
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recycling and valorization of LDPE: direct transformation into highly ordered doped-carbon materials and their application as electro-catalysts for the oxygen reduction reaction†
J.
Castelo-Quibén‡
 *,
E.
Bailón-García
*,
A. I.
Moral-Rodríguez
,
F.
Carrasco-Marín
and
A. F.
Pérez-Cadenas
*,
E.
Bailón-García
*,
A. I.
Moral-Rodríguez
,
F.
Carrasco-Marín
and
A. F.
Pérez-Cadenas
Carbon Materials Research Group, Department of Inorganic Chemistry, Faculty of Sciences, University of Granada, Campus Fuentenueva s/n, 18071, Granada, Spain. E-mail: estherbg@ugr.es; jesicacastelo@ugr.es
First published on 16th December 2021
Abstract
The energy demand and the environmental situation make the development of advanced catalysts for energy applications necessary. Considering the large volumes of plastic waste, the transformation of organic polymers into advanced carbon functional materials that are able to accomplish the oxygen reduction reaction via the desired 4-electron pathway is proposed as an integrated environmental remediation, in which plastic pollutants are converted into catalysts for fuel cells. Carbon-based electro-catalysts were obtained by pyrolyzing low-density polyethylene at low temperature by an easy one-step method that involves the generation of autogenous pressure which is the responsible for the spherical shape as well as the high degree of graphitization. The addition of transition metals (Fe, Co or Ni) modifies the carbonization process and CNFs emerge. The metal-free material leads to a purely 2e− pathway; however, the presence of metals improves all electrochemical parameters and the desired 4e−pathway is achieved. The graphitization degree, the metal dispersion and the presence of CNFs are the key factors for the ORR performance.
1. Introduction
The unstoppable growth of the energy demand, as well as environmental pollution, has intensely encouraged research on alternative and highly efficient energy conversion and storage systems and must be adequately addressed to maintain the sustainability of our environment. During the last few years, great efforts have been made to develop clean energy systems able to supply the current demand such as supercapacitors, metal–air batteries or fuel cells.1 In this regard, the production of electrical energy from chemical reactions by using fuel cells, especially the ones refueled with hydrogen from renewable sources, is generally considered one of the most promising solutions because of their competitive advantages, such as zero-emission, high efficiency, fast refueling, and low upfront cost.2In a typical proton-exchange membrane fuel cell (PEMFC), fuel molecules (e.g., hydrogen) are oxidized on the anode, and oxygen gas is reduced on the cathode, outputting electric energy with pure water and heat as the only by-products.3–5 Unfortunately, the difficulty in O2 activation and O–O bond cleavage causes sluggish kinetics of the oxygen reduction reaction (ORR) on the cathode; thus, great efforts are still being taken to improve the ORR kinetics.4
At present, platinum and its alloys are generally considered to be the most efficient ORR catalysts.6 Nevertheless, not only the high cost of Pt greatly hampers further large-scale adoption of PEMFCs but also its low tolerance to fuel molecules. Taking this into consideration, special attention has been paid during the last few decades to the development of low-Pt-content catalysts.7,8 Besides, alternative catalysts based on non-precious metals and metal-free materials are more interesting proposals and they are being actively studied.9,10 In this regard, carbon-based materials are considered as optimal candidates to accomplish the oxygen reduction reaction11,12 since carbon materials present numerous advantages such as high surface area, electrical conductivity, high mechanical strength and high electrochemical corrosion resistance. Moreover, their versatility facilitates the possibility of modifying on demand their textural, chemical and electrical properties by tuning the carbon precursor as well as by doping appropriately in accordance with the required applications.13,14 In this sense, H. Peng et al.15 prepared Mn, Fe, Co, Ni, and Cu-doped carbon materials derived from polyaniline and melamine and analyzed the effect of these transition metals on their structure and their performance as ORR catalysts. They found that transition metals enhance the ORR catalytic performance with decreasing activity in the order Fe > Co > Cu > Mn > Ni. This enhanced activity was ascribed to the joint effect of the active N content, metal residue, and the surface area and pore structure. A. Abdelwahab et al.16 synthesized carbon aerogels doped with Fe, Co and Ni by polymerization of resorcinol and formaldehyde in the presence of metal precursor salts. The metal nanoparticles well-dispersed on the carbon matrix catalyze the formation of graphitic clusters around them. The presence of these small and well-developed graphitic domains improves the electro-catalytic oxygen reduction. In turn, Y. Jing et al.17 prepared MOF-derived Co, Fe, and Ni co-doped N-enriched hollow carbon. They also observed that the addition of Fe and Ni enhances the graphitization degree and the specific surface area of the electrocatalyst. This high graphitization degree, the hollow carbon structure, and Co, Fe, and Ni co-doping effects contribute to the enhanced ORR performance. This demonstrates the potential of using transition metal doped carbon materials as efficient electrocatalysts for the ORR.
Additionally, obtaining these carbon materials from waste such as plastics would be ideal from an environmental and economic point of view. Nowadays, recycling plastic waste is not a bright scenario. In the 1990s in the USA, 2% of plastic waste was recycled, while 18% of it was burned with energy recovered but, nevertheless, 80% of plastic waste was landfilled. Even though the overall picture has been improving, the current data are far from encouraging. In 2017, the percentages increased to 8% for recycling, with 16% devoted to energy recovery but, similarly, most of the plastic waste (76%) has continued going into landfills.18 Unfortunately, it is still insufficient as a solution, especially taking into account that polymers need hundreds of years to degrade under normal environmental conditions. Because of their stability against the weather, microplastics are also present in oceans.19 In addition, it is estimated that in the US alone, around 100 tons of microplastics might enter the oceans annually.20 Not only that, but also recent studies have reported microplastics in the Antarctic marine system.21 These particles are ingested by a wide range of organisms in the flora and fauna of the marine environment developing toxicity.19 The present worrying situation encourages research on new methods for plastic recycling able to avoid the shortcomings of the current transformation.
Owing to this overview, developing advanced materials from plastic waste is certainly an attractive idea22,23 from an environmental and economic point of view. In this regard, we propose the direct transformation of polyethylene into carbon materials by a simple one-step pyrolysis method. Taking into account that the pyrolysis process is carried out in a closed reactor, it gives a better yield and also, no emissions are produced. Furthermore, the autogenous pressure generated during the process induces a high degree of graphitization. The addition of metals such as iron, nickel or cobalt also catalyzes the formation of carbon nanofibers which leads to a very positive impact on the electrochemical behavior.24 These metals are among the most abundant in the Earth's crust, and therefore, competitively priced. Hence, these properties make them excellent candidates for use as an electro-catalyst for the ORR. Therefore, in the present work, we demonstrate the high electrocatalytic performance in the oxygen reduction reaction of carbon nanocomposites with a high degree of graphitization obtained by the catalytic pyrolysis of plastic waste under high self-pressure conditions.
2. Experimental
2.1. Synthesis of carbon nanocomposites
Three different carbon (C)–metal composites (C–M) were obtained by pyrolysis of low-density polyethylene (LDPE) (Mw ∼76 kDa, Sigma-Aldrich, cat. N. 428043) catalyzed by the corresponding acetate of iron, cobalt or nickel. Also, a metal-free material was prepared under the same experimental conditions for comparative purposes. The samples thus obtained were labeled as C–Fe, C–Co, C–Ni, and C respectively. The average density and melting point of LDPE were 0.925 g cm−3 and 116 °C. More characterization details can be found elsewhere.25–27The used methodology was the following: firstly, 0.5 g of LDPE powder was placed and treated at 700 °C in a closed hand-made Hastelloy® reactor of 25 mL capacity in the absence of any solvent. The reactor was purged with Ar from the top of the vessel and then closed without any flux of inert gas and subjected to a thermal treatment in an oven. The heating rate was 10 °C min−1 with a dwell time of 2 hours at the target temperature. The yield percentage of this synthesis was used to calculate the theoretical amount of the corresponding metal precursor to obtain C–metal composites with a 10 wt% metal loading. The carbon–metal composites were obtained following the same procedure, except that the corresponding metal acetate was physically mixed with LDPE and placed into the reactor. The carbon yield obtained after the pyrolysis process was calculated as the weight of carbon obtained per weight of LDPE used.
2.2. Textural and chemical characterization
The porous texture was analyzed by N2 adsorption–desorption at −196 °C. Before measuring the gas adsorption isotherms, the samples were outgassed overnight at 110 °C under high vacuum (10−6 mbar). The BET equation was applied to the N2 adsorption data from which the specific surface area SBET was obtained. The Dubinin–Radushkevich (DR) equation was also applied to the adsorption data in order to obtain the corresponding micropore volume (W0) and the micropore mean width (L0). The total pore volume (V0.95) is obtained from the N2 adsorption isotherms at 0.95 relative pressure. Finally, the mesopore volume (VMESO) is calculated by applying the Gurvich rule.The texture and morphology of the samples were analyzed by scanning electron microscopy (SEM) and high-resolution electron microscopy (HRTEM) using an FEI microscope model Quanta 400 and an FEI Titan G2 microscope, respectively.
The crystalline phases of the obtained materials were analyzed by X-ray diffraction using a Bruker D8 Venture X-ray diffractometer with Cu Kα radiation. The XRD patterns were recorded in the 2θ range from 6° to 77°. The average crystal size was estimated by the application of the Debye–Scherrer equation.
Raman spectra were recorded using a micro-Raman JASCO NRS-5100 dispersive spectrophotometer with a 532 nm laser line.
The surface chemistry was studied by X-ray photoelectron spectroscopy (XPS) by using a Kratos Axis Ultra-DLD spectrometer equipped with a hemispherical electron analyzer connected to a DLD (delay-line detector) and an Al Kα monochromator with a power of 600 W. The X-ray source is a Mg/Al double anode with a power of 450 W.
The total metal content was determined by thermogravimetric analysis (TGA) and inductively coupled plasma optical emission spectrometry (ICP-OES) using a TGA/DSC1 thermogravimetric analyzer from Mettler-Toledo and an Optima 8300 ICP-OES from Perkin-Elmer, respectively.
2.3. Electro-chemical characterization
The carbon nanocomposites were electrochemically characterized with a Biologic VMP multichannel potentiostat using a standard three-electrode cell. The carbon nanocomposites, a platinum wire and Ag/AgCl were used as the cathode, the counter electrode and the reference electrode, respectively. To evaluate the electro-catalytic activity, a rotating disk electrode (RDE) (Metrohm Autolab RDE-2, 3 mm glassy carbon tip) was used as a working electrode, on which the carbon materials were deposited.In order to study the behavior of each sample in an electrolyte containing oxygen or in the absence of oxygen, cyclic voltammetry experiments (CV) were performed in O2-saturated 0.1 M KOH solution or a 0.1 M KOH solution completely degassed (by bubbling N2). The sweeping potential rate was 50 mV s−1, from 0.4 V to −0.8 V (vs. Ag/AgCl) while the RDE was rotating at 1000 rpm. To prepare the working electrode, 5 mg of sample were dispersed in 1 mL of a solution previously prepared which contains Nafion (5%) and distilled water in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (v:v) ratio and sonicated for 30 min until a homogeneous and stable ink was obtained; after that, 10 μL of this ink was loaded on the RDE tip and dried under infrared radiation. The glassy carbon tip was previously polished with alumina powder. Afterward, linear sweep voltammetry experiments (LSV) were performed in an O2-saturated 0.1 M KOH solution at different rotation rates from 500 rpm to 4000 rpm. The experiments were conducted from 0.4 V to −0.8 V (Ag/AgCl) at a sweep rate of 5 mV s−1.
:9 (v:v) ratio and sonicated for 30 min until a homogeneous and stable ink was obtained; after that, 10 μL of this ink was loaded on the RDE tip and dried under infrared radiation. The glassy carbon tip was previously polished with alumina powder. Afterward, linear sweep voltammetry experiments (LSV) were performed in an O2-saturated 0.1 M KOH solution at different rotation rates from 500 rpm to 4000 rpm. The experiments were conducted from 0.4 V to −0.8 V (Ag/AgCl) at a sweep rate of 5 mV s−1.
The experimental data were fitted to the Koutecky–Levich model to evaluate the electro-catalytic performance of the samples and to calculate the number of electrons transferred for each of them as well as the kinetic density current (jk) and the onset potential (EONSET).
3. Results and discussion
3.1. Morphological characterization
All materials were obtained as a solid powder and no presence of any liquid phase was found. Although the formation of gas products is probable, this was not analyzed during the synthesis. Cheng et al.28 studied the pyrolysis of polyethylene in a larger pressure range at different temperatures (from 380 °C to 470 °C) and they did not find a solid phase but rather a liquid fraction, composed mainly of paraffins and olefins. Also, they studied the gas phase whose analysis revealed the formation of short-chain hydrocarbons, primarily, ethane, ethylene, propane, and propylene. The product distribution changed accordingly with the increment of pressure and temperature. The higher the temperature, the higher the cracking degree. Similar findings were reported by Marcilla et al.,29 where the presence of a solid phase was also not found. Also, in the absence of a cracking catalyst, the temperature favors the breakage of the carbon chains to light hydrocarbons. On the other hand, Kong and Zhang have reported the catalytic pyrolysis of LDPE at 700 °C where the dark solid obtained was solely composed of carbon nanotubes with a low degree of graphitization.30 These findings demonstrate that the temperature for the polyethylene graphitization must be higher than 500 °C under pressured conditions.In the present work, smooth and highly graphitic carbon microspheres that are greatly homogeneous in size and shape were obtained in the absence of a catalyst after the pyrolysis of LDPE at 700 °C and under autogenous pressure (sample C, Fig. 1a and 2a). Nevertheless, in the presence of metals such as iron, cobalt or nickel (samples labeled as C–Fe, C–Co, and C–Ni, respectively), carbon nanofibers (CNFs) emerge (Fig. 1 and 2). Note that the morphology and CNF growth are deeply dependent on the doping metal used. Carbon spheres were also obtained in C–Fe (Fig. 1b and 2b) but short CNFs emerge on the surface of these spheres. A lower number of spheres are clearly obtained using Co as the doping metal but, in contrast, a higher amount of and longer CNFs are observed (Fig. 1c and 2c). Carbon spheres were not observed in C–Ni but agglomeration of nanofibers and fused carbon nanoparticles were obtained (Fig. 1d and 2d).
 | ||
| Fig. 1 SEM images of pure (C) and doped (C–Fe, C–Co and C–Ni) samples obtained by LDPE pyrolysis. | ||
 | ||
| Fig. 2 TEM and HRTEM images of pure (C) and doped (C–Fe, C–Co and C–Ni) samples obtained by LDPE pyrolysis. | ||
The carbon yield obtained was 36.1, 42.6, 47.7 and 49.3% for the non-catalytic and Ni, Co and Fe catalyzed pyrolysis, respectively. Consequently, the total metal content is, in all cases, lower than the theoretically expected (10 wt%) due to the higher yield of catalyzed pyrolysis and varies between each sample (7.8, 6.6 and 3.0, wt% for C–Ni, C–Co, and C–Fe, respectively, Table 1). This indicates that the introduction of a catalyst in the self-pressure pyrolysis of LDPE clearly favors carbon fixation, but this carbon fixation and CNF obtention depends on the metal used.
| Sample | S BET | W 0 (N2) | L 0 (N2) | V 0.95 | V MESO | M TGA | M ICP |
|---|---|---|---|---|---|---|---|
| m2 g−1 | cm3 g−1 | nm | cm3 g−1 | wt% | |||
| C | 7 | 0.001 | — | 0.010 | 0.008 | — | — |
| C–Fe | 30 | 0.015 | 2.55 | 0.080 | 0.065 | 3.0 | 2.8 |
| C–Co | 36 | 0.011 | 2.09 | 0.104 | 0.093 | 6.6 | 6.5 |
| C–Ni | 56 | 0.020 | 1.67 | 0.142 | 0.118 | 7.8 | 8.0 |
Therefore, the presence and type of metal seems to affect the LDPE cracking, carbon fixation and morphology and CNF growth. Generally, the catalytic pyrolysis of plastic waste increases the gaseous fraction yield and decreases the liquid fraction.31 Yu. V. Lugovoy et al.32 observed that the gaseous pyrolysis products (the C1–C3 hydrocarbon production) decrease in the following order of the iron-subgroup metals: Ni > Co > Fe > non-catalytic process and therefore, the solid fraction increases in this sense. Similar results were obtained in the present work, with the carbon yield decreasing in the order Fe > Co > Ni; however, the amount of carbon obtained from the catalytic pyrolysis was always higher than that obtained from the non-catalytic one and the morphology of the carbon fixed is also clearly different. Fe, Co and Ni favor the cracking of the plastic and increases the gas fraction, but they are also known as the most effective catalysts for catalytic chemical vapor deposition (CCVD) growth of carbon nanofibers and nanotubes.33,34 CCVD is a process which involves pyrolysis of a carbon source on catalyst nanoparticles in the temperature range of 600–1100 °C. Thus, the presence of metals in our system induces the deposition of the above-mentioned light hydrocarbons on the metal particles, developing the CNFs probably by in situ CCVD, and for this reason, the carbon matrix is developed in a large extent. However, the catalytic effect of Fe, Ni and Co on the CNF production is quite different. L. M. Hoyos-Palacio et al.35 observed that the morphological effects and efficiency of the CCVD of carbon nanostructures are dependent on the catalyst type. Low quality structures are mainly obtained with Fe nanoparticles, whereas cobalt promotes small diameters in carbon nanotubes and Ni promotes the best yield during the CCVD process.
In brief, Fe favors the LDPE cracking and increase of the gas fraction, but it is a catalyst for nanofiber growth, so a higher yield of carbon is obtained compared to non-catalyzed pyrolysis. The amount of the gaseous fraction increases using Co and Ni and therefore, a higher number of CNFs are obtained, losing the spherical morphology observed for the non-catalyzed and Fe-catalyzed samples (Fig. 1 and 2). However, due to this higher gasification, the carbon yield is lower than that obtained for the Fe-doped sample, and mostly nanofibers or agglomerated nanoparticles are observed. It is important to highlight that other studies did not report the presence of CNFs using a similar amount of cobalt, iron or nickel when the metals are dispersed into another polymeric carbon matrix such as carbon gels.16,36–38
Also note that highly graphitic carbon is obtained in all cases despite the different morphologies (spheres, CNFs or nanoparticles) as observed by HRTEM (Fig. 2) where the graphitization degree is appreciated in all samples. Details of microspheres' edges are depicted in Fig. 2a (inset), where the graphitic layers of the C sample can be observed. Fig. 2b displays, for C–Fe sample, the carbon microspheres, as well as the carbon nanofibers and the iron particles (responsible for having developed these CNFs) that are highly dispersed throughout the carbon material. Fast Fourier transform (FFT) analysis on one of the fibers (inset of Fig. 2b) shows a diffraction ring of 5.921 nm−1 in diameter, indicating a d-spacing equal to 0.33 nm that corresponds to the interplanar spacing of the (002) plane of the graphite hexagonal structure.39 Along the same lines, Fig. 2c shows the CNFs obtained by means of the cobalt particles. These nanofibers are significantly longer than the ones obtained with iron; such an observation was already noticed in the SEM images (Fig. 1). Additionally, here, the graphitization around the cobalt particles can be appreciated (Fig. 2c top inset), where the hexagonal phase (hcp) of Co0 (0.191 nm d-spacing) was also found. A good dispersion of the metal phase throughout the carbon matrix (Fig. 2d) is observed in C–Ni sample and also, it has developed nanofibers in which it can be appreciated the footprint generated during their growing (Fig. 2d top inset). Highly graphitic clusters are also observed in the fused carbon nanoparticles (Fig. 2d bottom inset). Finally, it is also important to highlight that most of the metal particles in the C–Ni and C–Fe samples are on the carbon surface whereas most of the Co particles are embedded in the carbon matrix (Fig. 2).
It is important to remark that an easy one-stage method is proposed in this manuscript to obtain CNF-based catalysts by the simple and direct pyrolysis at moderate temperature (700 °C) of a mixture of plastic waste and a transition metal precursor salt. Several authors40–42 have also obtained CNF/CNT-based catalysts. Nonetheless, a more complex two-stage process is required. In the first stage, plastic waste is pyrolyzed at 500–600 °C and, in the second stage, the non-condensable gases are converted into multi-walled CNTs over a transition metal-based catalyst at a higher temperature (800 °C).
3.2. Textural characterization
The textural properties were studied by analyzing the N2 adsorption–desorption isotherms (Fig. S1†), and the corresponding results are summarized in Table 1.The C sample shows a type II isotherm (IUPAC classification) characteristic of a non-porous solid. The low surface area arises just from the external surface of the microspheres. These data are in good agreement with the SEM and TEM analysis (Fig. 1 and 2) which show very smooth microspheres and the presence of pores was not perceptible. The presence of metals during the LDPE pyrolysis affects the morphology of the carbon samples and consequently, as can be observed from Fig. S1,† the textural properties. The metal-doped C samples present a type I–IV isotherm typical for micro–mesoporous materials, where the adsorption in micropores takes place at low relative pressures, but with the increase of P/P0, the adsorbed N2 also increases along with the formation of a hysteresis cycle that reveals the presence of mesopores. The mentioned increment in the surface area comes from the CNFs and their crosslinking which causes the formation of the pores. The development of carbon nanofibers on the carbon sphere surface in the C–Fe sample leads to the increment of surface area mainly by the generation of mesopores (Vmeso = 0.065 cm3 g−1). CNF growth and crosslinking are higher in the case of C–Co and, as a result, the wider porosity (meso and macroporosity) increases as denoted by the rapid increase of N2 adsorption at relative pressures close to 1.43 Nonetheless, in the case of C–Ni, the graphitic clusters (that mainly constitute the carbon matrix) better contribute to the porosity. Note that a well-defined hysteresis loop is observed in this sample denoting the development of mesoporosity, probably due to the interparticle pores created by fusion of carbon nanoparticles as observed by TEM and SEM, where the roughness, and therefore the pores, of the matrix are perceptible.
3.3. XRD and Raman spectroscopy
The graphitization degree of the samples observed by HRTEM is in good agreement with the XRD and Raman spectroscopy data (Fig. 3 and 4 and Table 2). X-Ray diffractograms present two intense peaks at 2θ = 26.0° and 43.8° for all samples which correspond to the graphite planes (002) and (100) respectively (JCPDS # 89-8487), denoting the high graphitization degree of the samples.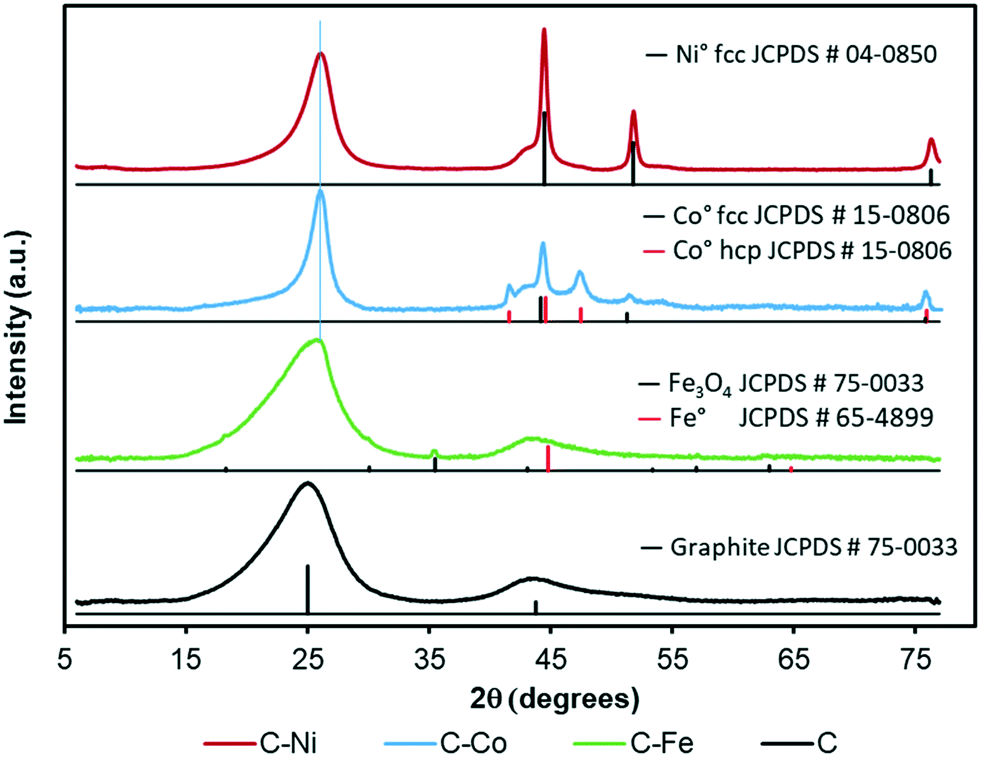 | ||
| Fig. 3 XRD patterns of the catalysts and the corresponding JCPDS card assignments. | ||
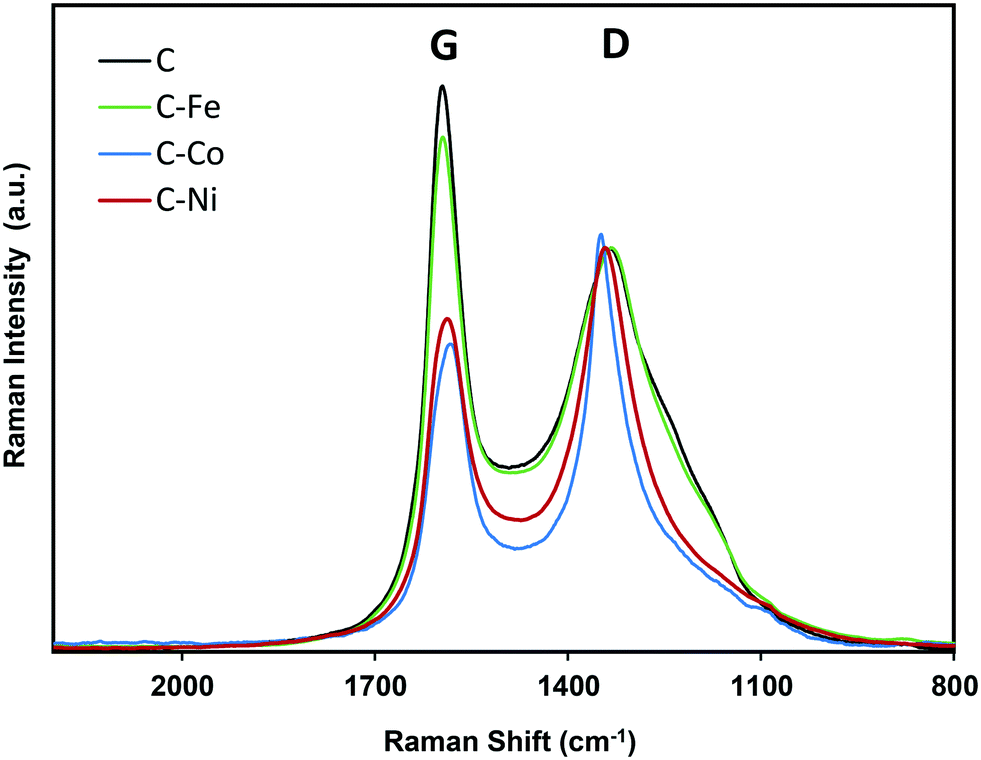 | ||
| Fig. 4 Raman spectra of pure (C) and doped (C–Fe, C–Co and C–Ni) samples obtained by LDPE pyrolysis. | ||
Note that the position and width of these peaks clearly depend on the metal used in the synthesis. Similar diffractograms are obtained for the C and C–Fe samples denoting the development of graphitic clusters of similar nature. Nevertheless, this (002) peak is slightly displaced in the C–Fe sample denoting the development of new structures with a lower d-spacing (probably from the development of CNFs on the surface of carbon spheres, see the TEM and SEM images). For the C–Ni and C–Co samples, mainly Co, the peak becomes sharper and shifts to higher angles denoting a higher graphitization degree.
For further details, the interlayer spacing (d002) and the stacking height (Lc) were determined using the Bragg (1) and Scherrer (2) equations, respectively:44
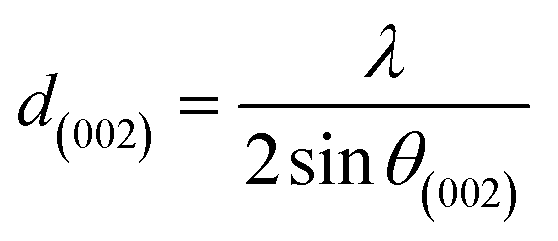 | (1) |
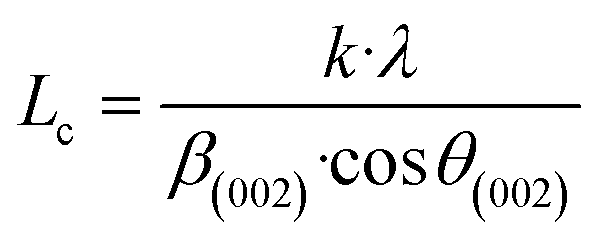 | (2) |
Regarding the metal phase, five new diffraction peaks emerge for the C–Co sample assigned to the face-centered cubic phase (fcc) and the hexagonal close packed phase (hcp) of reduced cobalt. The peaks at 44.2° (111), 51.3° (200) and 75.9° (220) are ascribed to the Co fcc phase (JCPDS # 15-0806), while the peaks at 41.6° (100), 44.6° (002), 47.5° (101) and 75.9° (110) are attributed to the Co hcp phase (JCPDS # 05-0727). The presence of zero oxidation state is attributable to the cobalt–carbon intimate contact, since it is well-dispersed throughout the carbon matrix in addition to being present inside the CNFs or covered by graphite layers. On the other hand, the larger production of hydrogen during the cracking of LDPE, especially at higher pressures,28 favors the metal reduction. Furthermore, in addition to the graphitic signals, the C–Ni diffractogram shows three intense peaks at 45.5° (111), 51.8 ° (200) and 76.3° (220) that perfectly match with the Ni0 pattern (JCPDS # 04-0850), and no presence of NiO was detected. Moreover, even in the surface, the reduced and oxidized phases coexist (see XPS, Fig. 5c). As for the C–Fe material, the small diffraction peaks (at 18.3° 30.1° and 35.5°) could be assigned to the formation of spinel Fe3O4 (JCPDS # 75-0033), whose existence has been confirmed by XPS (Fig. 5a). The presence of metallic Fe cannot be ruled out since the main diffraction peak of Fe0 (100) overlaps with that of the graphite (100) plane. All samples show a small metal crystal size (<15 nm, Table 2), implying that the metal phase is very well-dispersed thoughout the carbon matrix with the Ni-phases having the worst dispersion (see crystal size and TEM images). Since pyrolysis is performed at 700 °C and H2 can be produced during this pyrolysis, metallic phases are expected. Nonetheless, an oxidized phase could be also present due to the surface oxidation of metallic particles after exposure of the carbon-based samples to the atmosphere. In the case of the C–Ni and C–Co samples, only the metallic phase is detected by XRD whereas oxidized Fe is also detected in the Fe–C sample. This fact can be explained based on two factors: i) as pointed out by HRTEM, most of the metal particles are embedded in the carbon matrix in the Co–C and Ni–C samples due to the carbon growth, whereas Fe particles are mainly localized at the surface of carbon spheres or CNFs, and consequently, are easily oxidized during exposure to the atmosphere and ii) the reduction potential of Fe (−0.45 eV) is more negative than those of Ni (−0.26 eV) and Co (−0.28 eV) and thus, Fe particles can be more easily oxidized.
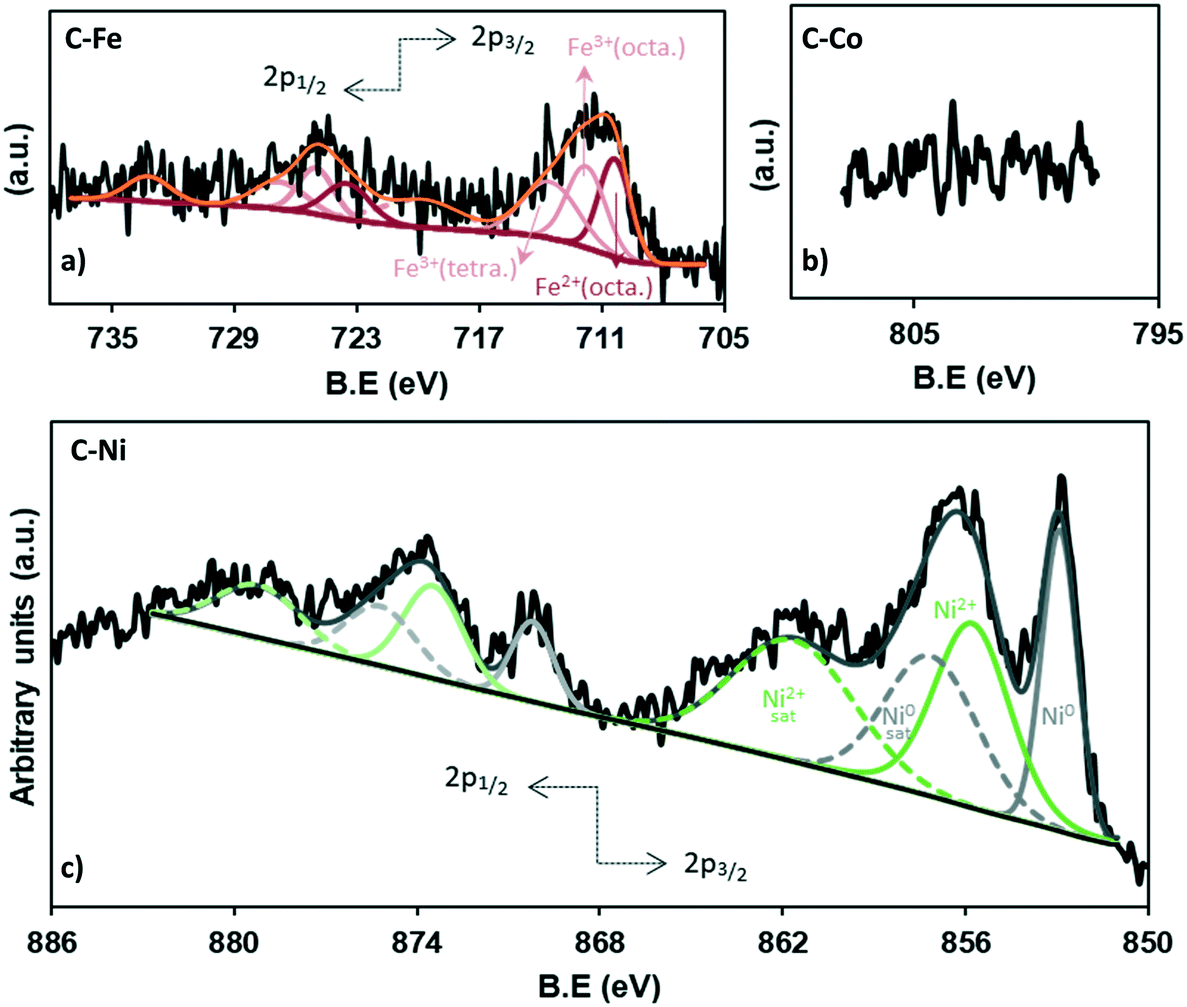 | ||
| Fig. 5 XPS patterns of the 2p metal region for a) C–Fe, b) C–Co (c) and C–Ni. | ||
The high degree of structural order of the samples was confirmed by Raman spectroscopy (Fig. 4). The ratio between the intensity of the defect band (D) and the graphitization band (G) is often used as an indicator of the structural order of carbon architectures.48 However, it has been reported that the ID/IG ratio is inversely proportional to the size of finite crystallites constituting the graphite-like carbons.49 Thus, the ID/IG ratio concerns not only the degree of graphitization but also the crystallite size. Therefore, for the characterization of the structure of graphite-like carbon materials, D and G band width (W) must be also considered and it be related with the degree of graphitization.50 Experimental results from graphitization studies of carbon materials showed that the band width of both the D and G bands are correlated with the structural state of the material. A high WD value indicates that its local structure is likely much more disordered (by disorientations and/or in-plane defects).50 Thus, a narrow WD and low ID/IG ratio (at similar crystallite sizes) means higher degree of graphitization.
Note that similar conclusions to those obtained by XRD are obtained from the Raman spectra. The Raman spectra of C and C–Fe are very similar; however, the D band becomes sharper and narrower for the Ni- and Co-doped samples, mainly for Co–C, denoting the increase of graphitization in these samples. Taking the ID/IG ratio into consideration, all materials present a high degree of graphitization, since the lower is the ID/IG ratio, the higher is the graphitization. Special attention should be paid to the C sample which presents a low intensity ratio (Table 2) in spite of being a metal-free material, so this ordering is not due to the presence of any graphitization catalyst. This demonstrates, once again, the effect of the generated pressure since other metal-free polymers carbonized at atmospheric pressure present higher ID/IG values even at higher temperatures.51,52 However, it should be noted that the ID/IG ratio linearly increases with increasing Lc; thus, to evaluate the modification of the graphitization degree by the metal doping, the D band width (WD) is considered. Note that as expected, the D band width (Table 2) decreases in the order C > C–Fe > C–Ni > C–Co, following the same trend observed for Lc and d(002) obtained from the XRD results, denoting an increase of the graphitization degree in this sense.
XPS was performed to deepen the analysis of the chemical nature of the carbon and metal phases of the materials. Fig. S2† and 5 and Table 3 show the XPS data. The C1s region spectra of samples are compared in Fig. S2a,† where a low amount of oxygen in the C sample is clearly observed since the two main peaks correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.6 eV) and C–C (285.6 eV), and there is almost no presence of oxygen contributions, in contrast to the C–M samples where peaks at binding energies higher than 286.0 eV, which correspond to oxygenated groups, become important. Additionally, the FWHM of the CC peak gives relevant information about the graphitic order, so a high crystallinity (low content of structural defects) is associated with a low FWHM, along with other factors such as the grain size, crystallite orientation and presence of heteroatoms within the surface, mainly oxygen, which induces the appearance of surface defects.53,54 In this case, the C sample presents the narrowest CC peak and the lowest oxygen content. Following this affirmation, Fig. S2c† suggests a linear trend between the oxygen content and the above mentioned FWHM, so, the higher the surface oxygen content, the broader the CC peak. Regarding the O1s region (Fig. S2b†), two peaks are required to fit this spectral region in C samples centered at 532.4 and 533.8 eV (Table 3) corresponding to CO and C–O bonds, respectively. In metal-doped carbons (C–M samples), a third peak is also required which is ascribed to metal–O bonds. Note that this contribution is less significant for the C–Co sample due to, as observed by HRTEM, most of the metal particles being embedded in the carbon matrix.
C (284.6 eV) and C–C (285.6 eV), and there is almost no presence of oxygen contributions, in contrast to the C–M samples where peaks at binding energies higher than 286.0 eV, which correspond to oxygenated groups, become important. Additionally, the FWHM of the CC peak gives relevant information about the graphitic order, so a high crystallinity (low content of structural defects) is associated with a low FWHM, along with other factors such as the grain size, crystallite orientation and presence of heteroatoms within the surface, mainly oxygen, which induces the appearance of surface defects.53,54 In this case, the C sample presents the narrowest CC peak and the lowest oxygen content. Following this affirmation, Fig. S2c† suggests a linear trend between the oxygen content and the above mentioned FWHM, so, the higher the surface oxygen content, the broader the CC peak. Regarding the O1s region (Fig. S2b†), two peaks are required to fit this spectral region in C samples centered at 532.4 and 533.8 eV (Table 3) corresponding to CO and C–O bonds, respectively. In metal-doped carbons (C–M samples), a third peak is also required which is ascribed to metal–O bonds. Note that this contribution is less significant for the C–Co sample due to, as observed by HRTEM, most of the metal particles being embedded in the carbon matrix.
C peak and the total metal content obtained by TGA (MTGA)
| Sample | C1s | O1s | M2p | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B.E. (eV) | Assign. | FWHM (eV) | Peak (%) | B.E. (eV) | Assign. | Peak (%) | O (%) | B.E. (eV) | Assign. | Peak (%) | M (%) | M TGA (%) | |
| C | 284.6 | CC |
0.95 | 75 | 532.4 | OC |
53 | 2.20 | — | — | |||
| 285.6 | C–C | 17 | 533.8 | O–C | 47 | ||||||||
| 286.6 | C–O | 3 | |||||||||||
| 287.7 | CO |
1 | |||||||||||
| 289.4 | COO− | 1 | |||||||||||
| 291.1 | π–π* | 2 | |||||||||||
| C–Fe | 284.6 | CC |
1.10 | 71 | 530.5 | O–Fe | 12 | 5.66 | 710.4 | Fe2+3/2(octa) | 22.0 | 0.84 | 3.03 |
| 285.5 | C–C | 13 | 532.5 | OC |
53 | 711.8 | Fe3+3/2(octa) | 22.4 | |||||
| 286.5 | C–O | 7 | 534.0 | O–C | 35 | 713.5 | Fe3+3/2(tetra) | 22.4 | |||||
| 287.7 | CO |
3 | 719.8 | sat. | — | ||||||||
| 289.3 | COO− | 4 | 723.5 | Fe2+1/2(octa) | 11.2 | ||||||||
| 291.0 | π–π* | 2 | 725.0 | Fe3+1/2(octa) | 11.7 | ||||||||
| 726.8 | Fe3+1/2(tetra) | 10.2 | |||||||||||
| 733.2 | sat. | — | |||||||||||
| C–Co | 284.6 | CC |
1.03 | 69 | 530.8 | O–Co | 3 | 4.77 | — | — | — | — | 6.65 |
| 285.6 | C–C | 13 | 532.7 | OC |
55 | ||||||||
| 286.5 | C–O | 8 | 534.1 | O–C | 42 | ||||||||
| 287.6 | CO |
3 | |||||||||||
| 289.3 | COO− | 4 | |||||||||||
| 291.0 | π–π* | 3 | |||||||||||
| C–Ni | 284.6 | CC |
1.01 | 69 | 530.8 | O–Ni | 13 | 4.53 | 852.9 | Ni03/2 | 31.2 | 2.02 | 7.84 |
| 285.5 | C–C | 14 | 532.3 | OC |
63 | 855.8 | Ni2+3/2 | 43.2 | |||||
| 286.5 | C–O | 6 | 533.9 | O–C | 21 | 857.1 | sat. | — | |||||
| 287.7 | CO |
4 | 861.6 | sat. | — | ||||||||
| 289.4 | COO− | 3 | 870.2 | Ni01/2 | 9.7 | ||||||||
| 291.1 | π–π* | 4 | 873.5 | Ni2+1/2 | 15.9 | ||||||||
| 875.2 | sat. | — | |||||||||||
| 879.2 | sat. | — | |||||||||||
The chemistry of the metal phase was also analyzed in depth (Fig. 5 and Table 3). The comparison between the total metal amounts by TGA versus XPS (Table 3) confirms that most of the metal particles are not on the external surface of carbon and because of the penetration depth of XPS, they cannot be measured, especially in the case of cobalt (Fig. 5b) where, as observed by HRTEM, most of the particles are embedded in the carbon matrix. Nonetheless, the high-resolution XPS analysis allows determining the iron and nickel species.
The deconvolution of the Fe2p region shows 8 peaks (Fig. 5a), three of which correspond to the 2p3/2 contribution (710.4, 711.8, 713.5 eV), while the 723.5, 725.0 and 726.8 eV peaks match with the 2p1/2 contribution. The peaks at 719.8 eV and 733.2 eV do not give information about the content but are assigned as satellites. The peak at 710.4 eV corresponds to the octahedral Fe2+ in Fe3O4 while the peaks at binding energies of 711.7 and 713.5 eV belong to the octahedral Fe3+ and tetrahedral Fe3+ in Fe3O4, respectively.55
Moreover, the equivalent percentage of each species is 32.9% (Fe2+(octa)), 33.5% (Fe3+(octa)) and 33.5% (Fe3+(tetra)), demonstrating the presence of Fe3O4 which presents a face-centered cubic spinel structure, where Fe(II) ions occupy half of the octahedral sites and the Fe(III) ions are located in the tetrahedral sites and the remaining octahedral sites with a 1:1:1 proportion.55 These results are consistent with the X-ray diffraction (Fig. 3).
On the other hand, the analysis of the Ni XPS spectra (Fig. 5c) demonstrates the existence of a high fraction of reduced nickel which represents 41% of the totalXPS Ni, while NiO constitutes 59%, which could be formed by surface reoxidation during storage in air before the analysis since only Ni0 reflections were found in the XRD data.
3.4. Electrocatalytic activity in alkaline media
The electrocatalytic ORR activity was studied by the RDE technique. CV experiments were performed while N2 or O2 was bubbled through a 0.1 M KOH solution (Fig. 6 and 7). As discussed in previous sections, the textural and chemical properties of the materials strongly depend on the metal used in the catalytic pyrolysis, and consequently, the electrochemical behaviour could be influenced by the presence of metal, presence of nanofibers/graphitization degree and textural properties. Therefore, the cyclic voltammograms with N2 bubbling of all samples were compared as shown in Fig. 6 in order to analyze the effect of the different chemical and textural properties on the electrochemical behavior of the samples. Since all samples present a low and similar % of oxygen (around 5 wt%, see XPS), the different electrochemical behaviours observed in Fig. 6 should be explained based on the conductivity/graphitization degree and textural properties of the samples. The narrow curves that enclose a little area, which means little capacitance, indicate the lower electrochemically active surface area of all samples.56 The CV area increases following the order C < C–Fe ≈ C–Co < C–Ni which agrees with the N2-adsorption data; note that the SBET increases in the same order. Thus, this improvement in the storage capacity by adding different metals can be directly attributed to the improvement of the textural properties since the graphitization degree in all samples is very high. It is also important to highlight that strong and well-defined redox peaks are observed within a potential window from −0.15 V to −0.25 V in the C–Co sample which are typical characteristics of faradaic reactions in alkaline electrolytes,57,58 indicating the strong pseudocapacitive nature of the Co-doped electrode, which is not observed for the other metal-doped samples. The C–Co sample is clearly coated with a thick carbon layer but is redox active in CV, which can be explained based on the graphitic nature of the carbon coating. The high electrical conductivity of the formed graphitic clusters around the Co particles favours electron transfer through the carbon layer and thus, the possibility of redox reactions. Similar observations were described by A. Abdelwahab and co-workers.59 They prepared carbon aerogels doped with nickel as electrodes for supercapacitors and observed an increase in capacity retention and pseudo-faradaic contributions, explained based on a better electrical conductivity due to the formation of graphitic clusters around the Ni nanoparticles.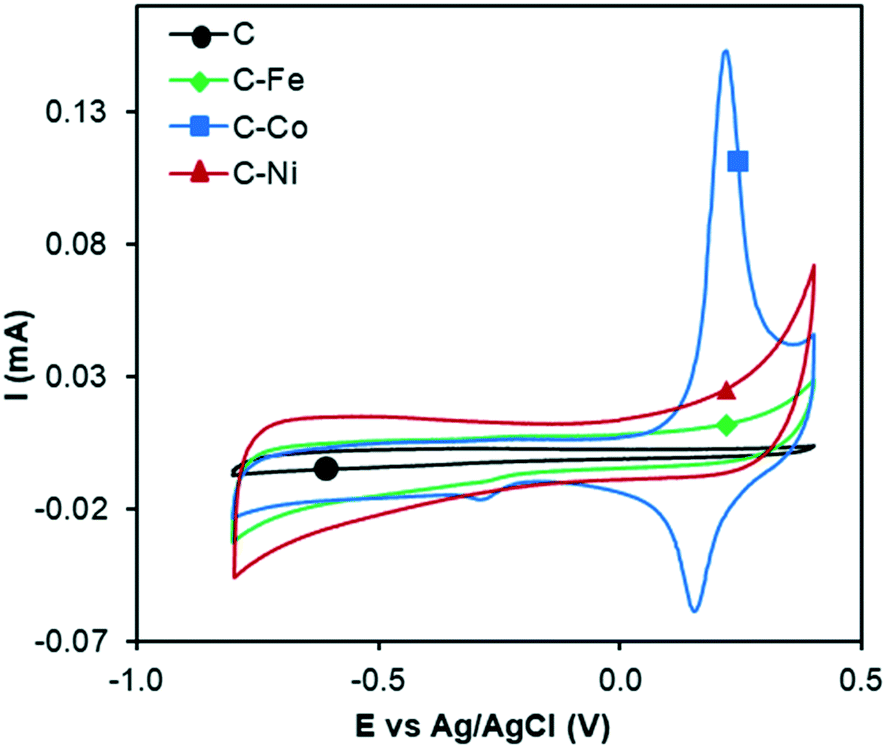 | ||
| Fig. 6 Cyclic voltammograms in N2-saturated KOH solution at 1000 rpm and 50 mV s−1 of C, C–Fe, C–Co and C–Ni. | ||
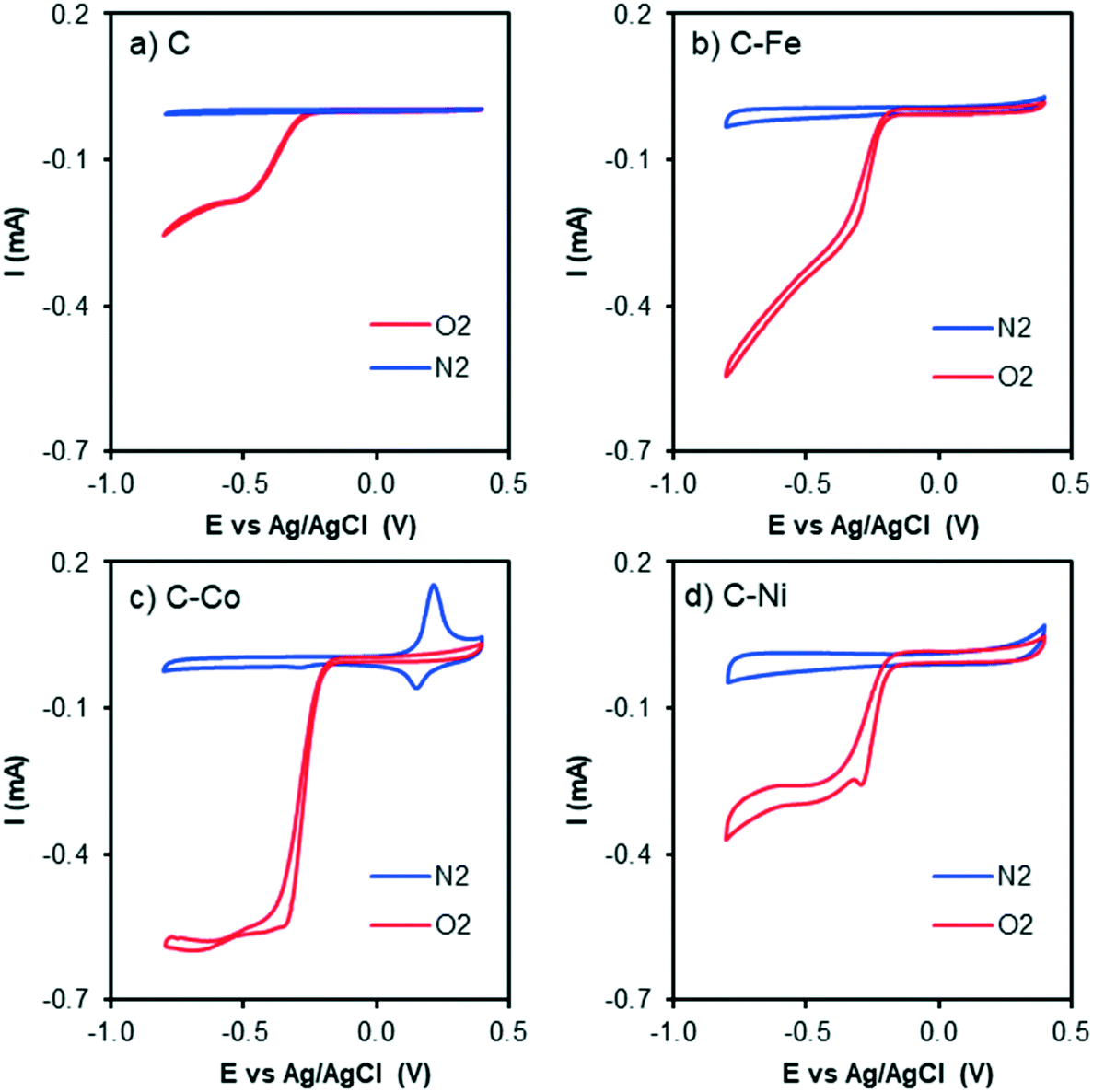 | ||
| Fig. 7 Cyclic voltammograms at 1000 rpm and 50 mV s−1 of a) C, b) C–Fe, c) C–Co and d) C–Ni with N2 bubbling (blue line) and O2 bubbling (red line) in KOH solution. | ||
The CV curves with N2 and O2 bubbling of all samples are compared in Fig. 7. An increment in the current intensity at values near −0.20 V (vs. Ag/AgCl) in the presence of O2 (red line) for all cases can be clearly observed, which indicates electrocatalytic activity in the oxygen electro-reduction reaction. Although from CV curves ORR activity is observed in all samples, C–Fe and C–Co are the ones which have achieved the highest current intensity at the same potential.
Therefore, to evaluate the electro-catalytic behaviour of the samples, the corresponding linear sweep voltammetry (LSV) curves in O2-saturated electrolyte at a sweeping rate of 5 mV s−1 from −0.8 V to 0.4 V (vs. Ag/AgCl) were obtained. The experiments were conducted at different rotation speeds from 500 rpm to 4000 rpm, in order to apply the Koutecky–Levich equation. Fig. 8 shows the LSV curves of C–Co at different rotation rates (Fig. 8a) and also a comparison of the curves of all samples at 4000 rpm (Fig. 8b). Data from the LSV curves were fitted to the K–L equation, obtaining K–L plots (Fig. 8c), and then, from the analysis of the K–L plots, the numbers of electrons transferred at different potentials were obtained (Fig. 8d) as well as the kinetic density current (jk) (Table 4). It is well known that the increment of the rotation speed favors the elimination of the diffusion limitations, so higher current intensities are achieved by increasing the rotation speed from 500 to 4000 rpm (Fig. 8a). The linearity of the K–L plots and near parallelism of the fitting lines denote a first-order kinetics toward the concentration of dissolved oxygen and an invariable electron transfer numbers with the applied potentials.60
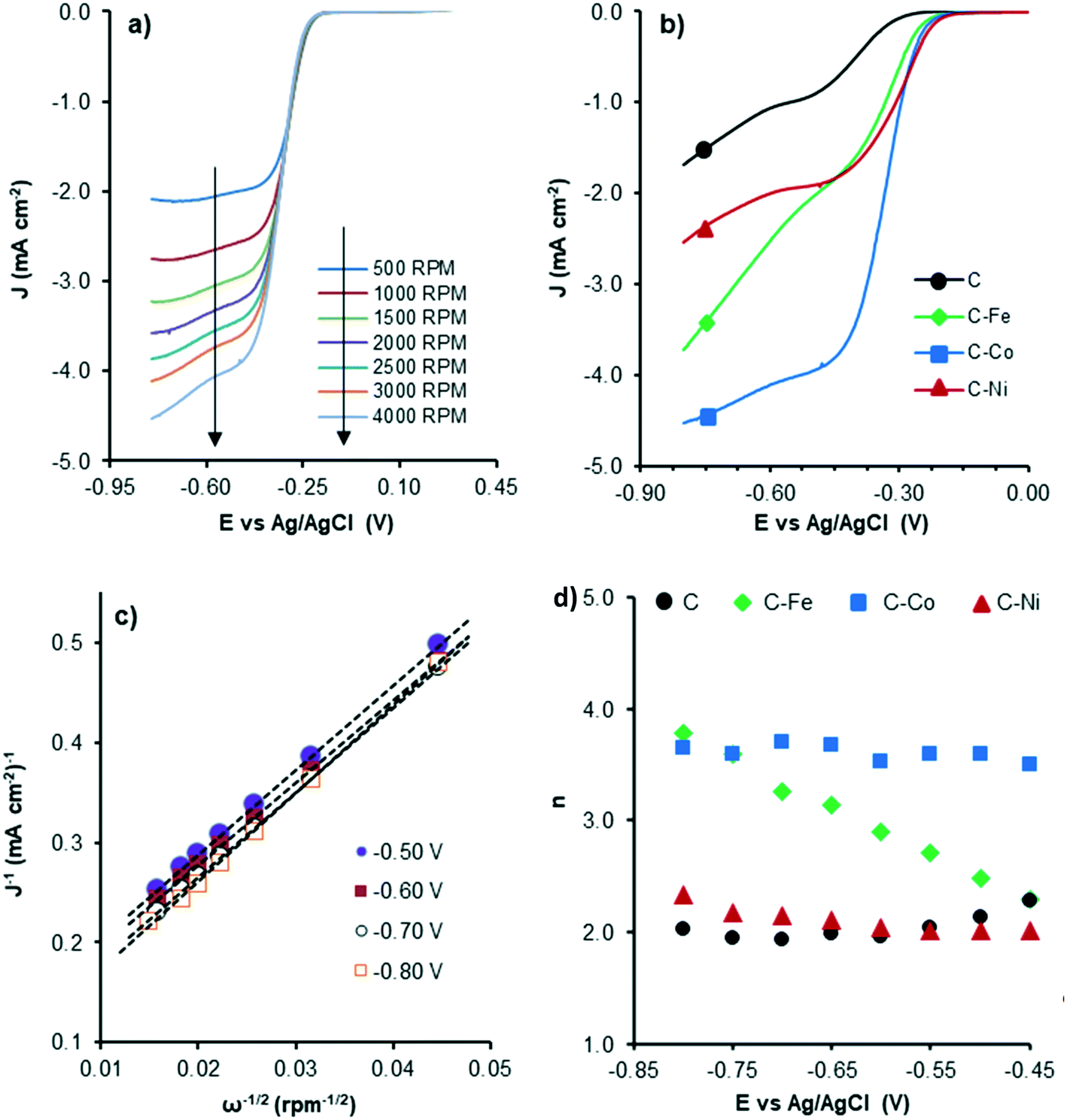 | ||
| Fig. 8 LSV curves of C–Co at different RDE rates (a), LSV curves at 4000 rpm of all samples (b), K–L plots of C–Co (c) and the number of electrons transferred at each potential for all samples (d). | ||
| Sample | E ONSET (V) | j k | n |
|---|---|---|---|
| mA cm−2 | |||
| a j k and n refer to K–L fitting at −0.7 V (vs. Ag/AgCl). | |||
| PE | −0.29 | 1.97 | 1.9 |
| PE-Fe | −0.20 | 5.64 | 3.3 |
| PE-Co | −0.18 | 10.37 | 3.7 |
| PE-Ni | −0.18 | 4.04 | 2.2 |
Although the C sample shows catalytic activity for the ORR, Fig. 8b shows the poorest performance not only because of the lower density current but also because the ORR starts at higher overpotentials. The metal-free material (C) is able to accomplish the oxygen reduction reaction purely via the 2-electron pathway (Fig. 8d) and the reaction starts at −0.29 V (vs. Ag/AgCl). Despite this material presents a high level of graphitization that contributes to the electronic transference, it has not a catalytic center which adsorbs and stabilizes the intermediates 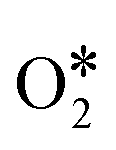 and OH* until the formation of OH– by the 4e-pathway, but it leads the reaction purely by the 2-electron pathway producing hydrogen peroxide. However, when some of the metals are present, the electro-catalytic activity is improved considerably. All the metal-doped carbon samples present a lower EONSET (−0.18 V vs. Ag/AgCl) and improved kinetics and they are able to transfer more electrons. Nonetheless, while C–Ni shows a similar n value to C, it exhibits improvements in the kinetic parameters, and C–Co and C–Fe stand out in all parameters, especially in the case of C–Co, which performs the ORR via the desired 4-electron pathway with high current density (jk, Table 4).
and OH* until the formation of OH– by the 4e-pathway, but it leads the reaction purely by the 2-electron pathway producing hydrogen peroxide. However, when some of the metals are present, the electro-catalytic activity is improved considerably. All the metal-doped carbon samples present a lower EONSET (−0.18 V vs. Ag/AgCl) and improved kinetics and they are able to transfer more electrons. Nonetheless, while C–Ni shows a similar n value to C, it exhibits improvements in the kinetic parameters, and C–Co and C–Fe stand out in all parameters, especially in the case of C–Co, which performs the ORR via the desired 4-electron pathway with high current density (jk, Table 4).
The critical factor controlling the activity and selectivity in ORR is the binding strength between the catalyst active sites and O species.61 The key to the ORR via the four-electron pathway is to achieve a strong enough interaction between the active site and O species, which gives easy O2 dissociation and favors the selectivity towards H2O, since a not too-strong interaction but strong enough to overcome the reaction barrier62 prevents O–O bond dissociation in the adsorbed *OOH, producing H2O2 as the main product. This active site/O species interaction and consequently, the ORR performance, is affected by several catalyst properties: the presence, type and dispersion of metal, graphitization degree/conductivity of carbon samples and textural properties. Regarding the metal, transition metals such as Fe, Co, Ni, Cu, Zn, and Mn display fairly strong oxygen adsorption in the ORR, which enhance the ORR efficiency and performance via the 4-electron pathway.63 Pan Xu et al.64 prepared pyrolyzed carbon supported metalaminopyrine electrocatalysts and observed that the catalytic activity increases in the order: Co ≫ Fe ≈ Cu > Mn ≫ Ni, whereas the electron transfer numbers (selectivity to H2O) followed the order: Fe > Mn > Co ≫ Cu > Ni. In this way, defects and edges in the C sample (Fig. 8d) can act as active sites but the interaction between these active sites and O species is not strong enough for the dissociation of O–O bonds and H2O2 is obtained as the product via purely the 2-electron pathway. The presence of metals in the C–M samples affects this interaction, favoring the 4-electron pathway, but the ORR performance depends on the metal nature. The activity trend is Co ≫ Fe > Ni and the trend of selectivity to H2O is Co > Fe ≫ Ni (Table 4), which agrees with the results obtained by Pan Xu et al.64
Other factors must be also considered to explain the different ORR performances of the samples such as metal accessibility and dispersion. The C–Ni sample presents the worst dispersion and the highest mesopore volume which can also affect the activity and selectivity, respectively. In mesoporous catalysts, the produced H2O2 can be easily transported in the mesoporous structure, being released within a relatively short contact time which leads to high H2O2 selectivity.65 This becomes more prominent in the Ni-doped carbon sample (C–Ni) since Ni has a higher tendency for H2O2 production than Fe and Co.15,64
The graphitization degree/conductivity also plays an important role in the ORR catalytic performance, especially in the C–Co sample where, as observed by TEM and XPS, most of the particles are embedded in the carbon matrix. Jaouen66 explained the ORR activity of metals encapsulated in single- (SWCNTs) and double-walled carbon nanotubes (DWCNTs) based on the effect of the promoted charge transfer from carbon to oxygen which favors both electrostatic and covalent bonding between them on the carbon layer encapsulating the metal. The improved electrical conductivity of the carbon support in the presence of a metal, mainly in the C–Co sample, guarantees faster charge transfer, which improves the ORR efficiency. The conductivity of carbon supports also influences the yield of H2O2; the 4-electron pathway is predominant if sufficient electrons are available in the catalyst–O2 system;67 thus, better electrical conductivity leads to a lower H2O2 yield.68 Therefore, the C–Co sample presents the highest graphitization degree which improves the ORR efficiency, stability at different potentials and H2O selectivity. Overall, the C–Co sample combined a high number of CNFs, high graphitization degree, high metal dispersion and optimized porous structure which improve the ORR efficiency via the 4-electron pathway. Therefore, in this manuscript, an easy one-step pyrolysis method at moderate temperatures is proposed by which waste plastics can be efficiently transformed into highly active ORR catalysts.
Reuse tests were performed to analyze the recyclability of the samples and data are presented in Fig. 9. A similar performance was observed after three reuse cycles, which could indicate that significant modification of the sample properties does not occur during the catalytic tests. To rule out any surface modification, HRTEM and SEM of the used sample was performed, and images are depicted in Fig. S3.† Note that significant modifications on the C–Co sample are not observed after the catalytic tests. CNFs are clearly observed by SEM (Fig. S3a†). Similar conclusions that the obtained for the C–Co fresh catalyst were obtained for the used one by HRTEM. It was observed that most of the Co particles are embedded on the carbon matrix, CNFs are obtained by means of the cobalt particles and the graphitization around the cobalt particles can be appreciated (inset Fig. S3b†), where the hexagonal phase (hcp) of Co0 was also found (0.191 nm d-spacing).
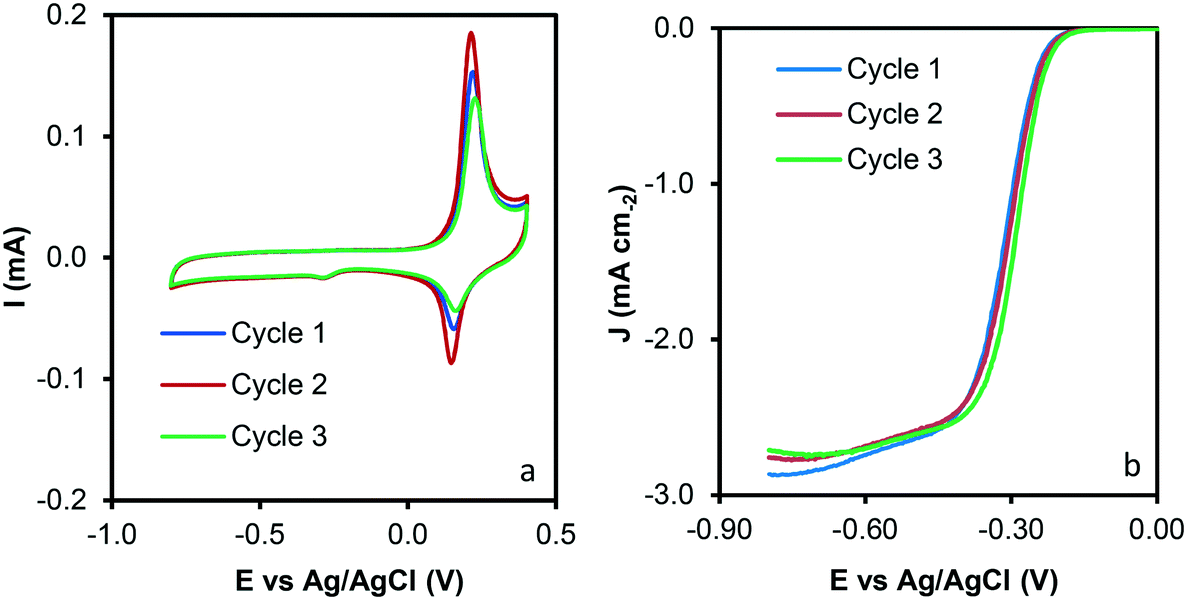 | ||
| Fig. 9 Reuse tests of the C–Co sample. a) Cyclic voltammograms at 1000 rpm and 50 mV s−1 and b) LSV curves at 1000 rpm and 5 mV s−1. | ||
Table 5 lists literature data obtained using Pt, Ni, Co and Fe supported on different carbon materials and metal-free catalysts (N and P-doped carbon materials) under similar experimental conditions. Pt/C catalysts present a good catalytic performance with the lowest Eonset potentials; however, a high amount of Pt (20%) is required to obtain considerable activity. Higher or similar jk values were obtained with our catalysts compared with those of other Pt or noble metal-based catalysts, despite our catalysts having very low metal loadings and not containing any noble metal. Moreover, the catalysts developed in this manuscript present similar or better catalytic performance than other electro-catalysts based on graphene or conductive polymers reported in the literature. It is also important to highlight that the materials presented in this manuscript have been obtained by a simple one step-method using plastic waste in comparison with other transition-metal/carbon catalysts obtained by more complicated and expensive sol–gel polymerization processes. Note also that similar Co/CNFs samples catalyze the ORR with lower n and jk values despite the higher content of metal in these samples (15 vs. 6.6 wt%). Therefore, the carbon-based materials prepared in this work are very promising non-noble metal catalysts obtained from plastic waste to be applied in the oxygen reduction reaction.
| Catalyst | Type of support | Metal content (%) | j k (mA cm−2) | E onset (V) vs. Ag/AgCl | n e− | Ref. |
|---|---|---|---|---|---|---|
| a Metal content obtained by XPS. b Potential values converted from the RHE; (N.R.) values not reported. | ||||||
| C–Co | CNF from plastic waste | 6.6 | 10.37 | −0.18 | 3.7 | Our work |
| Pt/vulcan | Carbon black | 21.2 | 8.50 | −0.08 | 3.9 | Our work |
| JT-Ni1 | MWCNTs | 2.6 | 17.7 | −0.21 | 2.9 | Previous work69 |
| JT-Ni2 | MWCNTs | 2.0 | 13.0 | −0.23 | 4.2 | Previous work69 |
| Co/N–C CNFs | Nitrogen-doped CNFs | 15.0 | ≈3 | −0.27b | 3.6 | 70 |
| CoCNF280 | CNFs | >11.1 | 4.0 | −0.04b | 3.6 | 71 |
| CoCNF370 | CNFs | >11.1 | 4.4 | −0.03b | 3.9 | 71 |
| Ni LDPE-800 | CNTs from plastic waste | 4.2 | 0.05 | −0.19 | N.R. | 41 |
| Ni PP-800 | CNTs from plastic waste | 5.8 | 0.05 | −0.18 | N.R. | 41 |
| Ni MP-800 | CNTs from plastic waste | 6.3 | 0.05 | −0.21 | N.R. | 41 |
| Pt/C | Carbon black | N.R. | N.R. | −0.10 | N.R. | 41 |
| PANI-Fe/MF-N1050 | Three-dimensional Fe, N-doped carbon nanosheets | N.R. | ≈5 | 0.03b | 3.7 | 72 |
| Ni-PANI | Polyaniline | 0.7a | 5.9 | −0.09b | 3.5 | 15 |
| Pt–Vulcan | Carbon black | 20.0 | 5.0 | −0.05b | 4.0 | 73 |
| Pt–C | Graphitic carbon | 20.0 | 5.0 | −0.05 | 3.9 | 74 |
| Co-NCX | Nitrogen-doped carbon xerogel | 0.1a | 3.1 | −0.21b | 3.5 | 75 |
| Fe-NCX | Nitrogen-doped carbon xerogel | 0.1a | 3.3 | −0.16b | 3.8 | 75 |
| CS | Carbon spheres | 0.0 | 8.7 | −0.23 | 2.4 | 10 |
| CSV2 | Carbon spheres–vanadium oxide | 3.0 | 14.5 | −0.17 | 3.6 | 10 |
| CSV5 | Carbon spheres–vanadium oxide | 6.5 | 19.0 | −0.16 | 3.4 | 10 |
| Pd/C | Carbon black | 20.0 | ≈20 | N.R. | 4.0 | 76 |
| PdCo/C | Carbon black | 20.0 | ≈13 | N.R. | 4.0 | 76 |
| Pd/rGO | Reduced graphene oxide | 20.0 | ≈5 | −0.14 | 2.8 | 76 |
| OCNT | MWCNTs | N.R. | N.R. | −0.14 | 2.6 | 76 |
| OCNT-Glu-900 | C/MWCNTs (glucose precursor) | N.R. | N.R. | −0.16 | 3.3 | 77 |
| OCNT-GN-900 | N-Doped C/MWCNTs (glucosamine hydrochloride precursor) | N.R. | N.R. | −0.10 | 3.6 | 77 |
| OCNT-Glu-Urea-900 | N-Doped C/MWCNTs (urea and glucose precursors) | N.R. | ≈29 | −0.04 | 3.6 | 77 |
| CA | Carbon aerogel | N.R. | 1.3 | −0.19 | 2.7 | 78 |
| CA-TC | P and N-doped carbon aerogels | N.R. | 1.7 | −0.13 | 3.3 | 78 |
| CK2 | KOH activated carbon | N.R. | 5.1 | −0.21 | 2.6 | 79 |
| CK2S4 | S-Doped activated carbon | N.R. | 10.1 | −0.17 | 3.5 | 79 |
| CK2S7 | S-Doped activated carbon | N.R. | 5.9 | −0.20 | 3.4 | 79 |
| ANi6 | Carbon aerogel | 5.8 | 28.1 | −0.21 | 4.2 | 16 |
| ACo6 | Carbon aerogel | 5.8 | 34.9 | −0.17 | 3.6 | 16 |
| AFe6 | Carbon aerogel | 6.1 | 26.2 | −0.22 | 4.1 | 16 |
| CoFe/NC | MOF derived N-enriched hollow carbon | N.R. | 2.0 | −0.15b | N.R. | 17 |
| CoFeNi/NC | MOF derived N-enriched hollow carbon | N.R. | 2.6 | −0.14b | N.R. | 17 |
4. Conclusions
In summary, highly graphitic carbon–metal composites were obtained under self-pressure conditions. All materials were obtained by the pyrolysis of low-density polyethylene at moderate temperature (700 °C) in a closed home-made Hastelloy® reactor. The synthesis methodology involves just one step where autogenous pressure is generated, modifying the chemistry and the morphology of the carbon materials to a great extent, and therefore, modifying the catalytic behavior. Moreover, the closed reactor used allows high mass yield, which is very important for efficient low-density polyethylene transformation.The presence of transition metals has triggered the growth of carbon nanofibers (CNFs) in all cases which differ in size and shape depending on the metal (Fe, Co or Ni). These CNFs have largely influenced the porous texture and graphitization degree, favoring electro-catalytic performance. The dispersion of the metal throughout the carbon matrix and the CNFs emerging seem to be key factors in the electro-catalytic behavior.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research has been supported by the Spanish projects from Junta de Andalucía P12-RNM-2892 and Grant ref. RNM-172. The authors also thank the “Unidad de Excelencia Química Aplicada a Biomedicina y Medioambiente” of the University of Granada (UEQ – UGR) for its technical assistance. E. Bailón-García is grateful to Junta de Andalucía for her postdoctoral fellowship (P18-RTJ-2974).References
- P. Liu, J. Yan, Z. Guang, Y. Huang, X. Li and W. Huang, J. Power Sources, 2019, 424, 108–130 CrossRef CAS.
- J. Lindorfer, D. C. Rosenfeld and H. Böhm, Future Energy, 2020, 495–517 CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- A. Gabe, J. García-Aguilar, Á. Berenguer-Murcia, E. Morallón and D. Cazorla-Amorós, Appl. Catal., B, 2017, 217, 303–312 CrossRef CAS.
- R. Engel, Int. J. Hydrogen Energy, 2012, 37, 16264 CrossRef CAS.
- F. B. Ometto, E. A. Carbonio, É. Teixeira-Neto and H. M. Villullas, J. Mater. Chem. A, 2019, 7, 2075–2086 RSC.
- T. Radhakrishnan, K. N. Nampoothiri and N. Sandhyarani, Electrochim. Acta, 2020, 340, 135840 CrossRef CAS.
- M. Song, Y. Song, W. Sha, B. Xu, J. Guo and Y. Wu, Catalysts, 2020, 10, 141 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed.
- A. Elmouwahidi, E. Bailón-García, A. F. Pérez-Cadenas, J. Castelo-Quibén and F. Carrasco-Marín, Carbon, 2019, 144, 289–300 CrossRef CAS.
- A. Elmouwahidi, J. F. Vivo-Vilches, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar and F. Carrasco-Marín, Chem. Eng. J., 2016, 306, 1109–1115 CrossRef CAS.
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Chem. Commun., 2018, 54, 4441–4444 RSC.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- C. Moreno-Castilla and F. J. Maldonado-Hódar, Carbon, 2005, 43, 455–465 CrossRef CAS.
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- A. Abdelwahab, J. Castelo-Quibén, J. F. Vivo-Vilches, M. Pérez-Cadenas, F. J. Maldonado-Hódar, F. Carrasco-Marín and A. F. Pérez-Cadenas, Nanomaterials, 2018, 8, 266 CrossRef PubMed.
- Y. Jing, Y. Cheng, L. Wang, Y. Liu, B. Yu and C. Yang, Chem. Eng. J., 2020, 397, 125539 CrossRef CAS.
- United State Environmental Protection Agency (EPA), Plastics: Material-Specific Data | Facts and Figures about Materials, Waste and Recycling.
- A. L. Andrady, Mar. Pollut. Bull., 2011, 62, 1596–1605 CrossRef CAS PubMed.
- R. C. Thompson, in Marine Anthropogenic Litter, ed. M. Bergmann, L. Gutow and M. Klages, Springer International Publishing, Cham, 2015, pp. 185–200 Search PubMed.
- A. Cincinelli, C. Scopetani, D. Chelazzi, E. Lombardini, T. Martellini, A. Katsoyiannis, M. C. Fossi and S. Corsolini, Chemosphere, 2017, 175, 391–400 CrossRef CAS PubMed.
- J. Castelo-Quibén, A. Elmouwahidi, F. J. Maldonado-Hódar, F. Carrasco-Marín and A. F. Pérez-Cadenas, Catalysts, 2018, 8, 198 CrossRef.
- A. Bazargan and G. McKay, Chem. Eng. J., 2012, 195, 377–391 CrossRef.
- J. Castelo-Quibén, E. Bailón-García, F. J. Pérez-Fernández, F. Carrasco-Marín and A. F. Pérez-Cadenas, Carbon, 2019, 155, 88–99 CrossRef.
- C. Huang, J. He, R. Narron, Y. Wang and Q. Yong, ACS Sustainable Chem. Eng., 2017, 5, 11770–11779 CrossRef CAS.
- M. R. Khan, H. Mahfuz and A. Kyriacou, Proc. ASME Int. Mech. Eng. Congr. Expo.--2009, 2009, 12, 451–458 Search PubMed.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1, 1422–1434 CrossRef CAS PubMed.
- L. Cheng, J. Gu, Y. Wang, J. Zhang, H. Yuan and Y. Chen, Chem. Eng. J., 2020, 385, 123866 CrossRef CAS.
- A. Marcilla, M. I. Beltrán and R. Navarro, J. Anal. Appl. Pyrolysis, 2009, 86, 14–21 CrossRef CAS.
- Q. Kong and J. Zhang, Polym. Degrad. Stab., 2007, 92, 2005–2010 CrossRef CAS.
- L. Quesada, M. C. de Hoces, M. A. Martín-Lara, G. Luzón and G. Blázquez, Sustainability, 2020, 12, 1–15 CrossRef.
- Y. V. Lugovoy, K. V. Chalov, O. P. Tkachenko, E. M. Sulman, J. Wärnå and D. Y. Murzin, RSC Adv., 2015, 5, 56460–56469 RSC.
- M. Ahmad and S. R. P. Silva, Carbon, 2020, 158, 24–44 CrossRef CAS.
- K. A. Shah and B. A. Tali, Mater. Sci. Semicond. Process., 2016, 41, 67–82 CrossRef CAS.
- L. M. Hoyos-Palacio, A. G. García, J. F. Pérez-Robles, J. González and H. V. Martínez-Tejada, IOP Conf. Ser.: Mater. Sci. Eng., 2014, 59, 012005 CAS.
- A. Elmouwahidi, E. Bailón-García, J. Castelo-Quibén, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar and F. Carrasco-Marín, J. Mater. Chem. A, 2018, 6, 633–644 RSC.
- A. F. Zainul Abidin, K. S. Loh, W. Y. Wong, A. B. Mohamad and I. Puspasari, Int. J. Hydrogen Energy, 2018, 43, 11047–11055 CrossRef CAS.
- Y. J. Lee, J. C. Jung, S. Park, J. G. Seo, S.-H. Baeck, J. R. Yoon, J. Yi and I. K. Song, Korean J. Chem. Eng., 2011, 28, 492–496 CrossRef CAS.
- L. Ci, H. Zhu, B. Wei, C. Xu, J. Liang and D. Wu, Mater. Lett., 2000, 43, 291–294 CrossRef CAS.
- D. Yao, H. Yang, Q. Hu, Y. Chen, H. Chen and P. T. Williams, Appl. Catal., B, 2021, 280, 119413 CrossRef CAS.
- J. G. S. Moo, A. Veksha, W. Da Oh, A. Giannis, W. D. C. Udayanga, S. X. Lin, L. Ge and G. Lisak, Electrochem. Commun., 2019, 101, 11–18 CrossRef CAS.
- J. C. Acomb, C. Wu and P. T. Williams, Appl. Catal., B, 2016, 180, 497–510 CrossRef CAS.
- K. Sing, Colloids Surf., A, 2001, 187–188, 3–9 CrossRef CAS.
- T. Qiu, J. G. Yang, X. J. Bai and Y. L. Wang, RSC Adv., 2019, 9, 12737–12746 RSC.
- C. J. Thambiliyagodage, S. Ulrich, P. T. Araujo and M. G. Bakker, Carbon, 2018, 134, 452–463 CrossRef CAS.
- F. J. Maldonado-Hódar, C. Moreno-Castilla, J. Rivera-Utrilla, Y. Hanzawa and Y. Yamada, Langmuir, 2000, 16, 4367–4373 CrossRef.
- M. Sevilla and A. B. Fuertes, Carbon, 2006, 44, 468–474 CrossRef CAS.
- Y. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 557–563 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- J. Vallerot, X. Bourrat, A. Mouchon and G. Chollon, Carbon, 2006, 44, 1833–1844 CrossRef CAS.
- S. J. Segovia-Sandoval, L. M. Pastrana-Martínez, R. Ocampo-Pérez, S. Morales-Torres, M. S. Berber-Mendoza and F. Carrasco-Marín, Sep. Purif. Technol., 2020, 237, 116341 CrossRef CAS.
- N. Rodríguez, Y. Y. Agámez-Pertuz, E. Romero, J. d. J. Díaz-Velásquez, J. A. Odriozola and M. Á. Centeno, J. Non-Cryst. Solids, 2019, 522, 119554 CrossRef.
- T. Takahagi and A. Ishitani, Carbon, 1988, 26, 389–395 CrossRef CAS.
- J. M. Jiménez Mateos and J. L. G. Fierro, Surf. Interface Anal., 1996, 24, 223–236 CrossRef.
- J. Lu, F. Fu, L. Zhang and B. Tang, Chem. Eng. J., 2018, 346, 590–599 CrossRef CAS.
- R. Zhao, W. Xia, C. Lin, J. Sun, A. Mahmood, Q. Wang, B. Qiu, H. Tabassum and R. Zou, Carbon, 2017, 114, 284–290 CrossRef CAS.
- X. Wang, C. Yan, A. Sumboja and P. S. Lee, Nano Energy, 2014, 3, 119–126 CrossRef CAS.
- V. Gupta, S. Gupta and N. Miura, J. Power Sources, 2008, 175, 680–685 CrossRef CAS.
- A. Abdelwahab, J. Castelo-Quibén, Ma. Pérez-Cadenas, F. J. Maldonado-Hódar, F. Carrasco-Marín and A. F. Pérez-Cadenas, Carbon, 2018, 139, 888–895 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- W. D. Wu, in Electrocatalysts for Fuel Cells and Hydrogen Evolution - Theory to Design, ed. A. Ray, I. Mukhopadhyay and R. K. Pati, IntechOpen, Rijeka, 2018, ch. 3 Search PubMed.
- P. Xu, W. Chen, Q. Wang, T. Zhu, M. Wu, J. Qiao, Z. Chen and J. Zhang, RSC Adv., 2015, 5, 6195–6206 RSC.
- J. Park, Y. Nabae, T. Hayakawa and M. A. Kakimoto, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS.
- F. Jaouen, J. Phys. Chem. C, 2009, 113, 15433–15443 CrossRef CAS.
- K. Qiu and Z. X. Guo, J. Mater. Chem. A, 2014, 2, 3209–3215 RSC.
- J. C. Li, D. M. Tang, P. X. Hou, G. X. Li, M. Cheng, C. Liu and H. M. Cheng, MRS Commun., 2018, 8, 1158–1166 CrossRef CAS.
- J. Castelo-Quibén, E. Bailón-García, F. J. Pérez-Fernández, F. Carrasco-marín and A. F. Pérez-Cadenas, Carbon, 2019, 155, 88–99 CrossRef.
- K. Yu, P. Shi, J. Fan, Y.-L. Min and Q.-J. Xu, J. Nanopart. Res., 2019, 21, 230 CrossRef CAS.
- I. T. Kim, M. J. Song, S. Shin and M. W. Shin, Appl. Surf. Sci., 2018, 435, 1159–1167 CrossRef CAS.
- J. Liu, Y. Zhu, X. Li, F. Du, R. Wang and L. Jiang, J. Alloys Compd., 2019, 788, 1274–1281 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- G. A. Ferrero, K. Preuss, A. B. Fuertes, M. Sevilla and M.-M. Titirici, J. Mater. Chem. A, 2016, 4, 2581–2589 RSC.
- S. Liu, C. Deng, L. Yao, H. Zhong and H. Zhang, J. Power Sources, 2014, 269, 225–235 CrossRef CAS.
- D.-S. Kim, T.-J. Kim, J.-H. Kim, E. F. A. Zeid and Y.-T. Kim, J. Electrochem. Sci. Technol., 2010, 1, 31–38 CrossRef CAS.
- B. Huang, L. Peng, F. Yang, Y. Liu and Z. Xie, J. Energy Chem., 2017, 26, 712–718 CrossRef.
- S. A. Wohlgemuth, R. J. White, M. G. Willinger, M. M. Titirici and M. Antonietti, Green Chem., 2012, 14, 1515–1523 RSC.
- A. Elmouwahidi, J. F. Vivo-Vilches, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar and F. Carrasco-Marín, Chem. Eng. J., 2016, 306, 1109–1115 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy02082j |
| ‡ Current address: Research, Development and Innovation Department, Financiera Maderera, S.A. (FINSA), 15707, Santiago de Compostela, Spain. |
| This journal is © The Royal Society of Chemistry 2022 |