
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic routes toward C3 products in copper-catalysed CO2 electroreduction†
Sergio
Pablo-García‡
 a,
Florentine L. P.
Veenstra‡
b,
Louisa Rui Lin
Ting‡
cd,
Rodrigo
García-Muelas
a,
Federico
Dattila
a,
Antonio J.
Martín
b,
Boon Siang
Yeo
*cd,
Javier
Pérez-Ramírez
*b and
Núria
López
*a
a,
Florentine L. P.
Veenstra‡
b,
Louisa Rui Lin
Ting‡
cd,
Rodrigo
García-Muelas
a,
Federico
Dattila
a,
Antonio J.
Martín
b,
Boon Siang
Yeo
*cd,
Javier
Pérez-Ramírez
*b and
Núria
López
*a
aInstitute of Chemical Research of Catalonia, ICIQ, The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: nlopez@iciq.es
bInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: jpr@chem.ethz.ch
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmyeos@nus.edu.sg
dSolar Energy Research Institute of Singapore, National University of Singapore, 7 Engineering Drive 1, 117574, Singapore
First published on 7th January 2022
Abstract
The electrocatalytic CO2 reduction (eCO2R) reaction powered by renewable electricity holds promise for the sustainable production of multi-carbon chemicals and fuels. On Cu-based catalysts, ethylene and ethanol (C2) have been produced in appreciable amounts. C3 products (mostly terminal oxygenates) have limited yields, whereas propylene is puzzlingly absent. Herein, we devise a divide-and-conquer strategy to explain the formation of the C3-backbone and elucidate the mechanism responsible for the observed selectivity by combining network graphs, density functional theory, and experiments to prune the network and benchmark the identified path. Our approach concludes that the most frequently reported products, propionaldehyde and 1-propanol, originate from the coupling of CH2CH with C(H)O. While propylene and 1-propanol share common intermediates, the former is barely produced due to the unfavourable formation of allyl alkoxy (CH2CHCH2O), whose crucial nature was confirmed experimentally. This work paves the way for tailoring selective routes towards C3 products via eCO2R.
Introduction
Developing functional catalysts for the electrochemical CO2 reduction (eCO2R) to complex products lies at the core of new efforts to develop sustainable technologies.1 Among available materials, copper-based electrocatalysts occupy a pivotal role due to their ability to form the C2+ backbone for high-value fuels and commodity chemicals.1,2 The type and amount of products formed are sensitive to the applied potential, electrolyte, and the preparation protocol of Cu.3–6 The established mechanism to the C2 fraction advocates that CO2 first reduces to CO, which dimerises to OCCO− and subsequently reduces to hydrocarbons and alcohols. Typically, the main C2 product is ethylene (up to ca. 74% Faradaic efficiency FE),7 although an exceptional ethanol selectivity (ca. 91% FE)8 has been reported on Cu clusters. For C4 products, the aldol condensation of acetaldehyde (C2) gives crotonaldehyde, which reduces to 1-butanol, albeit with low yields.9 Among C3 compounds, 1-propanol can be produced with appreciable yields (∼23% FE),10,11 whereas propylene (the corresponding C3 olefin; 0.36 eV less stable than 1-propanol, Table S1†) has only been detected as a trace product (<0.1% FE).9 This puzzling outcome contrasts with the vast formation of ethylene, which is less stable than ethanol by 0.47 eV, Table S1.† Furthermore, 2-propanol, which is the most thermodynamically stable C3 alcohol (0.17 eV lower than 1-propanol, Table S1†), has never been observed in eCO2R.12 Reports have indicated that the formation of the C3 backbone at high CO concentrations and relatively mild applied potentials (−0.36 to −0.56 V vs. RHE)11,13 requires asymmetric sites on oxide-derived Cu (OD-Cu) catalysts. Nonetheless, the reasons behind the low selectivity to C3 products in eCO2R at a molecular level is very limited. This is due to the large number of elementary steps (>103) that prevent the use of standard reaction sampling tools based on explicit density functional theory (DFT) and reaction profile analysis.Herein, we analyse electrocatalytic routes towards C3 products through a divide-and-conquer strategy based on the generation of the network graph and computational reaction profiles combined with key electrochemical experiments involving C1, C2, and C3 reagents. This new methodological approach enables us to (i) identify the most likely C1–C2 coupling steps towards C3 intermediates; (ii) elucidate the bifurcation points to different C3 products; and (iii) pinpoint kinetic bottlenecks hindering propylene production.
Experimental and computational methods
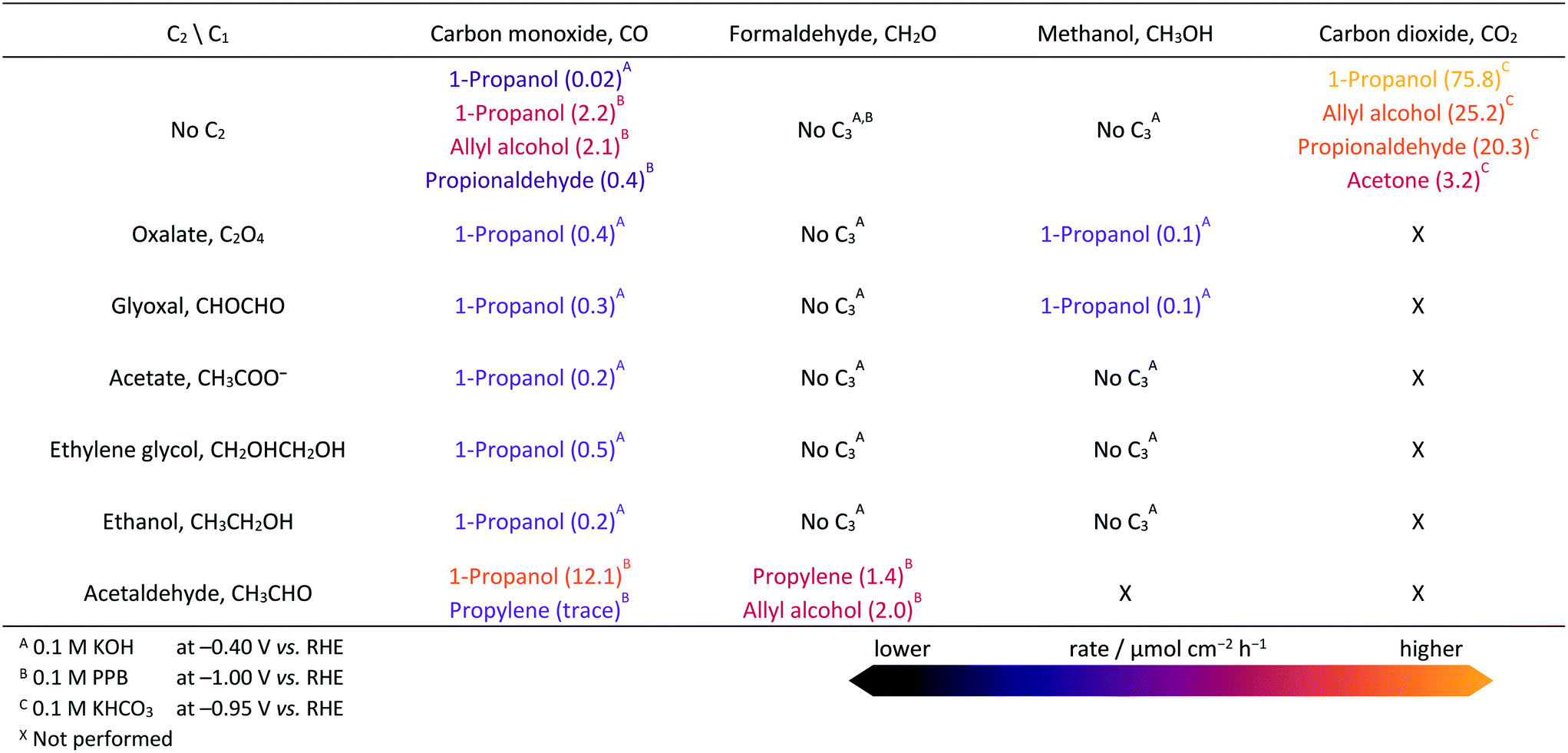
The electrocatalytic reactor used for our experiments is a gas-tight cell consisting of two compartments separated by a Nafion 211 membrane with gas-flow inlet and outlet ports. The cell has an OD-Cu working electrode, a gas diffusion layer (GDL) carbon paper counter electrode, and a leak-free Ag/AgCl (3 M KCl) reference electrode. Triplicate measurements were done, with the average values and standard deviations presented in the ESI.† Extended details about reagents, catalysts preparation, electrochemical measurements, detection limits, and product analyses are shown in Experimental and computational details in the ESI.† The OD-Cu catalyst was obtained from CuO (see Fig. S1† for XRD analysis). The physicochemical and catalytic properties of this material have been discussed elsewhere.9,14We initially reduced CO2 in 0.1 M KHCO3 at −0.95 V vs. RHE to maximise the production of multi-carbon products,14 as shown in Fig. 1 (see Fig. S2† for polarisation curves). C1 and C2 products account for 53% of the FE, whereas 7% corresponds to C3 products and the balance is H2. Our observed product distribution agrees with selectivity trends presented in the literature, which are summarised in Fig. S3,† and the paths to C2 products are shown in Fig. S4–S5.† To unravel the selectivity patterns observed from both the literature and our experiments, our workflow entails: (i) building the reaction network by encoding the corresponding structural graphs; (ii) sampling the intermediates by DFT; (iii) computing all C1–C2 backbone couplings by DFT; (iv) pruning the network of non-viable backbone formation routes by probing the products from CO, formaldehyde, and methanol co-reduction with C2 reactants (Table 1), with particular attention to missing products; (v) computing all routes from the C3 backbone to the final products using DFT and linear-scaling relationships (Table S15†) to identify the best routes towards propanol and propylene; (vi) experimental benchmarking of the main predicted routes via electrocatalytic tests with key intermediates.
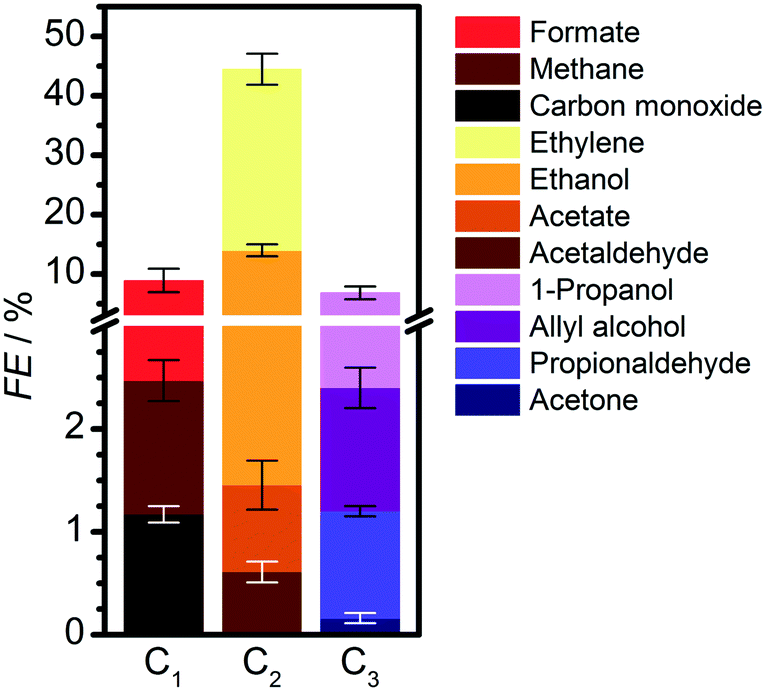 | ||
| Fig. 1 C1–C3 products formed from the electrocatalytic CO2 reduction on oxide-derived copper in 0.1 M KHCO3 at −0.95 V vs. RHE. Detailed results are presented in Table S2.† Hydrogen FE is 37.4%. | ||
|
While the routes to C1 and C2 products can be probed manually, as shown in the literature,15–20 the analysis of routes to C3 products demands automation. The full network containing all C1, C2, and C3 intermediates has 463 elements, represented here as nodes in a graph (see “Graph representation of the reaction network” in Experimental and computational details). The energies of intermediates (referenced to CO2, H2O, and H2) were evaluated with the computational hydrogen electrode (CHE)21,22 containing the DFT energy obtained with a PBE-D2 formulation23–25 (corrected for metal overbinding), our in-house developed implicit solvation model,26,27 and the polarisation term9,28 (see eqn S1–S4† in the Experimental and computational details). The D2 contribution on H* and CO* adsorption is small: 0.04 and 0.14 eV, respectively. Intermediates are linked by 2266 steps (edges linking the nodes in the graph): 55 C1–C1 and 636 C1–C2 couplings, 683 C–H and 305 O–H hydrogenations, and 587 C–O(H) cleavages. To ensure the desired accuracy, 586 out of all C1–C2 couplings (Tables S16–S19,† 92% of total), 10 C–O(H) breakings (Table S20†), and 8 hydrogenations (Table S21†) were explicitly obtained via nudged elastic band (DFT-NEB)29 and confirmed by vibrational analysis. Initial guesses for NEB were generated automatically (Experimental and computational details in ESI† and Notes S1–S4). Linear scaling relationships (LSR) were employed in the initial fast-sampling of C–H and O–H hydrogenations as they are reliable for these cases20,30,31 (Table S15†). The transition states for key hydrogenation steps in the main path were further refined with DFT-NEB. Heyrovsky-type reactions for C–OH breakings and C–H formations were considered (Note S5†). Tests on density functionals, LSR, and charge analysis are described in Note S6 and Fig. S6–S9,† and demonstrate that our strategy provides an excellent cost-efficiency balance.
Benchmark electrochemical experiments (Table 1) involving the reduction of selected C1 and C2 compounds and their mixtures were conducted at mild overpotentials (−0.4 V vs. RHE) in alkaline pH, where the production of multi-carbon products is expected to be boosted.32,33 In the case of aldehydes, which undergo side reactions in alkaline media,9 electrolysis was performed in neutral potassium phosphate buffer (PPB) at −1.0 V vs. RHE as the optimum condition for the production of propylene (Table S3†). To avoid interference of the parasitic hydrogen evolution reaction (HER) in assessing reactivity, we compared production rates, instead of Faradaic efficiencies, of the carbonaceous products formed under different conditions. Additional information on the experimental conditions can be found in Note S7.†
Results and discussion
Routes to main C1 and C2 products and precursors
The main outcome of previous computational studies on the manually-analysed network for eCO2R to C1 and C2 products over Cu(111) or Cu(100) can be summarised as follows:15–20 CO2 adsorbs on the metal and reduces to CO, a precursor for methanol and methane (C1). Alternatively, CO dimerises to OCCO−, which has been claimed to be the rate-determining step in the formation of the C2 backbone.15–19 On Cu(100), this process is endergonic by 0.84 eV at −0.4 V vs. RHE, (with an activation barrier of 1.42 eV), but can be promoted on defective surfaces.34 Additionally, OCCO− can be stabilised by electrolyte cations35,36 and the electric field at the cathode.28,36,37 OCCO− is further protonated and then dehydrated to CCO upon a proton-coupled electron transfer step. The latter intermediate is subsequently hydrogenated to CH2CHO.1,38 If hydrogenolysis occurs on the remaining O atom, CH2CH is formed, leading to ethylene. If hydrogenation occurs on the α-C (CαH2CHO), ethanol is formed (Fig. S4 and S5†).Exploring C1–C2 coupling reactions
To unravel the most likely reaction routes towards C3 backbone formation, a sequential analysis combining available reports, experiments and theory was applied. The possible C1–C2 couplings shown in Fig. 2a and Tables S16–S18† emerge from combining 10 C1 (–CHxO* or –CHx*) and 70 C2 precursors that can be hydrocarbons (CHyCHz*–) or oxygenates (CHyOCHzO*–, CHyOCHz*–, CHyCHzO*–). This gives rise to oxygenate-to-oxygenate, hydrocarbon-to-oxygenate, and hydrocarbon-to-hydrocarbon C–C coupling reactions. Thus, six families of intermediates, namely C3Hx, 1-C3OHx, 1,3-C3O2Hx, 2-C3OHx, 1,2-C3O2Hx and 1,2,3-C3O3Hx, x = 0–8 (families 1–6 in Fig. 2a) are formed, where the prefix numbers show the position of the oxygenated functional group, irrespective of it having alcohol, alkoxy, aldehyde, or ketone character. Molecular fragments with carboxylate, carboxylic acid, ethers, or cyclic backbones were not considered, as these functionalities have not been found experimentally in the pool of C3 products.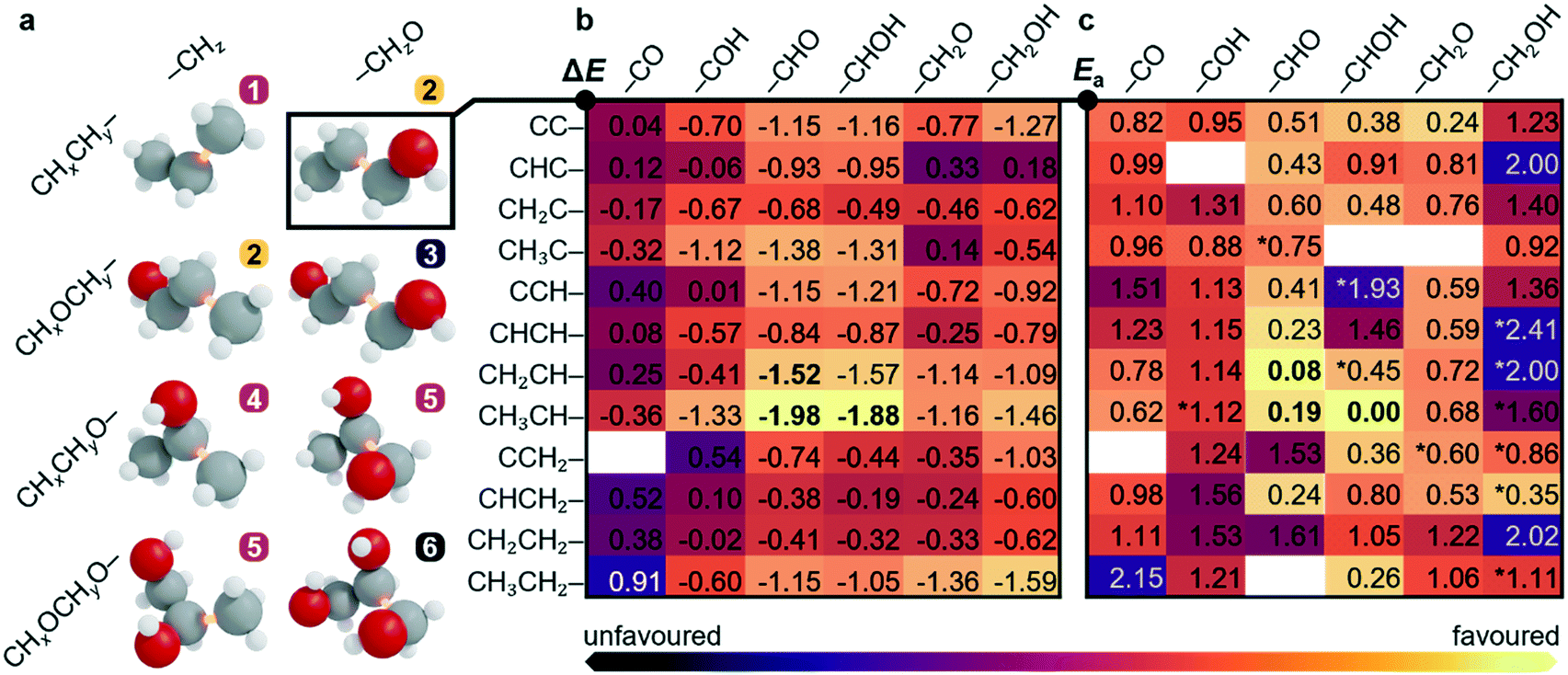 | ||
| Fig. 2 Screening process to narrow down the C1–C2 coupling steps. a CxHyOz backbones that can be obtained through C1–C2 couplings. Only the fully hydrogenated product is shown with the bond formed marked in orange. The numbers label six families of molecules and the colour stands for their abundance reported from CO2 reduction experiments, found either from the literature or this work. High, low, or zero relative abundance is shown in orange, purple, and black, respectively. Backbones 3 and 5 can be formed from two combinations of C1 and C2 intermediates. b and c Reaction and activation energies for CHxCHy–CHzO couplings between a C1 oxygenate and a C2 hydrocarbon, ΔE and Ea in eqn S3–S6.† Most likely steps indicated in bold. The full set of 10 C1 × 70 C2 coupling reactions is shown in Tables S16–S18.† Further details can be found in Notes S1–S2 and S8–S9.† | ||
We then compared the C3 products formed from the electrolysis of mixtures of CO with different C2 molecules (glyoxal, ethylene glycol, oxalate, acetate, ethanol, acetaldehyde) at open-circuit potential and −0.4 and −1.0 V vs. RHE (Tables 1 and S6 and S8–S9†). At −0.4 V vs. RHE, all these mixtures generate 1-propanol at rates much larger than the reduction of CO alone (Table S4†). Mixtures with ethylene glycol gave the highest yield, while those with ethanol and acetate gave the lowest. Propylene was not detected (detection limits of gaseous products are equivalent to 0.5 μmol cm−2 h−1, see ESI† Experimental and computational details). Overall, if a set of products with a given CxOy backbone is not observed experimentally, then such couplings can be considered unlikely, and the routes pruned from the network.
To further verify the nature of the active C1 fragment leading to 1-propanol, electrocatalytic tests of the C2 compounds with either methanol or formaldehyde (Table 1) were conducted. 1-Propanol was not detected in some of these experiments, though we observed allyl alcohol and propylene from the electrolysis of a formaldehyde and acetaldehyde mixture. Moreover, the reduction of CH2O itself produced only CH3OH and CH4 (Table 1 and S4†), but it does not produce C2 and C3 compounds as it is hardly broken into the more reactive CH2 and CHO species (Table S22†). CH3OH, on the other hand, was electrochemically inert (Tables S12 and S13†). The unique predominance of 1-propanol in experiments using CO indicates that *CO (or a derivative like *CHO) is instrumental in promoting 1-propanol formation. This is further confirmed by the absence of 1-propanol in experiments starting with CH2O or CH3OH (Table 1).
After considering the experimental input, we then switched to theory to explore the C1–C2 coupling reactions based on the reaction energies (ΔE, Table S16†), activation energies obtained by DFT-NEB (Ea, Table S17†), and complemented by the electrochemical driving force computed as the polarisation variation upon reaction (ΔΔQB in eqn S6, Table S18†). More favourable values are shown in brighter colours in Fig. 2b and c. The most likely candidates were then selected among all couplings, which reduced the set to CH2CH–CHO and CH3CH–CHO. In the following paragraphs, we describe how the different coupling families are retained or discarded during the analysis of the network based on abovementioned literature, experimental, and theoretical analyses:
 | ||
| Fig. 3 CH2CHO–CO coupling on OD-Cu models.34 a–c Initial, transition, and final state respectively. A surface cavity with high oxygen affinity assists the C–O bond breaking of the CH2CHO* precursor. A neighbouring polarised site, Cuδ+, weakly adsorbs CO. | ||
The activation barriers of the transition states associated with the formation of key C3 intermediates are sensitive to surface geometry and ensembles. Since defective Cu surfaces have been reported as selective to propanol formation,39,44 we assessed the role of defects on OD-Cu models34 for the concerted coupling of CH2CH(O)*–CO* to yield the simplest C3 precursor, CH2CHCO, Fig. 3. The Cu and CuOx structures were optimised for 10 ps through ab initio molecular dynamics and recurrent morphological motifs occured upon surface reconstruction.34 Out of twelve surface motifs assessed (Fig. S11†),34,46 an active site consisting of a surface cavity (Fig. 3a, centre) and a neighbouring Cuδ+ atom (Fig. 3a, right) is the most suitable for promoting the coupling. While CH2CHO* is trapped at the surface cavity, the high oxygen affinity of this site leads to the breaking of its C–O bond to give CH2CH* (Fig. 3b). On the other hand, CO adsorption is almost thermoneutral on the polarised copper site. Thus, the CH2CH* fragment can easily couple to the weakly bound CO* to form the C3 backbone (exergonic by 0.13 eV; Fig. 3c). In absence of polarized Cu sites (Fig. S11†), this step is endergonic by at least 0.6 eV, thus confirming the instrumental role of surface polarization. This coupling mechanism may explain the high selectivity toward 1-propanol (FE = 23% at −0.44 V vs. RHE) achieved on highly defective Cu surfaces containing a large number of surface cavities.11,39,44 This concept can also be extended to other key C1–C2 coupling reactions from moieties directly derived from CH2CHO and CO, such as CH2CH–CHO and CH3CH–CHO. Going beyond pure copper catalysts, we propose that intermetallic alloys containing high oxygen affinity elements coupled with weak CO binding sites could be highly selective to C3 as well. For instance, Cu–Ag alloys exhibited enhanced propanol selectivity depending on the silver atomic ratio,13,47 suggesting a CO spillover mechanism48 from Ag domains to facilitate the formation of CH2CH–CO.
Routes to C3 products
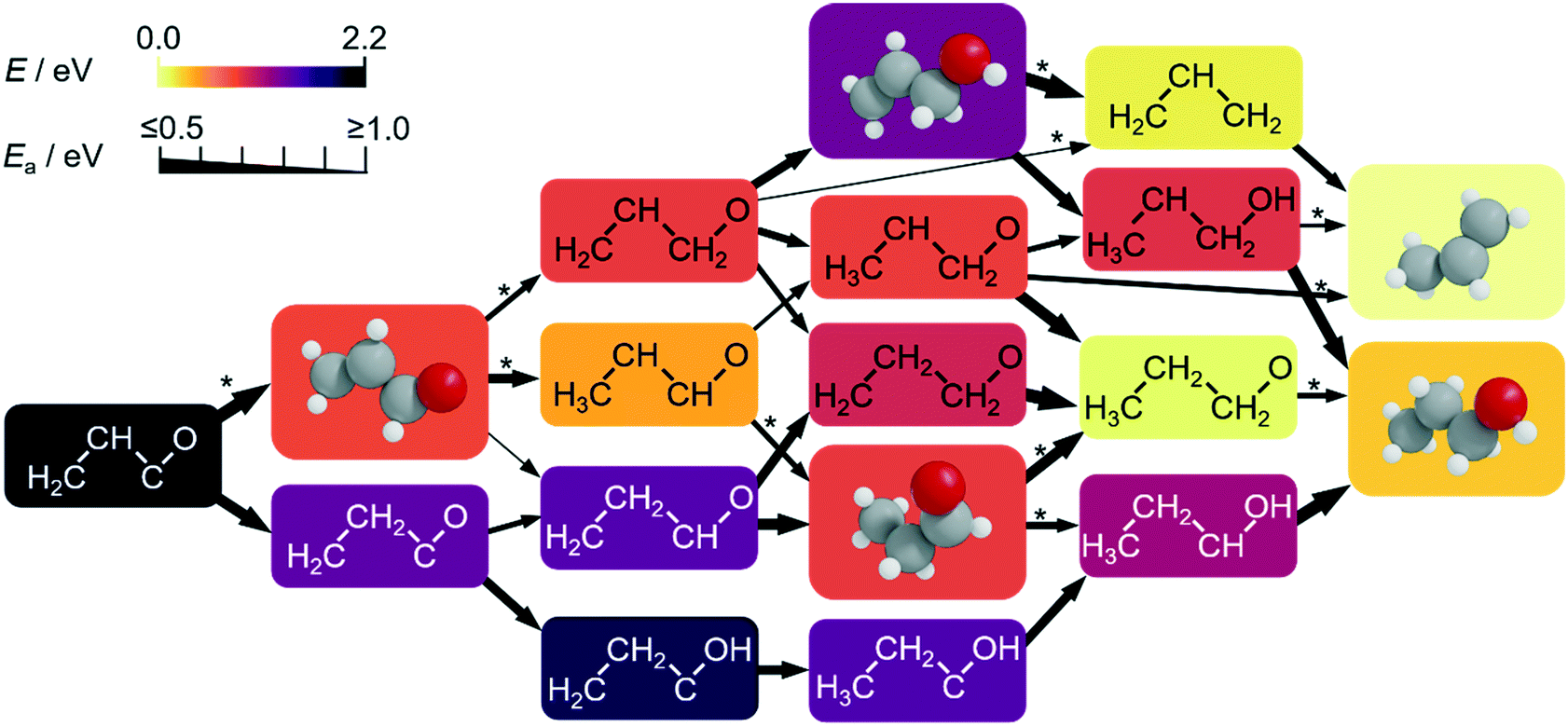 | ||
| Fig. 4 Computed subnetwork for CH2CHCO and CH2CHCHO conversion to propylene (C3H6) and 1-propanol (C3H7OH) at −0.4 V vs. RHE (full network in Fig. S12–S14†). The colours of the boxes scale with the relative DFT energy of their intermediates (eqn S3†). Relevant intermediates which can desorb and be used as probe molecules are drawn in 3D. We used allyl alcohol (C3H5OH) and propionaldehyde (C3H6O) as reactants in our experiments to confirm the pathways predicted by the network (Tables S23–S24†). The thickness of the arrows connecting the intermediates account for the activation energies (Ea, obtained by LSR. Those obtained explicitly by DFT are denoted by *). | ||
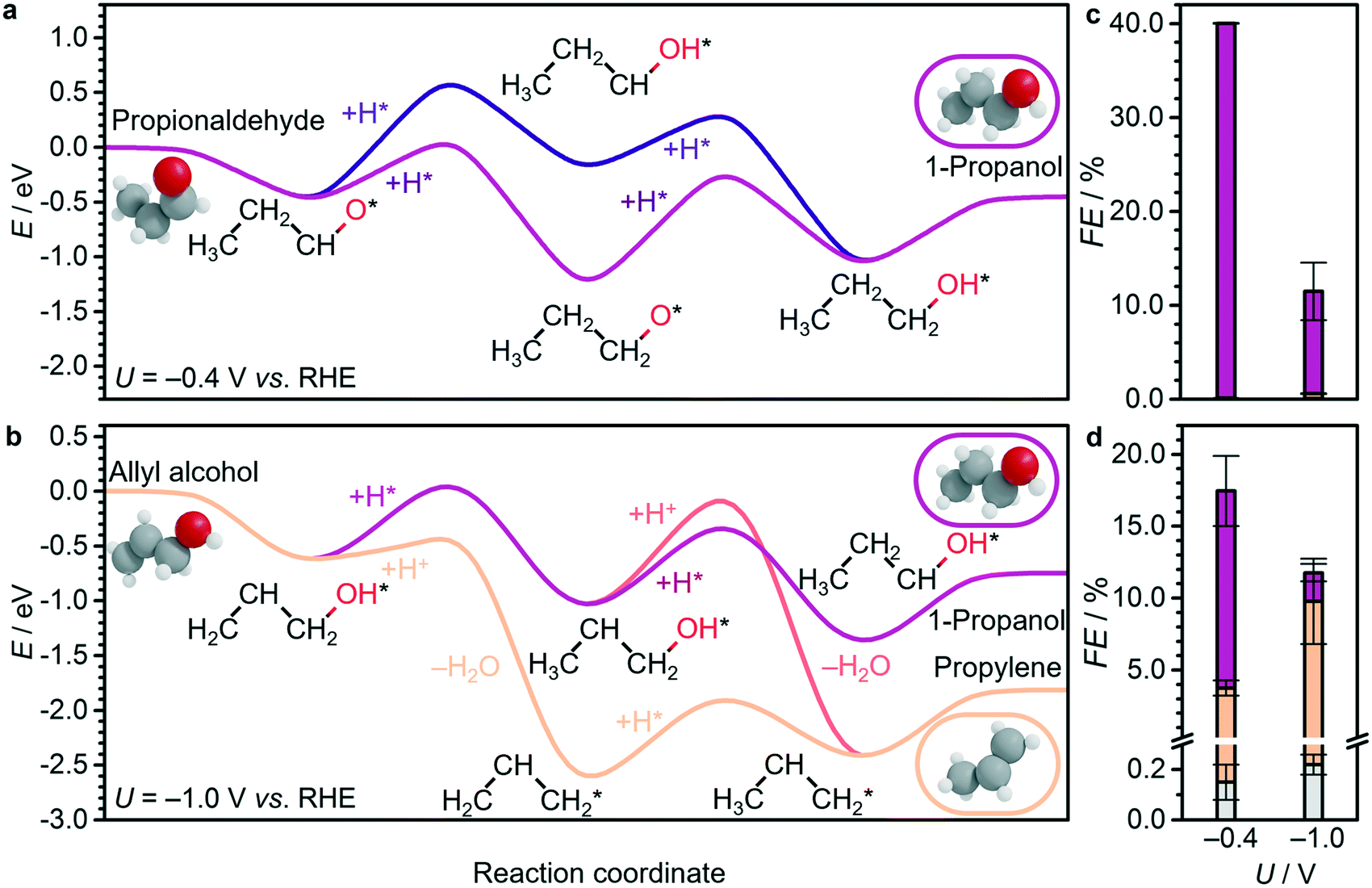 | ||
| Fig. 5 a and b Energy profiles for electrocatalytic reduction of key C3 compounds on Cu(100), using H2, CO2, and H2O as thermodynamic sinks, and shifting the energy reference to make a propionaldehyde and b allyl alcohol the zero. Corresponding products for experimental electroreduction of c propionaldehyde and d allyl alcohol at −0.4 V and −1.0 V vs. RHE on OD-Cu: propane (grey), propylene (orange), and 1-propanol (purple). Full product distributions are shown in Tables S23 and S24.† Other energy profiles at 0.0 V, −0.4 V, and −1.0 V vs. RHE are shown in Fig. S15-S17.† The (*) symbol refers to species adsorbed on the surface. Detailed DFT values can be found in Tables S20–S21.† | ||
Therefore, the production of propylene could be traced to the allyl alkoxy (CH2CHCH2O) intermediate, which is also a direct precursor of allyl alcohol. This proposition was verified experimentally by reducing allyl alcohol on OD-Cu (Fig. 5d), which gave noticeable amounts of propylene as theoretically predicted. Moreover, allyl alcohol (1.97 μmol cm−2 h−1) was detected alongside propylene (1.44 μmol cm−2 h−1) from the reduction of a mixture of acetaldehyde and formaldehyde (Table 1). In this combination, the most likely path occurs when acetaldehyde loses an acidic α-hydrogen (Hα–CH2CHO)9 to form CH2CHO, which dehydrates to form CH2CH. The latter compound reacts with CH2O to form CH2CHCH2O* (Fig. 4 and S12–S14†) which is mainly selective towards allyl alcohol and propylene, but not 1-propanol (Table 1). Interestingly, we note that CO2 reduction produced 1-propanol (FE = 4.4%) and allyl alcohol (FE = 1.2%) (Fig. 1), while propylene was absent. This can be rationalised by a mild eCO2R interface alkalinisation, which occurs under reaction conditions,45 favouring the desorption of allyl alkoxy (protonated in solution into allyl alcohol) and thus preventing propylene synthesis. Overall, these observations strongly suggest the key role of allyl alcohol in the route to propylene.
From a broader perspective, the low activity of Cu catalysts for eCO2R to C3 compounds, particularly propylene, could be improved through engineering at different scales. Currently, the most explored approaches to promote multi-carbon products include engineering catalyst surfaces with a high density of defects to improve activity, and optimizing the electrolyte and reactor conditions to alleviate mass transport limitations and tuning the environment at the electrode-electrolyte interface. Modifications at the process level could benefit from three different approaches: (i) one single reactor recycling C2 (or C1) eCO2R products to ensure a high concentration of active intermediates; (ii) independently optimised catalysts and reactors to produce C1 (Cu or Ag-based catalysts) and C2 (on an oxide-derived Cu catalyst) intermediates, which mix in a third reactor dedicated to the coupling to form the C3 backbone or alloys containing close domains of both; (iii) a process able to produce the relevant intermediate allyl alcohol (for which an effective catalyst is not yet known) which is then converted to propylene in a second unit. Although there are some experimental indications in the literature of the potential of strategies (i) and (ii),13 the detailed understanding of the reaction network and the elucidation of key intermediates presented in this work ultimately serve to direct future works toward realizing these solutions.
Conclusions
In conclusion, we have performed an integrated mechanistic analysis of the eCO2R to C3 products with all the individual steps available in our open database.49 Methodological implementations including structural graph network generation, fast energy screenings, and network pruning of irrelevant paths through experimental input allow the effective sampling of the complex C3 network pointing out the difficulties found when only a part of the reaction network is sampled.50 C2 and C3 products were found to share a common precursor, CH2CHO*. Our findings rationalise the generally observed low selectivity of eCO2R toward C3 products, as well as their enhancement on nanostructured Cu catalysts: (i) C3 backbones are formed via the sluggish coupling of CO or CHO with CH2CH*, preferentially at defects and (ii) all C3 precursors end up containing at least one O atom, i.e., CHx + C2Hy couplings are highly unlikely. The most stable mono-oxygenated intermediate CH2CHCHO* gives access to propylene, propionaldehyde, and 1-propanol. The inaccessible allyl alkoxy intermediate is identified as the most likely kinetic trap preventing propylene production as indicated by simulations and further reinforced with the electrolysis of allyl alcohol leading to propylene. Our mechanistic understanding paves the way towards the development of advanced electrocatalysts that promote C3 products, particularly alkenes.Author contributions
S. P.-G., Software, methodology, data curation, investigation, visualisation, writing – original draft. F. L. P. V., L. R. L. T., Methodology, data curation, investigation, visualisation, writing – original draft. R. G.-M., A. J. M., methodology, data curation, investigation, visualisation, validation, supervision, writing – original draft. F. D., Data curation, investigation, visualisation, writing – review & editing. B. S. Y., J. P.-R., N. L., Conceptualisation, funding acquisition, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financed by the Spanish Ministry of Science and Innovation (RTI2018-101394-B-I00, Severo Ochoa Centre of Excellence CEX2019-000925-S 10.13039/501100011033), ETH Research Grant (ETH-47 19-1), the National University of Singapore Flagship Green Energy Program (R143-000-A64-114, R143-000-A55-733 and R143-000-A55-646), and Ministry of Education of Singapore (R143-000-B52-114). This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation. The Barcelona Supercomputing Centre – MareNostrum (BSC-RES) is acknowledged for providing generous computational resources. The authors thank Mavis Pei Lin Kang from NUS for assisting with XRD analyses.Notes and references
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050 RSC.
- B. Schmid, C. Reller, S. Neubauer, M. Fleischer, R. Dorta and G. Schmid, Catalysts, 2017, 7, 161 CrossRef.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C. T. Dinh, T. T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C. J. Sun, T. Li, J. V. Muntean, R. E. Winans, D. J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. P. Veenstra, S. T.-J. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López, J. Pérez-Ramírez and B. S. Yeo, Angew. Chem., Int. Ed., 2020, 59, 21072–21079 CrossRef CAS PubMed.
- D. Gao, I. Sinev, F. Scholten, R. M. Arán-Ais, N. J. Divins, K. Kvashnina, J. Timoshenko and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2019, 58, 17047–17053 CrossRef CAS PubMed.
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. De Luna, C. T. Dinh, T. T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S. H. Hsieh, Y. S. Liu, G. A. Botton, W. F. Pong, X. Du, J. Guo, T. K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef PubMed.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS PubMed.
- X. Wang, Z. Wang, T. T. Zhuang, C. T. Dinh, J. Li, D. H. Nam, F. Li, C. W. Huang, C. S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S. C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- D. Ren, J. Fong and B. S. Yeo, Nat. Commun., 2018, 9, 925 CrossRef PubMed.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- Y. Huang, Y. Chen, T. Cheng, L.-W. Wang and W. A. Goddard, ACS Energy Lett., 2018, 3, 2983–2988 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- Q. Li, R. García-Muelas and N. López, Nat. Commun., 2018, 9, 526 CrossRef PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- N. Almora-Barrios, G. Carchini, P. Błoński and N. López, J. Chem. Theory Comput., 2014, 10, 5002–5009 CrossRef CAS PubMed.
- M. Garcia-Ratés and N. López, J. Chem. Theory Comput., 2016, 12, 1331–1341 CrossRef PubMed.
- M. Garcia-Ratés, R. García-Muelas and N. López, J. Phys. Chem. C, 2017, 121, 13803–13809 CrossRef.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- M. García-Mota, B. Bridier, J. Pérez-Ramírez and N. López, J. Catal., 2010, 273, 92–102 CrossRef.
- S. Pablo-García, M. Álvarez-Moreno and N. López, Int. J. Quantum Chem., 2021, 121, e26382 CrossRef.
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- J.-J. Lv, M. Jouny, W. Luc, W. Zhu, J.-J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef PubMed.
- F. Dattila, R. García-Muelas and N. López, ACS Energy Lett., 2020, 5, 3176–3184 CrossRef CAS.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- Y. Pang, J. Li, Z. Wang, C. S. Tan, P. L. Hsieh, T. T. Zhuang, Z. Q. Liang, C. Zou, X. Wang, P. De Luna, J. P. Edwards, Y. Xu, F. Li, C. T. Dinh, M. Zhong, Y. Lou, D. Wu, L. J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS.
- H. Xiao, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans., 1989, 85, 2309–2326 RSC.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS PubMed.
- T. Zhuang, Y. Pang, Z. Liang, Z. Wang, Y. Li, C. Tan, J. Li, C. T. Dinh, P. De Luna, P. L. Hsieh, T. Burdyny, H. H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D. H. Nam, Z. Y. Wu, Y. R. Zheng, A. H. Ip, H. Tan, L. J. Chen, S. H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- F. L. P. Veenstra, N. Ackerl, A. J. Martín and J. Pérez-Ramírez, Chem, 2020, 1–16 Search PubMed.
- F. Dattila, R. García-Muelas and N. López, Dataset associated to: Active and Selective Ensembles in Oxide- Derived Copper Catalysts for CO2 Reduction, ioChem-BD, 2020, DOI:10.19061/iochem-bd-1-165.
- A. Herzog, A. Bergmann, H. S. Jeon, J. Timoshenko, S. Kühl, C. Rettenmaier, M. Luna Lopez, F. T. Haase and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2021, 60, 7426–7435 CrossRef CAS PubMed.
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, ACS Catal., 2021, 11, 4456–4463 CrossRef CAS.
- R. García-Muelas, S. Pablo-García and N. López, Dataset associated to: Mechanistic Routes toward C3 products in Copper-Catalyzed CO2 Electroreduction, 2020, DOI:10.19061/iochem-bd-1-175.
- M. T. Tang, H.-J. Peng, J. H. Stenlid and F. Abild-Pedersen, J. Phys. Chem. C, 2021, 125, 26437–26447 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, supplementary notes, figures, and tables. See DOI: 10.1039/d1cy01423d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |