
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ferromagnetically coupled pseudo-calixarene [Co16] wheel that self-assembles as a tubular network of capsules†
Pinelopi A.
Tsami
a,
Thomais G.
Tziotzi
a,
Angelos B.
Canaj
 b,
Mukesh K.
Singh
b,
Scott J.
Dalgarno
*c,
Euan K.
Brechin
*b and
Constantinos J.
Milios
*a
b,
Mukesh K.
Singh
b,
Scott J.
Dalgarno
*c,
Euan K.
Brechin
*b and
Constantinos J.
Milios
*a
aDepartment of Chemistry, The University of Crete, Voutes, 71003 Herakleion, Greece. E-mail: komil@uoc.gr
bEaStCHEM School of Chemistry, The University of Edinburgh, Edinburgh, EH9 3FJ, Scotland, UK. E-mail: ebrechin@ed.ac.uk
cInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, UK. E-mail: S.J.Dalgarno@hw.ac.uk
First published on 9th September 2022
Abstract
Reaction of Co(OAc)2·4H2O and Hsal in a basic MeCN solution affords the hexadecanuclear wheel [Co16(sal)16(OAc)16]·16MeCN (1·16MeCN) that displays ferromagnetic nearest neighbour exchange and has pseudo-calixarene character. Symmetry equivalent wheels self-assemble to form remarkable tubular networks of capsules in the extended structure.
Molecular wheels attract continued attention, partly due to beautiful structural aesthetics, but also because some show fascinating and potentially useful physical properties.1 In the field of molecular magnetism, molecular wheels of 3d transition metals came into prominence in the late 1980s and early 1990s with the publication of [CrIII8] and [FeIII10].2 The former has inspired the development of a large family of homo- and heterometallic Cr wheels including the first examples of odd-numbered wheels that display topological spin frustration, with potential as quantum bits in information processing.3 The latter, and other antiferromagnetically coupled even-membered homometallic wheels are generally characterised by a diamagnetic spin ground state and display interesting quantum phenomena and spin dynamics, including tunneling of the Néel vector,4 spin-multiplet mixing effects5 and magnetic level repulsions.6 The intervening years have witnessed the publication of wheels of all the 3d metals with nuclearities up to eighty four.7 In CoII chemistry8 early examples of wheels included a dodecanuclear cluster built with a substituted pyridone9 and an heptanuclear Anderson wheel stabilised by tripodal alcohols10 which both display ferromagnetic exchange interactions.
The reaction between Co(OAc)2·4H2O and Hsal (salicylaldehyde) in a basic MeCN solution (see ESI† for full details) leads to the formation of pink single crystals after 3 days upon diffusion of Et2O into the mother liquor. Crystals of [Co16(sal)16(OAc)16]·16MeCN (1·16MeCN) were in a tetragonal cell and structure solution was performed in the P4/n space group. The asymmetric unit (ASU) contains one quarter of the formula, and symmetry expansion affords the wheel shown in Fig. 1 (Fig. S1 and S2†).
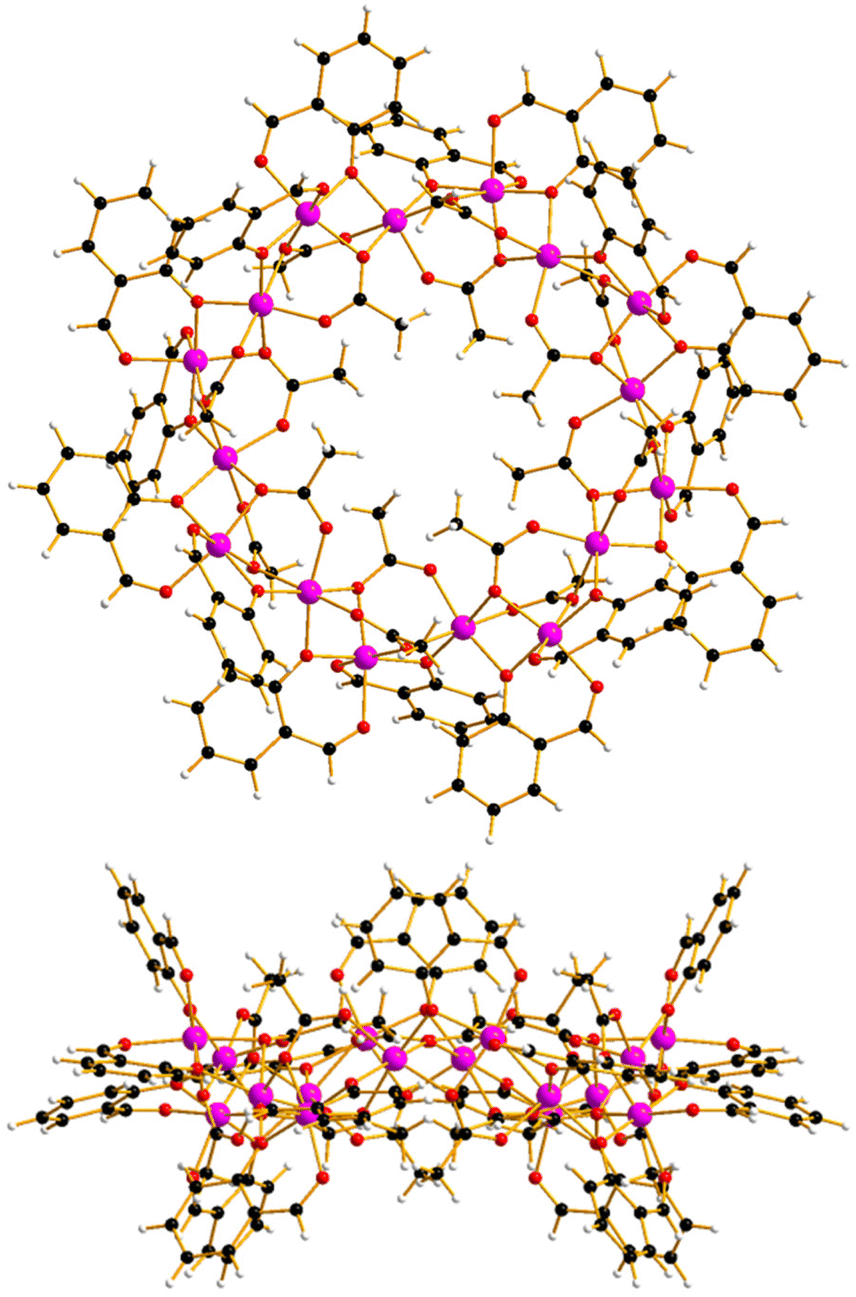 | ||
| Fig. 1 Orthogonal views of the molecular structure of complex 1 viewed perpendicular (top) and parallel to the [Co16] ‘plane’. Colour code: Co = pink, O = red, C = black, H = white. | ||
The metallic skeleton (Fig. 2) describes a single-stranded, sinusoidal [CoII16] wheel of approximate diameter, Co1⋯Co1′ = ∼14.5 Å. The ‘inside’ of the wheel is stabilised by sixteen μ3-, syn, syn, anti-OAc ligands. Eight lie in the metal plane, with four above and four below the plane. The sixteen μ-sal ligands are arranged in a similar manner on the ‘outside’ of the wheel. Eight μ-sal ligands are in a belt around the periphery of the wheel, whilst four are above the plane, and four below (Fig. 1). These out of plane μ-sal ligands form an interesting arrangement that is reminiscent of calixarenes,11 presenting a shallow hydrophobic pocket as a result. The magnetic unit between nearest neighbours contains one μ-O(alkoxide), one μ-O(carboxylate) and one syn, syn-O–C–O(carboxylate). The interaction between next nearest neighbours is mediated by the syn, anti-O–C–O(carboxylate) bridge. The Co–O–Co angles subtended by the μ-O atoms are in the range ∼92.1–99.5°, with the CoII ions all being in distorted octahedral {CoO6} geometries. There are multiple close intermolecular interactions between neighbouring wheels mediated by the sal/OAc ligands with C(H)⋯C distances ≥3.3 Å.
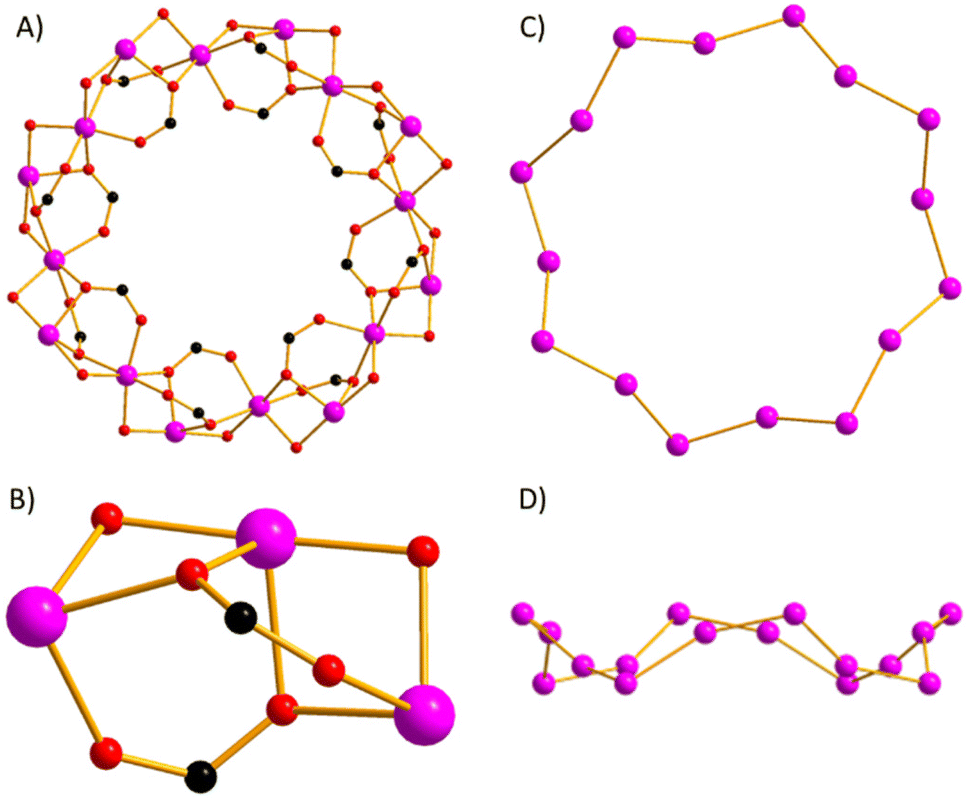 | ||
| Fig. 2 (A) The magnetic core of 1. (B) Close-up of the bridging between neighbouring CoII ions. The metallic core viewed perpendicular (C) and parallel (D) to the [Co16] ‘plane’. Colour code: Co = pink, O = red, C = black. | ||
A search of the Cambridge Structural Database (CSD) reveals approximately twenty [Co16] structures, with more than half being squares and tetrahedra stabilised by thia- and sulfonyl-calix[4]arenes.12 There are two other wheels, one comprising linked squares and cubes built with a bis-benzimidazole-diol ligand, and one incorporating four linear {Co4} subunits constructed with polytriazolate ligands.13
Further symmetry expansion of the new pseudo-calixarene structure presented by 1 reveals a remarkable arrangement in which symmetry equivalent (s.e.) wheels pack to form tubular networks of capsules, and channels between the tubules. Inspection of the ring structure of 1 in space filling representation (Fig. 3A, in the ab plane) shows a very small channel through the ring as a result of the syn, syn, anti-OAc ligands on the inside of the molecule. Symmetry expansion along the c axis gives rise to a dimer with the next s.e. wheel as shown in Fig. 3B (in the ac plane). These pseudo-calixarene wheels lock together in the solid state through inter-digitation of the μ-sal ligands, with some OAc ligands also forming part of the space filling belt. This gives rise to an encapsulated space with a volume of ∼272 Å3 that is occupied by disordered solvent molecules (Fig. 3C).14 It was not possible to resolve this disorder given the high symmetry and diffuse nature of the electron density. Inter-digitation continues along the c axis, forming infinite tubular stacks of capsules, and these pack as shown in Fig. 3D to form solvent filled channels that run parallel through the extended structure. Single crystals of 1 are solvent dependent, but the nature of the extended structure will be studied further with a view to forming more stable analogues that can be desolvated and explored for potential guest transport to the interior of the cages.
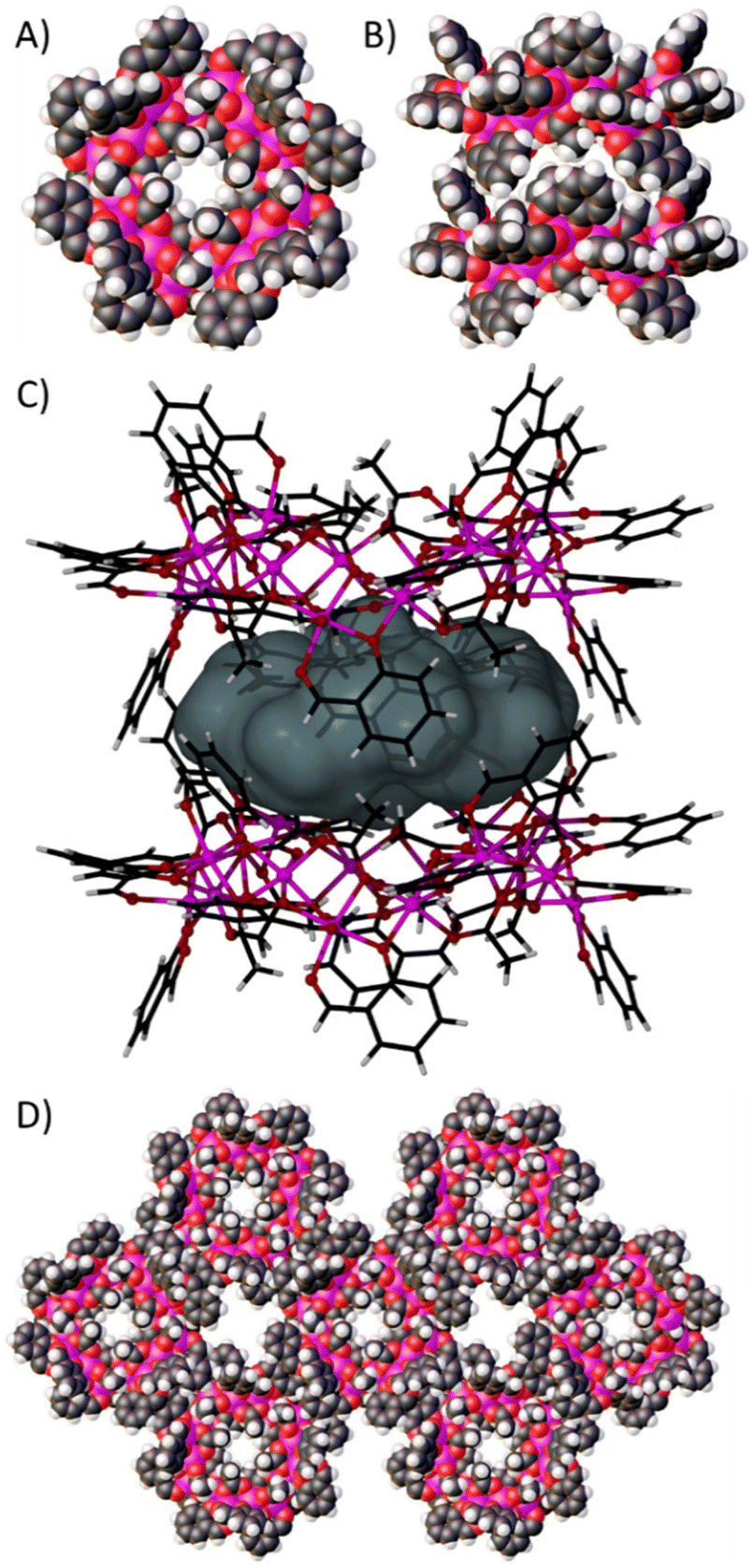 | ||
| Fig. 3 Space filling representations of 1 (A, in the ab plane) and two symmetry equivalents interdigitating to form a cage (B, in the ac plane). (C) Representation of the encapsulated space between [Co16] dimers with a volume of ∼272 Å3 occupied by disordered solvent molecules. (D) The extended structure showing the formation of channels by packing of infinite stacks of s.e. of 1 (in the ab plane). Colour code: Co = pink, O = red, C = black, H = white. | ||
DC magnetic susceptibility (χ) and magnetisation (M) measurements of 1 were taken in the T = 300–2.00 K, B = 0.1 T and T = 2.0–10 K and B = 0.5–9.0 T temperature and field ranges, respectively. These are plotted as the χT product versus T and M versus B in Fig. 4. The T = 300 K value of χT = 43.2 cm3 K mol−1 is equal to the value expected for sixteen non-interacting S = 3/2 CoII ions with g = 2.40. Upon cooling the χT value decreases slowly to ∼41.7 cm3 K mol−1 at 38 K before rising sharply to a maximum of ∼52.8 cm3 K mol−1 at T = 6 K, and then falling to ∼10.9 cm3 K mol−1 at T = 2 K. The initial drop in χT is due to the magnetic anisotropy of the octahedral CoII ions,15 while the increase at low temperature is due to weak ferromagnetic interactions. The drop in value between 6–2 K is most likely due to intermolecular antiferromagnetic interactions. This behaviour is similar to that observed for the pyridone-stabilised [Co12] wheel.9 The M vs. B data is in agreement with this interpretation, with the magnetisation increasing rapidly with increasing field, not saturating and reaching a value of M = 33.4 μB at T = 2 K and B = 9 T. There are no out-of-phase (χ′′) signals in ac susceptibility measurements down to T = 2 K and frequencies up to 3000 Hz. The ferromagnetic exchange in 1 is consistent with magneto-structural correlations developed for O-bridged CoII clusters where the sign and magnitude of the interaction is dictated by the Co–O–Co angle, with the exchange becoming more ferromagnetic with decreasing angle.16 Here, the Co–O–Co angles are all ≤99.5° and are therefore expected to mediate weak ferromagnetic exchange.
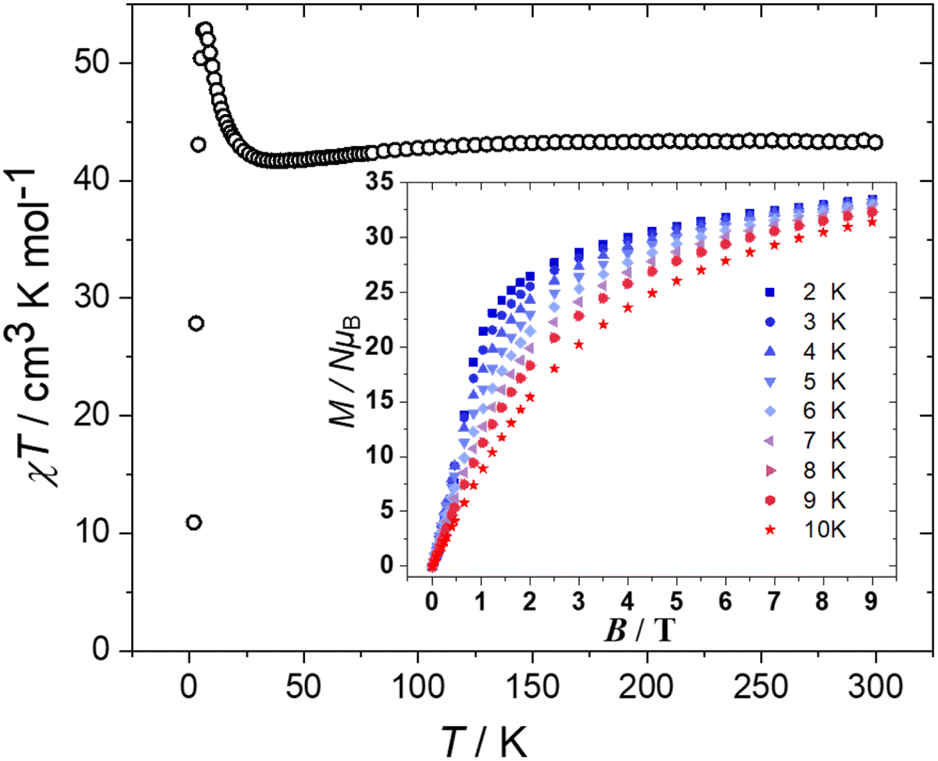 | ||
| Fig. 4 Plot of the χT product versus T in the range T = 300–2 K in an applied field, B = 0.1 T. Inset: plot of the M versus B data in the T = 2.0–10 K and B = 0.5–9.0 T temperature and field ranges, respectively. | ||
In order to probe the nature and magnitude of the magnetic exchange and anisotropy of the CoII ions further, we now turn to theory (see the computational details in the ESI† for full details).17 We have performed DFT calculations on a model of complex 1 (Model 1) based on its ASU, i.e. one [CoII4] moiety plus one linking CoII ion (Fig. S4†). Based on symmetry and structure there are four unique nearest neighbour magnetic exchange interactions with J values ranging between +1.7 cm−1 ≤ J ≤ +3.8 cm−1 (Table S2†). The narrow range of ferromagnetic exchange interactions found can be attributed to the similarity of the structural parameters present, including the average Co–μO–Co angles.16 We have also performed overlap integral calculations17 using the singly occupied molecular orbitals (SOMOs) of the CoII ions (Fig. S5†). These help to elucidate the magnitude and sign of magnetic interactions since their magnitude is directly proportional to the magnitude of the antiferromagnetic interaction, i.e. the larger the overlap the larger the antiferromagnetic interaction and vice versa. For 1, there are three intermediate and six small overlap interactions, resulting in a small ferromagnetic interaction overall. Interestingly, replacement of the phenoxide group in Model 1 with a point charge changes the sign of the magnetic interaction from ferromagnetic to antiferromagnetic (+4.2 cm−1 to −3.4 cm−1), highlighting the importance of this moiety for obtaining ferromagnetic exchange. We have also calculated the next-nearest neighbour exchange mediated via the syn, anti-O–C–O(carboxylate). These are weak and antiferromagnetic, with J < −0.2 cm−1 (Fig. S6†).
All the CoII ions in 1 are in distorted octahedral geometries (Table S3†), with previous magneto-structural studies suggesting such ions would possess large easy-plane anisotropy.18Ab initio NEVPT2 calculations on each CoII ion in 1 confirms this, with values in the range +41.2 ≤ D ≤ +87.1 cm−1. The dominant contribution to D arises from the dxz/yz → dxy electronic transition (Table S4, Fig. S7†). Positive axial zero-field splitting can be attributed to electronic transitions between orbitals with different mL values and the magnitude correlated to the energy separation between the orbitals involved in the electronic transition.19 The computed Dzz axes for Co1–Co4 are shown in Fig. S8† and are non-collinear, primarily due to the sinusoidal arrangement of metal centres. This is likely also the reason 1 is not an SMM. We also note that |D| ≫ |J| and in such scenarios an isotropic description of the exchange should be regarded with some caution.20
In summary, the reaction of Co(OAc)2·4H2O and Hsal affords the aesthetically pleasing [Co16(μ-sal)16(μ3-OAc)16] (1) wheel, which displays weak ferromagnetic nearest neighbour exchange interactions, in agreement with DFT calculations. Ab initio NEVPT2 studies suggest the presence of large single ion easy-plane anisotropy. In the extended structure the wheels form dimeric capsules via inter-digitation of the sal/acetate ions and this extends to form infinite tubular stacks of capsules upon symmetry expansion. Attempts to make analogues of 1 with different MII ions, different carboxylates and derivatised salicylaldehyde ligands are in progress, with a view to also examining guest transport to the interior of the cages.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project Number: 400). We thank the Leverhulme Trust (RPG-2021-176) and the European Union Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 832488. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission.References
- See for example: (a) O. Poncelet, L. G. Hubert-Pfalzgraf, J.-C. Daran and R. Astier, J. Chem. Soc., Chem. Commun., 1989, 1846–1848 RSC; (b) H. Barrow, D. A. Brown, N. W. Alcock, H. J. Clase and M. G. H. Wallbridge, J. Chem. Soc., Chem. Commun., 1995, 1231–1232 RSC; (c) J. S. Anderson, Nature, 1937, 140, 850 CrossRef CAS; (d) A. Müller, M. Koop, H. Bögge, M. Schmidtmann and C. Beugholt, J. Chem. Soc., Chem. Commun., 1998, 1501–1502 RSC.
- (a) N. V. Gerbeleu, A. S. Batsanov, G. A. Timko, Yu. T. Struchkov, K. M. Indrichan and G. A. Popovich, Patent SU1299116(A1), 1989-12-15. Bull. 1989, 46. Priority numbers: SU19853940102 date 1985-08-07 Search PubMed; (b) N. V. Gerbeleu, Y. T. Struchkov, G. A. Timko, A. S. Batsanov, K. M. Indrichan and G. A. Popovich, Dokl. Akad. Nauk SSSR, 1990, 313, 1459–1462 CAS; (c) K. L. Taft and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 9629–9630 CrossRef CAS; (d) K. L. Taft, C. D. Delfs, G. C. Papaefthymiou, S. Foner, D. Gatteschi and S. J. Lippard, J. Am. Chem. Soc., 1994, 116, 823–832 CrossRef CAS.
- See for example: (a) R. J. Woolfson, G. A. Timco, A. Chiesa, I. J. Vitorica-Yrezabal, F. Tuna, T. Guidi, E. Pavarini, P. Santini, S. Carretta and R. E. P. Winpenny, Angew. Chem., 2016, 128, 9002–9005 CrossRef; (b) M. Baker, T. Lancaster, A. Chiesa, G. Amoretti, P. J. Baker, C. Barker, S. J. Blundell, S. Carretta, D. Collison, H. U. Güdel, T. Guidi, E. J. L. Mcinnes, J. S. Moeller, J. H. Mutka, J. Ollivier, F. L. Pratt, P. Santini, F. Tuna, O. L. Tregenna-Piggott, I. Vitorica-Yrezabal, G. A. Timco and R. E. P. Winpenny, Chem. – Eur. J., 2016, 22, 1779–1788 CrossRef CAS PubMed.
- F. Meier and D. Loss, Phys. Rev. Lett., 2001, 86, 5373–5376 CrossRef CAS.
- S. Carretta, J. van Slageren, T. Guidi, E. Liviotti, C. Mondelli, D. Rovai, A. Cornia, A. L. Dearden, F. Carsughi, M. Affronte, C. D. Frost, R. E. P. Winpenny, D. Gatteschi, G. Amoretti and R. Caciuffo, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 094405 CrossRef.
- M. Affronte, A. Cornia, A. Lascialfari, F. Borsa, D. Gatteschi, J. Hinderer, M. Horvatić, A. G. M. Jansen and M.-H. Julien, Phys. Rev. Lett., 2002, 88, 167201 CrossRef CAS.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed.
- M. Murrie, Coord. Chem. Rev., 2010, 39, 1986–1995 CAS.
- E. K. Brechin, O. Cador, A. Caneschi, C. Cadiou, S. G. Harris, S. Parsons, M. Vonci and R. E. P. Winpenny, Chem. Commun., 2002, 1860–1861 RSC.
- M. Moragues-Canovás, C. E. Talbot-Eeckelaers, L. Catala, F. Lloret, W. Wernsdorfer, E. K. Brechin and T. Mallah, Inorg. Chem., 2006, 45, 7038–7040 CrossRef PubMed.
- (a) C. D. Gutsche, in Calixarenes: An Introduction, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008, ch. 2, pp. 27–160 Search PubMed; (b) S. T. Meally, C. McDonald, G. Karotsis, G. S. Papaefstathiou, E. K. Brechin, P. W. Dunne, P. McArdle, N. P. Power and L. F. Jones, Dalton Trans., 2010, 39, 4809–4816 RSC.
- See for example: (a) M. Liu and W. Liao, CrystEngComm, 2012, 14, 5727–5729 RSC; (b) X. Hang, S. Wang, X. Zhu, H. Han and W. Liao, CrystEngComm, 2016, 18, 4938–4943 RSC.
- (a) W.-Q. Lin, J.-D. Leng and M.-L. Tong, Chem. Commun., 2012, 48, 4477–4470 RSC; (b) Y.-Q. Hu, M.-H. Zeng, K. Zhang, S. Hu, F.-F. Zhou and M. Kurmoo, J. Am. Chem. Soc., 2013, 135, 7901–7908 CrossRef CAS.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141–1146 CrossRef CAS.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Dover Publications, New York, 1970 Search PubMed.
- X.-J. Song and X.-M. Xue, ACS Omega, 2020, 5, 8347–8354 CrossRef CAS PubMed.
- (a) M. K. Singh, A. Etcheverry-Berríos, J. Vallejo, S. Sanz, J. Martínez-Lillo, G. S. Nichol, P. J. Lusby and E. K. Brechin, Dalton Trans., 2022, 51, 8377–8381 RSC; (b) D. J. Cutler, M. Coletta, M. K. Singh, A. B. Canaj, L. J. McCormick, S. J. Coles, J. Schnack and E. K. Brechin, Dalton Trans., 2022, 51, 8945–8948 RSC; (c) M. Coletta, T. G. Tziotzi, M. Gray, G. S. Nichol, M. K. Singh, C. J. Milios and E. K. Brechin, Chem. Commun., 2021, 57, 4122–4125 RSC; (d) D. J. Cutler, M. K. Singh, G. S. Nichol, M. Evangelisti, J. Schnack, L. Cronin and E. K. Brechin, Chem. Commun., 2021, 57, 8925–8928 RSC; (e) M. K. Singh and G. Rajaraman, Inorg. Chem., 2019, 58, 3175–3188 CrossRef CAS; (f) C. McDonald, S. Sanz, E. K. Brechin, M. K. Singh, G. Rajaraman, D. Gaynor and L. F. Jones, RSC Adv., 2014, 4, 38182 RSC; (g) S. Hazra, S. Bhattacharya, M. K. Singh, L. Carrella, E. Rentschler, T. Weyhermueller, G. Rajaraman and S. Mohanta, Inorg. Chem., 2013, 52, 12881–12892 CrossRef CAS PubMed.
- S. Gómez-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 379 CrossRef.
- (a) M. K. Singh, P. Shukla, M. Khatua and G. Rajaraman, Chem. – Eur. J., 2020, 26, 464–477 CrossRef CAS PubMed; (b) Y.-F. Deng, M. K. Singh, D. Gan, T. Xiao, Y. Wang, S. Liu, Z. Wang, Z. Ouyang, Y.-Z. Zhang and K. R. Dunbar, Inorg. Chem., 2020, 59, 7622–7630 CrossRef CAS.
- (a) L. F. Chibotaru, L. Ungur, C. Aronica, H. Elmoll, G. Pilet and D. Luneau, J. Am. Chem. Soc., 2008, 130, 12445 CrossRef CAS PubMed; (b) O. Waldmann, M. Ruben, U. Ziener, P. Müller and J. M. Lehn, Inorg. Chem., 2006, 45, 6535–6540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, X-ray data and computational methodology. CCDC 2178060. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02554j |
| This journal is © The Royal Society of Chemistry 2022 |