
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lewis acid Sn-Beta catalysts for the cycloaddition of isoprene and methyl acrylate: a greener route to bio-derived monomers†
Philipp
Treu
 a,
Philipp
Huber
a,
Philipp N.
Plessow
a,
Felix
Studt
ab and
Erisa
Saraçi
*ab
a,
Philipp
Huber
a,
Philipp N.
Plessow
a,
Felix
Studt
ab and
Erisa
Saraçi
*ab
aInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
bInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Kaiserstraße 12, 76137 Karlsruhe, Germany. E-mail: erisa.saraci@kit.edu
First published on 4th November 2022
Abstract
Sn-Beta zeolite catalysts were unprecedently used for the Diels–Alder cycloaddition reaction between bio-available methyl acrylate and isoprene affording intermediates to bio-terephthalates. The use of the solid Lewis acid Sn-Beta allows for a greener process which is also more feasible for industrial implementation, when compared to the presently used homogeneous and often hazardous catalysts. Incorporating SnIV in the zeolite beta framework by dealumination followed by solid-state ion-exchange, resulted in an efficient cycloaddition catalyst with a selectivity favoring the para-adduct. Detailed characterization combined with computational DFT studies revealed that the tetrahedral framework Sn-sites in Sn-Beta zeolites are responsible for the superior catalytic activity and selectivity, which is maintained even at elevated temperatures.
Introduction
Plastics are some of the largest outputs of the chemical industry, with polyethylene terephthalate (PET), in particular, having an annual production volume of over 60 million tons.1 Shifting only 20% of the feedstock from fossil to renewable resources, i.e. biomass, for the worldwide production of PET would lead to an absorption of 17.2 Mt of CO2, which is equivalent to 40 million barrels of oil savings.2 Conventionally, PET is produced through polymerization of fossil-derived ethylene glycol (EG) and purified terephthalic acid (PTA). While the EG monomer in commercial “bio”-PET is already produced renewably from the dehydration of bio-ethanol, the PTA used is still majorly produced from the AMOCO oxidation of fossil-derived para-xylene, resulting in an only 30% renewable PET product.3 For a 100% renewable PET with a lower carbon footprint, the aromatic PTA monomer also needs to be derived from renewable resources, e.g. biomass.4 In this context, there have been several routes to renewable PTA, among which Diels–Alder (DA) cycloaddition of bio-based dienes and dienophiles is a particularly interesting strategy to synthesize PTA precursors. The routes using the DA chemistry go either through p-xylene as an intermediate,5–8 or through other intermediates to the final PTA.3,9–13 The p-xylene intermediate necessitates a subsequent 2-step oxidation to introduce the carboxylic groups present in PTA. One of the characteristics of bio-derived dienes/dienophiles is that they contain concomitant oxygen groups, which can be retained through the Diels–Alder transformation and can preclude the oxidation steps to bio-PTA. For example, the trans,trans-muconic acid (t,t-MA) isomer obtained either from fermentation of sugars,14 or cross-metathesis of other bio-derived intermediates,15 can react with ethylene to give a cycloadduct which can be subsequently dehydrogenated to terephthalates. When developing a sustainable process, atom economy is crucial. In this respect, the cycloaddition of isoprene with acrylic acid is a promising approach to PTA, as it provides an excellent atom efficiency. Moreover, both reactants can be derived from biomass; isoprene can be obtained renewably through hydrogenation and decyclization of itaconic acid16 or from fermentation processes,17 while acrylic acid can be derived from biomass platform molecules, e.g. glycerol18 and lactic acid,19via catalytic routes. The main challenge in the DA reaction between isoprene and acrylic acid is preventing isoprene polymerization, while achieving high activity and selectivity towards the para-cycloadduct, 4-methylcyclohex-2-ene-1-carboxylic acid.20 Both the para- and meta-adducts can be dehydrogenated over Pd-based catalysts providing p-toluic acid,21 which can then be oxidized to terephthalic acid, or its esters.22 Frost et al.23 reported the cycloaddition of isoprene and acrylic acid in high yield of 94% (para/meta ratio 23, with no solvent) by homogeneously catalyzing the reaction with TiCl4. However, this type of catalyst is highly toxic, corrosive and, as is typically for homogeneous catalytic systems, it is difficult to retrieve from the reaction solution. Therefore, developing a strategy to catalyze this reaction heterogeneously would make the process more sustainable and more feasible to industrial implementation. Zeolites offer the aptitude of having a high surface area and strong acid sites, which can be tuned between Brønsted and Lewis acidity by modifying the metal in the framework.24 In this respect, Bernardon et al.25 tried to optimize the DA reaction between methyl acrylate and isoprene by using zeolite catalysis with various Lewis- and/or Brønsted acidity. The para to meta-ratio achieved was high (up to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0), however, their isolated yield was very low (7%). It was also observed that Lewis acids catalyze fewer undesirable side reactions in DA reactions compared to Brønsted acid zeolites.26 The isolated framework metal sites in Beta zeolites display Lewis acid properties.27,28 Among Lewis-acid zeolites the Sn substituted beta (Sn-Beta) is a promising catalyst for the cycloaddition of isoprene and acrylates, due to its strong Lewis-acidity, high stability and easy preparation procedure.6,29
:0), however, their isolated yield was very low (7%). It was also observed that Lewis acids catalyze fewer undesirable side reactions in DA reactions compared to Brønsted acid zeolites.26 The isolated framework metal sites in Beta zeolites display Lewis acid properties.27,28 Among Lewis-acid zeolites the Sn substituted beta (Sn-Beta) is a promising catalyst for the cycloaddition of isoprene and acrylates, due to its strong Lewis-acidity, high stability and easy preparation procedure.6,29
In addition, calculation of the reaction rates and energy barriers of the elementary steps using density functional theory (DFT) have given further important insights into the reaction mechanism of the DA reaction. Research has particularly focused on the cycloaddition of furanic molecules with other dienophiles over zeolites,30–32 where computed energy barriers suggest that Brønsted acid sites do not particularly influence the reaction rate.33 However, Lewis acid zeolites, i.e. Sn-/Zr-/Hf-Beta28 or alkali containing zeolites,34 preferentially bind the dienophile, instead of the furanic molecule, enhancing its electrophilic character and the reaction rate relative to the uncatalyzed reaction. These results are however limited and activation energies have not been previously computed for the cycloaddition of i.e. isoprene and acrylates.
In this study, we have investigated the application of the solid Lewis acid Sn-Beta on the catalytic cycloaddition between bio-derived isoprene and methyl acrylate as an attractive route to terephthalate precursors (Fig. 1, red box). The cycloadducts can be easily transformed into para and meta-terephthalates via further dehydrogenation and oxidation (Fig. 1).
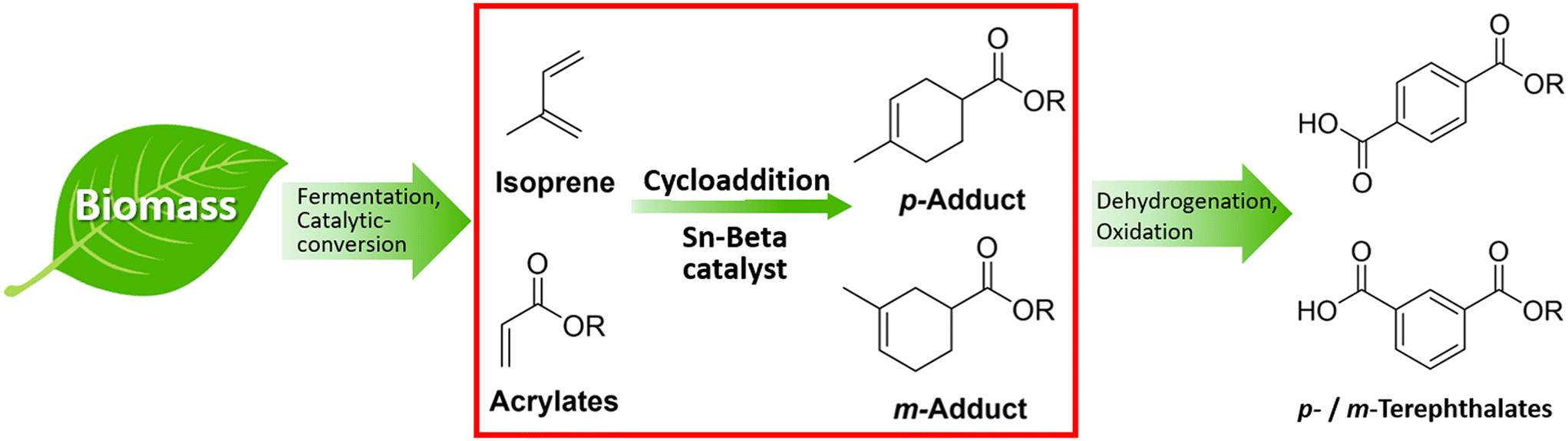 | ||
| Fig. 1 Approach to bio-PTA via intermediates obtained from the cycloaddition of bio-derivable isoprene and acrylates over solid Lewis acid Sn-Beta catalysts. | ||
By using different characterization techniques combined with DFT calculations, we studied the role of the tetrahedral framework Sn-sites in the zeolite Beta, their Lewis acid properties and their ability to catalyze the DA cycloaddition between isoprene and methyl acrylate.
Experimental section
Preparation of catalysts
Zeolite beta was used as a parent material. This zeolite is an intergrowth of the left handed and right handed polymorph A as well as polymorph B.35 Sn-substituted Beta catalysts (Sn-Beta) were obtained by dealumination of Al-containing parent zeolite (Al-Beta) and a subsequent solid state ion exchange (SSIE) with Sn (Fig. 2), according to a procedure by Hammond et al.29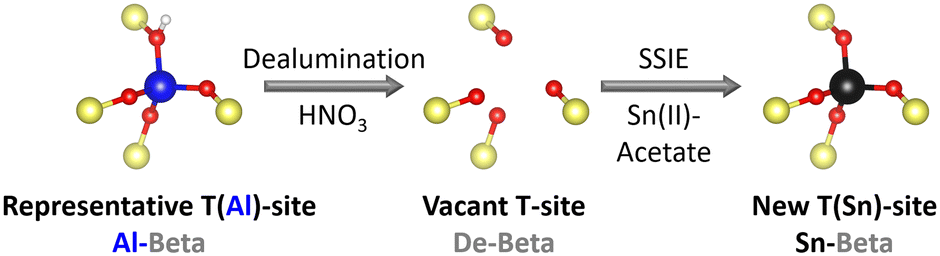 | ||
| Fig. 2 Scheme of the two step preparation procedure of Sn-Beta zeolites. SSIE = solid state ion exchange. | ||
Characterization of the catalysts
The crystalline phase of the zeolite catalysts was examined by powder X-ray diffraction (XRD) on a diffractometer (X'Pert Pro, PANalytical) scanning from 5° to 80°. Al and Sn content could be followed via inductively coupled plasma atomic emission spectroscopy (ICP-AES). For this, zeolite samples were suspended in a mixture of aqua regia and hydrofluoric acid and treated with microwave radiation (2 h, 500 W). The specific surface area, as well as pore volume of the zeolites were examined by nitrogen-sorption isotherms. Samples were first preheated at 350 °C for 4 h in vacuum. Afterwards, nitrogen was adsorbed at −196 °C according to the Brunauer–Emmett–Teller (BET) method via multipoint measurements using a NOVATouch (Anton Paar). Dealumination, correct incorporation of Sn into the framework of beta, as well as undesired formation of extra-framework SnO2 was examined by ultraviolet-visible (UV-vis) spectroscopy. For this, the samples were measured with a LAMBDA 650 UV/vis spectrometer (PerkinElmer) between the wavelengths of 180 nm to 800 nm. Spectralon was used as a reference and a scan velocity of 0.5 s nm−1 was applied. For X-ray photoelectron spectra (XPS) the samples were uniformly distributed on a carbon tape and fixed on a molybdenum sample holder and recorded in an ultra-high vacuum chamber (base pressure 10−8 Pa) equipped with an unmonochromated Al K alpha X-ray source and a Phoibos 150 analyzer (manufacturer SPECS). The angle between the analyzer and the X-ray source was 45°. The electrons originating from the samples were detected along the surface normal of the sample (sample area: 2 mm diameter). The samples were analyzed in the Sn 3d region from 500 to 475 eV and the energy scale calibrated using the adventitious C 1s peak. Pyridine adsorbed Fourier transform infrared (Py-FTIR) spectra were collected on a Bruker VERTEX 70 instrument in 4000–650 cm−1 range, applying attenuated total reflectance (ATR). Prior to the measurements, the powdered samples were treated with a stream of pyridine saturated Ar gas at RT for 45 min, and subsequently treated at 250 °C for 1 h.Catalytic activity measurements
Computational details
All structures were optimized using periodic density function theory with the dispersion-corrected PBE-D3 (ref. 36 and 37) functional and a geometric convergence criterion of 10−3 eV Å−1. The projector-augmented-wave method, an energy cut-off of 400 eV, and an energy convergence criterion of 10−8 eV were applied. The Brillouin zone was sampled only at the Γ-point using Gaussian smearing with a width of 0.1 eV. Calculations were carried out with the Vienna ab initio simulation package (VASP38,39) in version 5.4.1 and the Atomic Simulation Environment (ASE40). Transition states were identified using the Automated Relaxed Potential Energy Surface Scan (ARPESS41) method and verified by having only one imaginary frequency, which connects educts and products by distortion along the normal mode. For vibrations, only adsorbates and the active center (including the aluminium or tin atom and adjacent oxygen, hydrogen, and silicon atoms) were considered. Free energies were obtained using the harmonic oscillator approximation and for gas phase molecules additionally the rigid rotator and free translator approximation were used. Since the harmonic approximation often leads to inaccurate entropies for low-frequency modes, all obtained frequencies have been raised to 12 cm−1 if they were below this value, as described in detail in earlier work.42,43 The lattice constants of beta are a = 12.700 Å, b = 12.700 Å, and c = 26.600 Å as used in previous work.43 The Si:Al and Si:Sn ratios were 63:1.
Results and discussion
Catalysts physiochemical properties
We prepared a range of Sn-Beta catalysts with nominal loadings of 1, 2 and 5 wt% Sn. The influence of Sn incorporation on the structure of the zeolites is summarized in Table 1. N2 physisorption isotherms can be found in the ESI† (Fig. S1). After the dealumination, ICP-AES showed Al-content of 3.20 wt% in the parent zeolite was reduced to below detection limit in the dealuminated D-Beta. The Sn-content in the Sn-substituted zeolites was found to be at the aimed values of 1, 2 and 5 wt%.| Catalyst | ω Al or Sn [wt%] | A BET [m2 g−1] | A micro [m2 g−1] | V micro [m3 g−1] | V total [m3 g−1] |
|---|---|---|---|---|---|
| a Determined via ICP-AES. b Below detection limit. | |||||
| Al-Beta | 3.20 | 629 | 432 | 0.170 | 0.705 |
| D-Beta | n.d.b | 583 | 387 | 0.165 | 0.810 |
| 1Sn-Beta | 1.01 | 582 | 395 | 0.161 | 0.557 |
| 2Sn-Beta | 1.88 | 599 | 399 | 0.163 | 0.588 |
| 5Sn-Beta | 4.96 | 610 | 427 | 0.173 | 0.666 |
The BET surface area of all materials remain high and close to that of the parent zeolite (629 m2 g−1), indicating preservation of the zeolite structure after all the treatments. Small variations in surface area occur depending on the changes to the structure. The removal of Al from the beta framework resulted in a lower BET and micropore specific surface area (ABET and Amicro). The total volume also increased (Vtotal) at the expense of the micropore volume (Vmicro). These structural changes result from the creation of vacancies in the zeolite T-sites and partial framework deconstruction during the dealumination process.44,45 After the SSIE of D-Beta with Sn the surface area as well as micropore volume increase with Sn-loading, indicating the restoration of the zeolite framework by incorporation of Sn into the vacant T-sites.
Collected XRD spectra of the parent Al-Beta and Sn-samples (Fig. 3) revealed that the characteristic beta diffraction pattern could be preserved after all modification steps without damage to the zeolite structure.46 A shift of the diffraction peak (302) from 2θ = 22.5° (Al-Beta) to 22.9° (D-Beta) occurs after dealumination, suggesting that there is a contraction of the unit cell following the removal of Al from the framework (see also Fig. S2†). Upon incorporation of SnIV into the D-Beta, a recovery of the unit cell volume can be observed from the d302 spacing, which increases with Sn-content to corresponding 2θ values of 22.8°, 22.7° and 22.6° for 1Sn-Beta, 2Sn-Beta and 5Sn-Beta, respectively. This 2θ shift indicates that Sn indeed occupies and heals the vacant T-sites of Beta zeolite.47 Furthermore, no indication of extra-framework SnO2 formation could be detected by XRD, suggesting that either SnO2 is not present in the materials, or that any extra-framework Sn domains are too small (<2 nm) to generate detectable diffraction reflexes.
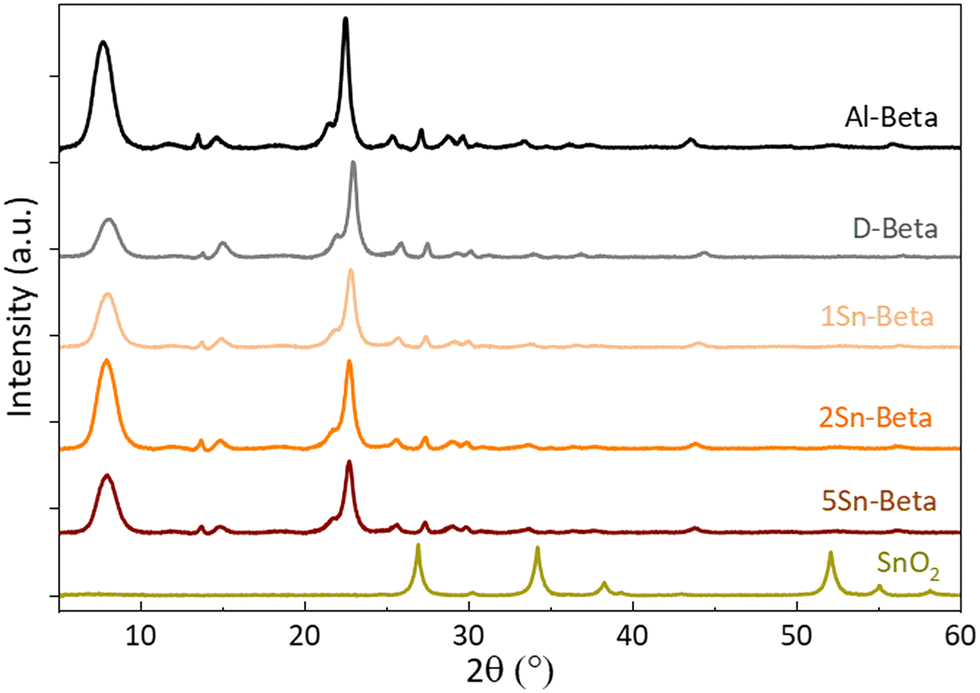 | ||
| Fig. 3 X-ray diffractograms of parent (Al-Beta), dealuminated (D-Beta) and Sn-substituted (1, 2 and 5Sn-Beta) zeolites. | ||
In order to further investigate the state of Sn in the zeolite structure, diffuse reflectance UV-vis analysis as well as XPS spectra were collected (Fig. 4). The UV-vis spectra of all materials shown in Fig. 4a were corrected with corresponding D-Beta spectra to better identify the absorption signals related to the presence of Sn species (see uncorrected spectra in Fig. S3†).44 Bulk SnO2, exhibits a very broad absorbance around 230–350 nm. For all Sn-substituted zeolites a UV absorbance band around 200 nm can be observed, which increases with Sn-loading. This band is assigned to ligand-to-metal charge transfer (O2−–Sn4+), corresponding to isolated tetrahedrally coordinated Sn species within the zeolite framework, recognized as the catalytically active Sn species.29 Although, SnO2 could not be detected by XRD (Fig. 3), UV-vis shows an absorption signal from 250 to 300 nm, which is more pronounced for Sn-Beta zeolites with higher Sn-loading. The signals around the position of 250–300 nm, similar to the absorption maximum of our SnO2 reference, have previously been ascribed to extra-framework Sn or polymeric Sn–O–Sn type species,45,47,48 possibly formed within the zeolite. The incorporation of Sn in the zeolite framework can also be confirmed by XPS spectra shown in Fig. 4b. The exclusively octahedrally coordinated Sn in SnO2 show binding values of 495.0 eV (3d5/2) and 486.5 eV (3d3/2).49 All Sn-Beta samples show two peaks, with the 3d5/2 signal at 496.5 eV and the 3d3/2 signal at 488.0 eV, which increase in intensity with Sn-loading.49 The signal shift of SnIV towards higher binding energies in the Sn-Beta samples as compared to SnO2 indicates that Sn detected by XPS is in tetrahedral coordination in the zeolite structure.47,49 Moreover, CD3CN adsorbed DRIFTS also show that the Sn incorporated into framework positions is present in the form of a mixture of open and closed Sn-sites (Fig. S4†).50 Further studies are needed to explore the correlation of the nature of framework Sn sites in zeolite beta (open, closed, hydrolysed- or defect-open sites) with the corresponding catalytic activity in the cycloaddition reaction. These spectroscopic results suggest successful incorporation of SnIV in the zeolite T-sites, although some extra-framework SnO2 may still be formed, especially at higher loadings.
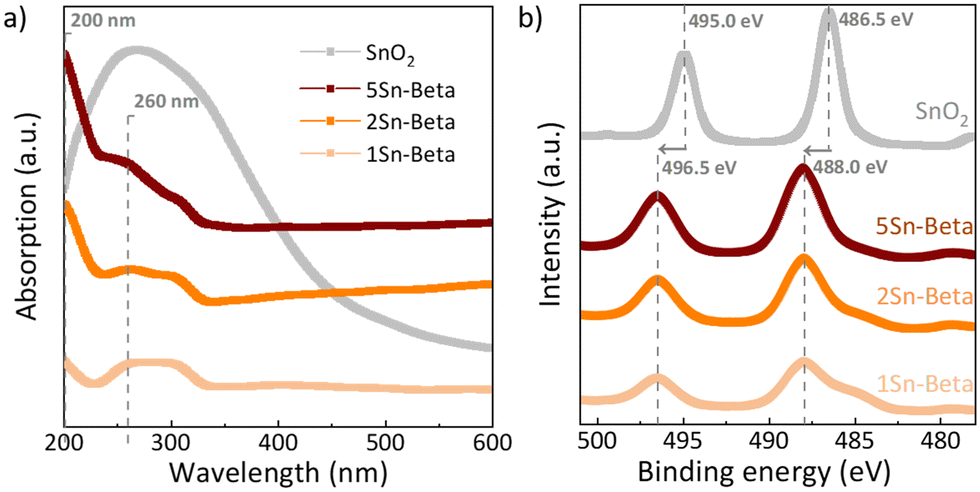 | ||
| Fig. 4 a) UV-vis of parent Al-Beta and Sn-Betas corrected with the D-Beta as a background reference; b) XPS spectra of Sn-Betas and SnO2 in Sn 3d region. | ||
Acid properties of the zeolites
The nature of the zeolite acid sites was identified by pyridine adsorption monitored by FTIR (Py-FTIR) (Fig. 5a). In the 1575–1425 cm−1 region, the vibrational bands of pyridine can be found. In particular the vibrational modes 8a, 8b, 19b, and 19a, are distinctively perturbed upon the interaction with Brønsted (BAS) or Lewis acid (LAS) sites.51 Being a Lewis base, pyridine interacts with the BAS forming pyridinium ions with FTIR bands 1525–1565 cm−1 and with the isolated electron pair of LAS, characterized by bands in the FTIR spectrum 1445–1465 cm−1.52 The parent Al-Beta sample showed mainly contributions of BAS at around 1549 cm−1 and the band at 1490 cm−1, the latter being characteristic for both Brønsted and Lewis acid sites.51,53,54 The spectra for the dealuminated D-Beta show no distinct absorption bands, confirming the removal of any acidity from the zeolite via dealumination. For the Sn-Beta samples, a band at 1453 cm−1 and 1491 cm−1 appear, which, in this case, correspond to pyridine adsorption on LAS.54,55 The intensity of these bands increases with Sn content in the zeolites, implying stronger LAS density for higher Sn-loading.56 In addition, a moderate broad band at 1552 cm−1 is also present in the Sn-Beta spectra, indicating some weak acidity which might reside in the structural silanol defects of the samples,53 or due to water molecules adsorbed at the tetrahedral SnIV-sites during the measurement.57 These results confirm the Lewis acid nature of the Sn-Beta zeolites. | ||
| Fig. 5 a) FTIR spectra of pyridine adsorbed on beta samples. Pyridine was desorbed for 1 h at 250 °C before spectra were taken. BA = Brønsted site, LA = Lewis site; b) optimized structures of adsorbed pyridine on beta samples. The calculated vibration of the 19b mode is given for each adsorption geometry. | ||
Theoretical calculations of pyridine adsorptions have been shown to reproduce the shift in the 19b vibration of pyridine58 and were conducted here to validate the Py-FTIR measurements (Fig. 5b). While the 8a, 8b, 19a and 19b ring vibrational modes are commonly considered to distinguish pyridine adsorption at LAS and at BAS, the main difference in the vibrations between BAS*Py and LAS*Py lies in the shift observed for the 19b mode.59,60 We computed this mode for Al-Beta*pyrdine to 1543 cm−1 (shift to the calculated 19b vibration mode of pyridine in the gas phase: Δvib = 116 cm−1), which fits to the experimental data (Fig. 5a, Table S5†). For Sn-Beta*pyridine, this mode is shifted to 1436 cm−1 (Δvib = 9 cm−1) in our computations which corresponds to the experimental band observed at 1453 cm−1. The less indicative 19a vibration mode is calculated to be at 1475 cm−1 and 1470 cm−1 for Al-Beta*pyridine and Sn-Beta*pyridine, respectively. The 1470 cm−1 band is indeed slightly visible for Sn-Beta samples in the experimental spectra (Fig. 5a). To explain the experimental band around 1552 cm−1 at Sn-Beta*pyridine, we also investigated the co-adsorption of water and pyridine. In this case, the 19b vibration mode is located at 1557 cm−1 (Δvib = 130 cm−1), very close to the experimental value. These findings are in line with other IR experiments.57,61–63 The calculated Gibbs free energies for adsorption at 20 °C are −103 kJ mol−1 and −81 kJ mol−1 for the co-adsorbed system and for the single adsorbed pyridine, respectively. This shows that co-adsorption is favourable thermodynamically and thus supports the possibility that water was present in the zeolites during the FTIR experiment.
Catalytic Diels–Alder reaction
The catalytic DA reaction between isoprene and methyl acrylate was systematically investigated over the parent, dealuminated and the Sn-containing zeolites. Lewis acids are known to catalyze this reaction.64,65 The reaction was performed at a mild temperature of 70 °C with the aim to perform the reaction, while still investigating the effect of the catalysts without significant thermodynamic DA activity that would occur at higher temperatures.23,25 Control experiments without any catalyst and with dealuminated Beta showed similar low yields of cycloadducts (5.7% and 5.8%, respectively), confirming that the Al-free zeolite has no influence on the reaction. The parent Al-Beta already shows a decent catalytic activity (Fig. 6a), which can be attributed to its Brønsted and some Lewis acidity being active in the DA.25,66 The cycloaddition activity improves for Sn-Beta zeolites as the Sn-content increases. Yields of cycloadducts increase steadily from 8.20 → 11.8 → 13.2 and para:meta-ratios from 3.8 → 4.0 → 4.3 with increasing Sn-loading (Fig. 6a).
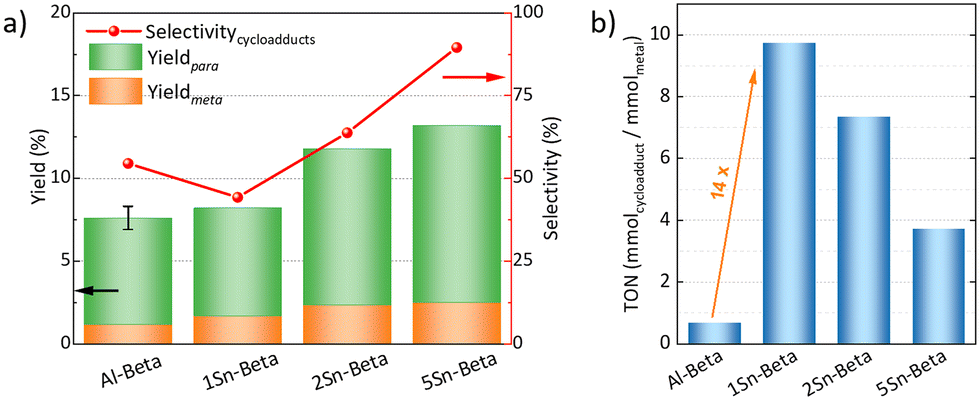 | ||
| Fig. 6 a) Catalytic conversion and yields of para and meta cycloadduct over different catalysts as well as without any catalyst, at 70 °C, 5 h, nisoprene:nmethyl acrylate = 3, 100 mg cat. Error bar for Al-Beta represents experimental uncertainty. b) Turnover numbers (TON) of parent and Sn-Betas per basis of the metal content. | ||
As observed from the different characterization techniques above, the LAS density progressively increases with Sn-content in the zeolites. As Lewis acidity is responsible for promoting DA cycloaddition,28 the Sn-substituted beta zeolites show superior activity and selectivity as compared to the parent Al-Beta zeolite.28 The carbon balance is always >90%, with other side products resulting from reactions of the cycloaddition products with isoprene,66 and isoprene di-/oligomerization mainly on Al-Beta, owing to its Brønsted acidity (Fig. S5†).67 The vacant silanol groups still present in the low-loaded Sn-Beta samples (1 and 2 wt%) may trap methyl acrylate via formation of hydrogen bonds to the carbonyl group, accounting for the lower selectivity of these materials.68 Indeed the selectivity significantly increases the more Sn is introduced into those vacant Si–OH sites. This suggests that the Lewis Sn-sites are necessary to catalyze the DA cycloaddition in a selective way.
Furthermore, turn over numbers (TON) were also calculated for all metal containing zeolites on a metal basis (Fig. 6b). All Sn-containing catalysts show higher TON activity compared to the standard Al-Beta. 1Sn-Beta in particular scores a TON of 9.72, which is 14 times higher than the one for Al-Beta. An apparent decrease of the TON is present as we go to higher Sn-loadings. We now know that not all Sn introduced during the synthesis goes to the empty T-sites of the zeolite to form the desired framework Sn sites. Some of it, especially when higher loadings are aimed, ends up in formation of extra-framework SnO2 during SSIE.69 This SnO2 is not only inactive in the reaction, as we have tested in control experiments, but it may also sit on the zeolite surface, possibly even blocking some of the active sites.48 Therefore, the TON which is normalized against the total amount of metal in the zeolites decreases with increasing Sn-loading. Since we have investigated the reaction at a mild temperature of 70 °C, we could use that as an optimization parameter for increasing the reaction productivity. The influence of the reaction temperature on the Diels–Alder reaction was investigated for Al-Beta and 5Sn-Beta and compared to the uncatalyzed reaction (Fig. 7). As expected, conversion and yield increase with temperature for all cases. However, for Al-Beta, the selectivity deteriorates significantly at higher temperatures. This is due to a stronger activation of isoprene at higher temperatures over Brønsted sites,26,66,70 which may lead to higher isoprene dimerization/oligomerization and other side products, as identified by GC-MS analysis (Fig. S6†). The formation of side products was hampered when Lewis acid 5Sn-Beta was used, even at 130 °C. The LAS in 5Sn-Beta have the ability to activate methyl acrylate by interaction with the carbonyl oxygen,64 which is also evidenced by the low amount of dimerization products of isoprene (Fig. S6†). These results imply that by using the Lewis acid Sn-Beta we can optimize the DA reaction yield by increasing the temperature without compromising selectivity.
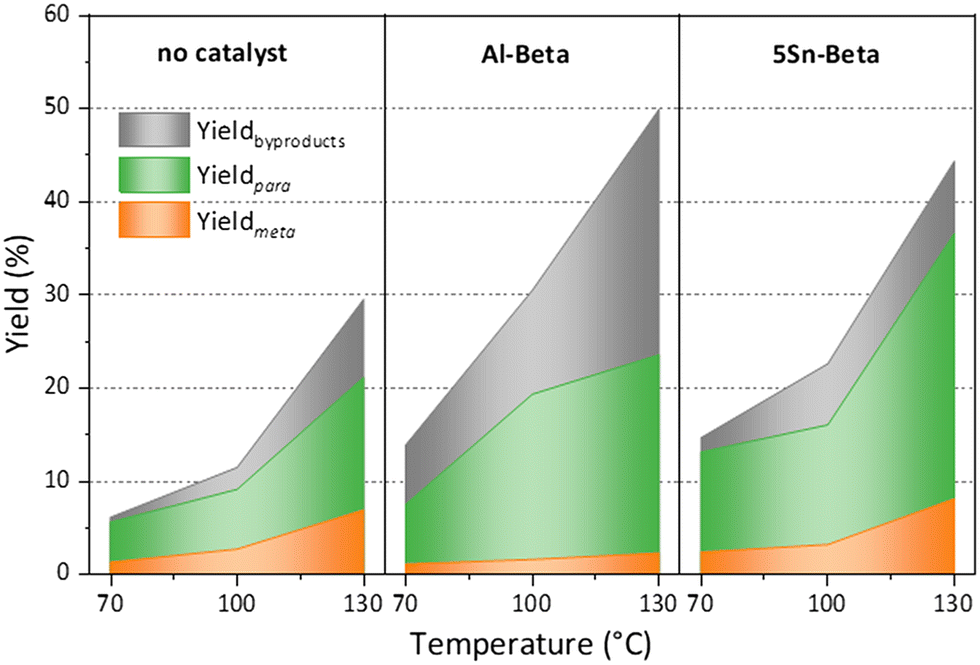 | ||
| Fig. 7 Temperature dependence of the Diels–Alder reaction, when using no catalyst, Al Beta or 5Sn-Beta. Reaction conditions: nisoprene:nmethyl acrylate = 3, 5 h, 100 mg cat. | ||
Computational DA activity on Sn-Beta
To gain insight into the effect of the catalyst structure on the reactivity and selectivity of the DA reactions, DFT-calculations at the PBE-D3 (ref. 36 and 37) level of theory were performed, for the uncatalyzed gas phase reaction, the reaction at the Brønsted acidic Al-site in Al-Beta and at the Lewis acidic site (dehydrated closed site) of the Sn-substituted zeolite. Al and Sn were studied only at one T-site (T8 site, see Fig. S7,† Si:Sn ratio of 63:1) in right handed Beta polymorph A, which was also considered for Al-substitution in previous studies due to its accessibility.43 Transition states were computed for the isomers arising from the possible combinations of endo/exo and meta/para position of the methyl group of isoprene (Fig. 8a). Depending on which side isoprene approaches the adsorbed methyl acrylate, the R or S enantiomer is formed. Thus, eight transition states were computed for the Sn substituted right handed Beta polymorph A T8 site, considering all combinations of endo/exo, meta/para, and R/S (Table S1†). For the Al containing zeolite, transition states located at all four adjacent oxygen atoms within the active center were considered and only transition states leading to the S enantiomer were computed (Table S2†). For Sn-Beta, judgements on isomer selectivity were based on the lower energy barriers that were calculated for each chiral pair of transition states, which are depicted in Fig. 8a. Likewise, for the four different oxygen atoms in the active center of Al-Beta, the lowest energy barriers are depicted only, for the comparison with Sn-Beta and the gas-phase reaction (Fig. 8b). A table with all calculated energy barriers is provided in the ESI† (Table S3). Apparent enantioselective effects, calculated for the T-site in right handed Beta polymorph A, disappear when considering a real Sn-Beta sample, as right handed and left handed Beta polymorph A exist in equal amounts in the Beta zeolite.35 Therefore, we expect racemic mixtures for both, the para and meta adduct in the real reaction.
 | ||
| Fig. 8 a) Computed transition states for the Diels–Alder reaction of isoprene and methyl acrylate on Sn-Beta. Calculations were made at a Sn-site in the T8 position of right handed Beta polymorph A. Among the enantiomeric pairs of transition states that were calculated on Sn-Beta, only the ones with lower energy barrier were depicted; b) computed internal activation energies (kJ mol−1) for the Diels–Alder reaction of isoprene and methyl acrylate. Barriers are given relative to the most stable pre-adsorbed complex formed by the two reactants in gas phase and T8 site of right handed Beta polymorph A, respectively. For comparison, only the lowest energy barriers for the four combinations of endo/exo and meta/para are depicted. | ||
The obtained reaction barriers, for the reaction with and without Sn-Beta, which are internal activation energies given relative to the pre-adsorbed reactants, are reported in Fig. 8b. For the reaction in the gas phase without the presence of a catalyst, pre-adsorption between the two reactants is weak (−5 kJ mol−1). The interaction of the reactants with the zeolite, on the other hand, is very strong leading to an adsorption enthalpy of −156 kJ mol−1 and −187 kJ mol−1 for Sn-Beta and Al-Beta, respectively. Note, that all transition states were referenced to the same, most stable computed pre-adsorption state on the T8 site in right handed beta polymorph A. Any calculated enantioselective effects apply only to the computed model and not to a real Sn-Beta sample, which contains both right handed and left handed beta polymorph A.35 Importantly, this study does not include solvation effects. Including the presence of solvent molecules typically leads to significantly weaker adsorption since the reactants need to displace solvent molecules to access the active site.43 The computed activation barriers in the uncatalyzed gas phase are all fairly similar, only deviating by 4 kJ mol−1. We note that this is clearly within the error-range of DFT, such that we cannot predict intrinsic kinetic selectivity for one of the reaction channels from our computational study.71,72
For the reaction in the Sn-Beta zeolite, the barriers for transition states with the methyl group in the meta position are 26 and 20 kJ mol−1 for the endo and exo transition state, respectively, thus being relatively similar. For the formation of the para isomer our calculations indicate larger variations, with the exo transition state being more stable (13 kJ mol−1) than the endo transition state (41 kJ mol−1). Overall, the calculations predict a moderate energy difference between the isomers, indicating a slight preference for the para adduct. This is also observed in our experimental data, where the para:meta-ratio for the uncatalyzed reaction is 3, while for the Sn-Beta catalyzed reactions this is increased to 3.8–4.3. The fact that large variations are observed for para/endo vs. para/exo suggests that selectivity towards specific isomers could in principle be achieved within the confinement in the zeolite pores.28,31
When the DA reaction is catalyzed by a Brønsted acidic site on Al-Beta, barriers for transition states show, also in agreement with the experimental data, a slight preference to the para-adduct. The barriers for the para/exo and para/endo transition state are 11 kJ mol−1 and 33 kJ mol−1 with the first being similar to the corresponding transition state catalyzed by Sn-Beta. The transition states for meta/exo and meta/endo are 16 kJ mol−1 and 35 kJ mol−1. The difference between para and meta of 5 kJ mol−1 is small and might be within the error-range of DFT calculations.71 Experiment and calculations show that Al-Beta also is an active, while unselective, catalyst for the DA. Sn-Beta seems to provide active Lewis acidic Sn-sites, which mainly lead to the desired products, without catalyzing side reactions, like it is the case for Brønsted acidic Al-sites in Al-Beta.
Conclusion
We have applied Sn-Beta zeolites as catalyst for the DA reaction between isoprene and methyl acrylate, typically homogeneously catalyzed by chloride salts. The preparation of Sn-Beta zeolites with a two-step method, i.e. dealumination and SSIE, introduced tetrahedrally coordinated Sn sites into the zeolite framework, which provide the Lewis acidity. Catalytic activity tests showed that Sn-Beta can selectively catalyze the DA cycloaddition between methyl acrylate and isoprene towards the para-cycloadduct, especially at high Sn-loadings. The DA activity is due to the isolated SnIV sites, tetrahedrally coordinated in the zeolite framework, which act as the Lewis acid sites for this reaction. The TON of these framework SnIV sites outperform the Al-Beta, with 1Sn-Beta having TON 14 times that of parent zeolite. Extra-framework SnO2 species formation should be avoided during synthesis, as they act as spectators, or even block some of the active sites. The yield of the highly selective 5Sn-Beta could be enhanced with increasing the temperature, without compromising the selectivity, unlike the parent zeolite Al-Beta where side reactions were prevalent. Additionally, DFT calculations emphasized experimental findings by calculating lower energy barriers for transition states and selectivity towards the para adduct on Sn-Beta compared to the gas-phase reaction. These findings, assert the potential of using Sn-Beta catalysts for the DA cycloaddition of isoprene with methyl acrylate, and possibly other combinations of dienes and dienophiles derivable from renewable resources. Further catalyst and reaction optimization opens possibilities towards easier process scale up and/or continuous operation, making the process more feasible for industrial application. This work demonstrates the effective application of solid catalysts for important catalytic processes for obtaining aromatic bio-monomers, addressing the high demand for this monomers in the packaging industry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. T. and E. S. gratefully acknowledge Prof. Dr. Jan-Dierk Grunwaldt (discussion and support), Prof. Dr. Silke Behrens (XRD), Armin Lautenbach (ICP-AES), Dr. Anna Zimina, Dr. Bärbel Krause (XPS), Nikolaj Slaby and Dr. Thomas Otto (BET). P. H., P. N. P. and F. S. acknowledge support by the state of Baden-Württemberg through bwHPC (bwunicluster and JUSTUS, RV bw17D011). Financial support from the Helmholtz Association (MTET, Subtopic-Nr. 38.03.02) is gratefully acknowledged. P. H. is funded by the German Research Foundation (DFG) – Projektnummer 434253773.References
- F. Neaţu, G. Culică, M. Florea, V. I. Parvulescu and F. Cavani, ChemSusChem, 2016, 9, 3102–3112 CrossRef PubMed.
- M. Volanti, D. Cespi, F. Passarini, E. Neri, F. Cavani, P. Mizsey and D. Fozer, Green Chem., 2019, 21, 885–896 RSC.
- J. Pang, M. Zheng, R. Sun, A. Wang, X. Wang and T. Zhang, Green Chem., 2015, 18, 342–359 RSC.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- C.-C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., 2012, 124, 1416–1419 CrossRef.
- C. Xue, J.-B. Zhao, L.-J. Chen, F.-W. Bai, S.-T. Yang and J.-X. Sun, Appl. Microbiol. Biotechnol., 2014, 98, 3463–3474 CrossRef CAS PubMed.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- R. Lu, F. Lu, J. Chen, W. Yu, Q. Huang, J. Zhang and J. Xu, Angew. Chem., 2016, 128, 257–261 CrossRef.
- M. Colonna, C. Berti, M. Fiorini, E. Binassi, M. Mazzacurati, M. Vannini and S. Karanam, Green Chem., 2011, 13, 2543–2548 RSC.
- M. Koehle, E. Saraçi, P. Dauenhauer and R. F. Lobo, ChemSusChem, 2017, 10, 91–98 CrossRef CAS PubMed.
- J. J. Pacheco, J. A. Labinger, A. L. Sessions and M. E. Davis, ACS Catal., 2015, 5, 5904–5913 CrossRef CAS.
- N.-Z. Xie, H. Liang, R.-B. Huang and P. Xu, Biotechnol. Adv., 2014, 32, 615–622 CrossRef CAS PubMed.
- E. Saraçi, L. Wang, K. H. Theopold and R. F. Lobo, ChemSusChem, 2018, 11, 773–780 CrossRef PubMed.
- O. A. Abdelrahman, D. S. Park, K. P. Vinter, C. S. Spanjers, L. Ren, H. J. Cho, K. Zhang, W. Fan, M. Tsapatsis and P. J. Dauenhauer, ACS Catal., 2017, 7, 1428–1431 CrossRef CAS.
- A. R. C. Morais, S. Dworakowska, A. Reis, L. Gouveia, C. T. Matos, D. Bogdał and R. Bogel-Łukasik, Catal. Today, 2015, 239, 38–43 CrossRef CAS.
- X. Li and Y. Zhang, ACS Catal., 2016, 6, 2785–2791 CrossRef CAS.
- X. Zhang, L. Lin, T. Zhang, H. Liu and X. Zhang, Chem. Eng. J., 2016, 284, 934–941 CrossRef CAS.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. Martin Alonso, R. Mariscal and M. López Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- T. Pfennig, R. L. Johnson and B. H. Shanks, Green Chem., 2017, 19, 3263–3271 RSC.
- S. Bérard, C. Vallée and D. Delcroix, Ind. Eng. Chem. Res., 2015, 54, 7164–7168 CrossRef.
- K. K. Miller, P. Zhang, Y. Nishizawa-Brennen and J. W. Frost, ACS Sustainable Chem. Eng., 2014, 2, 2053–2056 CrossRef CAS.
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS PubMed.
- C. Bernardon, B. Louis, V. Bénéteau and P. Pale, ChemPlusChem, 2013, 78, 1134–1141 CrossRef CAS PubMed.
- P. T. M. Do, J. R. McAtee, D. A. Watson and R. F. Lobo, ACS Catal., 2013, 3, 41–46 CrossRef CAS PubMed.
- W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, J. Phys. Chem. C, 2016, 120, 28533–28544 CrossRef CAS PubMed.
- T. Salavati-fard, S. Caratzoulas, R. F. Lobo and D. J. Doren, ACS Catal., 2017, 7, 2240–2246 CrossRef CAS.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- Y.-P. Li, M. Head-Gordon and A. T. Bell, ACS Catal., 2016, 6, 5052–5061 CrossRef CAS.
- N. Nikbin, P. T. Do, S. Caratzoulas, R. F. Lobo, P. J. Dauenhauer and D. G. Vlachos, J. Catal., 2013, 297, 35–43 CrossRef CAS.
- R. E. Patet, N. Nikbin, C. L. Williams, S. K. Green, C.-C. Chang, W. Fan, S. Caratzoulas, P. J. Dauenhauer and D. G. Vlachos, ACS Catal., 2015, 5, 2367–2375 CrossRef CAS.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- R. Y. Rohling, E. Uslamin, B. Zijlstra, I. C. Tranca, I. A. W. Filot, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2018, 8, 760–769 CrossRef CAS PubMed.
- J. M. Newsam, M. M. J. Treacy, W. T. Koetsier and C. B. De Gruyter, Proc. R. Soc. A, 1988, 420, 375–405 CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002–273032 CrossRef PubMed.
- P. N. Plessow, J. Chem. Theory Comput., 2018, 14, 981–990 CrossRef CAS PubMed.
- R. Y. Brogaard, C.-M. Wang and F. Studt, ACS Catal., 2014, 4, 4504–4509 CrossRef CAS.
- T. J. Goncalves, U. Arnold, P. N. Plessow and F. Studt, ACS Catal., 2017, 7, 3615–3621 CrossRef CAS.
- J. Dijkmans, D. Gabriëls, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- William N. P. van der Graaff, G. Li, B. Mezari, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2015, 7, 1152–1160 CrossRef CAS.
- A. Omegna, M. Vasic, J. van Anton Bokhoven, G. Pirngruber and R. Prins, Phys. Chem. Chem. Phys., 2003, 6, 447–452 RSC.
- B. Tang, W. Dai, G. Wu, N. Guan, L. Li and M. Hunger, ACS Catal., 2014, 4, 2801–2810 CrossRef CAS.
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS.
- M. P. Pachamuthu, K. Shanthi, R. Luque and A. Ramanathan, Green Chem., 2013, 15, 2158–2166 RSC.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- M. E. Z. Velthoen, S. Nab and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2018, 20, 21647–21659 RSC.
- Y. Yue, J. Fu, C. Wang, P. Yuan, X. Bao, Z. Xie, J.-M. Basset and H. Zhu, J. Catal., 2021, 395, 155–167 CrossRef CAS.
- C.-C. Chang, H. J. Cho, Z. Wang, X. Wang and W. Fan, Green Chem., 2015, 17, 2943–2951 RSC.
- V. Zholobenko, C. Freitas, M. Jendrlin, P. Bazin, A. Travert and F. Thibault-Starzyk, J. Catal., 2020, 385, 52–60 CrossRef CAS.
- V. S. Marakatti, A. B. Halgeri and G. V. Shanbhag, Catal. Sci. Technol., 2014, 4, 4065–4074 RSC.
- E. Peeters, G. Pomalaza, I. Khalil, A. Detaille, D. P. Debecker, A. P. Douvalis, M. Dusselier and B. F. Sels, ACS Catal., 2021, 11, 5984–5998 CrossRef CAS.
- V. L. Sushkevich, P. A. Kots, Y. G. Kolyagin, A. V. Yakimov, A. V. Marikutsa and I. I. Ivanova, J. Phys. Chem. C, 2019, 123, 5540–5548 CrossRef CAS.
- P. Huber, F. Studt and P. N. Plessow, J. Phys. Chem. C, 2022, 126, 5896–5905 CrossRef CAS.
- M. Castellà-Ventura, Y. Akacem and E. Kassab, J. Phys. Chem. C, 2008, 112, 19045–19054 CrossRef.
- J. A. Boscoboinik, X. Yu, E. Emmez, B. Yang, S. Shaikhutdinov, F. D. Fischer, J. Sauer and H.-J. Freund, J. Phys. Chem. C, 2013, 117, 13547–13556 CrossRef CAS.
- V. L. Sushkevich, I. I. Ivanova and A. V. Yakimov, J. Phys. Chem. C, 2017, 121, 11437–11447 CrossRef CAS.
- S. M. Maier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 8005–8013 CrossRef CAS.
- J. Yu, J. Luo, Y. Zhang, J. Cao, C.-C. Chang, R. J. Gorte and W. Fan, Microporous Mesoporous Mater., 2016, 225, 472–481 CrossRef CAS.
- P. Vermeeren, T. A. Hamlin, I. Fernández and F. M. Bickelhaupt, Angew. Chem., 2020, 132, 6260–6265 CrossRef.
- U. Pindur, G. Lutz and C. Otto, Chem. Rev., 1993, 93, 741–761 CrossRef CAS.
- M. Onaka, N. Hashimoto, Y. Kitabata and R. Yamasaki, Appl. Catal., A, 2003, 241, 307–317 CrossRef CAS.
- Z. Ma, X. Ma, H. Liu, Y. He, W. Zhu, X. Guo and Z. Liu, Chem. Commun., 2017, 53, 9071–9074 RSC.
- N. Rai, S. Caratzoulas and D. G. Vlachos, ACS Catal., 2013, 3, 2294–2298 CrossRef CAS.
- E. Peeters, I. Khalil, P. Eloy, S. Calderon-Ardila, J. Dijkmans, P. Ferrini, D. P. Debecker, R. A. Taylor, A. P. Douvalis, M. Dusselier and B. F. Sels, Chem. Mater., 2021, 33, 9366–9381 CrossRef CAS.
- M. Arruebo, J. L. Falconer and R. D. Noble, J. Membr. Sci., 2006, 269, 171–176 CrossRef CAS.
- P. N. Plessow and F. Studt, J. Phys. Chem. Lett., 2020, 11, 4305–4310 CrossRef CAS PubMed.
- J. Sauer, Acc. Chem. Res., 2019, 52, 3502–3510 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data for physisorption, UV-vis, XRD and GC-MS. For computational chemistry, PBE-D3, corresponding D3 contribution, ZPVE for transition states and harmonic frequencies, computed and experimental 19b frequencies for pyridine adsorption. See DOI: https://doi.org/10.1039/d2cy01337a |
| This journal is © The Royal Society of Chemistry 2022 |