
Porphyrinoids, a unique platform for exploring excited-state aromaticity
Jinseok
Kim
a,
Juwon
Oh
*b,
Atsuhiro
Osuka
*c and
Dongho
Kim
 *a
*a
aDepartment of Chemistry, Yonsei University, Seoul 03722, Korea. E-mail: dongho@yonsei.ac.kr
bDepartment of Chemistry, Soonchunhyang University, Asan-si 31538, Korea. E-mail: juwoh933@sch.ac.kr
cDepartment of Chemistry, Graduate School of Science, Kyoto University, Kyoto 606-8502, Japan. E-mail: oosuka.atsuhiro.46c@st.kyoto-u.ac.jp
First published on 8th December 2021
Abstract
Recently, Baird (anti)aromaticity has been referred to as a description of excited-state (anti)aromaticity. With the term of Baird's rule, recent studies have intensively verified that the Hückel aromatic [4n + 2]π (or antiaromatic [4n]π) molecules in the ground state are reversed to give Baird aromatic [4n]π (or Baird antiaromatic [4n + 2]π) molecules in the excited states. Since the Hückel (anti)aromaticity has great influence on the molecular properties and reaction mechanisms, the Baird (anti)aromaticity has been expected to act as a dominant factor in governing excited-state properties and processes, which has attracted intensive scientific investigations for the verification of the concept of reversed aromaticity in the excited states. In this scientific endeavor, porphyrinoids have recently played leading roles in the demonstration of the aromaticity reversal in the excited states and its conceptual development. The distinct structural and electronic nature of porphyhrinoids depending on their (anti)aromaticity allow the direct observation of excited-state aromaticity reversal, Baird's rule. The explicit experimental demonstration with porphyrinoids has contributed greatly to its conceptual development and application in novel functional organic materials. Based on the significant role of porphyrinoids in the field of excited-state aromaticity, this review provides an overview of the experimental verification of the reversal concept of excited-state aromaticity by porphyrinoids and the recent progress on its conceptual application in novel functional molecules.
 Jinseok Kim | Jinseok Kim received his BS (2017) degree from the Department of Chemistry at Yonsei University. He is currently a PhD student in Prof. Dongho Kim's laboratory. |
 Juwon Oh | Juwon Oh received his BS (2013) and PhD (2019) degrees from the Department of Chemistry at Yonsei University. After postdoctoral research with Prof. Dongho Kim at Yonsei University and Prof. Stephen R. Leone at the University of California Berkeley, he joined the Department of Chemistry at Soonchunhyang University as an Assistant Professor (2020). |
 Atsuhiro Osuka | Atsuhiro Osuka received his BS (1977) and PhD (1982) from Kyoto University. In 1979, he started his academic carrier at Ehime University as an Assistant Professor. In 1984, he came back to Kyoto University, where he has been a Professor since 1996. His research interests cover many aspects of synthetic approaches toward novel porphyrin-related compounds with intriguing structures, properties, and functions, which were recognized with the Chemical Society of Japan Award in 2010. Representative molecules explored in his laboratory include artificial photosynthetic reaction center models, meso–meso-linked porphyrin arrays, porphyrin tapes, expanded porphyrins, subporphyrins, and Möbius aromatic and antiaromatic molecules. |
 Dongho Kim | Dongho Kim received his BS (1980) from Seoul National University and PhD (1984) from Washington University. After postdoctoral research at Princeton University, he joined the Korea Research Institute of Standards and Science (1986). In 2000, he moved to Yonsei University as a Professor of Chemistry and is now an Underwood Distinguished Professor. He received the Scientist of the Month Award (1999), the Sigma-Aldrich Award (2005), the Korea Science Award in Chemistry (2006), the Star Faculty Award (2006), the National Science Achieving Excellence Award (2010), FILA Basic Science Award (2017), Academic Excellence Prize in the Korean Chemical Society (2018), JPA Honda–Fujishima Award (2019), Hans Fisher Award (2020), and Sudang Science Award (2020). His research activity has been focused on the ground- and excited-state aromaticity and excited-state properties and dynamics of various π-conjugated systems, including porphyrinoids. |
1. Introduction
Porphyrins are composed of four tetrapyrrolic macrocycles bridged by sp2-carbons. The rich metalation and redox chemistry arising from the pyrrole-based composition of porphyrinoids display various key roles in vital activities, such as respiration, photosynthesis, and electron-transfer/redox processes.1–6 Moreover, the macrocyclic π-conjugation and its easy tunability have offered additional scope for applications of porphyrinoids in dyes, anion sensors, organic semiconductors, optical-electronic materials, and photodynamic therapy.7 This nature of porphyrinoids has further motivated the extension of scientific interest to their structural analogues, such as contracted, isomeric, and expanded porphyrins in recent decades, because these size-transformations lead to significant changes in their physicochemical properties and increase their applicability.8For porphyrinoids, aromaticity is one of the most widely studied issues. Aromaticity is one of the most popular underlying concepts in the field of chemistry9,10 after the discovery of benzene by Michael Faraday in 182511 and the proposal of its resonance structure by August Kekulè in 1865.12 The significant energetic stabilization from the aromatic effect plays a critical role in governing the properties and reactivities of a wide variety of molecules, which was rationalized by the Hückel molecular orbital (HMO) theory by Erich Hückel in 1931.13 His [4n + 2]/[4n]π rule provides insight into the molecular stability in a surprisingly simple manner. Much attention regarding aromaticity has continuously been extended to the 3D structure and electronically excited state with its pivotal role in chemistry. In 1964, Edgar Heilbronner suggested the realization of aromaticity in Möbius strip topology with [4n]π electrons.14 After a while, Colin Baird introduced a modulation of electronic interactions in the lowest triplet state,15 which proposed the reversal of aromaticity in the excited states and its crucial role in photophysics and chemistry.
Over the past few decades, due to their aromaticity and distinct characteristics depending on their aromatic and antiaromatic nature, porphyrinoids have been the center of attention. The meso-methine-bridged pyrrole (and pyrrolic analogues including thiophene, furan, and selenophene) arrangement of porphyrinoids (Fig. 1) shows very effective π-conjugation.16–26 Notwithstanding the strong aromaticity of five-membered subunit rings, porphyrinoids have stable exocyclic π-conjugation over the entire molecule.27 This enables porphyrinoids to have a distinct aromatic or antiaromatic nature, depending on their [4n + 2] or [4n]π electrons, respectively, which dominates their molecular properties, such as diatropic (paratropic) ring current,28–30 conformation,31–33 absorption/emission,34 and nonlinear optical properties.35–39 In particular, expanded porphyrins have been highlighted with their highly flexible conformation by the increased ring size as well as the superior π-electron delocalization, which further facilitates the realization of distinct Möbius aromaticity and antiaromaticity in expanded porphyrins, as well as Hückel aromaticity and antiaromaticity.40–42 Their superior exocyclic π-conjugation enables the control of the number of electrons on the electronic circuit by redox reaction without the need for additional chemical changes26,43–50 and enables a distinct aromatic switching nature between Hückel and Möbius (anti)aromaticity by controlling the surrounding environment, such as controlling the temperature,51–53 solvent polarity,54,55 and (de)protonation.26,47,56–63
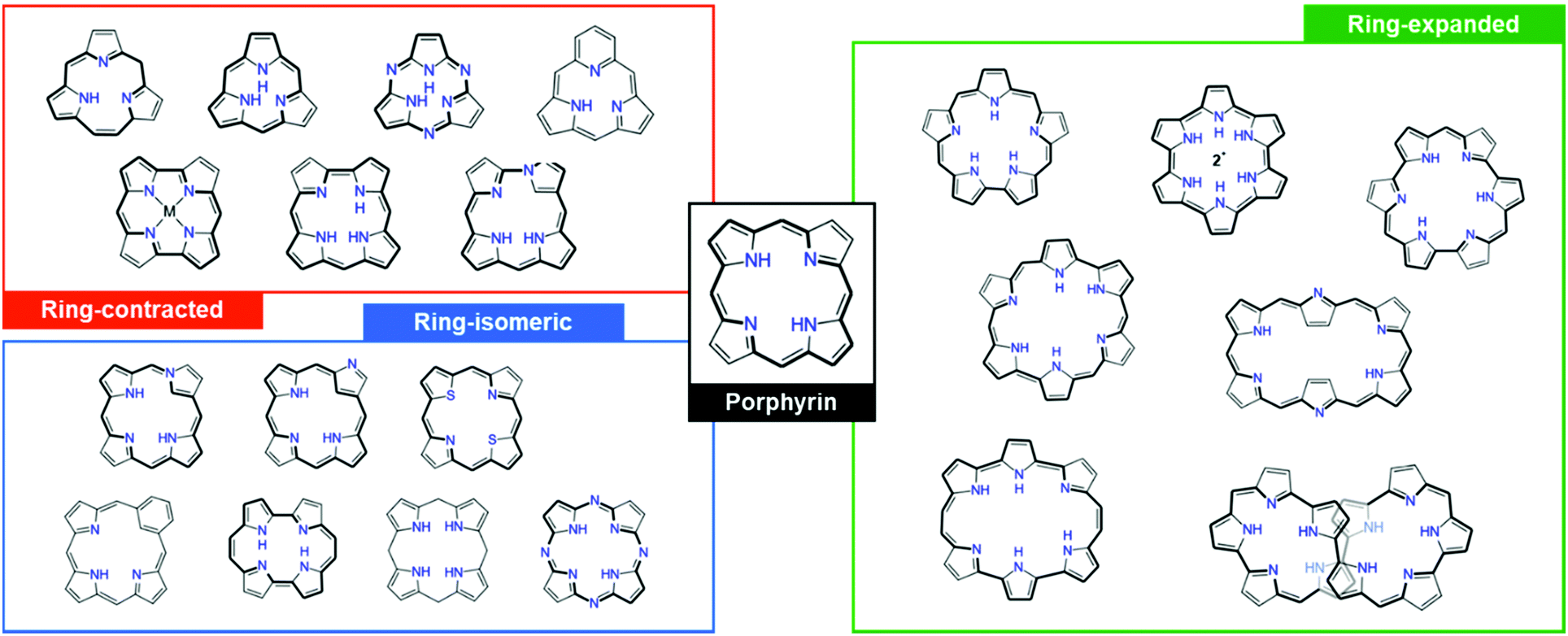 | ||
| Fig. 1 Various ring-contracted, -isomeric, and -expanded porphyrinoids as compared to the standard four tetrapyrrolic macrocycle, porphyrins. | ||
The distinct aromatic nature of porphyrinoids allows further studies on the fundamental characteristics of the aromaticity of oligomeric macrocycles. Because of the large flexibility of annulenes precluding the maintenance of aromaticity,64–67 the macrocyclic aromaticity has been realized mainly in cyclic oligomeric systems composed of aromatic subunits. In this regard, the contribution of local aromaticity from cyclic subunits and macrocyclic aromaticity from exo-cyclic π-conjugation to global aromaticity have been primary issues for the comprehensive understanding of their physicochemical properties. Here, the porphyrinoids are representative of fully π-conjugated oligomeric macrocycles and their intense aromatic nature serves as an effective test-bed. The experimentally observed similar features in 1H NMR and single-crystal X-ray diffraction results between porphyrinoids and annulenic counterparts ruled in favor of the major effect of macrocylic aromaticity.68–71 On the other hand, it was suggested by the energetic analysis of porphyrinoids that the local aromaticity of pyrrolic units contributes much more to their global aromaticity.72 According to evaluations based on the magnetically induced current, it is suggested that the local and macrocyclic aromaticity both mainly affect the global ring current.73–81
Recently, these merits of porphyrinoids for studying aromaticity in the ground state have been extended to the excited states. Following Baird's pioneering suggestion for aromaticity in the lowest triplet state, and the reversed Hückel rule for [4n]π aromatic and [4n + 2]π antiaromatic molecules,15 enormous scientific endeavors have been dedicated to revealing the excited-state aromaticity reversal, with the expectation of its vital role in photochemistry and excited-state behaviors.64–66,82–84 The reversed Hückel's counting rule for the lowest triplet state has been intensively verified with semi-empirical and ab initio levels of computational analyses.85–91 This has further led to the conceptual evolution of aromaticity to the transition and excited-singlet states and provided crucial insight into understanding photochemical processes and photophysical properties.66,92–94 Significant scientific endeavors have been dedicated to revealing the excited-state aromaticity reversal, with the expectation of its vital role in photochemistry and excited-state behaviors.82–84 However, most studies have been biased toward theoretical approaches. Experimental substantiation of excited-state aromaticity has been limited to only a few examples that are associated with reaction intermediates and mechanisms.95–98 The practical cause of the difficulty in experimental approaches is the restriction to direct measurements of typical magnetic, geometric, and chemical indices for evaluating aromaticity in the transient excited states. The distinct aromaticity-dependent properties of porphyrinoids, particularly their optical spectroscopic features, provide breakthroughs to these constraints for the experimental investigation of excited-state aromaticity.99,100 A simple redox reaction on porphyrinoids can provide aromatic and antiaromatic congener pairs with the same molecular framework but a different number of π-electrons and hydrogen atoms, which are suitable for investigating the pure effect of aromaticity.43,63,101 According to these features, porphyrinoids are an ideal platform for experimentally exploring excited-state aromaticity.
This review focuses mainly on the most recent progress in the verification of aromaticity reversal in the singlet and triplet states and their application with an emphasis on porphyrinoids. We begin by highlighting the concepts of aromaticity and introducing the unique nature of porphyrinoids, depending on aromaticity. We then present the research endeavors to reveal the reversal of aromaticity in porphyrinoids based on spectroscopic evidence. Lastly, we discuss the recent studies on the concept of excited-state aromaticity, which investigated the practical application of the concept to functional molecular systems such as diradicals, multiple exciton and charge generation, and proton transfer.
2. Aromaticity and the aromaticity-dependent nature of porphyrinoids
2.1. Concepts of aromaticity from the ground state to the excited states
Studies for revealing the relationships between molecular structures and properties are major issues in the field of chemistry because structures are closely related to the properties of chemical compounds. In this respect, aromaticity is one of the most important characteristics contributing to the physicochemical properties and structures of organic molecules. The formation of aromaticity by cyclic π-conjugation brings about energetic stabilization, which serves as a key for the underlying principle to rationalize a diverse range of structures, properties, and processes of organic molecules. The discovery of benzene by Faraday in 1825 marks the beginning of this important concept in chemistry.11 Hofmann classified benzene and its analogues as aromatic compounds in 1855 based on their properties, which distinguished them from unsaturated cyclic hydrocarbon compounds.102 In 1865, Kekulè proposed revolutionary cyclic resonance structures of benzene,12 alternating single/double-bonded structures, motivating numerous scientific endeavors devoted to the investigation of aromaticity and its effects.103–105 During this continuous effort, Hückel reported an innovative concept for aromaticity, the so-called [4n + 2]π rule for Hückel (anti)aromaticity (Fig. 2).13 The approximate linear combination of atomic π-orbitals, HMO theory, accounts for aromatic stabilization and antiaromatic destabilization effects remarkably well. Despite the embryonic level of quantum chemical approaches, this achievement plays a critical role in gaining crucial insight into molecular structures, properties, and reactivities readily based on Hückel (anti)aromaticity. Subsequently, with the advancement of experimental and computational technologies, continuous efforts have been devoted to exploring aromaticity and its principles, which extend the significance of the role of aromaticity and Hückel's rule to a wide variety of organic materials, and numerous studies have been focused on revealing the underlying effects of aromaticity.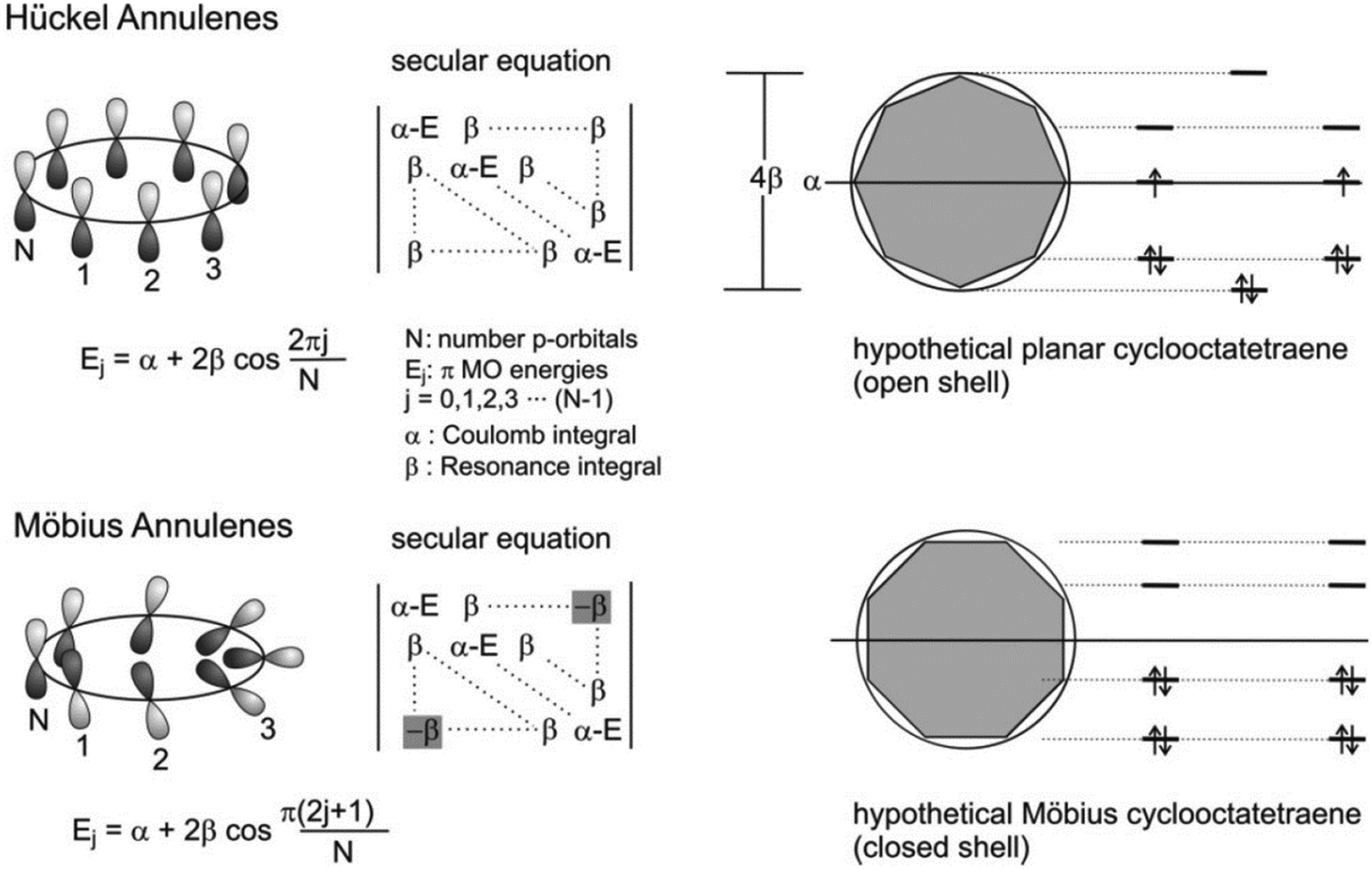 | ||
| Fig. 2 Frost-Musulin mnemonic for the Hückel and Zimmerman device for Möbius annulenes. Note that there is a -β in the secular equation of the Möbius annulene, representing the sign inversion. Adapted from ref. 177 with permission. Copyright 2006 American Chemical Society. | ||
With the crucial role of aromaticity in the field of chemistry, its concept has been extended to various molecular systems, such as Möbius,14 Craig,106 spherical,107,108 and Baird15 aromaticity. Among them, the most representative one is Möbius aromaticity, the extension of the aromaticity concept from the planar π-conjugation geometry to 3D Möbius-strip topology. Möbius aromaticity was first proposed by Heilbronner in his report,14 “Hückel molecular orbitals of Möbius-type conformations.” To apply HMO theory to the one-surface Möbius-strip conformation, two different boundary conditions are considered as compared to two-surface planar Hückel systems: (1) reduced p-orbital overlap, and (2) antiphase at an inversion point. These indicate that the counting rule for Hückel (anti)aromaticity is reversed in Möbius (anti)aromatic systems, [4n]π and [4n + 2]π for aromaticity and antiaromaticity, respectively. It is further rationalized that the conformational twist is overcome with Möbius aromatic stabilization (Fig. 2). However, because the π-orbital overlap and structural twist for Möbius-strip topology is conflicting, the Möbius aromatic compound has not yet been discovered in nature. The first synthetic Möbius aromatic molecule was achieved by Herges and colleagues109 three decades after the first conceptual announcement using an ingenious synthetic strategy of a connection of normal and in-plane(belt-like) conjugation. Since this pioneering synthetic work, various Möbius aromatic and antiaromatic molecules have been realized with porphyrinoids.110–126
In addition to the spatial extension of the concept of aromaticity from the planar Hückel to 3D Möbius-strip structures, recently, much of the attention on aromaticity has shifted to another topic, excited-state aromaticity.100,127 In general, aromaticity is considered for the singlet ground state. Since its pivotal role is in the chemical nature of organic molecules in the singlet ground state, aromaticity and antiaromaticity in the excited states have been expected to provide considerable insight into an understanding of photochemistry and excited-state processes. The conceptual foundation of excited-state aromaticity began with independent studies by Dewar and Zimmerman in the 1960s,128–130 where they mentioned aromaticity in the transition state to understand the progress of cyclic ring closure reactions. In 1971, Dougherty further developed this concept, accounting for the feasibility of pericyclic reactions with the term transition-state aromaticity, which highlighted the reversal of Hückel's counting rule in the transition state: [4n + 2]π for aromaticity and [4n]π for antiaromaticity.131
Aromaticity in the electronically excited state, the lowest triplet (T1) state, (π,π*)3, was first perceived by Baird in 1972.15 He analyzed the (anti)aromaticity of benzene and a series of hydrocarbons in the T1 state by the perturbation molecular orbital (PMO) theory (Fig. 3 and 4). More specifically, energetic stabilization and destabilization resulting from a π-electronic interaction between two decomposed polyenyl monoradicals was deduced as a model for T1-state aromatic molecules.82,127 Considering the T1 state of benzene, π-electronic interactions of two decomposed allyl radicals can be classified into two types of interactions (Fig. 3): the π-electronic interaction between (1) singly occupied π-MOs (π-SOMOs, Type-I interaction) and (2) π-SOMO and doubly-occupied and vacant π-MOs (Type-II interaction). Since the electronic interaction is determined by orbital symmetry and the energy level, the Type I interaction in T1 benzene is antibonding (out-of-phase) and destabilized based on the PMO theory. Here, there is no Type II interaction in T1 benzene. Thus, T1 benzene is more destabilized than the two allyl radicals, representing nonaromaticity. On the other hand, the Type I and II interactions for T1-state cyclooctatetraene are stabilized in the same manner (Fig. 4). This is known as ‘Baird's rule,’ and directly describes the reversal of Hückel's counting rule. The qualitative analysis of the reversal of aromaticity in T1 state was demonstrated with a semi-empirical level of calculations,15,85 where Aihara extended the concept of aromaticity reversal to Möbius aromaticity with a comparative analysis of the resonance energy of Möbius topology annulenes with the planar Hückel counterparts.
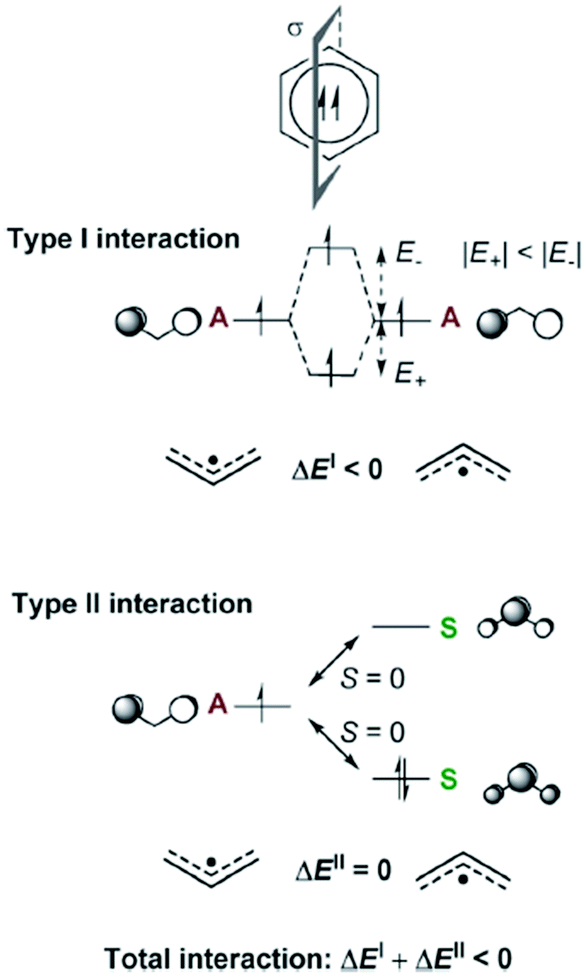 | ||
| Fig. 3 The Type I and Type II interactions between the π-MOs of two allyl monoradical fragments leading to triplet biradical benzene. For the Type II interaction, only one of the two equivalent interactions is displayed. Fragment orbitals that are symmetric with regard to the bisecting mirror plane are labelled S and those that are antisymmetric are labelled A. Zero orbital overlap between fragment orbitals of different symmetries is denoted as S = 0. Adapted from ref. 83 with permission. Copyright 2015 Royal Society of Chemistry. | ||
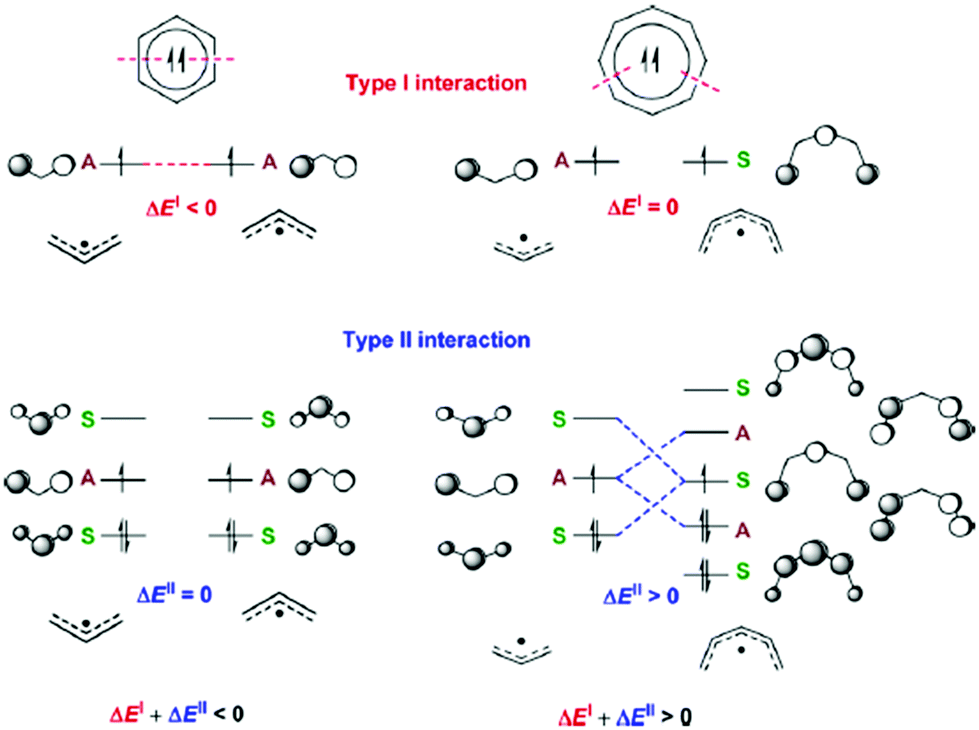 | ||
| Fig. 4 Schematic drawing that displays the Types I and II interactions (ΔEI and ΔEII, respectively) between the π-orbitals of suitable polyenyl radical fragments which when combined yield triplet biradical benzene and triplet biradical COT. The nonzero Types I and II interactions are marked with dashed lines in red and blue, respectively. The symmetry or antisymmetry of a π-MO with respect to a bisecting mirror plane is labeled with an S and an A, respectively. A total interaction ΔEI + ΔEII > 0 corresponds to a stabilization upon cyclization and a triplet state aromatic ring, whereas a total interaction ΔEI + ΔEII < 0 corresponds to a destabilization upon cyclization and a triplet state antiaromatic ring. Adapted from ref. 82 with permission. Copyright 2014 American Chemical Society. | ||
With advancements in computational technology and quantum mechanical tools, extensive efforts have been made to verify this pioneering concept more exactly with ab initio high-level quantum chemical calculations.84,86,98,132–137 Along with the triplet-state aromaticity, the scientific concern regarding aromaticity has expanded into singlet-excited states. Since most molecules exist in the singlet ground state, molecular excitation inevitably produces species in the singlet-excited states, and species in the triplet state are also formed through the singlet-excited state, a process known as intersystem crossing (ISC).138 The broader significance and universality of the singlet-excited states impart significance to singlet-excited-state aromaticity for understanding photochemistry and excited-state dynamics. Aromaticity and magnetic characteristics of benzene in the singlet- and triplet-excited states were investigated by Kataoka in 2004,139 where the strong antiaromatic nature was estimated with a semi-empirical calculation analysis. The extension of aromaticity to singlet-excited states was further verified with high-level ab initio quantum chemical calculations.86,140–143 In 2008, Karadakov demonstrated reversed (anti)aromaticity in the lowest singlet-excited state by magnetic index analyses with CASSCF calculations.144,145 Feixas et al. manifested this concept of excited-state aromaticity with analyses of excited-state π-electron delocalization.87,146,147
In conjunction with theoretical investigations, this concept of excited-state aromaticity has been proved experimentally with analyses of various chemical reactions of aromatic hydrocarbon compounds. Wan and Krogh proved that photosolvolysis of 9-fluorenol is driven by [4]π aromatic intermediates.95 After this seminal work, the formation of [4n]π aromatic species in photochemical reaction routes has been intensively scrutinized.96,97,148–160 Along the same line, the photochemical reaction routes, photoreactivity, and photophysical properties of aromatic arene derivatives have been rationalized based on the concept of excited-state aromaticity (Fig. 5).161–163 The destabilization induced by excited-state antiaromaticity triggers photochemical rearrangements for alleviating instability in the excited states, which governs the photochemical reaction routes and products (Fig. 6).83,164–172 In addition, the excited-state antiaromaticity of arene derivatives increases photoreactivity, which was applied in the photosilylation of polycyclic aromatic hydrocarbons and graphene.173 Correspondingly, transition-state aromaticity has been investigated with the direct observation of the photoinduced ring-inversion process of chiral thiophene-fused [4n]π annulenes based on the concept of aromaticity reversal, where the formation of planar [8]π aromatic cyclooctatetraene in the transition state reduces the activation enthalpy and facilitates the ring-inversion process (Fig. 7).94 Recently, the observation of interconvertible absorption spectra of aromatic and antiaromatic bis-rhodium hexaphyrins between the S0 and T1 states provided direct experimental evidence for aromaticity reversal in the T1 state.99
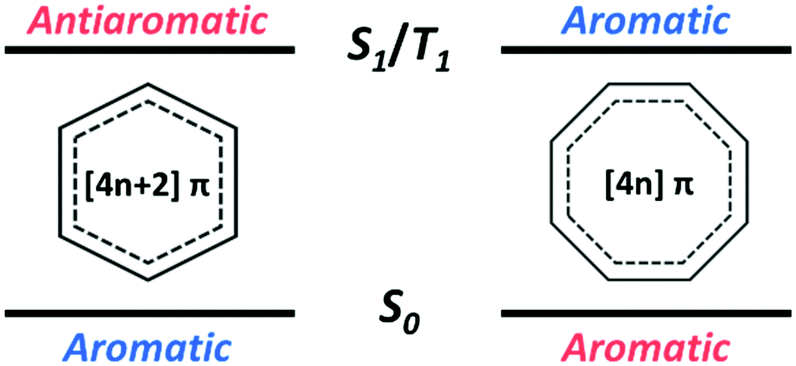 | ||
| Fig. 5 The reversed (anti)aromaticity in the excited states as the concept for excited-state (anti)aromaticity. | ||
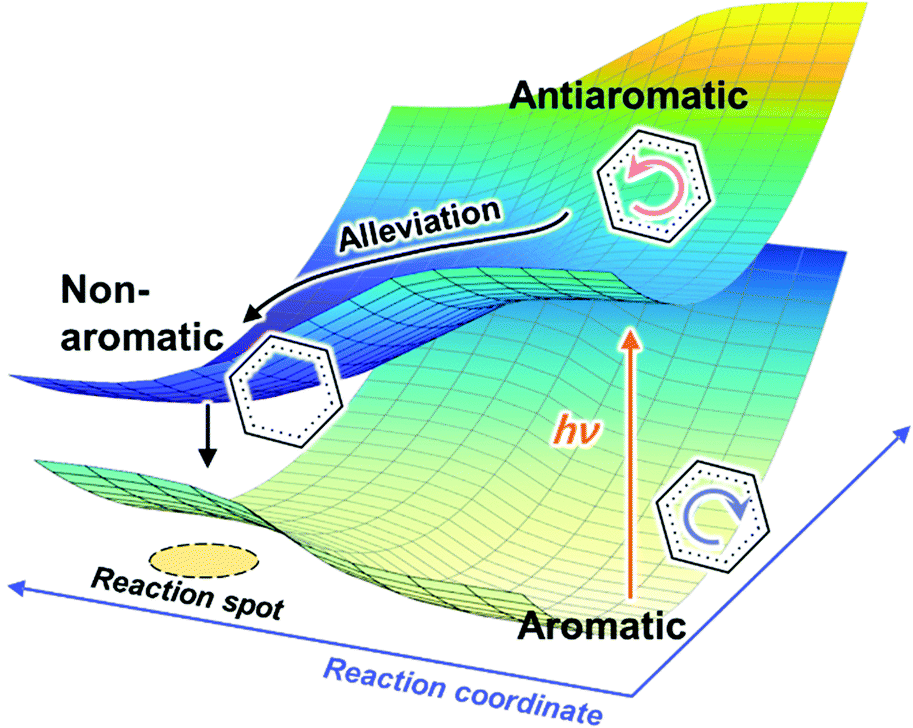 | ||
| Fig. 6 Schematic description of the effect of destabilization of antiaromaticity by aromaticity reversal on the excited state dynamics and photochemistry. | ||
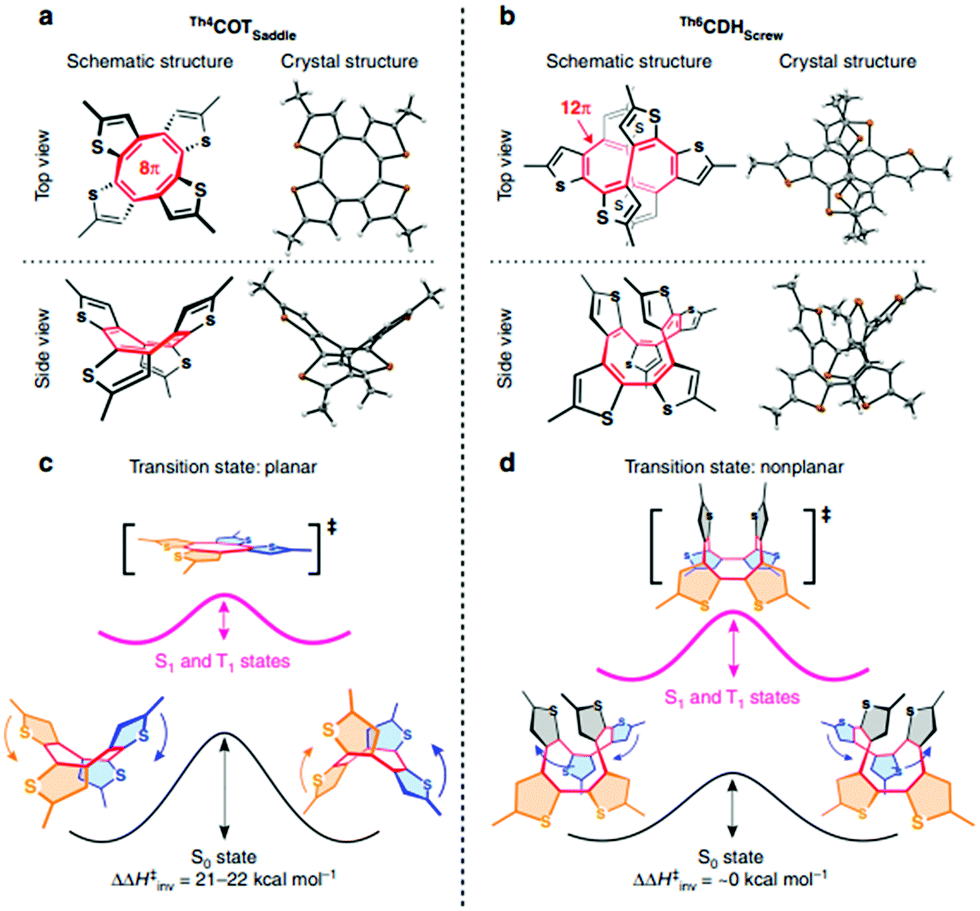 | ||
| Fig. 7 [4n]Annulene derivatives with and without Baird aromaticity upon photo-excitation. (a and b) Molecular structures and ORTEP drawings (50% ellipsoid probability) of Th4COTSaddle (a) and Th6CDHScrew (b), which have 8π-electron and 12π-electron annulene cores (red colored), respectively. (c and d) Schematic illustrations of the energy barriers for the ring inversion events of Th4COTSaddle (c) and Th6CDHScrew (d), where colored arrows represent the movement directions of the thiophene rings of the same color. Th4COTSaddle and Th6CDHScrew inverted through planar and nonplanar transition states, respectively, as shown in the square brackets. Black and pink-colored solid curves represent energy barriers in the ground and photoexcited states, respectively. Upon photoexcitation, the activation enthalpy for the ring inversion (ΔH‡inv) of Th4COTSaddle is lowered by 21–22 kcal mol−1 but that of Th6CDHScrew remains unchanged. Adapted from ref. 94 with permission. Copyright 2017 Springer Nature. | ||
2.2. Aromaticity-dependent characteristics of porphyrinoids and the evaluation of excited-state aromaticity
As previously mentioned, aromaticity is one of the most frequently used concepts to explain a wide range of underlying principles of molecular properties and chemical processes. Starting with a study of the resonance structure of benzene, annulenes have been privileged molecular systems for the in-depth investigation of aromaticity theoretically and experimentally.103,174–176 Even if annulenes are the most intuitive molecules for aromaticity, annulenic molecules larger than COT are inherently flexible and their π-conjugation is fragile. Thus, for macrocyclic annulenes, their large flexibility is practically detrimental to entire cyclic π-electron delocalization, resulting in localized π-conjugation and nonaromatic nature,177 practically restricting experimental investigation of molecular aromaticity.In recent decades, extensive interest in molecular aromaticity has focused on porphyrinoids due to their distinct and stable aromatic nature arising from macrocyclic π-conjugation.121,125,178–184 Compared to the sp2-carbon-based composition of annulenes, sp2-carbon-bridged pyrrolic macrocycles of porphyrins afford additional structural rigidity without perturbing exocyclic π-electron delocalization by pyrrole subunits, which results in stable heteroaromatic diaza [18]annulenic π-circuits comparable to that of planar symmetric C18H18. Based on these structural characteristics of porphyrinoids, expanded porphyrins, such as nonaphyrin, decaphyrin and dodecaphyrin121,184,185 show effective π-electron delocalization over the whole main macrocyclic pathway and solid aromaticity even for their substantially large sizes (Fig. 8).
 | ||
| Fig. 8 The fully π-conjugated aromatic-expanded porphyrins composed of more than seven pyrrole subunits. | ||
The superior π-electron delocalization and adequate conformational flexibility of porphyrinoids, in particular for expanded porphyrins, also allow the realization of various types of aromaticity and antiaromaticity in porphyrinoids (Fig. 9). [4n]Porphyrinoids, such as [16]norcorrole186 [20]orangarin,29,33 [24]pentaphyrin,187,188 [24]rosarin,189 and [28]hexaphyrin190–192 show a distinct paratropic ring current under planar geometry, indicating their antiaromatic nature. In addition to Hückel aromaticity and antiaromaticity, the concept of aromaticity under Möbius-strip topologies has been proved prolifically with porphyrinoids. After the first synthetic Möbius aromatic molecules by Herges and colleagues,109 the first example of Möbius aromatic porphyrinoids, A,D-di-p-benzihexaphyrin, was reported by Latos-Grazynski in 2007 (Fig. 10).193 Subsequently, Möbius aromaticity has been accomplished in various porphyrinoids, such as phosphorous-coordinated hexaphyrins and heptaphyrins,119 with their advantages of π-electron delocalization and flexible structures. Going one step forward, stable Möbius antiaromatic porphyrinoids have even been synthesized with vacataporphyrins,194 hexaphyrins,115 and heptaphyrins119 showing distinct paratropic ring currents under Möbius geometry. Because of these properties of porphyrinoids, aromaticity research has become central to porphyrinoid research. Particularly, based on an unexpectedly stable exocyclic π-conjugation of porphyrinoids with transformable flexible structures, extensive efforts have revealed the switching of their aromaticity by solvent and temperature control,51–54 redox reaction,117 and (de)protonation.56–59,62,195
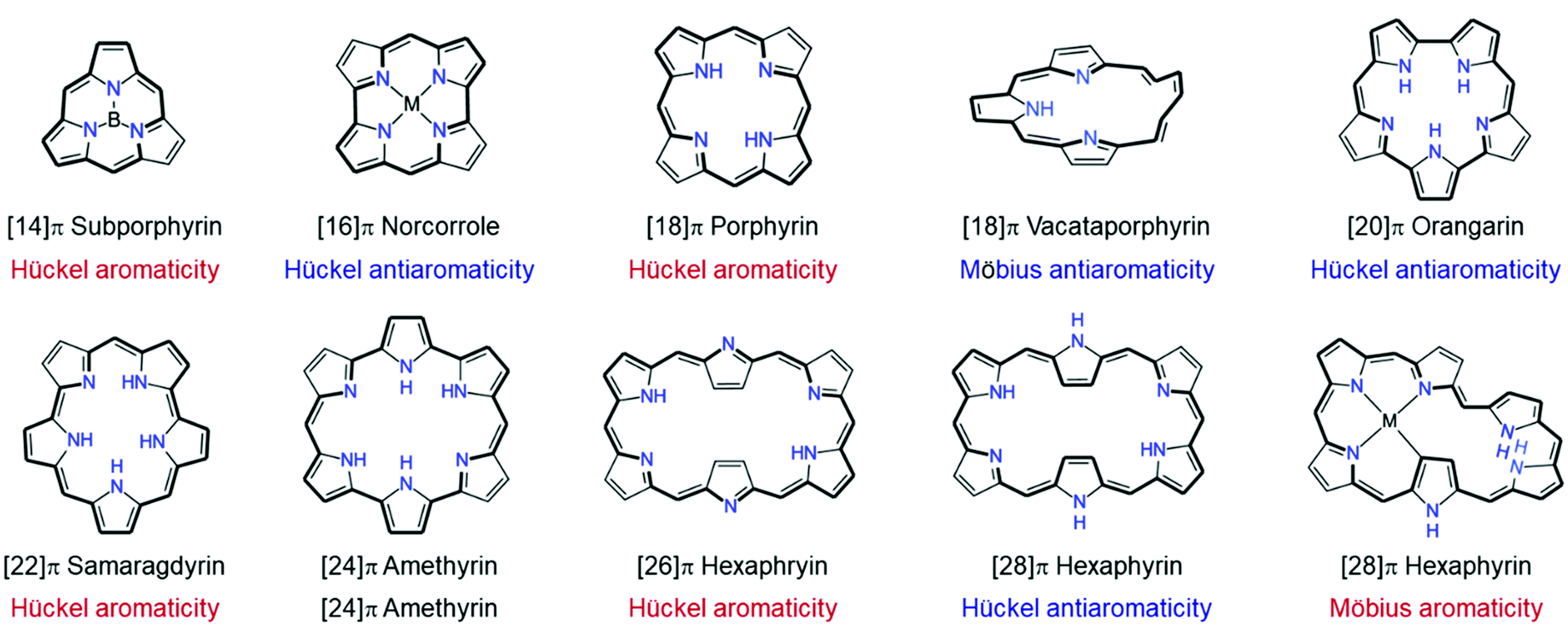 | ||
| Fig. 9 Representative Hückel and Möbius aromatic and antiaromatic porphyrinoids. | ||
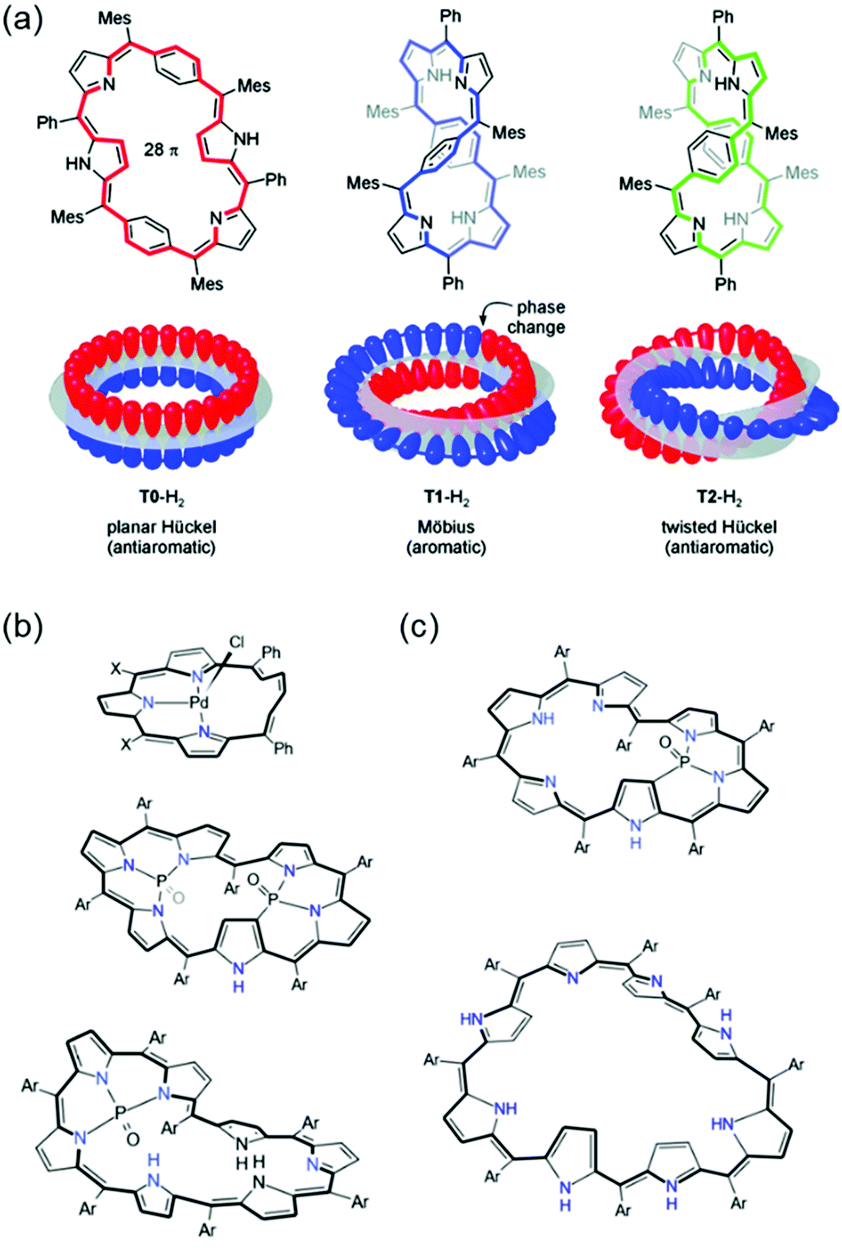 | ||
| Fig. 10 (a) The first realized Möbius aromatic system, A,D-di-p-benzihexaphyrin. Adapted from ref. 116 with permission. Copyright 2010 American Chemical Society. The representative Möbius antiaromatic (b) and aromatic (c) porphyrinoids. | ||
In addition to the typical aromaticity-dependent changes in the ring current, bond length alternation, and conformation, the aromaticity and antiaromaticity of porphyrinoids are distinctively reflected in the optical spectroscopic features, particularly for electronic absorption spectra (Fig. 11).34,35,38 This further renders the porphyrinoids effective for the investigation of aromaticity. Aromaticity covers the collective properties so as the macrocycles become larger, the evaluation of their aromaticity becomes more complicated experimentally and computationally. Recent papers on the aromaticity of cyclic porphyrin oligomers suggest the importance of multifaceted analyses for aromaticity in marocyclic molecules, not just a singular criterion.179,196,197 In this regard, the aromaticity-dependent optical spectroscopic features of porphyrinoids allow for more accurate investigation of their aromaticity.
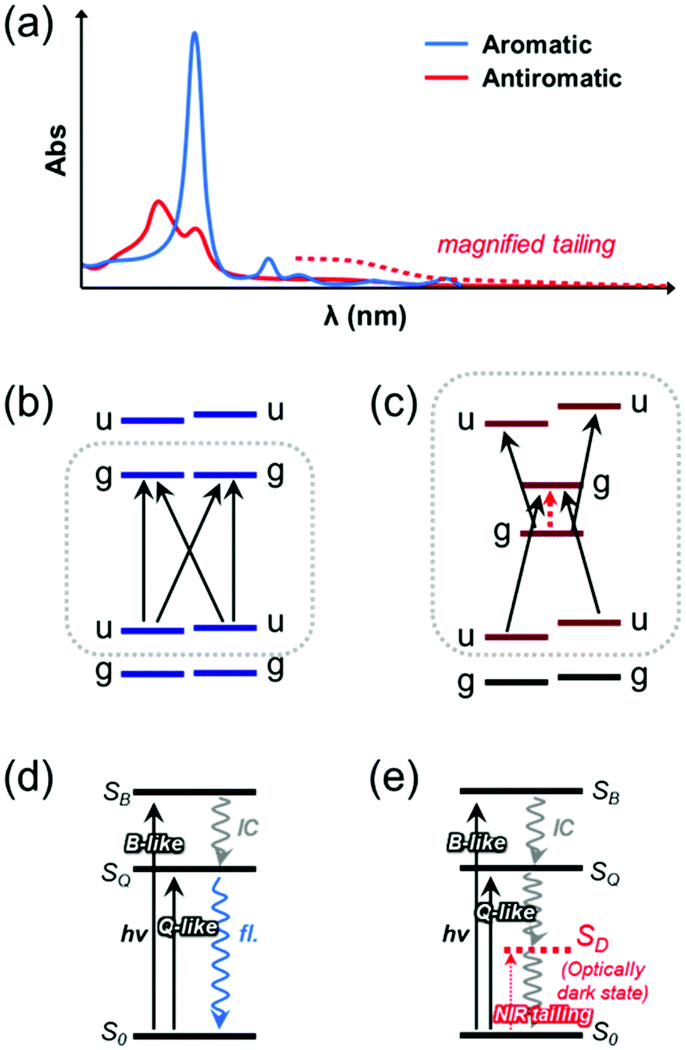 | ||
| Fig. 11 Aromaticity-dependent absorption spectral characteristics and π-electronic structures of porphyrinoids. (a) Representative electronic absorption spectra of aromatic and antiaromatic porphyrinoids. Scheme showing frontier molecular orbital structures of aromatic (b) and antiaromatic (c) porphyrinoids and electronic structures of aromatic (d) and antiaromatic (e) porphyrinoids. | ||
Aromatic porphyrinoids show intense B bands and weak Q bands in lower- and higher-wavelength regions, respectively. On the other hand, antiaromatic porphyrinoids exhibit overall attenuated absorption signals with a characteristic tailing feature over the near-infrared (NIR) region. These peculiar aromaticity-dependent optical spectral features of porphyrinoids arise from their distinguished electronic structures according to their aromatic and antiaromatic nature. The electronic structure of aromatic porphyrins was first explained with Gouterman's four-orbital model.172,198 In this model, a comparison of D4h diaza[18]annulene in porphyrins with a highly symmetric D16h C16H162− accounts for the two degenerate LUMO/LUMO+1 and quasi-degenerate HOMO/HOMO−1, which are the frontier MOs (FMOs). In these FMOs, their configuration interactions and the selection rule result in the optically allowed intense B band and forbidden weak Q bands in the steady-state electronic absorption spectra. Michl further developed the underlying principle with perimeter models, where the main electronic structures of aromatic and antiaromatic porphyrinoids were explained with four and six frontier MOs, respectively.199 Along with Gouterman's four-orbital model, the four degenerate FMOs and configuration interaction, producing optically allowed intense B band at higher-energy regions, optically forbidden weak Q bands at lower-energy regions, and their long-lived nanosecond-order fluorescence, are rationalized in more detail with his 4N + 2 perimeter model using the C16H162− perimeter.199–201
The electronic structure of antiaromatic porphyrinoids was described based on the configurational interaction of six FMOs with Michl's 4N perimeter model.38,101 In this concept, the FMOs of antiaromatic all-aza bis-gold hexaphyrin are comparatively analyzed with highly symmetric C24H244−. Compared to a degenerate singly occupied MO (SOMO) structure of the ideal cyclic annulenic counterpart, the inherently lowered molecular symmetry and structural distortion of the antiaromatic hexaphyrin, for alleviating antiaromatic destabilization, results in nondegenerate HOMO and LUMO with a considerably small energy gap. Here, HOMO and LUMO have the same gerade symmetry, which causes the lowest electronic transition between them to be optically forbidden by the selection rule.202,203 Thus, the small energy gap with the forbidden nature gives rise to the absorption tailing in the longer wavelength (NIR) region without fluorescence. Meanwhile, the degenerate HOMO−1/HOMO−2 and LUMO+1/LUMO+2 evolve into the energetically close structure with lowered symmetry lowering, where transitions from HOMO and LUMO to these MOs are optically allowed. Thus, relatively strong absorption signals are observed in the shorter-wavelength (UV-Vis) region. These unique aromaticity-dependent electronic structures of porphyrinoids have been intensively investigated with MCD spectroscopic analyses.51,204–209 Here, the geometrical complexity induced by additional ring-expension, -contraction, and -modification leads to a further symmetry-lowering and subsequently perturbs the characteristic four- and six-FMO structure of aromatic and antiaromatic porphyrinoids, respectively. In some core-modified expanded porphyrinoids, such as pentaphyrins and octaphyrins, the antiaromatic molecules show more intense absorption bands compared to their aromatic counterparts.203,210–212 In this review, we will focus on the relatively high-symmetric all-aza porphyrinoids for their aromaticity-dependent optical spectroscopic features.
Based on these MO analyses, π-electronic structures of aromatic and antiaromatic porphyrinoids have been comprehensively analyzed via transient absorption (TA) spectroscopy by Kim, Osuka and colleagues.20,35–38 TA spectroscopy is a type of time-resolved pump–probe spectroscopy for measuring photogenerated excited-state absorption and dynamics (Fig. 12). Because the TA spectrum is made up of absorbance difference (ΔA) signals, including excited-state absorption, induced by photoexcitation, it is mainly composed of three signals, namely, ground-state bleaching (GSB) from the decreased ground-state population, excited-state absorption (ESA), and stimulated emission (SE) from the excited population by photoexcitation. The temporal changes in these signals reflect the excited-state dynamics. Since the ground-state absorption spectra of porphyrinoids show obvious aromaticity-dependent features, the temporal changes in their TA spectra are also significantly affected by aromaticity-dependent electronic structures. For aromatic porphyrins, the configuration interactions of four FMOs generate the degenerate singlet excited states, the SB and SQ states, where a transition to the lowest excited state (degenerate SQ states), causing the weak Q-band absorption, is optically forbidden. Thus, its lifetime is relatively long, in the order of several nanoseconds, and aromatic porphyrins emit weak fluorescence (Q.Y. < 0.1). On the other hand, the significantly small energy gap between gerade HOMO and gerade LUMO in the six FMOs of antiaromatic porphyrins resulted in the energetically low dark excited singlet state (SD state), triggering the distinctive NIR absorption tailing. Here, the photoexcitation of antiaromatic porphyrinoids typically provokes the transitions to the higher singlet excited states (SB and SQ states, arising from the configuration interaction of HOMO−1/HOMO−2 with LUMO and HOMO with LUMO+1/LUMO+2) and subsequent internal conversion (IC) to the lowest dark state (SD state) occurs. This electronic nature triggers much shorter double exponential decay features in the orders of sub-picoseconds and tens of picoseconds without fluorescence emission. In addition to the temporal changes in the TA spectra, the TA spectral shape exhibits aromaticity-dependent features. The TA spectra of aromatic porphyrinoids show strong negative GSB signals with weak positive ESA signals on both sides of GSB signals. The sharp contrasting spectral features are observed in the TA spectra of antiaromatic porphyrinoids. They display relatively weak GSB signals with intense ESA signals in the TA spectra. These spectral features demonstrate that the ESA is also largely governed by the aromaticity-dependent electronic structures, implying that the ground-state absorption spectral features for aromaticity (or antiaromaticity) are reversed in the excited states.99
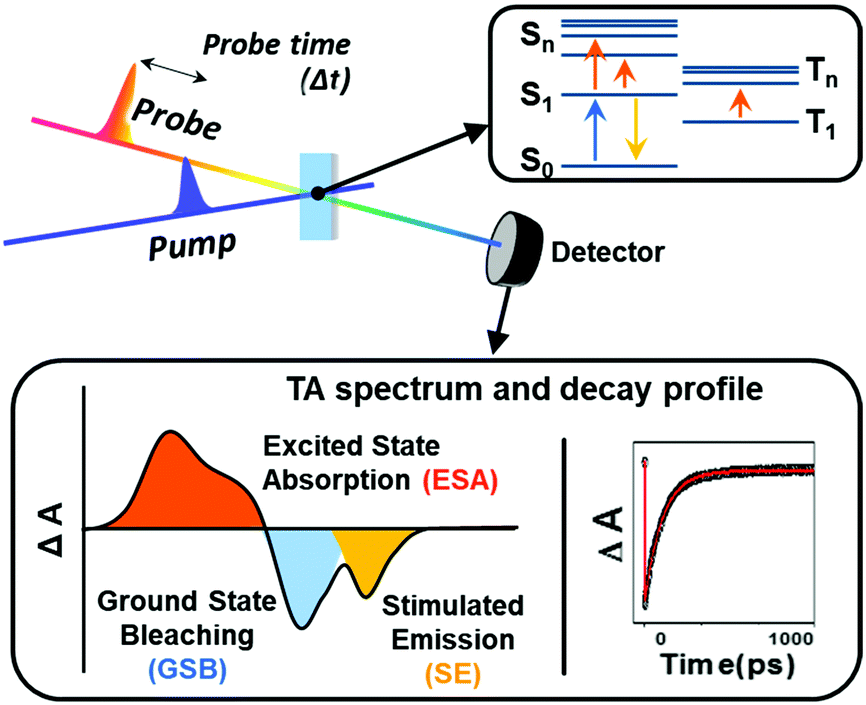 | ||
| Fig. 12 Schematic representation of transient absorption spectroscopy and measured spectra and decay profiles. | ||
These optical spectroscopic features of porphyrinoids play a key role in the direct experimental evaluation of excited-state aromaticity. Although there have been extensive theoretical studies on the aromaticity reversal in the excited states, its experimental demonstration has only been attained with scant examples about reaction intermediates and isomerization that show an indirect correlation between molecular properties and aromaticity in the excited states.95–98 This large difficulty in the experimental investigation of excited-state aromaticity lies in the practical constraints to measure typical indicators for evaluating excited-state aromaticity in the transient excited states, such as nuclear magnetic resonance and X-ray crystal measurements. Recent studies on the excited-state aromaticity of porphyrinoids have clarified that the aromaticity-dependent optical spectroscopic features of porphyrinoids serve as a direct index to evaluate their excited-state (anti)aromaticity and further provide a new perspective that the pump–probe spectroscopic measurements can act as an effective method for the evaluation of aromaticity in the excited states.100
Besides the distinctive optical spectroscopic features, porphyrinoids are one of the most relevant molecules for studying excited-state aromaticity based on their aromatic and antiaromatic congener sets.101 In general, the synthesis of a comparable set of [4n + 2]π and [4n]π molecules is arduous. The preparation of stable antiaromatic molecules is a challenging task213,214 however, for porphyrinoids, various antiaromatic cases have been reported.36,44,215 Moreover, the stable exocyclic π-conjugation pathway of porphyrinoids allows for a redox-induced aromaticity-switching nature, where the redox reaction only alters the number of π-electrons over the main macrocyclic π-conjugation without causing significant chemical changes.26,43–50 These molecular properties eventually facilitate the synthesis of various sets of aromatic and antiaromatic porphyrinoid congeners (Fig. 13). These congeners, with the same molecular framework but different numbers of π electrons and hydrogen atoms enable the pure effect of aromaticity to be unveiled free from other influences and allow the concept of excited-state aromaticity, aromaticity reversal, to be directly proved. With these advantages, excited-state aromaticity has recently been thoroughly demonstrated using porphyrinoids, which has further enabled the application of this concept to functional organic materials.
 | ||
| Fig. 13 The series of congener porphyrinoids: bis-rhodium Hückel aromatic and antiaromatic hexaphyrins (1 and 2, respectively), phenylene-bridged Hückel aromatic and antiaromatic hexaphyrins (3 and 4, respectively), palladium Hückel and Möbius aromatic hexaphyrins (5 and 6, respectively), N-confused rhodium Hückel and Möbius aromatic pentaphyrins (7 and 8, respectively), bispalladium Möbius aromatic and Hückel antiaromatic octaphyrins (9 and 10, respectively), bis-gold Hückel aromatic and antiaromatic hexaphyrins (11 and 22, respectively), and nickel-rhodium metalated Möbius aromatic and antiaromatic nonaphyrins (13 and 14, respectively). | ||
3. Verification of aromaticity reversal in the excited states with porphyrinoids
As previously mentioned, porphyrinoids exhibit unique aromaticity-dependent absorption spectral features, such as the magnitude of absorbance and spectral shape, as direct results of their distinguished electronic structures relying on (anti)aromaticity. The flexible structures of porphyrinoids also facilitate the determination of (anti)aromaticity based on their molecular geometries. Optical electronic and vibrational spectroscopies can measure these distinct characteristics of porphyrinoids in the excited states as well as the ground state.The formation of congeneric pairs of aromatic and antiaromatic porphyrinoids plays a decisive role in the verification of the concept of excited-state aromaticity reversal.63,100 Because the congener pairs are only different in the number of π-electron and hydrogen atoms, they allow for direct comparative analyses of features in the excited state induced by the aromaticity reversal. Here, the different numbers of hydrogen atoms may affect the molecular properties of porphyrinoids because their intramolecular hydrogen bonds arising from the multiple pyrrolic compositions have a significant influence on the molecular conformation and aromaticity.57,58,61,101,216–220 However, in this section, we discuss the research on metallated and bridged porphyrinoids, which show the negligible effects of the intramolecular hydrogen bond by the inner cavity modifications. The optical spectroscopic analyses of these aromatic and antiaromatic porphyrinoid congeners provide clear evidence for excited-state (anti)aromaticity.
3.1. Excited-state aromaticity reversal of porphyrinoids with electronic absorption analyses
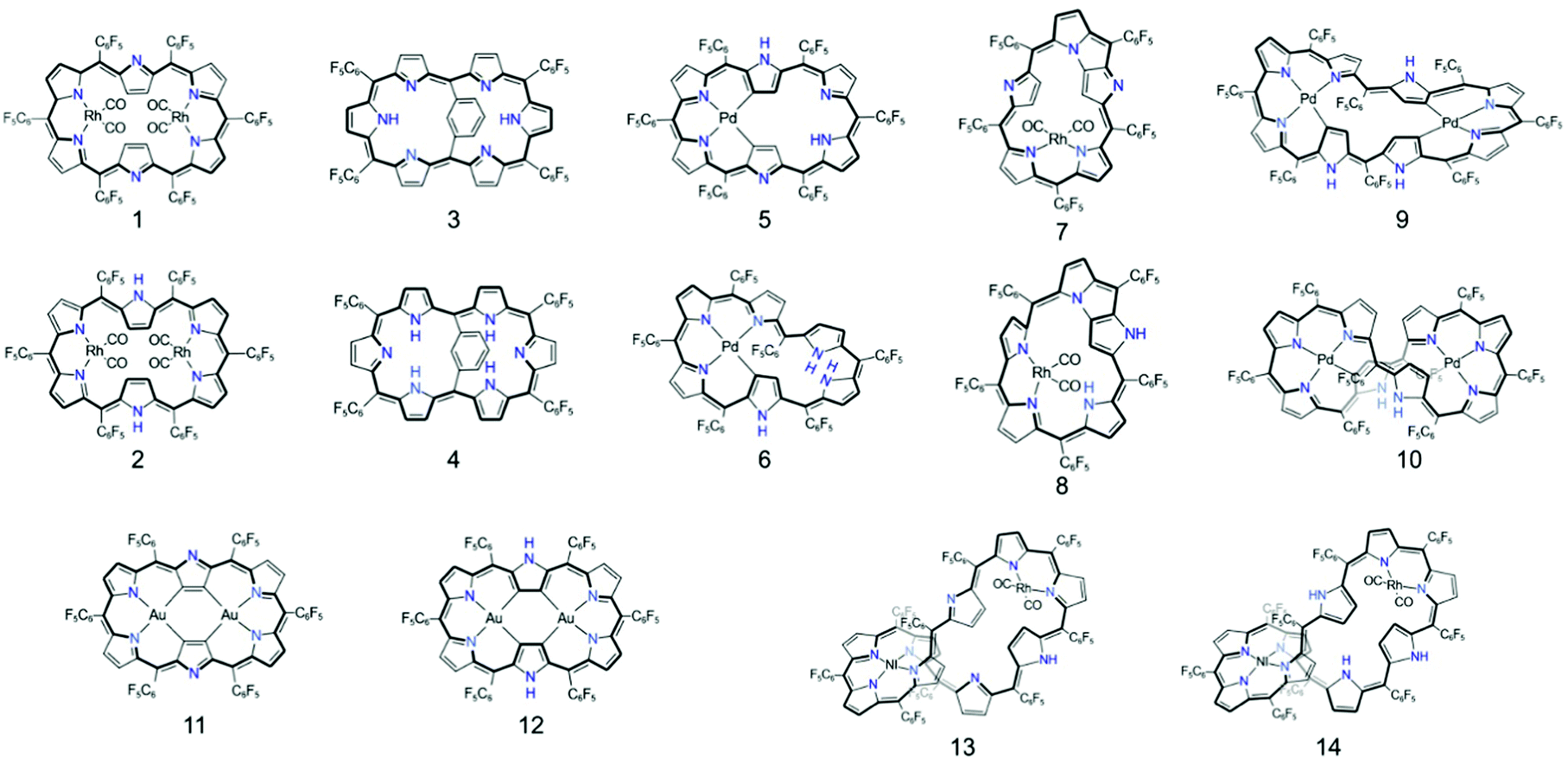
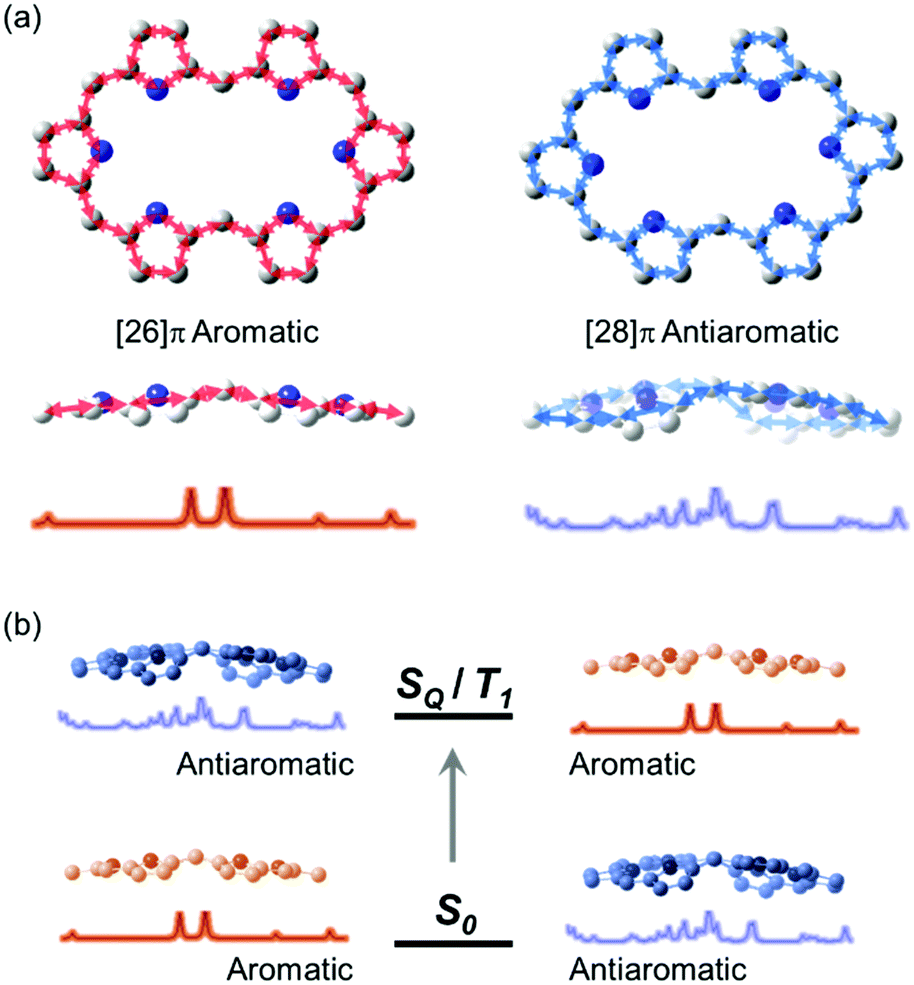
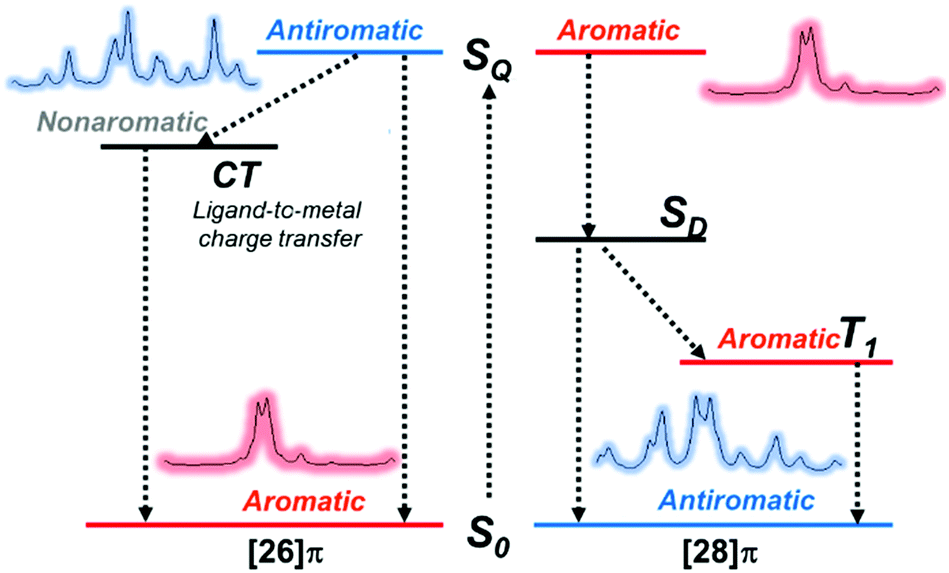
The first spectroscopic demonstration of Baird's rule, the reversal of aromaticity in the lowest triplet state, was achieved with bis-rhodium hexaphyrins (1 and 2).99 As described in Section 2.2, these two hexaphyrins are aromatic and antiaromatic congener sets, showing only a difference in two hydrogen atoms in the molecular configuration but leading to the apparent [26]π-aromatic 1 and [28]π-antiaromatic 2. In addition, coordinated rhodium metal ions in these molecules provoke negligible electronic perturbation on the π-electronic structure of hexaphyrin ligands and only promote ISC to the triplet states through the heavy metal effect. Thus, these porphyrinoids were regarded as ideal test-beds for verifying aromaticity reversal in the T1 state by analyzing their absorption spectral changes and electronic structures between the S0 and T1 states because their congeneric structures exclude other physicochemical effects.The ground-state electronic absorption (S0-absorption) spectra of 1 and 2 manifest well the characteristic spectral features of aromatic and antiaromatic porphyrinoids (Fig. 14). Here, 1 shows typical absorption features of aromatic porphyrins, such as intense B and weak Q bands. On the contrary, 2 exhibits a broad and weak absorption with a distinctive tailing signal in the NIR region, well reflecting the dark electronic transition with a significantly small energy gap from the antiaromatic nature. The pump–probe-type electronic absorption measurement, femtosecond-TA measurement, of 1 and 2 allows for the monitoring of the absorption spectral change in the T1 state and extracting the T1-state electronic absorption (T1-absorption) spectra. Because the triplet states are typically non-radiative, the simple composition of TA spectra for T1-state molecules, i.e., the sum of negative GSB and positive ESA signals, facilitates the extraction of T1-absorption spectra with high accuracy (Fig. 15). The T1-absorption spectra of 1 and 2 show undeniably interconvertible features to their S0-absorption spectra; the intense B-like and weak Q-like bands are displayed in the T1-absorption spectrum of 2 and the overall attenuated absorption with tailing features is confirmed in that of 1. The density functional theory (DFT) calculations rationalize the interconvertible spectra of 1 and 2 between the S0 and T1 states with the characteristic four- and six-FMO π-electronic structures. The main T1-state vertical excitations of 2, producing B- and Q-like bands, result from the configurational interaction of four degenerate FMOs, indicating the aromatic nature. Meanwhile, the calculation results for the T1-state vertical excitation of 1 showed the major participation of six FMOs, which is a characteristic of antiaromatic porphyrinoids, causing the distinctive tailing absorption.
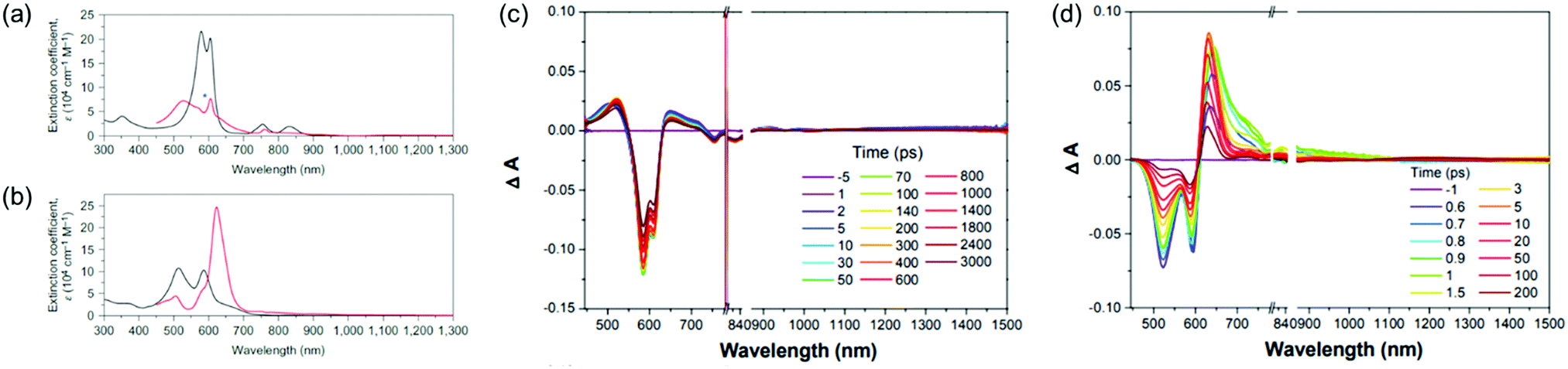 | ||
| Fig. 14 The ground- and triplet-state and transient absorption spectra of 1 and 2. The ground- (black lines) and triplet-state (red lines) absorption spectra of (a) 1 and (b) 2 plotted as a function of extinction coefficients. The red spectra are obtained by the summation of the ground-state absorption and scaled decay-associated TA spectra. The asterisk (*) indicates an experimental error peak due to different spectral resolutions between the two spectrometers used in the ground-state absorption and TA measurements. The transient absorption spectra of (c) 1 and (d) 2 in toluene with the photoexcitations at 590 and 505 nm, respectively. Adapted from ref. 99 with permission. Copyright 2015 Springer Nature Limited. | ||
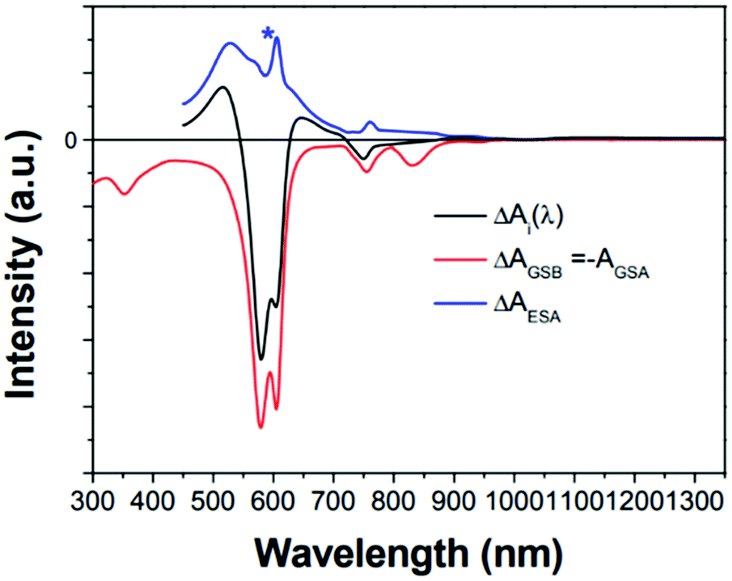 | ||
| Fig. 15 GSB (red line), and ESA (blue line) components calculated from the decay-associated spectrum for the T1 state (black line) of 1. Intensity ratio of [ ΔAESA(λmax)/ΔAGSB(λmax)] equals the ratio of extinction coefficients [εES(λ)/(−εGS(λ))]. The asterisk (*) indicates the experimental error peak due to different spectral resolutions between the two spectrometers used in ground-state absorption and transient absorption measurements. Adapted from ref. 99 with permission. Copyright 2015 Springer Nature Limited. | ||
Additional quantum chemical analyses confirmed the reversed aromaticity of 1 and 2 in the T1 state. As mentioned above, aromaticity is not a single measurable feature but collective properties. This requires multifaceted investigations to evaluate the multidimensional character of aromaticity, which have led to various criteria for estimating aromaticity theoretically as well as experimentally, such as magnetic, geometrical, energetic, and electronic bases. These computational analyses with different criteria are complementary to each other for the accurate assessment of aromaticity. Computational estimations of magnetic indices, such as the magnetic shielding effect for the nucleus-independent chemical shift (NICS)221 and visualization of ring current for anisotropy of the induced current density (ACID), and gauge including magnetically induced current (GIMIC),222–224 quantify and qualify the aromaticity-dependent ring current effect and they are frequently exploited, in that their results are well correlated with experimental NMR spectroscopic results. However, these magnetic quantum chemical methods tend to overestimate the delocalization and coupling of the adjacent atomic magnetic field. With DFT optimization results, bond length alternation (BLA), harmonic oscillator model of aromaticity (HOMA), and aromatic fluctuation index (FLU) act as topological aromaticity indices, which need reference values for estimation and show a restriction for intrinsic structural complexity, such as tight bonding and fused structures.103,225–227 The energetic stabilization effect of aromaticity can be evaluated with isomerization stabilization energy (ISE) and aromaticity stabilization energy (ASE) methods by a comparative analysis with reference model systems.64,228,229 Unfortunately, these methods are not applicable to porphyrinoids because breaking the π-conjugation of porphyrinoids at one specific position inevitably accompanies another broken conjugation. Recently, as electronic aromaticity criteria, multicenter indices (MCI), AV1245 and AVmin, were proposed for assessing the extent of electron sharing and transferability of the delocalized electrons.230–233 These methods directly approach electron delocalization, which indicates that they have great potential for applicability to π-conjugation and aromaticity of macromolecules. With these various computational methods for aromaticity, the collective analyses provide convincing and complementary pieces of evidence for the verification of aromaticity.
In this regard, cooperative computational analyses of 1 and 2 based on magnetic and geometrical bases advocate for the T1-state reversed (anti)aromaticity of 1 and 2. The inverse NICS and ACID results of 1 and 2 between the S0 and T1 states support well the experimental results for the reversed (anti)aromaticity, antiaromatic 1 and aromatic 2, in the T1 state. This is further supported by HOMA analyses that show a decrease (and increase) in the HOMA value, 0.62 to 0.58 (and 0.53 to 0.64), of 1 (and 2) from the S0 to T1 state. Therefore, the interconvertible absorption spectra of 1 and 2 between the S0 and T1 states most directly demonstrate the aromaticity reversal in the T1 state.
The aromaticity-dependent absorption spectral features of porphyrinoids also allow for the detection of the change in aromaticity in the singlet excited (Sn) states. The Sn states have attracted much attention in the field of photochemistry owing to their large universality; i.e., because most molecules are in the singlet ground state, the Sn states are inevitable in photochemical reactions. With this significance of Sn states, intensive theoretical studies have suggested an extension of the reversal concept of aromaticity to the Sn state based on the similarity between the singlet and triplet configurations.144,145 However, restrictions in experimental and even computational assessment to the Sn states have resulted in significant difficulties in investigating singlet-excited-state aromaticity, and only a few examples associated with photochemical reactions have been studied.161,162,234 In this context, the Sn-absorption spectral analysis of aromatic and antiaromatic porphyrinoid congeners affords an important experimental clue to the investigation of the singlet excited-state (anti)aromaticity.
The phenylene-bridged hexaphyrin congener set, aromatic 3 and antiaromatic 4, was highlighted for revealing singlet-excited-state aromaticity.235 As in 1 and 2, the meta-bridged phenylene produces a negligible perturbation on π-electronic structures of hexaphyrins, which sustains the aromaticity-dependent absorption spectral features in 3 and 4; the intense B and weak Q absorption bands for aromatic 3 and the weakened absorption with a NIR tailing for antiaromatic 4 (Fig. 16). Meanwhile, the absorption spectral changes in the singlet excited states can be observed with TA spectroscopy. Here, because excited singlet species can be fluorescent, the negative SE signals should be considered along with the GSB and ESA signals in TA spectra. Similar to the T1-absorption analysis, the comparative analysis of the three signals enables the extraction of the SQ-absorption spectra of 3 and 4. The SQ-absorption spectrum of 4 is comparable to the S0-absorption spectrum of 3, exhibiting B- and Q-like bands. In contrast, the analogous spectral features are displayed in the SQ-absorption spectrum of 3 and S0-absorption spectrum of 4. Although quantum calculation analyses of this characteristic SQ-absorption is limited and challenging in computational chemistry, the DFT-based geometry optimization analyses, such as the harmonic oscillator model of aromaticity (HOMA)236 and mean-plane-deviation (MPD),58,237 support the reversed (anti)aromaticity, antiaromatic 3 and aromatic 4, in the SQ state. Thus, the interconvertible absorption spectral features of 3 and 4 between S0 and SQ states provide reliable and direct experimental evidence for the aromaticity reversal in the singlet excited state.
 | ||
| Fig. 16 Ground-state (black lines) and the lowest singlet excited-state absorption (red lines) spectra of (a) 3 and (b) 4. The asterisk (*) indicates the instrumental error peak due to different spectral resolutions of the spectrometers employed in absorption and TA spectra. Adapted from ref. 235 with permission. Copyright 2015 American Chemical Society. | ||
The singlet excited-state (anti)aromaticity was also verified with cyclic multiple porphyrinoid arrays based on their fluorescent nature (Fig. 17).238 Although each porphyrin can have strong exocyclic π-conjugation, recent studies revealed that the diacetylene-bridged multiple porphyrin nanorings ([70]π c-P5, [84]π c-P6, [98]π c-P7, [112]π c-P8, and [126]π c-P9) possess distinct aromatic and antiaromatic natures from the global macrocyclic π-conjugation.179,181,182 Here, the fluorescent nature of the molecular ring was the focus. Because fully π-conjugated circular molecules are slightly fluorescent, inherently due to the selection rule,210,239–243 the symmetry-forbidden nature, these nanorings show weak fluorescence (Q.Y. < 0.05). Based on this underlying principle of molecular fluorescence, the radiative rates and quantum yields of the series of nanorings were correlated with their reversed (anti)aromaticity in the S1 state. The ring-symmetry of the nanorings in the emitting state, the S1 state, is largely affected by their global (anti)aromaticity. The global aromaticity leads to a planar and symmetric molecular geometry, where the improvement of the overall ring-symmetry suppresses the fluorescence emission. In contrast, the distorted and less symmetric structure of globally antiaromatic porphyrin nanorings enhances their fluorescence intensity. In this regard, the increased radiative rates and quantum yields of [4n + 2]π c-P5, c-P7, and c-P9 well reflect the ring distortion by their S1-state antiaromaticity (Fig. 18). On the other hand, the decreased values for [4n]π c-P6, and c-P8 are rationalized by their symmetrized circular geometries driven by the S1-state aromaticity. Therefore, the fluorescent nature of the porphyrin nanorings reinforces the aromaticity reversal in the singlet excited state.
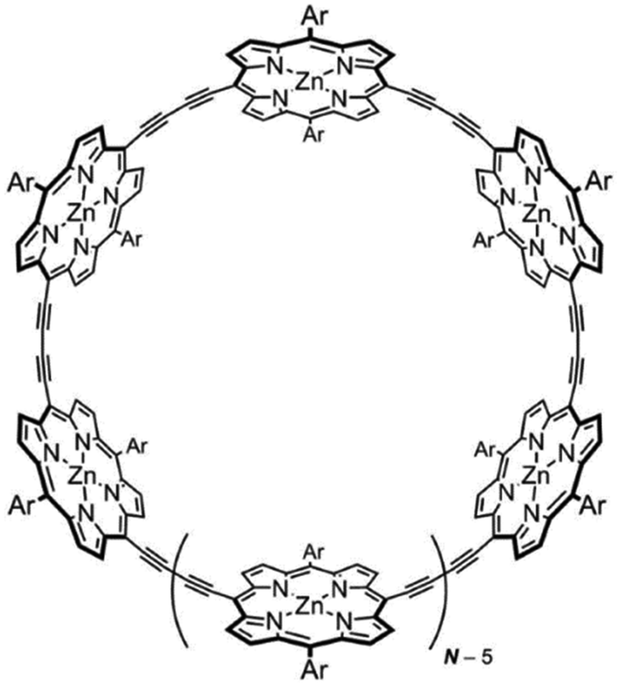 | ||
| Fig. 17 The molecular structure of diacetylene-bridged multiple porphyrin nanorings, c-PN (N = 5, 6, 7, 8, and 9). | ||
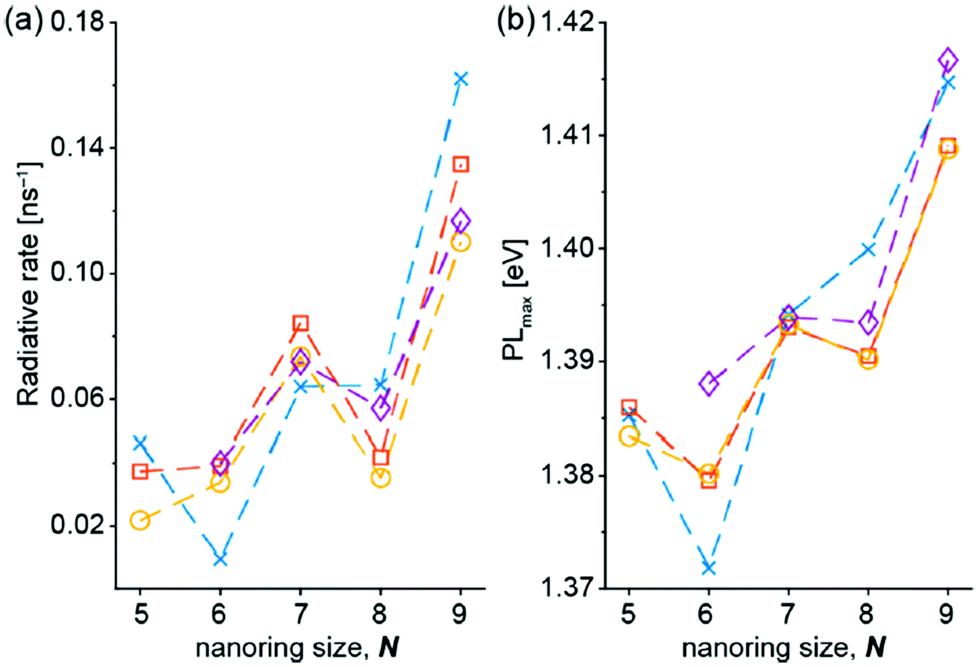 | ||
| Fig. 18 (a) Radiative rates and (b) energy of the photoluminescence peak maximum, measured in toluene containing 1% pyridine, referenced to l-P6 (1.47 eV) as a function of ring size N for c-P5–c-P9. Lines connect measurements from the same experimental replicate. λex = 500 nm. Adapted from ref. 238 with permission. Copyright 2020 American Chemical Society. | ||
3.2. Excited-state aromaticity reversal of Möbius aromatic porphyrinoids
Electronic structure models for porphyrins by Gouterman and Michl have been applied to the absorption spectra of Möbius aromatic and antiaromatic porphyrinoids.51,201,202 Although the molecular geometries of the Hückel and Möbius (anti)aromaticities are significantly different, both can be rationalized with HMO theory (Fig. 2). In particular, even though the phase inversion in Möbius-strip geometry changes the highest and lowest levels of π-electronic HMOs and eventually alters the [4n + 2]π rule into the [4n]π rule by Heilbronner's model, there is no significant difference in the degenerate structure of the middle-level MOs corresponding to FMOs. This has been well demonstrated with the experimental absorption spectra of Hückel and Möbius aromatic porphyrinoids and recent ab initio calculation results. Thus, the aromaticity-dependent optical spectroscopic features are commonly observed in aromatic and antiaromatic porphyrinoids and serve as an effective index for evaluating the excited-state aromaticity in Möbius (anti)aromatic porphyrinoids.In recent decades, porphyrinoids have been privileged molecular systems for the realization of Möbius (anti)aromaticity. The structural flexibility from the sp2-carbon-bridged cyclic oligopyrroles well accommodates Möbius-strip twisting without a break in the π-conjugation, which allows various Möbius aromatic and even antiaromatic porphyrinoids to be achieved since the first synthesis of a Möbius aromatic annulenic derivative.40,41 Thus, together with their aromaticity-dependent absorption spectral features, porphyrinoids act as ideal molecules to verify the concept of excited-state aromaticity, the aromaticity reversal, in Möbius (anti)aromatic systems.
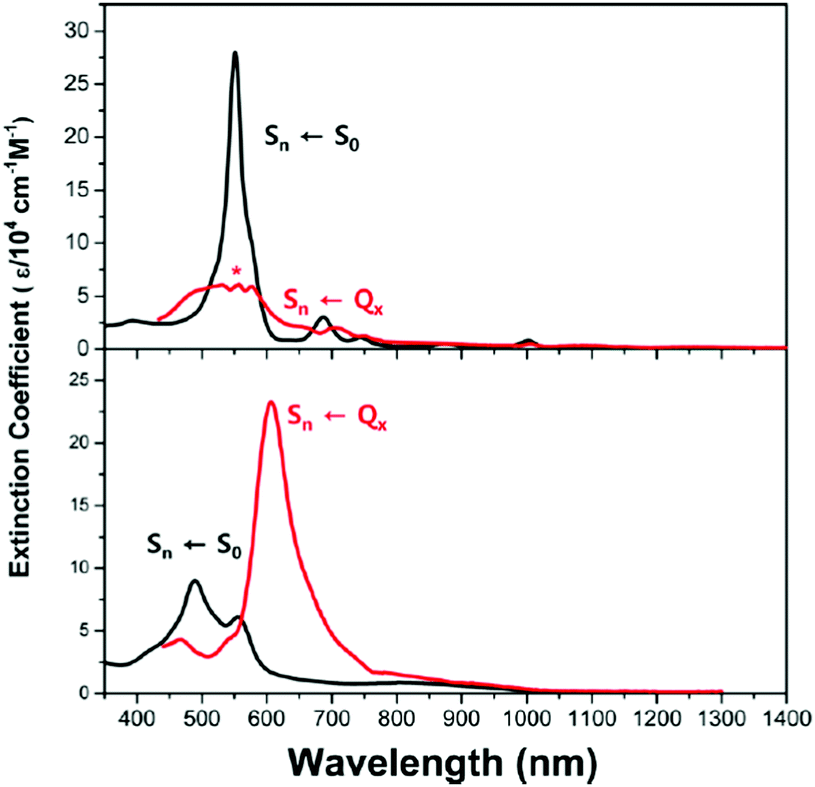
The excited-state Möbius (anti)aromaticity was investigated with a set of Hückel and Möbius aromatic porphyrinoids (palladium [28]hexaphyrin, 6, and N-fused rhodium [22]- and [24]pentaphyrin, 7 and 8, respectively).244 Their X-ray crystal structures show the planar conformation of 7 and one-surfaced Möbius-strip topology of 6 and 8, in which their distinct diatropic ring currents represent well the Hückel aromatic 7 and Möbius aromatic 6 and 8. Moreover, because 7 and 8 are Hückel and Möbius aromatic congeners, respectively, their comparative analysis demonstrates that the same Baird's rule can apply to the excited-state aromaticity. Here, because the coordinated metal ions facilitate ISC without significant perturbation on the ligand π-conjugation as in 1 and 2, their (anti)aromaticity is unambiguously reflected in the S0-absorption spectra. According to their distinct aromatic nature, 6, 7, and 8 show the conspicuous intense B and weak Q bands in the S0-absorption spectra (Fig. 19). On the other hand, their T1-absorption spectra extracted from the TA results exhibited obvious contrasting features. In these spectra, a reduction of absorbance and absorption tailing over the NIR region are commonly observed, indicating the antiaromatic nature of 6, 7, and 8 in the T1 state. The vertical excitation energy calculation results rationalize the contrasting spectral changes in 6, 7, and 8 with their altered π-electronic structures induced by the reversed T1-state antiaromaticity. The S0-absorption spectra mainly result from a configuration interaction of the degenerate four FMOs, the characteristic π-electronic structures of aromatic porphyrinoids. For the T1-absorption, the participation of six FMOs dominantly gives rise to the broad and weak signals with a tailing feature. Moreover, the quantum chemical analyses, such as NICS, HOMA, and MPD, show consistent results for the antiaromaticity of 6, 7, and 8 in the T1 state. Thus, the contrasting absorption spectra of 6, 7, and 8 between the S0 and T1 states provide a solid corroboration of the reversal of the Möbius (anti)aromaticity in the T1 state.
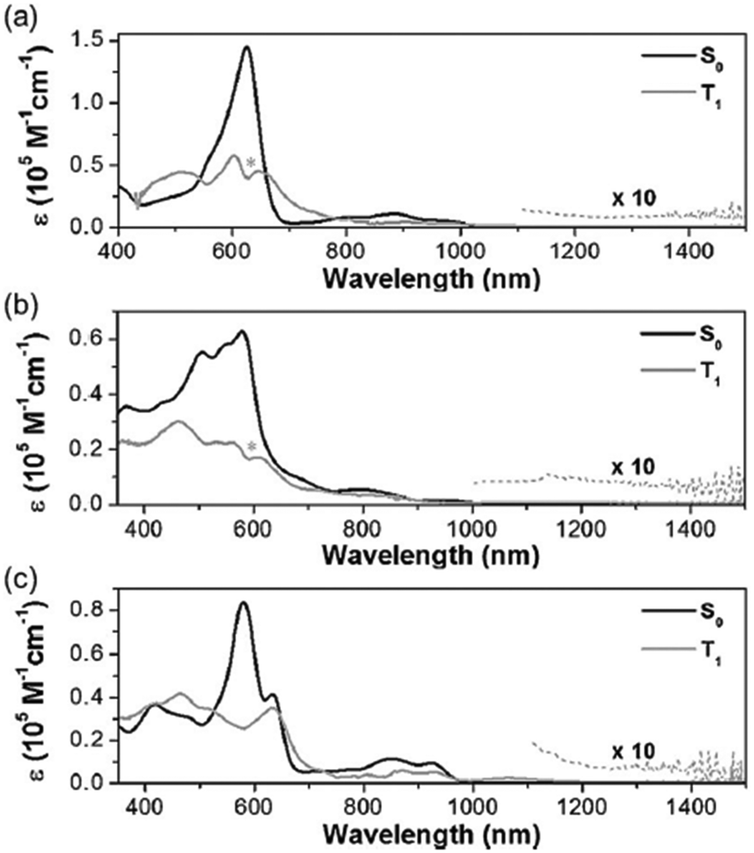 | ||
| Fig. 19 Ground-state (gray line) and lowest-triplet-state (red line) absorption spectra of (top) 6, (middle) 7, and (bottom) 8 with vertical energy transition plots for the ground states (gray columns) and triplet states (red columns). Adapted from ref. 244 with permission. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Aside from the Möbius (anti)aromaticity, the structural advantage of porphyrinoids allows more complex conformations, such as doubly twisted figure-of-eight and quadruply twisted structures48,54,178 without a break in the macrocyclic π-conjugation. In this regard, the concept of triplet-state aromaticity, the aromaticity reversal, was studied with complex-topology porphyrinoids, i.e., a set of bis-palladium [36] octaphyrin congeners (9 and 10).245 These two octaphyrins have the same number of π-electrons on the main macrocyclic π-circuit, but differ by only one hydrogen atom and molecular topology (single-sided Möbius and double-sided figure-of-eight geometries for 9 and 10, respectively). These different structures under the same number of π-electrons trigger Möbius aromaticity for 9 and Hückel antiaromaticity for 10, which are well reflected in their S0-absorption spectra with the intense B and weak Q bands for 9 and the weak and tailing absorption for 10 (Fig. 20). As in 1 and 2, the estimated T1-absorption spectra of 9 and 10 are interconvertible with their S0-absorption spectra, and these contrasting spectral changes are well explained by the characteristic four and six FMO structures. Thus, the verification of T1-state reversed (anti)aromaticity in these complicated-topology porphyrinoids well illustrates the versatility of this excited-state aromaticity rule to various types of (anti)aromatic systems.
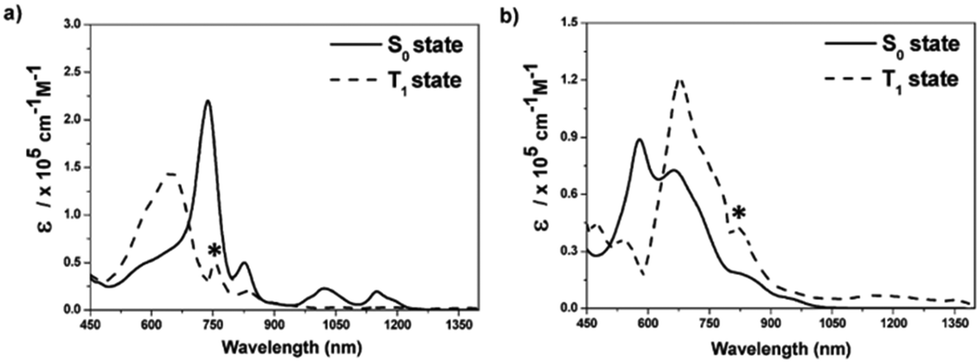 | ||
| Fig. 20 The S0- and T1-state absorption spectra of (a) 9 and (b) 10 in toluene. The asterisks indicate experimental errors that are due to the different spectral resolutions of the spectrometers used for ground-state absorption and TA measurements. Adapted from ref. 245 with permission. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
3.3. Excited-state aromaticity reversal of porphyrinoids with vibrational analyses
As mentioned above, intensive efforts for the study of aromaticity have established various indices for evaluating molecular aromaticity.38,63,195 Among the various indices, the molecular geometry can be considered as one of the most representative indices to determine molecular aromaticity. The equalized C–C bond length under the perfect planar geometry of benzene, a privileged molecule for aromaticity, has offered significant insight into the geometrical criteria for assessing aromaticity. However, molecular aromaticity should be determined by multidimensional characterization, not just by geometrical evaluation. The equalized bond length of benzene is not solely affected by aromaticity but a result of the cooperative effects of s- and p-orbitals.246 Moreover, some nonaromatic molecules show equalized bond lengths.247 In particular, because geometry-based indices, such as HOMA and FLU, are defined based on the geometrical features of fully and locally π-conjugated aromatic and nonaromatic reference molecules, respectively, they should be applied with caution for the nature of bond-length equalization and inherent structural effects such as fused ring structures.216,226,227,233,248–250 Although the geometry-based evaluation for aromaticity is insufficient in these respects, the molecular geometry, either planar or distorted conformations, can be directly associated with p-orbital overlap, i.e., the origin of aromaticity. The p-orbital overlap on the perfectly planar geometry can maximize the aromatic stabilization effect, whereas destabilization from antiaromaticity can be alleviated by structural distortion, reducing the p-orbital overlap. Thus, the evaluation of molecular geometry is very intuitive for the determination of (anti)aromaticity.103Porphyrinoids are advantageous for determining (anti)aromaticity based on their molecular structures. Various aromatic and antiaromatic porphyrinoid congeners allow for the disclosure of the pure effect of (anti)aromaticity on molecular structures.20,30 Furthermore, the flexible structures of porphyrinoids are nonintrusive to aromaticity-driven changes in molecular structures.42,63,251–253 Thus, aromatic porphyrinoids show planar and symmetric structures for effective π-conjugation, and antiaromatic porphyrinoids possess distorted and less symmetric structures for the alleviation of antiaromatic destabilization. These characteristics of aromatic and antiaromatic porphyrinoids have been intensively studied, and clarify the evaluation of (anti)aromaticity based on quantitative analyses of molecular geometries such as bond-length alternation (BLA), HOMA, and MPD.20,30,101
This merit of porphyrinoids for evaluating S0-state aromaticity can be equally applied to the excited states. As described in the study with the multiple porphyrin nanorings,180,238 aromaticity-driven conformations can be directly measured by optical spectroscopy, particularly with vibrational spectroscopy. Because molecular vibrations are the motions of atoms bound by chemical bonds, the collective vibrations are closely related to molecular structures.254–257 Moreover, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds are elementary constituents of π-conjugation. Thus, collective CC stretching vibrational modes show a close correlation with π-electron delocalization and further (anti)aromaticity.258–263 On account of these points, the aromaticity-dependent structural features of porphyrinoids can be detected with the IR-activity of their CC stretching modes (Fig. 21).100 The IR-activity of vibrational modes is inherently governed by the vibrational selection rule, i.e., changes in the dipole moment. The planar and symmetric structures of aromatic porphyrinoids result in effective π-conjugation and ultimately large aromatic stabilization. In this geometry, collective CC stretching motions along the main macrocyclic π-conjugation pathway cause symmetric and in-plane vibrations. Because these vibrations accompany a small change in the dipole moment, their IR-activity is weak and aromatic porphyrinoids show a monotonous infrared (IR) spectrum in the CC stretching region (1300–1700 cm−1). Meanwhile, the twisted and less symmetric structure of antiaromatic porphyrinoids triggers asymmetric collective CC stretching vibrations along their out-of-plane bond alignment. These vibrations are IR-active due to the induced significant change in the dipole moment, which subsequently leads to abundant IR spectral features in the 1300–1700 cm−1 region. Thus, the IR spectral features of porphyrinoids in the 1300–1700 cm−1 region show distinctive aromaticity-dependent features and the excited-state IR measurements can convey important information for evaluating the excited-state aromaticity.
C bonds are elementary constituents of π-conjugation. Thus, collective CC stretching vibrational modes show a close correlation with π-electron delocalization and further (anti)aromaticity.258–263 On account of these points, the aromaticity-dependent structural features of porphyrinoids can be detected with the IR-activity of their CC stretching modes (Fig. 21).100 The IR-activity of vibrational modes is inherently governed by the vibrational selection rule, i.e., changes in the dipole moment. The planar and symmetric structures of aromatic porphyrinoids result in effective π-conjugation and ultimately large aromatic stabilization. In this geometry, collective CC stretching motions along the main macrocyclic π-conjugation pathway cause symmetric and in-plane vibrations. Because these vibrations accompany a small change in the dipole moment, their IR-activity is weak and aromatic porphyrinoids show a monotonous infrared (IR) spectrum in the CC stretching region (1300–1700 cm−1). Meanwhile, the twisted and less symmetric structure of antiaromatic porphyrinoids triggers asymmetric collective CC stretching vibrations along their out-of-plane bond alignment. These vibrations are IR-active due to the induced significant change in the dipole moment, which subsequently leads to abundant IR spectral features in the 1300–1700 cm−1 region. Thus, the IR spectral features of porphyrinoids in the 1300–1700 cm−1 region show distinctive aromaticity-dependent features and the excited-state IR measurements can convey important information for evaluating the excited-state aromaticity.
 | ||
| Fig. 21 Schemes for (a) the dependence of CC stretching vibrational motions of porphyrinoids and their IR-activity on their aromaticity and antiaromaticity; (b) the related IR-spectral changes according to the aromaticity reversal in the excited states. | ||
The IR spectral features of 1 and 2, particularly from the CC stretching motions were intensively analyzed.187,264 Both Fourier-transform IR (FT-IR) spectra of 1 and 2 exhibited two exceptionally intense bands in the CC stretching region (1400–1550 cm−1), which were due to the common meso-pentafluorophenyl (C6F5) substituents (Fig. 22). The non-uniform charge distribution of the C6F5 substituents by electronegative fluorine atoms triggered the greatly IR-active CC stretching IR bands, which served as a standard for the comparison of IR spectral features. The comparison of the CC stretching IR bands of 1 and 2 based on the common IR bands of C6F5 substituents showed contrasting spectral features. Negligible IR bands were observed in the FT-IR spectrum of 1. On the other hand, distinct IR absorptions were displayed for 2. The sharp contrasting IR spectral features in the CC stretching region describe the distinctive molecular geometries of 1 and 2. The CC stretching motions along the planar and symmetric structure of aromatic 1 are IR-inactive, whereas the distorted structure of antiaromatic 2 causes the CC stretching motions to accompany the significant change in dipole moment, enhancing the IR-activity. In the same way, the excited-state molecular geometry of 1 and 2 can be estimated with the time-resolved IR (TR-IR) measurement. Apart from the intense GSB signal from the C6F5 substituents, there are opposite features, conspicuous excited-state IR absorption bands, in the TRIR spectra of 1. Conversely, 2 shows distinct ground-state IR bleaching signals with meager excited-state IR absorption bands. These contrasting TRIR spectra of 1 and 2 in the CC stretching region are strongly reminiscent of their interconvertible electronic absorption spectral features between the S0 and T1 states and illustrate well the geometrical change driven by the aromaticity reversal in the T1 state, the relatively distorted and planar structure of 1 and 2 in the T1 state due to their reversed antiaromaticity and aromaticity, respectively.
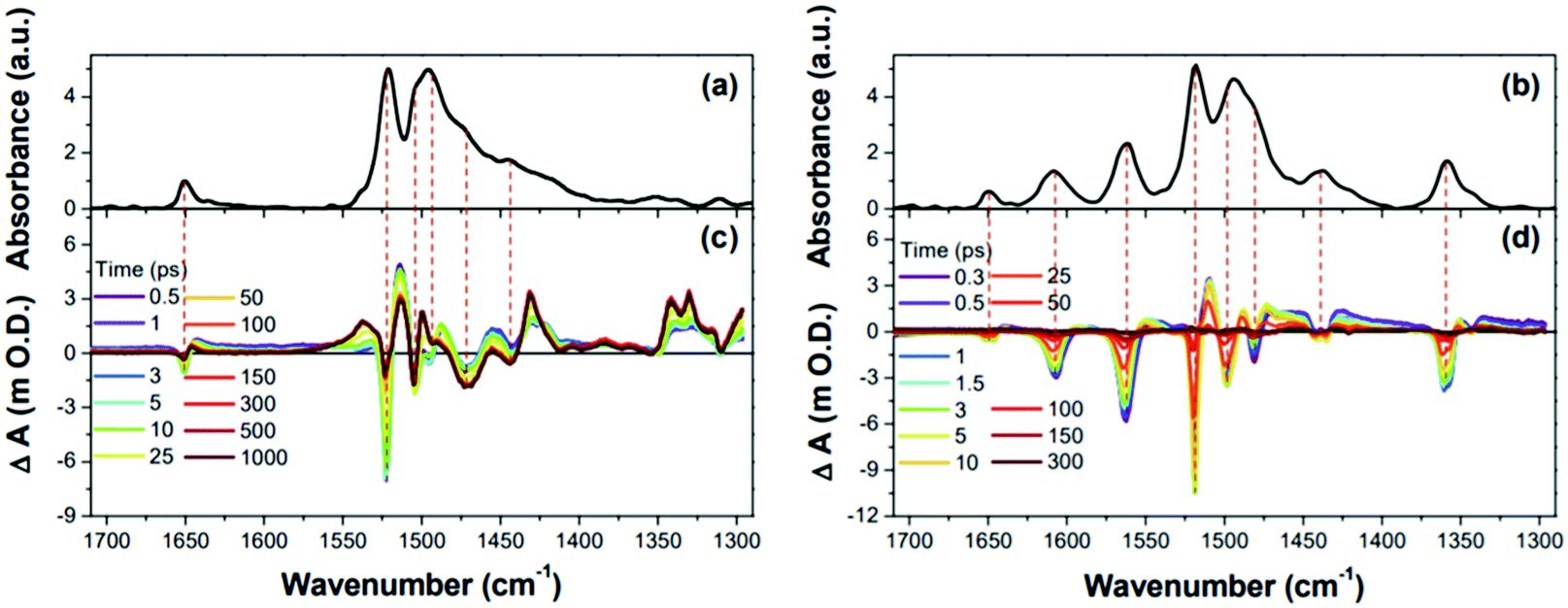 | ||
| Fig. 22 Fourier-transform IR spectra and time-resolved IR spectra of 1 and 2 in the 1710–1290 cm−1 region. (a) The black line shows the Fourier-transform IR spectrum of 1. (b) The black line shows the Fourier-transform IR spectrum of 2. (c) Time-resolved IR spectra of 1. (d) Time-resolved IR spectra of 2. Adapted from ref. 264 with permission. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
In addition to the T1-state aromaticity, the IR-activity of CC stretching modes of 3 and 4 corroborate the aromaticity reversal in the singlet excited states (Fig. 23).258 The FT-IR and TR-IR spectra of 3 and 4 show similar IR spectral features to those of 1 and 2. Because 3 and 4 have the same hexaphyrin molecular framework with meso-C6F5 substituents as 1 and 2, the spectral similarity between 1, 2, 3, and 4 denotes their aromaticity-dependent molecular geometries. In other words, the IR spectral features of 3 and 4 in the 1300–1700 cm−1 region are interpreted in the same way, in terms of the IR-activity of CC stretching modes, as in 1 and 2. The featureless IR absorption bands of 3 in the S0 state and 4 in the SQ state describe their aromaticity-driven planar and symmetric structures. On the other hand, the abundant IR absorption bands of 3 in the SQ state and 4 in the S0 state reflect their out-of-plane and asymmetric CC stretching modes resulting from their distorted structures, which describe well the geometrical transformation of 3 and 4 driven by their antiaromaticity and reinforce the aromaticity reversal in the excited singlet states.
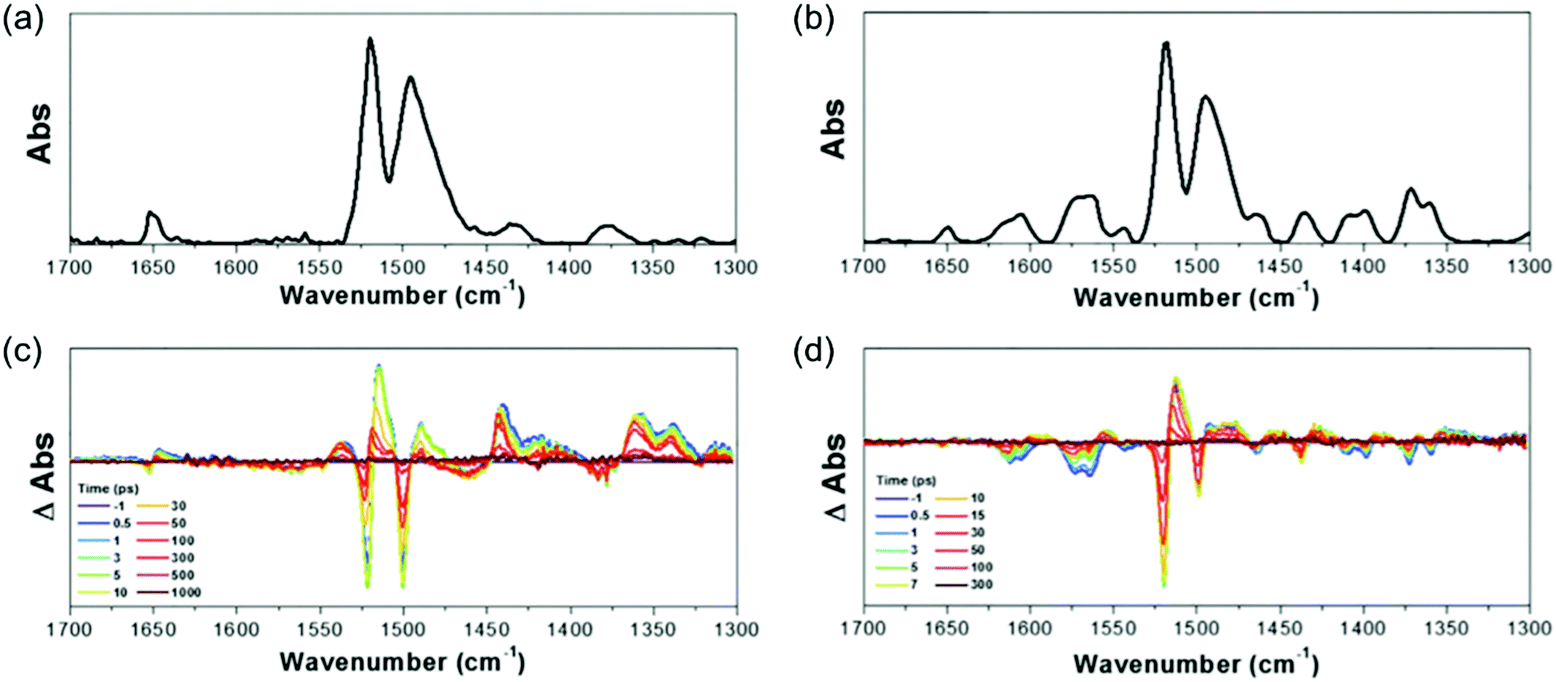 | ||
| Fig. 23 The Fourier-transform and time-resolved IR spectra of 3 and 4. (a and b) The Fourier-transform IR spectra of 3 and 4 in the solid state. (c and d) The time-resolved IR spectra of 3 and 4 in CH2Cl2 upon photoexcitation at 550 nm. Adapted from ref. 258 with permission. Copyright 2017 Cell press. | ||
In addition to the verification of the aromaticity reversal in the excited states, the IR spectral analyses allow for the effects of excited-state processes on the (anti)aromaticity. The ideal metalated aromatic and antiaromatic congener sets (1, 2, 7, 8, 9, and 10) for revealing the T1-state aromaticity show considerably efficient ISC by the strong heavy atom effect and weak electronic perturbation of metal ions. However, the effects of metalation are not restricted to ISC. The coordinated metal ions can significantly perturb the electronic structures of entire organometallic compounds and alter their excited-state dynamics. Indeed, various metalloporphyrinoids show effective metal–ligand interactions, where their derived rich redox chemistry enables the tunability of physicochemical properties and consequently results in various catalytic behaviors and pivotal enzymatic functions in nature.107,265–269 Thus, the excited-state aromaticity can also be affected by metalation effects and metal-associated excited behaviors.
Recent IR spectroscopic investigation of the excited-state aromaticity of the gold hexaphyrin congener set (aromatic 11 and antiaromatic 12) revealed the aromaticity reversal in the SQ state and the effects of subsequent processes on the excited-state (anti)aromaticity.270 Except for the coordinated Au ions, because 11 and 12 also possess the same molecular scaffold, the meso-C6F5 hexaphyrin, the comparative analysis of their IR spectral features from the characteristic CC stretching motions between the S0 and SQ state well represents the reversed aromaticity in the SQ state, antiaromaticity for 11 and aromaticity for 12 (Fig. 24). Here, although the set of 11 and 12 are analogous to the aromatic and antiaromatic congener sets (1, 2, 7, 8, 9, and 10), the electronic structure of aromatic 11 is perturbed by the coordinated Au ions. As a result, the photoexcitation of 11 triggers a ligand-to-metal charge transfer (LMCT) process instead of ISC by the heavy atom effect from the Au ions. This LMCT process of 11 is directly proved by the shift of the CC stretching IR band from meso-C6F5 substituents, indicating that the antiaromatic destabilization of 11 in the excited state is relieved. This suggests that the excited-state aromaticity can be modulated by the subsequent excited-state processes, further implying the role of the destabilization of excited-state antiaromaticity as a facilitator in the excited-state dynamics and photochemistry (Fig. 6).
 | ||
| Fig. 24 Schematic illustration of excited-state dynamics and the related change of aromaticity for 11 and 12. | ||
4. Functional molecular systems designed based on the concept of excited-state aromaticity
Baird's rule is referred to as a description of the excited-state aromaticity, in which [4n]π (or [4n + 2]π) systems become Baird aromatic (or antiaromatic) in the excited triplet states in contrast to the ground-state Hückel (anti)aromaticity. This aromaticity rule for the excited states, termed as “aromaticity reversal”, has great significance because an inversion of aromaticity in the excited states furnishes crucial insight into the behaviors in the excited states and involves photo-physical and chemical principles, which have attracted intensive theoretical and experimental efforts for revealing excited-state aromaticity based on the reversal concept. Recent studies on porphyrinoids have overcome the difficulties of conventional approaches to the study of aromaticity and have provided prima facie evidence for the reversal of (anti)aromaticity in the excited states.These scientific demonstrations of excited-state aromaticity further lead to developments in new scientific issues dealing with a comprehensive understanding of its effect on the excited states for its practical application beyond the satisfaction of simple curiosity. In other words, understanding the role of excited-state (anti)aromaticity enables a mechanistic elucidation of excited-state molecular properties, such as conductivity, photostability and photoreactivity.82,83,100 In particular, Baird's rule suggests the possibility that the excited-state energy surface is directly affected by the reversed (anti)aromaticity (Fig. 6 and 25). The energy levels of S1 and T1 states can be stabilized by the aromaticity reversal or aromatization in the excited states. Conversely, antiaromaticity in the S1 and T1 states lift their energy levels. This effect of excited-state aromaticity can eventually lead to its application in photosynthetic mechanisms and photoactive materials.92,271–274 Based on this feasibility, much of the attention on the excited-state aromaticity has been drawn to photoactive materials. The photoactive materials are materials in which their functionalities are dominated by photoinduced excited-state processes. In this regard, singlet fission (SF) has attracted much interest based on the T1-state stabilization by Baird aromaticity.275–278 SF is the excited-state process that generates two triplet excitons from a high-energy singlet exciton through intermolecular interactions.279,280 Because this multiple exciton generation process has a great advantage in energy harvesting, SF has been intensively studied for a decade. The prerequisite of SF is energy conservation (ES1 > 2ET1). This is a challenging condition to achieve in typical organic molecules and it is typically required to lower the energy level of the T1 state. Recent scientific attempts with indolonapththyridine thiophene derivatives showed the possibility to manipulate the singlet–triplet energy gap via Baird aromaticity in the T1 state (Fig. 26).281 The formation of [4]π Baird aromaticity on the five-membered heterocycle stabilized the energy level of the T1 state relative to that of the S1 state in the indolonapththyridine thiophene derivatives, which subsequently triggered the SF process in its spin-coated film environment with excellent stability. With this study, various molecular systems containing five-membered rings have been actively investigated as potential candidates for unconventional SF-active materials.275
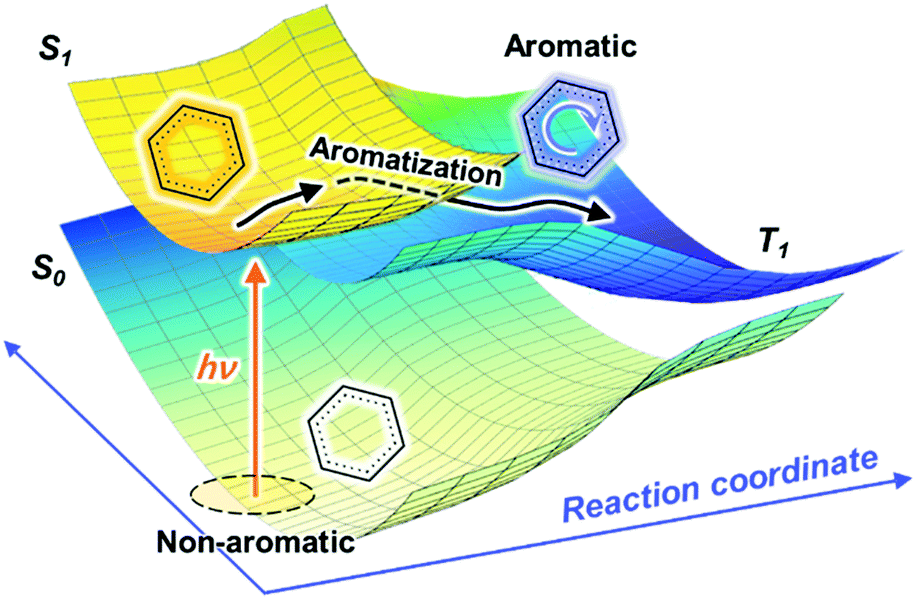 | ||
| Fig. 25 Schematic diagram of the effect of excited-state aromaticity, particularly for the effects of Baird aromatic stabilization on the energy level of T1, (π,π*)3 state. | ||
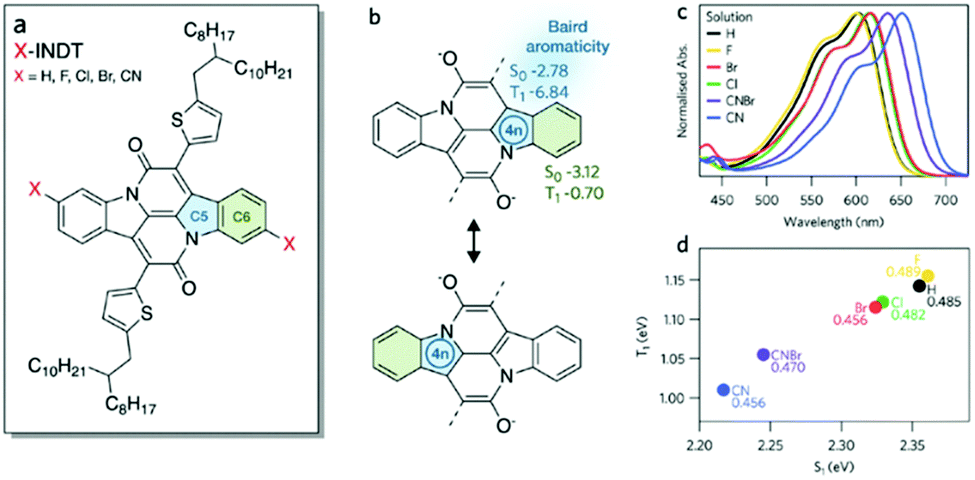 | ||
| Fig. 26 Optical properties and characteristics. (a) The polycyclic aromatic hydrocarbon indolonaphthyridine thiophene (INDT) with labeled rings of interest. (b) Resonance forms of INDT showing the origin of the Baird [4n] ring (C5, blue). Values of NICS(1) calculations for CN-INDT are shown for S0 and T1, where a more negative value denotes greater aromaticity. (c) Solution (chloroform) steady-state absorbance spectra of the investigated family of materials. (d) Theoretical energies (eV, TD-DFT/TDA, B3LYP/6-311++G**) of S1 and T1 relative to the ground state. The ratio of these energies is given inset. Adapted from ref. 281 with permission. Copyright 2019 American Chemical Society. | ||
In addition to the SF process, the T1-state stabilization effect of Baird aromaticity implies that aromatization in the T1 state can facilitate triplet-associated processes with an energetic advantage. A recent study on excited-state charge transfer (CT) processes in a dicyanomethyl-thiophenyl 1,6-methano[10]annulene derivative (TMTQ) illustrated the changes in the excited-state aromaticity (Fig. 27),282 excited-state aromatization, and the effects on the multiple charge generation process. TMTQ consists of a central 1,6-methano[10]annulene and two peripheral 5-dicyanomethyl-thiophene acceptors.283 Under this acceptor–donor–acceptor geometry, TMTQ displayed the photoinduced bidirectional CT processes accompanying the shifts of two equivalents of π-electrons, producing a Baird aromatic [8]π core annulene in the excited multi-excitonic state. This multiple CT process suggests that a shift of electronic charge in only the excited states is one of the effective strategies to modulate the excited-state aromaticity, and the T1-state aromatization plays a key role in stabilizing the energetically unfavorable doubly-charge separated species.
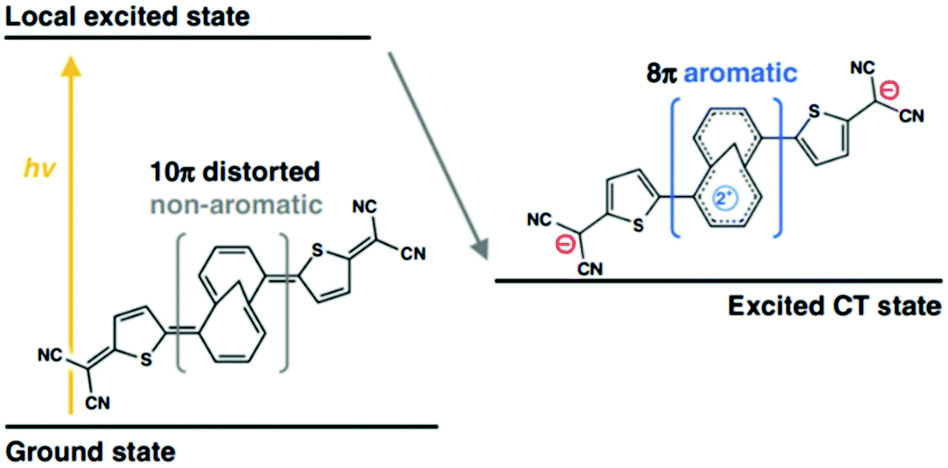 | ||
| Fig. 27 Charge-transfer-induced excited-state aromatization. The molecular structure of TMTQ and a schematic illustration of an excited-state aromatization induced by an intramolecular CT process. Adapted from ref. 282 with permission. Copyright 2019 Springer Nature Limited. | ||
In addition to the stabilization effect, the destabilization effect of Baird antiaromaticity is applied to excited-state processes.83,173,284 It is known that DNA base pairs are relatively strong against ultraviolet light by their efficient relaxation processes accompanying electron-driven proton transfer (EDPT). The EDPT in DNA base pairs is extremely fast as compared to other reactions.285 The efficient EDPT of DNA base pairs can be rationalized with the aromaticity reversal of purine-type DNA bases (adenine (A) and guanine (G)) in the excited states (Fig. 28). The π-conjugation in the heterocyclic ring of pyrimidine-type bases (cytosine (C), thymine (T), and uracil (U)) is largely perturbed by their carbonyl groups, leading to weak aromaticity in the S0 state. On the other hand, the cyclic π-conjugation in A and G is in fair preservation and results in distinct S0-state aromaticity. Thus, the purine-type bases become antiaromatic and highly reactive in the S1 state. This triggers the excited-state transfer of a proton and electron in DNA base pairs, from the purine-type bases to the pyrimidine-type bases, for alleviating the energetic destabilization from the excited-state antiaromaticity. Likewise, a recent study with o-salicylic acid and its derivatives also showed that their S1-state antiaromaticity promoted excited-state proton transfer to alleviate antiaromatic destabilization.286
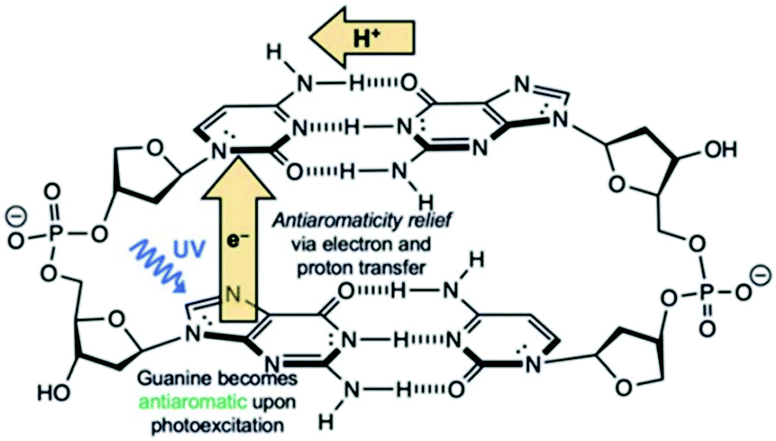 | ||
| Fig. 28 Schematic illustration of electron-driven proton transfer in a stacked G-C: G-C DNA duplex. Adapted from ref. 286 with permission. PNAS copyright. | ||
With this captivating subject on the excited-state aromaticity, research on porphyrinoids, particularly for expanded porphyrins, has also shown that the T1-state stabilization effect of Baird aromaticity facilitates the formation of stable organic radicaloids. Dithienothiophene-bridged [34]octaphyrin shows a unique dual aromatic nature from its large π-conjugation over the entire molecule (Fig. 29).287,288 The adoption of a π-conjugated bridge to the inside of [34]octaphyrin resulted in two possible [26]π and [34]π macrocyclic conjugations, which allowed the realization of bicycloaromaticity for the first time. While the two [26]π- and [34]π constituents are competing in the neutral bicyclic octaphyrin, its chemical oxidations permit access to distinctive oxidation states as a function of the two different aromatic subsystems. The one-electron oxidation results in the loss of π-electrons from the larger [36]π electronic circuit and the additional oxidation leads to the loss of π-electrons from the smaller [24]π electronic circuit based on their size-dependent chemical stability. Here, the two-electron oxidized bicyclic octaphyrin with 40 π-electrons globally is stabilized by Baird aromaticity and generates the triplet-ground-state biradicals arising from each monoradical nature of the dual π-conjugated circuits.
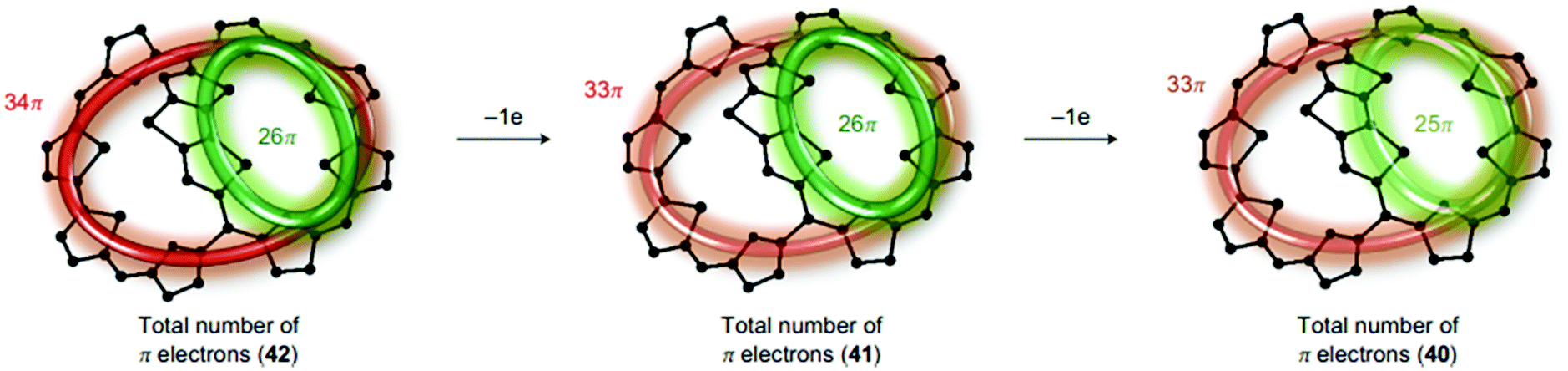 | ||
| Fig. 29 Bicycloaromaticity composed of [26]π- and [34]π cyclic conjugations in the dithienothiophene-bridged [34]octaphyrin and its neutral, one-electron, and two-electron oxidized forms. Adapted from ref. 287 with permission. Copyright 2017 Springer Nature Limited. | ||
Recent studies on diradicaloid molecular cages with expanded porphyrin analogues additionally prove the role of Baird aromatic stabilization in the development of organic radicals (Fig. 30).107,108,289 The sp2-carbon bridged conjugated oligothiophene (c-T12) shows efficient π-electron delocalization over the entire cage based on the relatively weak local aromatic nature of thiophene units. The quinoidal character from the full π-conjugation nature of c-T12 causes its neutral form to have the open-shell singlet state with Hückel aromaticity from the [38]π monocyclic conjugation. c-T122+, the dicationic diradical compound produced by two-electron oxidation is stabilized in the triplet ground state by Baird aromaticity from its monocyclic [36]π-conjugation. Furthermore, its tetracationic and hexacationic forms show [52]π and [50]π 3D global antiaromaticity and aromaticity in the open-shell and closed-shell singlet ground-state, respectively. Together with the radical nature of the bicyclic octaphyrins, these different kinds of diradicaloids, depending on the number of π-electrons, show how important the aromatic stabilization effect is for the formation of organic radicals. This further suggests that the open-shell triplet ground state, i.e., the diradical character, can be effectively stabilized (or destabilized) by Baird aromaticity (antiaromaticity), which has motivated various attempts for stable diradicals with this perspective.283,290–293
 | ||
| Fig. 30 Theoretical calculations and π-electron delocalization diagrams of c-T12, c-T122+, c-T124+ and c-T126+ in the ground state. (a) Calculated ACID plots are shown, with a magnetic field along the z-axis (vertically pointing out of the paper); the red and blue arrows indicate the diatropic and paratropic ring current flow, respectively. (b) Schematic π-electron delocalization pathways and the applied aromaticity rules are shown. The dominant conjugation pathways are highlighted in purple and blue for aromatic and antiaromatic systems, respectively. The arrows indicate open-shell diradical character, and the positive charge signs are placed at the positions closest to the counter ions. This does not mean that the spins and charges are localized, as they are actually delocalized along the backbone. All calculations were performed based on the single-crystal structures. Adapted from ref. 289 with permission. Copyright 2020 Springer Nature. | ||
5. Conclusions and outlook
Aromaticity, as one of the central principles for rationalizing molecular properties, is almost as old as modern chemistry. Aromaticity and antiaromaticity have been associated with some of the greatest areas of progress in organic chemistry. Recently, the scientific concern regarding aromaticity has advanced to the excited states. Since the S0-state (anti)aromaticity largely affects molecular properties, the (anti)aromaticity in the excited states is anticipated to explain excited-state processes and photochemistry. The recent studies with porphyrinoids have marked a milestone for the field of excited-state aromaticity. The characteristic features of porphyrinoids being largely dependent on their aromaticity and antiaromaticity have broken through the conventional adversities to assess (anti)aromaticity in the transient excited states. The aromaticity-dependent absorption spectral and geometrical features of porphyrinoids play a crucial role in the determination of excited-state (anti)aromaticity. Particularly, the optical spectroscopic observations of porphyrinoids serve as key indices to evaluate the reversed (anti)aromaticity in the excited states, where the interconvertible spectral features directly verify the reversal of aromaticity in the excited states.Furthermore, these studies on porphyrinoids have paved the way for the practical application and development of the concept of excited-state aromaticity. Understanding the role of excited-state aromaticity (or antiaromaticity) allows for the stabilization (or destabilization) of species in each excited state and enables the mechanistic elucidation of excited-state processes and photochemistry, which will eventually guide the design and development of novel functional molecular systems such as photoactive materials and organic radicaloids. In this regard, the unique nature of porphyrinoids, such as their superior π-electron delocalization and structural flexibility, can consistently act as a guideline to study excited-state (anti)aromaticity and further develop and enlarge its concept and applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2021R1A2C300630811 and 2021R1A6A1A03039503). The work was also supported by the Soonchunhyang University Research Fund.Notes and references
- A. R. Battersby, C. J. Fookes, G. W. Matcham and E. McDonald, Nature, 1980, 285, 17–21 CrossRef CAS PubMed.
- D. Dolphin, The Porphyrins, Academic Press, New York, 1978–1979, vol. 1–8 Search PubMed.
- K. M. Smith, Porphyrins and Metalloporphyrins, Elsevier, Amsterdam, 1975 Search PubMed.
- K. M. Kadish, K. M. Smith and R. Guilard, Chlorophylls and Bilins: Biosynthesis, Synthesis, and Degradation, The Porphyrin Handbook, Academic Press, San Diego, 1999, vol. 13 Search PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley-Blackwell, Oxford, UK, 2nd edn, 2014 Search PubMed.
- K. M. Kadish, K. M. Smith and R. Guilard, Biochemistry and Binding: Activation of Small Molecules, The Porphyrin Handbook, Academic Press, San Diego, 1999, vol. 4 Search PubMed.
- Z. Zhou and Z. Shen, Isr. J. Chem., 2016, 56, 119–129 CrossRef CAS.
- J. L. Sessler and S. J. Weghorn, Expanded, Contracted & Isomeric Porphyrins, Tetrahedron Organic Chemistry Series, Elsevier, Oxford, 1997, vol. 15 Search PubMed.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- P. R. Schleyer, Chem. Rev., 2001, 101, 1115–1118 CrossRef CAS PubMed.
- M. Faraday, Philos. Trans. R. Soc., 1825, 115, 440–466 CrossRef.
- F. A. Kekulé, Bull. Soc. Chim. Paris, 1865, 3, 98–110 Search PubMed.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- E. Heilbronner, Tetrahedron Lett., 1964, 5, 1923–1928 CrossRef.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- A. Srinivasan, V. R. G. Anand, S. K. Pushpan, T. K. Chandrashekar, K.-I. Sugiura and Y. Sakata, J. Chem. Soc., Perkin Trans. 2, 2000, 1788–1793 RSC.
- V. J. Bauer, D. L. J. Clive, D. Dolphin, J. B. Paine, F. L. Harris, M. M. King, J. Loder, S. W. C. Wang and R. B. Woodward, J. Am. Chem. Soc., 1983, 105, 6429–6436 CrossRef CAS.
- J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef CAS PubMed.
- T. D. Lash and S. T. Chaney, Angew. Chem., Int. Ed. Engl., 1997, 36, 839–840 CrossRef CAS.
- M. C. Yoon, R. Misra, Z. S. Yoon, K. S. Kim, J. M. Lim, T. K. Chandrashekar and D. Kim, J. Phys. Chem. B, 2008, 112, 6900–6905 CrossRef CAS PubMed.
- T. K. Chandrashekar, V. Prabhuraja, S. Gokulnath, R. Sabarinathan and A. Srinivasan, Chem. Commun., 2010, 46, 5915–5917 RSC.
- G. Karthik, M. Sneha, V. P. Raja, J. M. Lim, D. Kim, A. Srinivasan and T. K. Chandrashekar, Chem. – Eur. J., 2013, 19, 1886–1890 CrossRef CAS PubMed.
- G. Karthik, A. Srinivasan and T. K. Chandrashekar, Org. Lett., 2014, 16, 3472–3475 CrossRef CAS PubMed.
- A. Mallick, J. Oh, D. Kim and H. Rath, Chem. – Eur. J., 2016, 22, 8026–8031 CrossRef CAS PubMed.
- G. Karthik, W. Y. Cha, A. Ghosh, T. Kim, A. Srinivasan, D. Kim and T. K. Chandrashekar, Chem. – Asian J., 2016, 11, 1447–1453 CrossRef CAS PubMed.
- T. Sarma, G. Kim, S. Sen, W. Y. Cha, Z. Duan, M. D. Moore, V. M. Lynch, Z. Zhang, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 12111–12119 CrossRef CAS PubMed.
- T. D. Lash, J. Porphyrins Phthalocyanines, 2012, 15, 1093–1115 CrossRef.
- M. G. P. M. S. Neves, R. M. Martins, A. C. Tome, A. J. D. Silvestre, A. M. S. Silva, V. Felix, M. G. B. Drew and J. A. S. Cavaleiro, Chem. Commun., 1999, 385–386 RSC.
- E. Steiner and P. W. Fowler, Org. Biomol. Chem., 2004, 2, 34–37 RSC.
- Z. S. Yoon, S. B. Noh, D. G. Cho, J. L. Sessler and D. Kim, Chem. Commun., 2007, 2378–2380 RSC.
- V. G. Anand, S. Saito, S. Shimizu and A. Osuka, Angew. Chem., Int. Ed., 2005, 44, 7244–7248 CrossRef CAS PubMed.
- J. Aihara and H. Horibe, Org. Biomol. Chem., 2009, 7, 1939–1943 RSC.
- J. Aihara and M. Makino, Bull. Chem. Soc. Jpn., 2009, 82, 675–682 CrossRef CAS.
- J. L. Sessler, D. G. Cho, M. Stepien, V. Lynch, J. Waluk, Z. S. Yoon and D. Kim, J. Am. Chem. Soc., 2006, 128, 12640–12641 CrossRef CAS PubMed.
- Z. S. Yoon, J. H. Kwon, M. C. Yoon, M. K. Koh, S. B. Noh, J. L. Sessler, J. T. Lee, D. Seidel, A. Aguilar, S. Shimizu, M. Suzuki, A. Osuka and D. Kim, J. Am. Chem. Soc., 2006, 128, 14128–14134 CrossRef CAS PubMed.
- S. Mori, K. S. Kim, Z. S. Yoon, S. B. Noh, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 11344–11345 CrossRef CAS PubMed.
- Z. S. Yoon, D. G. Cho, K. S. Kim, J. L. Sessler and D. Kim, J. Am. Chem. Soc., 2008, 130, 6930–6931 CrossRef CAS PubMed.
- J. M. Lim, Z. S. Yoon, J. Y. Shin, K. S. Kim, M. C. Yoon and D. Kim, Chem. Commun., 2009, 261–273 CAS.
- S. Cho, Z. S. Yoon, K. S. Kim, M.-C. Yoon, D.-G. Cho, J. L. Sessler and D. Kim, J. Phys. Chem. Lett., 2010, 1, 895–900 CrossRef CAS.
- A. Osuka and S. Saito, Chem. Commun., 2011, 47, 4330–4339 RSC.
- Z. S. Yoon, A. Osuka and D. Kim, Nat. Chem., 2009, 1, 113–122 CrossRef CAS PubMed.
- S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 CrossRef CAS PubMed.
- S. Mori and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8030–8031 CrossRef CAS PubMed.
- H. Rath, N. Aratani, J. M. Lim, J. S. Lee, D. Kim, H. Shinokubo and A. Osuka, Chem. Commun., 2009, 3762–3764 RSC.
- T. Koide, K. Youfu, S. Saito and A. Osuka, Chem. Commun., 2009, 6047–6049 RSC.
- S. Sugawara, Y. Hirata, S. Kojima, Y. Yamamoto, E. Miyazaki, K. Takimiya, S. Matsukawa, D. Hashizume, J. Mack, N. Kobayashi, Z. Fu, K. M. Kadish, Y. M. Sung, K. S. Kim and D. Kim, Chem. – Eur. J., 2012, 18, 3566–3581 CrossRef CAS PubMed.
- M. Ishida, S. J. Kim, C. Preihs, K. Ohkubo, J. M. Lim, B. S. Lee, J. S. Park, V. M. Lynch, V. V. Roznyatovskiy, T. Sarma, P. K. Panda, C. H. Lee, S. Fukuzumi, D. Kim and J. L. Sessler, Nat. Chem., 2013, 5, 15–20 CrossRef CAS.
- S. I. Ishida, J. Kim, D. Shimizu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 5876–5880 CrossRef CAS PubMed.
- M. Umetani, J. Kim, T. Tanaka, D. Kim and A. Osuka, Chem. Commun., 2019, 55, 10547–10550 RSC.
- D. Xie, Y. Liu, Y. Rao, G. Kim, M. Zhou, D. Yu, L. Xu, B. Yin, S. Liu, T. Tanaka, N. Aratani, A. Osuka, Q. Liu, D. Kim and J. Song, J. Am. Chem. Soc., 2018, 140, 16553–16559 CrossRef CAS PubMed.
- J. Sankar, S. Mori, S. Saito, H. Rath, M. Suzuki, Y. Inokuma, H. Shinokubo, K. S. Kim, Z. S. Yoon, J. Y. Shin, J. M. Lim, Y. Matsuzaki, O. Matsushita, A. Muranaka, N. Kobayashi, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 13568–13579 CrossRef CAS.
- K. S. Kim, Z. S. Yoon, A. B. Ricks, J. Y. Shin, S. Mori, J. Sankar, S. Saito, Y. M. Jung, M. R. Wasielewski, A. Osuka and D. Kim, J. Phys. Chem. A, 2009, 113, 4498–4506 CrossRef CAS PubMed.
- M. C. Yoon, P. Kim, H. Yoo, S. Shimizu, T. Koide, S. Tokuji, S. Saito, A. Osuka and D. Kim, J. Phys. Chem. B, 2011, 115, 14928–14937 CrossRef CAS PubMed.
- M. C. Yoon, J. Y. Shin, J. M. Lim, S. Saito, T. Yoneda, A. Osuka and D. Kim, Chem. – Eur. J., 2011, 17, 6707–6715 CrossRef CAS PubMed.
- A. Jana, M. Ishida, K. Cho, S. K. Ghosh, K. Kwak, K. Ohkubo, Y. M. Sung, C. M. Davis, V. M. Lynch, D. Lee, S. Fukuzumi, D. Kim and J. L. Sessler, Chem. Commun., 2013, 49, 8937–8939 RSC.
- S. Saito, J. Y. Shin, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9657–9660 CrossRef CAS.
- J. M. Lim, J. Y. Shin, Y. Tanaka, S. Saito, A. Osuka and D. Kim, J. Am. Chem. Soc., 2010, 132, 3105–3114 CrossRef CAS.
- W.-Y. Cha, J. M. Lim, M.-C. Yoon, Y. M. Sung, B. S. Lee, S. Katsumata, M. Suzuki, H. Mori, Y. Ikawa, H. Furuta, A. Osuka and D. Kim, Chem. – Eur. J., 2012, 18, 15838–15844 CrossRef CAS.
- W. Y. Cha, T. Yoneda, S. Lee, J. M. Lim, A. Osuka and D. Kim, Chem. Commun., 2014, 50, 548–550 RSC.
- S. Ishida, T. Higashino, S. Mori, H. Mori, N. Aratani, T. Tanaka, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 3427–3431 CrossRef CAS PubMed.
- K. Naoda, H. Mori, J. Oh, K. H. Park, D. Kim and A. Osuka, J. Org. Chem., 2015, 80, 11726–11733 CrossRef CAS PubMed.
- W. Y. Cha, T. Soya, T. Tanaka, H. Mori, Y. Hong, S. Lee, K. H. Park, A. Osuka and D. Kim, Chem. Commun., 2016, 52, 6076–6078 RSC.
- Y. M. Sung, J. Oh, W. Y. Cha, W. Kim, J. M. Lim, M. C. Yoon and D. Kim, Chem. Rev., 2017, 117, 2257–2312 CrossRef CAS.
- C. S. Wannere, D. Moran, N. L. Allinger, B. A. Hess, L. J. Schaad and P. V. R. Schleyer, Org. Lett., 2003, 5, 2983–2986 CrossRef CAS.
- C. Gellini and P. R. Salvi, Symmetry, 2010, 2, 1846–1924 CrossRef CAS.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Molecules, 2020, 25, 711 CrossRef CAS PubMed.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, in Aromaticity, ed. I. Fernandez, Elsevier, 2021, pp. 235–259 Search PubMed.
- F. Sondheimer, R. Wolovsky and Y. Amiel, J. Am. Chem. Soc., 1962, 84, 274–284 CrossRef.
- E. Vogel, Angew. Chem., Int. Ed., 2011, 50, 4278–4287 CrossRef CAS PubMed.
- T. D. Lash, S. A. Jones and G. M. Ferrence, J. Am. Chem. Soc., 2010, 132, 12786–12787 CrossRef CAS PubMed.
- T. D. Lash, J. Porphyrins Phthalocyanines, 2011, 15, 1093–1115 CrossRef CAS.
- J. I. Wu, I. Fernandez and P. V. R. Schleyer, J. Am. Chem. Soc., 2013, 135, 315–321 CrossRef CAS PubMed.
- A. L. Sargent, I. C. Hawkins, W. E. Allen, H. Liu, J. L. Sessler and C. J. Fowler, Chem. – Eur. J., 2003, 9, 3065–3072 CrossRef CAS.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- P. V. R. Schleyer, H. Jiao, N. J. v. E. Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- M. K. Cyrañski, T. M. Krygowski, M. Wisiorowski, N. J. van Eikema Hommes and P. V. R. Schleyer, Angew. Chem., Int. Ed., 1998, 37, 177–180 CrossRef.
- J. Jusélius and D. Sundholm, J. Org. Chem., 2000, 65, 5233–5237 CrossRef PubMed.
- J. Jusélius and D. Sundholm, Phys. Chem. Chem. Phys., 2000, 2, 2145–2151 RSC.
- N. Otero, S. Fias, S. Radenkovic, P. Bultinck, A. M. Graña and M. Mandado, Chem. – Eur. J., 2011, 17, 3274–3286 CrossRef CAS PubMed.
- H. Fliegl and D. Sundholm, J. Org. Chem., 2012, 77, 3408–3414 CrossRef CAS PubMed.
- R. Pino-Rios, G. Cárdenas-Jirón and W. Tiznado, Phys. Chem. Chem. Phys., 2020, 22, 21267–21274 RSC.
- M. Rosenberg, C. Dahlstrand, K. Kilsa and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- R. Papadakis and H. Ottosson, Chem. Soc. Rev., 2015, 44, 6472–6493 RSC.
- F. Fratev, V. Monev and R. Janoschek, Tetrahedron, 1982, 38, 2929–2932 CrossRef CAS.
- J.-i. Aihara, Bull. Chem. Soc. Jpn., 1978, 51, 1788–1792 CrossRef CAS.
- S. Zilberg and Y. Haas, J. Phys. Chem. A, 1998, 102, 10851–10859 CrossRef CAS.
- F. Feixas, J. Vandenbussche, P. Bultinck, E. Matito and M. Solà, Phys. Chem. Chem. Phys., 2011, 13, 20690–20703 RSC.
- J. Zhu, K. An and P. V. R. Schleyer, Org. Lett., 2013, 15, 2442–2445 CrossRef CAS PubMed.
- P. B. Karadakov, P. Hearnshaw and K. E. Horner, J. Org. Chem., 2016, 81, 11346–11352 CrossRef CAS.
- P. B. Karadakov and S. Saito, Angew. Chem., Int. Ed., 2020, 59, 9228–9230 CrossRef CAS PubMed.
- S. Yadav, O. El Bakouri, K. Jorner, H. Tong, C. Dahlstrand, M. Solà and H. Ottosson, Chem. – Asian J., 2019, 14, 1870–1878 CrossRef CAS PubMed.
- K. Jorner, A. Dreos, R. Emanuelsson, O. El Bakouri, I. F. Galván, K. Börjesson, F. Feixas, R. Lindh, B. Zietz and K. Moth-Poulsen, J. Mater. Chem. A, 2017, 5, 12369–12378 RSC.
- H. Löfås, B. Jahn, J. Wärnå, R. Emanuelsson, R. Ahuja, A. Grigoriev and H. Ottosson, Faraday Discuss., 2014, 174, 105–124 RSC.
- M. Ueda, K. Jorner, Y. M. Sung, T. Mori, Q. Xiao, D. Kim, H. Ottosson, T. Aida and Y. Itoh, Nat. Commun., 2017, 8, 346 CrossRef PubMed.
- P. Wan and E. Krogh, J. Chem. Soc., Chem. Commun., 1985, 1207–1208 RSC.
- E. Krogh and P. Wan, Tetrahedron Lett., 1986, 27, 823–826 CrossRef CAS.
- E. Gaillard, M. A. Fox and P. Wan, J. Am. Chem. Soc., 1989, 111, 2180–2186 CrossRef CAS.
- H. J. Wörner and F. Merkt, Angew. Chem., Int. Ed., 2006, 45, 293–296 CrossRef PubMed.
- Y. M. Sung, M.-C. Yoon, J. M. Lim, H. Rath, K. Naoda, A. Osuka and D. Kim, Nat. Chem., 2015, 7, 418–422 CrossRef CAS PubMed.
- J. Oh, Y. M. Sung, Y. Hong and D. Kim, Acc. Chem. Res., 2018, 51, 1349–1358 CrossRef CAS PubMed.
- J. Y. Shin, K. S. Kim, M. C. Yoon, J. M. Lim, Z. S. Yoon, A. Osuka and D. Kim, Chem. Soc. Rev., 2010, 39, 2751–2767 RSC.
- A. W. Hofmann, Proc. R. Soc. London, 1856, 8, 1–3 Search PubMed.
- T. M. Krygowski and M. K. Cyranski, Chem. Rev., 2001, 101, 1385–1419 CrossRef CAS.
- H. S. Rzepa, Chem. Rev., 2005, 105, 3697–3715 CrossRef CAS PubMed.
- M. K. Cyrański, Chem. Rev., 2005, 105, 3773–3811 CrossRef PubMed.
- D. P. Craig and N. L. Paddock, Nature, 1958, 181, 1052–1053 CrossRef CAS.
- A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2000, 39, 3915–3917 CrossRef CAS PubMed.
- M. Bühl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1184 CrossRef PubMed.
- D. Ajami, O. Oeckler, A. Simon and R. Herges, Nature, 2003, 426, 819–821 CrossRef CAS PubMed.
- Y. Tanaka, S. Saito, S. Mori, N. Aratani, H. Shinokubo, N. Shibata, Y. Higuchi, Z. S. Yoon, K. S. Kim, S. B. Noh, J. K. Park, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 681–684 CrossRef CAS PubMed.
- J. K. Park, Z. S. Yoon, M. C. Yoon, K. S. Kim, S. Mori, J. Y. Shin, A. Osuka and D. Kim, J. Am. Chem. Soc., 2008, 130, 1824–1825 CrossRef CAS PubMed.
- N. Jux, Angew. Chem., Int. Ed., 2008, 47, 2543–2546 CrossRef CAS PubMed.
- S. Tokuji, J. Y. Shin, K. S. Kim, J. M. Lim, K. Youfu, S. Saito, D. Kim and A. Osuka, J. Am. Chem. Soc., 2009, 131, 7240–7241 CrossRef CAS PubMed.
- M. Inoue, K. S. Kim, M. Suzuki, J. M. Lim, J. Y. Shin, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2009, 48, 6687–6690 CrossRef CAS.
- T. Higashino, J. M. Lim, T. Miura, S. Saito, J. Y. Shin, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 4950–4954 CrossRef CAS PubMed.
- M. Stepien, B. Szyszko and L. Latos-Grazynski, J. Am. Chem. Soc., 2010, 132, 3140–3152 CrossRef CAS PubMed.
- M. Inoue and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 9488–9491 CrossRef CAS PubMed.
- J. M. Lim, M. Inoue, Y. M. Sung, M. Suzuki, T. Higashino, A. Osuka and D. Kim, Chem. Commun., 2011, 47, 3960–3962 RSC.
- T. Higashino, B. S. Lee, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2012, 51, 13105–13108 CrossRef CAS PubMed.
- K. Naoda, Y. M. Sung, J. M. Lim, D. Kim and A. Osuka, Chem. – Eur. J., 2014, 20, 7698–7705 CrossRef CAS PubMed.
- T. Yoneda, Y. M. Sung, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 13169–13173 CrossRef CAS PubMed.
- S. Ishida, T. Tanaka, J. M. Lim, D. Kim and A. Osuka, Chem. – Eur. J., 2014, 20, 8274–8278 CrossRef CAS PubMed.
- T. Higashino, T. Soya, W. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 5456–5459 CrossRef CAS PubMed.
- S. I. Ishida, J. O. Kim, D. Kim and A. Osuka, Chem. – Eur. J., 2016, 22, 16554–16561 CrossRef CAS.
- T. Soya, H. Mori, Y. Hong, Y. H. Koo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 3232–3236 CrossRef CAS PubMed.
- M. Izawa, T. Kim, S. I. Ishida, T. Tanaka, T. Mori, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 3982–3986 CrossRef CAS PubMed.
- H. Ottosson, Nat. Chem., 2012, 4, 969–971 CrossRef CAS PubMed.
- M. J. S. Dewar, Tetrahedron, 1966, 22, 75–82 CrossRef.
- H. E. Zimmerman, J. Am. Chem. Soc., 1966, 88, 1564–1565 CrossRef CAS.
- H. E. Zimmerman, J. Am. Chem. Soc., 1966, 88, 1566–1567 CrossRef CAS.
- R. C. Dougherty, J. Am. Chem. Soc., 1971, 93, 7187–7201 CrossRef CAS.
- O. Chapman, C. McIntosh and J. Pacansky, J. Am. Chem. Soc., 1973, 95, 614–617 CrossRef CAS.
- V. Gogonea, P. v. R. Schleyer and P. R. Schreiner, Angew. Chem., Int. Ed., 1998, 37, 1945–1948 CrossRef CAS.
- H. J. Wörner and F. Merkt, J. Chem. Phys., 2007, 127, 034303 CrossRef PubMed.
- H. J. Wörner and F. Merkt, Angew. Chem., Int. Ed., 2009, 48, 6404–6424 CrossRef PubMed.
- C. J. Kastrup, S. P. Oldfield and H. S. Rzepa, Chem. Commun., 2002, 642–643 RSC.
- P. B. Karadakov, M. Di and D. L. Cooper, J. Phys. Chem. A, 2020, 124, 9611–9616 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of molecular photochemistry: An introduction, University Science Books, Sausalito, California, 2009 Search PubMed.
- M. Kataoka, J. Chem. Res., 2004, 2004, 573–574 CrossRef.
- A. Soncini and P. Fowler, Chem. Phys. Lett., 2008, 450, 431–436 CrossRef CAS.
- I. S. Ulusoy and M. Nest, J. Am. Chem. Soc., 2011, 133, 20230–20236 CrossRef CAS PubMed.
- K. R. Asmis, M. Allan, O. Schafer and M. Fülscher, J. Phys. Chem. A, 1997, 101, 2089–2095 CrossRef CAS.
- L. Madden, A. Mebel, M.-C. Lin and C. Melius, J. Phys. Org. Chem., 1996, 9, 801–810 CrossRef CAS.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 12707–12713 CrossRef CAS PubMed.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 7303–7309 CrossRef CAS.
- F. Feixas, E. Matito, M. Sola and J. Poater, J. Phys. Chem. A, 2008, 112, 13231–13238 CrossRef CAS PubMed.
- F. Feixas, E. Matito, M. Solà and J. Poater, Phys. Chem. Chem. Phys., 2010, 12, 7126–7137 RSC.
- P. Wan, E. Krogh and B. Chak, J. Am. Chem. Soc., 1988, 110, 4073–4074 CrossRef CAS.
- I. McAuley, E. Krogh and P. Wan, J. Am. Chem. Soc., 1988, 110, 600–602 CrossRef CAS.
- P. Wan and E. Krogh, J. Am. Chem. Soc., 1989, 111, 4887–4895 CrossRef CAS.
- P. Wan, D. Budac and E. Krogh, J. Chem. Soc., Chem. Commun., 1990, 255–257 CAS.
- P. Wan, D. Budac, M. Earle and D. Shukla, J. Am. Chem. Soc., 1990, 112, 8048–8054 CrossRef CAS.
- D. Budac and P. Wan, J. Org. Chem., 1992, 57, 887–894 CrossRef CAS.
- E. Krogh and P. Wan, J. Am. Chem. Soc., 1992, 114, 705–712 CrossRef CAS.
- P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef CAS.
- D. Shukla and P. Wan, J. Am. Chem. Soc., 1993, 115, 2990–2991 CrossRef CAS.
- D. Budac and P. Wan, Can. J. Chem., 1996, 74, 1447–1464 CrossRef CAS.
- D. Budac and P. Wan, J. Photochem. Photobiol., A, 1996, 98, 27–37 CrossRef CAS.
- D. Brousmiche, D. Shukla and P. Wan, Chem. Commun., 1997, 709–710 RSC.
- D. Shukla and P. Wan, J. Photochem. Photobiol., A, 1998, 113, 53–64 CrossRef CAS.
- H. Ottosson, K. Kilså, K. Chajara, M. C. Piqueras, R. Crespo, H. Kato and D. Muthas, Chem. – Eur. J., 2007, 13, 6998–7005 CrossRef CAS PubMed.
- M. Rosenberg, H. Ottosson and K. Kilså, Phys. Chem. Chem. Phys., 2011, 13, 12912–12919 RSC.
- H. Möllerstedt, M. C. Piqueras, R. Crespo and H. Ottosson, J. Am. Chem. Soc., 2004, 126, 13938–13939 CrossRef PubMed.
- P. Seiler and J. Wirz, Helv. Chim. Acta, 1972, 55, 2693–2712 CrossRef CAS.
- W. J. Feast and W. E. Preston, J. Chem. Soc., Chem. Commun., 1974, 985–986 RSC.
- W. R. Dolbier, K. Matsui, H. J. Dewey, D. V. Horák and J. Michl, J. Am. Chem. Soc., 1979, 101, 2136–2139 CrossRef CAS.
- J. Li and T. L. Lowary, Org. Lett., 2008, 10, 881–884 CrossRef CAS.
- H. Kato, M. Brink, H. Möllerstedt, M. C. Piqueras, R. Crespo and H. Ottosson, J. Org. Chem., 2005, 70, 9495–9504 CrossRef CAS PubMed.
- S. Villaume and H. Ottosson, J. Phys. Chem. A, 2009, 113, 12304–12310 CrossRef CAS PubMed.
- R. K. Mohamed, S. Mondal, K. Jorner, T. F. Delgado, V. V. Lobodin, H. Ottosson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 15441–15450 CrossRef CAS PubMed.
- R. Ayub, R. Papadakis, K. Jorner, B. Zietz and H. Ottosson, Chem. – Eur. J., 2017, 23, 13684–13695 CrossRef CAS.
- T. Slanina, R. Ayub, J. Toldo, J. Sundell, W. Rabten, M. Nicaso, I. Alabugin, I. Fdez Galvan, A. K. Gupta, R. Lindh, A. Orthaber, R. J. Lewis, G. Gronberg, J. Bergman and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 10942–10954 CrossRef CAS.
- R. Papadakis, H. Li, J. Bergman, A. Lundstedt, K. Jorner, R. Ayub, S. Haldar, B. O. Jahn, A. Denisova and B. Zietz, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- T. M. Krygowski and M. Cyranski, Aromatic Character of Carbo- cyclic π-Electron Systems Deduced from Molecular Geometry, in Advances in Mocular Structure Research, ed. I. M. Hargittai, JAI Press, London, 1997, vol. 3, p. 227 Search PubMed.
- E. D. Bergmann, Aromaticity, Pseudoaromaticity and Antiaromaticity, Proceedings of an International Symposium held in Jerusalem 1970, ed. E. D. Bergmann, B. Pullman, Israel Academy of Sciences and Humanities, Jerusalem, 1971, p. 386.
- V. I. Minkin, M. N. Glukhovtsev and B. Y. Simkin, Aromaticity and Antiaromaticity. Electronic and Structural Aspects, Wiley, New York, 1994 Search PubMed.
- R. Herges, Chem. Rev., 2006, 106, 4820–4842 CrossRef CAS PubMed.
- T. Soya, H. Mori and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 15882–15886 CrossRef CAS PubMed.
- M. D. Peeks, T. D. Claridge and H. L. Anderson, Nature, 2017, 541, 200–203 CrossRef CAS PubMed.
- G. Bressan, M. D. Peeks, H. L. Anderson, S. R. Meech and I. A. Heisler, J. Phys. Chem. C, 2019, 123, 27222–27229 CrossRef CAS.
- S. M. Kopp, H. Gotfredsen, J. R. Deng, T. D. W. Claridge and H. L. Anderson, J. Am. Chem. Soc., 2020, 142, 19393–19401 CrossRef CAS PubMed.
- M. Rickhaus, M. Jirasek, L. Tejerina, H. Gotfredsen, M. D. Peeks, R. Haver, H. W. Jiang, T. D. W. Claridge and H. L. Anderson, Nat. Chem., 2020, 12, 236–241 CrossRef CAS PubMed.
- M. Jirasek, M. Rickhaus, L. Tejerina and H. L. Anderson, J. Am. Chem. Soc., 2021, 143, 2403–2412 CrossRef CAS PubMed.
- T. Soya, W. Kim, D. Kim and A. Osuka, Chem. – Eur. J., 2015, 21, 8341–8346 CrossRef CAS PubMed.
- S. Shimizu, R. Taniguchi and A. Osuka, Angew. Chem., Int. Ed., 2005, 44, 2225–2229 CrossRef CAS PubMed.
- T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., 2012, 124, 8670–8673 CrossRef.
- J.-Y. Shin, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2001, 40, 619–621 CrossRef CAS.
- S. Mori, J.-Y. Shin, S. Shimizu, F. Ishikawa, H. Furuta and A. Osuka, Chem. – Eur. J., 2005, 11, 2417–2425 CrossRef CAS PubMed.
- J. L. Sessler, S. J. Weghorn, T. Morishima, M. Rosingana, V. Lynch and V. Lee, J. Am. Chem. Soc., 1992, 114, 8306–8307 CrossRef CAS.
- T. K. Ahn, J. H. Kwon, D. Y. Kim, D. W. Cho, D. H. Jeong, S. K. Kim, M. Suzuki, S. Shimizu, A. Osuka and D. Kim, J. Am. Chem. Soc., 2005, 127, 12856–12861 CrossRef CAS PubMed.
- T. Yoneda, T. Kim, T. Soya, S. Neya, J. Oh, D. Kim and A. Osuka, Chem. – Eur. J., 2016, 22, 4413–4417 CrossRef CAS PubMed.
- A. Nakai, J. Kim, D. Kim and A. Osuka, Chem. – Asian J., 2019, 14, 968–971 CrossRef CAS.
- M. Stepien, L. Latos-Grazynski, N. Sprutta, P. Chwalisz and L. Szterenberg, Angew. Chem., Int. Ed., 2007, 46, 7869–7873 CrossRef CAS PubMed.
- E. Pacholska-Dudziak, J. Skonieczny, M. Pawlicki, L. Szterenberg, Z. Ciunik and L. Latos-Grażyński, J. Am. Chem. Soc., 2008, 130, 6182–6195 CrossRef CAS PubMed.
- J. Kim, G. Kim and D. Kim, J. Porphyrins Phthalocyanines, 2020, 24, 1278–1299 CrossRef CAS.
- R. Guerrero-Avilés, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Angew. Chem., Int. Ed., 2021, 60, 24080–24088 CrossRef PubMed.
- M. Jirásek, H. L. Anderson and M. D. Peeks, Acc. Chem. Res., 2021, 54, 3241–3251 CrossRef PubMed.
- M. Gouterman, G. H. Wagnière and L. C. Snyder, J. Mol. Spectrosc., 1963, 11, 108–127 CrossRef CAS.
- J. Michl, J. Am. Chem. Soc., 1978, 100, 6801–6811 CrossRef CAS.
- J. Michl, J. Am. Chem. Soc., 1978, 100, 6812–6818 CrossRef CAS.
- J. Michl, Pure Appl. Chem., 1980, 52, 1549–1563 CAS.
- J. Mack, Chem. Rev., 2017, 117, 3444–3478 CrossRef CAS PubMed.
- T. Woller, P. Geerlings, F. De Proft, B. Champagne and M. Alonso, J. Phys. Chem. C, 2019, 123, 7318–7335 CrossRef CAS.
- A. Gorski, B. Lament, J. Davis, J. Sessler and J. Waluk, J. Phys. Chem. A, 2001, 105, 4992–4999 CrossRef CAS.
- J. Mack, M. J. Stillman and N. Kobayashi, Coord. Chem. Rev., 2007, 251, 429–453 CrossRef CAS.
- J. Mack, M. Bunya, Y. Shimizu, H. Uoyama, N. Komobuchi, T. Okujima, H. Uno, S. Ito, M. J. Stillman and N. Ono, Chem. – Eur. J., 2008, 14, 5001–5020 CrossRef CAS PubMed.
- A. Muranaka, O. Matsushita, K. Yoshida, S. Mori, M. Suzuki, T. Furuyama, M. Uchiyama, A. Osuka and N. Kobayashi, Chem. – Eur. J., 2009, 15, 3744–3751 CrossRef CAS PubMed.
- Z. Zhou, Y. Chang, S. Shimizu, J. Mack, C. Schütt, R. Herges, Z. Shen and N. Kobayashi, Angew. Chem., Int. Ed., 2014, 53, 6563–6567 CrossRef CAS PubMed.
- X. Liang, J. Mack, L.-M. Zheng, Z. Shen and N. Kobayashi, Inorg. Chem., 2014, 53, 2797–2802 CrossRef CAS.
- J. Oh, H. Joung, W. Kim, J. Yang and D. Kim, J. Phys. Chem. C, 2021, 125, 1947–1953 CrossRef CAS.
- T. Higashino, A. Kumagai, S. Sakaki and H. Imahori, Chem. Sci., 2018, 9, 7528–7539 RSC.
- T. Woller, P. Geerlings, F. De Proft, B. Champagne and M. Alonso, Molecules, 2018, 23, 1333 CrossRef PubMed.
- J. L. Sessler, S. J. Weghorn, Y. Hiseada and V. Lynch, Chem. – Eur. J., 1995, 1, 56–67 CrossRef CAS.
- C. H. Hung, J. P. Jong, M. Y. Ho, G. H. Lee and S. M. Peng, Chem. – Eur. J., 2002, 8, 4542–4548 CrossRef CAS.
- H. Mori, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 12997–13001 CrossRef CAS PubMed.
- M. Alonso, P. Geerlings and F. De Proft, Chem. – Eur. J., 2012, 18, 10916–10928 CrossRef CAS.
- M. Toganoh and H. Furuta, J. Org. Chem., 2010, 75, 8213–8223 CrossRef CAS PubMed.
- M. Toganoh and H. Furuta, J. Org. Chem., 2013, 78, 9317–9327 CrossRef CAS PubMed.
- M. Alonso, P. Geerlings and F. De Proft, Chem. – Eur. J., 2013, 19, 1617–1628 CrossRef CAS PubMed.
- J. Oh, H. Mori, Y. M. Sung, W. Kim, A. Osuka and D. Kim, Chem. Sci., 2016, 7, 2239–2245 RSC.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- J. Kruszewski and T. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- E. Matito, M. Duran and M. Sola, J. Chem. Phys., 2005, 122, 014109 CrossRef PubMed.
- E. Matito, M. Duran and M. Sola, J. Chem. Phys., 2006, 125, 59901 CrossRef.
- P. V. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4, 2873–2876 CrossRef CAS PubMed.
- W. C. Herndon and N. S. Mills, J. Org. Chem., 2005, 70, 8492–8496 CrossRef CAS PubMed.
- F. Feixas, E. Matito, J. Poater and M. Solà, J. Comput. Chem., 2008, 29, 1543–1554 CrossRef CAS PubMed.
- F. Feixas, J. O. C. Jiménez-Halla, E. Matito, J. Poater and M. Sola, J. Chem. Theory Comput., 2010, 6, 1118–1130 CrossRef CAS.
- E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
- I. Casademont-Reig, T. Woller, J. Contreras-García, M. Alonso, M. Torrent-Sucarrat and E. Matito, Phys. Chem. Chem. Phys., 2018, 20, 2787–2796 RSC.
- K. Jorner, R. Emanuelsson, C. Dahlstrand, H. Tong, A. V. Denisova and H. Ottosson, Chem. – Eur. J., 2014, 20, 9295–9303 CrossRef CAS PubMed.
- Y. M. Sung, J. Oh, W. Kim, H. Mori, A. Osuka and D. Kim, J. Am. Chem. Soc., 2015, 137, 11856–11859 CrossRef CAS PubMed.
- T. M. Krygowski and M. K. Cyrański, Tetrahedron, 1999, 55, 11143–11148 CrossRef CAS.
- M. Suzuki and A. Osuka, Chem. Commun., 2005, 3685–3687 RSC.
- M. D. Peeks, J. Q. Gong, K. McLoughlin, T. Kobatake, R. Haver, L. M. Herz and H. L. Anderson, J. Phys. Chem. Lett., 2019, 10, 2017–2022 CrossRef CAS PubMed.
- M. Matsushita, M. Ketelaars, A. M. van Oijen, J. Köhler, T. J. Aartsma and J. Schmidt, Biophys. J., 2001, 80, 1604–1614 CrossRef CAS.
- R. J. Cogdell and J. Köhler, Biochem. J., 2009, 422, 193–205 CrossRef CAS PubMed.
- J. K. Sprafke, D. V. Kondratuk, M. Wykes, A. L. Thompson, M. Hoffmann, R. Drevinskas, W.-H. Chen, C. K. Yong, J. Karnbratt and J. E. Bullock, J. Am. Chem. Soc., 2011, 133, 17262–17273 CrossRef CAS PubMed.
- K. H. Park, P. Kim, W. Kim, H. Shimizu, M. Han, E. Sim, M. Iyoda and D. Kim, Angew. Chem., Int. Ed., 2015, 54, 12711–12715 CrossRef CAS PubMed.
- K. H. Park, W. Kim, J. Yang and D. Kim, Chem. Soc. Rev., 2018, 47, 4279–4294 RSC.
- J. Oh, Y. M. Sung, W. Kim, S. Mori, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2016, 55, 6487–6491 CrossRef CAS PubMed.
- Y. Hong, J. Oh, Y. M. Sung, Y. Tanaka, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2017, 56, 2932–2936 CrossRef CAS PubMed.
- S. Shaik, A. Shurki, D. Danovich and P. C. Hiberty, Chem. Rev., 2001, 101, 1501–1540 CrossRef CAS PubMed.
- A. Stanger, Chem. Commun., 2009, 1939–1947 RSC.
- S. Ostrowski and J. C. Dobrowolski, RSC Adv., 2014, 4, 44158–44161 RSC.
- J. C. Dobrowolski, ACS Omega, 2019, 4, 18699–18710 CrossRef CAS PubMed.
- H. Fliegl, D. Sundholm, S. Taubert and F. Pichierri, J. Phys. Chem. A, 2010, 114, 7153–7161 CrossRef CAS PubMed.
- J.-Y. Shin, H. Furuta, K. Yoza, S. Igarashi and A. Osuka, J. Am. Chem. Soc., 2001, 123, 7190–7191 CrossRef CAS PubMed.
- R. Misra and T. K. Chandrashekar, Acc. Chem. Res., 2008, 41, 265–279 CrossRef CAS PubMed.
- B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839–2909 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- R. Karoui, G. Downey and C. Blecker, Chem. Rev., 2010, 110, 6144–6168 CrossRef CAS PubMed.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- E. Wiercigroch, E. Szafraniec, K. Czamara, M. Z. Pacia, K. Majzner, K. Kochan, A. Kaczor, M. Baranska and K. Malek, Spectrochim. Acta, Part A, 2017, 185, 317–335 CrossRef CAS PubMed.
- J. Oh, Y. M. Sung, H. Mori, S. Park, K. Jorner, H. Ottosson, M. Lim, A. Osuka and D. Kim, Chem, 2017, 3, 870–880 CAS.
- S. L. Georgopoulos, R. Diniz, M. I. Yoshida, N. L. Speziali, H. F. Dos Santos, G. M. A. Junqueira and L. F. de Oliveira, J. Mol. Struct., 2006, 794, 63–70 CrossRef CAS.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2014, 118, 223–237 CrossRef CAS PubMed.
- D. Setiawan, E. Kraka and D. Cremer, J. Org. Chem., 2016, 81, 9669–9686 CrossRef CAS PubMed.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CrossRef CAS PubMed.
- J. C. Dean, T. Mirkovic, Z. S. Toa, D. G. Oblinsky and G. D. Scholes, Chem, 2016, 1, 858–872 CAS.
- Y. M. Sung, J. Oh, K. Naoda, T. Lee, W. Kim, M. Lim, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2016, 55, 11930–11934 CrossRef CAS PubMed.
- T. Shimidzu, H. Segawa, T. Iyoda and K. Honda, J. Chem. Soc., Faraday Trans. 2, 1987, 83, 2191–2200 RSC.
- A. M. Brun, A. Harriman, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8657–8663 CrossRef CAS.
- I. M. Dixon, J.-P. Collin, J.-P. Sauvage, F. Barigelletti and L. Flamigni, Angew. Chem., Int. Ed., 2000, 39, 1292–1295 CrossRef CAS.
- S. Preiß, A. Päpcke, L. Burkhardt, L. Großmann, S. Lochbrunner, M. Bauer, T. Opatz and K. Heinze, Chem. – Eur. J., 2019, 25, 5940–5949 CrossRef PubMed.
- Y. Wang, H. Kai, M. Ishida, S. Gokulnath, S. Mori, T. Murayama, A. Muranaka, M. Uchiyama, Y. Yasutake, S. Fukatsu, Y. Notsuka, Y. Yamaoka, M. Hanafusa, M. Yoshizawa, G. Kim, D. Kim and H. Furuta, J. Am. Chem. Soc., 2020, 142, 6807–6813 CrossRef CAS PubMed.
- J. Kim, J. Oh, T. Soya, T. Yoneda, S. Park, M. Lim, A. Osuka and D. Kim, Angew. Chem., Int. Ed., 2020, 59, 5129–5134 CrossRef CAS PubMed.
- S. Escayola, C. Tonnelé, E. Matito, A. Poater, H. Ottosson, M. Solà and D. Casanova, Angew. Chem., Int. Ed., 2021, 60, 10255–10265 CrossRef CAS PubMed.
- T. S. Slanina, R. Ayub, J. Toldo, J. Sundell, W. Rabten, M. Nicaso, I. Alabugin, I. F. Galván, A. K. Gupta and R. Lindh, J. Am. Chem. Soc., 2020, 142, 10942–10954 CrossRef CAS PubMed.
- K. Jorner, W. Rabten, T. Slanina, N. P. Vedin, S. Sillén, J. W. Ludvigsson, H. Ottosson and P.-O. Norrby, Cell Rep. Phys. Sci., 2020, 1, 100274 CrossRef.
- K. Jorner, F. Feixas, R. Ayub, R. Lindh, M. Solà and H. Ottosson, Chem. – Eur. J., 2016, 2793–2800 CrossRef CAS PubMed.
- O. El Bakouri, J. R. Smith and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 5602–5617 CrossRef CAS PubMed.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- W. Zeng, O. El Bakouri, D. W. Szczepanik, H. Bronstein and H. Ottosson, Chem. Sci., 2021, 12, 6159–6171 RSC.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485 RSC.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- K. J. Fallon, P. Budden, E. Salvadori, A. M. Ganose, C. N. Savory, L. Eyre, S. Dowland, Q. Ai, S. Goodlett, C. Risko, D. O. Scanlon, C. W. M. Kay, A. Rao, R. H. Friend, A. J. Musser and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 13867–13876 CrossRef CAS PubMed.
- J. Kim, J. Oh, S. Park, J. L. Zafra, J. R. DeFrancisco, D. Casanova, M. Lim, J. D. Tovar, J. Casado and D. Kim, Nat. Commun., 2019, 10, 4983 CrossRef PubMed.
- B. C. Streifel, J. L. Zafra, G. L. Espejo, C. J. Gómez-García, J. Casado and J. D. Tovar, Angew. Chem., Int. Ed., 2015, 54, 5888–5893 CrossRef CAS PubMed.
- Z. Wen, L. J. Karas, C.-H. Wu, I. Judy and C. Wu, Chem. Commun., 2020, 56, 8380–8383 RSC.
- L. J. Karas, C.-H. Wu, H. Ottosson and J. I. Wu, Chem. Sci., 2020, 11, 10071–10077 RSC.
- C.-H. Wu, L. J. Karas, H. Ottosson, I. Judy and C. Wu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20303–20308 CrossRef CAS PubMed.
- W. Y. Cha, T. Kim, A. Ghosh, Z. Zhang, X. S. Ke, R. Ali, V. M. Lynch, J. Jung, W. Kim, S. Lee, S. Fukuzumi, J. S. Park, J. L. Sessler, T. K. Chandrashekar and D. Kim, Nat. Chem., 2017, 9, 1243–1248 CrossRef CAS PubMed.
- C. A. Gould, J. Marbey, V. Vieru, D. A. Marchiori, R. David Britt, L. F. Chibotaru, S. Hill and J. R. Long, Nat. Chem., 2021, 1–5 Search PubMed.
- Y. Ni, T. Y. Gopalakrishna, H. Phan, T. Kim, T. S. Herng, Y. Han, T. Tao, J. Ding, D. Kim and J. Wu, Nat. Chem., 2020, 12, 242–248 CrossRef CAS PubMed.
- J. R. DeFrancisco, G. Lopez-Espejo, J. L. Zafra, S. Yadav, R. E. Messersmith, C. J. Gomez-Garcia, H. Ottosson, J. Casado and J. D. Tovar, J. Phys. Chem. C, 2018, 122, 12148–12157 CrossRef CAS.
- C. Shu, H. Zhang, A. Olankitwanit, S. Rajca and A. Rajca, J. Am. Chem. Soc., 2019, 141, 17287–17294 CrossRef CAS PubMed.
- C. Liu, Y. Ni, X. Lu, G. Li and J. Wu, Acc. Chem. Res., 2019, 52, 2309–2321 CrossRef CAS PubMed.
- C. Liu, M. E. Sandoval-Salinas, Y. Hong, T. Y. Gopalakrishna, H. Phan, N. Aratani, T. S. Herng, J. Ding, H. Yamada and D. Kim, Chem, 2018, 4, 1586–1595 CAS.
| This journal is © The Royal Society of Chemistry 2022 |