
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Performance of chemical structure string representations for chemical image recognition using transformers
Kohulan
Rajan
 a,
Christoph
Steinbeck
a and
Achim
Zielesny
*b
a,
Christoph
Steinbeck
a and
Achim
Zielesny
*b
aInstitute for Inorganic and Analytical Chemistry, Friedrich-Schiller-University Jena, Lessingstr. 8, 07743 Jena, Germany
bInstitute for Bioinformatics and Chemoinformatics, Westphalian University of Applied Sciences, August-Schmidt-Ring 10, D-45665 Recklinghausen, Germany. E-mail: achim.zielesny@w-hs.de
First published on 15th January 2022
Abstract
The use of molecular string representations for deep learning in chemistry has been steadily increasing in recent years. The complexity of existing string representations, and the difficulty in creating meaningful tokens from them, lead to the development of new string representations for chemical structures. In this study, the translation of chemical structure depictions in the form of bitmap images to corresponding molecular string representations was examined. An analysis of the recently developed DeepSMILES and SELFIES representations in comparison with the most commonly used SMILES representation is presented where the ability to translate image features into string representations with transformer models was specifically tested. The SMILES representation exhibits the best overall performance whereas SELFIES guarantee valid chemical structures. DeepSMILES perform in between SMILES and SELFIES, InChIs are not appropriate for the learning task. All investigations were performed using publicly available datasets and the code used to train and evaluate the models has been made available to the public.
Introduction
Deep learning in chemistry is increasingly used to address problems in chemistry and cheminformatics.1 One of these problems is Optical Chemical Structure Recognition (OCSR), which aims to decode a 2D bitmap image of a chemical structure into a computer-readable file or string representation. OCSR techniques are necessary, for example, to extract chemical structure information buried graphically in the chemical literature and patents2 and store it in publicly available databases to enable their comprehensive retrieval with chemical structure, substructure, or similarity searches. In a recent review paper, we surveyed the available OCSR tools, most of which rely on rule-based approaches,3–5 and proposed deep learning solutions as a promising alternative.6OCSR approaches with deep learning utilize complex neural networks that require appropriate representations of chemical structures to encode and decode molecular information. Commonly, a 2D bitmap image of a chemical structure depiction is converted back into a textual representation – a character string – of that same structure. The human-readable SMILES7 representation is one of the most widely used molecular string formats. But for deep learning purposes this line notation was shown to consist of several problems8 which are primarily caused by the tokenization of its character string. As an example, structural branches are introduced with an opening bracket “(” and closed at a subsequent string position with a closing bracket “)”. The same holds for ring openings and closures which are marked by a number where a ring opens or closes. However, once SMILES strings are partitioned into tokens based on characters, the precise placement of these markers at potentially distant positions within the text string causes problems for many deep neural networks. Due to these apparent inefficiencies new textual representations of chemical structures like DeepSMILES8 and SELFIES9 have recently been developed to overcome the sketched problems. The DeepSMILES string representation aims at avoiding the problems due to branches in SMILES by using closing brackets only for branches where the number of brackets indicates the branch length. For ring closures a single symbol at the ring-closure location is used instead of two symbols at the ring-opening and ring-closing locations. In contrast to SMILES and DeepSMILES which must be partitioned into single character tokens, the SELFIES representation defines separate enclosed tokens within square brackets “[…]” so that discrete meaningful tokens are provided by the representation itself (see Fig. 1).
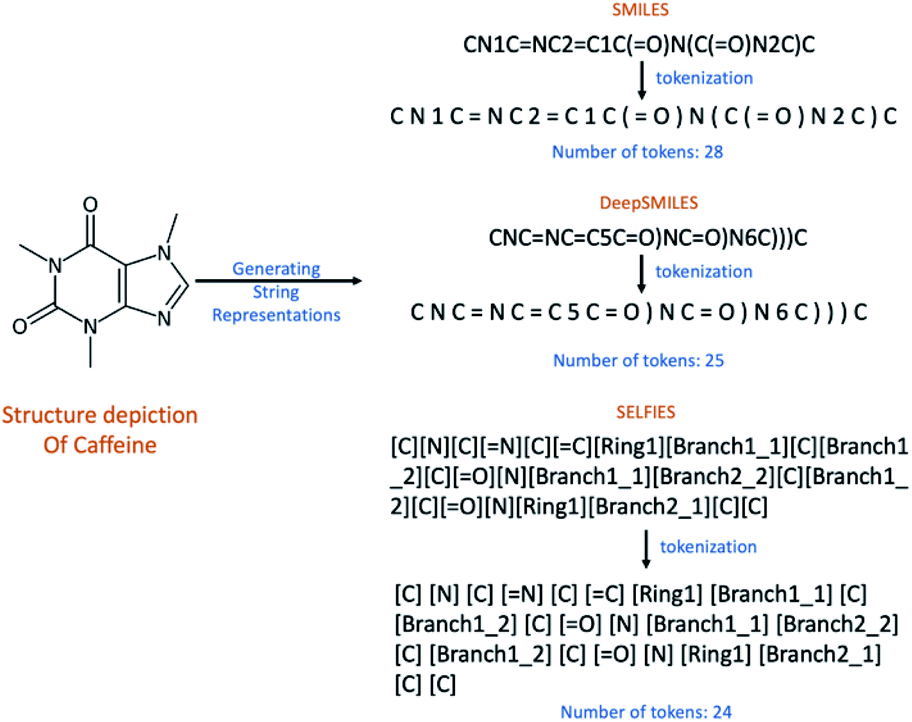 | ||
| Fig. 1 SMILES, DeepSMILES, and SELFIES are divided into tokens which are separated with spaces. | ||
In a recent OCSR study,10 we encountered similar problems with SMILES representations which eventually led to a SELFIES based implementation. By using SELFIES as the output representation, a predicted SELFIES string always converts into a valid molecule due to the SELFIES decoding algorithm. In contrast, predicted SMILES may be invalid due to syntax errors such as mismatched binding symbols, branching, or ring closure. Other recent OCSR approaches11–14 that used SMILES strings for output representation did not specifically address their inherent problems.
To further support OCSR development this work reports findings of a comparative case study for chemical image to chemical structure translation with SMILES, DeepSMILES and SELFIES. In addition, InChIs are included as an output which was proposed by a recent Kaggle competition.15
Methods
Data
In this study, all data were taken from ChEMBL16 and PubChem17 databases. The data was originally downloaded in SDF format. Using the Chemistry Development Kit (CDK)18 the chemical structures were converted into SMILES strings with and without stereochemistry information. After the SMILES conversion, the DECIMER filtering rules10 were applied to obtain a balanced dataset. Then two datasets were created, one containing SMILES without stereochemistry and one with stereochemistry information.The filtering rules for the datasets without stereochemistry included the following,
• have a molecular weight of fewer than 1500 Daltons,
• not possess counter ions,
• only contain the elements C, H, O, N, P, S, F, Cl, Br, I, Se and B,
• not contain isotopes of hydrogens (D, T),
• have 3–40 bonds,
• only contain implicit hydrogens, except in functional groups,
• have less than 40 SMILES tokens,
• no stereochemistry was allowed.
After filtering, a total of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 655225 molecules were obtained from ChEMBL. Dataset partitioning into training and test datasets is a challenging task: with a simple random partitioning, the test dataset may not cover the relevant chemical space which could lead to biased results. To avoid this problem, the RDKit19 MaxMin20 algorithm was applied, so that equally diverse training and test subsets were created which cover a similar chemical space.
655225 molecules were obtained from ChEMBL. Dataset partitioning into training and test datasets is a challenging task: with a simple random partitioning, the test dataset may not cover the relevant chemical space which could lead to biased results. To avoid this problem, the RDKit19 MaxMin20 algorithm was applied, so that equally diverse training and test subsets were created which cover a similar chemical space.
A set of 3 million molecules from PubChem was used to investigate whether the network performs better with more data. Here, the dataset was twice as large as the ChEMBL dataset. The PubChem dataset was filtered using the same rules as above, and the RDKit MaxMin algorithm was again applied to create the test set.
For the datasets with stereochemistry, a total of 1653833 molecules were obtained from ChEMBL and 3 million molecules from PubChem. Again, the RDKit MaxMin algorithm was used to select diverse training and test subsets. Table 1 provides an overview of the datasets.
| Database name | ChEMBL | PubChem | ||
|---|---|---|---|---|
| Dataset name | Dataset 1 | Dataset 2 | Dataset 3 | Dataset 4 |
| Dataset description | Without stereochemistry | With stereochemistry | Without stereochemistry | With stereochemistry |
| Train dataset size | 1536000 |
1536000 |
3072000 |
3072000 |
| Test dataset size | 119225 |
117833 |
250000 |
250000 |
The dataset with stereochemistry obtained from ChEMBL was a little smaller than the corresponding dataset without stereochemistry since stereochemistry adds new characters to SMILES, thereby lowering the number of available molecules due to the applied ruleset. With PubChem, however, the dataset size can be managed, since PubChem is much larger than ChEMBL.
Textual data
The generated molecule sets were then converted into different textual representations of the chemical structures: SMILES, DeepSMILES, SELFIES and InChIs21 and then split into tokens. For SELFIES this was a straightforward process since they already inherit a token-like word representation. Thus, SELFIES were split into tokens by using a space between the squared brackets “] [”.For splitting SMILES, DeepSMILES and InChIs into tokens another set of rules had to be applied. They were split after,
• every heavy atom,
• every open bracket and close bracket “(”,“)”,
• every bond symbol “=”,”#”,
• every single-digit number and
• all the characters inside the squared brackets were retained as-is.
The “InChI=1S/” token was kept as one single token. As it is common in all InChIs, it was not used as a token during training but was later added to the predicted strings during post-processing to evaluate the results.
In addition to the token count, the maximum string length found in the datasets was calculated. This refers to the length of the longest string available in each dataset and plays a role during training and testing. During training, the input vocabulary size which the network can handle was determined by comparing the number of tokens with the maximum length. In cases where the maximum length found in a dataset was smaller than the number of tokens available in the dataset, the input vocabulary size would be the number of tokens, otherwise, it would be the maximum length. During testing, the maximum length was used to determine when to stop predicting a structure if the end token is not met. Table 2 summarizes the number of tokens and the maximum string length found in each dataset. Datasets with stereochemistry information contain more tokens than datasets without. SELFIES representation led to more tokens than SMILES or DeepSMILES representation. InChIs had the lowest number of tokens but the largest maximum length of the longest string. With datasets 1 and 2, it became clear that InChIs perform significantly worse than the other string representations, so they were omitted in training and testing datasets 3 and 4.
| Database name | ChEMBL | PubChem | ||||||
|---|---|---|---|---|---|---|---|---|
| Dataset 1 (without stereochemistry) | Dataset 2 (with stereochemistry) | Dataset 3 (without stereochemistry) | Dataset 4 (with stereochemistry) | |||||
| Number of tokens | Maximum length of string | Number of tokens | Maximum length of string | Number of tokens | Maximum length of string | Number of tokens | Maximum length of string | |
| SMILES | 52 | 81 | 104 | 81 | 73 | 87 | 125 | 83 |
| SELFIES | 69 | 80 | 187 | 88 | 98 | 84 | 205 | 90 |
| DeepSMILES | 76 | 93 | 127 | 101 | 97 | 93 | 148 | 96 |
| InChI | 32 | 236 | 41 | 273 | — | — | — | — |
Image data
A production-quality bitmap image of each molecule was generated with the CDK Structure Diagram Generator (SDG) at a resolution of 300 × 300 pixels. Each molecule was rotated by a random angle ranging from 0 to 360° and depicted. The generated images were saved in 8 bit PNG format. Each image contains a single structure only.The features from these images were extracted as vectors by using the pre-trained weights of the ‘noisy student’22 trained EfficientNet-B3 (ref. 23) model. The extracted image features were then saved into NumPy arrays.24 These topics were discussed in detail in our previous publication.25
The extracted image features combined with the tokenized textual data were then converted into TFRecords.26 TFRecords are binary records that can be used to train a model faster using Cloud Tensor Processing Units (TPUs)27 on the Google Cloud Platform (GCP).
For training purposes, each TFRecord contains 128 data points consisting of 128 image feature vectors accompanied by 128 tokenized string representations. The TFRecords were generated on an in-house server and then moved into a Google Cloud Storage bucket.
Each dataset contains the same image data but different string representations.
Network, training and testing
In this work, we use the same network as in DECIMER Image-Transformer,25 a transformer-based network model similar to the “Base model” as explained in Google's publication, Attention Is All You Need.28 This network uses four encoder–decoder layers and eight attention heads. Attention has a dimension size of 512 and feed-forward networks have a dimension size of 2048. The columns and rows here correspond to the image features we extracted as vectors, which are 10 × 10 × 1536. A dropout rate of 10% is used to prevent overfitting. According to the publication “Attention Is All You Need” the network is trained using the Adam optimizer with a custom learning rate scheduler. The loss is calculated by using sparse categorical cross-entropy between the real and predicted SELFIES. The network was coded with Python 3 using TensorFlow 2.3 (ref. 29) on the backend.Throughout the training process, all models were trained on TPU v3-8 devices in the Google cloud. When comparing the training speed and network performance, a batch size of 1024 was found to be an adequate choice. The models were trained until the training loss had converged. In total, we trained eight models on datasets 1 and 2, and six models on datasets 3 and 4.
Once the models were fully converged, they were tested on an in-house server equipped with a GPU. To determine how many of the predictions were identical, the predictions were compared to the original strings. After the identical prediction calculations, all the predictions were converted to SMILES.
An analysis of the Tanimoto30 similarity index was conducted between the original and predicted SMILES using PubChem fingerprints available in the CDK. The Tanimoto similarity indices help to understand how well the network was able to learn chemical string representations since sometimes the predictions were not identical but only similar to the original structures and even for isomorphic structures, there can be many different SMILES.
Results and discussion
The purpose of this study was to examine different chemical string representations that are available for deep learning in chemistry by their performance on chemical image to string translation using transformer networks. Predictions were valid if the images could be translated into structures correctly.All the test results were assessed as following,
• Valid DeepSMILES/SELFIES/InChI: the predicted DeepSMILES, SELFIES and InChIs that could decode back into SMILES strings. The rest were deemed invalid.
• Valid SMILES: predicted SMILES and decoded SMILES which could be parsed to calculate the Tanimoto similarity calculations. The rest were classified as invalid SMILES.
• Identical predictions: this calculation identified how many predictions matched the original string representations. This was accomplished by using a one-to-one character string match. If a single character was wrong in the predicted string, it was considered as a wrong prediction.
• Average Tanimoto: the Tanimoto similarity between the original and predicted SMILES was calculated from the valid SMILES and the average Tanimoto similarity index was calculated against the entire test dataset.
• Tanimoto 1.0 Percentage: the percentage of molecule pairs (original and predicted) with a Tanimoto similarity index of 1.0, which was calculated from the valid SMILES of the entire test dataset.
Results for the ChEMBL dataset
From ChEMBL two datasets were obtained to train and test, one with stereochemistry (dataset 1) and one without stereochemistry (dataset 2). Table 3 summarizes the test results obtained with training on images from dataset 1.| SMILES | DeepSMILES | SELFIES | InChI | |
|---|---|---|---|---|
| Test dataset size | 119225 |
119225 |
119225 |
119225 |
| Invalid DeepSMILES/SELFIES/InChI | 0.00% | 0.07% | 0.00% | 30.79% |
| Valid DeepSMILES/SELFIES/InChI | 100.00% | 99.93% | 100.00% | 69.21% |
| Invalid SMILES | 0.35% | 0.10% | 0.00% | 0.00% |
| Valid SMILES | 99.65% | 99.83% | 100.00% | 69.21% |
| Identical predictions (string match) | 80.87% | 78.67% | 68.85% | 64.28% |
| Tanimoto 1.0 percentage (not identical) | 86.30% | 84.11% | 73.88% | 65.53% |
| Average Tanimoto | 0.97 | 0.97 | 0.95 | 0.69 |
SMILES performed best in comparison to the other representations. Comparing the identical predictions and the Tanimoto 1.0 count, SMILES based models were more accurate. This could be due to fewer tokens in the SMILES language space. Additionally, the maximum SMILES string was shorter than the rest. As a result, the model learns the representations better. Even though the InChIs have fewer tokens compared to the other representations, having a lesser number of tokens increases the maximum length of each string compared to the other representations, which ultimately creates more errors for learning and predicting. In addition, valid InChI predictions were predominantly identical to the original string.
Even though SELFIES has the most valid structures, the overall predictivity of the SELFIES-based model was lower than that of SMILES and DeepSMILES. Overall, SMILES were simpler to learn – but for guaranteed valid structures, SELFIES were the best option.
To estimate the impact of stereochemistry, the same procedure was repeated with dataset 2 where the models were trained from scratch. The results are summarized in Table 4.
| SMILES | DeepSMILES | SELFIES | InChI | |
|---|---|---|---|---|
| Test dataset size | 117833 |
117833 |
117833 |
117833 |
| Invalid DeepSMILES/SELFIES/InChI | 0.00% | 0.11% | 0.00% | 32.99% |
| Valid DeepSMILES/SELFIES/InChI | 100.00% | 99.89% | 100.00% | 67.01% |
| Invalid SMILES | 0.81% | 0.64% | 0.08% | 0.00% |
| Valid SMILES | 99.19% | 99.25% | 99.92% | 67.01% |
| Identical predictions (string match) | 78.16% | 77.07% | 66.59% | 59.10% |
| Tanimoto 1.0 percentage (not identical) | 85.02% | 83.89% | 72.07% | 63.49% |
| Average Tanimoto | 0.97 | 0.97 | 0.94 | 0.66 |
Inclusion of stereochemistry information led to a lowered accuracy. For DeepSMILES and InChIs, the number of invalid predictions increased. Additionally, the fraction of invalid SMILES increased for all representations except InChIs. After parsing all InChIs, there were only valid SMILES.
SMILES with stereochemistry reduced the overall predictability and accuracy due to the new artefacts added to the images. In addition, one should consider that the overall token count in these datasets increased due to stereochemistry with additional tokens being introduced.
SMILES were overall best to get the most accurate predictions. Since InChIs showed a significantly inferior performance, it was decided to restrict further investigations to SMILES, DeepSMILES and SELFIES.
Results for the PubChem dataset
In order to determine model improvement with increasing data size, the training and test data were doubled by utilizing data from PubChem. As pointed out above, InChIs were omitted in subsequent testing.The number of molecules available in PubChem is currently a 110 million. For this work, 3 million molecules for training and 250000 molecules for testing were obtained by random selection: the resulting tokens were carefully compared to those in the ChEMBL dataset to ensure a similar token set. Using the PubChem derived datasets with (datasets 3) and without stereochemistry (datasets 4), the same training and testing procedures were repeated, and the same evaluation procedure was used as before. For the dataset without stereochemistry (dataset 3) the results are summarized in Table 5.
| SMILES | DeepSMILES | SELFIES | |
|---|---|---|---|
| Test dataset size | 250000 |
250000 |
250000 |
| Invalid DeepSMILES/SELFIES/InChI | 0.00% | 0.08% | 0.00% |
| Valid DeepSMILES/SELFIES/InChI | 100.00% | 99.92% | 100.00% |
| Invalid SMILES | 0.22% | 0.08% | 0.00% |
| Valid SMILES | 99.78% | 99.84% | 100.00% |
| Identical predictions (string match) | 88.62% | 87.52% | 82.96% |
| Tanimoto 1.0 percentage (not identical) | 92.19% | 91.08% | 86.42% |
| Average Tanimoto | 0.98 | 0.98 | 0.97 |
By comparison of Table 5 with Table 3, it can be concluded that the data increase improved the model's performance in general. Again, SMILES show the best accuracy on the test results and SELFIES still retain 100% valid structures.
DeepSMILES falls somewhere between these two. Although DeepSMILES has more valid structures than SMILES, when considering overall accuracy, the DeepSMILES format falls behind: comparing DeepSMILES to SELFIES, DeepSMILES has a better accuracy because of its SMILES like representation, but its overall number of valid structures lags behind SELFIES (see Fig. 2).
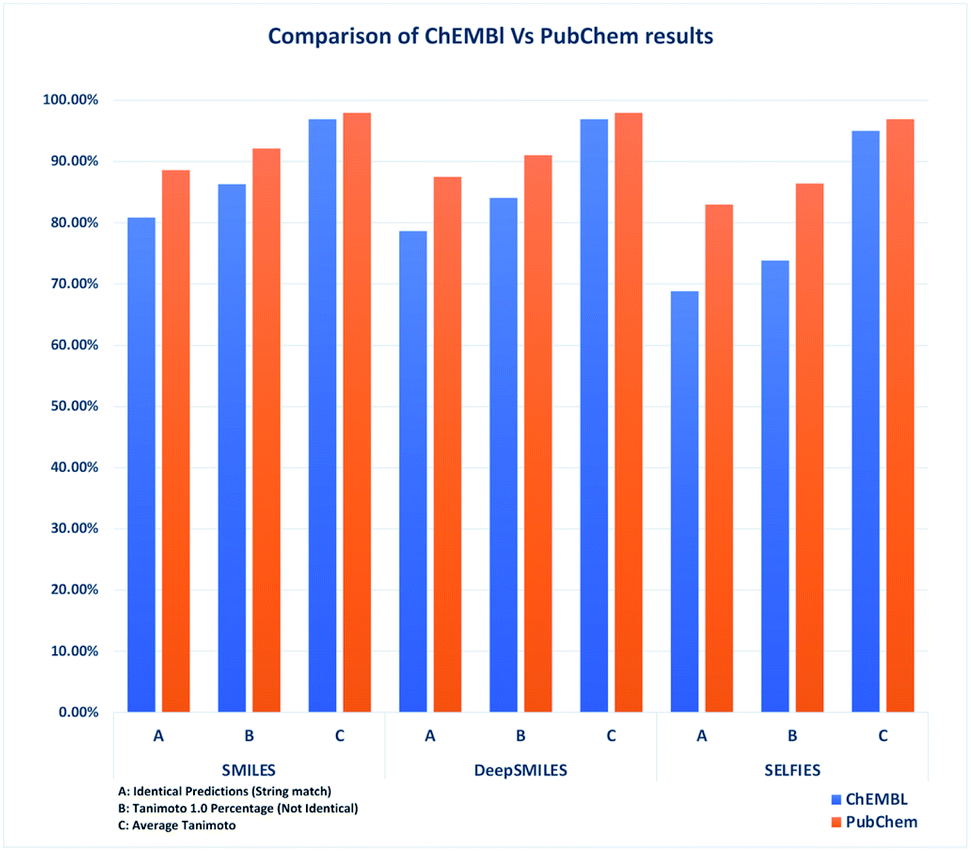 | ||
| Fig. 2 Comparison of identical predictions, Tanimoto 1.0 count and average Tanimoto of ChEMBL vs. PubChem datasets (without stereochemistry). | ||
A summary of the results for dataset 4 with stereochemistry can be found in Table 6.
| SMILES | DeepSMILES | SELFIES | |
|---|---|---|---|
| Test dataset size | 250000 |
250000 |
250000 |
| Invalid DeepSMILES/SELFIES/InChI | 0.00% | 0.06% | 0.00% |
| Valid DeepSMILES/SELFIES/InChI | 100.00% | 99.94% | 100.00% |
| Invalid SMILES | 0.34% | 0.05% | 0.00% |
| Valid SMILES | 99.66% | 99.88% | 100.00% |
| Identical predictions (string match) | 85.80% | 83.80% | 79.73% |
| Tanimoto 1.0 percentage (not identical) | 91.69% | 90.60% | 86.00% |
| Average Tanimoto | 0.98 | 0.98 | 0.97 |
Compared to Table 4, the results in Table 6 showed that increasing the dataset size also increased the overall accuracy. Datasets with stereochemistry did not perform as well as datasets without. However, the overall accuracy did increase compared to the datasets derived from ChEMBL. In addition, all of the SELFIES predictions which were decoded back into SMILES were valid, providing 100% valid structures in comparison with Table 4. SMILES again performed best in terms of predictability and accuracy, see Fig. 3.
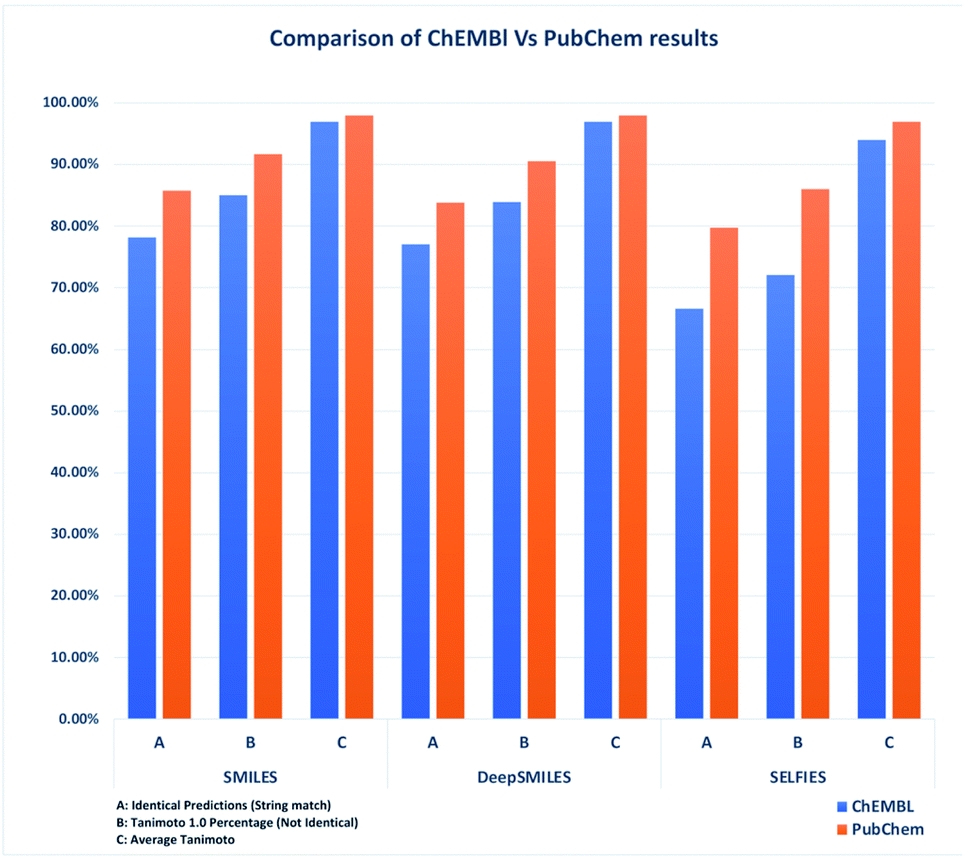 | ||
| Fig. 3 Comparison of identical predictions, Tanimoto 1.0 count and average Tanimoto of ChEMBL vs. PubChem datasets (with stereochemistry). | ||
Conclusion
The performance of different textual chemical structure representations for the chemical image to structure translation using transformers was investigated. The most accurate models were obtained by using the SMILES representation. Using SELFIES, however, we were able to produce models that led to predictions with fewer invalid structures. DeepSMILES models always fell between SMILES and SELFIES. To ensure that the models improve similarly with more data, the datasets were scaled up: the results showed the same comparative performance. For most accurate predictions, models should be trained using SMILES, for maximizing valid structures SELFIES should be used.The valid structures generated after decoding from SELFIES and DeepSMILES showed that the SELFIES decoding was superior to DeepSMILES decoding. SMILES and DeepSMILES should always be used with a set of rules on how to split them into meaningful tokens. SELFIES do not require this. There were fewer tokens in DeepSMILES than in SELFIES because the representation was similar to that in SMILES.
Since SELFIES encoding is a promising endeavor under active development, improved SELFIES variants could reach or even surpass the SMILES predictivity with the additional advantage of a 100% structural validity.
Abbreviations
| CDK | Chemistry development kit |
| DECIMER | Deep learning for chemical image recognition |
| GCP | Google cloud platform |
| GPU | Graphical processing unit |
| InChI | International chemical identifier |
| PCA | Principal component analysis |
| SDF | Structure data file |
| SDG | Structure diagram generator |
| SELFIES | Self-referencing embedded strings |
| SMILES | Simplified molecular-input line-entry system |
| TPU | Tensor processing units |
Funding
The authors acknowledge funding by the Carl-Zeiss-Foundation. Open Access funding is enabled and organized by Project DEAL.Data availability
(1) The code for Performance of chemical structure string representations for chemical image recognition using transformers can be found at https://github.com/Kohulan/DECIMER_Short_Communication. The version of the code employed for this study is version 1.0.(2) Data and processing scripts for this paper, including in SMILES format are available at Zenodo at DOI: http://10.5281/zenodo.5155037.
Author contributions
KR designed, developed the software, performed the data analysis, and wrote the paper. CS and AZ conceived the project and supervised the work. All authors contributed to and approved the manuscript.Conflicts of interest
AZ is co-founder of GNWI – Gesellschaft für naturwissenschaftliche Informatik mbH, Dortmund, Germany.Acknowledgements
The authors like to thank Google for free computing time on their TensorFlow Research Cloud infrastructure. The support of the Open Access Publication Fund of the Westphalian University of Applied Sciences is gratefully acknowledged.References
- A. C. Mater and M. L. Coote, J. Chem. Inf. Model., 2019, 59, 2545–2559 CrossRef CAS PubMed.
- I. V. Tetko, O. Engkvist and H. Chen, Future Med. Chem., 2016, 8, 1801–1806 CrossRef CAS PubMed.
- I. V. Filippov and M. C. Nicklaus, J. Chem. Inf. Model., 2009, 49, 740–743 CrossRef CAS PubMed.
- T. Peryea, D. Katzel, T. Zhao, N. Southall and D.-T. Nguyen, Abstracts of Papers of The American Chemical Society, 2019, p. 258 Search PubMed.
- V. Smolov, F. Zentsev and M. Rybalkin, TREC, 2011 Search PubMed.
- K. Rajan, H. O. Brinkhaus, A. Zielesny and C. Steinbeck, J. Cheminf., 2020, 12, 60 CAS.
- D. Weininger, J. Chem. Inf. Model., 1988, 28, 31–36 CrossRef CAS.
- N. O'Boyle and A. Dalke, ChemRxiv, 2018, DOI:10.26434/chemrxiv.7097960.v1.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Mach. Learn.: Sci. Technol., 2020, 1, 045024 CrossRef.
- K. Rajan, A. Zielesny and C. Steinbeck, J. Cheminf., 2020, 12, 65 CAS.
- D.-A. Clevert, T. Le, R. Winter and F. Montanari, Chem. Sci., 2021, 12, 14174–14181 RSC.
- I. Khokhlov, L. Krasnov, M. Fedorov and S. Sosnin, ChemRxiv, 2021, DOI:10.26434/chemrxiv.14602716.v1.
- J. Staker, K. Marshall, R. Abel and C. M. McQuaw, J. Chem. Inf. Model., 2019, 59, 1017–1029 CrossRef CAS PubMed.
- H. Weir, K. Thompson, A. Woodward, B. Choi, A. Braun and T. J. Martínez, Chem. Sci., 2021, 12, 10622–10633 RSC.
- Bristol-Myers Squibb – molecular translation, https://www.kaggle.com/c/bms-molecular-translation Search PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2019, 47, D1102–D1109 CrossRef PubMed.
- C. Steinbeck, Y. Han, S. Kuhn, O. Horlacher, E. Luttmann and E. Willighagen, J. Chem. Inf. Comput. Sci., 2003, 43, 493–500 CrossRef CAS PubMed.
- G. Landrum, RDKit: Open-Source Cheminformatics Software, http://rdkit.org Search PubMed.
- M. Ashton, J. Barnard, F. Casset, M. Charlton, G. Downs, D. Gorse, J. Holliday, R. Lahana and P. Willett, Quant. Struct.-Act. Relat., 2002, 21, 598–604 CrossRef CAS.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, J. Cheminf., 2015, 7, 23 Search PubMed.
- Q. Xie, M.-T. Luong, E. Hovy and Q. V. Le, 2020, arXiv pre-print server, arxiv:1911.04252.
- M. Tan and Q. Le, presented in part at the Proceedings of the 36th International Conference on Machine Learning, Proceedings of Machine Learning Research, 2019 Search PubMed.
- S. Van Der Walt, S. C. Colbert and G. Varoquaux, Comput. Sci. Eng., 2011, 13, 22–30 Search PubMed.
- K. Rajan, A. Zielesny and C. Steinbeck, J. Cheminf., 2021, 13, 61 Search PubMed.
- TensorFLow, TFRecord and tf.train.Example, https://www.tensorflow.org/tutorials/load_data/tfrecord, accessed October 08, 2021 Search PubMed.
- T. Norrie, N. Patil, D. H. Yoon, G. Kurian, S. Li, J. Laudon, C. Young, N. Jouppi and D. Patterson, IEEE Micro, 2021, 41, 56–63 Search PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, 2017, arXiv pre-print server, arxiv:1706.03762.
- M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean, M. Devin, S. Ghemawat, I. Goodfellow, A. Harp, G. Irving, M. Isard, Y. Jia, R. Jozefowicz, L. Kaiser, M. Kudlur, J. Levenberg, D. Mane, R. Monga, S. Moore, D. Murray, C. Olah, M. Schuster, J. Shlens, B. Steiner, I. Sutskever, K. Talwar, P. Tucker, V. Vanhoucke, V. Vasudevan, F. Viegas, O. Vinyals, P. Warden, M. Wattenberg, M. Wicke, Y. Yu and X. Zheng, 2016, arXiv pre-print server, arxiv:1603.04467.
- T. T. Tanimoto, An Elementary Mathematical Theory of Classification and Prediction, International Business Machines Corporation, 1958 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |