
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Indium(III)/2-benzoylpyridine chemistry: interesting indium(III) bromide-assisted transformations of the ligand†
Christina
Stamou
a,
Zoi G.
Lada
 b,
Christos T.
Chasapis
c,
Dionissios
Papaioannou
*a,
Pierre
Dechambenoit
*d and
Spyros P.
Perlepes
*ab
b,
Christos T.
Chasapis
c,
Dionissios
Papaioannou
*a,
Pierre
Dechambenoit
*d and
Spyros P.
Perlepes
*ab
aDepartment of Chemistry, University of Patras, 26504 Patras, Greece. E-mail: dapapaio@upatras.gr; perlepes@upatras.gr
bInstitute of Chemical Engineering Sciences (ICE-HT), Foundation for Research and Technology-Hellas (FORTH), P.O. Box 1414, Platani, 26504 Patras, Greece
cNMR Facility, Instrumental Analysis Laboratory, School of Natural Sciences, University of Patras, 26504 Patras, Greece
dCentre de Recherche Paul Pascal, UMR 5031, CNRS, University of Bordeaux, 33600 Pessac, France. E-mail: pierre.dechambenoit@u-bordeaux.fr
First published on 28th September 2022
Abstract
Reactions of 2-benzoylpyridine, (py)(ph)CO, with InX3 (X = Cl, Br) in EtOH at room temperature have been studied. The InCl3/(py)(ph)CO system has provided access to complex [InCl3{(py)(ph)CO}(EtOH)]·{(py)(ph)CO} (1) and the byproduct {(pyH)(ph)CO}Cl (2). The reaction of InBr3 with (py)(ph)CO has led to a mixture of (L)[InBr4{(py)(ph)CO}] (3) and [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (4), where L+ is the 9-oxo-indolo[1,2-a]pyridinium cation and (py)(ph)CH(O)− is the anion of (pyridin-2-yl)methanol. Based on solubility and crystallisation time differences between the two components of the mixture, complex 4 was isolated in pure form, i.e. free from 3. The formations of the counterion L+ and the coordinated (py)(ph)CH(O)− anion represent clearly InBr3-promoted/assisted transformations. Reaction mechanisms have been proposed for the formation of 2, 3 and 4. Complex 4 could also be isolated by the reaction of InBr3 and pre-formed (py)(ph)CH(OH) in EtOH. The solid-state structures of 1, 3 and 4 were determined by single-crystal X-ray crystallography, while the identity of the salt 2 was confirmed by microanalyses and a variety of spectroscopic techniques, including ESI-MS spectra. In the indium(III) complexes, the metal ions are 6-coordinate with a distorted octahedral geometry. The halogeno groups (Cl−, Br−) in the three complexes are terminal. The (py)(ph)CO molecule behaves as a N,O-bidentate (1.11) ligand in 1 and 3. A terminal EtOH ligand completes the coordination sphere of InIII in 1. The alkoxo oxygen atoms of the two 2.21 (py)(ph)CH(O)− ligands doubly bridge the InIII centers in 4 creating a {InIII2(μ-OR)2}4+ core; a nitrogen atom of one reduced organic ligand, two bromo ions and one terminal EtOH molecule complete the 6-coordination at each metal centre. Complexes 1, 3 and 4 were characterised by IR and Raman spectroscopies, and the data were discussed in terms of their known solid-state structures. Molar conductivity data and 1H NMR spectra were used in an attempt to probe the behaviour of the complexes in DMSO. The to-date observed metal ion-assisted/promoted transformations of (py)(ph)CO are also discussed.
Introduction
The renaissance of inorganic chemistry in the early 1960s was a consequence of the discovery and study of transition-metal organometallic chemistry. The great advantage of organometallic chemistry is the ability of scientists to isolate and subsequently utilise unusual organic fragments bonded to a transition metal ion. In the last 30 years or so, alternative methods to ligand reactivity have been developed that involve classical coordination complexes (i.e. of Werner type) as opposed to organometallic compounds. The stimulus for such research activities came from the study of natural biological systems. It is well known that several types of enzymes bind or require metal ions to perform their roles. Since enzymes can utilise metal ions to perform complicated organic reactions under aerobic conditions in aqueous environments at room temperature and ambient pressure, it was logical for scientists to think that such reactions can be carried out on the bench, providing, for example, a better approach to homogeneous catalysis. Today there are many and varied “organic” reactions that proceed in the presence of metal ions, the latter promoting, catalysing or initiating the reactions. Thus, metal ion-directed chemistry and the use of metal ions to control the stereochemical course of reactions are a “hot” research theme in contemporary inorganic chemistry.1–7The principle of the reactivity of coordinated ligands is simple. Any chemical reaction involves movement of electrons (either complete transfer or sharing). Thus, anything that changes the distribution, movement or availability of electrons can affect the reactivity. When the coordination bond forms, the electronic arrangement in parts of the ligand is perturbed to some extent. Given the fact that the reactivity is based upon the electronic structure, it is obvious that the reactivity of coordinated ligands is different from that of the free ligands. The mechanisms with which a metal ion may alter the chemical behaviour of a coordinated ligand involve conformational, polarisation and π-bonding changes.1
Several reactions of carbonyl compounds, which are often significant in C–C bond formation, display sensitivity upon complexation with metal ions.7 The coordination of a carbonyl molecule to a metal ion through the oxygen atom is expected to modify the reactivity of the carbonyl group. The commonest type of reaction associated with carbonyl compounds involves attack of the carbonyl carbon atom by a nucleophile. Since both the carbonyl carbon atom and the metal ion normally have electrophilic character, a nucleophile could initially attack at either the carbon atom of the carbonyl group or at the metal ion. This means that the products of the reaction can arise either by direct attack at the carbon by the free nucleophile or by attack at the metal centre followed by attack at carbon by the coordinated nucleophilic agent. There has been a debate about the predominant mechanism, i.e. attack by a free or by a coordinated nucleophile.1,8,9
In the last 25 years our groups have had an intense interest in the area of the reactivity of carbonyl-containing coordinated ligands of the general types A-CO-A,10,11 A-CO-CO-A,10,12 A-CO-A-CO-A,12 where A is a donor group. We have concentrated mainly on di-2-pyridyl ketone, (py)2CO, and discovered more than ten metal ion-assisted transformations.10,13 Due to the polarisation effect, the already existing electrophilicity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbon is increased dramatically by coordination of the oxygen atom in solution. The active character of the carbonyl group is further enhanced by the strong electron-attractive property of the 2-pyridyl rings. Thus, upon coordination to a metal centre, several nucleophiles can attack the carbonyl carbon (Scheme 1) leading to exciting coordinated ligands which in most cases can not been stabilized in the absence of metal ions. Single deprotonation of the ligands (deprotonation may be double in the case of H2O as nucleophile), combined with the presence of donor atom(s) in the negatively charged nucleophile give a flexible character in the ligand with variable coordination modes, leading to coordination clusters and polymers with aesthetically beautiful structures and interesting properties (magnetic, optical, …).
O carbon is increased dramatically by coordination of the oxygen atom in solution. The active character of the carbonyl group is further enhanced by the strong electron-attractive property of the 2-pyridyl rings. Thus, upon coordination to a metal centre, several nucleophiles can attack the carbonyl carbon (Scheme 1) leading to exciting coordinated ligands which in most cases can not been stabilized in the absence of metal ions. Single deprotonation of the ligands (deprotonation may be double in the case of H2O as nucleophile), combined with the presence of donor atom(s) in the negatively charged nucleophile give a flexible character in the ligand with variable coordination modes, leading to coordination clusters and polymers with aesthetically beautiful structures and interesting properties (magnetic, optical, …).
 | ||
| Scheme 1 General reactivity pattern of coordinated di-2-pyridyl ketone, (py)2CO. The neutral nucleophile is represented by nucH and can be H2O, alcohols, Me2CO, MeCN, pyrazoles, secondary amino acids, etc. The attack of the nucleophile on the carbonyl carbon atom requires its deprotonation, which is often achieved by an external base. Mn+ is the metal ion (n = 2, 3). When nucH is H2O, the product is the gem-diol derivative of (py)2CO, (py)2C(OH)2, whereas the addition of alcohols (ROH) produces the hemiacetal form of (py)2CO, (py)2C(OR)(OH). Note that compounds (py)2C(nuc)(OH) and their anionic forms do not exist as free species, but only in the presence of metal ions (i.e. as ligands in their respective metal complexes). Important note: in the abbreviations of the ligands, the carbonyl oxygen atom is written without parenthesis, whereas the oxygen atoms which form single bonds with the central carbon are written within parenthesis. | ||
Based on the experience gained from our research efforts with (py)2CO, we started to study the metal ion-involving chemistry of (py)CO(B), where B is a non-donor group. We were interested in investigating if the latter ligands could undergo metal ion-assisted reactivity on the carbonyl group; if yes, we anticipated different identities of the products and possibly new reactivity pathways. This work describes some aspects of In(III)/(py)(ph)CO chemistry with emphasis on the reactivity characteristics of its carbonyl group; (py)(ph)CO is 2-benzoylpyridine (Scheme 2), which normally behaves as a N(ring), O(carbonyl)-chelating ligands.14 This ligand does not possess the second ring-N atom that is present in (py)2CO. The two ligands have the same size, while the phenyl group has also an electron-withdrawing character (albeit weaker than that of the 2-pyridyl group). In addition, both ligands lack acidic α-hydrogens (i.e. C–H bonds adjacent to the carbonyl group), present, for example, in 2-acetylpyridine where the C–H bonds of the methyl group are polar and potential reaction sites. The rather little investigated reactivity of (py)(ph)CO towards transition metal ions (NiII, CuI, CuII, RuII, ReV) has been studied (vide infra) by few groups15 – including our group16 – with impressive results; the general conclusion of those studies is that there is space for further research in this area. This belief was the main stimulus of the present report.
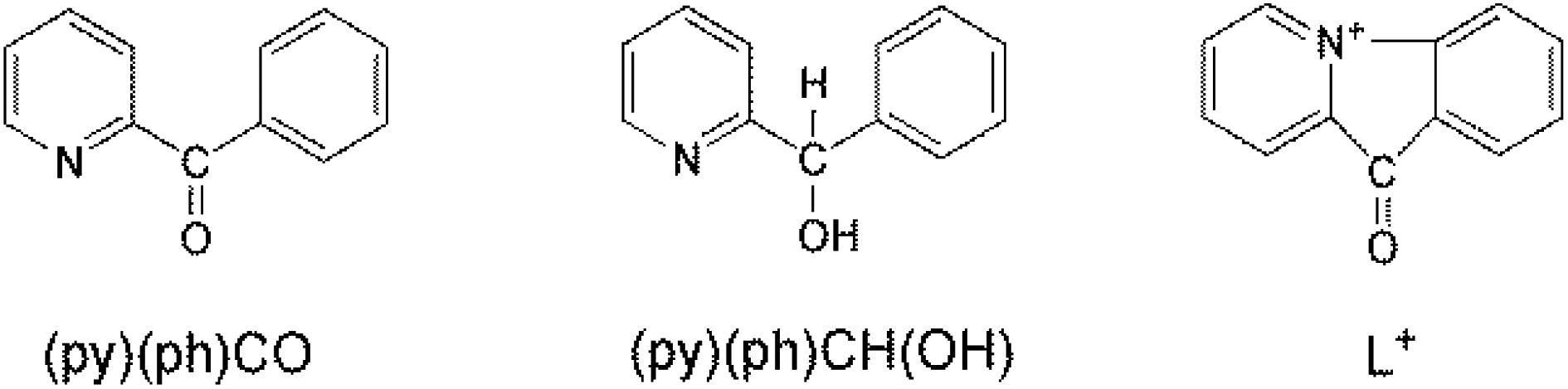 | ||
| Scheme 2 The free 2-benzoylpyridine ligand, (py)(ph)CO, whose In(III) chemistry has been studied in the present work, its reduced analogue (pyridin-2-yl)methanol, (py)(ph)CH(OH), and the 9-oxo-indolo[1,2-a]pyridinium cation (L+); all these species are discussed in the text. The important note mentioned in the caption of Scheme 1 is valid also for L+ and the ligands illustrated in the schemes that follow. | ||
Contrary to previous work which involved 3d-, 4d- and 5d-metals,15,16 we selected to work with the group 13 In(III) ion. The main reason of our choice was the recently reported unusual reactivity of (py)2CO towards this metal ion.17 In addition to the well established importance of indium in various aspects of material science (e.g. development of LCD monitors and television sets,18a applications of indium-tin oxide thin films,18b perovskites,18c,d optical materials,18e …), In(III) coordination complexes continue to attract the interest of many inorganic chemistry groups around the world due to their involvement in MOF chemistry (e.g. as sensors19a and efficient photocatalysts for hydrogen evolution19b), homogeneous (e.g. ring-opening polymerization of cyclic ethers19c) and heterogeneous (e.g. cycloaddition of CO2 with epoxides19d) catalysis, medicinal chemistry,19g NMR spectroscopy (115In with 95.7% natural abundance and nuclear spin 9/2 is a valuable NMR-active nucleus in solid-state research17,19h) and synthetic inorganic chemistry.19i,j
Experimental section
Chemicals and instrumentation
All manipulations were performed in the normal laboratory atmosphere under aerobic conditions. The materials (reagent grade) and solvents were used as received. Phenyl(pyridin-2-yl)methanol [an alternative name is (2-pyridine)(phenyl)methanol] (Scheme 2), (py)(ph)CH(OH), was synthesized by the reduction of (py)(ph)CO with NaBH4 as previously reported;20 its purity was checked by microanalyses, IR and IH NMR spectra. Elemental microanalyses (C, H, N) were performed by the University of Patras Instrumental Analysis Laboratory. Melting points were determined with an electrothermal apparatus and are uncorrected. Conductivity measurements were performed in DMSO at 25 ± 1 °C with a Metrohm-Herisau E-527 bridge and a cell of standard constant; the concentration of the solutions was ∼10−3 M. IR spectra were recorded using a PerkinElmer 16 PC FT-IR spectrometer. For the Raman measurements, the T64000 Horiba-Jobin Yvon micro-Raman setup was used. The excitation wavelength was 514.5 nm emitted from a DPSS laser (Cobolt Fandango TMISO laser, Norfolk, UK). The laser power on the samples was 2 mW. The backscattered radiation was collected from a single configuration of the monochromator after passing through an appropriate edge filter (LP02-633RU-25, laser2000, UK, Ltd, Huntingdon, Cambridgeshire, UK). The calibration of the instrument was achieved via the standard Raman peak position of Si at 520.5 cm−1. The spectral resolution was 5 cm−1. An attempt to obtain the Raman scattering of [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (vide infra) was also made. However, it was not possible to record the Raman spectrum since fluorescence of the samples was detected, overlapping the Raman effect. With the goal to bypass the fluorescence of the compound, we used a different wavelength (632.8 nm) of the Raman excitation laser line for the measurements; our attempts to overcome the fluorescence effect were again unsuccessful. The inherent fluorescence of the sample has been further supported through photoluminescence measurements (Fig. S1†). 1H and 13C NMR spectra were obtained at 600 and 150 MHz, respectively, on a Bruker Avance III HD spectrometer; the signal of the undeuterated portion of the solvent (d6-DMSO) was used as reference. Electron-spray ionization (ESI) mass spectra were recorded at 30 eV on a Waters Micromass ZQ spectrometer using HPLC grade MeOH as solvent, in the positive and negative modes.Synthetic procedures
Single-crystal X-ray crystallography
Crystallographic data (Table S1†) were collected with a Bruker APEX II Quasar diffractometer, equipped with a graphite monochromator centred on the path of Mo Kα radiations. Single crystals of 1, 3 and 4, taken directly from the reaction solution, were coated with Cargille™ NHV immersion oil and mounted on a fiber loop, followed by data collection at 120 K. The program SAINT was used to intergrate the data, which were thereafter corrected using SADABS.21 The structures were solved using SHELXT22a and refined by full-matrix least-squares technique on F2 using SHELXL-2018.22b All non-H atoms were refined with anisotropic displacement parameters, whereas H atoms were assigned to ideal positions and refined isotropically using a riding model, except those on the O atom of coordinated EtOH molecules in 1 and 4 which were located on the difference density map and introduced using DFIX constraints.The crystals of 3 are very thin needles. The small size makes the diffraction very poor, and the anisotropic shape makes the diffraction strongly dependent on the orientation of the crystal. Therefore, the data were cut at 1.05 Å as there is no diffraction above and Rint becomes larger. The result is a low θmax value, as well as a poor resolution and data/parameters ratio. SIMU restraints were applied to prevent nearly 2D thermal ellipsoids, as a probable consequence of the low resolution.
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication with the deposition numbers 2203979–2203981.†
Results and discussion
Synthetic comments
Our approach to activate the proligand (py)(ph)CO by indium(III) for further reactivity was to treat InX3·nH2O (X = Cl, n = 3; X = Br, n = 0) with the ligand in the absence of external base (addition of Et3N or NaOMe resulted in amorphous solids which could not be characterized). At the outset of our efforts we realized that the InX3·nH2O/(py)(ph)CO reaction systems are complicated, and the reactivity patterns of the chloride and bromide “salts” are completely different. For this reason, we kept as many as possible synthetic (reaction time and temperature, molar ratio of the reactants, solvent, concentrations in some experiments) and crystallization (storage of the reaction solutions at room temperature or 5 °C) parameters constant for comparable results. The discussion that follows represents simplified mechanisms, and it is supported by X-ray structural studies and strong spectroscopic evidence of the main products (1, 3, 4) and the side product (2), see the next two parts of this section (“Description of structures” and “Spectroscopic discussion in brief”). Treatment of InCl3·4H2O with (py)(ph)CO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) in EtOH at room temperature gave a colourless solution which became slowly yellow upon storage at 5 °C for a period of 2–3 days; from this solution was isolated a mixture of colourless and dark yellow crystals of [InCl3{(py)(ph)CO}(EtOH)]·{(py)(ph)CO} (1; yield ∼65%) and {(pyH)(ph)CO}Cl (i.e. the hydrochloride salt of 2-benzoylpyridine, 2; yield <15%), respectively. Due to the sharp colour and shape difference, the crystals were separated manually and independently characterised by microanalyses, IR, Raman and NMR spectra; in addition, the structure of 1 was unambiguously determined by single-crystal X-ray crystallography, while the identity of 2 was further supported by ESI-MS spectra. The dark yellow compound is clearly a byproduct of the reaction since its yield never exceeded 15%. Since the reaction solution remains colourless during the first hours of the crystallisation, we suspected that there is a kinetic effect in the formation of 1 and 2 in solution. Thus, we stopped the crystallisation after 12 h and collected the precipitated crystals of pure 1 from an almost colourless solution. To avoid coprecipitation of 2, we used a more dilute reaction solution (compared with the solution that gave the mixture) because we had observed that 2 is more soluble in EtOH than 1. A consequence of these two modifications is the low yield for the isolation of pure 1.
:2) in EtOH at room temperature gave a colourless solution which became slowly yellow upon storage at 5 °C for a period of 2–3 days; from this solution was isolated a mixture of colourless and dark yellow crystals of [InCl3{(py)(ph)CO}(EtOH)]·{(py)(ph)CO} (1; yield ∼65%) and {(pyH)(ph)CO}Cl (i.e. the hydrochloride salt of 2-benzoylpyridine, 2; yield <15%), respectively. Due to the sharp colour and shape difference, the crystals were separated manually and independently characterised by microanalyses, IR, Raman and NMR spectra; in addition, the structure of 1 was unambiguously determined by single-crystal X-ray crystallography, while the identity of 2 was further supported by ESI-MS spectra. The dark yellow compound is clearly a byproduct of the reaction since its yield never exceeded 15%. Since the reaction solution remains colourless during the first hours of the crystallisation, we suspected that there is a kinetic effect in the formation of 1 and 2 in solution. Thus, we stopped the crystallisation after 12 h and collected the precipitated crystals of pure 1 from an almost colourless solution. To avoid coprecipitation of 2, we used a more dilute reaction solution (compared with the solution that gave the mixture) because we had observed that 2 is more soluble in EtOH than 1. A consequence of these two modifications is the low yield for the isolation of pure 1.
A possible mechanistic scheme accounting for the formation of 1 and 2 is shown in Scheme 3. According to this scheme, (py)(ph)CO initially forms the chelate I. Due to steric or/and electronic reasons, a second chelating ligand cannot be coordinated to InIII to yield the anticipated 6-coordinate complex. Instead, the much less sterically demanding solvent molecule is coordinated to the metal ion increasing the coordination number of InIII from five to six. This complexation leads to increased acidity of the ethanol hydroxyl proton which is then associated to the N atom of a second (py)(ph)CO molecule through an intermolecular H bond giving rise to the crystalline complex 1. On standing at ambient temperature in solution, complex 1 is slowly converted to another crystalline compound which precipitates out of the reaction mixture, and proved by a combination of spectroscopic techniques to be {(pyH)(ph)CO}Cl (2); the formation of the latter salt involves the base-induced decomposition of the initial complex 1 with formation of the new species II (or a dimer of it formed through bridging of the deprotonated ethoxido groups) which is obviously soluble in EtOH. The precipitation of the salt 2 from an ethanol solution might be attributed to a strong intramolecular N–H⋯Ocarbonyl H bond forming a five-membered ring, which increases the hydrophobicity of the hydrochloride salt.
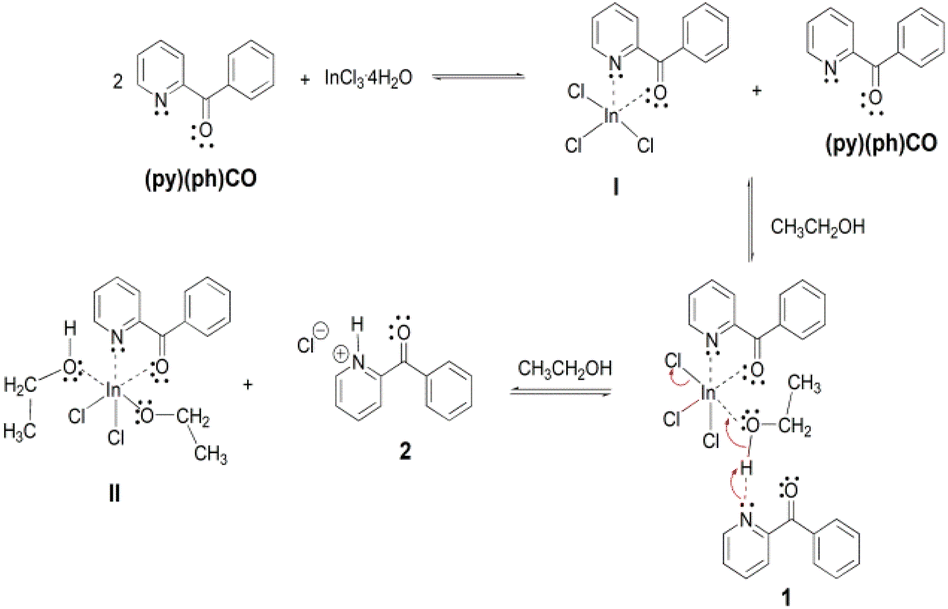 | ||
| Scheme 3 The proposed mechanism for the formation of complex [InCl3{(py)(ph)CO}(EtOH)]·{(py)(ph)CO} (1) and 2-benzoylpyridinium chloride, {(pyH)(ph)CO}Cl (2). | ||
The InBr3/(py)(ph)CO reaction pattern is completely different. Treatment of InBr3 with (py)(ph)CO (1:2) in EtOH at room temperature gave a colourless solution which rapidly turned to yellow upon stirring. Storage of the reaction solution at room temperature for ∼15 days resulted in the precipitation of a mixture of yellow crystals of (L)[InBr4{(py)(ph)CO}] (3; yield ∼40%) and colourless crystals of [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (4; yield ∼30%), where L+ is the cation shown in Scheme 2 and (py)(ph)CH(O)− is the monoanion of the reduced form of 2-benzoylpyridine whose structural formula (in the form of the neutral molecule) is also shown in Scheme 2. Due to the colour and shape difference, the crystals were separated manually and their solid-state structures were solved by single-crystal X-ray crystallography; they were also characterised by spectroscopic methods and microanalyses. Since we noticed that the amount of the yellow crystals decreased as a function of crystallisation time, probably due to their dissolution in EtOH (an indication of this is the color change of the supernatant solution to more intense, i.e. dark yellow, with time), we devised a method to isolate pure 4. The same procedure was followed, but with two modifications. The reaction solution was more dilute (to facilitate dissolution of 3) and the crystallisation period increased to ∼2 months; these modifications lead to the isolation of pure 4 (albeit with a low yield) from a dark yellow reaction mixture. The formation of 3 and 4, that contain L+ and the ligand (py)(ph)CH(O)−, using the above mentioned procedures, is clearly InIII-promoted/assisted. “Blind” experiments, i.e. without addition of InBr3, under identical conditions resulted in the isolation of pure (ph)(ph)CO (microanalyses, IR and 1H NMR evidences) after slow evaporation of EtOH at room temperature. As anticipated, pure 4 can also be prepared by the 1:1 reaction between InBr3 and performed20 (py)(ph)CH(OH) in a good yield (∼70%). Although we performed more than 300 reactions changing several reaction and crystallisation conditions, we have not been able to prepare 3 in pure form.
A possible mechanistic scheme accounting for the formation of 3 and 4 is shown in Scheme 4. According to this proposal, (py)(ph)CO initially forms the chelate III, similar to the one formed with InCl3 (I in Scheme 3). This molecule takes up a second (py)(ph)CO ligand which is coordinated in a monodentate manner through its carbonyl oxygen atom, forming the intermediate IV. Then a nucleophilic aromatic substitution with ring closure takes place through an intramolecular nucleophilic attack (a Michael-type reaction) of the uncoordinated pyridyl nitrogen atom on the adjacent phenyl ring, which is facilitated by the conjugated carbonyl group being activated through coordination to InIII. The thus obtained enolate intermediate V re-establishes the carbonyl functionality with concomitant rearomatization of the phenyl ring and simultaneous expulsion of a hydride (H−) ion, which is used to reduce the carbonyl group of the other (i.e. coordinated) (py)(ph)CO molecule. That way two new intermediates are formed, namely the coordinatively unsaturated new complex VI and the cyclized ammonium-type salt (L)+Br−. Finally, the initial chelate III is combined with the salt (L)+Br− providing the product (L)[InBr4{(py)(ph)CO}] (3). On the other hand, the intermediate complex VI (in which the coordination number of InIII is four) dimerizes and is further associated with two solvent (EtOH) molecules to form the other product 4 which has the normal coordination number six at each InIII centre.
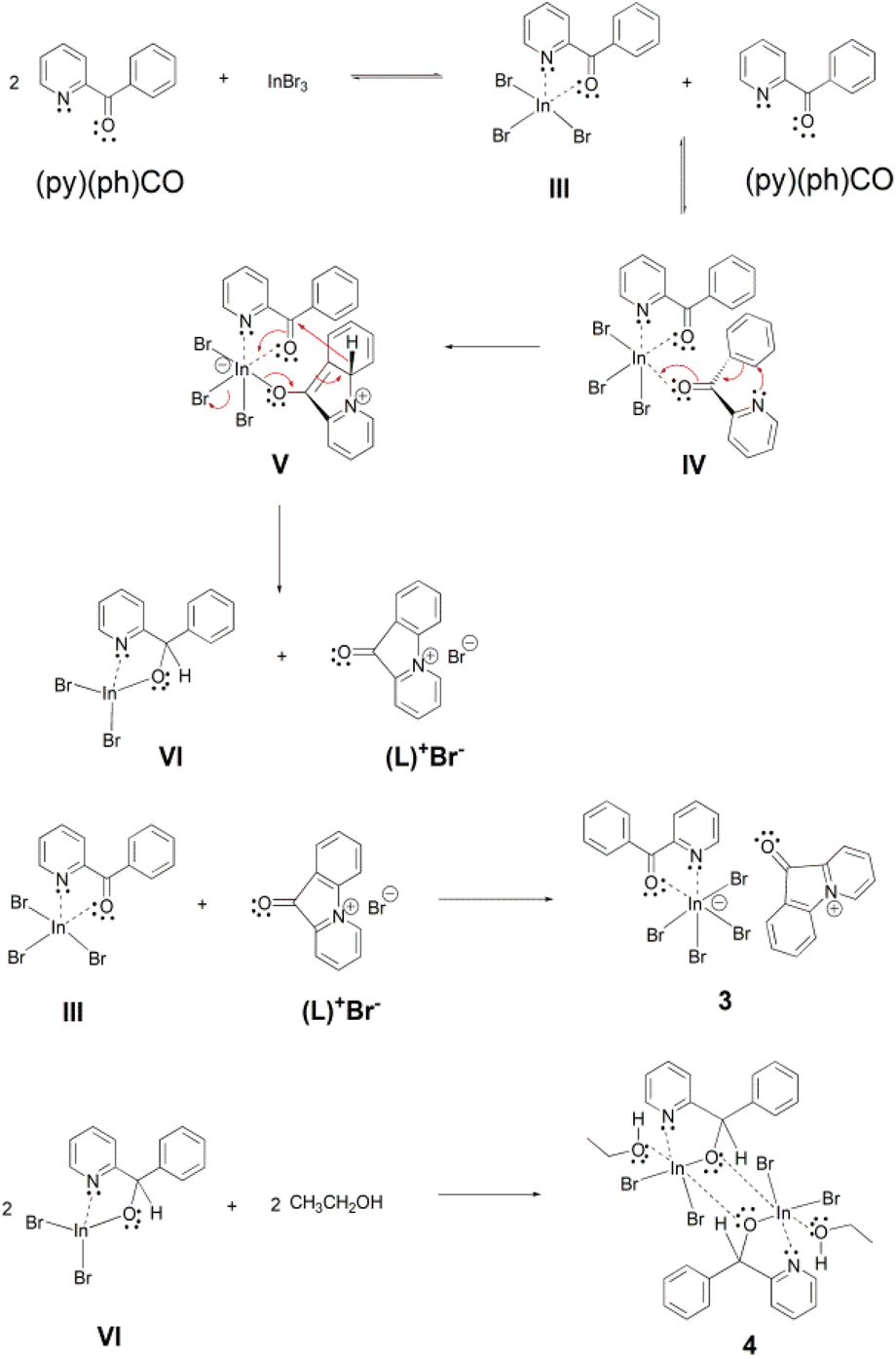 | ||
| Scheme 4 The proposed mechanism for the formation of complexes (L)[InBr4{(py)(ph)CO}2] (3) and [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (4). | ||
Compounds 1 and 4 are slightly soluble in EtOH, whereas 2 and 3 have a moderate solubility. All compounds are readily soluble in DMF and DMSO, but the solid-state structures of 1, 3 and 4 are not retained (at least in DMSO) as proven by 1H NMR spectroscopy (vide infra). Compound 2 has a limited solubility in H2O, whereas complexes 1, 3 and 4 are not stable as evidenced by the IR spectra of the collected solid materials.
As mentioned in Introduction, the reactivity of coordinated (py)(ph)CO has been investigated by few groups,15 including our group.16 The ligation modes of the resulting ligands are shown in Scheme 5, which also includes the structural formula of the cation L+ which counterbalances the charges of the anionic polymers {[CuIX2]}nn− (X = I, SCN)15a,b and [InBr4{(py)(ph)CO}]− in complex 3; a mechanism of its for mation is proposed in this work (Scheme 4). In complex [RuIICl{(py)CO2}(CO)(PPh3)2],15c the ligand has been oxidized to the coordinated picolinate(−) ion. Complexes [ReVOX2{(py)(ph)CH(O)}(PPh3)] (X = Cl, Br),15d,g [SnII(L′){(py)(ph)CH(O)}],15h where (L′)− is HC{CMeN(2,6-iPr2C6H3)2}−, and [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (4) contain the anionic ligand (py)(ph)CH(O)− which is the 2-electron reduced form of (py)(ph)CO. Complexes [CuII2{(py)(ph)C(OH)(O)}2{(py)(ph)CO}2(H2O)](ClO4)2,15e,f [CuII2{(py)(ph)C(OMe)(O)}2{(py)(ph)CO}2](ClO4)2,16a [CuII4(OMe)2(NO3)4{(py)(ph)C(OMe)(O)}2{(py)(ph)CO}2]16a and [CuII2(NO3)2{(py)(ph)C(OEt)(O)}2(EtOH)]16a contain the bridging monoanion of the gem-diol (the first one) and the hemiacetal (the next three ones) forms of (py)(ph)CO, formed in situ by the nucleophilic addition of H2O and alcohols (MeOH, EtOH) on the electrophilically activated (through coordination of the carbonyl oxygen atom and possibly the pyridyl ring) carbonyl carbon atom and subsequent deprotonation. For the formation of complexes [CuII2(NO3)2{(py)(ph)C(CH2NO2)(O)}2]16a and [NiII{(py)(ph)C(CH2CN)(O)}2],16b which were prepared under strongly basic conditions, the OH− ion abstracts one of the methyl hydrogens of the solvent (CH3NO2, CH3CN); once the carbanion (−CH2NO2, −CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) is formed, it attacks the positive (δ+) carbonyl carbon atom of (py)(ph)CO. The metal ion polarises further the carbonyl group of (py)(ph)CO, making it more susceptible to nucleophilic attack by −CH2NO2 or −CH2CN and stabilises the final ligand.
N) is formed, it attacks the positive (δ+) carbonyl carbon atom of (py)(ph)CO. The metal ion polarises further the carbonyl group of (py)(ph)CO, making it more susceptible to nucleophilic attack by −CH2NO2 or −CH2CN and stabilises the final ligand.
 | ||
| Scheme 5 Structural formulae, abbreviations and crystallographically established coordination modes of ligands derived from the to-date metal ion-assisted reactivity of (py)(ph)CO. The bridging behaviour of (py)(ph)CH(O)− from such reactivity studies has been observed for the first time in [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (4). The cation L+ has also resulted from (py)(ph)CO and it behaves as counterion in anionic polymers of Cu(I) and in complex (L)[InBr4{(py)(ph)CO}] (3), also described in this work. A list of the relevant complexes appears in Table 1. | ||
Description of structures
Partially labelled molecular structures and the supramolecular networks of complexes 1, 2 and 3 are shown in Fig. 1–4 and S2–S5.† Selected interatomic distances and angles are listed in Tables S2–S4.† | ||
| Fig. 1 Partially labelled plots of the structures of the molecules [InCl3{(py)(ph)CO}(EtOH)] and lattice (py)(ph)CO (upper part) that are present in complex 1. The dark green dashed line indicates the intramolecular H bond. | ||
 | ||
| Fig. 2 Partially labelled plots of the structures of the anion [InBr4{(py)(ph)CO}]− (right) and the cation L+ (left) that are present in complex 3. H atoms are omitted for clarity. Colour code: C, grey; N, blue; O, red; H, white; Br, orange; In, brown. | ||
 | ||
| Fig. 3 Partially labelled plot of the structure of the molecule [In2Br4{(py)(ph)CH(O)}2(EtOH)2] that is present in complex 4. H atoms are omitted for clarity. Colour code: C, grey; N, blue; O, red; Br, orange; In, brown. | ||
 | ||
| Fig. 4 Stick representation of the crystal structure of 4 along the (ac) plane, showing the packing of the dinuclear molecules and the weak Caromatic–H⋯Br H-bonding interactions (dashed orange lines) that form an 1D network along the α crystallographic axis. Colour code: C, grey; N, blue; O, red; Br, orange; In, brown. | ||
Complex 1 crystallises in the monoclinic space group P21/c. Its structure consists of mononuclear molecules [InCl3{(py)(ph)CO}(EtOH)] and lattice (py)(ph)CO molecules in an 1:1 ratio. In the complex molecule the InIII atom is 6-coordinate. The ligands are three terminal chloro (or chlorido) atoms (Cl1, Cl2, Cl3), one EtOH molecule and one bidentate chelating (η1:η1 or 1.11 adopting the Harris notation23) (py)(ph)CO molecule. The metal coordination geometry is distorted octahedral, the trans angles being in the range 158.0(1)–166.0(1)°. The distortion is primarily a consequence of the small bite angle [69.0(1)°] of the bidentate ligand. The three chloro ligands occupy cis (or fac) positions in the octahedron. The In1–Cl, In1–O and In–N bond lengths are typical for complexes with octahedral indium(II).17,19d,24 The carbonyl C6–O1 bond of the coordinated (py)(ph)CO molecule is longer than that of the free (i.e. uncoordinated) molecule25 [1.234(2) vs. 1.213(2) Å] due to coordination which weakens the carbonyl bond. This bond length is 1.217(2) Å in the lattice (ph)(ph)CO molecule of the compound, similar with that of the free one.25 Another consequence of the formation of the chelating ring in 1 is the position of the pyridyl nitrogen and carbonyl group; these are on opposite sides in the free molecule25 and on the same side in the coordinated (ph)(ph)CO.
The In(III) complex in 1 is H-bonded with the lattice (py)(ph)CO molecule through a O–H⋯N H bond involving the oxygen atom of the coordinated EtOH molecule (O2) as donor and the free pyridyl nitrogen atom (N2) as acceptor, the O⋯N distance being 2.675(4) Å. Weaker intermolecular C–H⋯O H bonds between the aromatic rings and the carbonyl groups are also present; together with Caromatic–H⋯Cl H bonds, all these weak interactions form a 3D lattice in the crystal (Fig. S2†). Compound 1 is the first structurally characterized In(III) complex containing (ph)(ph)CO as ligand.
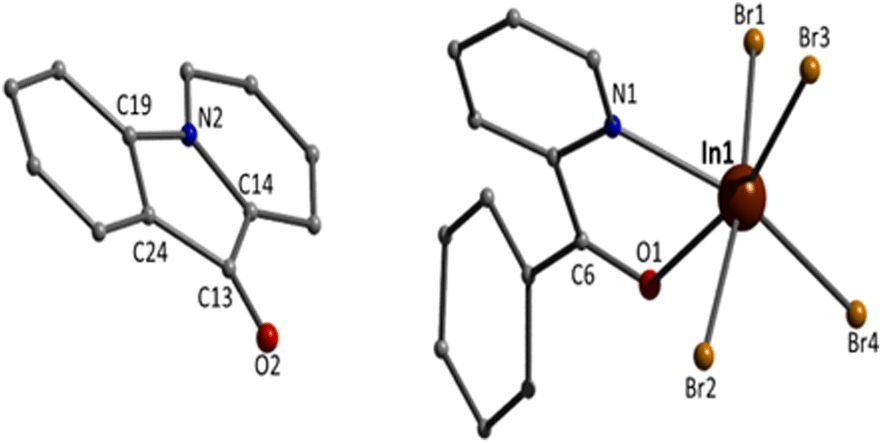
Although the quality of the crystal structure of 3 is not high, the basic structural features are clearly visible. The complex crystallises in the monoclinic space group C2/c. Its structure consists of mononuclear [InBr4{(py)(ph)CO}]− anions and L+ cations in an 1:1 ratio. In the anion, the 6-coordinate InIII atom is bonded to four bromo (or bromido) atoms and to one 1.11 (py)(ph)CO ligand. The InIII–Br bond lengths are in the narrow 2.563(2)–2.647(2) Å. Again the carbonyl bond of the coordinated (py)(ph)CO molecule is weaker than that of the free ligand25 [1.251(19) vs. 1.213(2) Å] due to the coordination, but similar with the corresponding bond length of the coordinated (py)(ph)CO molecule in 1. The metal coordination geometry is distorted octahedral, with the trans angles in the range 158.2(4)–169.8(1)°. The L+ cation is planar the C13–O2 and C19–N2 bond distances are 1.18(2) and 1.41(2) Å, indicative of double carbon–oxygen and single carbon–nitrogen bonds, respectively, in agreement with its formulation (Scheme 2). The carbonyl bond length is similar with the corresponding bond distances in the free ligand25 and in the lattice (py)(ph)CO molecule in 1. This bond length is also close with that in the Cu(I) complexes that contain the L+ cation (Table 1).
| Compounda,b | Ref. |
|---|---|
| a Lattice solvent molecules have been omitted. b For the structural formulae of the transformed ligands and their coordination modes, see Scheme 5. c L+ is the uncoordinated counterion 9-oxo-indolo[1,2-a]pyridinium, whose structural formula is also shown in Scheme 5. d X = Cl, Br. (L′)− is HC{CMeN(2,6- iPr2C6H3)2}− with iPr representing the isopropyl group. | |
| {(L)[CuII2]}nc |
15a |
| {(L)[CuI(SCN)2]}nc |
15b |
| [RuIICl{(py)CO2}(CO)(PPh3)2] | 15c |
| [ReVOX2{(py)(ph)CH(O)}(PPh3)]d | 15d and g |
| [CuII2{(py)(ph)C(OH)(O)}2{(py)(ph)CO}2(H2O)] (ClO4)2 | 15e and f |
| [SnII(L′){(py)(ph)CH(O)}] | 15h |
| [CuII2{(py)(ph)C(OMe)(O)}2{(py)(ph)CO}2] (ClO4)2 | 16a |
| [CuII4(OMe)2(NO3)4{(py)(ph)C(OMe)(O)}2{(py)(ph)CO}2] | 16a |
| [CuII2(NO3)2{(py)(ph)C(OEt)(O)}2(EtOH)] | 16a |
| [CuII2(NO3)2{(py)(ph)C(CH2NO2)(O)}2] | 16a |
| [NiII{(py)(ph)C(CH2CN)(O)}2] | 16b |
| [In2Br4{(py)(ph)CH(O)}2(EtOH)2] (4) | This work |
| (L)[InBr4{(py)(ph)CO}] (3)c | This work |
The supramolecular packing of the anions and cations in the crystal structure of 3 involve weak interionic Caromatic–H⋯Ofree carbonyl and Caromatic–H⋯Br H-bonding interactions, forming a 3D network in the crystal (Fig. S4†).
The L+ cation is also present in the structures of the anionic polymers {(L)[CuII2]}n15a and {(L)[CuI(SCN)2]}n,15b which were isolated from CuI/(py)(ph)CO and Cu(SCN)/(py)(ph)CO reaction mixtures in EtOH, respectively.
Complex 4 crystallises in the monoclinic space group P21/c. Its structure contains dinuclear [In2Br4{(ph)(ph)CH(O)}2(EtOH)2] molecules. The complex contains both enantiomeric forms of the (py)(ph)CH(O)− ligand, with S and R chiralities on C6 and C18, respectively, placing the phenyl rings in trans positions. Slightly different metrics (see ESI†) and a different conformation of the coordinated EtOH make the nearly meso complex not exactly centrosymmetric. However, the space group is centrosymmetric with inversion centres residing between molecules. The deprotonated alkoxo (or alkoxido) oxygen atoms of the two 2.21 (py)(ph)CH(O)− ligands doubly bridge the two metal centres. The In1⋯In2 distance is relatively short [3.459(1) Å] due to the presence of two monoatomic bridges. Each bridge is nearly symmetrical; for example the In1–O2 and In2–O2 distances are 2.134(3) and 2.161(3) Å, respectively. The In–O–In angles are ∼107°. The central {InIII2(μ-OR)2}4+ core [R- = (py)(ph)CH-] can be described as approximately rhombic. However, the four sides of the “rhombus” are not equal [2.134(3)–2.184(3) Å]; furthermore, the two InIII and the two bridging alkoxo oxygen atoms are not strictly coplanar with torsion angles of ∼1.9°. The large diagonal of the “rhombus” is 3.459(1) Å, while the short one is 2.565(1) Å. At each InIII atom, two terminal bromo atoms, the pyridyl nitrogen atom of one ligand and the oxygen atom of one terminal EtOH molecule complete 6-coordination. The InIII–Br bond lengths [2.532(1)–2.622(1) Å] are similar with those in 3 which also contains only terminal bromides. The InIII–Oalkoxo bonds are stronger than the InIII–OEtOH ones, as expected. The indium(III) coordination geometries are distorted octahedral, the trans angles being in the ranges 144.4(1)–172.6(1)° and 145.2(1)–173.6(1)° for In1 and In2, respectively. For both metal ions, the trans pairs of donor atoms are defined by Br, Oalkoxo, Br, OEtOH and Oalkoxo, N. There are two weak intramolecular H bonds (Fig. S5†). The donor atoms are the EtOH oxygens O3 and O4, and the acceptors are the bromo groups Br4 and Br2, respectively; the donor and acceptor atoms of each H bond belong to the coordination sphere of different metal ions [for example, O4⋯Br2 = 3.279(2) Å and O4–H(O4)⋯Br2 = 178.8°]. A consequence of these H bonds is that the InIII–Br bond lengths involving the H-bonded bromo atoms [In1–Br2 = 2.614(1) and In2–Br4 = 2.622(1) Å] are longer (i.e. weaker) than those involving the “free” ones [In1–Br1 = 2.537(1) and In2–Br3 = 2.532(1) Å]. The C6–O1 and C18–O2 bond lengths are identical [1.406(6/5) Å] and this value is typical17 for single carbon–oxygen bonds, confirming the reduction at the carbonyl group of (py)(ph)CO to (py)(ph)CH(O)−. These bond distances are almost identical with those seen in complexes containing the (py)(ph)CH(O)− ligand;15d,g,h for example, this bond length is 1.40(2) Å in complex [SnII(L′){(py)(ph)CH(O)}],15h where (L′)− is HC{CMeN(2,6-iPr2C6H3)2}−.
Weak intermolecular Caromatic–H⋯Br interactions are present in the structure of 4 (C⋯Br = 3.80–3.83 Å), forming an 1D network in the crystal (Fig. 4).
Complex 4 is the first structurally characterised In(III) complex with the neutral or anionic form of (py)(ph)CH(OH) as ligand. It joins a small family of metal complexes with (py)(ph)C(H)(OH)26 or (py)(ph)CH(O)−;15d,g,h,26,27 three of them (Table 1) have been derived by reactivity studies of coordinated (py)(ph)CO.15d,g,h The neutral molecule behaves as a N,O-chelating ligand (1.11),26 while the anionic ligand acts in the 1.11,15d,g 1.0115h or 2.2126,27 manner.
Spectroscopic discussion in brief
Data are shown in Fig. 5, 6 and S1, S6–S17.† In the IR spectra of 1 (Fig. S6†) and 4, the weak-to-medium intensity bands at ∼3440 and ∼3320 cm−1, respectively, are assigned to the ν(OH) vibration of the coordinated EtOH molecules;16a their relatively low wavenumber and broadness are both indicative of H bonding (established by crystallography). The presence of EtOH is manifested by the appearance of the signals at δ ∼1.1 (–CH3), ∼3.5 (–CH2) and ∼4.35 (–OH) ppm (Fig. S7†). The IR spectra of 1 and 3 (Fig. S8†) exhibit a strong-to-medium intensity band at ∼1625 cm−1 attributed to the ν(CO) of the ligated (py)(ph)CO;16a due to coordination, this band has been shifted to a lower wavenumber compared with the corresponding vibration in the spectrum of free (py)(ph)CO at 1668 cm−1 (Fig. S9†).28 The corresponding Raman peak appears at 1626 (1), 1628 (3) and 1665 [free (py)(ph)CO] cm−1 (Fig. 5 and S10†). The IR spectra of 1 and 3 show an additional carbonyl stretching vibration at 1668 cm−1. These can be undoubtedly assigned to the ν(CO) vibration of the lattice (py)(ph)CO molecule that is present in 1 and the L+ (Scheme 2) ion which counterbalances the charge of [InBr4{(py)(ph)CO}]− in compound 3. The corresponding Raman peaks appear at 1671 (1) and 1670 (3) cm−1. Thus, vibrational spectroscopy can easily reflect the two different carbonyl groups in each of these two complexes. The IR spectrum of 4 does not display a band in the carbonyl stretching vibration region (1730–1620 cm−1) region, in accordance with the reduction of the carbonyl group (i.e. absence of 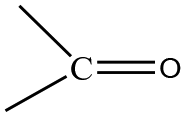 ) in this dinuclear complex; the nearest band at 1608 cm−1 in the spectrum of 4 is due to an aromatic stretching vibration. The medium intensity IR band at 1082 cm−1 can be assigned29,30 to the stretching vibration of the single carbon–oxygen bond, ν(C–O), of the ligand. The Raman peaks of 1 at 233 cm−1 and 3 at 196 cm−1 are attributed to a stretching vibration of the terminal InIII–Cl, ν(In–Cl)t, and InIII–Br, ν(In–Br)t, respectively;17,31 both peaks are absent from the spectrum of free (py)(ph)CO, as expected.
) in this dinuclear complex; the nearest band at 1608 cm−1 in the spectrum of 4 is due to an aromatic stretching vibration. The medium intensity IR band at 1082 cm−1 can be assigned29,30 to the stretching vibration of the single carbon–oxygen bond, ν(C–O), of the ligand. The Raman peaks of 1 at 233 cm−1 and 3 at 196 cm−1 are attributed to a stretching vibration of the terminal InIII–Cl, ν(In–Cl)t, and InIII–Br, ν(In–Br)t, respectively;17,31 both peaks are absent from the spectrum of free (py)(ph)CO, as expected.
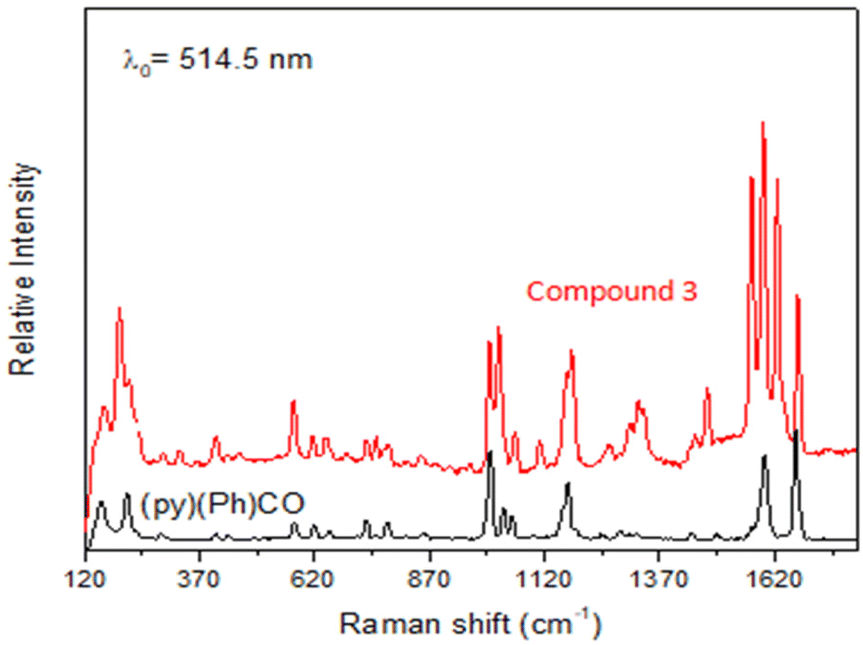 | ||
| Fig. 5 The Raman spectra (cm−1) of free (py)(ph)CO (bottom) and compound (L)[InBr4{(py)(ph)CO}] (3). | ||
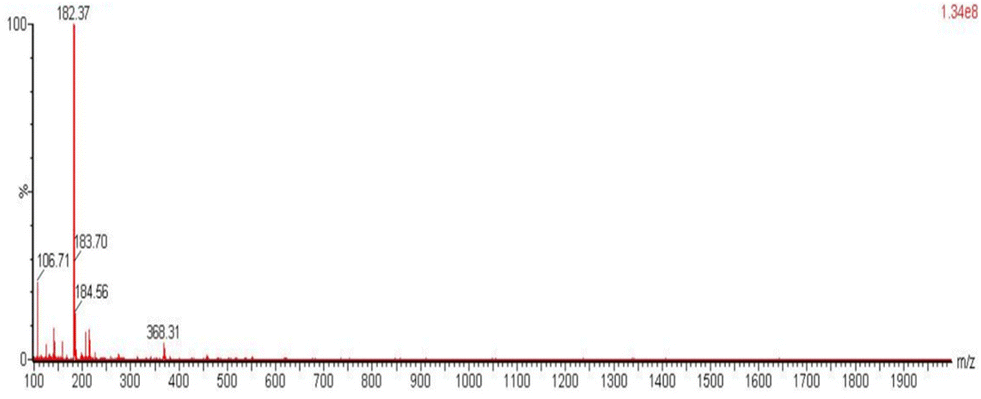 | ||
| Fig. 6 The ESI-MS spectrum of {(pyH)(ph)CO}Cl (2) in the positive mode. | ||
The behavior of 1, 3 and 4 in solution (DMSO) was probed by 1H NMR spectroscopy. The spectrum of 1 in d6-DMSO (Fig. S7†) shows, in addition to the EtOH signals, all the signals of free (py)(ph)CO (Fig. S11†) at exactly the same δ values. This, together with the negligible value of the molar conductivity (ΛM) of 1 in DMSO (3 S cm2 mol−1),32 indicates decomposition of the complex in solution to release free (py)(ph)CO, eqn (1).
 | (1) |
The 1H NMR spectrum of 3 in d6-DMSO is a sum of signals arising from the cation L+ and free (py)(ph)CO. The ΛM value in DMSO (36 S cm2 mol−1) is indicative of an 1:1 electrolyte, again suggesting decomposition in solution, eqn (2).
 | (2) |
In addition to the typical EtOH signals, the 1H NMR spectrum of 4 (Fig. S12†) shows the typical signals of free (py)(ph)CO. This was a surprise because the complex contains the reduced form of 2-benzoylpyridine, i.e. the anionic ligand (py)(ph)CH(O)−. We anticipated the appearance of a singlet in the δ range 4.5–6.5 ppm attributable14e,15h,33 to the quaternary CH proton of the reduced ligand; such a signal could not be seen in more than ten samples of 4. The only explanation we can offer is the aerobic oxidation of (py)(ph)CH(O)−, catalyzed by In(III), in the presence of oxygen. This reaction with Co(O2CMe)2·4H2O as catalyst is well documented.27b It is interesting to note that these catalytic aerobic oxidation reactions are very selective to pyridine-based secondary alcohols, such as (py)(ph)CH(OH), over primary alcohols and that they take place without any external ligand, base or additive.27b Compound 2 (vide supra) was identified as the hydrochloride salt of 2-benzoylpyridine, {(pyH)(ph)CO}Cl, by microanalyses, physical techniques and spectroscopic methods. A AgNO3 test for an acidic sample of the compound is positive for Cl− ions. Its melting/decomposition point is 168 °C, much higher than that of the free (py)(ph)CO (42–43 °C). The ΛM value (38 S cm2 mol−1) in DMSO indicates an 1:1 electrolyte in solution. In the IR spectrum (Fig. S13†), the bands at ∼2850 and 1738 cm−1 are assigned to the ν(NH+) and ν(CO) vibrations, respectively. The high wavenumber of ν(CO) reflects the –I inductive effect of the positive N atom on the carbonyl group. The 1H NMR spectrum of 2 (Fig. S14†) is pretty similar with that of free (py)(ph)CO, with the only essential difference being the doublet at δ 8.49 ppm in the spectrum of the former which is attributed to the NH+ proton. The aromatic proton signals appear at the same δ values in the two spectra; this is most probably due to the existence of a strong five-membered intramolecular N–H⋯Ocarbonyl H bond, mentioned in the explanation of the mechanism detailed in Scheme 3. For the same reason, the 13C{1H} NMR spectra of (py)(ph)CO (Fig. S15†) and 2 (Fig. S16†) are also very similar. A definite proof for the identity of this compound comes from mass spectrometry (Fig. 6 and S17†). The ESI-MS spectrum of the compound in the positive mode shows the expected [M + H]+ quasi-molecular ion at m/z 184.56 and an associated fragment at m/z 106.70 ([PhCHO]+), but also the [M]+ molecular ion at m/z 183.70. However, the strongest peak by far is at m/z 182.37 attributed to [M − 1]+, whose structure is probably identical with that of the cation L+ of the salt (L)+Br− (Scheme 4); this is formed from the molecular ion (radical cation) with a H atom loss. On the other hand, the ESI-MS spectrum of the same compound in the negative mode provides strong evidence in favour of the hydrochloride salt 2. Indeed, the quasi-molecular ion is observed at m/z 259.22, 257.23 and 255.23, which corresponds to the cluster [2 + HCl]− formed by an one-electron reduction of 2 and uptake of one HCl molecule.34
Concluding comments
In this work, we have reported that the employment of (py)(ph)CO in reactions with InBr3 has resulted in interesting transformations of the ligand, demonstrating that metal ion-driven reactivity of 2-pyridyl ketones remains an interesting area of research. The complex salt 3 is the third structurally characterised compound containing the cation L+ (Table 1), while compound 4 is the fourth structurally characterised complex possessing the reduced anionic ligand (py)(ph)CH(O)− arising from (py)(ph)CO (Table 1). Mechanistic schemes have been proposed for the observed transformations (Scheme 4) in 3 and 4, and for the byproduct 2. Since all the previous examples on the reactivity of coordinated (py)(ph)CO were with transition metals, our results show that transformations are also possible with p-block elements. Although compound (py)(ph)CH(OH) can be prepared by metal ion-free reduction of (py)(ph)CO,20 the observed (py)(ph)CO → 4 (this work) and (py)(ph)CO → [ReVOX2{(py)(ph)CH(O)}(PPh3)]15d,g transformations show that this reaction can be also metal ion-assisted. Overall compounds 1, 3 and 4 are the first In(III) complexes derived from (py)(ph)CO or (py)(ph)CO-based species.A comparison between the reactivity of coordinated (py)(ph)CO (Scheme 2) and (py)2CO (Scheme 1) reveals that the formation of L+ has been observed only for the former (ref. 15d, g and this work). On the contrary, the ligand (py)2CH(O)− has been derived from the NiII-promoted reduction of (py)2CO under solvothermal conditions.35
The question of why InCl3 does not provide L+ or (py)(ph)CH(O)− (whereas InBr3 does), under identical reaction conditions, is difficult to be answered without DFT calculations. Differences in the products of reactions of InCl3 and InBr3 with simple ligands are often observed, even from the early years of the development of In(III) chemistry.36 Lattice energies of the products have been proposed as the reason of the differences. Based on this assumption, it is possible that transformed species are present in the reaction InCl3/(py)(ph)CO solutions in EtOH and that the more insoluble compounds 1 and 2 are preferentially precipitated. Using a more chemical basis, it is well known37 that ligand reactivity is affected by three factors: (a) the electron-acceptor/donor properties of the metal centre; (b) the electron-donor/acceptor properties of the ligands; and (c) the nature of the co-ligands. In our case, only factor c is variable. We believe that the slightly different net electron donor/acceptor abilities of chlorides and bromides are an explanation for the observed differences. Perhaps, the Br− co-ligand has a net electron donor/acceptor ability more opposite (compared to the Cl− one) to that of (py)(ph)CO, thus assisting In(III) towards activation of the primary organic ligand;37 if this hypothesis is valid, the chloride and (py)(ph)CO have similar electronic ability, they complete each other and feel a reduced activation by the metal centre.
We do believe that this particular research topic has more interesting results to give. We continue to investigate the reactivity of other activated compounds containing carbonyl groups (ketones and aldehydes) and metal-mediated synthesis, and we expect further advances not only in stoichiometric reactions but also, more importantly, in catalysis. Our current efforts involve: (i) reactions of the other trivalent metals of group 13, e.g. Al(III) and Ga(III), with (py)(ph)CO; and (ii) replacement of the phenyl ring of (py)(ph)CO with a methyl group (i.e., use of 2-acetylpyridine) which is less bulky, has an opposite inductive effect and contains polar C–H bonds adjacent to the carbonyl group (i.e. weakly acidic α-hydrogens), all these characteristics giving hopes16a for interesting reactivity patterns with group 13 and other p-block metals. Preliminary results with Ga(III) concerning point (ii) reveal the formation of the coordinated ligand (py)C(Me)(OH)CH2C(OMe)(O)(py)−, a transformation which has been reported previously in Cu(II) chemistry.16a As a final note, we plan to include advanced theoretical calculations to more rigorously explain the experimental facts and the observed differences in the reactivity patterns of similar systems.
Author contributions
CS: investigation, data curation, formal analysis. ZGL: data curation, formal analysis, validation. CTC: data curation, formal analysis. DP: conceptualisation, supervision, writing – review and editing, writing manuscript. PD: data curation, investigation, formal analysis, writing review and editing, writing manuscript. SPP: conceptualisation, supervision, project management, writing manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Research Director George Voyiatzis for permitting us to use the Raman facilities at ICE-HT/FORTH and Dr Vlasoula Bekiari for recording the emission spectrum of complex 4 at the Department of Crop Science of the University of Patras (Messolonghi campus).References
- E. C. Constable, Metals and Ligand Reactivity, VCH, Weinheim, Germany, 1996, pp. 22–62 Search PubMed.
- G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson, Boston, USA, 5th edn, 2014, pp. 468–470 Search PubMed.
- D. Sun, D.-F. Wang, N. Zhang, F.-J. Liu, H.-J. Hao, R.-B. Huang and L.-S. Zheng, Dalton Trans., 2011, 40, 5677–5679 RSC.
- M. Riaz, R. K. Gupta, H.-F. Su, Z. Jagličic, M. Kurmoo, C.-H. Tung, D. Sun and L.-S. Zheng, Inorg. Chem., 2019, 58, 14331–14337 CrossRef CAS.
- Y.-Q. Zhao, M.-X. Fang, Z.-H. Xu, X.-P. Wang, S.-N. Wang, L.-L. Han, X.-Y. Li and D. Sun, CrystEngComm, 2014, 16, 3015–3019 RSC.
- B. Liu, F. Yu, M. Tu, Z.-H. Zhu, Y. Zhang, Z.-W. Ouyang, Z. Wang and M.-H. Zeng, Angew. Chem., Int. Ed., 2019, 58, 3748–3753 CrossRef CAS PubMed.
- Z.-H. Zhu, Q. Hu, H.-L. Pan, Y. Zhang, H. Xu, M. Kurmoo, J. Huang and M.-H. Zeng, Sci. China: Chem., 2019, 62, 719–726 CrossRef CAS.
- R. W. Hay, in Reactions of Coordinated Ligands, ed. P. S. Braterman, Plenum, New York, USA, 1989, vol. 2 Search PubMed.
- R. W. Hay, in Comprehensive Coordination Chemistry I, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, vol. 6 Search PubMed.
- For older reviews, see: (a) G. S. Papaefstathiou and S. P. Perlepes, Comments Inorg. Chem., 2002, 23, 249–274 CrossRef CAS; (b) A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC (perspective); (c) T. C. Stamatatos, C. G. Efthymiou, C. C. Stoumpos and S. P. Perlepes, Eur. J. Inorg. Chem., 2009, 3361–3391 CrossRef CAS (microreview).
- T. C. Stamatatos, S. P. Perlepes, C. P. Raptopoulou, A. Terzis, C. S. Patrickios, A. J. Tasiopoulos and A. K. Boudalis, Dalton Trans., 2009, 3354–3362 RSC.
- Representative examples: (a) G. S. Papaefstathiou, C. P. Raptopoulou, A. Tsohos, A. Terzis, E. G. Bakalbassis and S. P. Perlepes, Inorg. Chem., 2000, 39, 4658–4662 CrossRef CAS; (b) T. C. Stamatatos, S. P. Perlepes, C. P. Raptopoulou, V. Psycharis, C. S. Patrickios, A. J. Tasiopoulos and A. K. Boudalis, Inorg. Chem. Commun., 2009, 12, 402–405 CrossRef CAS.
- Representative examples from our group: (a) C. C. Stoumpos, O. Roubeau, G. Aromi, A. J. Tasiopoulos, V. Nastopoulos, A. Escuer and S. P. Perlepes, Inorg. Chem., 2010, 49, 359–361 CrossRef CAS PubMed; (b) C. G. Efthymiou, C. Papatriantafyllopoulou, G. Aromi, S. J. Teat, G. Christou and S. P. Perlepes, Polyhedron, 2011, 30, 3022–3025 CrossRef CAS; (c) C. G. Efthymiou, I. Mylonas-Margaritis, C. P. Raptopoulou, V. Psycharis, A. Escuer, C. Papatriantafyllopoulou and S. P. Perlepes, Magnetochemistry, 2016, 2, 30 CrossRef; (d) Z. G. Lada, Y. Sanakis, C. P. Raptopoulou, V. Psycharis, S. P. Perlepes and G. Mitrikas, Dalton Trans., 2017, 46, 8458–8475 RSC; (e) C. C. Stoumpos, P. Danelli, G. Zahariou, M. Pissas, V. Psycharis, C. P. Raptopoulou, Y. Sanakis and S. P. Perlepes, Polyhedron, 2021, 207, 115350 CrossRef CAS; (f) K. Skordi, A. Anastasiades, A. D. Fournet, R. Kumar, M. Schulze, W. Wernsdorfer, G. Christou, V. Nastopoulos, S. P. Perlepes, C. Papatriantafyllopoulou and A. J. Tasiopoulos, Chem. Commun., 2021, 57, 12484–12487 RSC.
- For example, see: (a) B. Machura, J. Palion, J. Mrozinski, B. Kalinska and R. Kruszynski, Polyhedron, 2013, 49, 216–222 CrossRef CAS; (b) B. Machura, I. Nawrot, K. Michalik and Z. Drzazga, Polyhedron, 2011, 30, 2294–2302 CrossRef CAS; (c) C. J. Milios, T. C. Stamatatos, P. Kyritsis, A. Terzis, C. P. Raptopoulou, R. Vicente, A. Escuer and S. P. Perlepes, Eur. J. Inorg. Chem., 2004, 2885–2901 CrossRef CAS; (d) A. Schneider and H. Vahrenkamp, Z. Anorg. Allg. Chem., 2003, 629, 2122–2126 CrossRef CAS; (e) C. Sudbrake and H. Vahrenkamp, Inorg. Chim. Acta, 2001, 318, 23–30 CrossRef CAS.
- (a) M. A. S. Goher, A. E. H. Abdou, B.-S. Luo and T. C. W. Mak, J. Coord. Chem., 1995, 36, 71–80 CrossRef CAS; (b) M. A. S. Goher, R.-J. Wang and T. C. W. Mak, J. Coord. Chem., 1996, 38, 151–154 CrossRef CAS; (c) J. G. Malecki and R. Kruszynski, Polyhedron, 2007, 26, 2686–2694 CrossRef CAS; (d) B. Machura, J. Milek, R. Kruszynski, J. Kusz and J. Mrozinski, Polyhedron, 2008, 27, 1262–1269 CrossRef CAS; (e) S. Dey, S. Sarkar, E. Zangrando, H. Stoeckli Evans, J.-P. Sutter and P. Chattopadhyay, Inorg. Chim. Acta, 2011, 367, 1–8 CrossRef CAS; (f) Y. Li and L. Jin, J. Cluster Sci., 2011, 22, 41–47 CrossRef CAS; (g) N. C. Yumata, G. Habarurema, J. Mukiza, T. I. A. Gerber, E. Hosten, F. Taherkhani and M. Nahali, Polyhedron, 2013, 62, 89–103 CrossRef CAS; (h) A. Jana, H. W. Roesky, C. Schulzke and A. Doring, Angew. Chem., Int. Ed., 2009, 48, 1106–1109 CrossRef CAS PubMed.
- (a) A. A. Kitos, C. G. Efthymiou, M. J. Manos, A. J. Tasiopoulos, V. Nastopoulos, A. Escuer and S. P. Perlepes, Dalton Trans., 2016, 45, 1063–1077 RSC; (b) A. A. Kitos, D. P. Giannopoulos, C. Papatriantafyllopoulou, L. Cunha-Silva and S. P. Perlepes, Inorg. Chem. Commun., 2016, 64, 53–55 CrossRef CAS.
- C. Stamou, W. Papawassiliou, J. P. Carvalho, K. F. Konidaris, V. Bekiari, P. Dechambenoit, A. J. Pell and S. P. Perlepes, Inorg. Chem., 2021, 60, 4829–4840 CrossRef CAS PubMed.
- (a) F. J. Alguacil, Molecules, 2020, 25, 5238 CrossRef CAS PubMed; (b) C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson, Harlow, UK, 5th edn, 2018, p. 391 Search PubMed; (c) R. Kaiukov, G. Almeida, S. Marras, Z. Dang, D. Baranov, U. Petralanda, I. Infante, E. Mugnaioli, A. Griesi, L. De Trizio, M. Gemmi and L. Manna, Inorg. Chem., 2020, 59, 548–554 CrossRef CAS PubMed; (d) J. Liang, X. Han, J.-H. Yang, B. Zhang, Q. Fang, J. Zhang, Q. Ai, M. M. Ogle, T. Terlier, A. A. Marti and J. Lou, Adv. Mater., 2019, 31, 1903448 CrossRef CAS; (e) Y. Huang, T.-K. Jiang, B.-P. Yang, C.-L. Hu, Z. Fang and J.-G. Mao, Inorg. Chem., 2022, 61, 3374–3378 CrossRef CAS PubMed.
- (a) Y.-Z. Li, G.-D. Wang, Y.-K. Lu, L. Hou, Y.-Y. Wang and Z. Zhu, Inorg. Chem., 2020, 59, 15302–15311 CrossRef CAS PubMed; (b) C. Lu, D. Xiong, C. Chen, J. Wang, Y. Kong, T. Liu, S. Ying and F.-Y. Yi, Inorg. Chem., 2022, 61, 2587–2594 CrossRef CAS PubMed; (c) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef CAS; (d) G.-M. Liang, P. Xiong, K. Azam, Q.-L. Ni, J.-Q. Zeng, L. C. Gui and X.-J. Wang, Inorg. Chem., 2020, 59, 1653–1659 CrossRef CAS PubMed; (e) M. Gut and J. P. Holland, Inorg. Chem., 2019, 58, 12302–12310 CrossRef CAS PubMed; (f) M. R. Gill, M. G. Walker, S. Able, O. Tietz, A. Lakshminarayanan, R. Anderson, R. Chalk, A. H. El-Sagheer, T. Brown, J. A. Thomas and K. A. Vallis, Chem. Sci., 2020, 11, 8936–8944 RSC; (g) D. V. Wagle, S. P. Kelley, G. A. Baker, K. Sikligar and J. L. Atwood, Angew. Chem., Int. Ed., 2020, 59, 8062–8065 CrossRef CAS PubMed; (h) F. Chen, G. Ma, G. M. Bernard, R. G. Cavell, R. McDonald, M. J. Ferguson and R. E. Wasylishen, J. Am. Chem. Soc., 2010, 132, 5479–5493 CrossRef CAS PubMed; (i) S. Banerjee, S. Dutta, S. Kumar Sarkar, N. Graw, R. Herbst-Irmer, D. Koley, D. Stalke and H. W. Roesky, Dalton Trans., 2020, 49, 14231–14236 RSC; (j) K. Zeckert and D. Fuhrmann, Inorg. Chem., 2019, 58, 16736–16742 CrossRef CAS.
- H. Kim and S. K. Kang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o947 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, ver. 2.03, Bruker Analytical X-Ray Systems, Madison, WI, USA, 2000 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349–2355 RSC.
- C. Birnara, V. G. Kessler and G. S. Papaefstathiou, J. Coord. Chem., 2017, 70, 1–20 CrossRef.
- M. Sievert, R. Dinelt and H. Bock, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 674–676 CrossRef.
- F. Di Salvo, M. Y. Tsang, F. Teixidor, C. Viñas, J. G. Planas, J. Crassous, N. Vanthuyne, N. Aliaga-Alcalde, E. Ruiz, G. Coquerel, S. Clevers, V. Dupray, D. Choquesillo-Lazarte, M. Light and M. B. Hursthouse, Chem. – Eur. J., 2014, 20, 1081–1090 CrossRef CAS PubMed.
- (a) Z. Hao, N. Li, X. Yan, Y. Li, S. Zong, H. Liu, Z. Han and J. Lin, New J. Chem., 2018, 42, 6968–6975 RSC; (b) I. Karthikeyan, S. K. Alamsetti and G. Sekar, Organometallics, 2014, 33, 1665–1671 CrossRef CAS; (c) W.-Q. Chen, Y.-M. Chen, T. Lei, W. Liu and Y. Li, Inorg. Chem. Commun., 2012, 19, 4–9 CrossRef CAS; (d) A. Schneider and H. Vahrenkamp, Z. Anorg. Allg. Chem., 2004, 630, 1059–1061 CrossRef CAS.
- T. M. Kolev and P. Bleckmann, Spectrosc. Lett., 1989, 22, 1215–1227 CrossRef CAS.
- L. J. Bellamy, The Infra-red Spectra of Complex Molecules, Methuen, London, UK, 2nd edn, 1966, pp. 108–111 Search PubMed.
- C. N. R. Rao, Chemical Applications of Infrared Spectroscopy, Academic Press, New York, USA, 1963, pp. 175–188 Search PubMed.
- D. M. Adams, Metal-Ligand and Related Vibrations, Arnold, London, UK, 1967, pp. 26–79 Search PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- Z. Hao, N. Li, X. Yan, X. Yue, K. Liu, H. Liu, Z. Han and J. Lin, J. Organomet. Chem., 2018, 870, 51–57 CrossRef CAS.
- N. B. Lentz and R. S. Houk, J. Am. Soc. Mass Spectrom., 2007, 18, 285–293 CrossRef CAS.
- C. G. Efthymiou, T. C. Stamatatos, C. Papatriantafyllopoulou, A. J. Tasiopoulos, W. Wernsdorfer, S. P. Perlepes and G. Christou, Inorg. Chem., 2010, 49, 9737–9739 CrossRef CAS.
- A. J. Carty, Can. J. Chem., 1967, 45, 345–351 CrossRef.
- A. J. L. Pombeiro and V. Yu. Kukushkin, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Amsterdam, The Netherlands, 2004, vol. 1, pp. 585–594 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data (Table S1–S4) and various structural plots of complexes 1, 3 and 4, and spectroscopic (IR, Raman, 1H and 13C NMR, ESI-MS, fluorescence) material for the compounds. CCDC 2203979–2203981. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02851d |
| This journal is © The Royal Society of Chemistry 2022 |