
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic palladium(II)-indenyl complexes bearing phosphines as ancillary ligands: synthesis, and study of indenyl amination and anticancer activity†
Enrica
Bortolamiol
a,
Francesco
Fama
a,
Ziyun
Zhang
b,
Nicola
Demitri
 c,
Luigi
Cavallo
b,
Isabella
Caligiuri
d,
Flavio
Rizzolio
*ad,
Thomas
Scattolin
*e and
Fabiano
Visentin
*a
c,
Luigi
Cavallo
b,
Isabella
Caligiuri
d,
Flavio
Rizzolio
*ad,
Thomas
Scattolin
*e and
Fabiano
Visentin
*a
aDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari, Campus Scientifico Via Torino 155, 30174 Venezia-Mestre, Italy. E-mail: fvise@unive.it
bDepartment KAUST Catalysis Centre, KCC, King Abdullah University of Science and Technology, Thuwal-23955-6900, Saudi Arabia
cElettra – Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, 34149 Basovizza, Trieste, Italy
dPathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCS, via Franco Gallini 2, 33081, Aviano, Italy. E-mail: flavio.rizzolio@unive.it
eDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: thomas.scattolin@unipd.it
First published on 28th June 2022
Abstract
The reactivity of palladium(II) indenyl derivatives and their applications are topics relatively less studied, though in recent times these compounds have been used as pre-catalysts able to promote challenging cross-coupling processes. Herein, we propose the first systematic study concerning the nucleophilic attack on the palladium(II) coordinated indenyl fragment and, for this purpose, we have prepared a library of new Pd-indenyl complexes bearing mono- or bidentate phosphines as spectator ligands, developing specific synthetic strategies. All novel compounds are thoroughly characterized, highlighting that the indenyl ligand presents always a hapticity intermediate between η3 and η5. Secondary amines have been chosen as nucleophiles for the present study and indenyl amination has been monitored by UV-Vis and NMR spectroscopies, deriving a second order rate law, with dependence on both complex and amine concentrations. The rate-determining step of the process is the initial attack of the amine to the coordinated indenyl fragment, and this conclusion has been supported also by DFT calculations. The determination of second order rate constants has allowed us to assess the impact of the phosphine ligands on the kinetics of the process and identify the steric and electronic descriptors most suitable for predicting the reactivity of these systems. Finally, in vitro tests have proven that these organometallic compounds promote antiproliferative activity towards ovarian cancer cells better than cisplatin and possibly by adopting a different mechanism of action.
Introduction
The Pd-indenyl organometallic functional group has received relatively less attention, especially when considering the huge number of publications devoted to palladium-allyl derivatives.1 This is rather odd because some peculiar structural and bonding motifs of indenyl fragments could give these systems a reactivity of great interest. In this context, the central issue concerns the indenyl hapticity that may theoretically be η1, η3 or η5, with the further option of intermediate configurations and the possibility of easily switching from one to another. It is well known that electron-poor complexes of early and middle transition metals generally prefer the highest hapticity to maximize the formal electron count and ensure electronic saturation. Conversely, more electron-rich metal centres can accept lower hapticity with consequent slip-fold distortion of the indenyl ligand. The latter feature, which is mainly attributable to benzenoid resonance stabilization, makes the indenyl ligand more versatile than the cyclopentadienyl congener, which hardly changes its η5 configuration (indenyl effect).2 An up-to-date overview of the literature on palladium indenyl compounds highlights that these systems seem to generally prefer η3 hapticity although some notable examples of Pd-η1-indenyl complexes are well-known.2–4 In most cases they are neutral mononuclear or dinuclear Pd(II) complexes, with structures influenced by the nature of ancillary ligands. Another important class of compounds is represented by dinuclear Pd(I) species, in which the indenyl fragment is μ2 bridging4–7 (Scheme 1). | ||
| Scheme 1 Significant examples of Pd(II)- and Pd(I)-indenyl complexes.4–7 | ||
Remarkably, all complexes are characterized by a formal electron count of 16 and somehow, this feature ends up determining the hapticity of the indenyl ligand.
However, it should be stressed that the η3-binding mode of the complexes reported in Scheme 1 has a certain percentage of η5-character, which can be quantified by well-defined structural2,8 and spectroscopic9 parameters. Generally speaking, an increase of the degree of pentahapto coordination mode is associated with approaching to the planarity of the five-membered ring with its five carbons tending to have very similar distances from the metal centre.
Seminal works of the Zargarian group have shown the capability of some Pd(II)-indenyl derivatives to catalyse a wide range of organic processes such as isomerization, oligomerization and polymerization of olefins and Mizoroki–Heck coupling.3,10,11 More recently, Hazari and Nolan have developed a series of precatalysts of the type [PdCl(1-tBu-indenyl)(L)] (with L = NHC or Buchwald-type phosphine) able to promote a number of challenging cross-coupling processes such as α-arylation of methyl ketones, and Suzuki–Miyaura and Buchwald–Hartwig reactions with deactivated substrates.12 The key to success of these derivatives stems from the presence of the 1-tBu-indenyl scaffold which facilitates the selective formation of active Pd(0)-monoligated species disfavouring the production of inactive Pd(I) dimers.
The literature provides much less information on cationic palladium-indenyl species of the type [Pd(indenyl)(L2)]+,11,13 and above all no specific studies of their reactivity can be found. This is particularly striking in relation to the large number of publications dealing with their η3-allyl counterparts. For example, it is well-known that these derivatives are key-intermediates in the Tsuji–Trost catalytic cycle during which they are subjected to nucleophilic attack on the allyl fragment by many suitable substrates resulting in allyl substituted products.14 In the past, our research group has studied the kinetics and reaction mechanisms of palladium-allyl amination, assessing the influence of spectator ligands, allyl substituents, the type of amine and solvent.15
In this work we aim at filling the knowledge gap concerning the nucleophilic attack on palladium(II) coordinated indenyl. The choice of an amine as a nucleophilic agent allows us to take advantage of the information obtained from our previous studies on allyl derivatives and, at the same time, compare the behaviour of these two organometallic systems. In this regard, we have opted to prepare an array of Pd(II)-indenyl complexes equipped with aryl(hetero)phosphines as supporting ligands, since they have been shown to be efficient for promoting allyl amination and also, as is well-known, their steric/electronic features can be easily fine-tuned.
Finally, encouraged by the recent results obtained on the antiproliferative activity of cationic palladium-η3-allyl derivatives towards cancer cells,17 we wanted to see if these promising properties could also be extended to these novel Pd(II)-indenyl derivatives.
Results and discussion
Synthesis of neutral palladium-indenyl complexes
Firstly, we have prepared a series of neutral complexes of the type [PdCl(indenyl)(PAr3)], adopting the original procedure described by Zargarian and co-workers and based on the dimer [Pd(μ-Cl)(indenyl)]2 as a starting material.3 The library of the selected phosphines includes p-substituted phenyl and heteroaromatic derivatives, as shown in Scheme 2. All reactions proceed smoothly, and even the more deactivated phosphines afforded the target products in good yields. NMR analysis of the compounds confirms unambiguously their identity. The coordination of entering phosphine is validated by the presence of a single peak in 31P{1H} NMR spectra, shifted 30–40 ppm downfield from the free phosphine, with the only noteworthy exception of a 2-trifurylphosphine derivative (in this case Δδ = −17 ppm) but the peculiar behaviour of this phosphine has already been observed in complexes with other metal centres.16 Consistently, the asymmetry induced by the two different ancillary ligands differentiates all the signals ascribable to the protons and carbons of the indenyl fragment. In more detail, it can be noted that carbon C3 and its associated proton H3 (labelling shown in Scheme 2) exhibit lower chemical shifts than C1 and H1, (Δδ of 20 and 2 ppm, respectively). This effect reflects a non-symmetrical Pd–C interaction and can be easily explained by the higher trans-influence of phosphine compared to chloride. | ||
| Scheme 2 Synthesis of neutral and cationic Pd(II)-indenyl complexes bearing monodentate phosphines. | ||
Further information about the palladium-indenyl interaction can be deduced by the chemical shift of junction carbons C3a and C7a. As a matter of fact, according to the Baker and Marder empirical protocol,9 the magnitude of Δδ (13C) = δ (C3a/7a of M-Ind) − δ (C3a/7a of Na+Ind−) is correlated with the hapticity of the indenyl fragment in solution. In this series of compounds Δδ (13C) is always between +2.8 and +6.2 ppm, (Table S7 in the ESI†) basically indicating intermediate hapticity between η3 and η5.
Synthesis of cationic palladium-indenyl complexes bearing two monodentate phosphines
The simple addition of PAr3 to a solution of the generic neutral complex [PdCl(indenyl)(PAr3)] does not lead to the formation of the desired cationic derivative [Pd(indenyl)(PAr3)2]Cl, but induces the reduction of indenyl hapticity to η1, affording the intermediate species [PdCl(η1-indenyl)(PAr3)2] in the first instance. This chemical event triggers cascade reactions with the final production of the bis-indene, [PdCl2 (PAr3)2] and the dimer [(μ-η3-ind)(μ-Cl) Pd2 (PAr3)2], which are, in addition to the starting complex, the only palladium species detectable in solution, when the reaction is monitored by NMR spectroscopy (Scheme 3). To overcome this issue, a good option is to preventively remove the chloride ligand from the palladium coordination sphere. As an alternative to the “classical” dehalogenation with silver derivatives,6 we have used less expensive sodium salt which, in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloromethane/methanol mixture, is able to induce the precipitation of sodium chloride after the addition of a second equivalent of phosphine (Scheme 2).
:1 dichloromethane/methanol mixture, is able to induce the precipitation of sodium chloride after the addition of a second equivalent of phosphine (Scheme 2).
 | ||
| Scheme 3 Cascade reactions in the absence of a dechlorinating agent. | ||
Of course, it is possible to operate without isolating the neutral complex [PdCl(indenyl)(PAr3)], starting directly from [Pd(μ-Cl)(indenyl)]2 and adding in succession one equivalent of phosphine, the sodium salt in methanol solution and finally the second equivalent of phosphine. With this synthetic protocol we were able to isolate all the final products, except for the complex bearing two tris(4-trifluoromethylphenyl)phosphine, which decomposes readily during the synthesis.
The only peak exhibited by the 31P{1H} NMR spectra of the new complexes [Pd(indenyl)(PAr3)2]ClO4 is always localised at chemical shifts slightly lower than that of the corresponding neutral species [PdCl(indenyl)(PAr3)], (except for the compound coordinating two tri(2-furyl)phosphines). Moreover, the higher symmetry of these complexes greatly simplifies 1H and 13C{1H} NMR spectra, where the coincident H1–H3 and C1–C3 resonate at 5.5 and 96 ppm, respectively. From the position of C3a/7a, it is possible to obtain Δδ (13C) values which, being always very close to zero, (see Table S7 in the ESI†) indicate that the indenyl ligand exhibits a hapticity intermediate between η3 and η5 also in these cationic species. This bonding motif is further confirmed by the structure obtained by single crystal X-ray diffraction of complex 3b (Fig. 1), from which it was possible to determine the structural parameters ΔM–C,18 hinges and fold angles (HA19 and FA20). Their values (0.298 Å, 13.24° and 11.65°, respectively) are consistent with an intermediate coordination between η3 and η5 of the indenyl fragment.2
 | ||
| Fig. 1 X-ray molecular structure of 3b, showing thermal displacement ellipsoids at the 50% probability level with the hydrogen atoms, counterion and solvent molecules omitted for clarity. | ||
Finally, the presence of two intense bands at about 1080 and 620 cm−1 in the IR spectra, attributable to νClO4 and δClO4 respectively, indirectly certifies the cationic nature of the synthesized complexes.
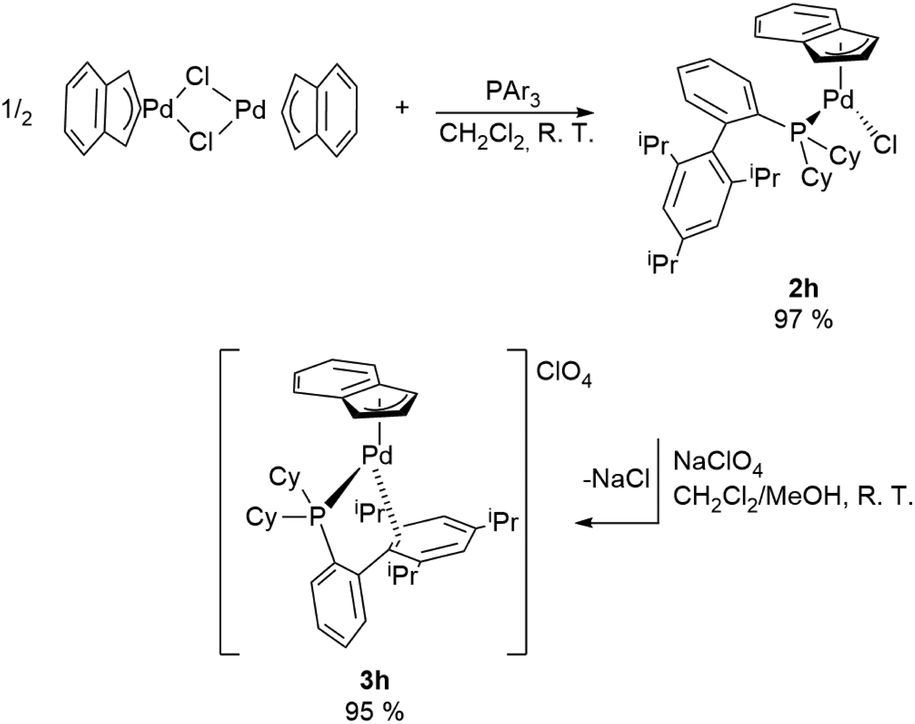
A special case is represented by XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) which interacts with the Pd-indenyl fragment in a different way from the other monodentate phosphines investigated so far. This is largely due to its peculiar structure and high steric hindrance.21
Indeed, using the same synthetic protocol described above the coordination of only one XPhos molecule can be obtained (Scheme 4). The definitive structure of the final cationic complex [Pd(indenyl)(XPhos)]ClO4 (3h) was determined by single crystal X-ray analysis and a view of that is shown in Fig. 2.
 | ||
| Scheme 4 Synthesis of neutral and cationic Pd(II)-indenyl complexes (2h and 3h) bearing XPhos. | ||
 | ||
| Fig. 2 X-ray molecular structure of 3h, showing thermal displacement ellipsoids at the 50% probability level with the hydrogen atoms, counterion and solvent molecules omitted for clarity. | ||
From this study it emerges that XPhos is anchored to the metal centre with the phosphorus donor atom and via a η2-coordination of non-phosphine containing an aryl-ring as witnessed by Pd–C19_3 and Pd–C20_3 distances (2.367 and 2.490 Å, respectively). This binding motif is quite unusual but has already been observed in other palladium(II) complexes.22
NMR studies confirm that the structure in solution is the same as that in the solid state. As a matter of fact, the integration of the signals in the 1H NMR spectrum proves that the ratio between the indenyl fragment and coordinated phosphine is 1:1. Moreover, seven and nine different signals attributable to indenyl fragment spectra are traceable in 1H and 13C{1H} NMR, respectively, as a direct consequence of the described coordination mode of XPhos. Finally, the only signal present in 31P{1H}NMR spectra resonates at a chemical shift significantly higher than that in free XPhos (Δδ = 65 ppm), certifying the coordination of the phosphine to a more electron-poor metal centre than those of the bis-phosphine-cationic complexes described above.
Synthesis of cationic palladium-indenyl complexes bearing bidentate phosphines
The same synthetic strategy followed for complexes described in the previous paragraph could not be used for the title compounds. In fact, the presence of two phosphorus donor atoms in the designated ligands activates the unwanted reaction reported in Scheme 3, immediately after the mixing of bidentate phosphine with the dimeric precursor [Pd(μ-Cl)(indenyl)]2. This undesired side-reaction cannot be avoided even if a solution of NaClO4 is added in advance. Therefore, we were forced to opt for the preliminary treatment of [Pd(μ-Cl)(indenyl)]2 with silver triflate that ensured exhaustive dechlorination and the formation in situ of [Pd(μ-OTf)(indenyl)]2. The successive addition of bidentate phosphine, followed by the removal of AgCl, afforded the five desired complexes [Pd(indenyl)(P–P)]OTf, with satisfactory yields (Scheme 5). | ||
| Scheme 5 Synthesis of cationic Pd(II)-indenyl complexes bearing bidentate phosphines. | ||
As usual, NMR analysis helps us to confirm the identity of the isolated compounds. As a natural consequence of the coordination of a symmetric chelate ligand on the metal centre, all 31P{1H} NMR spectra are characterised by the presence of a single peak resonating at chemical shifts significantly higher than that in the uncoordinated diphosphine. Among the signals detectable in 1H and 13C{1H} NMR spectra, it is possible to easily distinguish those belonging to the indenyl moiety, among which H1/H3 and C1/C3 can be found at 5.3–6.3 and 90–95 ppm, respectively. Also, in these derivatives the hapticity of the indenyl ligand is intermediate between η3 and η5, as inferred from the small values of Δδ(13C) and ΔM–C, and HA and FA parameters (see Tables S2–7 in the ESI†). The latter were derived from the X-ray structures of complexes 4, 5 and 6 (Fig. 3).
 | ||
| Fig. 3 X-ray molecular structures of 4 (left) and 5 (right) and 6 (down) are presented, showing thermal displacement ellipsoids at the 50% probability level with hydrogen atoms, counterions and solvent molecules omitted for clarity. | ||
Finally, the single peak present in 19F{1H} NMR spectra, detectable at a typical resonance frequency of −78 ppm, certifies the presence of the triflate counterion.
Kinetic and computational studies on indenyl amination
Nucleophilic attack on the Pd-indenyl fragment is surprisingly an unexplored topic considering a large amount of information available on the closely related Pd-allyl group. A kinetic and mechanistic study of this reaction could prove especially useful to better understand the distinctive features of the Pd-indenyl organometallic function in view of its use in synthesis, catalysis and medicinal chemistry. Moreover, it can allow us to assess how the electronic and steric characteristics of the phosphine ligands23 affect this kind of reactivity. A reliable model reaction for probing the susceptibility of the Pd-allyl substrates to nucleophilic attack is typically an amination reaction. In this regard, several studies have amply proved that, in the absence of ligand substitution processes, amine carries out its exclusive attack on the terminal allyl carbon, following a second order kinetic law, with dependence on both complex and amine concentrations.15 Therefore, in the first instance, a series of preliminary tests have been carried out in order to verify whether this also applied to Pd-indenyl substrates. Piperidine was chosen as a reagent probe for its high reactivity due to its strong nucleophilicity and low steric hindrance. The reaction was studied in excess of amine and in the presence of dimethyl fumarate for stabilizing the resulting palladium(0) derivative. As a matter of fact, after the formation of the Pd-η2-indenyl-ammonium complex, the excess of amine promotes its deprotonation and dimethyl fumarate replaces the ensuing indenyl-amine in the Pd(0) coordination sphere. These last two steps are fast and do not affect the reaction rate that is insensitive to the concentration of dimethyl fumarate (Scheme 6). This way of proceeding is particularly useful for preventing the decomposition process that could especially prejudice the spectrophotometric study of the reaction (vide infra). | ||
| Scheme 6 Reaction of indenyl amination with piperidine ([complex] = 5 × 10−5–1 × 10−4 mol dm−3, [dimetylfumarate] = 1.5 × 10−4–3 × 10−4 mol dm−3, [piperidine] ≥ 10[complex]; in CHCl3, T = 298 K). | ||
The reaction monitoring by 1H NMR spectroscopy shows that the initially formed 1-(1H-inden-1-yl)piperidine slowly isomerizes to the more thermodynamically stable 1-(1H-inden-3-yl)piperidine (using DFT calculations the difference amounts to 9.1 kcal mol−1). This process, which is possibly catalysed by basic conditions24 (excess of piperidine), proceeds, in most cases, much more slowly than the indenyl amination of palladium complexes. It is important to stress that the nature of the final organic product 1-(1H-inden-3-yl)piperidine is attested as well as by the presence of its expected signals in the 1H NMR spectra (in good agreement with those of the simulated spectrum) and by GC-MS analysis of the final reaction mixture (Fig. S96 in the ESI†). The identity of the final complexes [Pd(η2-dmfu)(PAr3)2] and [Pd(η2-dmfu)(P–P)] can be unambiguously ascertained by comparing the signals present in the final reaction mixture with those of the 1H and 31P{1H} NMR spectra of these Pd(0) derivatives obtained independently in the past by another synthetic route (e.g. reacting [Pd2(dba)3]·CHCl3 with dmfu and the respective phosphine).25
These preliminary studies also provide us with general information about the rate of the process. Under the experimental conditions used (298 K, CDCl3, [Pd-indenyl complex] = 1 × 10−2 mol dm−3 and [piperidine] = 5 × 10−2 mol dm−3), all reactions are completed in a few minutes, with the only exceptions represented by complexes [Pd(DPPE)(indenyl)]OTf, [Pd(DPPP)(indenyl)]OTf and [Pd(DPPBz)(indenyl)]OTf which need a few hours to reach completion. Excluding the last cases, for which we have reserved a separate treatment (vide infra), a general kinetic study has been carried out taking advantage of UV-Vis spectroscopy. As matter of fact, all our Pd-indenyl derivatives are strongly coloured (deep-orange) and show a strong absorption band at around 400 nm that gradually decreases during the reaction. The presence of an isosbestic point, localized in all cases at around 380 nm, suggests the absence of intermediate species at significant concentrations.
For quantitative kinetic measurements, the course of reactions of CHCl3 solutions of Pd-indenyl complexes (1 × 10−4 or 5 × 10−5 mol dm−3) in the presence of dimethyl fumarate (3 × 10−4 or 1.5 × 10−4 mol dm−3), upon addition of variable aliquots of a large excess of piperidine, was monitored at a wavelength of 400 nm. Under such pseudo-first order conditions the reaction proceeded smoothly to completion in a few minutes, and over this period of time, as proved by previous NMR studies, the isomerization from 1-(1H-inden-1-yl)piperidine to 1-(1H-inden-3-yl)piperidine can be neglected.
The conversion of Pd-indenyl complexes to the final Pd(0) derivatives appears to obey the customary mono-exponential absorbance (At) vs. time (t) relationship (Fig. 4):
| At = A∞ + (A0 − A∞)exp(−kobst) |
 | ||
| Fig. 4 Fit of absorbance to time for reaction 3e + piperidine in the presence of dimethyl fumarate (λ = 400 nm, solvent CHCl3, T = 298 K, [3e] = 1 × 10−4 mol dm−3, [piperidine] = 1 × 10−3 mol dm−3, [dimethyl fumarate] = 3 × 10−4 mol dm−3). The inset shows the linear regression analysis of kobs (s−1) to [piperidine], for the same reaction. | ||
The pseudo first order rate constants kobs were derived by nonlinear regression of absorbance At data to time.
For all reactions examined, kobs values fit the simple linear relationship (inset Fig. 4):
| kobs = k2 [piperidine] |
Therefore, it can be concluded that the kinetic law governing this process is of the second order, with dependence on both the Pd-indenyl complex and amine concentrations:
| rate = k2 [complex][piperidine] |
This result is in accordance with a mechanism where the initial bimolecular attack of amine to the coordinated indenyl fragment represents the rate-determining step of the process, as already observed for the corresponding Pd-allyl complexes (Scheme 6).
The values of second-order constants relative to complexes bearing monodentate phosphines are listed in Table 1.
| Compound | k 2 (mol−1 dm3 s−1) |
|---|---|
| 3a | 55.5 ± 1.9 |
| 3b | 420 ± 20 |
| 3c | 250 ± 20 |
| 3e | 6.4 ± 0.1 |
| 3f | 13.3 ± 0.4 |
| 3g | 410 ± 10 |
| 3h | 13.1 ± 0.7 |
In the light of these data, we can propose the following observations:
(a) Considering the four complexes equipped with monodentate aryl-phosphines 3a–c and 3e, the influence of the different substituents in the para-position is apparent, with the k2 values increasing with an increase in the electron-withdrawing nature of the substituents. This observation is in full agreement with the proposed mechanism whereby a more electron-poor metal centre makes the coordinated indenyl fragment more susceptible to nucleophilic attack. This effect can be somewhat systematized recurring to the Hammett equation: fitting logk2 values vs. tabulated para-substituent σ constants, we can obtain the value of the reaction constant ρ amounting to 3.7 ± 0.7 (Fig. S95 in the ESI†).
(b) The particularly high reactivity of [Pd(indenyl)(P(2-furyl)3)2]ClO4 (3g) is also due to the high electron-withdrawing nature of hetero-aryl phosphines.
(c) Intriguingly, the complex [Pd(indenyl)(P(Ph)2(2-py))2]ClO4 (3f) shows lower reactivity than the very structurally similar [Pd(indenyl)(PPh3)2]ClO4 (3a) as opposed to what would be expected considering that the replacement of a phenyl with a more electron withdrawing pyridyl ring should have an activating effect. We can therefore assume that there is some form of stabilization of the starting complex or destabilization of the transition state of the rate-determining step, promoted by pyridyl-nitrogen.
Limited to complex [Pd(indenyl)(PPh3)2]ClO4 (3a), we have also determined the rate constants k2, when diethylamine and morpholine are used as nucleophiles. The choice of these two different amines was made to better assess the weight of steric and electronic effects on this kind of reaction. As a matter of fact, morpholine has the same steric hindrance as piperidine but less basicity, whereas diethylamine has the same basicity but is more sterically encumbered. Consistently, both resulted in a k2 value lower than that of piperidine (23 ± 1 and 4.9 ± 0.1 vs. 56 ± 2, respectively), but the decrease of one order of magnitude recorded for diethylamine seems to indicate that the steric factor plays a more important role than the electronic one.
The complex [Pd(indenyl)(XPhos)]ClO4 (3h), which differs from those examined so far for the presence of an only coordinated phosphine, reacts with piperidine more slowly than expected considering that the indenyl fragment is bound to a more electron-poor metal centre. This fact is possibly due to more steric congestion around the indenyl ligand (see Fig. 2).
For what concerns the complexes equipped with bidentate phosphines, only two of the prepared compounds (6 and 8) are reactive enough to determine the relative k2 rate constant at the concentrations adopted for spectrophotometric studies. For the remaining three complexes (4, 5, and 7), the reactions have been monitored by 31P{1H} NMR spectroscopy, which allows working at higher concentrations thus making the process faster. A series of preliminary tests have helped us to define the best conditions to carry out the experiments (see the Experimental section). The two equations used to determine the k2 rate constants are in this case:
| d[complex]/dt = k2[complex][piperidine] |
| [piperidine] = [piperidine]0–2[complex] |
| Compound | k 2 (mol−1 dm3 s−1) | Bite angle (°) |
|---|---|---|
| 4 | (4.0 ± 0.2) × 10−2 | 86 |
| 5 | (7.1 ± 0.3) × 10−2 | 91 |
| 6 | 1.81 ± 0.9 | 99 |
| 7 | (2.08 ± 0.06) × 10−2 | 83 |
| 8 | 8.50 ± 0.4 | 104 |
| 9 | 0.607 ± 0.002 | 86 |
It may finally be interesting to compare the reactivity of Pd-indenyl with that of the Pd-allyl fragment. To this end, we have chosen the two complexes coordinating 1,2 bis(diphenylphosphine)ethane [Pd(DPPE)(indenyl)]OTf (4) and [Pd(DPPE)(η3-allyl)]OTf (9). The latter has been synthesized with the same method of its indenyl congener, starting from the dimeric precursor [Pd(μ-Cl)(η3-allyl)]2. The first preliminary test carried out in the NMR tube has shown the higher reactivity of the Pd-allyl complex 9 with piperidine compared to 4, completing the reaction after a few minutes. A more detailed UV-Vis spectrophotometric study has allowed determining the k2 rate constant relative to this substrate that, amounting to 0.607 ± 0.002 mol−1 dm3 s−1, is about one order of magnitude higher than that of the corresponding indenyl derivative 4. This effect can be attributed to the partial η5-character adopted by an indenyl fragment in this type of complex, which strengthens its bond with the palladium centre, and disadvantages the nucleophilic attack.
With these data in hand, DFT calculations were performed. X-ray molecular structures (when available) were used as a starting point for geometry optimizations.
Initial efforts were made to compare the trend in the rate constants with the energy barriers of the rate-determining step. To this end we selected the two most active systems, 3b and 3g, and the two of the least active ones, 3a and 3f. As shown in Fig. 5, the calculated energy barriers for the initial nucleophilic attack of piperdine to the indenyl fragment are 13.0 and 12.7 kcal mol−1 for 3b and 3g (high reactivity) and 13.8 and 15.4 for 3a and 3f (lower reactivity), respectively. This is in agreement with the experimentally observed trend of rate constants.
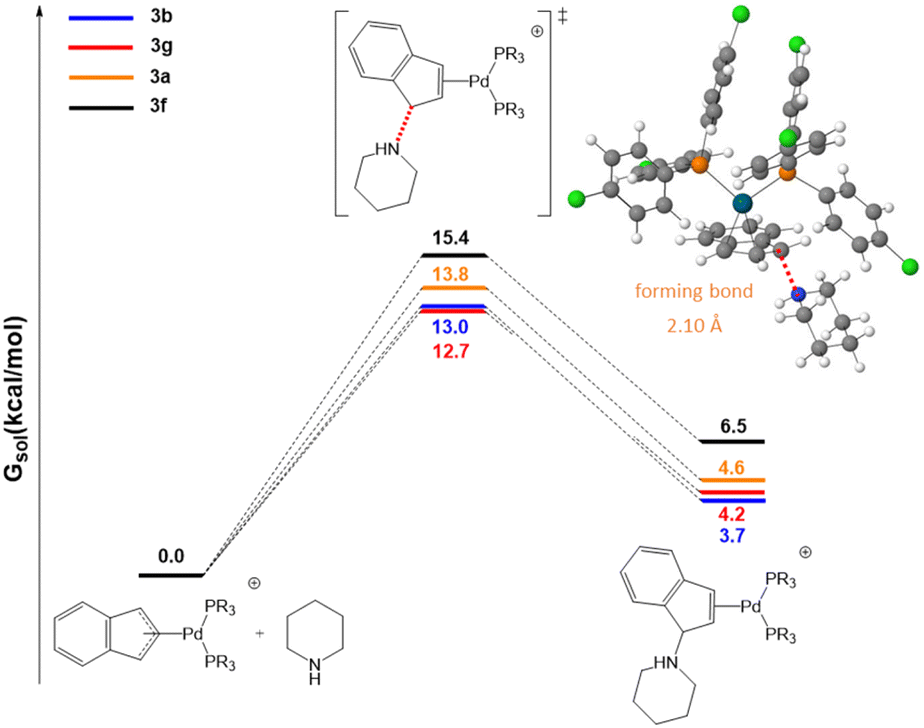 | ||
| Fig. 5 Free energy profiles in chloroform for piperidine attack on the indenyl moiety. The inset shows the optimized geometry of transition state 3b. | ||
Having reproduced the experimental trends, we performed an analysis of the steric and electronic properties of the various systems to shed light on the structural parameters impacting most of the reactivity behavior. Focusing on steric features, a comparison of the %Vbur and the bite angle results in a strong correlation, R2 = 0.81, for 11 Pd-indenyl complexes (3a–3g of Table 1, bearing two monodentate phosphines, and 4–8 of Table 2, bearing a bidentate phosphine and an indenyl moiety). This indicates that %Vbur and the bite angles can be interchanged as descriptors capturing steric pressure on the active center. As for electronic descriptors, we focused on the charge from natural population analysis (NPA) on the indenyl group, the Pd atom, and the ligand. In addition, we also considered the HOMO and LUMO energies.
Considering the small number of systems, 11, that mono- and bidentate phosphines are intrinsically different in flexibility and that the corresponding complexes contain different counterions, attempts to build a statistically robust regression model failed. We will thus provide qualitative analysis of trends for the 6 and 5 complexes bearing a mono- or bidentate phosphine. The R2 values from univariate linear regression between the single descriptors and the experimental rate constants are reported in Table 3.
| Descriptor | Monodentate | Bidentate |
|---|---|---|
| %VBur | 0.17 | 0.47 |
| Bite angle | 0.20 | 0.52 |
| E(HOMO) | 0.70 | 0.09 |
| E(LUMO) | 0.65 | 0.01 |
| E(LUMO) − E(HOMO) | 0.02 | 0.09 |
| NPA indenyl | 0.59 | 0.54 |
| NPA Pd | 0.38 | 0.49 |
| NPA Phosphine | 0.35 | 0.66 |
| NPA indenyl-Pd | 0.76 | 0.01 |
| NPA ind − (Pd + ligand) | 0.63 | 0.54 |
| E diss (indenyl) | 0.67 | 0.95 |
Starting with monodentate systems, poor correlation is observed between the two steric descriptors and the rate constants, with R2 values of approximately 0.2. This poor correlation could either be due to the flexibility of mono-phosphines, impossible to capture with a single geometry, or the fact that four out of the six mono-phosphines differ by the nature of the para substituents. This should minimize differences in steric hindrance while impacting electronic properties. Indeed, a stronger correlation is observed between the rate constants and the HOMO and LUMO energies, as well as the NPA charges, especially of the indenyl ligand, with R2 values of approximately 0.6–0.7. Higher activity corresponds to less electron-rich indenyls, or complexes having lower energy LUMOs, which would be more inclined to undergo nucleophilic attack. Consistently, the LUMO of 3b reported in Fig. 6 is clearly localized on these C atoms. A strong correlation is also achieved using the difference between the indenyl and Pd NPA charges, which can be taken as a measure of the polarization of the Pd-indenyl interaction, with an R2 value of 0.76. Higher rate constants correspond to more polarized Pd-indenyl bonds.
 | ||
| Fig. 6 Graphic representation of the LUMO of 3b. | ||
Moving to diphosphine based complexes, a reasonable correlation is observed between the steric descriptors and the rate constants, with R2 values of approximately 0.5. The stronger impact of steric hindrance is in line with the clearly different steric requirements of the four bidentate phosphines, with larger %VBur and bite angles corresponding to higher rate constants. The topographic steric maps in Fig. 7 show how the hindrance in 8 results in a smaller reactive pocket compared to that of 7. A reasonable correlation also results from using NPA charges as electronic descriptors, with R2 values of approximately 0.5. A reasonable correlation is again calculated with the NPA charge on the indenyl moieties, with higher constant rates again corresponding to more electron deficient indenyls.
 | ||
| Fig. 7 Steric maps of complexes 7 and 8. | ||
A comparison between R2 values of the considered mono- and bidentate phosphines seems to indicate that the rate constants of complexes with monodentate phosphines are mostly determined by electronic features, whereas those of complexes with bidentate phosphines are determined by both steric and electronic features. Interestingly, the rate constants of both mono- and bidentate-based complexes correlate well with the indenyl dissociation energies. This can be understood considering that dissociation energies are influenced by both steric and electronic effects, and thus could be considered as a versatile descriptor.
Antiproliferative activity towards ovarian cancer and normal cells
At the end of this work, we propose potential for application of this class of compounds, a study of their antiproliferative activity towards cancer cells. In this regard, it is pointed out that in the literature only our recent contribution describes the behaviour of a particular class of Pd-indenyl derivatives in a biological environment,26 and therefore this issue is worth further investigating. The tumour cell lines chosen for in vitro assays were ovarian cancer cells OVCAR-5, and A2780 and its cisplatin resistant clone A2780cis, all being particularly sensitive to treatment with organopalladium complexes. Our previous studies have highlighted the effectiveness of Pd-allyl derivatives, which present, as stated before, some common characteristics with Pd-indenyl substrates.17The list of the tested compounds includes only those complexes that have shown no significant decomposition after 24 hours in 1:1 DMSO-d6/D2O solution. Unfortunately, all complexes with para-substituted aryl phosphines 3b–e do not meet this requirement.
To this series of Pd-indenyl compounds we have also added the complex [Pd(DPPE)(η3-allyl)]OTf (9), in order to compare, on equal terms, the behaviours of Pd-indenyl and Pd-allyl fragments.
The cytotoxicity data of our compounds on the three ovarian cancer cell lines are summarised in Table 4, in terms of IC50 values (half inhibitory concentrations). In the same table, data referred to a line of normal cells (MRC-5, human lung fibroblasts) are also reported for comparison. Moreover, all the tests have also been extended to cisplatin as a positive control.
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| A2780 | A2780cis | OVCAR-5 | MRC-5 | |
| Data after 96 h of incubation. Stock solutions in DMSO for all complexes; stock solutions in H2O for cisplatin. A2780, cisplatin-sensitive ovarian cancer cells; A2780cis, cisplatin-resistant ovarian cancer cells; OVCAR-5, high-grade serous ovarian cancer cells; and MRC-5, normal lung fibroblasts. | ||||
| Cisplatin | 0.9 ± 0.1 | 32 ± 9 | 1.3 ± 0.1 | 3.48 ± 0.09 |
| 3a | 0.27 ± 0.02 | 0.26 ± 0.04 | 0.34 ± 0.08 | 2.5 ± 0.4 |
| 3f | 0.26 ± 0.03 | 0.30 ± 0.04 | 0.48 ± 0.02 | 0.9 ± 0.2 |
| 3g | 0.13 ± 0.05 | 3.0 ± 0.2 | 4.6 ± 0.5 | 2.27 ± 0.09 |
| 3h | 0.73 ± 0.07 | 1.2 ± 0.2 | 1.9 ± 0.2 | 3.7 ± 0.3 |
| 4 | 0.122 ± 0.007 | 0.22 ± 0.07 | 0.10 ± 0.01 | 0.7 ± 0.1 |
| 5 | 0.12 ± 0.03 | 0.19 ± 0.08 | 0.075 ± 0.002 | 0.51 ± 0.03 |
| 6 | 0.18 ± 0.02 | 0.17 ± 0.03 | 0.13 ± 0.02 | 0.32 ± 0.03 |
| 7 | 0.062 ± 0.009 | 0.17 ± 0.05 | 0.15 ± 0.02 | 0.306 ± 0.003 |
| 8 | 0.29 ± 0.02 | 0.29 ± 0.07 | 0.336 ± 0.008 | 2.4 ± 0.4 |
| 9 | 0.020 ± 0.004 | 0.051 ± 0.008 | 0.03 ± 0.02 | 0.15 ± 0.06 |
On the basis of the obtained data, the following remarks may be proposed:
(a) All complexes show high antiproliferative activity towards all three types of ovarian cancer cells, with IC50 values always significantly lower than cisplatin (often of about one order of magnitude in the cases of cisplatin sensitive A2780 and OVCAR-5 cell lines). In this respect, we emphasize that OVCAR-5 cells are high-grade serous ovarian cancer (HGSOC) cells, the most common and deadly form of the disease.
(b) With the only exception of the complex coordinating the trifurylphosphine (3g), all assayed Pd-indenyl derivatives exhibit the same degree of toxicity towards cisplatin-sensitive A2780 and cisplatin-resistant A2780cis cell lines. This suggests that our complexes might operate with a mechanism of action different from that of cisplatin (classical DNA metalation). Remarkably, the fact that their IC50 values are lower by about two orders of magnitude than cisplatin qualifies these palladium compounds as potential metallodrugs toward forms of cancer resistant to cisplatin.
(c) The cytotoxicity of all diphosphine Pd-indenyl complexes against tumour cells appears basically independent of the nature of the phosphine used (monodentate or chelate). However, the complex coordinating only one phosphine ([Pd(indenyl)(XPhos)]ClO4 (3h) seems to be slightly less active.
(d) Unfortunately, in most cases our complexes have the same toxicity on tumour and normal cells (MRC-5). The only noteworthy exceptions have been represented by complexes [Pd(indenyl)(PPh3)2]ClO4 (3a) and [Pd(DPEphos)(indenyl)]OTf (8) that show significant selectivity (ca. one order of magnitude) towards cancer cells.
(e) From the comparison between the IC50 values of [Pd(DPPE)(indenyl)]OTf (4) and [Pd(DPPE)(η3-allyl)]OTf (9) it is apparent that the latter complex is more cytotoxic than the former on all tested cell lines. This could make us think that the susceptibility of the polyenyl fragment to undergo nucleophilic attack plays a key role in the mechanism of action of this kind of complex.
Conclusions
The results achieved in this work contribute to deepening the knowledge of palladium(II)-indenyl compounds, highlighting some of their features and potential. In particular, our attention has focused on cationic derivatives bearing phosphines as ancillary ligands. First, we have refined the synthetic protocols for the preparation of complexes with mono- and bidentate phosphines, providing detailed information on the structural and spectroscopic characteristics of the products. A characteristic common to all complexes is the hapticity of the indenyl fragment that is always intermediate between η3 and η5.These compounds have proven to be ideal for a systematic study of nucleophilic attacks on coordinated-indenyl, an issue that had never been addressed to date. For the case in which the designed nucleophile is an amine, an in-depth kinetic analysis has allowed us to establish the rate law and the consequent reaction mechanism. DFT calculations have confirmed that the rate-determining step of the process is the initial nucleophilic attack of amine on the coordinated indenyl-fragment. This bimolecular step is favoured by the decrease of electron density on the indenyl-fragment and the steric hindrance of ligands. A systematic study, based on experimental data, made it possible to select the most suitable steric and electronic descriptors to predict the susceptibility of the Pd-indenyl complexes to nucleophilic attack. Among steric descriptors, the possibility of using the bite angle of bidentate phosphines should be emphasised, in addition to the most predictable %Vbur.
An interesting and never before attempted application of these new compounds concerns their behaviour in the biological environment. As a matter of fact, a good number of the synthesized Pd-indenyl complexes have shown significant antiproliferative activity towards three different ovarian cancer cell lines. Their cytotoxicity is nearly always significantly higher than cisplatin, used as a positive control. Furthermore, with their effectiveness being basically the same on cisplatin-sensitive (A2780) and cisplatin-resistant (A2780cis) cells, it is reasonable to suppose that these novel complexes present a mechanism of action different from the classical DNA metalation of platinum chemotherapeutics drugs. Complexes 3a and 8 deserve a special mention, coordinating PPh3 and DPEphos respectively that exhibit significant selectivity towards tumor cells.
Experimental
Solvents and reagents
All syntheses were carried out under an inert atmosphere using standard Schlenk techniques. The solvent CH2Cl2 was distilled over P2O5 and stored under a N2 atmosphere. CHCl3 was distilled and stored in silver foil. All other solvents and chemicals were commercial grade products and used as purchased. [Pd(μ-Cl)(indenyl)]2 and Pd(II)-allyl complex 9 were synthesized according to the published protocols.27,28NMR, UV-Vis and IR measurements
1D-NMR and 2D-NMR spectra were recorded on Bruker 300 or 400 Advance spectrometers. Chemical shift values (ppm) are given relative to TMS (1H and 13C), H3PO4 (31P) and CCl3F (19F).IR spectra were recorded on a PerkinElmer Spectrum One spectrophotometer and UV-Vis spectra were recorded on a PerkinElmer Lambda 40 spectrophotometer equipped with a PerkinElmer PTP 6 (Peltier temperature programmer) apparatus.
Computational details
Geometries were optimized with the Gaussian0929 package using the PBE0-D3 functional.30–32 The electronic configuration of the system was described with the split-valence SVP basis set33 for main group atoms (C, H, N, P, F, Cl and O) and the relativistic Stuttgart–Dresden effective core potential with the associated valence triple-ζ basis set for Pd and Fe.34 All geometries were confirmed as the minimum or transition state through frequency calculations. The reported free energies were built through single point energy calculations on the PBE0-D3 geometries using the M06 functional and the triple-ζ TZVP basis set for main group atoms.35–37 Solvent effects were included with the PCM model using chloroform as the solvent.38,39 To this M06/TZVP electronic energy in solvent, thermal corrections were added from the gas-phase frequency calculations at the PBE0-D3/SVP level.Crystal structure determination
3b, 3h, 4, 5 and 6 crystal data were collected at the XRD1 and XRD2 beamlines of the Elettra Synchrotron, Trieste (Italy),40 using a monochromatic wavelength of 0.620 Å, at 100 K (298K for 5). The data sets were integrated, scaled and corrected for Lorentz, absorption and polarization effects using the XDS package.41 Data from two random orientations of the same crystals have been merged to obtain complete datasets for the triclinic 3h crystal form, using the CCP4-Aimless code.42,43 The structures were solved by direct methods using the SHELXT program44 and refined using full-matrix least-squares implemented in SHELXL-2018/3.45 Thermal motions for all non-hydrogen atoms have been treated anisotropically and hydrogens have been included on calculated positions, riding on their carrier atoms. Geometric restrains (SAME, SADI, DFIX, DANG) have been applied to disordered fragments (triflate counterion in 5 and solvent in 3h). The Coot program was used for structure building.46 The crystal data are given in the ESI.† Pictures were prepared using Ortep347 and Pymol48 software. Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre and allocated the deposition numbers CCDC 2173702, 2173705, 2173701, 2173703, and 2173704 for 3b, 3h, 4, 5, and 6, respectively.†Preliminary studies and kinetic measurements
All the reactions were firstly studied by 1H and 31P{1H} NMR spectroscopy by preparing a CDCl3 solution of the selected complex at a concentration range of 0.008–0.010 mol dm−3 in the presence of dimethyl fumarate (0.012–0.015 mol dm−3), in order to stabilize the final Pd(0) complex formed after a nucleophilic attack at the indenyl fragment. An appropriate amount of piperidine (0.040–0.050 mol dm−3) was added and the reaction was followed monitoring the disappearance of the Pd(II) complex and the concomitant appearance of the Pd(0) olefin complex and the final organic product.In the case of complex 3a morpholine and diethylamine were also employed as nucleophilic agents.
The kinetic studies of complexes 3a, 3b, 3c, 3e, 3f, 3g, 3h, 5, and 8 were carried out by the UV-Vis technique. A series of preliminary tests were performed in order to evaluate the wavelength of the highest absorbance change. The complex of interest was dissolved in CHCl3 ([complex] = 1 × 10−4–5.0 × 10−5 mol dm−3) in the presence of dimethyl fumarate (3.0 × 10−4–5.0 × 10−5 mol dm−3) and was placed in a thermostated cell compartment at 298 K in a spectrophotometer. An adequate excess of piperidine was added ([piperidine] ≥ 10[complex]) and the reactions were monitored by recording spectra as a function of time at a wavelength interval of 366 to 600 nm. The kinetics of the nucleophile attack was studied by recording spectra with a fixed wavelength for each complex evaluated from 378 to 398 nm under pseudo-first order conditions. To a solution of the complex in 3 mL of CHCl3 ([complex] = 10−4–5.0 × 10−5 mol dm−3) in the presence of dimethyl fumarate (3.0 × 10−4–5.0 × 10−5 mol dm−3), known aliquots of piperidine were added using fresh mother solution in CHCl3 ([piperidine] = 0.06–0.14 mol dm−3) (Fig. S80–S91†).
In the case of complexes 4, 5, and 7 the reactions were too slow for UV-Vis conditions. Therefore, the kinetics of the nucleophilic attack was studied by 1H NMR spectroscopy following the integration change of one characteristic peak related to the initial complex. All spectra were recorded at 298 K in CDCl3 where [complex]0:[dmfu]:[piperidine]0 = 0.01:0.015:0.04 mol dm−3 (Fig. S92–S94†).
Synthesis of neutral Pd-indenyl complexes
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 4.58 (ddd, J = 2.7, 2.0, 0.8 Hz, 1H, H3), 6.38 (d, J = 7.5 Hz, 1H, H4), 6.48–6.54 (m, 1H, H1), 6.71–6.74 (m, 1H, H2), 6.87 (t, J = 7.5 Hz, 1H, H5), 7.07 (t, J = 7.5 Hz, 1H, H6), 7.26 (d, J = 7.5 Hz, 1H, H7), 7.34–7.52 (m, 15H, Ar–H).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 27.5.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 4.63–4.65 (m, 1H, H3), 6.44 (d, J = 7.5 Hz, 1H, H4), 6.54–6.60 (m, 1H, H1), 6.69–6.72 (m, 1H, H2), 6.91 (t, J = 7.5 Hz, 1H, H5), 7.10 (t, J = 7.5 Hz, 1H, H6), 7.28 (d, J = 7.5 Hz, 1H, H7), 7.42–7.31 (m, 12H, Ar–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 80.3 (CH, d, JC–P = 3.8 Hz, C3), 97.7 (CH, d, JC–P = 23.2 Hz, C1), 111.4 (CH, d, JC–P = 5.8 Hz, C2), 116.5 (CH, C4), 120.0 (CH, C7), 126.8 (CH, C5), 127.7 (CH, C6), 129.3 (CH, d, JC–P = 11.5 Hz, Ar–CH), 129.8 (C, d, JC–P = 45.6 Hz, ipso-Ar–C), 134.8 (C, d, JC–P = 1.7 Hz, C3a), 135.1 (CH, d, JC–P = 13.6 Hz, Ar–CH), 135.7 (C, d, JC–P = 4.9 Hz, C7a), 137.9 (C, d, JC–P = 2.6 Hz, p-Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 26.0.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 4.64 (pseudo t, 1H, H3), 6.44 (d, J = 7.5 Hz, 1H, H4), 6.55–6.59 (m, 1H, H1), 6.72–6.76 (m, 1H, H2), 6.92 (t, J = 7.5 Hz, 1H, H5), 7.12 (td, J = 8.6, 1.6 Hz, 7H, H6, Ar–H), 7.30 (d, J = 7.5 Hz, 1H, H7), 7.42–7.50 (m, 6H, Ar–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 79.8 (CH, d, JC–P = 3.7 Hz, C3), 97.3 (CH, d, JC–P = 23.6 Hz, C1), 111.3 (CH, d, JC–P = 5.9 Hz, C2), 116.1 (CH, dd, JC–F, C–P = 21.4, 12.0 Hz, m-Ar–CH), 116.4 (CH, C4), 119.8 (CH, C7), 126.51 (CH, C5), 127.3 (C, dd, JC–P, C–F = 47.6, 3.5 Hz, ipso-Ar–C), 127.4 (CH, C6), 134.7 (C, d, JC–P = 1.5 Hz, C3a), 135.7 (C, d, JC–P = 5.0 Hz, C7a), 135.8 (CH, dd, JC–P, C–F = 14.2, 8.5 Hz, o-Ar–CH), 164.3 (C, dd, JC–F, C–P = 253.5, 2.5 Hz, p-Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 25.3 (q, JP–F = 2.5 Hz).
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −108.0 (d, JF–P = 2.5 Hz).
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 4.71 (pseudo t, 1H, H3), 6.39 (d, J = 7.4 Hz, 1H, H4), 6.60–6.67 (m, 1H, H1), 6.72 (pseudo t, 1H, H2), 6.92 (t, J = 7.5 Hz, 1H, H5), 7.14 (t, J = 7.5 Hz, 1H, H6), 7.31 (d, J = 7.4 Hz, 1H, H7), 7.54–7.76 (m, 12H, Ar–H).
13C{1H} NMR (100.0 MHz, CDCl3, T = 298 K, ppm) δ: 80.9 (CH, d, JC–P = 3.6 Hz, C3), 98.2 (CH, d, JC–P = 23.1 Hz, C1), 111.6 (CH, d, JC–P = 5.9 Hz, C2), 116.5 (CH, C4), 120.2 (CH, C7), 123.5 (C, d, JC–F = 272.8 Hz, CF3–C), 125.8–126.0 (CH, o,m-Ar–CH), 127.2 (CH, C5), 128.1 (CH, C6), 133.4 (C, dd, JC–P, C–F = 32.9, 2.6 Hz, p-Ar–C), 134.3–134.6 (CH, o,m-Ar–CH), 134.8 (C, JC–P = 1.5 Hz, C3a), 135.2 (C, d, JC–F = 42.5 Hz, ipso-Ar–C), 135.6 (C, d, JC–P = 5.0 Hz, C7a).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 28.0.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −63.2.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.82 (s, 9H, OCH3) 4.57 (pseudo t, 1H, H3), 6.46 (d, J = 7.8 Hz, 2H, H1, H4), 6.72 (pseudo t, 1H, H2), 6.89 (d, J = 8.7 Hz, 7H, H5, Ar–H), 7.07 (t, J = 7.5 Hz, 1H, H6), 7.25 (d, J = 7.8 Hz, 1H, H7), 7.38 (dd, J = 11.3, 8.7 Hz, 6H, Ar–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 55.5 (CH3, OCH3), 78.9 (CH, d, JC–P = 3.9 Hz, C3), 96.9 (CH, d, JC–P = 23.2 Hz, C1), 111.2 (CH, d, JC–P = 5.9 Hz, C2), 114.2 (CH, d, JC–P = 11.9 Hz, o-Ar–CH), 116.8 (CH, C4), 119.6 (CH, C7), 123.7 (C, d, JC–P = 50.9 Hz, ipso-Ar–C), 126.1 (CH, C5), 127.0 (CH, C6), 135.1 (C, d, JC–P = 1.9 Hz, C3a), 135.5 (CH, d, JC–P = 13.7 Hz, m-Ar–CH), 136.2 (C, d, JC–P = 4.7 Hz, C7a), 161.5 (C, d, JC–P = 2.4 Hz, p-Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 23.5.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 4.82 (pseudo t, 1H, H3), 6.39 (d, J = 7.5 Hz, 1H, H4), 6.52–6.58 (m, 1H, H1), 6.67–6.70 (m, 1H, H2), 6.84 (t, J = 7.5 Hz, 1H, H5), 7.06 (t, J = 7.5 Hz, 1H, H6), 7.25 (d, J = 7.5 Hz, 1H, H7) 7.28–7.74 (m, 13H, Ar–H, Py–H), 7.79 (d, J = 4.2 Hz, 1H, 6-Py–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 78.5 (CH, C3), 97.7 (CH, d, JC–P = 23.0 Hz, C1), 111.4 (CH, d, JC–P = 5.9 Hz, C2), 116.9 (CH, C4), 119.6 (CH, C7), 124.3 (CH, d, JC–P = 2.4 Hz, 3-Py–C), 126.4 (CH, C5), 127.2 (CH, C6), 128.5 (CH, d, JC–P = 10.8 Hz, Ar–CH), 128.6 (CH, d, JC–P = 10.8 Hz, Ar–CH), 130.8–131.5 (C, CH, Ar–C, Py–CH), 132.1 (C, C3a) 134.2 (CH, d, JC–P = 12.1 Hz, Ar–CH), 134.5 (CH, d, JC–P = 12.1 Hz, Ar–CH), 134.9 (C, C7a) 136.0 (CH, d, JC–P = 4.6 Hz, Py–CH), 136.1 (CH, d, JC–P = 9.5 Hz, Py–CH), 150.4 (CH, d, JC–P = 14.6 Hz, 6-Py), 156.8 (C, d, JC–P = 65.4 Hz, 2-Py).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 29.3.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 5.52 (pseudo t, 1H, H3), 6.49–6.44 (m, 3H, Furyl–H), 6.64 (bd, 1H, H1), 6.73–6.68 (m, 2H, H2, H4), 6.91 (t, J = 7.5 Hz, 1H, H5), 6.97 (t, J = 2.9 Hz, 3H, Furyl–H), 7.07 (t, J = 7.5 Hz, 1H, H6), 7.25 (d, J = 6.5 Hz, 1H, H7), 7.71 (s, 3H, Furyl–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 76.2 (CH, d, JC–P = 3.6 Hz, C3), 98.9 (CH, d, JC–P = 25.3 Hz, C1), 111.4 (CH, d, JC–P = 5.7 Hz, C2), 111.6 (CH, d, JC–P = 8.7 Hz, Furyl–CH), 118.3 (CH, C4), 119.8 (CH, C7), 124.7 (CH, d, JC–P = 23.3 Hz, Furyl–CH), 126.8 (CH, C5), 127.8 (CH, C6), 135.2 (C, d, JC–P = 1.5 Hz, C3a), 136.3 (C, d, JC–P = 5.5 Hz, C7a), 143.5 (C, d, JC–P = 71.9 Hz, Furyl–C) 148.7 (CH, d, JC–P = 5.3 Hz, Furyl–CH).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: −28.5.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 0.94 (d, J = 6.7 Hz, 6H, iPr–CH3–H), 1.01 (d, J = 6.7 Hz, 6H, iPr–CH3–H), 0.86–1.12 (m, 2H, Cy–CH–H), 1.28 (dt, J = 7.0, 4.2 Hz, 6H, iPr–CH3–H), 1.23–1.50 (br, 16H, Cy–CH2–H), 1.58 (br, 4H, Cy–CH2–H), 2.46 (p, J = 6.8 Hz, 1H, iPr–CH–H), 2.66 (p, J = 6.8 Hz, 1H, iPr–CH–H), 2.93 (p, J = 6.8 Hz, 1H, iPr–CH–H), 4.98 (bs, 1H, H3), 6.22–6.27 (m, 1H, H1), 6.59–6.63 (m, 1H, H2), 6.83 (d, J = 7.4 Hz, H4), 6.91 (t, J = 7.4 Hz, H5), 6.95–7.10 (m, 4H, H6, Ar–H), 7.20 (d, J = 7.4 Hz, 1H, H7), 7.34–7.39 (m, 2H, Ar–H), 7.79–7.89 (m, 1H, Ar–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 22.4–28.2 (CH3, CH2, CH, iPr–CH3, Cy–CH2, Cy–CH), 30.9 (CH, iPr–CH), 31.0 (CH, iPr–CH), 34.6 (CH, iPr–CH), 73.3 (CH, d, JC–P = 3.7 Hz, C3), 94.8 (CH, d, JC–P = 22.5 Hz, C1), 111.5 (CH, d, JC–P = 6.0 Hz, C2), 117.2 (CH, C4), 119.5 (CH, C7), 121.2 (CH, d, JC–P = 3.9 Hz, Ar–CH), 125.3 (CH, C5), 126.0 (CH, JC–P = 13.8 Hz, Ar–CH), 126.8 (CH, C6), 128.5 (CH, d, JC–P = 2.4 Hz, Ar–CH), 132.3 (C, d, JC–P = 27.1 Hz, ipso-Ar–C), 133.8 (CH, d, JC–P = 6.4 Hz, Ar–CH), 136.4 (C, d, JC–P = 1.5 Hz, C3a), 136.5–136.6 (C, Ar–C), 137.4 (C, d, JC–P = 4.7 Hz, C7a), 137.6–138.1 (CH, Ar–CH), 141.4 (C, Ar–C), 146.4 (C, d, JC–P = 7.3 Hz, Ar–C), 149.2 (C, Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 57.9.
All neutral complexes [PdCl(indenyl)(PAr3)] isolated contain always small traces of the respective Pd(I) dimers [(μ-η3-ind)(μ-Cl)Pd2(PAr3)2], and for this reason the elemental analyses were not helpful for their characterization.
Synthesis of cationic Pd-indenyl complexes
[Pd(indenyl)(PPh3)2]ClO4 (3a). The neutral complex 2a was obtained by the reaction between 0.0200 g (0.0389 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0204 g (0.0778 mmol) of PPh3 using 4 mL of anhydrous CH2Cl2. 0.0208 g (0.1483 mmol) of NaClO4·H2O dissolved in 2 mL of methanol and a solution of 0.0204 g (0.0778 mmol) of PPh3 in 2 mL of CH2Cl2 (CH2Cl2/MeOH ≈ 3/1) were added. The mixture was stirred at room temperature for 15 min and the solvent was removed at low pressure. 5 mL of CH2Cl2 was added, the mixture was filtered on Millipore apparatus and the solution was concentrated under vacuum. The title complex is precipitated by the addition of diethyl ether and dried under vacuum. 0.0600 g (yield 96%) was obtained as a dark orange powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 5.48–5.51 (m, 2H, H1, H3), 6.18–6.28 (m, 2H, H4, H7), 7.06–7.12 (m, 14H, H5, H6, Ar–H), 7.30 (t, J = 7.3 Hz, 13H, H2, Ar–H), 7.21 (t, J = 7.4 Hz, 6H, p-Ar–H).
13C{1H} NMR (100.0 MHz, CDCl3, T = 298 K, ppm) δ: 95.6 (CH, t, JC–P = 11.7 Hz, C1, C3), 114.0 (CH, t, JC–P = 5.3 Hz C2), 118.9 (CH, C4, C7), 127.9 (CH, C5, C6), 129.1 (CH, t, JC–P = 5.8 Hz, Ar–CH), 130.2 (C, dd, JC–P = 24.0, 23.1 Hz, ipso-Ar–C), 131.4 (CH, p-Ar–CH), 132.0 (C, t, JC–P = 3.2 Hz, C3a, C7a), 133.6 (CH, t, JC–P = 6.1 Hz, Ar–CH).
31P{1H} NMR (162.0 MHz, CDCl3, T = 298 K, ppm) δ: 26.4.
IR (KBr pellet, cm−1): νClO = 1091, δClO = 623.
Elemental analysis calcd (%) for C45H37ClO4P2Pd: C, 63.92, H, 4.41; found: C, 63.64, H, 4.58.
HRMS calcd for [C45H37P2Pd]+: 745.1405; found: 745.1440.
[Pd(indenyl)(P(p-Cl-C6H4)3)2]ClO4 (3b). The neutral complex 2b was obtained by the reaction between 0.0504 g (0.0980 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0717 g (0.1961 mmol) of tris(4-Cl-phenyl)phosphine using 5 mL of anhydrous CH2Cl2. The cationic complex 3b was obtained in the same manner as complex 3a using 0.0551 g (0.3922 mmol) of NaClO4·H2O dissolved in 3 mL of methanol and a solution of 0.0717 g (0.1961 mmol) of tris(4-Cl-phenyl)phosphine in 4 mL of CH2Cl2. 0.1733 g (yield 84%) of complex 3b was obtained as a pale orange powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 5.50–5.53 (m, 2H, H1, H3), 6.47–6.49 (m, 2H, H4, H7), 7.03–7.09 (m, 12H, o-Ar–H), 7.14–7.17 (m, 2H, H5, H6), 7.33 (d, J = 8.0 Hz, 12H, m-Ar–H), 7.56 (t, J = 3.3 Hz, H2).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 97.0 (CH, t, JC–P = 11.7 Hz, C1, C3), 116.2 (CH, t, JC–P = 5.6 Hz C2), 118.9 (CH, C4, C7), 128.0 (C, dd, JC–P = 23.8, 23.7 Hz, ipso-Ar–C), 128.2 (CH, C5, C6), 129.7 (CH, t, JC–P = 5.8 Hz, m-Ar–CH), 132.3 (C, C3a, C7a), 134.7 (CH, t, JC–P = 6.7 Hz, o-Ar–CH), 138.8 (C, p-Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 24.4.
IR (KBr pellet, cm−1): νClO = 1089, δClO = 622.
Elemental analysis calcd (%) for C45H31Cl7O4P2Pd: C, 51.37, H, 2.97; found: C, 51.69, H, 2.80.
HRMS calcd for [C45H31Cl6P2Pd]+: 952.9042; found: 952.9018.
[Pd(indenyl)(P(p-F-C6H4)3)2(indenyl)]ClO4 (3c). The neutral complex 2c was obtained by the reaction between 0.0507 g (0.0986 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0624 g (0.1972 mmol) of tris(4-F-phenyl)phosphine using 6 mL of anhydrous CH2Cl2. The cationic complex 3c was obtained in the same way as complex 3a using 0.0554 g (0.3944 mmol) of NaClO4·H2O dissolved in 3 mL of methanol and a solution of 0.0624 g (0.1972 mmol) of tris(4-F-phenyl)phosphine in 3 mL of CH2Cl2. 0.1580 g (yield 80%) of complex 3c was obtained as a pale orange powder.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 5.49–5.53 (m, 2H, H1, H3), 6.41–6.44 (m, 2H, H4, H7), 7.00–7.21 (m, 26H, H5, H6, Ar–H) 7.54 (t, J = 3.3 Hz, H2).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 96.4 (CH, t, JC–P = 11.9 Hz, C1, C3), 116.0 (CH, d, JC–P = 4.6 Hz C2), 116.8 (CH, td, JC–F, C–P = 21.6, 6.2 Hz, m-Ar–CH), 118.9 (CH, C4, C7), 125.9 (C, dd, JC–P, C–P, C–F = 26.4, 24.0, 3.4 Hz, ipso-Ar–C), 128.0 (CH, C5, C6), 132.3 (C, t, JC–P = 3.2 Hz, C3a, C7a), 135.8 (CH, dt, JC–P = 7.6 Hz, JC–F = 7.6 Hz, o-Ar–CH), 164.6 (C, d, JC–F = 255.8 Hz, p-Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 24.0.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −106.0
IR (KBr pellet, cm−1): νClO = 1091, δClO = 623.
Elemental analysis calcd (%) for C45H31ClF6O4P2Pd: C, 56.68, H, 3.28; found: C, 57.01, H, 3.10.
HRMS calcd for [C45H31F6P2Pd]+: 853.0840; found: 853.0895.
[Pd(indenyl)(P(p-OCH3-C6H4)3)2]ClO4 (3e). The neutral complex 2e was obtained by the reaction between 0.0495 g (0.0962 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0679 g (0.1925 mmol) of tris(4-OCH3-phenyl)phosphine using 4 mL of anhydrous CH2Cl2. The cationic complex 3e was obtained in the same way as complex 3a using 0.0541 g (0.3848 mmol) of NaClO4·H2O dissolved in 3 mL of methanol and a solution of 0.0679 g (0.1925 mmol) of tris(4-OCH3-phenyl)phosphine in 5 mL of CH2Cl2. 0.1940 g (yield 98%) of complex 3e were obtained as a pale orange powder.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 3.83 (s, 18H, o-CH3–H), 5.37–5.41 (m, 2H, H1, H3), 6.41–6.44 (m, 2H, H4, H7), 6.80 (d, J = 8.1 Hz, 12H, Ar–H), 6.90–7.00 (m, 12H, Ar–H), 7.11–7.15 (m, 3H, H2, H5, H6).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm) δ: 55.7 (CH, OCH3), 94.5 (CH, t, JC–P = 12.0 Hz, C1, C3), 113.3 (CH, C2), 114.5 (CH, t, JC–P = 6.0 Hz, o, m-Ar–CH), 118.9 (CH, C4, C7), 122.0 (C, dd, JC–P = 26.7, 26.4 Hz, ipso-Ar–C), 127.4 (CH, C5, C6), 132.0 (C, t, JC–P = 3.1 Hz, C3a, C7a), 135.1 (CH, t, JC–P = 6.9 Hz, o, m-Ar–CH), 161.8 (C, p-Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 23.1.
IR (KBr pellet, cm−1): νClO = 1090, δClO = 623.
Elemental analysis calcd (%) for C45H49ClO10P2Pd: C, 59.72, H, 4.82; found: C, 59.38, H, 5.04.
HRMS calcd for [C51H49O6P2Pd]+: 925.2039; found: 925.2135.
[Pd(indenyl)(P(Ph)2(2-py))2]ClO4 (3f). The neutral complex 2f was obtained by the reaction between 0.0503 g (0.0978 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0515 g (0.1957 mmol) of (diphenyl-2-pyridylphosphine) using 5 mL of anhydrous CH2Cl2. The cationic complex 3f was obtained in the same way as complex 3a using 0.0550 g (0.3914 mmol) of NaClO4·H2O dissolved in 3 mL of methanol and a solution of 0.0515 g (0.1957 mmol) of (diphenyl-2-pyridylphosphine) in 4 mL of CH2Cl2. 0.1605 g (yield 97%) of complex 3f was obtained as a pale orange powder.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm) δ: 5.57–6.62 (m, 2H, H1, H3), 6.19–6.22 (m, 2H, H4, H7), 6.87–6.98 (m, 3H, H2, Py–H), 7.01–7.05 (m, 2H, H5, H6), 7.07–7.54 (m, 24H, Ar–H, Py–H,), 8.25 (d, J = 4.4 Hz, 2H, 6-Py–H).
13C{1H} NMR (100.0 MHz, CDCl3, T = 298 K, ppm) δ: 94.6 (CH, t, JC–P = 11.6 Hz, C1, C3), 112.0 (CH, t, JC–P = 5.2 Hz C2), 118.7 (CH, C4, C7), 124.7 (CH, Py–CH), 128.0 (CH, C5, C6), 128.1–136.7 (CH, C, Ar–CH, Ar–C, Py–CH, Py–C), 131.1 (C, t, JC–P = 3.2 Hz, C3a, C7a), 150.2 (CH, t, JC–P = 9.0 Hz, 6-Py–CH), 154.7–156.3 (CH, C, Ar–CH, Ar–C, Py–CH, Py–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 27.5.
IR (KBr pellet, cm−1): νClO = 1093, δClO = 620.
Elemental analysis calcd (%) for C43H35ClN2O4P2Pd: C, 60.93, H, 4.16, N, 3.31; found: C, 60.65, H, 4.32, N, 3.57.
HRMS calcd for [C43H32N2P2Pd]+: 747.1310; found: 747.1315.
[Pd(indenyl)(P(2-furyl)3)2]ClO4 (3g). The neutral complex 2g was obtained by the reaction between 0.0504 g (0.0980 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0455 g (0.1961 mmol) of tris(2-furyl)phosphine using 4 mL of anhydrous CH2Cl2. The cationic complex 3g was obtained in the same way as complex 3a using 0.0551 g (0.3992 mmol) of NaClO4·H2O dissolved in 3 mL of methanol and a solution of 0.0455 g (0.1961 mmol) of tris(2-furyl)phosphine in 5 mL of CH2Cl2. 0.1142 g (yield 74%) of complex 3g was obtained as a pale brown powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 6.47 (dd, J = 3.5, 1.7 Hz, 6H, Furyl–H), 6.50–6.58 (m, 8H, H1, H3, Furyl–H), 6.73–6.81 (m, 2H, H4, H7), 6.83 (t, J = 3.2 Hz, 1H, H2), 7.15–7.17 (m, 2H, H5, H6), 7.61–7.63 (m, 6H, Furyl–H).
13C{1H} NMR (100 MHz, CDCl3, T = 298 K, ppm) δ: 93.9 (CH, t, JC–P = 12.1 Hz, C1, C3), 111.1 (CH, t, JC–P = 6.3 Hz, C2), 112.1 (CH, t, JC–P = 4.0 Hz, Furyl–C), 120.1 (CH, C4, C7), 124.2 (CH, t, JC–P = 9.7 Hz, Furyl–C), 128.6 (CH, C5, C6), 131.1 (C, t, JC–P = 3.7 Hz, C3a, C7a), 140.9–142.6 (C, Furyl–C), 149.5 (CH, t, JC–P = 3.2 Hz, Furyl–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: −27.1.
IR (KBr pellet, cm−1): νClO = 1108, δClO = 622.
Elemental analysis calcd (%) for C33H25ClO10P2Pd: C, 50.47, H, 3.21; found: C, 50.80, H, 3.01.
HRMS calcd for [C33H25O6P2Pd]+: 685.0161; found: 685.0206.
[Pd(indenyl)(XPhos)]ClO4 (3h). The neutral complex 2h was obtained by the reaction between 0.0299 g (0.0581 mmol) of [Pd(μ-Cl)(indenyl)]2 and 0.0555 g (0.1162 mmol) of XPhos using 4 mL of anhydrous CH2Cl2 and after 15 min 0.0327 g (0.2328 mmol) of NaClO4·H2O dissolved in 2 mL of methanol was added. The mixture was stirred at room temperature for 15 min and the solvents were removed at low pressure. 5 mL of CH2Cl2 was added and the mixture was filtered on Millipore apparatus and the solution was concentrated under vacuum. The title complex is precipitated by the addition of diethyl ether and n-pentane and dried under vacuum. 0.0880 g (yield 95%) of complex 3g was obtained as an orange powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 0.55 (d, J = 6.7 Hz, 3H, iPr–CH3), 0.76 (d, J = 6.7 Hz, 3H, iPr–CH3), 0.86 (d, J = 6.7 Hz, 3H, iPr–CH3), 1.13–1.45 (m, 8H, Cy–CH2), 1.46–1.50 (m, 8H, iPr–CH3, Cy–CH2), 1.54 (d, J = 6.9 Hz, 3H, iPr–CH3), 1.65–1.98 (m, 8H, Cy–CH2), 2.08 (p, J = 6.7 Hz, 1H, iPr–CH), 2.11–2.25 (m, 3H, iPr–CH, Cy–CH2), 2.33–2.42 (m, 1H, Cy–CH), 2.48–2.57 (m, 1H, Cy–CH), 3.18 (p, J = 6.9 Hz, 1H, iPr–CH), 4.58–4.63 (m, 1H, H3), 6.23–6.24 (m, 1H, H1), 6.62–6.64 (m, 1H, H2), 6.69 (dd, J = 7.3, 2.7, Hz, 1H, Ar–H), 6.83 (d, J = 7.5 Hz, 1H, H7), 6.96 (t, J = 7.5 Hz, 1H, H5), 7.08 (t, J = 7.5 Hz, 1H, H6), 7.20 (s, 1H, Ar–H), 7.28 (d, J = 7.5 Hz, 1H, H4), 7.44 (tt, J = 7.5, 1.7 Hz, 1H, Ar–H), 7.50 (s, 1H, Ar–H), 7.53 (tt, J = 7.5, 1.7 Hz, 1H, Ar–H), 7.70 (t, J = 7.1 Hz, 1H, Ar–H).
13C{1H} NMR (100.0 MHz, CDCl3, T = 298 K, ppm) δ: 23.6–25.9 (CH, iPr–CH3), 26.0–30.2 (CH2, Cy–CH2), 32.0–34.3 (CH, iPr–CH), 37.3 (CH, d, JC–P = 18.9 Hz, CH–Cy), 37.6 (CH, d, JC–P = 21.4 Hz, CH–Cy), 71.1 (CH, d, JC–P = 3.3 Hz, C3), 113.8 (CH, d, JC–P = 6.9 Hz, C2), 116.9 (CH, d, JC–P = 21.4 Hz, C1), 118.3 (C, d, JC–P = 4.4 Hz, Ar–C), 119.4 (CH, C7), 121.0 (CH, C4), 125.8 (CH, Ar–CH), 126.4 (CH, Ar–CH), 127.6 (CH, C5), 128.9 (CH, d, JC–P = 5.5 Hz, Ar–CH), 129.3 (CH, C6), 131.8–132.4 (CH, Ar–CH), 132.9 (C, d, JC–P = 38.9 Hz, Ar–C), 134.8 (C, d, JC–P = 2.2 Hz, C3a), 138.8 (C, d, JC–P = 5.1 Hz, C7a), 145.1 (C, d, JC–P = 20.3 Hz, Ar–C), 146.0 (C, Ar–C), 150.3 (C, Ar–C), 152.3 (C, Ar–C).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 54.9.
IR (KBr pellet, cm−1): νClO = 1089, δClO = 623.
Elemental analysis calcd (%) for C42H56ClO4PPd: C, 63.24, H, 7.08; found: C, 62.87, H, 7.26.
HRMS calcd for [C42H56PPd]+: 697.3154; found: 697.3209.
[Pd(DPPE)(indenyl)]OTf (4). To 0.0503 g (0.0978 mmol) of [Pd(μ-Cl)(indenyl)]2 dissolved in 15 mL of anhydrous CH2Cl2, 0.0528 mg (0.2054 mmol) of AgOTf were added and the mixture was stirred at room temperature for 5 min. A solution of 0.0779 g (0.1956 mmol) of 1,2-bis(diphenylphosphino)ethane in 5 mL of anhydrous CH2Cl2 was added under an inert atmosphere (N2). The resulting red solution was stirred for 30 min at room temperature and filtered through a small pad of Celite to remove the AgCl. From the concentrated dark solution, the title complex was precipitated by the addition of diethyl ether and dried under vacuum. 0.1200 g (yield 80%) was obtained as an orange powder.
1H NMR (300 MHz, CDCl3, T = 298 K, ppm,) δ: 2.50 (d, J = 19.1 Hz, 4H, PCH2), 6.33 (pseudo q, 2H, H1, H3), 6.60 (t, J = 3.2 Hz, 1H, H2), 6.63–6.70 (m, 2H, H4, H7), 7.03–7.11 (m, 2H, H5, H6), 6.80–7.80 (bm, 20H, Ar–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 28.0 (CH2, t, JC–P = 24.3 Hz, PCH2), 88.4 (CH, t, JC–P = 11.9 Hz, C1, C3), 109.5 (CH, bt, C2), 118.9 (CH, C4, C7), 127.1 (CH, C5, C6), 128.9 (C, C3a, C7a).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 61.0.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −78.1.
IR (KBr pellet, cm−1): νCF = 1230, νSO = 1026, δSO = 633.
Elemental analysis calcd (%) for C36H31F3O3P2PdS: C, 56.22, H, 4.06, S, 4.17; found: C, 56.60, H, 3.91, S, 4.34.
HRMS calcd for [C42H56P2Pd]+: 619.0936; found: 619.0972.
[Pd(DPPP)(indenyl)]OTf (5). Complex 5 was obtained by the same synthetic route of complex 4 using 0.0301 g (0.0586 mmol) of [Pd(μ-Cl)(indenyl)]2 dissolved in 10 mL of anhydrous CH2Cl2, 0.0316 mg (0.1230 mmol) of AgOTf and a solution of 0.0483 g (0.1172 mmol) of 1,3-bis(diphenylphosphino)propane in 3 mL of anhydrous CH2Cl2. 0.0777 g (yield 85%) was obtained as an orange powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 1.50–3.00 (bm, 6H, CH2), 5.29 (pseudo q, 2H, H1, H3), 6.81 (t, J = 3.1 Hz, 1H, H2), 6.82–6.86 (m, 2H, H4, H7), 6.94 (bs, 4H, Ar–H), 7.13–7.19 (m, 2H, H5, H6), 7.33–7.60 (bm, 16H, Ar–H).
13C{1H} NMR (75.0 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 18.5 (CH2, CH2), 24.6 (CH2, t, JC–P = 17.5 Hz, PCH2), 92.5 (CH, t, JC–P = 12.0 Hz, C1, C3), 111.6 (CH, t, JC–P = 5.8 Hz, C2), 118.5 (CH, C4, C7), 126.5 (CH, C5, C6), 129.2 (CH, bs, Ar–CH), 129.8 (C, t, JC–P = 5.8 Hz, C3a, C7a).
31P{1H} NMR (162.0 MHz, CDCl3, T = 298 K, ppm) δ: 8.2.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −78.0.
IR (KBr pellet, cm−1): νCF = 1221, νSO = 1152, δSO = 635, 512.
Elemental analysis calcd (%) for C37H33F3O3P2PdS: C, 56.75, H, 4.25, S, 4.09; found: C, 56.40, H, 4.43, S, 4.48.
HRMS calcd for [C36H33P2Pd]+: 633.1092; found: 633.1133.
[Pd(DPPF)(indenyl)]OTf (6). Complex 6 was obtained by the same synthetic route of complex 4 using 0.0301 g (0.0586 mmol) of [Pd(μ-Cl)(indenyl)]2 dissolved in 9 mL of anhydrous CH2Cl2, 0.0316 mg (0.1230 mmol) of AgOTf and a solution of 0.0649 g (0.1172 mmol) of 1,1′-ferrocenediyl-bis(diphenylphosphine) in 2 mL of anhydrous CH2Cl2. 0.0777 g (yield 90%) was obtained as a red powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 4.03 (s, 2H, Fc–H), 4.22 (s, 2H, Fc–H), 4.43 (d, J = 15.8 Hz, 4H, Fc–H), 5.45 (pseudo q, 2H, H1, H3), 6.16–6.22 (m, 2H, H4, H7), 6.95 (t, J = 3.1 Hz, 1H, H2), 7.00–7.05 (m, 2H, H5, H6), 7.15–7.26 (m, 4H, Ar–H), 7.52 (t, J = 7.5 Hz, 4H, p-Ar–H), 7.58–7.68 (m, 12H, Ar–H).
13C{1H} NMR (100.0 MHz, CDCl3, T = 298 K, ppm) δ: 72.5 (C, dd, JC–P = 31.4, 30.2 Hz, Fc–C), 74.2 (CH, t, JC–P = 3.9 Hz, Fc–CH), 74.4 (CH, t, JC–P = 3.7 Hz, Fc–CH), 76.1 (CH, dt, JC–P = 12.0, 5.8 Hz, Fc–CH), 94.2 (CH, t, JC–P = 11.8 Hz, C1, C3), 112.5 (CH, t, JC–P = 5.4 Hz, C2), 118.5 (CH, C4, C7), 127.9 (CH, C5, C6), 129.2 (CH, t, JC–P = 5.5 Hz, Ar–CH), 129.7 (CH, t, JC–P = 5.5 Hz, Ar–CH), 130.5 (C, t, JC–P = 3.1 Hz, C3a, C7a), 131.6 (C, Ar–C), 132.4–133.5 (CH, Ar–CH), 134.1 (CH, JC–P = 6.6 Hz, Ar–CH).
31P{1H} NMR (162.0 MHz, CDCl3, T = 298 K, ppm) δ: 30.1.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −77.9.
IR (KBr pellet, cm−1): νSO = 1312, νCF = 1224, 1160, νSO = 1031, δSO = 637.
Elemental analysis calcd (%) for C44H35F3FeO3P2PdS: C, 57.13, H, 3.81, S, 3.47; found: C, 56.84, H, 3.98, S, 3.80.
HRMS calcd for [C43H35FeP2Pd]+: 775.0598; found: 775.0649.
[Pd(DPPBz)(indenyl)]OTf (7). Complex 7 was obtained by the same synthetic route of complex 4 using 0.0319 g (0.0621 mmol) of [Pd(μ-Cl)(indenyl)]2 dissolved in 10 mL of anhydrous CH2Cl2, 0.0335 mg (0.1304 mmol) of AgOTf and a solution of 0.0554 g (0.1241 mmol) of 1,2-bis(diphenylphosphino)benzene in 5 mL of anhydrous CH2Cl2. 0.0759 g (yield 75%) was obtained as an orange powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 6.30 (pseudo q, 2H, H1, H3), 6.48–6.54 (m, 2H, H4, H7), 6.66 (t, J = 3.2 Hz, 1H, H2), 6.89–6.96 (m, 2H, H5, H6), 6.80–7.68 (bm, 24H, Ar–H).
13C{1H}-NMR (100.0 MHz, CDCl3, T = 298 K, ppm, selected peaks) δ: 89.6 (CH, t, JC–P = 11.9 Hz, C1, C3), 110.0 (bt, C2), 118.6 (CH, C4, C7), 127.3 (CH, C5, C6), 128.6 (C, t, JC–P = 3.1 Hz, C3a, C7a).
31P{1H} NMR (162.0 MHz, CDCl3, T = 298 K, ppm) δ: 57.7.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −78.0.
IR (KBr pellet, cm−1): νSO = 1313, νCF = 1223, 1149, νSO = 1030, δSO = 636.
Elemental analysis calcd (%) for C40H31F3O3P2PdS: C, 58.80, H, 3.82, S, 3.92; found: C, 58.54, H, 3.99, S, 3.78.
HRMS calcd for [C39H31P2Pd]+: 667.0936; found: 667.0989.
[Pd(DPEphos)(indenyl)]OTf (8). Complex 8 was obtained by the same synthetic route of complex 4 using 0.0296 g (0.0576 mmol) of [Pd(μ-Cl)(indenyl)]2 dissolved in 9 mL of anhydrous CH2Cl2, 0.0311 mg (0.1210 mmol) of AgOTf and a solution of 0.0620 g (0.1241 mmol) of (oxydi-2,1-phenylene)bis(diphenylphosphine) in 2 mL of anhydrous CH2Cl2. 0.0902 g (yield 87%) was obtained as an orange powder.
1H NMR (400 MHz, CDCl3, T = 298 K, ppm) δ: 5.59 (pseudo q, 2H, H1, H3), 5.72–5.79 (m, 2H, H4, H7), 6.64 (q, J = 6.8 Hz, 4H, Ar–H), 6.86–7.02 (m, 9H, H2, H5, H6, Ar–H), 7.27–7.64 (m, 18H, Ar–H).
13C{1H} NMR (100.0 MHz, CDCl3, T = 298 K, seleckted peaks, ppm) δ: 95.0 (CH, C1, C3), 112.6 (CH, t, JC–P = 5.6 Hz, C2), 118.5 (CH, C4, C7), 128.0 (CH, C5, C6), 131.5 (C, C3a, C7a).
31P{1H} NMR (121.5 MHz, CDCl3, T = 298 K, ppm) δ: 15.8.
19F{1H} NMR (377.2 MHz, CDCl3, T = 298 K, ppm) δ: −77.9.
IR (KBr pellet, cm−1): νCF = 1222, 1146, νSO = 1031, δSO = 636.
Elemental analysis calcd (%) for C46H35F3O4P2PdS: C, 60.77, H, 3.88, S, 3.53; found: C, 61.03, H, 3.70, S, 3.39.
HRMS calcd for [C45H35OP2Pd]+: 759.1198; found: 759.1264.
Cell viability assay
Three cancer cell lines (A2780, A2780cis, OVCAR-5) and one normal cell line (MRC-5) were employed and were grown in accordance with the supplier's instructions and maintained at 37 °C in a humidified atmosphere of 5% CO2. One thousand cancer cells and eight thousand MRC-5 cells were placed in a 96 well plate and treated after 24 h with six different concentrations of Pd(II) complexes (0.001, 0.01, 0.1, 1, 10, 100 μM). After 96 h of treatment, the cell viability was measured using a CellTiter-Glo assay (Promega, Madison, WI, USA) with a Tecan M1000 instrument. IC50 values were calculated from logistical dose–response curves. Averages were obtained from triplicate and error bars are standard deviations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding: this research was funded by Fondazione AIRC per la Ricerca sul Cancro, IG23566.Notes and references
- (a) J. Tsuji, in Handbook of Organopalladium Chemistry Organic Synthesis, ed. E. Negishi, Wiley-Interscience, New York, 2002, p. 1669 Search PubMed; (b) P. Espinet and A. C. Albeniz, in Comprehensive Organometallic Chemistry III, ed. E. R. Crabtree and D. M. P. Mingos, Elesevier, 2007, vol. 8, p. 358 Search PubMed; (c) P. S. Pregosin and R. Salzmann, Coord. Chem. Rev., 1996, 155, 35 CrossRef CAS; (d) K. J. Szabó, Chem. Soc. Rev., 2001, 30, 136 RSC; (e) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863 CrossRef CAS; (f) R. A. Fernandes and J. L. Nallasivam, Org. Biomol. Chem., 2019, 17, 8647 RSC; (g) T. Scattolin and S. P. Nolan, Trends Chem., 2020, 2, 712 Search PubMed.
- D. Zargarian, Coord. Chem. Rev., 2002, 233–234, 157 CrossRef CAS.
- C. Sui-Seng, G. D. Enright and D. Zargarian, Organometallics, 2004, 23, 1236 CrossRef CAS.
- (a) M. J. Chalkley, L. M. Guard, N. Hazari, P. Hofmann, D. P. Hruszkewycz, T. J. Schmeier and M. K. Takase, Organometallics, 2013, 32, 4223 CrossRef CAS; (b) E. A. Bielinski, W. Dai, L. M. Guard, N. Hazari and M. K. Takase, Organometallics, 2013, 32, 4025 CrossRef CAS.
- (a) T. Tanase, T. Nomura, Y. Yamamoto and K. J. Kobayashi, Organomet. Chem., 1991, 410, C25 CrossRef CAS; (b) T. Tanase, T. Nomura, T. Fukushima, Y. Yamamoto and K. J. Kobayashi, Inorg. Chem., 1993, 32, 4578 CrossRef CAS.
- C. Sui-Seng, G. D. Enright and D. Zargarian, J. Am. Chem. Soc., 2006, 128, 6508 CrossRef CAS PubMed.
- N. Hazari and D. P. Hruszkewycz, Chem. Soc. Rev., 2016, 45, 2871 RSC.
- (a) T. Foo and R. G. Bergman, Organometallics, 1992, 11, 1801 CrossRef CAS; (b) T. Foo and R. G. Bergman, Organometallics, 1992, 11, 1811 CrossRef CAS; (c) P. Caddy, M. Green, J. A. K. Howard, J. M. Squire and N. J. White, J. Chem. Soc., Dalton Trans., 1981, 400 RSC.
- (a) F. H. Köhler, Chem. Ber., 1974, 107, 570 CrossRef; (b) R. T. Baker and T. H. Tulip, Organometallics, 1986, 5, 839 CrossRef CAS; (c) S. A. Westcott, A. K. Kakkar, G. Stringer, N. J. Taylor and T. B. Marder, J. Organomet. Chem., 1990, 394, 777 CrossRef CAS.
- C. Sui-Seng, A. Castonguay, Y. Chen, D. Gareau, L. F. Groux and D. Zargarian, Top. Catal., 2006, 37, 81 CrossRef CAS.
- C. Sui-Seng, L. F. Groux and D. Zargarian, Organometallics, 2006, 25, 571 CrossRef CAS.
- (a) P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari and D. P. Hruszkewycz, ACS Catal., 2015, 5, 3680 CrossRef CAS; (b) Y. Liu, T. Scattolin, A. Gobbo, M. Belis, K. Van Hecke, S. P. Nolan and C. S. J. Cazin, Eur. J. Inorg. Chem., 2022, e202100840 CAS; (c) S. Ostrowska, T. Scattolin and S. P. Nolan, Chem. Commun., 2021, 57, 4354 RSC.
- (a) J. Vicente, J. A. Abad, R. Bergs, P. G. Jones and M. C. R. de Arellano, Organometallics, 1996, 15, 1422 CrossRef CAS; (b) J. Vicente, J. A. Abad, R. Bergs, M. C. R. de Arellano, E. Martinez-Viviente and P. G. Jones, Organometallics, 2000, 19, 5597 CrossRef CAS.
- B. M. Trost and D. L. Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed.
- (a) B. Crociani, S. Antonaroli, G. Bandoli, L. Canovese, F. Visentin and P. Uguagliati, Organometallics, 1999, 18, 1137 CrossRef CAS; (b) F. Visentin and A. Togni, Organometallics, 2007, 26, 3746 CrossRef CAS; (c) L. Canovese, F. Visentin, C. Santo, G. Chessa and V. Bertolasi, Organometallics, 2010, 29, 3027 CrossRef CAS; (d) L. Canovese, F. Visentin, C. Levi and A. Dolmella, Dalton Trans., 2011, 40, 966 RSC; (e) L. Canovese, F. Visentin, T. Scattolin, C. Santo and V. Bertolasi, Polyhedron, 2016, 119, 377 CrossRef CAS.
- (a) S. Karmaker, S. Gosh, S. E. Kabir, D. T. Haworth and S. V. Lindeman, Inorg. Chim. Acta, 2012, 382, 199 CrossRef CAS; (b) U. Monkowius, S. Nogai and H. Schmidbaur, Z. Naturforsch., 2003, 58, 751 CrossRef CAS.
- (a) T. Scattolin, V. A. Volonshki, F. Visentin and S. P. Nolan, Cell Rep. Phys. Sci., 2021, 2, 100446 CrossRef CAS; (b) T. Scattolin, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, C. Santo, F. Rizzolio, I. Caligiuri and F. Visentin, Dalton Trans., 2018, 47, 13616 RSC; (c) T. Scattolin, E. Bortolamiol, I. Caligiuri, F. Rizzolio, N. Demitri and F. Visentin, Polyhedron, 2020, 186, 114607 CrossRef CAS; (d) T. Scattolin, E. Bortolamiol, F. Rizzolio, N. Demitri and F. Visentin, Appl. Organomet. Chem., 2020, e5876 CAS; (e) T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 1 CrossRef PubMed; (f) T. Scattolin, A. Piccin, M. Mauceri, F. Rizzolio, N. Demitri, V. Canzonieri and F. Visentin, Polyhedron, 2021, 207, 115381 CrossRef CAS; (g) T. Scattolin, G. Andreetta, M. Mauceri, F. Rizzolio, N. Demitri, V. Canzonieri and F. Visentin, J. Organomet. Chem., 2021, 952, 122014 CrossRef CAS.
- ΔM–C = 0.5{(M–C(3a) + M–C(7a)} − 0.5{M–C(1) + M–C(3)}.
- The hinge angle HA is the angle between the planes formed by the atoms C(1), C(2), C(3) and C(1), C(3), C(3a), C(7a).
- The fold angle FA is the angle between the planes formed by the atoms C(1), C(2), C(3) and C(3a), C(4), C(5), C(6), C(7), and C(7a).
- (a) S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871 CrossRef CAS PubMed; (b) C. H. Burgos, T. E. Barder, X. Huang and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 4321 CrossRef CAS PubMed; (c) B. T. Ingoglia, C. C. Wagen and S. L. Buchwald, Tetrahedron, 2019, 75, 4199 CrossRef CAS PubMed.
- P. Kocovsky, S. Vyskocil, I. Cisarova, G. C. Lloyd-Jones, S. C. Stephen and C. P. Butts, J. Am. Chem. Soc., 1999, 121, 7714 CrossRef CAS.
- (a) C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS; (b) T. Scattolin and S. P. Nolan, in Comprehensive Organometallic Chemistry IV, 2022, in press Search PubMed.
- T. E. Ready, J. C. W. Chien and M. D. Rausch, J. Organomet. Chem., 1996, 519, 21 CrossRef CAS.
- R. Fernández-Galán, F. A. Jalón, B. R. Manzano, J. Rodríguez-de la Fuente, M. Vrahami, B. Jedlicka, W. Weissensteine and G. Jogl, Organometallics, 1997, 16, 3758 CrossRef.
- T. Scattolin, I. Pessotto, E. Cavarzerani, V. Canzonieri, L. Orian, N. Demitri, C. Shmidt, A. Casini, E. Bortolamiol, F. Rizzolio, F. Visentin and S. P. Nolan, Eur. J. Inorg. Chem., 2022, e202200 Search PubMed.
- P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari, D. P. Hruszkewycz, H. P. Shah and M. T. Tudge, ACS Catal., 2015, 5, 3680 CrossRef CAS.
- O. Piechaczyk, C. Thoumazet, Y. Jean and P. le Floch, J. Am. Chem. Soc., 2006, 128, 14306 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- U. Häussermann, M. Dolg, H. Stoll, H. Preuss, P. Schwerdtfeger and R. M. Pitzer, Mol. Phys., 1993, 78, 1211 CrossRef.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535 CrossRef.
- T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- A. Lausi, M. Polentarutti, S. Onesti, J. R. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi, D. Lamba, A. Pifferi, S. C. Mande, D. D. Sarma, S. M. Sharma and G. Paolucci, Eur. Phys. J. Plus, 2015, 130(43), 1 Search PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(2), 125 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235 CrossRef CAS PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- P. Emsley, B. Lohkamp, W. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45(4), 849 CrossRef CAS.
- L. Schrodinger, 2015, https://www.pymol.org.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2173701–2173705. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01821g |
| This journal is © The Royal Society of Chemistry 2022 |