
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical carbon capture processes for mitigation of CO2 emissions
Mohammad
Rahimi
 *ab,
Aliza
Khurram
c,
T. Alan
Hatton
*d and
Betar
Gallant
*c
*ab,
Aliza
Khurram
c,
T. Alan
Hatton
*d and
Betar
Gallant
*c
aDepartment of Civil and Environmental Engineering, University of Houston, Houston, TX 77204, USA. E-mail: mrahimi@uh.edu
bMaterials Science and Engineering Program, University of Houston, Houston, TX 77204, USA
cDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: bgallant@mit.edu
dDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: tahatton@mit.edu
First published on 30th September 2022
Abstract
Carbon capture and storage (CCS) is essential if global warming mitigation scenarios are to be met. However, today's maturing thermochemical capture technologies have exceedingly high energy requirements and rigid form factors that restrict their versatility and limit scale. Using renewable electricity, rather than heat, as the energy input to drive CO2 separations provides a compelling alternative to surpass these limitations. Although electrochemical technologies have been extensively developed for energy storage and CO2 utilization processes, the potential for more expansive intersection of electrochemistry with CCS is only recently receiving growing attention, with multiple scientific proofs-of-concept and a burgeoning pipeline with numerous concepts at various stages of technology readiness. Here, we describe the emerging science and research progress underlying electrochemical CCS processes and assess their current maturity and trajectory. We also highlight emerging ideas that are ripe for continued research and development, which will allow the impact of electrochemical CCS to be properly assessed in coming years.
 Mohammad Rahimi | Dr Mohammad (Mim) Rahimi is an Assistant Professor of Environmental Engineering at the Department of Civil & Environmental Engineering at the University of Houston (UH). He also holds an affiliate appointment with the Materials Science and Engineering Program. Before joining UH, Dr Rahimi was a postdoctoral associate at the Department of Chemical Engineering at Massachusetts Institute of Technology (MIT) (2018–2021). Dr Rahimi obtained his PhD in chemical engineering from Pennsylvania State University in 2017. His research group at UH develops electrochemical processes for climate change mitigation, including a wide range of technologies for carbon capture from air and point sources. |
 Aliza Khurram | Aliza Khurram is a Postdoctoral Associate in the Trancik Lab at the Institute for Data, Systems and Society (IDSS) at MIT. Her current research is focused on building data-informed, energy systems models for elucidating green hydrogen production and storage cost targets for enabling deeply-decarbonized, integrated energy systems. Aliza received her SM (’17) and PhD (’20) degrees in Mechanical Engineering from MIT where she developed new electrochemical approaches for CO2 management. She is a two-time recipient of the MIT Energy Initiative Fellowship (2019, 2021) for her work on integrated CO2 capture and electrochemical mineralization, and the economics of excess renewable energy-powered hydrogen. |
 T. Alan Hatton | T. Alan Hatton is the Ralph Landau Professor and Director of the David H. Koch School of Chemical Engineering Practice at the Massachusetts Institute of Technology. He obtained his BSc and MSc degrees in Chemical Engineering at the University of Natal, Durban, South Africa, and worked at the Council for Scientific and Industrial Research in Pretoria for three years before attending the University of Wisconsin, Madison, to obtain his PhD. He is currently Faculty Lead on Carbon Management in the MIT Energy Initiative Future Energy Systems Center. His research interests encompass self-assembly of surfactants and block copolymers, synthesis and functionalization of magnetic nanoparticles, and the exploitation of stimuli-responsive materials for chemical and environmental processing applications, with a particular current emphasis on molten oxide materials and electrochemically mediated operations for carbon dioxide capture. |
 Betar Gallant | Betar M. Gallant is an Associate Professor and the Class of ‘22 Career Development Professor in the Department of Mechanical Engineering at MIT. She obtained her SB (’08), SM (’10), and PhD (’13) degrees from the same department. Following her PhD, Dr Gallant was a Kavli Nanoscience Institute Prize Postdoctoral Fellow at Caltech in the Division of Chemistry and Chemical Engineering from 2013–2015. Her research group at MIT is working on, among other electrochemistry topics, CO2 capture and its integration with direct electrochemical conversion or mineralization in the captured state, including associated electrolyte design principles and electrochemical mechanism studies. |
1. Introduction
Human activity is causing the Earth's climate to change faster than it ever has throughout the history of modern civilization.1 Any plan of action to mitigate climate change must include methods for reducing greenhouse gas emissions, particularly carbon dioxide (CO2).2 In this context, the Intergovernmental Panel on Climate Change (IPCC) and International Energy Agency (IEA) developed scenarios to limit the global temperature rise to less than 2 °C (known as the “2 °C scenario”) or more aggressively to 1.5 °C (“beyond 2 °C scenario”).3–5 For the “2 °C scenario”, annual CO2 emissions, currently more than 35 Gt,1 need to be reduced by ∼75% by 2060, while for the “beyond 2 °C scenario”, technological advancements and deployment are pushed to their practical limits to reach net-zero emissions by 2060 and to maintain net zero or below thereafter.5 These emission reduction plans include a portfolio of options (Fig. 1) that involve diverse strategies ranging from implementation of nuclear and renewable energy, efficiency improvements in present-day energy sectors, and switching to fuels with low or zero carbon footprints. These three strategies emphasize changes in the nature of energy inputs and energy savings where possible and rely largely on existing technologies that face challenges of global scale-up. A fourth and vital option emphasizes deployment of less-mature technologies—such as carbon capture and storage (CCS)—which are assumed to become available at scale; CCS comprises ∼15% and ∼25% of the overall emission reduction by 2060 for the 2 °C scenario and beyond 2 °C scenario, respectively. Current CCS technologies face challenges, principal among which are high energy requirements: following CO2 capture, the thermal regeneration process consumes 20% to 30% of the power-generating capacity of a coal-powered plant,6 strongly disincentivizing its adoption and making it more cost-effective in coming years to simply continue emitting CO2 in the absence of a carbon cost or other regulations.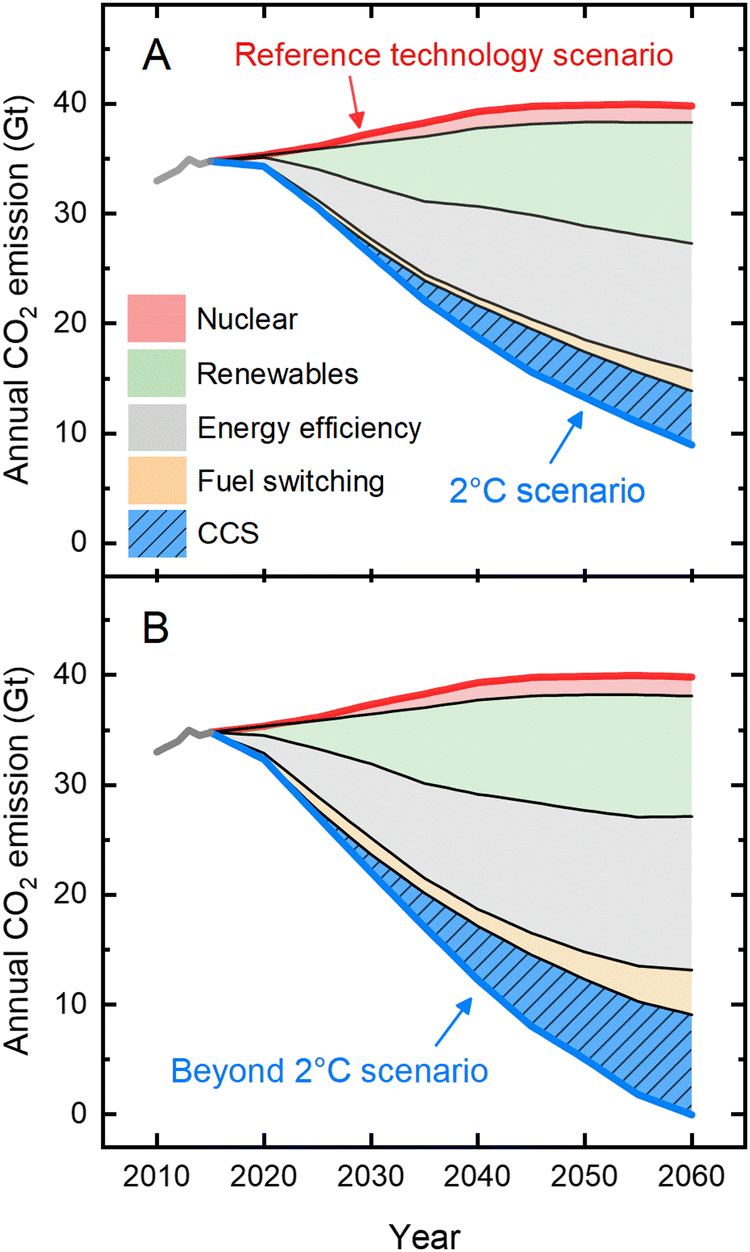 | ||
| Fig. 1 A portfolio of options to mitigate annual CO2 emissions. Detailed action plans were designed to arrive at the margin of a 2 °C global temperature rise and beyond. The portfolio was designed to further reduce the emissions from the “Reference technology scenario” which takes into account today's commitments by countries to limit emissions according to the Paris Agreement; if realized, it would still result in an average temperature increase of 2.7 °C. For the “2 °C scenario” (panel A), annual CO2 emissions are projected to be reduced by ∼75% (compared to that of the reference scenario), while for the “beyond 2 °C scenario” (panel B), net-zero emissions are expected to be needed by 2060. The figure highlights the importance of deployment of interim technologies such as carbon capture and storage (CCS) in the mitigation action plans. The data were adapted from Energy Technology Perspective5 by the International Energy Agency (IEA). | ||
In this context, there has been growing interest in developing electrochemical technologies that can replace conventional thermochemical CCS processes. Electrochemical technologies allow for direct and often more-efficient manipulation of CO2 in the bound (captured) state, achieving efficient separation of CO2 from the sorbent and obviating the need for inefficient steam heating. Electrochemical approaches also offer a wide-open design space to re-imagine how capture and separation processes may operate at a fundamental molecular level. The field has expanded significantly in recent years to include a multitude of technology concepts at varying stages of technology readiness. Given the urgency to identify solutions on a decadal and sub-decadal timescale, it is important to critically examine how technologies in the development pipeline stand to contribute to and potentially enhance the scenarios envisioned in Fig. 1.
The objective of this review is to provide a timely assessment of electrochemical CCS concepts along with, where appropriate at this stage, their current technology status and challenges for practical development. Although we refer to the approach as CCS, we largely emphasize the upstream separation step in the review, with the understanding that storage needs to be developed (though is outside the scope of the work). Two excellent recent reviews have contributed focused analyses pertaining mainly to one category of electrochemical CCS, pH-swing processes;7 as well as a cross-cutting thermodynamics assessment of electrochemical CO2 separations.8 In this work, in contrast, we comprehensively review the four broad categories of electrochemical CCS—(1) electrochemical generation of nucleophiles, (2) electrochemical modulation of proton concentration (also known as pH-swing processes), (3) electrochemical capacitive adsorption, and (4) electrochemically mediated amine regeneration—as well as some earlier-stage electrochemically based systems that are emerging (e.g., electrochemical mineralization by direct amine–CO2 reduction). We evaluate the current development status for each category and indicate suggestions for future research needs. We believe this approach offers clear context and indicates pathways for the community to further advance these emerging electrochemical CCS technologies.
Following capture, carbon must be sequestered and prevented from re-entering the environment. While geologic storage will be necessary for the permanent sequestration of the major proportion of captured CO2, there is also a strong interest in its potential utilization as a feedstock for chemical production, primarily by electrochemical routes (Fig. 2). Thus, for context and comparison, we begin by briefly describing potential roles and limitations of electron-competitive CO2-to-chemicals or CO2-to-fuels conversion (known as carbon capture and utilization; CCU), with a critical eye towards large-scale environmental impact. Given identified limitations, the remainder of the review discusses operating principles and current status of the growing set of technologies enabling coupling of electrochemistry into CO2 capture and storage processes. These processes take a CO2 mixture (e.g., CO2, N2, O2) as an input and have varying outputs, either as a separated stream of CO2 for subsequent storage or as a mineralized form (Fig. 2). Overall, electrochemical approaches to CO2 management are becoming more versatile and variable, which is broadening their potential for impact; meanwhile some have progressed beyond bench scale testing and stand ready to be implemented at pilot scales and beyond.
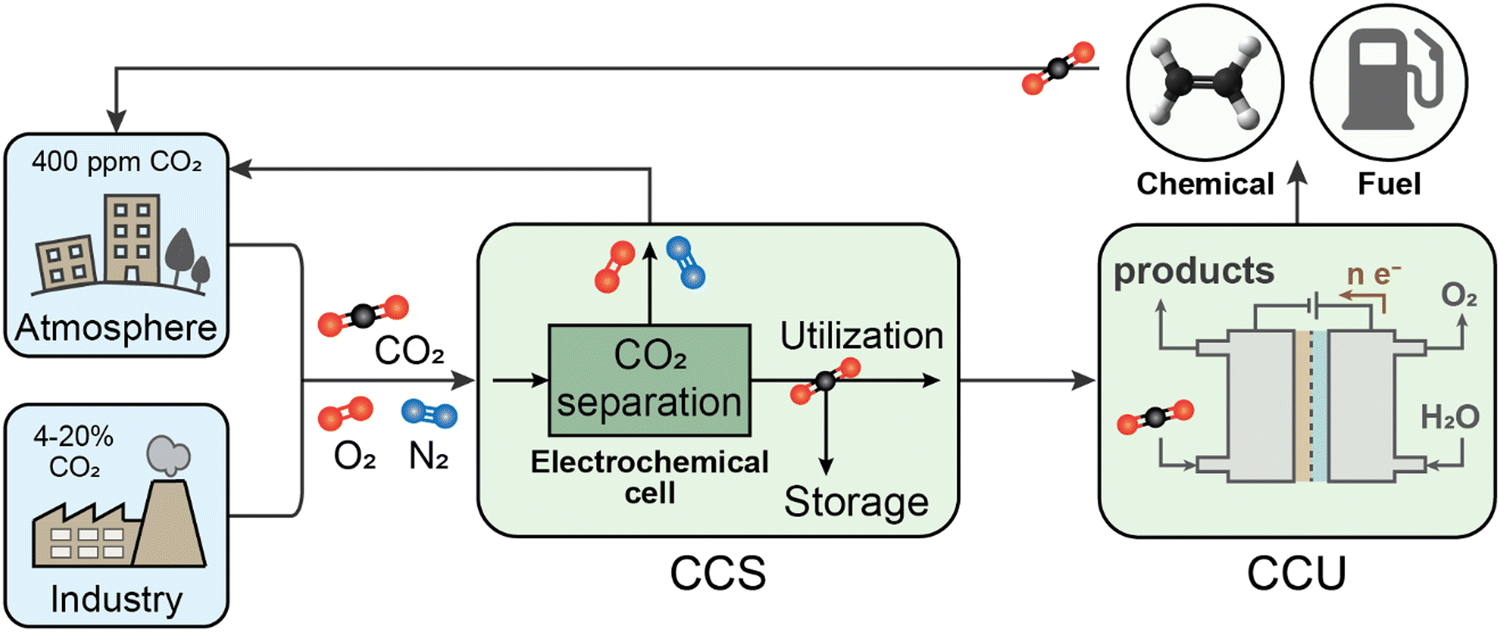 | ||
| Fig. 2 Roles of electrochemical interventions in facilitating CO2 separation, transformation, and storage. Electrochemical capture systems can replace current thermochemical separations of CO2 emitted from concentrated point-source emissions (e.g., industrial sectors such as coal- or gas-fired power plants, cement, steel, etc.) and distributed sources for direct-air capture. The captured CO2 can be permanently stored through an additional (separate or integrated) electrochemical process, or further electrochemically converted to value-added products such as chemicals and fuels. The former route is often known as carbon capture and storage (CCS), while the latter is referred to as carbon capture and utilization (CCU). The utilization scenario is not closed, emitting much of the CO2 back to the atmosphere upon consumption of the produced chemical or fuel. | ||
2. Electrochemical conversion of CO2 to chemicals or fuels – the “Utilization” scenario
Electrochemical technologies that directly manage CO2 as a mass flow input have conventionally focused on generating new products from CO2 with economic value. Electrochemical reduction of CO2 generally proceeds in an aqueous environment near neutral pH, which is maintained by a bicarbonate (HCO3−) buffer that results from hydration of CO2 dissolved in water. At the cathode, CO2 is electrochemically reduced (CO2 reduction reaction, or CO2RR) to form a range of possible C1, C2, and Cn (n > 2) products. The protons and electrons needed are supplied by water electrolysis at the anode side of the cell, which liberates O2(g) by the oxygen evolution reaction (OER), i.e., 2H2O → O2 + 4H+ + 4e−. Both the OER and CO2RR are kinetically sluggish and require additional applied electrode voltage (overpotential, an energy loss) to drive the reactions at reasonable rates; thus, catalysts are routinely needed. Excellent reviews9–11 have discussed materials and performance aspects of specific OER catalysts, which often rely on precious metals such as Pt, Ru, or Ir, to lower the overpotential to within several hundred millivolts, and are not further described here. At the cathode, catalysts are critical to achieve selectivity towards CO2 conversion over the competitive, kinetically-facile hydrogen evolution reaction (HER, 2H+ + 2e− → H2).9,12 Carbon monoxide (CO) and formate (HCOO−) can nowadays be made with attractive faradaic efficiencies (FE) exceeding 90% and low cathode overpotentials using appropriate catalysts such as Au or Ag (for CO)13,14 or Sn and In (for HCOO−).15,16 Formation of protonated formic acid remains significantly more challenging. The yields and faradaic efficiencies of further-reduced products are much lower than those of CO and HCOO− at similar cathode potentials; significantly higher overpotentials are required to achieve moderate faradaic yields of methanol (CH3OH), methane (CH4), ethylene (C2H4), and other products.17 At these high overpotentials, CO2 selectivity significantly decreases and a wide range of co-reduction products are generated, necessitating costly separations that are often not considered in detail. Putting together kinetic losses at both the CO2RR and OER sides of the cell, in addition to significant mass transport and ohmic losses due to membrane requirements, CO2 conversion systems for highly reduced products require energy inputs that are substantially higher than those needed to conduct water electrolysis alone.Consequently, the technologies that have progressed beyond the laboratory scale to prototype development have largely focused on the more-facile CO2-to-CO conversion reaction.18–20 CO is a versatile CO2 reduction product because it may be used either directly as a chemical feedstock, such as in Fischer–Tropsch synthesis of hydrocarbons, or can be further electrochemically reduced, e.g., to produce products such as methane, ethylene, and ethanol.21–23 As further electrochemical reduction will accrue additional purification steps, energy inputs and separations penalties from the baseline mentioned above, it is not discussed further here; the reader is referred to several excellent papers on the underlying electrochemistry and catalysis of these reactions.10,24,25 Because Fischer–Tropsch requires an H2 stream as input, some researchers have suggested capitalizing on co-production of CO and H2 at proper ratios, in which case the parasitic HER may be turned to advantage.26,27 In this scenario, potential drying and change in the properties of the electrolyte solutions must be carefully monitored.
Factors influencing potential for impact
Global emissions associated with current fossil production of CO and formic acid in the chemical industry are 3.6 and 1 Mt, respectively.20,28,29 These numbers, which represent an upper bound of decarbonization potential, are 3–4 orders of magnitude lower than the scale of overall industrial CO2 emissions (Fig. 1); thus even if deployed at scale, electrochemical processes for their production would have relatively minor environmental impact. Additionally, large amounts of electricity are required for electrosynthesis even if the reactions can proceed at thermodynamic limits, thus the current carbon intensity of the grid makes CO2 mitigation potential even lower. De Luna et al.,28 assuming current average European Union (EU) grid intensity of 0.295 kgCO2e kWh−1 and optimistic faradaic and energy conversion efficiencies, concluded that among CO, formic acid, ethanol and ethylene, only CO and formic acid could be electrosynthesized with marginally lower CO2 emissions (order of 1–2 Mt CO2 per year avoided each) than with today's fossil-based processes. Electrosynthesis of ethanol and ethylene under similarly optimistic conditions would, instead, increase emissions at current grid intensities by ∼3–4 times. Fig. 3 illustrates a schematic of the CO2-to-CO process and summarizes the Gibbs free energy of electrochemical reaction (ΔGr) to produce a certain product together with its current market size in the chemical industry and annual global CO2 emissions. An ideal product requires low energy to produce (low ΔGr) and offers high market size and mitigation potential. Unfortunately, no products meet all three metrics. Even more critically, the eventual utilization of CO2-derived products will result in them being emitted back to the environment on relatively short timescales (months-years).30–32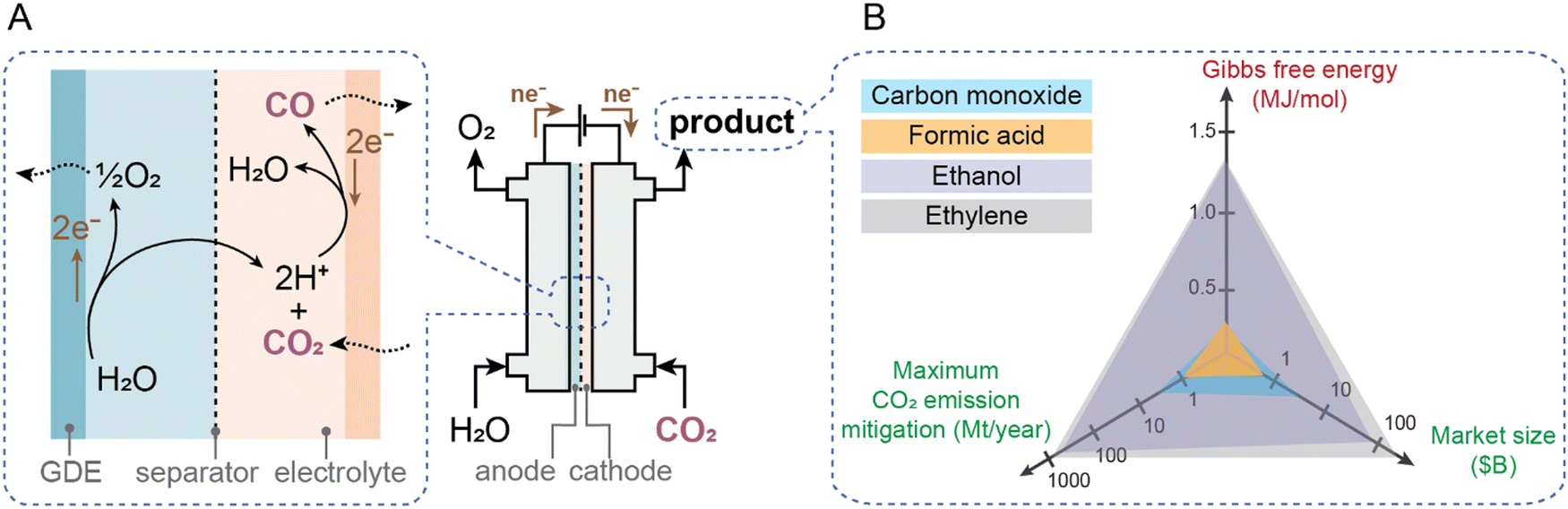 | ||
| Fig. 3 Electrochemical reduction of CO2 to produce value-added chemicals. (A) A schematic of the CO2-to-CO process at the cathode and oxygen evolution reaction at the anode in an aqueous environment. A two-compartment electrochemical cell with a separator and gas diffusion electrodes (GDE) is shown. (B) The Gibbs free energy to electrochemically produce a certain chemical together with its market size and annual CO2 emission mitigation potential28 is presented. | ||
An important consideration associated with global-scale electrosynthesis is the magnitude of electricity required. Katelhon et al.33 considered a broader portfolio of 20 large-volume chemicals that have potential to be electrosynthesized, including CO, ethylene and methanol, together accounting for approximately 75% of the chemical industries’ CO2 emissions. The authors concluded that CO2 utilization (also including electrolytic H2 production, a major feedstock which was not considered by De Luna et al.28) could achieve a higher mitigation value of 3.5 Gt CO2 per year of avoided emissions by 2030. However, achieving this target and ‘greening’ the chemical industry would require >18 PWh of low-carbon electricity, or 55% or more of the projected global electricity generation capacity in 2030 (Fig. 4) for chemicals alone. In these scenarios, the remaining renewable electricity must be partitioned among other uses including home and industrial use, charging of electric vehicles, transportation-scale H2 production, and so on. For comparison, we roughly estimate the electricity required for electrochemical CO2 capture and separation (Fig. 1), assuming that conventional thermal capture/regeneration processes are replaced with electricity-driven technologies. The calculations assume, for simplicity, the same capture energy penalty for these electrochemical processes as that of the conventional thermal process with a total energy requirement of 240 kJ molCO2−134,35 (this thermal work can be converted to electrical work, kJe, by assuming a Carnot efficiency of 25%; 60 kJe molCO2−1). Notably, the estimated electrical energy requirements for CO2 capture and separation are significantly lower than that for electrochemical CO2 conversion (e.g. 0.5–1 PWh in 2030; Fig. 4). This observation highlights the importance and attractiveness of the CCS processes from an energy, grid infrastructure, and environmental impact perspective. In particular, it emphasizes that integration of electrochemical capture processes with conventional fossil-based production methods of critical chemicals may be a compelling alternative to electrolysis-based production in the likely shorter-term scenarios where renewables production capacities cannot meet widespread decarbonization demands.
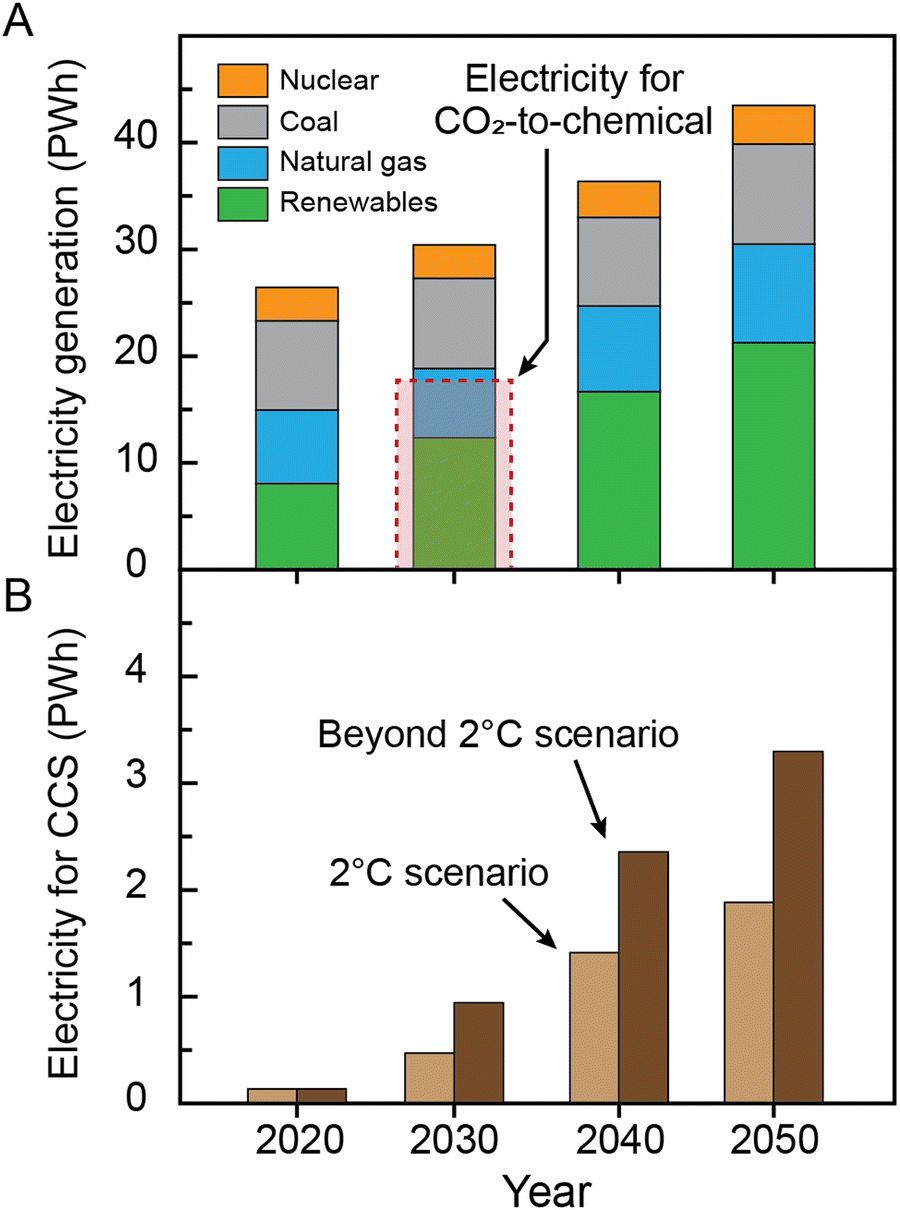 | ||
| Fig. 4 The electricity required for electrochemical reduction of CO2 to produce chemicals and CCS. (A) Comparison of the electricity required for electrochemical reduction of CO2 to produce chemicals and the projected global electricity generation. The electricity generation data were adapted from International Energy Outlook 202136 provided by the U.S. Energy Information Administration (EIA). (B) Projected electricity consumption needed to achieve the goal of mitigation through CCS, assuming full transition toward electricity-driven capture processes. In our calculations, we considered the same capture energy penalty for these electrochemical processes as that of the state-of-the-art thermal process. | ||
Overall, electrosynthesis should be considered as an economic incentive for industry to retrieve sunk costs from CO2 capture prior to eventual release to the atmosphere; and/or as a technological testbed to develop short-term CO2 demand while longer-term CCS technologies for permanent fixation are being developed. Both of these points have merit, but they are different from having climate impacts, which are assessed by us and others28,37 to be low. Therefore, additional strategies beyond CO2 conversion are essential if electrochemical technologies are to reach the levels of impact needed for the 2 °C scenario and beyond.
3. Electrochemistry for carbon capture and storage (CCS)
Carbon capture and storage (CCS) plays a key role in achieving the goals of the designated emission mitigation plan, with a reduction share of up to 5 Gt (2 °C scenario) to 9 Gt (beyond 2 °C scenario) of CO2 per year by 2060 (blue wedges in Fig. 1). Hundreds of CCS operations need to be constructed worldwide in the 2020s, increasing to thousands in 2030 and beyond.38,39 The development of scalable and cost-effective technologies to capture CO2 from either large-point sources or air is an immediate priority to achieve the mitigation goals of the action plan.CO2 separation from a mixed gas matrix is the most energy-intensive step of CCS, and much effort has gone into developing separation technologies with minimum energy penalty per unit of CO2 captured. Current CO2 capture technologies use thermal cycles where a nucleophilic agent absorbs CO2 from mixed gas streams (e.g., CO2 and N2 in flue gas) and pure CO2 is subsequently released on thermal regeneration of the nucleophilic agent.40–42 The most developed thermal-based capture system uses an amine absorbent such as monoethanolamine (MEA) which acts as a nucleophile.40,43 Despite their technological maturity, amine-based thermal scrubbing processes face several challenges that have hindered their deployment. The key concerns are the high regeneration energy penalty, degradation of amines at high temperature, corrosion, and high operational costs.41,44–46 Potassium carbonate (K2CO3) has been widely investigated as an absorbent to replace the amines in the thermal scrubbing process. It offers several advantages, including high capacity for CO2 absorption, low degradation rate, ease of regeneration, and low cost, toxicity, and corrosiveness.47–50 The major challenge of using K2CO3 as an absorbent is its low rate of reaction with CO2 which lowers the performance at the absorber stage.47,51
A fundamentally different approach employs electrochemical processes to drive the CO2 separation. Electrochemical-based CO2 separations have the advantage of being readily integrated as plug-and-play processes that do not require external sources of steam, high pressures, or vacuum to operate. Following, we describe four emerging electrochemical processes for CO2 capture, and discuss their working principles, potentials for emission mitigation, and challenges for future developments. Other earlier-stage electrochemically based systems such as electrochemical mineralization by direct amine–CO2 reduction are also discussed in detail.
Electrochemical generation of nucleophiles
By electrochemically reducing organic redox chemicals to produce nucleophiles that bind to the electrophilic carbon center in CO2, carbon dioxide can be selectively separated from other gases. The absorbent can then be electrochemically oxidized to regenerate the organic absorbent and release pure CO2.40,52–56 Quinone-based nucleophiles have been widely studied as the redox-active organic component that can effectively capture-release CO2 at different reduction potentials. Early works on employing quinone chemistry for CO2 capture mostly focused on understanding the mechanism involved during the electrochemical capture-release stages, investigating effective redox candidates, and optimizing the electrolyte media. Several quinone compounds including phenanthrenequinone,53 benzoquinone,57 and naphthoquinone58 dissolved in ionic liquid-based electrolytes were investigated. As an example, the process is illustrated for a naphthoquinone in Fig. 5(A). However, these early-stage systems required that the quinone be actively transported between two electrodes to switch between CO2-loaded and CO2-lean states, limiting their implementation in a number of applications where pumping and a large footprint are problematic. To address this, recently, an electrochemical device was demonstrated in which an anthraquinone (namely poly(1,4-anthraquinone)) was polymerized and immobilized onto a carbon mesh as the working electrode, with a polyvinylferrocene composite used as the counter electrode, and an ionic liquid employed as the electrolyte (Fig. 5(B)). The electrochemical cell demonstrated high capture and release of CO2 over 7000 cycles at 60–70% quinone utilization and 90% faradaic efficiency (i.e., electrons utilized to release CO2) with energy requirements of 40–90 kJe mol−1.59 The new recent results reported an exceptional retention of 80% quinone utilization after 215![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.60 In a different manifestation of using stabilized anthraquinone as a redox active molecule, the ionic liquid electrolyte was replaced by highly concentrated salts (lithium bis(trifluoromethanesulfonyl)imide; LiTFSI). The energy requirement to separate CO2 from a simulated flue gas (with 15% CO2) was experimentally demonstrated to be ∼56 kJe mol−1.61
000 cycles.60 In a different manifestation of using stabilized anthraquinone as a redox active molecule, the ionic liquid electrolyte was replaced by highly concentrated salts (lithium bis(trifluoromethanesulfonyl)imide; LiTFSI). The energy requirement to separate CO2 from a simulated flue gas (with 15% CO2) was experimentally demonstrated to be ∼56 kJe mol−1.61
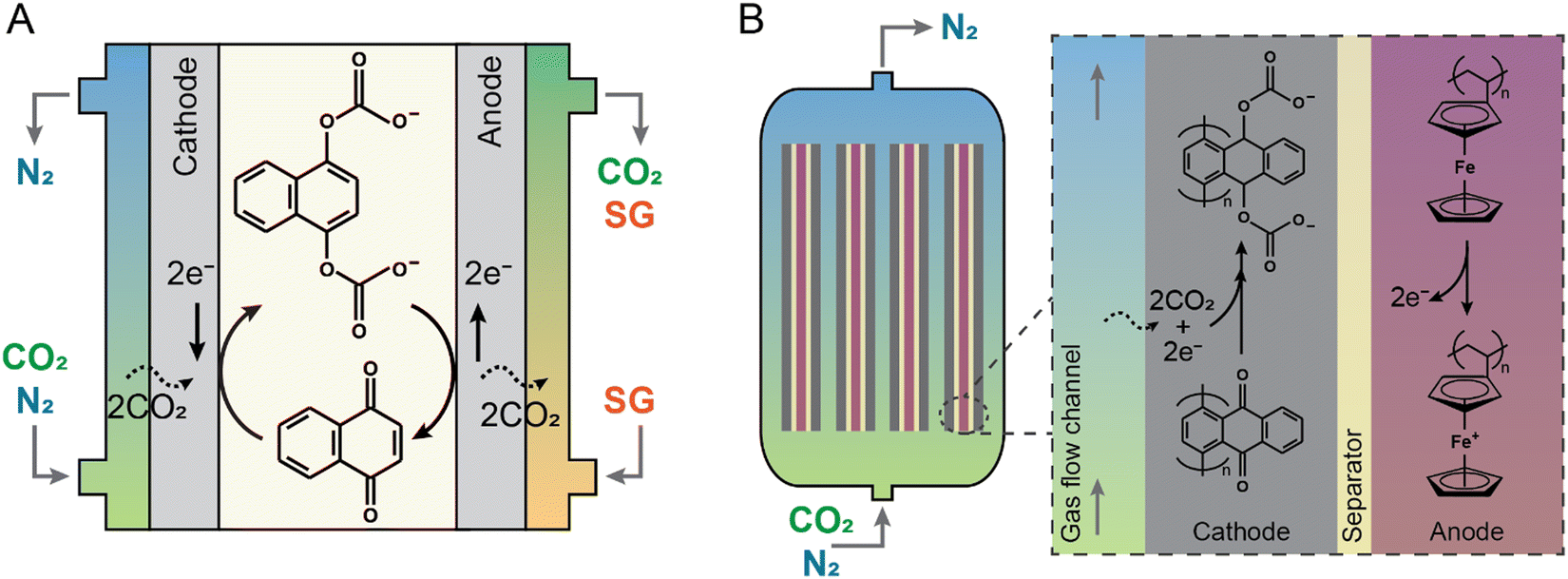 | ||
| Fig. 5 Schematic of two electrochemical generation of nucleophile processes for carbon capture. CO2 is separated from a mixed gas stream by (A) a static cell with 1,4-naphthoquinone and (B) a flow cell with poly(1,4-anthraquinone) as the redox active molecule. In the static cell, the gas mixture is introduced through a gas-breathing cathode, while the desorbed CO2 is removed by a sweep gas (SG) through a gas-breathing anode. In the flow cell, the gas mixture is introduced at the bottom of the electrochemical device and the treated gas is removed from the top. Each individual electrochemical cell consists of a cathode coated with poly-1,4-anthraquinone composite and a polyvinylferrocene-containing anode. Upon saturation of the cathode with CO2, the polarity is reversed to release CO2 from the cell. | ||
Recently, a liquid quinone sorbent, as a redox-active molecule that is liquid at room temperature, was employed to develop a flowing, electrochemically mediated carbon capture process. A good electrochemical stability and continuous capture and release of CO2 was achieved in a full bench scale process. Promising initial energetics between 50 and 200 kJ mol−1 CO2 are found.62
There still are opportunities and challenges associated with electrochemical capture-release of CO2 by redox active quinone compounds. While the early-stage systems were developed for proof-of-concept at laboratory scales, the recent immobilized, quinone-based systems were scaled up beyond that – for example, a stacked parallel passage electrochemical cell contactor was developed to enable continuous capture-release of CO2.59 Both the early-stage and recent quinone-based approaches were designed to effectively operate at a wide range of CO2 concentrations, enabling these technologies to be implemented for capture from large point sources (e.g., industrial flue gas) as well as potentially from air through a process known as direct air capture (DAC). When applying these technologies for CO2 separation from a gas mixture containing significant amounts of O2, such as air, the long-term stability of the quinones must be carefully investigated because O2 can oxidize some reduced quinones to form superoxide radical anions. This can be minimized by carefully tuning the electron density of the quinone to make it more selective towards CO2.57,63
Although quinone-based nucleophiles have been the most-studied, other organic redox compounds such as bipyridine64,65 and thiolate66 have also been investigated for electrochemical capture and release of CO2. The separation mechanisms are similar to that of the quinone because these compounds selectively capture CO2 in their reduced states and release the CO2 upon oxidation. Promising experimental and computational results with fast kinetics for both electron transfer and CO2 capture were observed for a bipyridine (e.g., 4,4′-bipyridine). The radical anion is created by a one-electron reduction of 4,4′-bipyridine, and it quickly forms a covalent bond with carbon dioxide to create an adduct. This adduct undergoes a one-electron oxidation, which liberates bipyridine and carbon dioxide.64 As with the quinones, in its reduced state this compound reacts rapidly with O2, rendering it unavailable for reaction with CO2 and destabilizing the compound to lower the efficiency of the process.40 Thiolates, such as benzylthiolate, were also investigated as nucleophiles that could potentially capture CO2;66 while it is capable of rapid CO2 capture, this process suffered from unwanted side reactions and irreversibility.40
Electrochemical modulation of proton concentration
CO2 can also be captured and released by electrochemical modulation of the absorbent proton concentration as the primary driver. These processes, normally referred to as electrochemical pH swing, rely on the reduction and oxidation of a proton-coupled electrochemical reaction (e.g., X + ne− + nH+ → XHn) to modulate the pH of the absorbent. These methods make use of the CO2 thermodynamic equilibrium speciation's sensitivity to pH in aqueous solutions. While the release of CO2 happens at the anode, where acidic conditions are formed, regenerating free CO2, an increase in pH at the cathode of an electrochemical cell furthers the capture of CO2 as either HCO3− or CO32−.40,54,67 In some manifestations, the metal carbonates that result from the mineralization of the carbonate ions with the presence of metal cations in solution, such as Ca2+, Sr2+, or Mn2+, precipitate out of solution.68,69 Such processes potentially offer a simple and scalable route to electrochemical CO2 capture-release or capture-mineralization at atmospheric pressure.Three different processes, membrane electrodialysis (MED), redox-mediated pH swing, and recently, an electrochemically-driven proton concentration process (PCP), have been developed to take use of the CO2 hydration's pH responsiveness for CO2 separation (Fig. 6). In most MEDs, a voltage is supplied across an alternating stack of anion-exchange membranes and water-dissociating bipolar membranes to trigger CO2 capture by hydroxide generation and release through proton generation (Fig. 6(A)). The MED system has been developed to separate CO2 potentially from both large point sources (namely power plant flue gas with 15% CO2)70 and air, through DAC71,72 with results indicating that the process could be successfully implemented at scale. Although a variety of different configurations of the membrane type and electrolyte were investigated,70–76 the cost of CO2 separation using MEDs was likely to be high because of the use of several bipolar and anion-exchange membranes. Additionally, as described previously, water splitting, which is the core electrochemical reaction driving MED, is energetically demanding, and results in a high energy penalty for CO2 capture and release.
 | ||
| Fig. 6 Schematics of three different electrochemical modulation of proton concentration processes developed for carbon capture. CO2 is separated from a mixed gas stream using (A) membrane electrodialysis (MED), (B) redox-mediated pH swing using the proton-coupled redox reaction of 2,6-dimethylbenzoquinone, and (C) an electrochemically-driven proton concentration process (PCP) with H+ intercalating electrodes. In MED, a voltage is supplied across an alternating stack of anion-exchange membranes (AEM) and water-dissociating bipolar membranes (BPM) to trigger CO2 capture by hydroxide (OH−) generation and release through proton (H+) generation. The electrolytes are circulated between the appropriate section of the cell and corresponding acid or base tanks. In the pH swing process, the gas mixture is introduced through a gas-breathing cathode, while the desorbed CO2 is removed by a sweep gas (SG) through a gas-breathing anode. In PCP, an AEM is used to separate the cell compartments, and CO2 is captured in an absorption column prior to release in the electrochemical cell. | ||
A redox-mediated pH swing process using quinone compounds has also been proposed for both CO2 capture from industrial flue gas and for DAC. In this approach, the reversible coupling of the quinone molecule with H+ upon reduction or oxidation is exploited to modulate the solution pH, rather than relying on reduction of the molecule itself to generate a nucleophilic site to capture CO2 (as discussed earlier). In an early-stage investigation, hydroquinone (HQ) and 2,6-dimethylbenzoquinone (DMBQ) were considered and the system was formulated around capturing CO2 from flue gas. In this approach, CO2 was captured in the form of HCO3− at a gas-breathing cathode, where reduction of quinone consumed H+, resulting in increased local pH. Subsequently, electromigration facilitated diffusional HCO3− transfer across the cell to the anode, where quinone oxidation released H+ to decrease the local pH. This pH decrease resulted in the dissociation of HCO3− to drive the release of CO2 which desorbed through the gas-breathing anode77 (Fig. 6(B)). The primary limitations of this approach are the complexity of the system due to the use of catalysts such as platinum, palladium, and ruthenium metals to improve the kinetics of the quinone redox reaction, and the low solubility of quinone as the active material. The quinone solubility is particularly important as it directly dictates the pH swing ability (and consequently CO2 capture capacity of the system).
In another manifestation, tiron, which has a higher solubility in aqueous solutions than either HQ or DMBQ, was investigated as an electrochemically active mediator to generate a pH gradient.78 A successful experimental separation of CO2 from a simulated flue gas in a batch-type cell was demonstrated; however, the developed system suffered from a reversibility issue because the alkalinity could not be recovered in subsequent cycles of operation. Recently, a flow configuration, similar to those employed for flow batteries, with phenazine-based organic mediators was developed to capture CO2 from flue gas. Phenazine derivatives such as 7,8-dihydroxyphenazine-2-sulfonic acid (DHPS) exhibited high solubilities and fast kinetics, significantly lowering the energy requirement for CO2 capture-release compared to that of tiron.79 The major concern in using DHPS as the active compound is its high sensitivity towards O2 which limits its application in CO2 capture from actual industrial flue gases (normally contain 3–7% O2) or DAC. An attempt was recently made to formulate an electrochemical process with a quinone-based proton-coupled reaction to capture and release CO2 from flue gas and from air. The results showed low theoretical and experimental energy penalties for both capture scenarios, ranging from 16 to 75 kJe mol−1.80
Recently, 1-aminopyridinium (1-AP) nitrate as a redox-active amine absorbent in an aqueous solution was employed for electrochemical capture and release of CO2 through proton modulation (i.e., capture as bicarbonate ions).81 Reversible electrochemical redox-active amine cycles were demonstrated, obtaining CO2 capture and release with electron utilization (i.e., mole of CO2 per mole of electrons) of up to 1.25 over a wide range of CO2 concentrations and, in particular, from ambient air. The developed redox-active amine showed high stabilities towards oxygen when implemented for DAC, with energy requirement as little as 162 kJe per mole of CO2,81 which is comparable to the target energy penalty of DAC of 100 kJe mol−1.82 This is a promising new redox chemistry for DAC applications, which is in early-stage of development.
A different type of CO2 capture system based on electrochemical modulation of proton concentration was recently introduced which relies on H+ exchange (i.e., intercalation/deintercalation) between a solid electrode and the electrolyte.83 This cell configuration contrasts with the previous scenario where the active molecule (e.g., quinone) is the proton carrier. Manganese oxide (MnO2) electrodes exhibit efficient proton intercalation/deintercalation behavior through a proton-coupled reaction: MnO2(s) + H+(aq) + e− ⇌ MnOOH(s). The process scheme, dubbed ‘proton concentration process’ (PCP), consists of a two-compartment symmetrical electrochemical cell with MnO2 electrodes and an absorber similar to those found in thermal scrubbing systems. The absorbent is a potassium carbonate (K2CO) solution in which CO2 is absorbed as bicarbonate (HCO3−) and carbonate (CO32−). After the absorber captures the CO2, the stream with a high CO2 loading is transferred to the anode compartment of the electrochemical cell, where deintercalation of protons from a MnO2 anode raises the proton concentration and shifts the equilibrium of CO2(aq)/HCO3−(aq)/CO32−(aq) toward CO2 production. In a flash tank that is positioned after the anode compartment, the desorbed CO2 is separated from the solution. The stream is then transferred to the cathode compartment to regenerate the absorbent, where intercalation causes the proton concentration to decrease. The regenerated solution is returned to the absorber column for further absorption (Fig. 6(C)). Based on insights from a process model to evaluate the energy penalty in the captured CO2 from flue gas streams (∼33 kJe mol−1),83 a bench-scale experimental setup was constructed in which a continuous CO2 desorption was achieved through reversible cycles.84 The system does not suffer from solubility constraints on the active component due to the nature of the proton-coupled reaction in PCP which relies on a solid electrode interaction (as opposed to a dissolved molecule). Rather, the CO2 capture-release is dictated by the capacity of the material to host proton ions. Future investigations on PCP should include development of electrodes with higher proton capacities.
Electrochemical capacitive adsorption
Electrochemical capacitive systems such as supercapacitors are widely used for electrical/electrochemical energy storage purposes. In a supercapacitor, charge is stored in an electrical double layer formed at the electrode–electrolyte interface in a non-faradaic manner—there is no redox-process associated with the capacitive charge storage.85 Supercapacitor concepts were recently adapted to design processes in which CO2 adsorption and desorption are accomplished by a reversible charge and discharge of capacitive electrodes. The initial work was done in a batch-type configuration with porous carbon electrodes and aqueous NaCl solution to separate CO2 from a simulated industrial flue gas, and was referred to as ‘supercapacitive swing adsorption’ (SSA; Fig. 7(A)).86 Despite selective and reversible cycles, the process energy requirement was high (∼100 kJe mol−1), and the kinetics of CO2 capture-release were slow. In addition, the cell prevented separation of gases from a continuous gas stream mixture, and the composition of the gas barely changed. To address these issues, a continuous flow system inspired by a coin-type supercapacitor was designed. It had thin electrode sheets for reduced electrical resistance and a gas diffusion layer that enabled gas flow through the module and increased the contact area between the gas and the electrode.87 SSA was also formulated to define metrics that allow for a quantitative evaluation of the process, including energy consumption to drive CO2 capture-release. Based on these metrics, the galvanostatic method (i.e., constant current) was identified to be the most favorable charge–discharge protocol for the SSA process as it consumed the least energy and time, and is the most energy efficient. At a charging current of 1 mA, the total energy requirement of the device was 57 kJ mol−1 to concentrate CO2 from 15% in the feed gas to 46% in the effluent gas.88 The charging protocol along with the new mechanism based on movement of CO2-derived species into and out of electrode micropores were investigated in details in a recent investigation.89 In the future developments, the effluent concentration needs to be further increased (close to 100%) to be able to generate a CO2 stream that is suitable for subsequent storage or utilization processes. | ||
| Fig. 7 Schematics of two electrochemical capacitive adsorption systems for carbon capture. (A) Supercapacitive swing adsorption (SSA) and (B) membrane capacitive deionization (MCDI). In a SSA, charge is stored in an electrical double layer formed at the electrode–electrolyte interface resulting in CO2 removal from the gas phase headspace. In MCDI, one electrode is covered by an anion exchange membrane (AEM), while the other by a cation exchange membrane (CEM), and CO2 is captured in an absorption column using water prior to release in the electrochemical cell. | ||
The SSA process was adapted in the design and construction of a hybrid system that could simultaneously capture and mineralize CO2 (a method to sequester CO2) in one cell. A dual-material anode made from a porous carbon (e.g., graphite) acting as supercapacitor and a sacrificial metal (e.g., aluminum) was used to mineralize the captured CO2.90 The gas was captured as HCO3− within the diffuse part of the electrical double layer and reacted with metal cations generated by oxidation of aluminum as a sacrificial metal to form mineralized carbon dioxide (e.g., aluminum hydroxycarbonate mineral). Water electrolysis occurred at the cathode, generating H2 and OH−, the latter of which facilitated CO2 absorption. The process was demonstrated experimentally to capture and mineralize CO2 from a dilute gas stream (5% CO2) while producing H2 as a valuable by-product. The cell configuration, especially the ratio of carbon to metal in the dual-material anode, was also optimized to maximize the efficiency and capacity in the capture and sequestration of CO2. The energy requirement for the optimized capture-mineralization processes was 230 kJemol−1.91 This estimation does not consider the energy cost for preparation of the metal electrode used in the process. In light of the current annual production rate for aluminum and steel and their recycling rate, it was predicted that the developed technology could annually capture and sequester 20–45 million tons of CO2 using aluminum or ∼800 million tons using steel.90
In another manifestation of CO2 capture by capacitive adsorption, a membrane capacitive deionization (MCDI) process was developed, which relies on the adsorption of ionic CO2 molecules (i.e., HCO3− and CO32−) within the electrical double layer established at a carbon-based anode surface. To satisfy charge neutrality, protons are simultaneously adsorbed on the cathode. The adsorbed ions are removed upon short-circuiting of the cell or switching of the polarity of the electrodes, leading to CO2 desorption from the solution to the gas phase.92 In a typical MCDI, one electrode is covered by an anion exchange membrane, while the other by a cation exchange membrane93 (Fig. 7(B)). CO2 was successfully captured from simulated flue gas through energy-efficient cycles ranged between 60% and 80% in an experimental demonstration of the process with total energy requirements of 40–50 kJe mol−1. The gas stream was sparged into deionized water as the absorbent, and was then pumped through the MCDI cell for capacitive adsorption of HCO3− and CO32− ions.92 Since deionized water was used as the absorbent, the system operated at a low CO2 cyclic capacity.
Compared to SSA, the developed MCDI offers higher adsorption efficiencies, mainly due to the use of ion exchange membranes. A theoretical investigation along with experimental validation demonstrated the necessity for the use of ion exchange membranes to ensure that only HCO3− and CO32− ions are delivered to the anode during the adsorption process, and to hinder the delivery of repulsed co-ions (i.e., H+) to the solution from the anode compartment.94,95 This controlled transport of ions resulted in high adsorption efficiencies and low energy consumption. However, the cost of CO2 separation with a MCDI cell is likely to be high because of the use of ion exchange membranes, which was shown in other systems to significantly impact the capital cost – in some cases, it accounted for more than 50% of the capital cost.96,97 In addition, the proposed MCDI configuration suffers from high ohmic resistances, mainly due to the low conductivity of deionized water used as the absorbent. Although MCDI is still in the early stages of development, such systems may present a chance to take advantage of knowledge in the area of capacitive deionization for carbon capture.
Electrochemical capacitive adsorption processes offer unique opportunities for CO2 separation. They can be charged within seconds or minutes, providing potential time advantages for such processes.98,99 In addition, these processes normally use inexpensive, available, and environmentally-friendly materials such as porous carbons and aqueous NaCl solutions. The biggest concern associated with the current capacitive-based technologies is the low capacity of the electrodes that eventually dictates the cyclic capacity for CO2 capture-release. The current carbon-based electrodes used in both SSA and MCDI exhibited capacities of ∼0.01–0.02 molCO2 per melectrode2. To put this into context, the capacity of anthraquinone immobilized onto a carbon mesh electrode discussed earlier is ∼0.1 molCO2 per melectrode2.59 For MCDI, the system also suffers from low absorption kinetics associated with slow CO2–water equilibrium. Future development of capacitive adsorption processes for CO2 capture should include optimization of the cell configuration and absorbent chemistry, as well as fabrication of electrodes with higher capacities.
Electrochemically mediated amine regeneration
Electrochemically mediated amine regeneration (EMAR) has been developed over the past decade as an efficient, low-energy, and potentially scalable method to capture CO2 from point sources such as flue gas.96,100,101 The EMAR process scheme is similar to that of the amine-based thermal approach, but a two-compartment electrochemical cell that can function at the same temperature as the absorber replaces the high-temperature scrubbing stage. The method depends on the competitive binding of CO2 and a suitable metallic ion, such as Cu2+, to an amine molecule, such as ethylenediamine (EDA), which serves as the absorbent. The amine in the absorption column absorbs CO2 in a manner similar to the conventional thermal method. However, this CO2-rich amine stream from the absorber is supplied to the anode compartment of the electrochemical cell, where Cu2+ ions are produced electrochemically by the oxidation at a Cu plate anode (i.e., Cu0 → Cu2+ + 2e−) to trigger amine–CO2 dissociation such that CO2 is formed (i.e., Cu2+ + 2 EDA-CO2 → Cu(EDA)22+ + 2CO2). This two-phase gas–liquid stream enters a flash tank that allows for a complete gas separation. The electrochemical plating of copper from the copper–amine complex onto the cathode then regenerates the CO2 lean (Cu-rich) stream (i.e., Cu(EDA)22+ + 2e− → Cu0 + 2EDA). The absorption column receives the regenerated amines to continue CO2 capture (Fig. 8).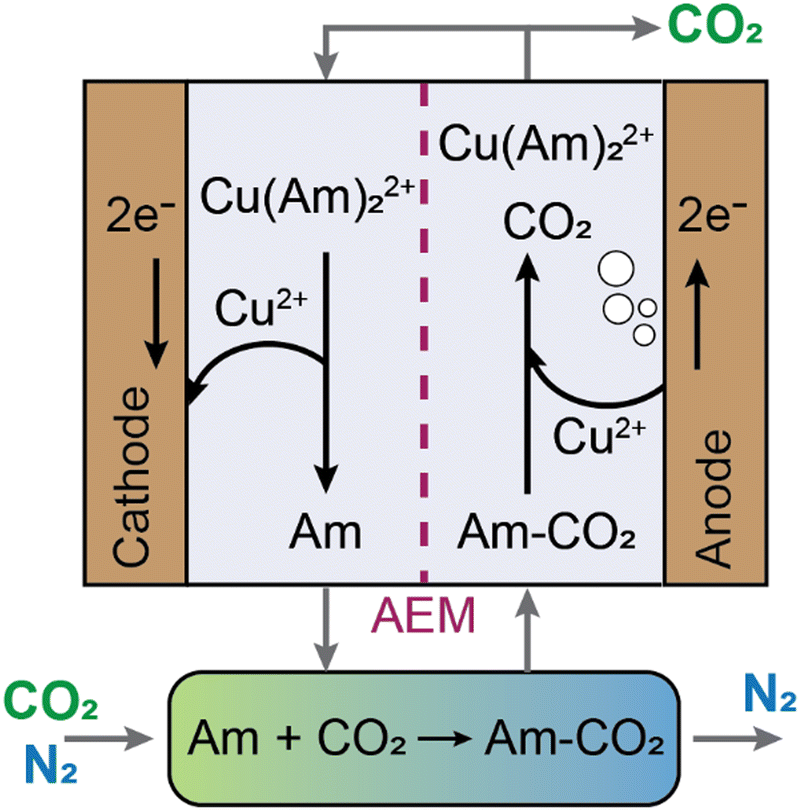 | ||
| Fig. 8 Schematics of an electrochemically mediated amine regeneration developed for carbon capture. An electrochemically mediated amine regeneration (EMAR) with copper electrodes and an anion exchange membrane (AEM) is demonstrated to separate CO2 from a gas mixture. In EMAR, CO2 is captured in an absorption column using an amine (noted as “Am”) prior to release in the electrochemical cell. | ||
A systematic analysis of several transition metals was performed along with monoamines, diamines, and polyamines to determine the best chemistry for the EMAR process.102,103 A combination of Cu as the metal and EDA as the amine was found to be an effective chemistry, owing to the high CO2 capacity (i.e., ∼0.5 molCO2 per molEDA), high stability constant of the copper complexation with EDA, suitable reduction potential of the complex, good reversibility, and low cost.100,102 Based on this chemistry and using the EMAR technique, continuous CO2 capture from flue gas was accomplished for 50 hours of operation, spanning 25 sucessive cycles of absorption and desorption. Additionally, it was demonstrated experimentally that the Cu-EDA combination can effectively transfer electrical energy to CO2 desorption with an electron utilization (defined as moles of CO2 desorbed per one mole of electrons transferred) of ∼0.8.104
To enable the EMAR process to be scaled up further, factors concerning the long-term operation of the process must be carefully considered. The high vapor pressure of EDA is one of the factors that could potentially complicate running the process long-term on a large scale. A mixture of EDA and aminoethylethanolamine (AEEA), which exhibited a significantly lower vapor pressure, was investigated to address this concern. The electrochemical performance of the mixed amines was evaluated, and it was shown that an equimolar mixture of EDA and AEEA can provide more energetically efficient operations relative to that with the single amine, EDA, while still maintaining comparable electron utilization. A continuous EMAR process to separate CO2 from a flue gas was run for over 100 hours (50 cycles of 2 hours each) using this mixture and achieved a steady CO2 gas output of ∼6 mL min−1. The desorbed gas was analyzed and found to be 100% CO2, confirming no evaporation of the amine during long-term operation.105
The EMAR approach offers several advantages over other thermally driven amine-based processes for capturing CO2. EMAR can be carried out at low temperatures (∼50 °C), in contrast to thermal procedures, which reduces the rate of thermal amine degradation—one of the main challenges in the thermal scrubbing processes.43,106,107 Additionally, it provides essentially no additional energy penalty for CO2 desorption at moderate to high pressures (1–10 bar),108 minimizing the downstream compression costs of CO2 storage.
A possible limitation of the EMAR process is, however, that the amines deployed must be able to form soluble complexes with metal ions (e.g., Cu2+) to displace and release the CO2. The number of amines possessing this property is limited compared to those that can be used for the traditional thermal processes. For example, MEA, which is the benchmark amine in the thermal scrubbing, cannot be easily used in an EMAR cycle as MEA does not complex copper; rather, Cu(OH)2 precipitates out of solution. Other absorbents including ammonia109 and imidazole110 were recently investigated in an EMAR-like process and the results were comparable to that of a system operating with EDA.
Cu electrode stability can potentially be a concern because electrochemical plating and stripping of metals may not be 100% efficient; the electrode will gradually lose Cu to precipitation with prolonged cycling, and eventually requires replacement.96 In an electrochemical cell similar to EMAR with Cu electrodes and EDA, it was found that the system suffers from high rate of stripping compared to that of the plating, resulting in a complete corrosion of the electrode after a few cycles.111 The unbalanced rates of plating and stripping in the EMAR cell are probably not that severe, as the cell successfully operated over 50 cycles (overall 100 hours) without performance decay or visual observation of any electrode instability.105 Ionic surfactants such as sodium dodecyl sulfate (SDS) were successfully implemented in the EMAR electrolyte to further improve the electrode stability.112 Nevertheless, a comprehensive detailed study on the electrode stability is necessary for future developments of the EMAR process.
Electrochemical mineralization by direct amine–CO2 reduction
It has recently been shown to be possible to transfer electrons to amine–CO2 complexes in solution and affect N–C bond cleavage,113–115 providing an alternative to thermal regeneration or Cu2+-driven separation. The initial proof-of-concept of electrochemical N–C bond cleavage was demonstrated in a non-aqueous environment within an electrochemical cell utilizing a Li metal anode (to provide an exemplar source of metal ions), carbon cathode and amine-containing electrolyte (Fig. 9). The cell was discharged under near-ambient temperatures and pressures (T = 25 °C and PCO2 = 1.4 bars). CO2 was first captured in a dimethyl sulfoxide (DMSO)-based electrolyte containing an amine-sorbent (2-ethoxyethylamine, EEA) and a Li+ salt (LiClO4). CO2 uptake by the amine (RNH2) resulted in the formation of a mixture of carbamic acid (RNH2 + CO2 → RNHCOOH) and Li carbamate/ammonium ions (2RNH2 + CO2 + Li+ → RNHCOO−Li+ + RNH3+), with Li carbamate determined to be the electroactive species. Compared to the inactive lean amine or physiosorbed CO2, Li carbamate discharge proceeded at high voltages (∼2.9 V vs. Li/Li+) indicating a dual role of the amine as both CO2 sorbent and activating agent for further electrochemistry. Solid inorganic carbonate (Li2CO3) and carbon were obtained as discharge products at high charge capacities (>1000 mA h gcarbon−1), providing evidence for electron-driven COO-detachment, and can be contrasted with previously-described processes yielding CO2(g) as an output. Isotopic labeling indicated that carbonate was derived from the CO2 rather than solvent or carbon cathode; no amine-derived products were observed in the cathodes, which confirmed that the reduction reaction entailed selective N–C bond cleavage. Importantly, evidence for amine regeneration under electrochemical reducing conditions was found via1H NMR; a turnover number of ∼10 was observed in initial measurements, limited by formation of passivating carbonate deposits on the electrode.113 Long-term studies are needed to investigate turnover limits.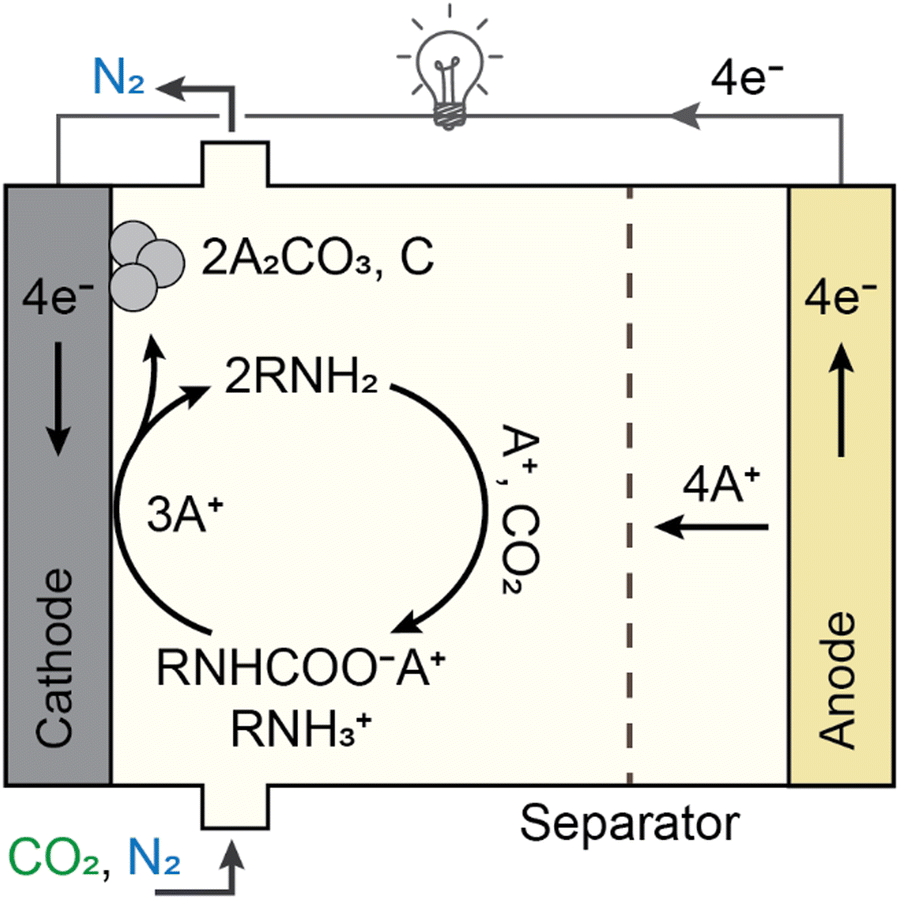 | ||
| Fig. 9 Schematic representation of an integrated electrochemical mineralization by direct amine–CO2 reduction. CO2 is captured in a non-aqueous, amine-containing electrolyte. Upon electrochemical reduction at a cathode, the amine–CO2 species undergo N–C bond cleavage and form CO2-derived solid phases (carbonate and carbon). In one type of cell realizing this cathode reaction, the anode is an alkali/alkaline earth metal cation source and the cathode is carbon. The figure shows a model process for an alkali cation (noted as “A+”). | ||
Subsequent follow-up studies exploring the translation of this initial model process to other electrolyte systems revealed new amine–solvent–salt combinations which may be promising for developing improved processes with higher electrochemical reaction rates.114 Altering the alkali salt cation, for example, had implications for both the carbamic acid/carbamate equilibria and the subsequent alkali carbamate reduction kinetics. Smaller cations – or harder Lewis acids – resulted in increased equilibrium populations of alkali carbamate (Li+ > Na+ > K+), ranging from ∼50% for Li+ to ∼11% for K+.114 Upon electrochemical polarization, however, the kinetics were largely determined by alkali cation desolvation energetics in nonaqueous solvents, which favored larger alkali cations (e.g.: altering the cation from Li+ to K+ resulted in a nearly two-fold increase in the electrochemical reaction rate). Furthermore, apart from EEA, additional amine structures such as diisopropylamine (DIPA) (in glyme-based electrolytes) were also found to be active for integrated capture-mineralization, and facilitated amine–CO2 conversion over a wider temperature range (up to 70 °C) reflective of practical capture conditions.112 A recent study reported that direct amine–CO2 reduction can also proceed in aqueous electrolytes with proper selection of electrolyte salt, catalyst, and cell conditions (temperature and flow rates), yielding CO with up to 70% FE using a Ag catalyst, providing a first indication that amine-facilitated CO2 conversion is viable in aqueous media as well.116
While these early studies have demonstrated a first step of scientific feasibility, amine-mediated CO2 electrochemistry is still in early stages of development. From a fundamental point of view, the rates of such reactions need to be significantly accelerated. Currently, the Li+-based process has the potential to sequester an estimated 2 tons CO2 per year per mstored3 (assuming complete utilization of active surface area); with K+, this mineralization rate could be further increased by nearly two-fold. 2 While these rates are significantly higher than the chemical carbonation rates attained either naturally (∼1 gram CO2 per year m−3 for olivine) and are comparable to in situ carbonation at elevated temperatures and pressures (∼1 ton CO2 per year m−3 at T = 185 °C and PCO2 = 150 bar), further improvements in current cathode architectures will be necessary to maximize active surface area utilization and realize attainable gains in mineralization rates. In terms of assessing practicality, an essential next step is to examine the potential to replace the scarce and CO2-intensive Li metal anode with more earth-abundant metal anode materials (e.g.: Na, K, Ca, Mg) or their mineral sources (e.g. silicates or oxides) as a source of cations in the mineralization reaction. Along these lines, a recent life-cycle analysis (LCA) of several proposed electrochemical mineralization processes employing amines found that Na is a leading contender for prospective sacrificial mineralization reactions given the low-CO2-intensity nature of its production.117 Estimated cradle-to-gate CO2 emissions reductions from a power plant using the Na-based system were found to be 30–70% compared to baseline business-as-usual operation of a power plant without CO2 mitigation, and assuming that the produced Na2CO3 can further displace current fossil-intensive production methods of this mineral feedstock. Further work is also needed to test tolerance of such processes to realistic flue gas contaminants, including O2 and H2O, and to examine feasibility of processes requiring harvesting of electrochemically-formed carbonate and physical replacement of metal ion-sourcing anodes to enable quasi-continuous operation. Ultimately, exploration of electrochemical mineralization reactions in aqueous media is also of interest and may exhibit scientific synergies with thermochemical efforts currently underway.118 Beyond LCA, techno-economic assessments of integrated CO2 capture-mineralization to carbonates are ongoing and will be critical to guide future research and development pathways in this area.
Other emerging electrochemical CCS processes
Although an extensive review is outside the scope of this article, other additional electrochemical methods have also received growing attention as means to facilitate sequestration of CO2 into solid phases (mineral carbonates and/or carbon) and are briefly described here. Under CO2-rich conditions, and at high temperature (>500 °C), molten carbonate electrolysis has been shown to achieve combined capture-electrochemical conversion of CO2 to value-added carbon and oxygen.119–121 In such a scheme, molten carbonates – either pure compounds (e.g.: Li2CO3, melting point (m.p.) = 730 °C) or eutectic mixtures with lower melting points (e.g.: LiNaKCO3, m.p. = 399 °C) – are typically reduced electrochemically to form amorphous and/or graphitic carbon alongside dissolved metal oxides and O2− anions (e.g.: Li2CO3 + 4e− → C(s) + Li2O(dissolved) + 2O2−).122 The resulting molten oxides can then readily absorb CO2 to regenerate the consumed carbonate electrolyte (Li2O(dissolved) + CO2 (g) → Li2CO3), whereas the oxide anions can be oxidized at an inert anode to evolve O2 gas (2O2− → O2(gas) + 4e−). While carbonate reduction to C can be carried out on inexpensive cathodes such as stainless steel or Ni with relatively high faradaic efficiencies (>80%), the high temperature requirement of this process may pose significant operational challenges (e.g., materials corrosion) which require careful consideration to evaluate the potential of this technology for CCS at scale. In a different manifestation, a new class of high-temperature CO2 transport membranes is recently developed, which offers several advantages in CO2 capture over previously-developed molten carbonate electrolyzers. Excellent reviews123–125 have discussed processes, materials, reactors, and performance aspects of these emerging technologies, and are not further described here.In addition to direct CO2 conversion to solids, indirect electrochemical mineralization of CO2 to solid carbonates has also been reported. In aqueous-based systems, a two-compartment cell containing an acidic anolyte and an alkaline catholyte with a Ca2+ or Mg2+ source (e.g.: silicates (CaSiO3)126 or chlorides (CaCl2)127) was employed for CO2 mineralization. Membrane-electrolysis was performed to form H+via H2 oxidation at the anode, and to generate OH− in the catholyte via alkaline water reduction at the cathode (2H2O + 2e− → 2OH− + H2). The hydroxide anions reacted with the metal cations to form metal hydroxides (e.g.: 2OH− + Mg2+ → Mg(OH)2), which subsequently absorbed CO2 to form insoluble metal carbonates (e.g.: CaCO3, MgCO3)127 that were easily separated from the bulk electrolyte. Overall, future efforts focusing on optimization of technical components such as membrane selectivity, acid/alkali recovery rates, and reactor design alongside economic factors and potential challenges such as diffusion limitation issues will be important to determine the scalability of electrochemically-mediated CO2 mineralization against more conventional CO2 mineralization routes.
4. Current status and future developments of electrochemical CCS
The above electrochemical CCS concepts have developed rapidly in recent years at various levels ranging from process formulations to lab scale plants both for CO2 capture from large point sources and for DAC (Fig. 10). Most of the investigations on electrochemical generation of nucleophiles (labeled as “EGN” in Fig. 10) are still at the proof-of-concept stage. Since the capture mechanism (e.g., using quinones) is relatively complex, significant efforts have been made to understand the reaction pathway at a molecular level. Systems have targeted CO2 separation from streams with both high (e.g., flue gas, ∼12–15%) and low (air, ∼400 ppm) concentrations. In contrast, the electrochemical modulation of proton concentration (labeled as “EMPC”) in CO2 capture processes has benefited from investigation at both proof-of-concept and lab-scale levels. The relative advancement of this technology is mainly due to the comparatively simple capture chemistry underlying this approach, which exploits the well-known phenomenon of pH sensitivity of CO2 hydration and established electrochemical technologies to provide sources/sinks for protons at electrodes. EMPCs allow separation of CO2 from both large point sources (using quinones to modulate the pH) and from air (using membrane electrolysis). The development of the electrochemical capacitive adsorption (ECA) processes is mostly progressed in the proof-of-concept category. Similar to EMPCs, the capture mechanism (which in this instance exploits well-known and robust electrical double layer storage) is relatively simple. Only capture from high concentrations, such as simulated industrial flue gas, has been reported so far. The electrochemically mediated amine regeneration (EMAR) process has received significant attention in both performing proof-of-concept type investigations and developing large lab-scale experimental setups. Despite efficient and stable performance for CO2 separation from simulated industrial flue gases, this approach has not been implemented for DAC purposes because EMAR energy penalty to separate CO2 from dilute streams is assumed to be high. This is mainly because absorbents with higher binding affinity with CO2 are needed for DAC applications, which results in higher energy to break the bond and regenerate the absorbent.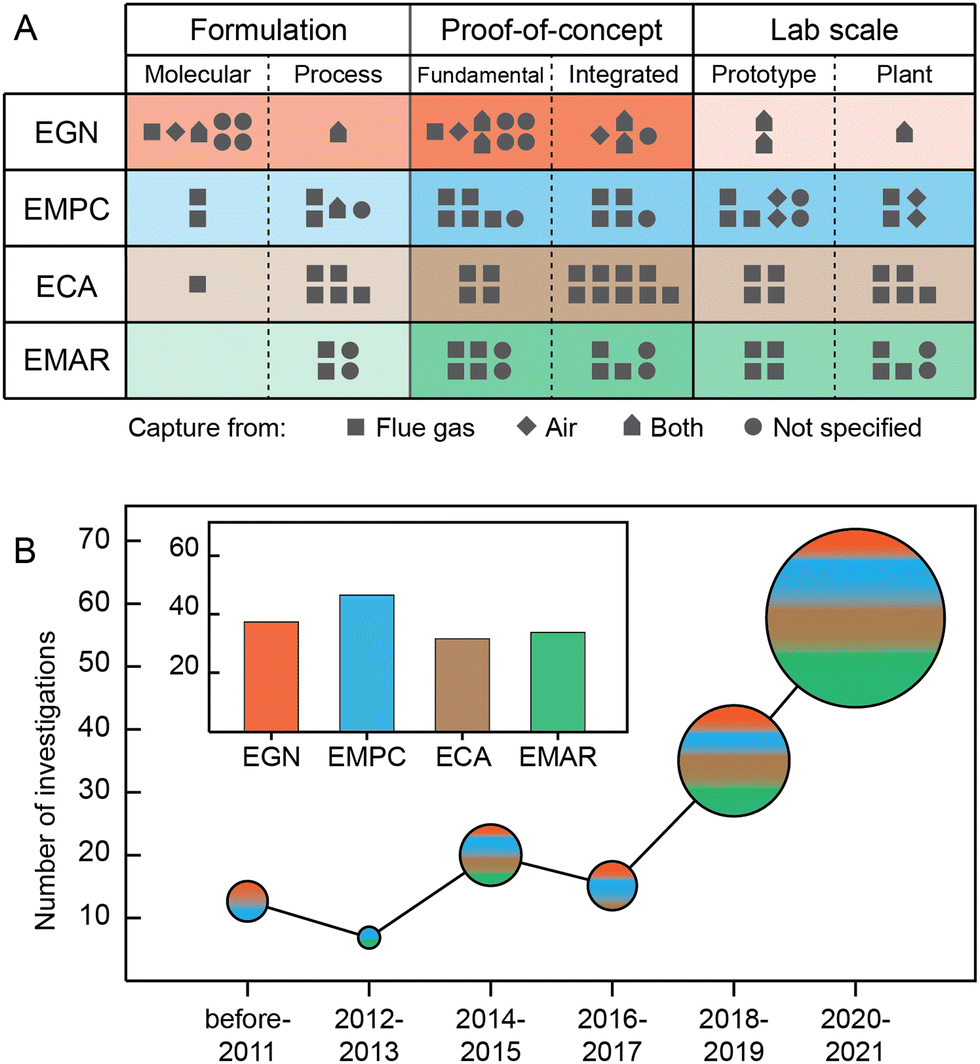 | ||
| Fig. 10 The current status of the technology development for electrochemical CCS processes. (A) A literature survey allows categorization of the development levels of electrochemical processes for CCS in three categories: formulation, proof-of-concept, and lab-scale demonstration. Various processes were considered including electrochemical generation of nucleophiles (EGN), electrochemical modulation of proton concentration (EMPC), electrochemical capacitive adsorption (ECA), and electrochemically mediated amine regeneration (EMAR). These systems were developed to separate CO2 from flue gas, air, or both—indicated here using different symbols. In some cases, the CO2 concentration was unspecified because either the system was at its early-stage of development and the focus was on the process formulation and proof-of-concept or because the absorption was done from a pure CO2 stream. Some studies can be included in multiple categories, depending on the scale of development reported. The processes are color coded, in which the color intensity is correlated with the number of studies in each category. (B) The trend in the number of investigations in the last years, which confirms electrochemical CCS processes are an emerging field of research. The magnitude of each circle represents the total investigations in that time period, while the fraction of each color indicates the share of each approach. Almost the same number of investigations have been conducted for each of the general approaches listed (inset panel). | ||
Several technical considerations must be carefully evaluated for future development of electrochemical CCS processes at the pilot scale and beyond. The oxygen sensitivity of various processes is a major concern; O2 can substantially degrade performance by either deactivating the active redox compound or by directly participating in electrochemical reactions, resulting in poor efficiencies. This issue needs to be fully addressed before any large-scale implementation for CO2 separation from actual point sources or DAC can be realized, given that industrial flue gas and air contain notable amounts of oxygen (3–7%, and 21%, respectively). In addition, the effect of other gaseous compounds such as nitrogen oxides (NOx) and sulfur oxides (SOx) that exist in industrial flue gases should be investigated, as they might reduce the system performance by deactivating redox compounds, poisoning the electrode, and/or impacting the CO2 capture mechanism. Operation of processes with actual flue gas streams in the future is essential to provide a more realistic picture of their performance.
Another key area that needs to be addressed is process stability. Most of the developed approaches are based on reversible electrochemical reactions to perform the capture-release cycles, mainly involving oxidation and reduction of a molecule or stripping and plating of an electrode. For a large scale, long-term operation, these electrochemical systems should be fully reversible to avoid unacceptable performance decay over time. The lab-scale experimental setups developed to date may not be sufficient for accurate evaluation of reversibility because they are designed to operate for shorter time periods with limited cycles, where minor irreversibilities will not be evident. Lab-scale setups should either be redesigned to allow for continuous operation, or investigations should be done at larger scales with longer operational times to evaluate realistic system reversibilities. Another important aspect of system stability is the durability of membranes in membrane-based systems such as proton concentration processes. It should be confirmed that the ion exchange membranes maintain selectivity under variable conditions and over relevant timescales. Potential problems commonly associated with membranes, e.g. fouling, should be carefully investigated.
Overall, electrochemical CCS processes are a relatively new class of separations as is clear from the fact that most of the reports reviewed here have been published only over the past four years (Fig. 10(B)). However, other electrochemical technologies, including batteries, have a long history of development on many frontiers. Although electrochemical CCS processes are still in the development stage, the scientific proofs-of-concept are now well within place such that they provide an opportunity for experts on batteries, electrochemical capacitors, and fuel cell/electrolyzer design to apply their considerable expertise to the carbon capture field. This could potentially lead to faster development of new chemistries, alternative electrodes, modified cell configurations, and even entirely new concepts. We believe the wave of research on developing electrochemical CCS processes that has recently begun is just a prelude to the exciting years ahead for these technologies.
5. Electrochemical CCS: short-term and long-term implementation
The implementation of electrochemical CCS processes to assist in the mitigation of CO2 emissions at various scales should be pursued with continuing technical advances. Here, we recommend short- and long-term implementation strategies for these electrochemical CCS systems. The implementation should be synchronized with the process technical advances and separation limits; some approaches are only effective when the input concentration of CO2 is high, while others can also be efficient at dilute concentrations, enabling capture directly from ambient air. In addition, the electrical energy requirement associated with the capture-release process plays an important role (specially in short-term implementations) because this energy is partially provided by fossil fuels before full transition to renewables is feasible.Electrochemical CCS processes are generally implemented for two purposes: capture of CO2 from large point sources, such as industrial flue gas, or from diluted streams, such as air. Based on the current status of the technology, electrochemical CCS processes generally perform better for CO2 separation from large point sources. In addition, fossil fuels are expected to remain a significant source of energy for decades to come, and rapid integration of CCS with fossil fuel power plants is a priority. Therefore, to be effective, short-term implementation of electrochemical CCS processes should be considered through retrofitting of these technologies into existing and future power plants to separate CO2 from flue gas streams before they are released to the atmosphere.
Electrochemical processes must be competitive in terms of energy demands and cost relative to state-of-the-art amine thermal scrubbing approaches to be an appealing investment. To evaluate the energy requirement and cost of the electrochemical CCS, a comprehensive analysis of the process flow diagram including all energy penalties (e.g., electrochemical cell, and operational elements such as pumps and compressors) is required to compare these systems fairly with thermal approaches. To date, such an analysis was reported only for the electrochemically mediated amine regeneration (EMAR) approach, where a complete process scheme from CO2 capture from flue gas to compression at 150 bar (similar to that of thermal processes) is modeled and evaluated.108 The analysis on the energy requirement indicated that EMAR with ethylenediamine (EDA) as the amine requires less energy compared to that of the EDA-based thermal process to desorb the same amount of CO2 from flue gas. The EMAR energy requirement was very competitive with that of the MEA-based process on the most advanced CCS technology, and also the piperazine-based thermal process that is still being developed (Fig. 11(A)).
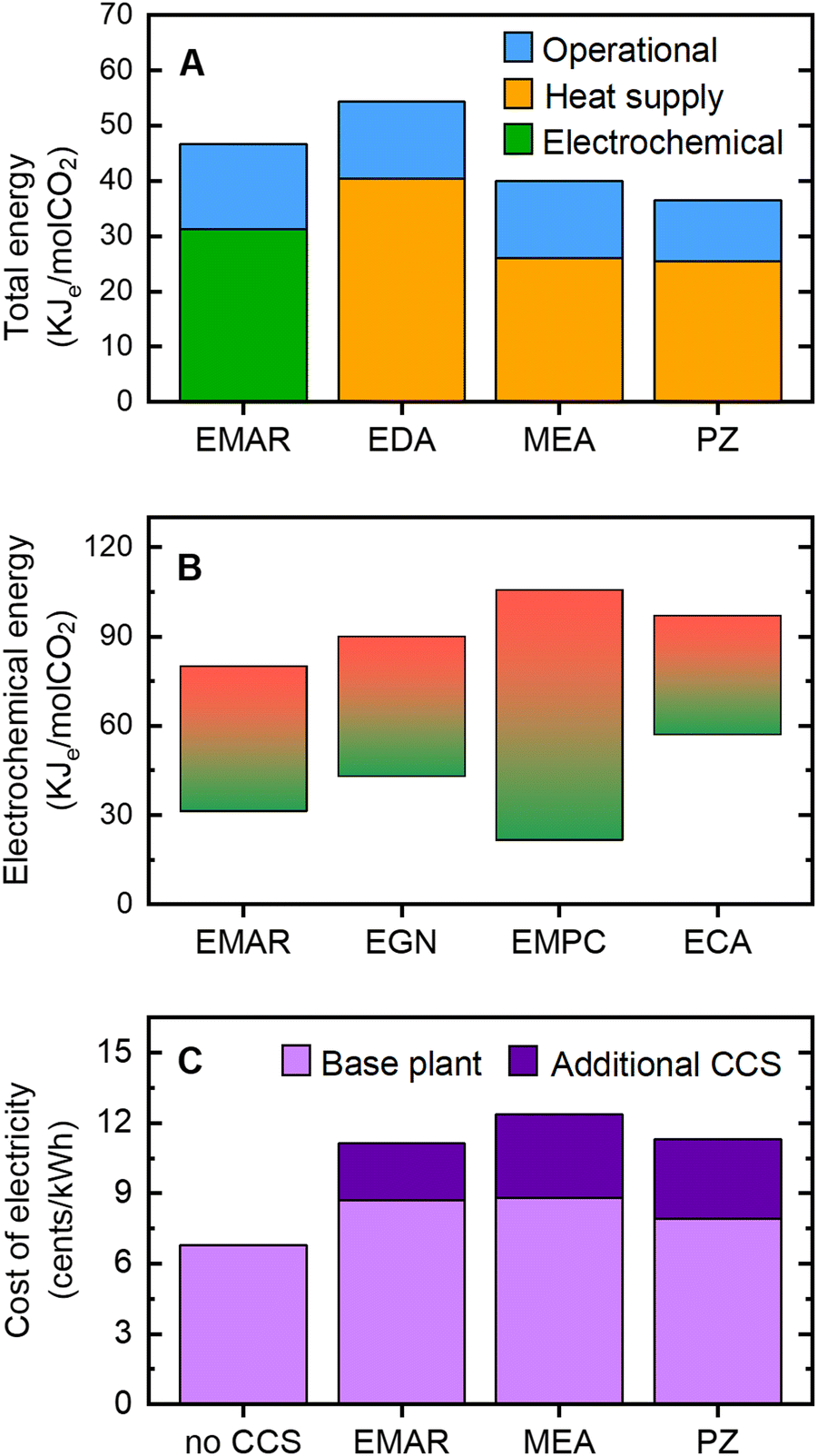 | ||
| Fig. 11 The energy requirement and cost of implementing CCS processes. (A) Comparison of the total energy requirements for CO2 capture with electrochemically mediated amine regeneration (EMAR) and thermal processes with ethylenediamine (EDA), monoethanolamine (MEA), or piperazine (PZ) amines.96,108 Various components of energy associated with the electrochemical process, heat supplied for the thermal process, and operational energy penalties such as pumps and compressor are indicated. For EMAR, the data was adapted from an investigation aimed to model an integrated process by including the absorption and electrochemical desorption stages. For thermal processes, equivalent electrical works, kJe, were estimated by assuming a Carnot efficiency of 25% (B) Comparison of the energy requirement of the electrochemical processes of EMAR104,108 with quinone-based electrochemical generation of nucleophiles (EGN),59 electrochemical modulation of proton concentration (EMPC) with a quinone,78,79 and electrochemical capacitive adsorption (ECA) using a supercapacitance.87,130 For each process, the graph was constructed based on the range of experimental energy values reported in the literature to separate CO2 from simulated industrial flue gas streams which varied based on the cell configuration, electrode and electrolyte used. (C) Comparison of the cost of electricity generated by a coal-fired power plant without a CCS unit (no CCS), with EMAR, and with thermal processes with MEA or PZ amines CCS units. The base plant cost scaled to its net generation capacity, and additional costs associated with the CCS unit include the capital expenditures and operating expenses. The data were adapted from the literature.96 | ||
Although other electrochemical CCS systems lack a comprehensive analysis of the energy requirements of a full system, their energy penalties related to the electrochemical process to desorb CO2 can be compared to that of the EMAR process. This comparison could be used to roughly predict their total energy requirements for CO2 capture, as the full process scheme developed for EMAR (which includes balance of plant such as pumps and compressors) can also be (to some extent) applied to these systems. Based on the values reported for the electrochemical desorption work of the developed systems, it can be predicted that they will be as competitive as EMAR if they are scaled up to the same level (Fig. 11(B)).
In terms of cost, implementation of any CCS process in power plants would raise the price of the electricity produced. A comparison of the final cost of electricity after implementing both EMAR and thermal processes led to the conclusion that retrofitting an EMAR unit for CCS would add almost the same cost to the final electricity price as would the other thermal processes (Fig. 11(C)). The reported cost values of EMAR are adapted from a comprehensive techno-economic analysis of a theoretically scaled-up system for post-combustion CO2 capture from a 550 MWe power plant capturing 3.1 MtCO2 annually.128,129 More than 50% of the total electricity cost is attributed to the base plant cost, which is further raised by 20% as a result of decreases in the power plant's net generation capacity when an EMAR unit is installed (still lower than the scenario in which a thermal process is implemented).96 Overall, electrochemical CCS processes, such as EMAR, have the ability to operate isothermally in a plug-and-play mode using simply electricity as an input without the requirement for complicated heat integration could potentially give a cost- and energy-efficient alternative to avoiding the drawbacks of thermally based CO2 release in the benchmark amine process.
It should be noted that most of the electricity required will be generated by renewable sources of energy in the future when designing a long-term implementation plan for the electrochemical CCS processes. This means that those technologies that are not yet practical due to their high energy penalty will be appealing in the future when they are connected to renewables as an inexpensive and abundant source of energy with minimum carbon footprint. Meanwhile, direct air capture of CO2 is increasingly expected to emerge as a key technology in the coming decades.131 DAC is essential for climate change mitigation as capture of CO2 from large point sources such as flue gas streams can only cut emissions but they cannot reduce the climate risk posed by the anthropogenic CO2 that has already been added to the air;132 DAC could be considered as a method to atone for past transgressions. However, the energy requirement and cost of capturing DAC have been the main limitations.133,134 This is because it is fundamentally energy intensive and relatively costly with the conventional technologies to capture CO2 from dilute streams. Therefore, there are unique opportunities for electrochemical processes to play a key role for future DAC, since the energy penalty and cost of these electricity-driven approaches will be minimized in the future when they are integrated with renewables. In addition to employment for DAC, electrochemical CCS processes should effectively continue to be used to capture CO2 from large point sources in the long-term implementation plan because a substantial amount of electricity will still be generated by fossil fuels (often through coal-fired power plants) in the future.
Electrochemical CCS processes could be implemented either as independent units or be integrated partially with a conventional technology for DAC purposes. To date, several electrochemical approaches such as electrochemical generation of nucleophiles by redox-active quinone molecules57,59,81,135 have been developed independently to capture CO2 from very dilute streams such as air (Fig. 10). An evaluation of the experimental energy penalties and a techno-economic analysis of CO2 capture from air indicate the great promise of these recently developed approaches for DAC, making them appealing for future implementation and investment. In addition, electrochemical CCS processes could be integrated with conventional DAC approaches in which the thermal regeneration step is replaced by an electrochemical cycle resulting in a capture-release process that relies exclusively on electricity. For example, in a recent attempt, a hybrid DAC process was proposed which captured CO2 using a wet scrubbing with an aqueous potassium hydroxide solution (as a conventional method) while the solvent regeneration and CO2 release were carried out through a membrane electrodialysis136—an electrochemical scheme previously developed with bipolar and ion exchange membranes.71 The results of the techno-economic analysis showed that the electrochemical-based regeneration process could be less energy intensive than the thermal. However, the system still suffered from the high costs of bipolar and ion exchange membranes. This integrated process could be particularly interesting in the future, when more affordable and improved membranes are available.136 Overall, the implementation of electrochemical processes (both independently and partially) for DAC purposes has been initiated, but it still requires future advancements, especially to further reduce the cost of capture.
A significant fraction of the total global CO2 emissions reports to the oceans, with the resulting increased acidification leading to destruction of coral reefs, and harming of shellfish and other marine life. Since the total amount of CO2 absorbed in the oceans is similar to that in the atmosphere (and at 100 mg L−1, is 140-fold more concentrated), effective means for its removal could contribute significantly to the overall reduction in the environmental burden imposed by this greenhouse gas. The oceans serve as a natural absorption medium, and thus only the regeneration step need be considered in liberating the CO2 and re-alkalizing the ocean waters. In the few studies reported to date, this has generally been accomplished through pH modulation in either bipolar membrane electrodialysis or electrodeionization processes,137–143 with the acidification and basification of separate ocean streams, accompanied by the release of molecular CO2 which can be removed by vacuum stripping, and, in some cases, also the precipitation of CaCO3.144
These technologies require expensive bipolar and anion and cation exchange membranes, and the preparation of electrolyte solutions for the terminal anode and cathode chambers bookending the bipolar stacks, either via addition of chemical reagents or by reverse osmotic deionization of seawater. An attractive alternative approach to the ocean water CO2 removal without water-splitting is to exploit pH modulation of feed streams through the release of protons on application of an appropriate voltage across an asymmetric membrane-less electrochemical cell comprised of intercalating electrodes, a process that is currently under development in our laboratories.
In addition to DAC and ocean removal applications, electrochemical CCS processes could be effectively implemented to separate CO2 from diverse mobile sources with dilute concentrations. For example, the transportation sector, where individual units, known as small emitters (e.g., car or airplane), produce a relatively low amount of CO2, but in aggregate they account for ∼20% of the global emissions, could benefit from the installation of electrochemical-based CCS units on board mobile platforms to effectively capture CO2 before it is released to the atmosphere. Due to the diversity and quantity of small emitters, there is a vast opportunity for flexible plug-and-play units (e.g., electrochemical processes) to play a vital role in mitigating the associated emission as it is challenging to neutralize these large quantities of CO2 by conventional capture technologies. These unique advantages of electrochemical CCS processes may compensate for their energy penalty and cost of capturing CO2 from dilute streams such as air or small emitters.
List of abbreviations
| AEM | Anion exchange membrane |
| BPM | Bipolar membrane |
| CCS | Carbon capture and storage |
| CCU | Carbon capture and utilization |
| CEM | Cation exchange membrane |
| DAC | Direct air capture |
| ECA | Electrochemical capacitive adsorption |
| EGN | Electrochemical generation of nucleophile |
| EIA | Energy information administration |
| EMAR | Electrochemically mediated amine regeneration |
| EMPC | Electrochemical modulation of proton concentration |
| EU | European union |
| FE | Faradaic efficiencies |
| GDE | Gas diffusion electrode |
| HER | Hydrogen evolution reaction |
| IEA | International energy agency |
| IPCC | Intergovernmental panel on climate change |
| LCA | Life-cycle analysis |
| MCDI | Membrane capacitive deionization |
| MED | Membrane electrodialysis |
| OER | Oxygen evolution reaction |
| PCP | Proton concentration process |
| SG | Sweep gas |
| SSA | Supercapacitive swing adsorption |
Conflicts of interest
T. Alan Hatton is a Co-Founder and Scientific Advisory Board member of Verdox, Inc., which is developing the electro-swing adsorption process for commercial applications.References
- IPCC, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, ed. V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2021 DOI:10.1017/9781009157896.
- G. Luderer, Z. Vrontisi, C. Bertram, O. Y. Edelenbosch, R. C. Pietzcker, J. Rogelj, H. S. De Boer, L. Drouet, J. Emmerling and O. Fricko, Nat. Clim. Change, 2018, 8, 626–633 CrossRef CAS.
- UNFCCC, 2015.
- R. K. Pachauri, M. R. Allen, V. R. Barros, J. Broome, W. Cramer, R. Christ, J. A. Church, L. Clarke, Q. Dahe and P. Dasgupta, Climate change 2014: synthesis report. Contribution of Working Groups I, II and III to the fifth assessment report of the Intergovernmental Panel on Climate Change, IPCC, 2014.
- I. IEA, Catalysing Energy Technology Transformations, 2017.
- K. Z. House, C. F. Harvey, M. J. Aziz and D. P. Schrag, Energy Environ. Sci., 2009, 2, 193–205 RSC.
- R. Sharifian, R. Wagterveld, I. Digdaya, C. Xiang and D. Vermaas, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- S. E. Renfrew, D. E. Starr and P. Strasser, ACS Catal., 2020, 10, 13058–13074 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS PubMed.
- S. Ma and P. J. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS PubMed.
- A. S. Agarwal, E. Rode, N. Sridhar and D. Hill, in Handbook of Climate Change Mitigation and Adaptation, ed. W.-Y. Chen, T. Suzuki and M. Lackner, Springer, New York, NY, 2014, pp.1–40 DOI:10.1007/978-1-4614-6431-0_86-1.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- B. Zhang, Y. Jiang, M. Gao, T. Ma, W. Sun and H. Pan, Nano Energy, 2021, 80, 105504 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364(6438), eaav3506 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- D. S. Ripatti, T. R. Veltman and M. W. Kanan, Joule, 2019, 3, 240–256 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Energy Environ. Sci., 2017, 10, 1180–1185 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- F. A. Marke, accessed January, 2018, 22, 2019.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243–249 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- S. J. Bennett, D. J. Schroeder and S. T. McCoy, Energy Procedia, 2014, 63, e7992 Search PubMed.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187–11194 CrossRef PubMed.
- T. Fout, T. Shultz, M. Woods, M. J. Turner, A. J. Zoelle and R. E. James III, Cost and Performance Baseline for Fossil Energy Plants, Volume 1: Bituminous Coal and Natural Gas to Electricity, Revision 4, NETL, 2018.
- R. Idem, T. Supap, H. Shi, D. Gelowitz, M. Ball, C. Campbell and P. Tontiwachwuthikul, Int. J. Greenhouse Gas Control, 2015, 40, 6–25 CrossRef CAS.
- E. I. A. U.S. Department of Energy, International Energy Outlook 2021, 2021.
- J. C. Abanades, E. S. Rubin, M. Mazzotti and H. J. Herzog, Energy Environ. Sci., 2017, 10, 2491–2499 RSC.
- V. Scott, S. Gilfillan, N. Markusson, H. Chalmers and R. S. Haszeldine, Nat. Clim. Change, 2013, 3, 105–111 CrossRef CAS.
- Y.-M. Wei, J.-N. Kang, L.-C. Liu, Q. Li, P.-T. Wang, J.-J. Hou, Q.-M. Liang, H. Liao, S.-F. Huang and B. Yu, Nat. Clim. Change, 2021, 11, 112–118 CrossRef.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo and L. A. Hackett, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- M. Rahimi, S. M. Moosavi, B. Smit and T. A. Hatton, Cell Rep. Phys. Sci., 2021, 2, 100396 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- R. T. Porter, M. Fairweather, C. Kolster, N. Mac Dowell, N. Shah and R. M. Woolley, Int. J. Greenhouse Gas Control, 2017, 57, 185–195 CrossRef CAS.
- T. N. G. Borhani, A. Azarpour, V. Akbari, S. R. W. Alwi and Z. A. Manan, Int. J. Greenhouse Gas Control, 2015, 41, 142–162 CrossRef CAS.
- K. A. Mumford, K. H. Smith, C. J. Anderson, S. Shen, W. Tao, Y. A. Suryaputradinata, A. Qader, B. Hooper, R. A. Innocenzi and S. E. Kentish, Energy Fuels, 2011, 26, 138–146 CrossRef.
- R. R. Bhosale, A. Kumar, F. AlMomani, U. Ghosh, A. AlNouss, J. Scheffe and R. B. Gupta, Ind. Eng. Chem. Res., 2016, 55, 5238–5246 CrossRef CAS.
- D. Grimekis, S. Giannoulidis, K. Manou, K. D. Panopoulos and S. Karellas, Int. J. Greenhouse Gas Control, 2019, 81, 83–92 CrossRef CAS.
- H. Thee, N. J. Nicholas, K. H. Smith, G. da Silva, S. E. Kentish and G. W. Stevens, Int. J. Greenhouse Gas Control, 2014, 20, 212–222 CrossRef CAS.
- D. H. Apaydin, E. D. Głowacki, E. Portenkirchner and N. S. Sariciftci, Angew. Chem., Int. Ed., 2014, 53, 6819–6822 CrossRef CAS PubMed.
- M. B. Mizen and M. S. Wrighton, J. Electrochem. Soc., 1989, 136, 941 CrossRef CAS.
- J. S. Kang, S. Kim and T. A. Hatton, Curr. Opin. Green Sustainable Chem., 2021, 100504 CrossRef CAS.
- F. Simeon, M. C. Stern, K. M. Diederichsen, Y. Liu, H. J. Herzog and T. A. Hatton, J. Phys. Chem. C, 2022, 126, 1389–1399 CrossRef CAS.
- A. T. Bui, N. A. Hartley, A. J. Thom and A. C. Forse, J. Phys. Chem. C, 2022, 126(33), 14163–14172 CrossRef CAS.
- P. Scovazzo, J. Poshusta, D. DuBois, C. Koval and R. Noble, J. Electrochem. Soc., 2003, 150, D91–D98 CrossRef CAS.
- B. Gurkan, F. Simeon and T. A. Hatton, ACS Sustainable Chem. Eng., 2015, 3, 1394–1405 CrossRef CAS.
- S. Voskian and T. A. Hatton, Energy Environ. Sci., 2019, 12, 3530–3547 RSC.
- S. Voskian and J. Boyson, Verdox's Electrochemical DAC, 2022, DOI: https://www.youtube.com/watch?v=Av_Sbd2cx3s.
- Y. Liu, H.-Z. Ye, K. M. Diederichsen, T. Van Voorhis and T. A. Hatton, Nat. Commun., 2020, 11, 1–11 CrossRef CAS.
- K. M. Diederichsen, Y. Liu, N. Ozbek, H. Seo and T. A. Hatton, Joule, 2022, 6, 221–239 CrossRef CAS.
- Q. Li, C. Batchelor-McAuley, N. S. Lawrence, R. S. Hartshorne and R. G. Compton, ChemPhysChem, 2011, 12, 1255–1257 CrossRef CAS PubMed.
- R. Ranjan, J. Olson, P. Singh, E. D. Lorance, D. A. Buttry and I. R. Gould, J. Phys. Chem. Lett., 2015, 6, 4943–4946 CrossRef CAS PubMed.
- H. Ishida, T. Ohba, T. Yamaguchi and K. Ohkubo, Chem. Lett., 1994, 905–908 CrossRef CAS.
- P. Singh, J. H. Rheinhardt, J. Z. Olson, P. Tarakeshwar, V. Mujica and D. A. Buttry, J. Am. Chem. Soc., 2017, 139, 1033–1036 CrossRef CAS.
- S. Jin, M. Wu, Y. Jing, R. G. Gordon and M. J. Aziz, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- O. Oloye and A. P. O’Mullane, ChemSusChem, 2021, 14, 1767–1775 CrossRef CAS.
- C. Zhou, J. Ni, H. Chen and X. Guan, Sustainable Energy Fuels, 2021, 5(17), 4355–4367 RSC.
- S. Datta, M. P. Henry, Y. J. Lin, A. T. Fracaro, C. S. Millard, S. W. Snyder, R. L. Stiles, J. Shah, J. Yuan and L. Wesoloski, Ind. Eng. Chem. Res., 2013, 52, 15177–15186 CrossRef CAS.
- M. D. Eisaman, L. Alvarado, D. Larner, P. Wang, B. Garg and K. A. Littau, Energy Environ. Sci., 2011, 4, 1319–1328 RSC.
- M. D. Eisaman, L. Alvarado, D. Larner, P. Wang and K. A. Littau, Energy Environ. Sci., 2011, 4, 4031–4037 RSC.
- R. J. Gilliam, B. K. Boggs, V. Decker, M. A. Kostowskyj, S. Gorer, T. A. Albrecht, J. D. Way, D. W. Kirk and A. J. Bard, J. Electrochem. Soc., 2012, 159, B627–B628 CrossRef CAS.
- A. Mehmood, M. I. Iqbal, J.-Y. Lee, J. Hwang, K.-D. Jung and H. Y. Ha, Electrochim. Acta, 2016, 219, 655–663 CrossRef CAS.
- K. A. Littau and F. E. Torres, System and method for recovery of CO2 by aqueous carbonate flue gas capture and high efficiency bipolar membrane electrodialysis, US Pat., 8535502, 2013 Search PubMed.
- M. D. Eisaman, D. E. Schwartz, S. Amic, D. Larner, J. Zesch, F. Torres and K. Littau, Energy-efficient electrochemical CO2 capture from the atmosphere, in Technical Proceedings of the 2009 Clean Technology Conference and Trade Show, 2009, pp. 3–7 Search PubMed.
- J. D. Watkins, N. S. Siefert, X. Zhou, C. R. Myers, J. R. Kitchin, D. P. Hopkinson and H. B. Nulwala, Energy Fuels, 2015, 29, 7508–7515 CrossRef CAS.
- C. Huang, C. Liu, K. Wu, H. Yue, S. Tang, H. Lu and B. Liang, Energy Fuels, 2019, 33, 3380–3389 CrossRef CAS.
- H. Xie, Y. Wu, T. Liu, F. Wang, B. Chen and B. Liang, Appl. Energy, 2020, 259, 114119 CrossRef CAS.
- S. Jin, M. Wu, R. G. Gordon, M. J. Aziz and D. G. Kwabi, Energy Environ. Sci., 2020, 13, 3706–3722 RSC.
- H. Seo, M. Rahimi and T. A. Hatton, J. Am. Chem. Soc., 2022, 144, 2164–2170 CrossRef CAS PubMed.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS PubMed.
- M. Rahimi, G. Catalini, S. Hariharan, M. Wang, M. Puccini and T. A. Hatton, Cell Rep. Phys. Sci., 2020, 1, 100033 CrossRef.
- M. Rahimi, G. Catalini, M. Puccini and T. A. Hatton, RSC Adv., 2020, 10, 16832–16843 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- B. Kokoszka, N. K. Jarrah, C. Liu, D. T. Moore and K. Landskron, Angew. Chem., Int. Ed., 2014, 53, 3698–3701 CrossRef CAS PubMed.
- C. Liu and K. Landskron, Chem. Commun., 2017, 53, 3661–3664 RSC.
- S. Zhu, K. Ma and K. Landskron, J. Phys. Chem. C, 2018, 122, 18476–18483 CrossRef CAS.
- T. B. Binford, G. Mapstone, I. Temprano and A. C. Forse, Nanoscale, 2022, 14, 7980–7984 RSC.
- K. J. Lamb, M. R. Dowsett, K. Chatzipanagis, Z. W. Scullion, R. Kröger, J. D. Lee, P. M. Aguiar, M. North and A. Parkin, ChemSusChem, 2018, 11, 137–148 CrossRef CAS PubMed.
- M. R. Dowsett, C. M. Lewis, M. North and A. Parkin, Dalton Trans., 2018, 47, 10447–10452 RSC.
- L. Legrand, O. Schaetzle, R. de Kler and H. V. Hamelers, Environ. Sci. Technol., 2018, 52, 9478–9485 CrossRef CAS PubMed.
- P. Biesheuvel and A. Van der Wal, J. Membr. Sci., 2010, 346, 256–262 CrossRef CAS.
- L. Legrand, Q. Shu, M. Tedesco, J. Dykstra and H. Hamelers, J. Colloid Interface Sci., 2020, 564, 478–490 CrossRef CAS PubMed.
- M. Suss, S. Porada, X. Sun, P. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 RSC.
- M. Wang, H. J. Herzog and T. A. Hatton, Ind. Eng. Chem. Res., 2020, 59, 7087–7096 CrossRef CAS.
- M. Rahimi, T. Kim, C. A. Gorski and B. E. Logan, J. Power Sources, 2018, 373, 95–102 CrossRef CAS.
- K. Landskron, Angew. Chem., Int. Ed., 2018, 57, 3548 CrossRef CAS PubMed.
- A. Rinberg, A. M. Bergman, D. P. Schrag and M. J. Aziz, ChemSusChem, 2021, 14(20), 4439–4453 CrossRef CAS.
- M. C. Stern, F. Simeon, H. Herzog and T. A. Hatton, Energy Environ. Sci., 2013, 6, 2505–2517 RSC.
- M. C. Stern and T. A. Hatton, RSC Adv., 2014, 4, 5906–5914 RSC.
- M. C. Stern, Electrochemically-mediated amine regeneration for carbon dioxide separations, PhD thesis, Massachusetts Institute of Technology, 2013 Search PubMed.
- X. Wu, H. Fan, M. Sharif, Y. Yu, K. Wei, Z. Zhang and G. Liu, Appl. Energy, 2021, 302, 117554 CrossRef CAS.
- M. Wang, M. Rahimi, A. Kumar, S. Hariharan, W. Choi and T. A. Hatton, Appl. Energy, 2019, 255, 113879 CrossRef CAS.
- M. Rahimi, K. M. Diederichsen, N. Ozbek, M. Wang, W. Choi and T. A. Hatton, Environ. Sci. Technol., 2020, 54, 8999–9007 CrossRef CAS.
- S. Zhou, X. Chen, T. Nguyen, A. K. Voice and G. T. Rochelle, ChemSusChem, 2010, 3, 913–918 CrossRef CAS PubMed.
- A. Hasanzadeh, S. G. Holagh and A. Chitsaz, J. CO2 Util., 2021, 54, 101758 CrossRef CAS.
- M. Wang, S. Hariharan, R. A. Shaw and T. A. Hatton, Int. J. Greenhouse Gas Control, 2019, 82, 48–58 CrossRef CAS.
- C. Wang, K. Jiang, T. W. Jones, S. Yang, H. Yu, P. Feron and K. Li, Chem. Eng. J., 2021, 131981 Search PubMed.
- J. Boualavong and C. A. Gorski, ACS ES&T Engg, 2021, 1(7), 1084–1093 Search PubMed.
- M. Rahimi, A. D’Angelo, C. A. Gorski, O. Scialdone and B. E. Logan, J. Power Sources, 2017, 351, 45–50 CrossRef CAS.
- M. Rahimi, F. Zucchelli, M. Puccini and T. Alan Hatton, ACS Appl. Energy Mater., 2020, 3, 10823–10830 CrossRef CAS.
- A. Khurram, M. He and B. M. Gallant, Joule, 2018, 2, 2649–2666 CrossRef CAS.
- A. Khurram, L. Yan, Y. Yin, L. Zhao and B. M. Gallant, J. Phys. Chem. C, 2019, 123, 18222–18231 CrossRef CAS.
- A. Khurram, H. Gao and B. M. Gallant, J. Phys. Chem. C, 2020, 124, 18877–18885 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. S. Rasouli, F. Li, M. Luo, A. H. Ip and Y.-C. Joo, Nat. Energy, 2020, 1–8 CAS.
- O. Pfeiffer, A. Khurram, E. A. Olivetti and B. M. Gallant, J. Ind. Ecol., 2022, 26, 1306–1317 CrossRef CAS.
- G. Gadikota, Nat. Rev. Chem., 2020, 4, 78–89 CrossRef CAS.
- H. Yin, X. Mao, D. Tang, W. Xiao, L. Xing, H. Zhu, D. Wang and D. R. Sadoway, Energy Environ. Sci., 2013, 6, 1538–1545 RSC.
- L. Hu, Y. Song, J. Ge, J. Zhu and S. Jiao, J. Mater. Chem. A, 2015, 3, 21211–21218 RSC.
- H. Wu, Z. Li, D. Ji, Y. Liu, L. Li, D. Yuan, Z. Zhang, J. Ren, M. Lefler and B. Wang, Carbon, 2016, 106, 208–217 CrossRef CAS.
- J. Ren, F.-F. Li, J. Lau, L. González-Urbina and S. Licht, Nano Lett., 2015, 15, 6142–6148 CrossRef CAS.
- G. A. Mutch, L. Qu, G. Triantafyllou, W. Xing, M.-L. Fontaine and I. S. Metcalfe, J. Mater. Chem. A, 2019, 7, 12951–12973 RSC.
- P. Zhang, J. Tong, K. Huang, X. Zhu and W. Yang, Prog. Energy Combust. Sci., 2021, 82, 100888 CrossRef.
- G. Chen, A. Feldhoff, A. Weidenkaff, C. Li, S. Liu, X. Zhu, J. Sunarso, K. Huang, X. Y. Wu and A. F. Ghoniem, Adv. Funct. Mater., 2022, 32, 2105702 CrossRef CAS.
- D. Shuto, H. Nagasawa, A. Iizuka and A. Yamasaki, RSC Adv., 2014, 4, 19778–19788 RSC.
- H. Xie, T. Liu, Z. Hou, Y. Wang, J. Wang, L. Tang, W. Jiang and Y. He, Environ. Earth Sci., 2015, 73, 6881–6890 CrossRef CAS.
- M. Wang, R. Shaw, E. Gencer and T. A. Hatton, Ind. Eng. Chem. Res., 2020, 59, 14085–14095 CrossRef CAS.
- M. Wang, H. J. Herzog and T. A. Hatton, Ind. Eng. Chem. Res., 2020, 59, 7087–7096 CrossRef CAS.
- S. Zhu, J. Li, A. Toth and K. Landskron, ACS Appl. Mater. Interfaces, 2019, 11, 21489–21495 CrossRef CAS PubMed.
- C. Breyer, M. Fasihi, C. Bajamundi and F. Creutzig, Joule, 2019, 3, 2053–2057 CrossRef.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- M. Fasihi, O. Efimova and C. Breyer, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- H. Azarabadi and K. S. Lackner, Appl. Energy, 2019, 250, 959–975 CrossRef.
- A. Hemmatifar, J. S. Kang, N. Ozbek, K.-J. Tan and T. A. Hatton, ChemSusChem, 2022, 15(6), e202102533 CrossRef CAS PubMed.
- F. Sabatino, M. Mehta, A. Grimm, M. Gazzani, F. Gallucci, G. J. Kramer and M. van Sint Annaland, Ind. Eng. Chem. Res., 2020, 59, 7007–7020 CrossRef CAS.
- M. D. Eisaman, K. Parajuly, A. Tuganov, C. Eldershaw, N. Chang and K. A. Littau, Energy Environ. Sci., 2012, 5, 7346–7352 RSC.
- C.-F. de Lannoy, M. D. Eisaman, A. Jose, S. D. Karnitz, R. W. DeVaul, K. Hannun and J. L. Rivest, Int. J. Greenhouse Gas Control, 2018, 70, 243–253 CrossRef CAS.
- M. D. Eisaman, J. L. Rivest, S. D. Karnitz, C.-F. de Lannoy, A. Jose, R. W. DeVaul and K. Hannun, Int. J. Greenhouse Gas Control, 2018, 70, 254–261 CrossRef CAS.
- H. D. Willauer, F. DiMascio, D. R. Hardy and F. W. Williams, Ind. Eng. Chem. Res., 2014, 53, 12192–12200 CrossRef CAS.
- H. D. Willauer, F. DiMascio, D. R. Hardy and F. W. Williams, Energy Fuels, 2017, 31, 1723–1730 CrossRef CAS.
- I. A. Digdaya, I. Sullivan, M. Lin, L. Han, W.-H. Cheng, H. A. Atwater and C. Xiang, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- L. Yan, J. Bao, Y. Shao and W. Wang, ACS Energy Lett., 2022, 7, 1947–1952 CrossRef CAS.
- R. Sharifian, L. Boer, R. Wagterveld and D. Vermaas, Chem. Eng. J., 2022, 438, 135326 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |