S. He, Y. Xia, X. Liu, W. Jiang, Y. Yu, H. Zhang and W. Chen, Talanta, 2018, 176, 444–449 CrossRef CAS PubMed.
- Z. Lu, E. Rey, S. Vemulapati, B. Srinivasan, S. Mehta and D. Erickson, Lab Chip, 2018, 18, 3865–3871 RSC.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210 RSC.
- W. Guo, J. Hansson and W. van der Wijngaart, Anal. Chem., 2020, 92, 6194–6199 CrossRef CAS PubMed.
- H. Li and A. J. Steckl, Anal. Chem., 2019, 91, 352–371 CrossRef CAS PubMed.
- D. M. Cate, J. A. Adkins, J. Mettakoonpitak and C. S. Henry, Anal. Chem., 2015, 87, 19–41 CrossRef CAS PubMed.
- E. Fu, S. A. Ramsey, P. Kauffman, B. Lutz and P. Yager, Microfluid. Nanofluid., 2011, 10, 29–35 CrossRef CAS PubMed.
- A. Ainla, M. M. Hamedi, F. Güder and G. M. Whitesides, Adv. Mater., 2017, 29, 1702894 CrossRef PubMed.
- J. R. Choi, Z. Liu, J. Hu, R. Tang, Y. Gong, S. Feng, H. Ren, T. Wen, H. Yang, Z. Qu, B. Pingguan-Murphy and F. Xu, Anal. Chem., 2016, 88, 6254–6264 CrossRef CAS PubMed.
- C. K. Koo, F. He and S. R. Nugen, Analyst, 2013, 138, 4998–5004 RSC.
- J. R. Choi, J. Hu, R. Tang, Y. Gong, S. Feng, H. Ren, T. Wen, X. Li, W. A. Wan Abas, B. Pingguan-Murphy and F. Xu, Lab Chip, 2016, 16, 611–621 RSC.
- J. T. Connelly, J. P. Rolland and G. M. Whitesides, Anal. Chem., 2015, 87, 7595–7601 CrossRef CAS PubMed.
- P. Chen, C. Chen, Y. Liu, W. Du, X. Feng and B.-F. Liu, Sens. Actuators, B, 2019, 283, 472–477 CrossRef CAS.
- K. Scida, B. Li, A. D. Ellington and R. M. Crooks, Anal. Chem., 2013, 85, 9713–9720 CrossRef CAS PubMed.
- Z. Yang, G. Xu, J. Reboud, S. A. Ali, G. Kaur, J. McGiven, N. Boby, P. K. Gupta, P. Chaudhuri and J. M. Cooper, ACS Sens., 2018, 3, 403–409 CrossRef CAS PubMed.
- H. Ahn, B. S. Batule, Y. Seok and M. G. Kim, Anal. Chem., 2018, 90, 10211–10216 CrossRef CAS PubMed.
- T. Komatsu, M. Maeki, A. Ishida, H. Tani and M. Tokeshi, ACS Sens., 2020, 5, 1287–1294 CrossRef CAS PubMed.
- B. A. Rohrman and R. R. Richards-Kortum, Lab Chip, 2012, 12, 3082–3088 RSC.
- E. A. Phillips, T. J. Moehling, K. F. K. Ejendal, O. S. Hoilett, K. M. Byers, L. A. Basing, L. A. Jankowski, J. B. Bennett, L. K. Lin, L. A. Stanciu and J. C. Linnes, Lab Chip, 2019, 19, 3375–3386 RSC.
- S. Ebnesajjad, Surface Treatment of Materials for Adhesive Bonding, William Andrew Publishing, Oxford, 2nd edn, 2014, pp. 39–75 DOI:10.1016/B978-0-323-26435-8.00004-6.
- A. Di Gianfrancesco, Materials for Ultra-Supercritical and Advanced Ultra-Supercritical Power Plants, Woodhead Publishing, 2017, pp. 197–245 DOI:10.1016/B978-0-08-100552-1.00008-7.
- A. S. Holik, Encyclopedia of Materials: Science and Technology, Elsevier, Oxford, 2001, pp. 6458–6463 DOI:10.1016/B0-08-043152-6/01142-6.
- J. W. Lichtman and J.-A. Conchello, Nat. Methods, 2005, 2, 910–919 CrossRef CAS PubMed.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS PubMed.
- B. G. de Grooth, M. van Dam, N. C. Swart, A. Willemsen and J. Greve, Cytometry, 1987, 8, 445–452 CrossRef CAS PubMed.
- D. J. Webb and C. M. Brown, Cell Imaging Techniques: Methods and Protocols, Humana Press, Totowa, NJ, 2013, pp. 29–59 DOI:10.1007/978-1-62703-056-4_2.
- S. W. Paddock, Mol. Biotechnol., 2000, 16, 127–149 CrossRef CAS PubMed.
- A. S. Maddox and P. S. Maddox, Methods in Cell Biology, Academic Press, 2012, vol. 107, pp. 1–34 Search PubMed.
- A. M. Larson, Nat. Photonics, 2011, 5, 1 CrossRef.
- P. T. C. So, C. Y. Dong, B. R. Masters and K. M. Berland, Annu. Rev. Biomed. Eng., 2000, 2, 399–429 CrossRef CAS PubMed.
- L. Li, W. Guo, Y. Yan, S. Lee and T. Wang, Light: Sci. Appl., 2013, 2, e104 CrossRef.
- C. G. Galbraith and J. A. Galbraith, J. Cell Sci., 2011, 124, 1607–1611 CrossRef CAS PubMed.
- G. Perinetti, T. Müller, A. Spaar, R. Polishchuk, A. Luini and A. Egner, Traffic, 2009, 10, 379–391 CrossRef CAS PubMed.
- J. M. Pullman, J. Nylk, E. C. Campbell, F. J. Gunn-Moore, M. B. Prystowsky and K. Dholakia, Biomed. Opt. Express, 2016, 7, 302–311 CrossRef CAS PubMed.
- K. I. Willig, S. O. Rizzoli, V. Westphal, R. Jahn and S. W. Hell, Nature, 2006, 440, 935–939 CrossRef CAS PubMed.
- S. K. Cool, K. Breyne, E. Meyer, S. C. De Smedt and N. N. Sanders, J. Fluoresc., 2013, 23, 909–920 CrossRef CAS PubMed.
- M. Lesser, High Performance Silicon Imaging, Woodhead Publishing, 2014, pp. 78–97 DOI:10.1533/9780857097521.1.78.
- B. Q. Spring, A. O. Abu-Yousif, A. Palanisami, I. Rizvi, X. Zheng, Z. Mai, S. Anbil, R. B. Sears, L. B. Mensah, R. Goldschmidt, S. S. Erdem, E. Oliva and T. Hasan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E933–E942 CrossRef CAS PubMed.
- G. Oh, E. Chung and S. H. Yun, Opt. Fiber Technol., 2013, 19, 760–771 CrossRef CAS.
- C. C. Horgan, M. S. Bergholt, A. Nagelkerke, M. Z. Thin, I. J. Pence, U. Kauscher, T. L. Kalber, D. J. Stuckey and M. M. Stevens, Theranostics, 2021, 11, 2006–2019 CrossRef CAS PubMed.
- W. Wang, J. Zhao, M. Short and H. Zeng, J. Biophotonics, 2015, 8, 527–545 CrossRef PubMed.
- S. H. Seo, B. M. Kim, A. Joe, H. W. Han, X. Chen, Z. Cheng and E. S. Jang, Biomaterials, 2014, 35, 3309–3318 CrossRef CAS PubMed.
- E. Cordero, I. Latka, C. Matthaus, I. Schie and J. Popp, J. Biomed. Opt., 2018, 23, 1–23 Search PubMed.
- X. Yi, F. Wang, W. Qin, X. Yang and J. Yuan, Int. J. Nanomed., 2014, 9, 1347–1365 CrossRef PubMed.
- J.-B. Li, H.-W. Liu, T. Fu, R. Wang, X.-B. Zhang and W. Tan, Trends Chem., 2019, 1, 224–234 CrossRef CAS PubMed.
- L. Wang, W. Du, Z. Hu, K. Uvdal, L. Li and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 14026–14043 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klan, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- W. Xu, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 7476–7487 CrossRef CAS PubMed.
- L.-J. Liu, W. Liu, G. Ji, Z.-Y. Wu, B. Xu, J. Qian and W.-J. Tian, Chin. J. Polym. Sci., 2019, 37, 401–408 CrossRef CAS.
- P. Zhao, K. He, Y. Han, Z. Zhang, M. Yu, H. Wang, Y. Huang, Z. Nie and S. Yao, Anal. Chem., 2015, 87, 9998–10005 CrossRef CAS PubMed.
- P. Zhao, Q. Xu, J. Tao, Z. Jin, Y. Pan, C. Yu and Z. Yu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2018, 10, e1483 Search PubMed.
- S. Chinnathambi and N. Shirahata, Sci. Technol. Adv. Mater., 2019, 20, 337–355 CrossRef CAS PubMed.
- M. Montalti, A. Cantelli and G. Battistelli, Chem. Soc. Rev., 2015, 44, 4853–4921 RSC.
- X. Bao, Y. Yuan, J. Chen, B. Zhang, D. Li, D. Zhou, P. Jing, G. Xu, Y. Wang, K. Hola, D. Shen, C. Wu, L. Song, C. Liu, R. Zboril and S. Qu, Light: Sci. Appl., 2018, 7, 91 CrossRef PubMed.
- P. Zheng and N. Wu, Chem. – Asian J., 2017, 12, 2343–2353 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- X. Shu, A. Royant, M. Z. Lin, T. A. Aguilera, V. Lev-Ram, P. A. Steinbach and R. Y. Tsien, Science, 2009, 324, 804–807 CrossRef PubMed.
- D. Yu, W. C. Gustafson, C. Han, C. Lafaye, M. Noirclerc-Savoye, W.-P. Ge, D. A. Thayer, H. Huang, T. B. Kornberg, A. Royant, L. Y. Jan, Y. N. Jan, W. A. Weiss and X. Shu, Nat. Commun., 2014, 5, 3626 CrossRef CAS PubMed.
- C. Li, L. Cao, Y. Zhang, P. Yi, M. Wang, B. Tan, Z. Deng, D. Wu and Q. Wang, Small, 2015, 11, 4517–4525 CrossRef CAS PubMed.
- L. Y. Huang, S. Zhu, R. Cui and M. Zhang, Anal. Chem., 2020, 92, 535–542 CrossRef CAS PubMed.
- D. E. Kim, D. Schellingerhout, F. A. Jaffer, R. Weissleder and C. H. Tung, J. Cereb. Blood Flow Metab., 2005, 25, 226–233 CrossRef PubMed.
- M. Zhang, J. Yue, R. Cui, Z. Ma, H. Wan, F. Wang, S. Zhu, Y. Zhou, Y. Kuang, Y. Zhong, D.-W. Pang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6590–6595 CrossRef CAS PubMed.
- J. Ando, K. Fujita, N. I. Smith and S. Kawata, Nano Lett., 2011, 11, 5344–5348 CrossRef CAS PubMed.
- S. Harmsen, R. Huang, M. A. Wall, H. Karabeber, J. M. Samii, M. Spaliviero, J. R. White, S. Monette, R. O’Connor, K. L. Pitter, S. A. Sastra, M. Saborowski, E. C. Holland, S. Singer, K. P. Olive, S. W. Lowe, R. G. Blasberg and M. F. Kircher, Nat. Biotechnol., 2015, 7, 271ra277 Search PubMed.
- J. Y. Huang, C. Zong, L. J. Xu, Y. Cui and B. Ren, Chem. Commun., 2011, 47, 5738–5740 RSC.
- T. Wei, H. Xing, H. Wang, Y. Zhang, J. Wang, J. Shen and Z. Dai, Talanta, 2020, 210, 120625 CrossRef CAS PubMed.
- A. Huefner, W. L. Kuan, R. A. Barker and S. Mahajan, Nano Lett., 2013, 13, 2463–2470 CrossRef CAS PubMed.
- J. Chang, A. Zhang, Z. Huang, Y. Chen, Q. Zhang and D. Cui, Talanta, 2019, 198, 45–54 CrossRef CAS PubMed.
- D. Ghosh, A. F. Bagley, Y. J. Na, M. J. Birrer, S. N. Bhatia and A. M. Belcher, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13948–13953 CrossRef CAS PubMed.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- J. Chen, Z. Sheng, P. Li, M. Wu, N. Zhang, X. F. Yu, Y. Wang, D. Hu, H. Zheng and G. P. Wang, Nanoscale, 2017, 9, 11888–11901 RSC.
- A. Jaworska, L. E. Jamieson, K. Malek, C. J. Campbell, J. Choo, S. Chlopicki and M. Baranska, Analyst, 2015, 140, 2321–2329 RSC.
- L. Puppulin, S. Hosogi, H. Sun, K. Matsuo, T. Inui, Y. Kumamoto, T. Suzaki, H. Tanaka and Y. Marunaka, Nat. Commun., 2018, 9, 5278 CrossRef CAS PubMed.
- J. Zhang, Z. Liu, P. Lian, J. Qian, X. Li, L. Wang, W. Fu, L. Chen, X. Wei and C. Li, Chem. Sci., 2016, 7, 5995–6005 RSC.
- B. Tang, F. Yu, P. Li, L. Tong, X. Duan, T. Xie and X. Wang, J. Am. Chem. Soc., 2009, 131, 3016–3023 CrossRef CAS PubMed.
- B. Dong, X. Song, C. Wang, X. Kong, Y. Tang and W. Lin, Anal. Chem., 2016, 88, 4085–4091 CrossRef CAS PubMed.
- J. Song, J. Zhou and H. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS PubMed.
- J. Yin, X. Kong and W. Lin, Anal. Chem., 2021, 93, 2072–2081 CrossRef CAS PubMed.
- Y. Zhou, S. Yu, J. Shang, Y. Chen, Q. Wang, X. Liu and F. Wang, Anal. Chem., 2020, 92, 15069–15078 CrossRef CAS PubMed.
- D. W. Hwang, H. Y. Kim, F. Li, J. Y. Park, D. Kim, J. H. Park, H. S. Han, J. W. Byun, Y.-S. Lee, J. M. Jeong, K. Char and D. S. Lee, Biomaterials, 2017, 121, 144–154 CrossRef CAS PubMed.
- W. Fu, C. Yan, Z. Guo, J. Zhang, H. Zhang, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2019, 141, 3171–3177 CrossRef CAS PubMed.
- R. Yan, Y. Hu, F. Liu, S. Wei, D. Fang, A. J. Shuhendler, H. Liu, H.-Y. Chen and D. Ye, J. Am. Chem. Soc., 2019, 141, 10331–10341 CrossRef CAS PubMed.
- X. Gao and N. Wu, Electrochem. Soc. Interface, 2016, 25, 79–81 CrossRef CAS.
- Point of Care/Rapid Diagnostics Market, https://www.marketsandmarkets.com/Market-Reports/point-of-care-diagnostic-market-106829185.html.
| This journal is © The Royal Society of Chemistry 2022 |