
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Secondary organic aerosol formation from gasoline and diesel vehicle exhaust under light and dark conditions†
Yu
Morino
 *a,
Ying
Li
b,
Yuji
Fujitani
a,
Kei
Sato
a,
Satoshi
Inomata
a,
Kiyoshi
Tanabe
a,
Shantanu H.
Jathar
c,
Yoshinori
Kondo
a,
Tomoki
Nakayama
d,
Akihiro
Fushimi
a,
Akinori
Takami
a and
Shinji
Kobayashi
a
*a,
Ying
Li
b,
Yuji
Fujitani
a,
Kei
Sato
a,
Satoshi
Inomata
a,
Kiyoshi
Tanabe
a,
Shantanu H.
Jathar
c,
Yoshinori
Kondo
a,
Tomoki
Nakayama
d,
Akihiro
Fushimi
a,
Akinori
Takami
a and
Shinji
Kobayashi
a
aNational Institute for Environmental Studies, Tsukuba, Ibaraki 305-8506, Japan. E-mail: morino.yu@nies.go.jp
bInstitute of Atmospheric Physics, Chinese Academy of Sciences, Beijing, 100029, China
cDepartment of Mechanical Engineering, Colorado State University, Fort Collins, CO 80523, USA
dFaculty of Environmental Science, Graduate School of Fisheries and Environmental Sciences, Nagasaki University, Nagasaki 852-8521, Japan
First published on 4th October 2021
Abstract
Secondary organic aerosol (SOA) formed from vehicle exhaust contributes substantially to the atmospheric particulate matter in urban air but there still remain uncertainties in the simulation of the SOA by air quality models. This omission is due partly to uncertainties about the differences in SOA formation between vehicle types, exhaust aftertreatment devices, and oxidation conditions in the air. In this work, smog chamber experiments were conducted to investigate SOA formation from diluted exhaust gases emitted by two light-duty gasoline vehicles and one passenger and one medium-duty diesel vehicles. Emissions were aged in the smog chamber under light (ultraviolet) and dark (with high O3) conditions. In these experiments, there were considerable emissions of elemental carbon and primary organic aerosol by a gasoline direct-injection vehicle and a diesel truck, but the corresponding emissions were relatively small from a gasoline vehicle with port fuel injection and a passenger diesel vehicle. However, significant amounts of SOA were produced from all four vehicles. Box model simulations indicated that OH exposure was higher and the ratio of NO to peroxy radicals (HO2 + RO2) was lower under dark conditions in the presence of high added O3 than under light conditions. The higher concentrations and yields of SOA under dark conditions than under light conditions could partly be explained by the lower NO/(HO2 + RO2) ratios under dark conditions. Simulations with a volatility basis-set model indicated that aromatic compounds were the dominant precursors of SOA from gasoline vehicles (64–84%), whereas unspeciated intermediate-volatility organic compounds (IVOC) were the dominant precursors for the production of SOA from diesel vehicles (75–87%). The observed O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratios were higher for gasoline SOA (0.7–0.8) than diesel SOA (0.3–0.4), and this difference was explained by the differences in their precursors. SOA formed under the light and dark conditions was similar in the dominant precursors and oxidants, as well as the O:C ratios, but different in the NO/(HO2 + RO2) ratios during the oxidation processes. Thus, we cannot conclude that SOA formed under the light and dark conditions has similar chemical composition, and their characteristics should be further examined in future studies.
:C ratios were higher for gasoline SOA (0.7–0.8) than diesel SOA (0.3–0.4), and this difference was explained by the differences in their precursors. SOA formed under the light and dark conditions was similar in the dominant precursors and oxidants, as well as the O:C ratios, but different in the NO/(HO2 + RO2) ratios during the oxidation processes. Thus, we cannot conclude that SOA formed under the light and dark conditions has similar chemical composition, and their characteristics should be further examined in future studies.
Environmental significanceSecondary organic aerosol (SOA) formed from vehicle exhaust contributes substantially to the atmospheric aerosol but there still remain uncertainties in the simulation of the SOA, particularly due to differences in SOA formation between vehicle types, exhaust aftertreatment devices, and oxidation conditions in the air. In this work, we conducted smog chamber experiments and box-model simulations to investigate controlling factors of yields, oxygen–carbon (O:C) ratios, and dominant precursors of SOA from gasoline and diesel vehicles under light and dark conditions. Differences in SOA yields and O:C ratios between gasoline and diesel SOA under the light and dark conditions were explained by the different precursors and oxidation conditions, which provides useful insight into the accurate modeling of SOA from vehicle exhaust.
|
1 Introduction
Vehicle exhaust contains gaseous and particulate atmospheric pollutants, and those pollutants have important impacts on human health, the Earth's radiative balance, and ecosystems.1–5 Carbonaceous aerosol, including elemental carbon (EC) and organic aerosol (OA), are major components of particulate matter (PM) from motor vehicles. PM directly emitted to the atmosphere (e.g., EC, primary organic aerosol (POA), and metallic compounds) from motor vehicles has been examined extensively since the 1990s.6–9 PM produced in the atmosphere (e.g., secondary organic aerosol (SOA)) has also been studied over the last 15 years.10–14 Those studies have revealed that SOA production is strongly affected, in addition to the vehicle characteristics, by the experimental setups, such as reactor types (smog chamber or flow reactor) and oxidation conditions (e.g., light versus dark conditions).15 The estimated contributions of secondary PM from motor vehicles are therefore associated with large uncertainties. Because the SOA generated from the vehicle exhaust may be more toxic than SOA formed from the individual volatile organic compounds (VOC) or POA from vehicle exhaust,16,17 there is a need to better understand the characteristics of SOA generated from vehicle exhaust.Both diesel and gasoline vehicles have been shown to be important sources of atmospheric SOA, and their relative contributions differ among studies and regions. In the United States (U.S.), most previous studies have indicated that gasoline vehicles make greater contributions to SOA.15,18–21 For example, contributions of gasoline-exhaust are ∼56–79% to vehicle SOA over the U.S.18 or ≥90% to vehicle SOA over the U.S. and Southern California.19,21 In contrast, Gentner, et al.22 have indicated that diesel vehicles make higher contributions (∼90%) to vehicle-SOA in the U.S. In Europe, Dunmore, et al.23 have shown from their comprehensive measurements of organic compounds in London that diesel vehicles make higher contributions to atmospheric SOA. The relative numbers of miles travelled by diesel vehicles versus gasoline vehicles are higher in Europe than in the U.S. For gasoline vehicles, the relative market share of gasoline direct injection spark ignition (GDI) vehicles versus conventional port-fuel injection (GPI) vehicles is increasing.24–26 In a GDI engine, gasoline fuel is sprayed directly into the cylinder, and the compression ratio is relatively high compared to a GPI engine, in which gasoline is injected into the intake port. As a result, GDI vehicles have the advantages of better fuel efficiency and lower CO2 emissions than GPI vehicles. However, primary PM emissions are higher from GDI vehicles than from GPI vehicles.9,24,25
Production yields and major precursors of SOA vary widely among different vehicle types (e.g., gasoline or diesel, GPI or GDI), emission control technologies, and running conditions.9,12,13,25,27–31 Understanding the SOA characteristics of different vehicle types is thus necessary for accurate estimation of the contributions of different sources to atmospheric SOA.
During the last decade, it has been shown that speciated VOC (e.g., single-ring aromatics and aliphatic compounds) cannot fully explain the production of SOA from vehicle exhaust and that the contributions of unspeciated precursors are critical.12,13,19,28,29,32,33 To estimate those contributions, emissions and formation rates of SOA from intermediate-volatility organic compounds (IVOC, with effective saturation concentrations (C*) of 104–106 μg m−3) have been evaluated. A large part of IVOC is chemically unresolved and thus is not explicitly included in emission inventories.34,35 Lu, et al.31 have estimated that unspeciated cyclic compounds make the predominant contributions to gasoline-exhaust IVOC, whereas unspeciated linear and branched alkanes make the greatest contributions to diesel-exhaust IVOC. In addition, the magnitude and chemical composition of the IVOC emitted from motor vehicles depends on the vehicle type and operating conditions,13,19,28,29,36 and contributions of IVOC to the formation of SOA from vehicle exhaust are still uncertain. Because the yields of SOA produced from IVOC are high,37,38 IVOC are important precursors of SOA from motor vehicles,28,29,35 Lu et al.39 have recently developed a parameterization of SOA formation from six lumped IVOC emitted from mobile sources; their model simulation has indicated that IVOC emitted from mobile sources make important contributions to the SOA simulated in Los Angeles. IVOC may make large contributions to ambient SOA, and thus, quantification of their contributions is important.
As analyses using numerical simulations are necessary to quantitatively evaluate the contributions of these emission sources to atmospheric PM, there have been attempts to simulate the production of SOA from motor vehicles.40 Robinson et al.10 have shown that semi-volatile organic compounds (SVOC) make important contributions to SOA from diesel vehicle exhaust by using a volatility basis-set model (VBS) that explicitly calculates the partitioning and multigenerational reactions of POA, SVOC, and IVOC.10,41 Recent modeling analyses have indicated that IVOC is a key precursor to atmospheric SOA from diesel exhaust.15,42,43
Previous experimental and modeling studies of SOA formation from vehicle exhaust have focused primarily on light conditions. Only a few studies have addressed the aging of vehicle SOA under dark conditions. Samy and Zielinska44 have conducted chamber experiments of diesel exhaust aging under dark conditions with high concentrations of N2O5 (for nitrate radical [NO3] production). They have found that there is less formation of SOA under dark conditions than under conditions where photooxidation is possible. However, to our knowledge, the yield of SOA formed under dark conditions with high O3 concentrations has not been well evaluated. Studies of the toxicity of SOA from vehicle exhaust have made use of SOA produced under dark conditions with high O3 concentrations, partly because the large amounts of SOA necessary for the toxicological studies could be prepared relatively simply without the light sources required for photooxidation.45,46 Because of the addition of high concentrations of O3, the main oxidants and the ratio of NO to peroxy radicals could be different between the light and dark experiments. Comparison of the formation mechanism and properties of SOA formed under light and dark conditions should provide insights into the toxicological properties of SOA from vehicle exhausts. For the analysis of SOA oxidation under dark conditions, simulations of gas-phase chemical mechanisms can provide useful insights into the processes involved in SOA formation. However, to our knowledge, no such analyses have been conducted.
In this study, SOA formation associated with dilute emissions from two light-duty gasoline vehicles as well as one light-duty and one medium-duty diesel vehicles was investigated under both light and dark conditions in a smog chamber. The aims of this study were (1) to compare the production of SOA and the oxygen:carbon (O:C) ratios of OA from different vehicle types and oxidation conditions (i.e., light and dark conditions), (2) to evaluate the ability of the VBS model to simulate the formation of SOA and O:C ratios, (3) to evaluate the contributions of different precursors to SOA formation. This experimental and modelling study allowed us to investigate the characteristics of SOA from both gasoline and diesel exhaust under light and dark conditions.
2 Methods
2.1. Experimental
Table 1 lists the four vehicles tested during this study. There were two light-duty gasoline vehicles, including one GDI vehicle and a GPI vehicle. The light-duty passenger diesel (Diesel-P) vehicle was equipped with a diesel particulate filter (DPF) and diesel oxidation catalyst (DOC). The medium-duty diesel truck (Diesel-T) was equipped with a DOC only. The loads of the test vehicles were consistent with Japanese emission regulation test protocols and equaled the vehicle weights +110 kg. The emission standards of the test vehicles were compliant with the 2009 emission regulations.47 The average vehicle age in Japan (8.7 year for passenger vehicles and 11.4 year for freight vehicles in 2020, Automobile Inspection and Registration Information Association) indicates that model year 2012 vehicles are still being used. The fuels and lubricating oils used in this study were those commonly used in Japan. The percentages of aromatics contained in the used regular gasoline fuel and low-sulfur-content (6 ppm) diesel fuel were measured to be 24.8% and 22.5%, respectively. The densities of the used gasoline and diesel fuels were 0.73 g cm−3 and 0.84 g cm−3, respectively. The driving conditions corresponded to the Japanese official transient mode (JC08 mode) under cold-start conditions.16,24,48 Controlled vehicle testing showed that during cold starts the DOC was not fully active and therefore unburned and incompletely combusted material passed through the exhaust after-treatment systems.27,49| Vehicle ID | Emission control | Model year | Eng. size (L) | Mileage (km) |
|---|---|---|---|---|
| GDI | Three-way catalyst | 2012 | 1.19 | 30970 |
| GPI | Three-way catalyst | 2012 | 1.19 | 15700 |
| Diesel-P | DOC + DPF | 2012 | 2.18 | 40600 |
| Diesel-T | DOC | 2004 | 2.95 | 34700 |
The experiments were carried out in a 6 m3 fluorinated ethylene propylene (FEP) film bag chamber (FBC). The dimensions of the FEP film bag were 1.4 × 1.4 × 2.4 m3, and the film thickness was 50 μm. Fig. 1 shows the experimental setup. All exhaust during the JC08 cycle was introduced directly into a 30 m3 exhaust-gas diffusion chamber (EDC) that was filled with clean air. The clean air was supplied by a clean-air generator (DAR-2200, Horiba Ltd.) and contained less than 0.1 ppm of total hydrocarbon (THC) and less than 0.1 ppm of NO. The duration of the exhaust injection period was about 20 min. After exhaust injection, there was a period of 60 min to allow mixing in the chamber and to characterize the composition of the fresh exhaust. The partially diluted exhaust gases in the EDC were then transferred by three pumps (OSP-90W, As One Corp.), and finally the reaction air in the 6 m3 FEP FBC was prepared with target concentrations. The total dilution factors from the raw exhaust to the 6 m3 FBC were ∼25 for gasoline vehicle exhausts and ∼10 for diesel vehicle exhausts.
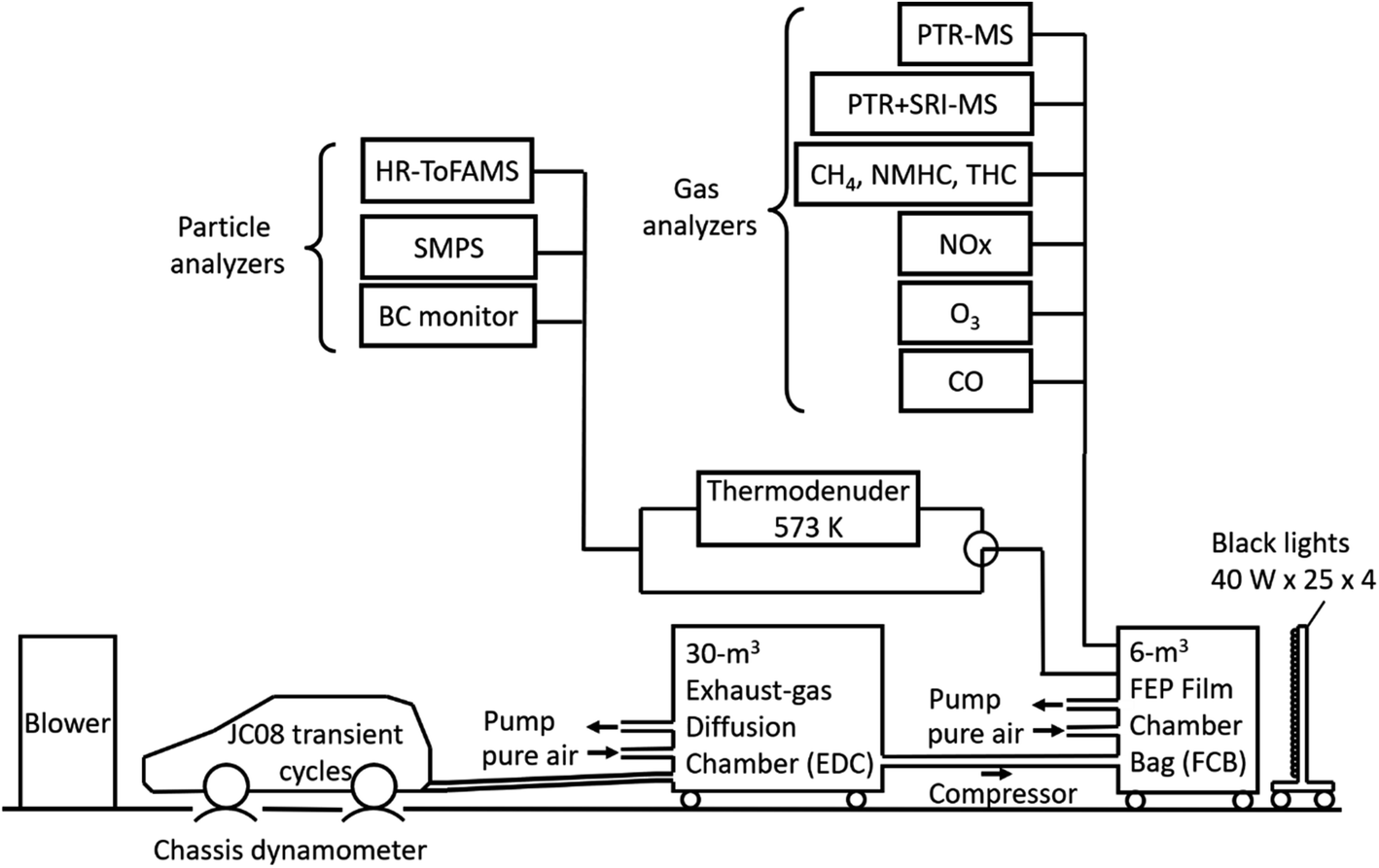 | ||
| Fig. 1 Schematic diagram of the experiments for the measurement of gas and particulate compounds from motor-vehicle exhaust (abbreviations: PTR + SRI-MS: proton transfer reaction + switchable reagent ions–mass spectrometry, NMHC: non-methane hydrocarbon, THC: total hydrocarbon, HR-Tof-AMS: high-resolution time-of-flight aerosol mass spectrometer, SMPS: scanning mobility particle sizer, BC: black carbon, FEP: fluorinated ethylene propylene). | ||
An array of instruments was used to characterize gas- and particulate-phase pollutants inside the 6 m3 FBC. Gas-phase organic species were measured with two proton transfer reaction mass spectrometers (PTR-MS, Ionicon), as detailed in our previous studies50,51 and Section S1 of the ESI.† The detection sensitivities are listed in Tables S1–S3 of the ESI.† Gas monitors were used to measure NO, NO2, CO, O3, non-methane hydrocarbons (NMHC), THC, and CH4. The monitors were zeroed daily and calibrated at least weekly. Concentrations of black carbon (BC) were measured with a black carbon monitor (Model AE51, Aethlabs). Nonrefractory PM1.0 concentrations and the chemical composition of PM1.0 were measured with a high-resolution time-of-flight aerosol mass spectrometer (HR-ToFAMS, Aerodyne, Inc.).52 For the estimate of O:C ratios, the contributions of gas–CO2 to the mass spectrum were subtracted, as in previous studies.53 The uncertainties of the OA concentrations and O:C ratios that were used in this study were 20–40% (ref. 54 and 55) and ∼20%,53 respectively. Particle number distributions were measured with scanning mobility particle sizers (SMPS Models 3034, 3936, TSI, Inc.).
The data shown in this study were obtained in the 6 m3 FBC. The ratio of the CO concentration in the 30 m3 EDC and 6 m3 FBC (i.e., chambers before and after passage through the pumps, respectively) was 0.183; the corresponding ratio of the THC concentrations was 0.194. As CO is unlikely to be lost in the pumps, the similar ratios for the CO and THC across the two chambers suggest that THC was not lost in the pumps either. In contrast, particle volume concentrations normalized to CO concentrations were reduced by a factor of ∼7 after passage through the pumps (i.e., ∼86% of particles were lost in the pumps). The fact that the geometric mean diameter of the volume concentration changed marginally (284.0 nm and 273.0 nm before and after passage through the pumps, respectively) suggested that evaporation reduced particle volume concentration only by 11% and particle loss was predominantly associated with wall loss. We should note that we analyzed POA in the 30 m3 EDC and SOA in the 6 m3 FBC. If we assume that the precursors of SOA were not lost in the pumps, then we can assume that the POA/SOA ratio was accurately estimated in this study. It is possible that some of the SVOC were deposited on the pump wall (materials of hard anodized aluminum), even though THC did not decrease after passage through the pumps. Thus, the observed concentrations of SOA might be underestimated because of the loss of SVOC.
For photoreaction experiments, the 6 m3 chamber was irradiated with 100 ultraviolet (UV) black-light bulbs (350BL, Hitachi Ltd.) from four sides (25 bulbs in each side). The wavelength of the peak intensity of the UV black lights was 350 nm, and the radiant flux of each bulb was 40 watts. The rate of NO2 photolysis was 0.198 ± 0.003 min−1. H2O2 (50% w/w) was injected as a radical precursor to boost the reaction for the diesel experiments; H2O2 was not injected for the gasoline experiments to maintain hydroxyl radical (OH) concentrations similar to those for the diesel experiments. In the diesel experiments without H2O2 injection, the estimated OH exposure was ∼5 times lower, and the produced SOA concentration was smaller by 94% (Diesel-P) or 50% (Diesel-T) than in the experiments with H2O2 injection. Hereafter, we analyzed the experimental results with H2O2 injection. The experiments without H2O2 injection will not be further discussed. The average OH levels in the 6 m3 FBC experiments were inferred from the decay of toluene and benzene measured with the PTR-MS.56 For the ozonolysis experiments, ozone was generated by a corona discharge ozone generator (Iwasaki Electric, model OP-20W). The UV lights were turned on or O3 was added at what is referred to as time t = 0 throughout this paper. The temperature in the room containing the 6 m3 FEP FBC was maintained at 25–28 °C. The relative humidity (RH) inside the 6 m3 FBC was estimated from the RH in the 30 m3 EDC (∼90%) and purified air (∼40%) to be around 50%.
Quantification of SOA production in the 6 m3 FBC required that losses of both particles and vapors to the chamber walls be corrected. In this study, the particle wall loss (PWL) rate was calculated from experimental data, whereas the volatility-dependent vapor wall loss (VWL) rate was estimated using a model calculation, as detailed in Section 2.2. The PWL was treated as a first-order process with a rate constant determined from the measured rate of decay of aerosol volume concentrations after the formation of SOA, as summarized in Table S4 of the ESI.† For the experiments of diesel vehicles, the geometric mean volume diameter of aerosol did not change after 2.5 h from time t = 0 (light on or O3 injection). The decay rates of aerosol volume concentrations measured by SMPS were thus used to determine the PWL rates (0.21–0.45 h−1). The maximum diameter of the aerosol that the SMPS could measure in the diesel-vehicle experiments was ∼790 nm, but for the gasoline vehicle experiments, the corresponding maximum diameter was only ∼470 nm. Because the diameters of some aerosol exceeded 470 nm in the gasoline vehicle experiments, the volume concentration of the SOAs could not be fully captured by the SMPS. The PWL rate was therefore estimated not from the SMPS data, but from the decay rate of the EC concentrations (0.19 h−1). For the GDI experiment under dark conditions, the number concentration decreased at a high rate of 1.4 h−1 in the first 1.5 h of the experiment. Because particle diameters increased during this experiment, the volume concentration could not be used to calculate the PWL rate; therefore, we made a second-best estimate by using number concentrations as alternatives to correct the PWL57 in the first 1.5 h of that experiment. Because the uncertainties associated with this estimate were undoubtedly large, the results of this experiment should be interpreted with caution (e.g., Fig. 2). After the first 1.5 h of that experiment, the PWL correction rate was the same rate that was used in the other experiments (0.19 h−1).
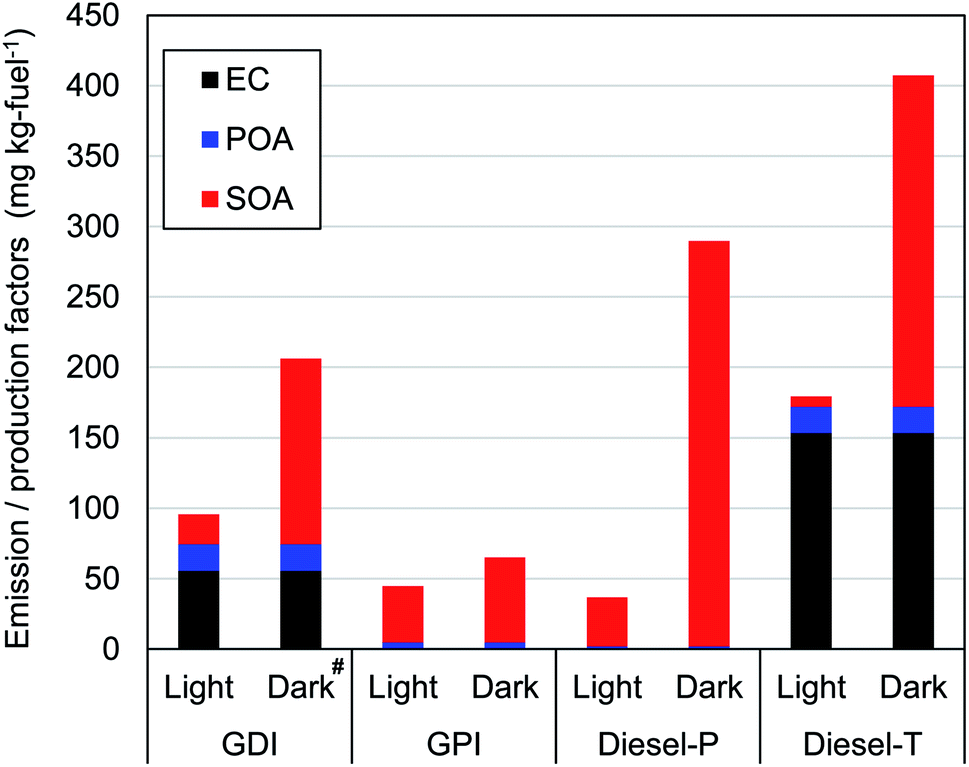 | ||
| Fig. 2 Emission factors of elemental carbon (EC) and primary organic aerosol (POA) and production factors of secondary organic aerosol (SOA) from two gasoline-powered and two diesel-fuel motor vehicles (GDI: gasoline-direct injection, GPI, gasoline port-fuel injection: Diesel-P, diesel-fuel passenger car: Diesel-T, diesel-fuel truck). #The SOA data in the GDI experiments under dark conditions include large uncertainties associated with the correction for particle wall loss (PWL), as noted in Section 2.1. | ||
Table 2 shows the initial experimental conditions. The NOx emitted from diesel vehicles was much higher than the NOx emitted from gasoline vehicles. Initial NOx concentrations were 0.27–0.36 ppm for gasoline vehicles, 4.4–4.5 ppm for the Diesel-P vehicle, and 16.7–17.9 ppm for the Diesel-T vehicle. In general, the NMHC/NOx ratio during the experiments for gasoline exhaust was >10 ppmC per ppm-NOx. Under those low-NOx conditions, peroxy radicals react primarily with HO2 radicals to form organoperoxy species.58,59 In contrast, the NMHC/NOx ratio during the experiments with diesel exhaust was <1 ppmC per ppm-NOx. Under those conditions, peroxy radicals react primarily with NO to form NO2 and an alkoxy radicals. Peroxy radicals can also react with NO to form organic nitrates, and nitrate-containing SOA species have been observed under high-NOx conditions.58
| Vehicle | Condition | Initial concentration | ||||
|---|---|---|---|---|---|---|
| H2O2 (ppm) | O3 (ppm) | NMHC (ppmC) | NOx (ppm) | NMHC/NOx (ppmC ppm−1) | ||
| GDI | Light | 3.53 | 0.29 | 12.17 | ||
| Dark | 8 | 6.64 | 0.36 | 18.44 | ||
| GPI | Light | 3.62 | 0.27 | 13.41 | ||
| Dark | 5 | 4.27 | 0.29 | 14.72 | ||
| Diesel-P | Light | 4 | 4.25 | 4.35 | 0.98 | |
| Dark | 11 | 4.48 | 4.52 | 0.99 | ||
| Diesel-T | Light | 4 | 4.12 | 16.66 | 0.25 | |
| Dark | 20 | 3.47 | 17.85 | 0.19 | ||
Background-corrected emission factors (EF in mg per kg-fuel) for EC and POA and the production factors for SOA were calculated on a fuel-consumption basis using a carbon mass balance as follows:
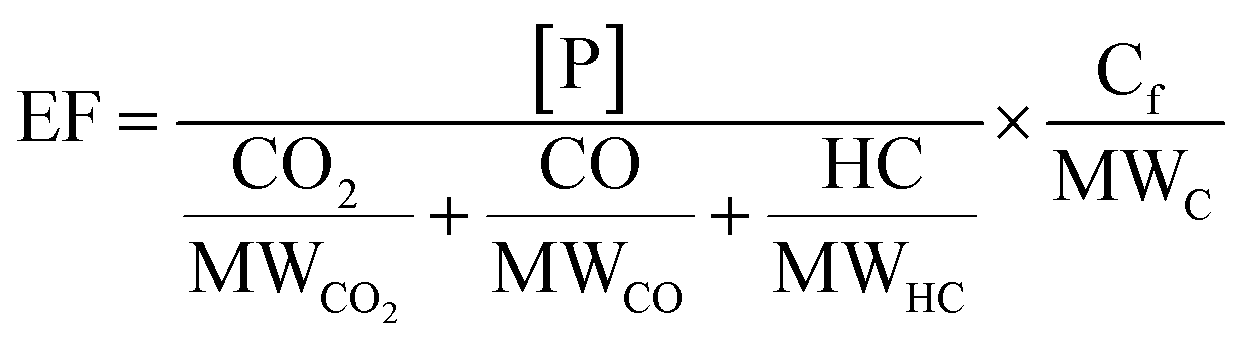 | (1) |
2.2. Model
In this study, we conducted box model simulations with the VBS framework41 coupled with the gas-phase chemistry mechanism SAPRC99.61Three types of SOA precursors were considered: VOC (C* ≥ 107 μg m−3), IVOC (C* = 104–106 μg m−3), and SVOC (C* = 10−1 to 103 μg m−3). Table S5 of the ESI† lists the lumped VOC classes used in SAPRC99. The organic compounds measured by PTR-MS in this study were then mapped to each lumped VOC class for the initial conditions of the model simulation (Table S6 of the ESI†), as is usually conducted for air quality simulations. Any alkane or alkene with a carbon number (NC) ≥ 12 and any single-ring aromatic with NC ≥ 11 measured by PTR-MS was not apportioned to the lumped VOC classes because these compounds are classified into IVOC. IVOC emissions were introduced into the model in proportion to NMHC emissions, as in previous studies.7,19,28,29 Initial concentrations of five lumped IVOC were calculated by using the ratio of IVOC to NMHC estimated by Lu, et al.39 (Table 1 of Lu, et al.39) and are summarized in Table S6 of the ESI.† The IVOC collectively accounted for 4.3% and 50.1% of NMHC emissions of gasoline and diesel exhaust, respectively. The mass fraction distribution of SVOC in C* bins (10−1 to 102 μg m−3) were taken from Zhao, et al.28 Zhao, et al.,29 and the initial SVOC concentration was normalized so that the initial OA concentration could be reproduced using the equation 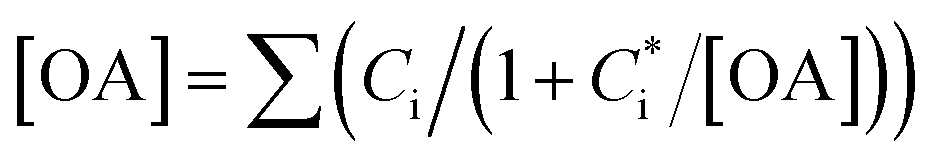 , where [OA] is the OA concentration and Ci is the total (gas + aerosol) concentration of compound i with a saturation concentration of
, where [OA] is the OA concentration and Ci is the total (gas + aerosol) concentration of compound i with a saturation concentration of 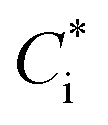 .
.
Products of VOC oxidation were represented as four surrogate compounds with saturation concentrations 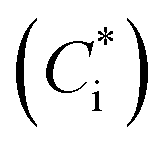 of 1, 10, 100, and 1000 μg m−3. NOx-dependent SOA mass yields of the oxidation products from C7–C11 alkanes (ALK5), monoaromatic compounds (ARO1 and ARO2), and isoprene were taken from Murphy and Pandis62 and are listed in Table S7 of the ESI.† SOA formation from alkenes has not been considered in recent air quality models.63–65 In this study, we considered SOA formation from OLE2 (internal alkenes), but not from OLE1 (terminal alkenes), as discussed in Section S2 of the ESI.† Products of IVOC oxidation were represented as four surrogate compounds with
of 1, 10, 100, and 1000 μg m−3. NOx-dependent SOA mass yields of the oxidation products from C7–C11 alkanes (ALK5), monoaromatic compounds (ARO1 and ARO2), and isoprene were taken from Murphy and Pandis62 and are listed in Table S7 of the ESI.† SOA formation from alkenes has not been considered in recent air quality models.63–65 In this study, we considered SOA formation from OLE2 (internal alkenes), but not from OLE1 (terminal alkenes), as discussed in Section S2 of the ESI.† Products of IVOC oxidation were represented as four surrogate compounds with 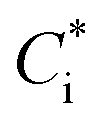 of 0.1, 1, 10, and 100 μg m−3, and the SOA mass yields were taken from chamber experiments under high-NOx conditions (Table S9 of the ESI†).39 SOA mass yields from alkane IVOC under high- and low-NOx conditions are similar,66 whereas those from aromatic IVOC are higher at low-NOx condition than at high-NOx condition.38 In our simulations, aromatic IVOC has a small contribution to SOA production (Section 3.3.2), and thus, NOx dependence of SOA yields from IVOC were not considered in this study. Formation of SOA from primary-SVOC followed Robinson, et al.10 Multi-generational aging reactions for products from VOC and IVOC were not considered as well. Gas-particle partitioning was calculated using absorptive partitioning theory67 assuming that all of the organics formed a quasi-ideal solution and that the bulk gas and particle phases were always in equilibrium. The temperature dependence of the saturation concentration was estimated with the previously reported vaporization enthalpy.39,68 We should note that SAPRC99 is a lumped mechanism; hence it cannot represent the detailed behavior of individual VOCs. However, SAPRC99 and the later version of the SAPRC mechanism have been among the most widely used chemical mechanisms in air quality models, and we therefore used the SAPRC99 mechanism for the evaluation of the experimental data.
of 0.1, 1, 10, and 100 μg m−3, and the SOA mass yields were taken from chamber experiments under high-NOx conditions (Table S9 of the ESI†).39 SOA mass yields from alkane IVOC under high- and low-NOx conditions are similar,66 whereas those from aromatic IVOC are higher at low-NOx condition than at high-NOx condition.38 In our simulations, aromatic IVOC has a small contribution to SOA production (Section 3.3.2), and thus, NOx dependence of SOA yields from IVOC were not considered in this study. Formation of SOA from primary-SVOC followed Robinson, et al.10 Multi-generational aging reactions for products from VOC and IVOC were not considered as well. Gas-particle partitioning was calculated using absorptive partitioning theory67 assuming that all of the organics formed a quasi-ideal solution and that the bulk gas and particle phases were always in equilibrium. The temperature dependence of the saturation concentration was estimated with the previously reported vaporization enthalpy.39,68 We should note that SAPRC99 is a lumped mechanism; hence it cannot represent the detailed behavior of individual VOCs. However, SAPRC99 and the later version of the SAPRC mechanism have been among the most widely used chemical mechanisms in air quality models, and we therefore used the SAPRC99 mechanism for the evaluation of the experimental data.
The O:C ratio of the POA was based on the individual experimental results. Murphy, et al.69 have assigned O:C ratios for all the “anthropogenic” SOA. In this study, we assigned different O:C ratios to different precursors by using experimental O:C ratios from Zhao, et al.28 for SOA from aromatic VOC, Chhabra, et al.70 for SOA from aromatic-IVOC, and Cappa and Wilson71 for SOA from alkane-IVOC (Table S10 of the ESI†).
As noted in Section 2.1, the PWL rate in the SOA reaction chamber was calculated from experimental data. The simulation model with the formulation of Krechmer, et al.72 were used to calculate the volatility-dependent VWL rate. Morino, et al.73 have also provided a formulation of the VWL calculation.
3 Results and discussion
3.1. Emission and production factors of carbonaceous aerosol
Fig. 2 and Table S11† summarize EF of primary particles (EC and POA) and production factors (PF) of SOA. For the evaluation of variability, two experiments were conducted to measure concentrations of for POA and EC emitted by the GDI and Diesel-P vehicles. The standard errors ([standard deviation]/[average]) of the POA and EC concentrations were 23% and 16%, respectively for the GDI vehicle. The relative error for the Diesel-P vehicle was 46% for POA, and the EC concentration was below the limit of detection.For gasoline vehicles, primary emissions of EC and POA from GPI were small (<4 mg per kg-fuel), whereas the EF of EC and POA from GDI were 55 mg per kg-fuel and 19 mg per kg-fuel (2.4 mg km−1 and 0.83 mg km−1), respectively. The higher emissions of primary particles from GDI vehicles were consistent with results of previous studies,9,14,25,74 though the reported contributions varied among vehicles. We should note that sample size of this study is small (one experiment for each condition and vehicle), and thus, this comparison with previous experiments is solely intended for the check of the consistency, but not for the demonstration of the representativeness of the tested vehicles. The ratio of EC to [EC + POA] of 0.75 from GDI in this study was within previous estimates of 0.20 in Du, et al.,25 0.80–0.86 in Platt, et al.,14 and 0.88 in Saliba, et al.9 As summarized in Table S12 of the ESI,† the variability of the EF of EC and POA has been large in previous studies (>2 orders of magnitude). The EF differ, for example, among model years, engine types, emission control technologies, and operating modes. Discussion of this point is beyond the scope of this study because of the small sample size, although the EF of the EC and POA in this study were within the range of those in previous studies, both for GDI and GPI vehicles. In contrast to the EF of the POA, the PF of the SOA were higher for the GPI vehicle (40 mg per kg-fuel) than for the GDI vehicle (21 mg per kg-fuel) in the light conditions. These PFs were within the range of previous research for gasoline vehicles under light conditions. Gordon, et al.12 have reported SOA PFs of exhaust from 6 gasoline vehicles with LEV2 emission standards (manufactured in 2004 or later) that ranged from 2 to 55 mg per kg-fuel with an average of 18.8 mg per kg-fuel at an OH exposure of 5 × 106 molecules per cm3 per h. Gordon, et al.12 have also found that SOA is much more efficiently generated during cold-start tests than during hot-start tests. Nordin, et al.75 have reported a higher SOA-PF (480 mg per kg-fuel) from an older regulation (2005) gasoline vehicle (Euro 4) with an OH exposure of around 5.0 × 106 molecules per cm3 per h under the cold-start conditions. Du, et al.25 have reported that the SOA-PF from GPI vehicle (with an emission standard equivalent to Euro 4) and GDI vehicle (with an emission standard equivalent to Euro 5) under light and cold-start conditions were 21 and 55 mg per kg-fuel, respectively. As already noted, SOA-PF should depend on the type of vehicle, emission control technology (e.g., aftertreatment devices), and driving conditions. In addition to these differences, oxidation conditions are also important determinants of SOA-PF. For both GDI and GPI vehicles, the SOA-PF were higher under dark conditions than under light conditions, even at similar levels of the OH exposure. This point is discussed in Section 3.3.1.
For diesel vehicles, the Diesel-P with DPF and DOC emitted negligible primary particles, whereas the EF of EC and POA from the Diesel-T with DOC were 153 mg per kg-fuel and 18.4 mg per kg-fuel, respectively. These findings are consistent with previous work, which has indicated a significant reduction of primary particles with the use of DPFs (Table S12 of the ESI†). For example, Gordon, et al.13 have shown that emission factors of primary particles from heavy-duty diesel vehicles (HDDV) with DPF were <12 mg per kg-fuel, whereas the EF of medium-duty diesel vehicles (MDDV) without DPF exceeded 100 mg per kg-fuel. In contrast to the primary particles, the SOA-PF from the Diesel-P was even higher under both light and dark conditions (35 mg per kg-fuel and 288 mg per kg-fuel, respectively) than that from the Diesel-T (7 mg per kg-fuel and 235 mg per kg-fuel). The SOA-PF from DPF-equipped vehicles varied among studies (0–85 mg per kg-fuel under light conditions, as in Table S12†). As will be shown in Section 3.3.1, production yields of SOA from diesel vehicle exhaust were higher under dark conditions than under light conditions. We should note that the SOA-PF of this study under light conditions was within the range of previous studies with DOC or DPF (Tables S11 and S12 of the ESI†).
3.2. Gas-phase reactions
Box model calculations with the chemical mechanism, SAPRC99, revealed the different characteristics of major chemical reaction pathways between vehicle types (gasoline or diesel vehicles) and between oxidation conditions (light or dark). Fig. 3 indicates the production pathways of ROx radicals (sum of OH, HO2, and organo-peroxy [RO2] radicals). Fig. S3 of the ESI† is the same figure on a linear timescale.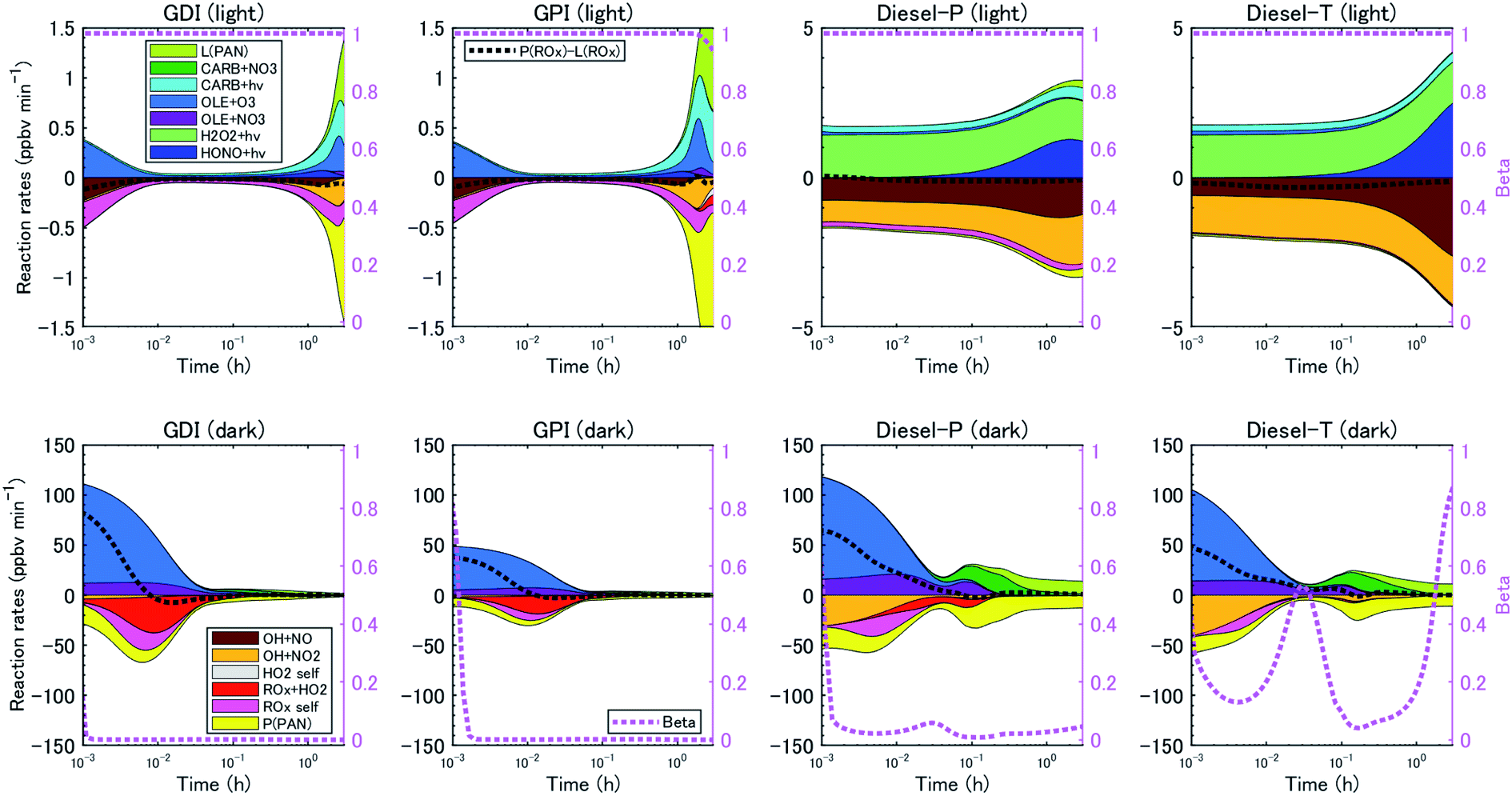 | ||
| Fig. 3 Budget of production and loss rates of the OH, HO2, and RO2 radicals (left axis) and branching ratio, β, estimated with eqn (3) (right axis) in the simulation on a logarithmic timescale. | ||
The box model calculations indicated that under the light conditions, the dominant radical production process in the case of the gasoline exhaust was photolysis of carbonyl compounds followed by ozonolysis of alkenes and HONO photolysis. Photolysis of carbonyl compounds has been reported to make large contributions to the production of radicals in the ambient air of urban areas76,77 and of an oil and gas basin.78 In the experiments with diesel exhaust under the light conditions, the NMHC/NOx ratio was low (Table 2), and because we expected that irradiation of the exhaust would not produce sufficient HOx radical to facilitate SOA formation, we added H2O2. The photolysis of H2O2 was therefore the dominant radical production pathway, followed by photolysis of HONO and carbonyl compounds.
With the addition of high concentrations of O3 under the dark conditions (5–20 ppmv, Table 2), reactions of alkenes and O3/NO3 were the major radical production pathways during the first several minutes of the experiments. Over the first half hour, reactions between alkenes and O3 made the highest contributions to radical production from gasoline exhaust, whereas reactions involving carbonyls and alkenes + NO3 made the highest contributions in the case of diesel exhaust. This contrast in the contributions of oxidants reflected the different initial NMHC/NOx ratios (Table 2). One hour after t = 0, reactions involving carbonyl and NO3 made the highest contributions to radical production, production and loss of peroxyacyl nitrates (PANs) balanced, and OH radical concentrations decreased (Fig. S4 of the ESI†). We should note again that these radical production pathways are not representative of the reactions under ambient conditions; the chemical reactions that occur under dark conditions are intended to simulate reactions that might occur during toxicological experiments, not chemical reactions that occur in the atmosphere at night.
The reaction pathways of RO2 radicals are critical to the determination of the SOA yields. RO2 radicals react with NO when NOx concentrations are high, whereas RO2 radicals react with other RO2 and HO2 radicals when NO concentrations are low. Previous studies have revealed that NOx concentrations significantly alter yields of SOA from biogenic VOC79,80 and aromatic VOC81 but perhaps not for alkane VOC.66 The difference between the SOA yields at high- and low-NOx conditions could be explained by the different product distributions. RO2 radicals react with NO to form RO at high-NOx conditions. Then, carbonyl or hydroxycarbonyl products are formed after RO reacts with O2, decomposes through C–C bond scission, or isomerizes through H-atom shift reactions.82 RO2 radicals also react with NO or NO2 to form organic nitrate or peroxynitrates. In contrast, RO2 radicals react with HO2 or other RO2 radicals at low NOx conditions, and generate hydroperoxide, carbonyls, hydroxycarbonyls, or alcohol products. The NOx dependence of the SOA yield, αi, is parameterized as follows:
| αi = αi,high NOxβ + αi,low NOx(1 − β) | (2) |
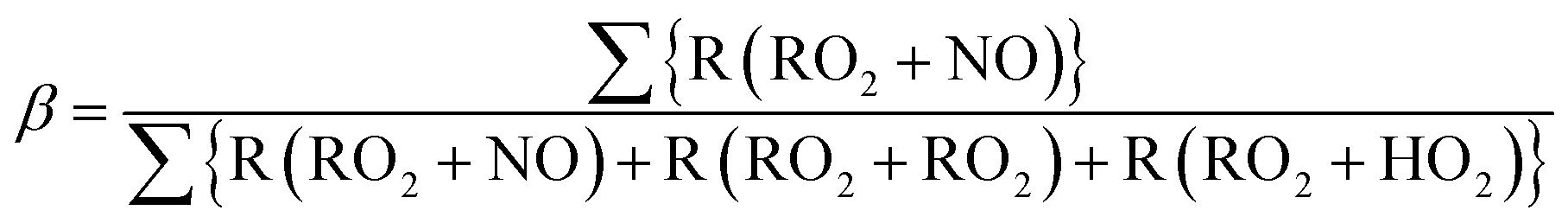 | (3) |
3.3. SOA formation
:C ratios of SOA as functions of OH exposure. Here, the OH exposure was calculated based on the average decay rates of C8 and C9 aromatic VOCs measured by the PTR-MS under both light and dark conditions 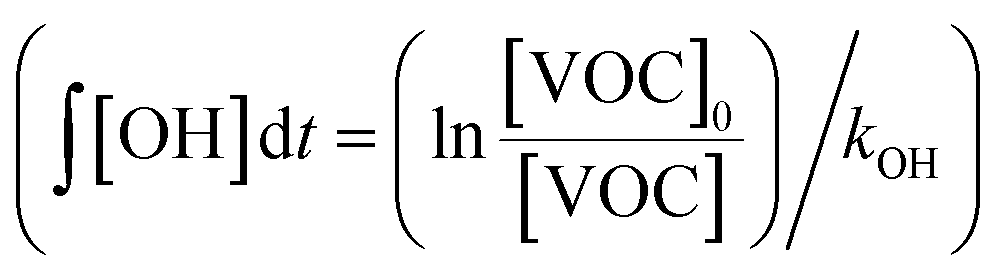 . Ratios of individual VOC compounds were not used because the temporal fluctuations of individual VOC compounds were large. Because the PTR-MS does not quantify isomers, the estimated kOH values of the C8 and C9 aromatic VOCs (1.45 ± 0.66 and 4.06 ± 1.39 molecules per cm3 per s, respectively56) calculated from the average kOH values of the VOC were sources of uncertainty in the estimate of OH exposure. In addition, Fig. S5 of the ESI† summarizes the observed temporal variations of OA concentrations and O:C ratios of OA.
. Ratios of individual VOC compounds were not used because the temporal fluctuations of individual VOC compounds were large. Because the PTR-MS does not quantify isomers, the estimated kOH values of the C8 and C9 aromatic VOCs (1.45 ± 0.66 and 4.06 ± 1.39 molecules per cm3 per s, respectively56) calculated from the average kOH values of the VOC were sources of uncertainty in the estimate of OH exposure. In addition, Fig. S5 of the ESI† summarizes the observed temporal variations of OA concentrations and O:C ratios of OA.
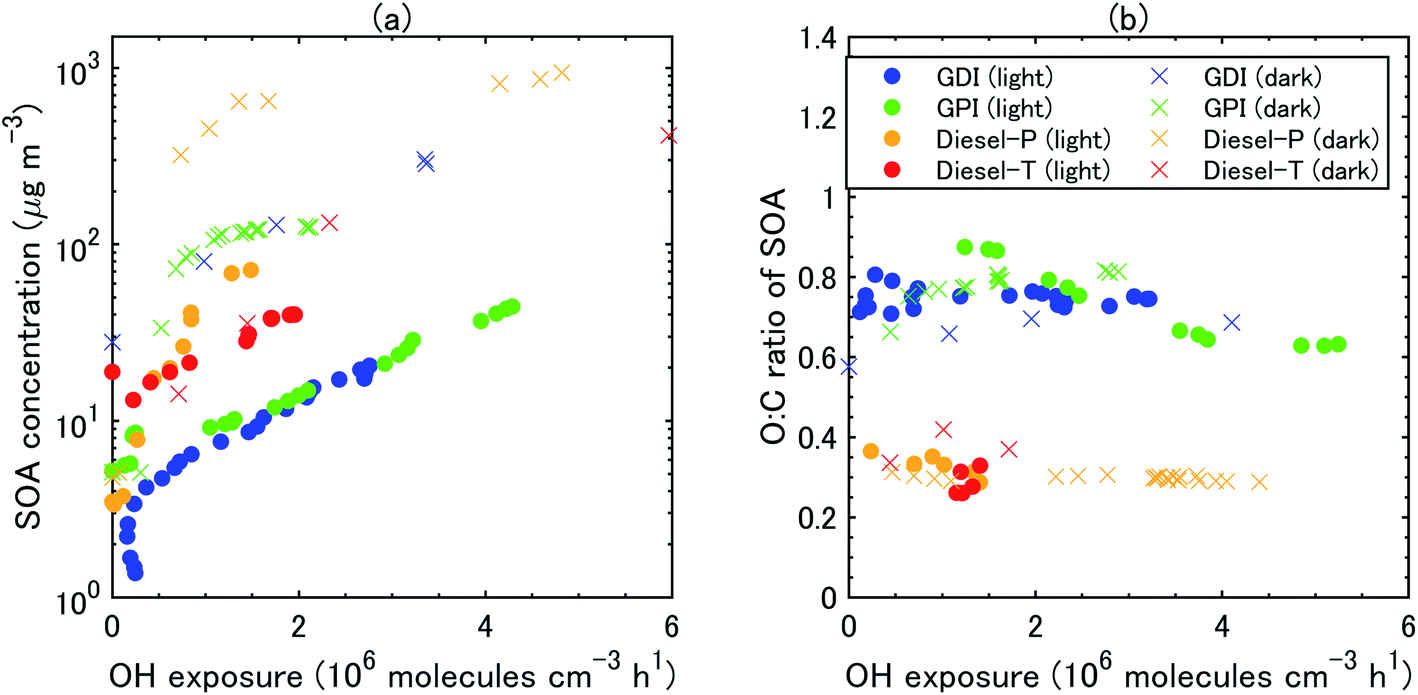 | ||
| Fig. 4 (a) Observed concentrations of secondary organic aerosol (OA) and (b) O:C ratio of SOA as a function of OH exposure under the light and dark conditions. Estimation of concentrations and O:C ratios of SOA and OH exposure is described in the main text. Observed SOA concentrations were corrected for particle wall loss but not for vapor wall loss, although vapor wall loss was considered in the simulation. | ||
During all the experiments, SOA concentration increased with OH exposure. For gasoline exhaust, the concentrations of SOA were clearly higher under the dark conditions than that under the light conditions at the same OH exposure level. The concentrations of SOA from gasoline exhaust under light conditions started to increase about 1–2 h after t = 0 (Fig. S5 of the ESI†). The delay of SOA formation in the case of the gasoline vehicle exhaust was consistent with results of previous experiments,75 which have shown that the formation of SOA from idling gasoline passenger vehicles is delayed by 30–45 min after lights are turned on. In contrast, under the dark conditions, SOA formation occurred immediately after t = 0 and continued for about 30 min. This contrast results not only from the different behavior of OH radical, but also from the different NOx regime, as already discussed in Section 3.2. These differences of SOA concentrations (in turn, OA concentrations) reflected the fact that SOA yields were also higher under dark conditions (20–35%) than under light conditions (2–15%) (Fig. 5). Our estimated SOA yields were within the range of those estimated for six gasoline vehicles with the LEV2 emission standard,12 although the SOA yields of the six vehicles were highly variable (3–46%).
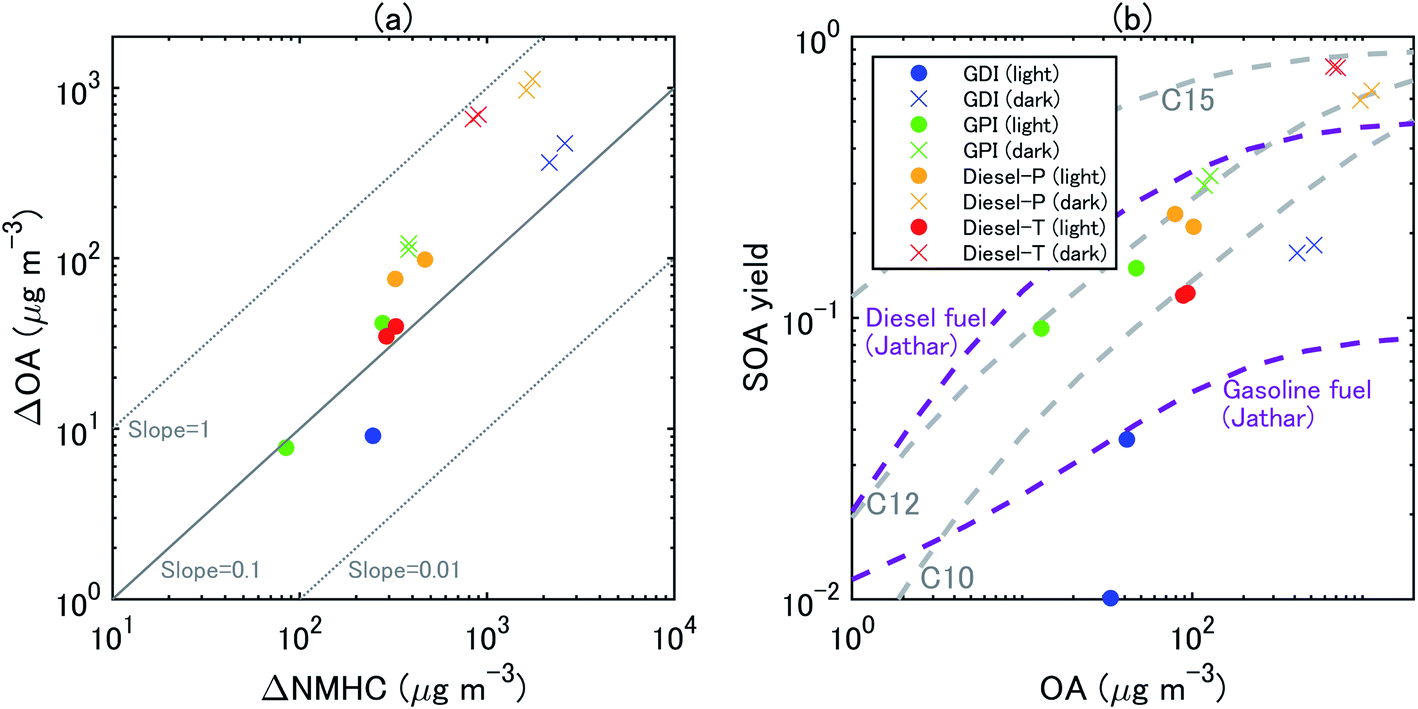 | ||
| Fig. 5 Scatterplots between (a) ΔOA and ΔNMHC and (b) SOA yields and OA concentrations at 1 h and 2 h from the start of the eight experiments. The SOA mass yields from unburned diesel and gasoline fuels in Jathar, et al.84 are also shown. | ||
The concentration of SOA was higher from diesel exhaust than from gasoline exhaust at similar levels of OH exposure (Fig. 4). The SOA mass yield from diesel exhaust was higher under the dark conditions (60–80%) than under the light conditions (10–20%) (Fig. 5). The high mass yields of SOA when OA concentrations were high reflected the semi-volatile nature of SOA (more oxidation products partition into the particle phase when OA concentrations are high). We should note that the NMHC monitor could not measure some portion of oxygenated VOC,83 and the estimated SOA yield from VOC could therefore have been overestimated. Fig. 5 shows the SOA mass yields from unburned diesel and gasoline fuels along with similar yields reported by Jathar, et al.84 The SOA mass yields for unburned fuel agreed with the SOA mass yields of the diesel engine exhaust both with and without aftertreatment devices reported by Jathar, et al.84 Our estimated SOA yields under the light conditions agreed within a factor of 2–3 with the yield curve of Jathar, et al.,84 except for the overestimation in the experiment of GPI. We should note that in the GDI experiment under light conditions, the SOA yield at t = 1 h was low compared to that of previous studies and to the yield at t = 2 h. This low yield at t = 1 h could be explained by the high NO concentrations, as detailed in the next section. The SOA yields estimated by Gordon, et al.13 for HDDV without DOC or DPF (hot-start experiments) were 4–19% at around 1–30 μg m−3 of OA concentrations. These SOA yields are also consistent with the yield curve of diesel fuel reported by Jathar, et al.84 It is possible that SOA produced during dark conditions is lost by the photolysis of organoperoxide compounds; thus, SOA yields in the dark experiments may be reduced after irradiation.85
:C ratios with the observations. The observed OA concentrations were generally reproduced by the simulation, particularly during the first hour of the experiments. As already noted, the SOA concentrations slowly increased during the experiments with gasoline exhaust under the light conditions, whereas SOA concentrations increased rapidly with both gasoline and diesel exhaust under the dark conditions. The simulation reproduced these trends. The simulation results indicated that this difference was caused by the different temporal variations of oxidant concentrations: the concentration of OH radical increased in the first two hours during the light experiments, whereas the OH concentration reached a maximum immediately after the start of the experiments and gradually decreased afterward during the dark experiments (Fig. S4 of the ESI†). We should note that the evolution of OA concentrations with diesel exhaust under the light conditions was not well captured by the simulation. The rate of increase of the observed OA concentrations slowed during the first hour of the experiments, whereas the simulated OA concentrations increased for ≥2 h after the start of the experiments. In the case of the experiments with a diesel truck, the decay rate of aromatic VOC was underestimated (Fig. S6 of the ESI†). This result suggests that the underestimation of SOA could be partly explained by the underestimation of the concentrations of the oxidants.
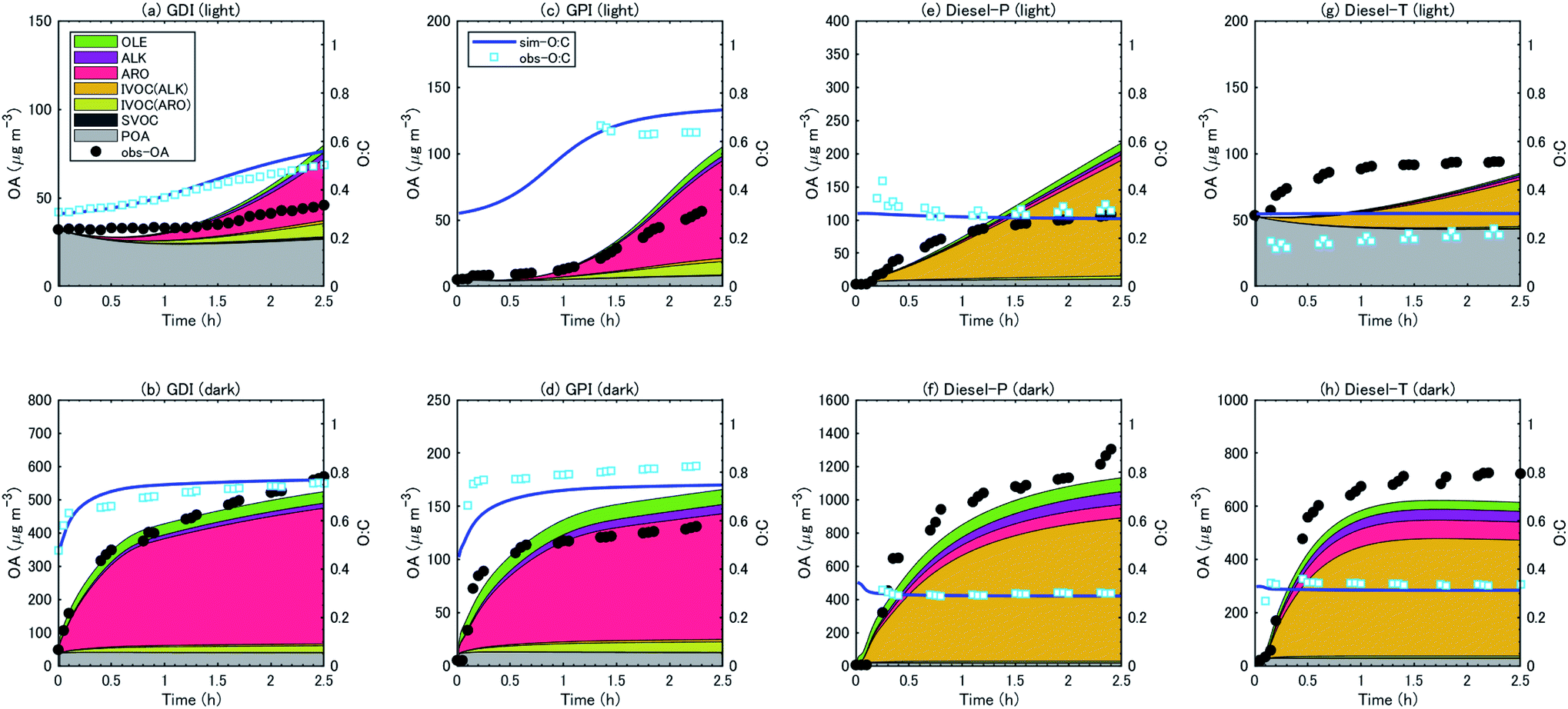 | ||
| Fig. 6 Time-series of OA concentrations (left axis) and O:C ratios (right axis) in the simulation. Contributions of precursor groups to the simulated OA concentrations are indicated by hatching with different colors. | ||
For gasoline vehicles, the largest contributor to SOA production was aromatic VOCs, followed by aromatic IVOC (light conditions) or alkene VOC (dark conditions). The simulated results indicated that aromatic VOCs accounted for 64–84% of the total SOA, and the sum of the alkane and alkene VOCs accounted for 11–15% of the SOA (Table 3). Unspeciated SVOC and IVOC accounted for 6–23% of the SOA. In contrast, the SOA from diesel exhaust was produced predominantly from the oxidation of IVOC (mostly alkane IVOC) (75–87%); the contribution of speciated VOCs (aromatics and alkanes) was less than 25%.
| VOC | VOC | VOC | IVOC | IVOC | SVOC | ||
|---|---|---|---|---|---|---|---|
| (% alkanes) | (% alkenes) | (% aromatics) | (% alkanes) | (% aromatics) | % | ||
| GDI | Light | 6.8 | 7.2 | 63.5 | 3.3 | 15.7 | 3.5 |
| Dark | 3.1 | 7.5 | 84.0 | 1.1 | 4.2 | 0.2 | |
| GPI | Light | 3.7 | 7.3 | 74.0 | 2.6 | 11.6 | 0.8 |
| Dark | 5.6 | 9.6 | 76.7 | 1.5 | 6.3 | 0.3 | |
| Diesel-P | Light | 2.7 | 6.1 | 4.1 | 84.4 | 2.3 | 0.4 |
| Dark | 6.6 | 7.7 | 7.3 | 77.3 | 0.9 | 0.1 | |
| Diesel-T | Light | 3.4 | 3.4 | 5.6 | 82.7 | 2.8 | 2.0 |
| Dark | 6.8 | 5.7 | 11.7 | 74.3 | 1.1 | 0.4 |
In previous studies, the contributions of aromatic VOCs to the SOA from gasoline exhaust were variable: several studies have estimated that single-ring aromatic VOCs are the dominant precursors of the SOA from gasoline exhaust; their contribution has been estimated to be 40–70%.25,29,75,86,87 Our results were similar to or slightly higher than these previous estimates. Previous estimates of IVOC contributions to SOA have been limited, though several studies have estimated that aromatic IVOC contributed to SOA production by 10–40%.86–88 Our estimate under light conditions (comparable with previous estimates) was 12–16%, which is similar to or less than previous estimates. For diesel vehicles, Eluri, et al.43 have estimated that IVOC accounts for 81–90% of SOA. Jathar, et al.19 and Zhao, et al.28 have also indicated that unspeciated compounds make predominant contributions to the SOA from diesel exhaust.
Under the light conditions, concentrations of SOA from diesel exhaust rapidly increased, whereas SOA formation from gasoline exhaust was slow, and there was a delay of 0.5–1.0 h. This delay of gasoline-SOA formation was consistent with previous findings of delays of SOA formation from aromatic VOC:73,81,89 the formation of SOA from aromatic VOC has been shown to be suppressed in the presence of NO, but the concentrations of SOA start to increase after NO has been consumed. As shown in Fig. S7,† concentrations of gasoline-SOA started to increase when NO concentration fell below 50 ppbv, while formation of diesel-SOA occurred even with high NO concentration (over 1 ppmv).
:C ratio of the SOA (Fig. 4) by assuming that the O:C ratio of the POA did not change during the course of oxidation. The O:C ratio of the SOA was clearly higher for gasoline exhaust than for diesel exhaust. The O:C ratio of SOA from the diesel exhaust was around 0.3–0.4. O:C ratio of SOA from gasoline exhaust 2.5 h after t = 0 was 0.7–0.8 both in the light and dark experiments. The O:C ratio of the SOA did not change during the course of oxidation, and it did not change significantly as a function of vehicle type (GDI vs. GPI, or Diesel-P vs. Diesel-T) or oxidation conditions (i.e., light vs. dark). For both gasoline and diesel exhaust, the O:C ratio of the OA increased as the SOA/OA ratio increased, and it was higher under the dark conditions than under the light conditions (Fig. S5†).
These O:C ratios are consistent with previous experimental results. For example, Pieber, et al.90 have shown from their experiments with a smog chamber and oxidation flow reactor that the O:C ratio of OA is 0.6–0.8 after photooxidation of gasoline exhaust with OH exposure of 8 × 106 to 3 × 107 molecules per cm3 per h. They excluded data with high NH4NO3 concentrations because these conditions are outside the AMS interference calibration and the O:C ratios in those cases may have been upward biased. Eluri, et al.43 have indicated that the O:C ratio of OA after photooxidation of diesel exhaust with aftertreatment devices (DPF and DOC) is around 0.2–0.4. Chirico, et al.11 have shown that the O:C ratios of OA after the photooxidation of diesel exhaust without a DPF are 0.1–0.3. The agreement of our experimental data with all of these results suggested that the differences of the O:C ratios between gasoline and diesel OA resulted from some common cause in addition to the differences in specific vehicles and oxidation conditions.
The observed O:C ratios, including the contrast between the gasoline- and diesel-SOA, was generally reproduced by the simulation (Fig. 6). The observed contrast of the O:C ratios between the gasoline- and diesel-OA was consistent with the O:C ratios of the simulated precursors. The O:C ratio was 0.5–0.8 in the gasoline-OA, wherein SOA from aromatic VOC was dominant, and it was 0.2–0.3 in the diesel-OA, wherein SOA from alkane IVOC was dominant (Table 3). This difference of the O:C ratios between gasoline- and diesel-SOA clearly suggested that the major precursors of SOA from gasoline and diesel exhaust differed. The O:C ratios of the SOA from a single precursor could occasionally change during the course of oxidation of the precursor,91,92 and the detailed evolution of O:C ratios during that oxidation has been simulated in some detailed SOA models.93,94 However, even our simple simulation could reproduce the contrast between the O:C ratios of the gasoline- and diesel-SOA.
Overall, the observed OA concentrations and O:C ratios were generally reproduced by the simulation. The normalized mean error during the first two hours of the individual experiments was <15% for both diesel and gasoline exhaust in the dark conditions, 35–43% for gasoline exhaust in the light conditions, and 18–97% for diesel exhaust in the light conditions (Table S13 of the ESI†). Because we used the aggregated chemical mechanism, SAPRC99, the simulation did not rigorously represent the reaction rates and product yields of individual VOC (Table S5 of the ESI†).
One unexplored consideration that may affect the chemical composition of SOA is organic nitrate. In our studies, the NO3 radical chemistry did not contribute much to SOA formation. However, the experiments were conducted with NOx in the vehicle exhaust, and the SOA formed during our experiments could thus have included organic nitrate. As noted in Section 3.2, SOA was formed in the high-NO conditions during the light experiments, and in the low-NO conditions during the dark experiments. In addition, the NOx/NMHC ratio was higher for diesel exhaust than for gasoline exhaust (see Section 2.1). A comparison of the fractions of organic nitrate in SOA between vehicle types and light and dark conditions might provide information about the factors that control the amount of organic nitrate in SOA.
Extensive research has been conducted on the contribution of organic nitrate to SOA from biogenic VOC.95–97 Organic nitrate in SOA from biogenic VOC (e.g., isoprene and monoterpenes) is produced via oxidation by the NO3 radical or via oxidation by OH radical or O3 followed by RO2 + NO reactions. Formation of organic nitrate could result in high rates of SOA production because the vapor pressure is lowered with the addition of nitrate functionality. The behavior of organic nitrate is thus one of the important considerations in understanding SOA production yields.
The fraction of organic nitrate has been estimated by using the NO2+/NO+ signal ratio measured with a high-resolution aerosol mass spectrometer (HR-AMS),98 because the NO2+/NO+ ratios differ between inorganic and organic nitrates.99,100 The NO2+/NO+ signal ratios of biogenic SOA101–103 and aromatic SOA99,104–106 are lower than the corresponding ratio of inorganic ammonium nitrate. These experimental data have been used for quantification of the organic nitrate fraction of ambient air in Europe and the U.S.96,107 However, to our knowledge, there has been no measurement of the NO2+/NO+ signal ratio in vehicle-exhaust SOA.
The NO2+/NO+ signal ratio of ammonium nitrate measured by the HR-AMS used in this experiment was 0.55. In contrast, the same ratio in aromatic SOA measured by the same HR-AMS was 0.17–0.26 (ref. 105) and was similar to the value reported for aromatic SOA in another study (0.13 for SOA from the photooxidation of 1,3,5-trymethylbenzene99). Fig. 7 summarizes the relationship between NO2+ and NO+ signals during our experiments. Fig. 7 indicates that the NO2+/NO+ ratio of the vehicle-exhaust SOA was significantly lower than that of ammonium nitrate, except for the gasoline-exhaust SOA during the first 1–2 h after the start of the dark experiments. The NO2+/NO+ ratio was 0.36–0.50 for the gasoline-exhaust SOA at more than 2 h after t = 0, and it was lower (0.17–0.36) for the diesel-exhaust SOA. To our knowledge, the NO2+/NO+ ratio of the SOA from IVOC has not been previously reported. If we assume that the NO2+/NO+ ratio of IVOC-SOA is similar to that of aromatic SOA, the fraction of organic nitrate was higher in diesel-SOA than in gasoline-SOA. This conclusion is consistent with the higher ratio of organic nitrate to total nitrate (=inorganic + organic nitrate) in diesel exhaust (Fig. 7(d)) versus gasoline exhaust (Fig. 7(c)): the organic-nitrate/total-nitrate ratio estimated from an ion balance of the AMS unit-mass signals was higher for diesel exhaust (0.6–0.8) than for gasoline exhaust (0.3–0.4). In the gasoline experiments, NH3 could also have been emitted, and the contribution of ammonium nitrate could thus be larger in gasoline exhaust than in diesel exhaust. In addition, as noted in Section 2.1, the NMHC/NOx ratio was higher in the gasoline exhaust than in the diesel exhaust, and it is thus possible that diesel-SOA contains more organic nitrate than does gasoline-SOA. The absolute concentration of NO3− determined from high-resolution mass spectrometric analysis of organic nitrate is inaccurate,98,106 and only relative comparisons were therefore made here. Nonetheless, significant amounts of organic nitrate appear to be present in both diesel-SOA and gasoline-SOA.
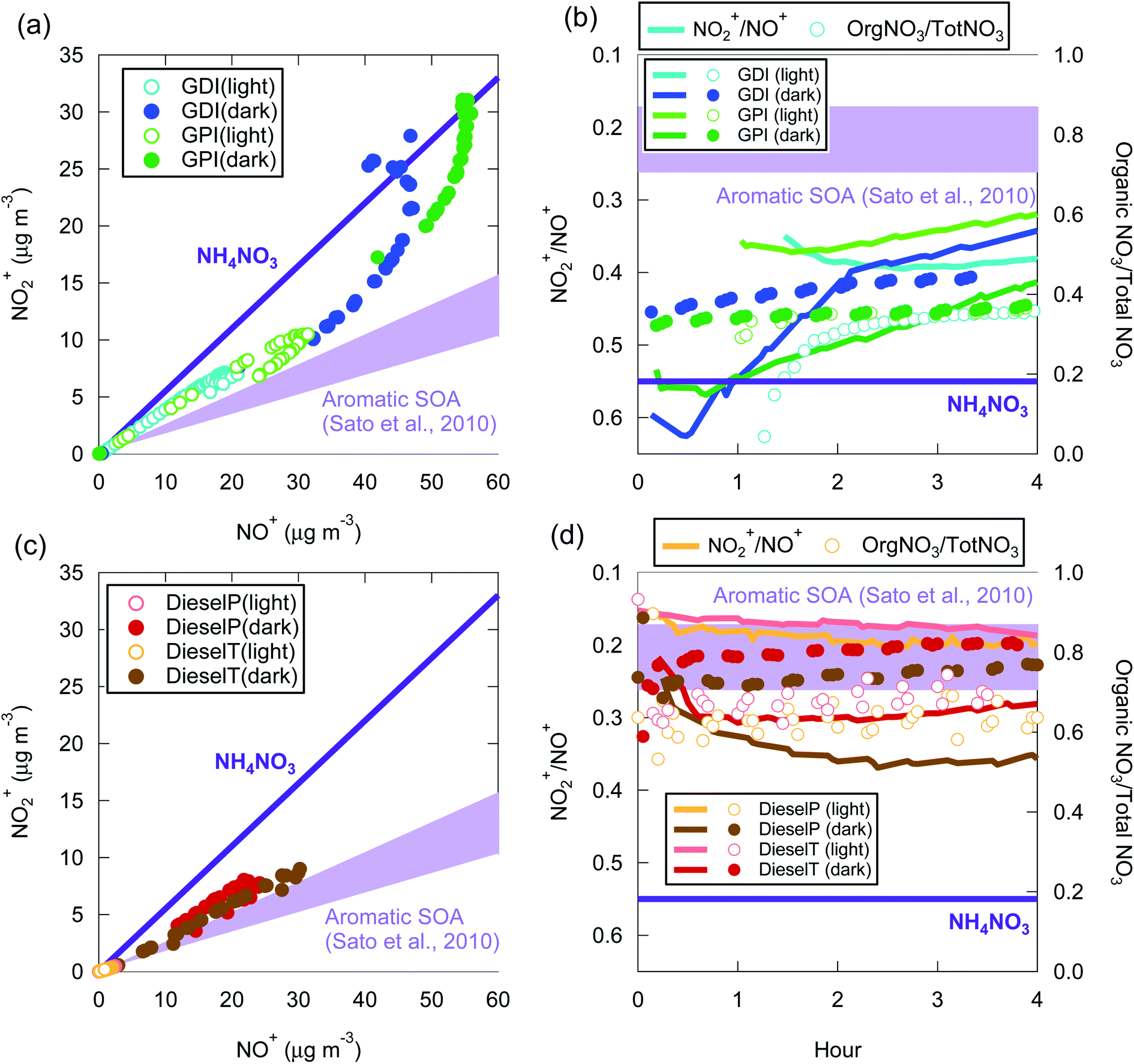 | ||
| Fig. 7 Relationship between NO2+ and NO+ signals (a and c) and time series of NO2+/NO+ and organic-nitrate/total-nitrate ratio (b and d) during the experiments with gasoline exhaust (a and b) and diesel exhaust (c and d). The NO2+/NO+ ratios for inorganic ammonium nitrate (NH4NO3) and aromatic SOA105 are also indicated. | ||
From the standpoint of oxidation conditions, the analysis of NO2+/NO+ ratios indicated that the fraction of organic nitrate was higher in the light than the dark in the OA from both gasoline exhaust and diesel exhaust (Fig. 7). The fraction of organic nitrate has also been estimated by assuming an ionic balance of inorganic compounds.96
| [Organic nitrate] = [nitrate] − [inorganic nitrate] = [nitrate] − 62([ammonium]/18–2[sulfate]/96 − [chloride]/35.5) | (4) |
In contrast, this analysis of ionic balance shows that the fraction of organic nitrate was lower in the light experiments. We could not conclude whether the chemical composition differed between the SOA formed in the light and dark conditions. For this evaluation, chemical analyses of the SOA formed under light and dark conditions will be required in future studies.
4 Conclusions and implications
Emission and production factors of OA from the exhaust of gasoline vehicles and diesel vehicles were analyzed in smog chamber experiments under light and dark conditions. POA and EC were emitted from a GDI vehicle and an old diesel truck, whereas relatively smaller amounts of POA and EC were emitted from a GPI vehicle and a later-model diesel passenger car. Significant amounts of SOA were produced from all four vehicles, and the SOA production factor was larger for the experiments in the dark conditions than in the light conditions. This difference was explained by the different SOA yields and may have been a result of the lower NO/(HO2 + RO2) ratio in the dark conditions.The observed OA concentrations and O:C ratios were reproduced by using a 1-D VBS module with a model setup that included an up-to-date IVOC module and the O:C ratios of individual SOA. In both the light and dark conditions, aromatic VOC was the predominant precursor for the SOA from gasoline vehicles (64–84%), and IVOC (unspeciated alkanes) was the major contributor to the SOA from diesel vehicles (75–87%). The observed O:C ratios of the SOA were clearly higher in the SOA from gasoline vehicles (0.7–0.8) versus diesel vehicles (0.3–0.4). This difference could be explained by the differences between the precursors, i.e., aromatic VOC in case of the gasoline exhaust and alkane IVOC in case of diesel exhaust.
Under both light and dark conditions, the dominant oxidant was the OH radical, and the dominant precursors for gasoline and diesel vehicles were similar between the light and dark conditions. In addition, the O:C ratio of the SOA was similar in the light and dark conditions. However, because the NO/(HO2 + RO2) ratios were different, SOA was formed under the high-NO condition in the light experiments, whereas under the low-NO condition in the dark experiments. The differences of the chemical compositions of the SOA formed in the light and dark conditions may have been caused by these differences. We could not resolve the differences between the chemical compositions of the SOA produced in the light and dark conditions based on an analysis of organic nitrate. Because the SOA produced in the dark condition with high concentrations of O3 has previously been used for toxicological studies, the differences of the chemical compositions of SOA should be further examined in future studies. Recent toxicological studies have indicated that naphthalene-SOA has higher oxidative potential than monoaromatic-SOA or other biogenic/anthropogenic SOA.108–110 Considering that the dominant SOA precursors differ between gasoline and diesel SOA, it is possible that the oxidative potentials of gasoline and diesel SOA are different. IVOC from diesel exhaust was dominated by alkanes:31,39 thus it is recommended that toxicological impact of alkane IVOC is assessed for an informed understanding of the toxicological impact of vehicle exhaust.
Author contributions
Yu Morino: conceptualization, data curation, funding acquisition, investigation, project administration, resources, software, writing – original draft, writing – review & editing. Ying Li: data curation, investigation, software, writing – original draft, writing – review & editing. Yuji Fujitani: conceptualization, data curation, funding acquisition, investigation, project administration, resources, writing – original draft, writing – review & editing. Kei Sato: conceptualization, data curation, funding acquisition, investigation, project administration, resources, writing – original draft, writing – review & editing. Satoshi Inomata: data curation, investigation, resources, writing – original draft, writing – review & editing. Kiyoshi Tanabe: conceptualization, investigation, supervision, writing – review & editing. Shantanu H. Jathar: software, writing – review & editing. Yoshinori Kondo: data curation, investigation, resources. Tomoki Nakayama: investigation, resources. Akihiro Fushimi: investigation, writing – review & editing. Akinori Takami: supervision. Shinji Kobayashi: supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the Environment Research and Technology Development Fund (JPMEERF20185001, JPMEERF20145008, JPMEERF14S11201, JPMEERF20155006) of the Environmental Restoration and Conservation Agency, the Grants-in-Aid for Scientific Research (JP16H06305) of the Japan Society for the Promotion of Science, and a competitive fund from the National Institute for Environmental Studies (2013–2015). The authors acknowledge Aiko Matsunaga (CARB) for providing useful information about the formation of SOA from alkenes. The authors gratefully acknowledge the technical assistance provided by Mr H. Konno, Mr T. Fujii (Horiba Techno Service), Ms. M. Ihara, Mr Y. Sugaya, and Ms. M. Chiba (NIES). KS and YF thanks Prof. David Cocker, III (UC Riverside), Dr Shunsuke Nakao (UC Riverside) and Dr Hiroyuki Hagino (JARI) for providing useful technical information on FEP film smog chamber system.References
- J. Lelieveld, J. S. Evans, M. Fnais, D. Giannadaki and A. Pozzer, The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 2015, 525, 367–371 CrossRef CAS PubMed.
- Y. Morino, K. Ueda, A. Takami, T. Nagashima, K. Tanabe, K. Sato, T. Noguchi, T. Ariga, K. Matsuhashi and T. Ohara, Sensitivities of Simulated Source Contributions and Health Impacts of PM2.5 to Aerosol Models, Environ. Sci. Technol., 2017, 51, 14273–14282 CrossRef CAS PubMed.
- K. L. Cheung, L. Ntziachristos, T. Tzamkiozis, J. J. Schauer, Z. Samaras, K. F. Moore and C. Sioutas, Emissions of Particulate Trace Elements, Metals and Organic Species from Gasoline, Diesel, and Biodiesel Passenger Vehicles and Their Relation to Oxidative Potential, Aerosol Sci. Technol., 2010, 44, 500–513 CrossRef CAS.
- J. Pavlovic, A. L. Holder and T. L. Yelverton, Effects of Aftermarket Control Technologies on Gas and Particle Phase Oxidative Potential from Diesel Engine Emissions, Environ. Sci. Technol., 2015, 49, 10544–10552 CrossRef CAS PubMed.
- M. R. Miller, Oxidative stress and the cardiovascular effects of air pollution, Free Radical Biol. Med., 2020, 151, 69–87 CrossRef CAS PubMed.
- M. Matti Maricq, Chemical characterization of particulate emissions from diesel engines: A review, J. Aerosol Sci., 2007, 38, 1079–1118 CrossRef.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Measurement of emissions from air pollution sources. 2. C-1 through C-30 organic compounds from medium duty diesel trucks, Environ. Sci. Technol., 1999, 33, 1578–1587 CrossRef CAS.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Measurement of emissions from air pollution sources. 5. C-1-C-32 organic compounds from gasoline-powered motor vehicles, Environ. Sci. Technol., 2002, 36, 1169–1180 CrossRef CAS PubMed.
- G. Saliba, R. Saleh, Y. Zhao, A. A. Presto, A. T. Lambe, B. Frodin, S. Sardar, H. Maldonado, C. Maddox, A. A. May, G. T. Drozd, A. H. Goldstein, L. M. Russell, F. Hagen and A. L. Robinson, Comparison of Gasoline Direct-Injection (GDI) and Port Fuel Injection (PFI) Vehicle Emissions: Emission Certification Standards, Cold-Start, Secondary Organic Aerosol Formation Potential, and Potential Climate Impacts, Environ. Sci. Technol., 2017, 51, 6542–6552 CrossRef CAS PubMed.
- A. L. Robinson, N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce and S. N. Pandis, Rethinking organic aerosols: Semivolatile emissions and photochemical aging, Science, 2007, 315, 1259–1262 CrossRef CAS PubMed.
- R. Chirico, P. F. DeCarlo, M. F. Heringa, T. Tritscher, R. Richter, A. S. H. Prevot, J. Dommen, E. Weingartner, G. Wehrle, M. Gysel, M. Laborde and U. Baltensperger, Impact of aftertreatment devices on primary emissions and secondary organic aerosol formation potential from in-use diesel vehicles: results from smog chamber experiments, Atmos. Chem. Phys., 2010, 10, 11545–11563 CrossRef CAS.
- T. D. Gordon, A. A. Presto, A. A. May, N. T. Nguyen, E. M. Lipsky, N. M. Donahue, A. Gutierrez, M. Zhang, C. Maddox, P. Rieger, S. Chattopadhyay, H. Maldonado, M. M. Maricq and A. L. Robinson, Secondary organic aerosol formation exceeds primary particulate matter emissions for light-duty gasoline vehicles, Atmos. Chem. Phys., 2014, 14, 4661–4678 CrossRef.
- T. D. Gordon, A. A. Presto, N. T. Nguyen, W. H. Robertson, K. Na, K. N. Sahay, M. Zhang, C. Maddox, P. Rieger, S. Chattopadhyay, H. Maldonado, M. M. Maricq and A. L. Robinson, Secondary organic aerosol production from diesel vehicle exhaust: impact of aftertreatment, fuel chemistry and driving cycle, Atmos. Chem. Phys., 2014, 14, 4643–4659 CrossRef.
- S. M. Platt, I. El Haddad, S. M. Pieber, A. A. Zardini, R. Suarez-Bertoa, M. Clairotte, K. R. Daellenbach, R. J. Huang, J. G. Slowik, S. Hellebust, B. Temime-Roussel, N. Marchand, J. de Gouw, J. L. Jimenez, P. L. Hayes, A. L. Robinson, U. Baltensperger, C. Astorga and A. S. H. Prevot, Gasoline cars produce more carbonaceous particulate matter than modern filter-equipped diesel cars, Sci. Rep., 2017, 7, 1–9 CrossRef CAS.
- D. R. Gentner, S. H. Jathar, T. D. Gordon, R. Bahreini, D. A. Day, I. El Haddad, P. L. Hayes, S. M. Pieber, S. M. Platt, J. de Gouw, A. H. Goldstein, R. A. Harley, J. L. Jimenez, A. S. Prevot and A. L. Robinson, Review of Urban Secondary Organic Aerosol Formation from Gasoline and Diesel Motor Vehicle Emissions, Environ. Sci. Technol., 2017, 51, 1074–1093 CrossRef CAS.
- Y. Fujitani, A. Furuyama, K. Tanabe and S. Hirano, Comparison of Oxidative Abilities of PM2.5 Collected at Traffic and Residential Sites in Japan. Contribution of Transition Metals and Primary and Secondary Aerosols, Aerosol Air Qual. Res., 2017, 17, 574–587 CrossRef CAS.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Frohlich-Nowoisky, Y. Fujitani, A. Furuyama, P. S. J. Lakey, J. Lelieveld, K. Lucas, Y. Morino, U. Poschl, S. Takaharna, A. Takami, H. J. Tong, B. Weber, A. Yoshino and K. Sato, Aerosol Health Effects from Molecular to Global Scales, Environ. Sci. Technol., 2017, 51, 13545–13567 CrossRef CAS PubMed.
- P. L. Hayes, A. G. Carlton, K. R. Baker, R. Ahmadov, R. A. Washenfelder, S. Alvarez, B. Rappenglück, J. B. Gilman, W. C. Kuster, J. A. de Gouw, P. Zotter, A. S. H. Prévôt, S. Szidat, T. E. Kleindienst, J. H. Offenberg, P. K. Ma and J. L. Jimenez, Modeling the formation and aging of secondary organic aerosols in Los Angeles during CalNex 2010, Atmos. Chem. Phys., 2015, 15, 5773–5801 CrossRef CAS.
- S. H. Jathar, T. D. Gordon, C. J. Hennigan, H. O. T. Pye, G. Pouliot, P. J. Adams, N. M. Donahue and A. L. Robinson, Unspeciated organic emissions from combustion sources and their influence on the secondary organic aerosol budget in the United States, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10473–10478 CrossRef CAS PubMed.
- R. Bahreini, A. M. Middlebrook, J. A. de Gouw, C. Warneke, M. Trainer, C. A. Brock, H. Stark, S. S. Brown, W. P. Dube, J. B. Gilman, K. Hall, J. S. Holloway, W. C. Kuster, A. E. Perring, A. S. H. Prevot, J. P. Schwarz, J. R. Spackman, S. Szidat, N. L. Wagner, R. J. Weber, P. Zotter and D. D. Parrish, Gasoline emissions dominate over diesel in formation of secondary organic aerosol mass, Geophys. Res. Lett., 2012, 39, L06805 CrossRef.
- S. H. Jathar, M. Woody, H. O. T. Pye, K. R. Baker and A. L. Robinson, Chemical transport model simulations of organic aerosol in southern California: model evaluation and gasoline and diesel source contributions, Atmos. Chem. Phys., 2017, 17, 4305–4318 CrossRef CAS.
- D. R. Gentner, G. Isaacman, D. R. Worton, A. W. Chan, T. R. Dallmann, L. Davis, S. Liu, D. A. Day, L. M. Russell, K. R. Wilson, R. Weber, A. Guha, R. A. Harley and A. H. Goldstein, Elucidating secondary organic aerosol from diesel and gasoline vehicles through detailed characterization of organic carbon emissions, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18318–18323 CrossRef CAS PubMed.
- R. E. Dunmore, J. R. Hopkins, R. T. Lidster, J. D. Lee, M. J. Evans, A. R. Rickard, A. C. Lewis and J. F. Hamilton, Diesel-related hydrocarbons can dominate gas phase reactive carbon in megacities, Atmos. Chem. Phys., 2015, 15, 9983–9996 CrossRef CAS.
- A. Fushimi, Y. Kondo, S. Kobayashi, Y. Fujitani, K. Saitoh, A. Takami and K. Tanabe, Chemical composition and source of fine and nanoparticles from recent direct injection gasoline passenger cars: Effects of fuel and ambient temperature, Atmos. Environ., 2016, 124, 77–84 CrossRef CAS.
- Z. Du, M. Hu, J. Peng, W. Zhang, J. Zheng, F. Gu, Y. Qin, Y. Yang, M. Li, Y. Wu, M. Shao and S. Shuai, Comparison of primary aerosol emission and secondary aerosol formation from gasoline direct injection and port fuel injection vehicles, Atmos. Chem. Phys., 2018, 18, 9011–9023 CrossRef CAS.
- N. Zimmerman, J. M. Wang, C. H. Jeong, J. S. Wallace and G. J. Evans, Assessing the Climate Trade-Offs of Gasoline Direct Injection Engines, Environ. Sci. Technol., 2016, 50, 8385–8392 CrossRef CAS PubMed.
- S. H. Jathar, B. Friedman, A. A. Galang, M. F. Link, P. Brophy, J. Volckens, S. Eluri and D. K. Farmer, Linking Load, Fuel, and Emission Controls to Photochemical Production of Secondary Organic Aerosol from a Diesel Engine, Environ. Sci. Technol., 2017, 51, 1377–1386 CrossRef CAS PubMed.
- Y. Zhao, N. T. Nguyen, A. A. Presto, C. J. Hennigan, A. A. May and A. L. Robinson, Intermediate Volatility Organic Compound Emissions from On-Road Diesel Vehicles: Chemical Composition, Emission Factors, and Estimated Secondary Organic Aerosol Production, Environ. Sci. Technol., 2015, 49, 11516–11526 CrossRef CAS PubMed.
- Y. Zhao, N. T. Nguyen, A. A. Presto, C. J. Hennigan, A. A. May and A. L. Robinson, Intermediate Volatility Organic Compound Emissions from On-Road Gasoline Vehicles and Small Off-Road Gasoline Engines, Environ. Sci. Technol., 2016, 50, 4554–4563 CrossRef CAS PubMed.
- Y. Zhao, A. T. Lambe, R. Saleh, G. Saliba and A. L. Robinson, Secondary Organic Aerosol Production from Gasoline Vehicle Exhaust: Effects of Engine Technology, Cold Start, and Emission Certification Standard, Environ. Sci. Technol., 2018, 52, 1253–1261 CrossRef CAS PubMed.
- Q. Lu, Y. Zhao and A. L. Robinson, Comprehensive organic emission profiles for gasoline, diesel, and gas-turbine engines including intermediate and semi-volatile organic compound emissions, Atmos. Chem. Phys., 2018, 18, 17637–17654 CrossRef CAS.
- D. S. Tkacik, A. T. Lambe, S. Jathar, X. Li, A. A. Presto, Y. Zhao, D. Blake, S. Meinardi, J. T. Jayne, P. L. Croteau and A. L. Robinson, Secondary organic aerosol formation from in-use motor vehicle emissions using a potential aerosol mass reactor, Environ. Sci. Technol., 2014, 48, 11235–11242 CrossRef CAS PubMed.
- R. Xu, M. S. Alam, C. Stark and R. M. Harrison, Behaviour of traffic emitted semi-volatile and intermediate volatility organic compounds within the urban atmosphere, Sci. Total Environ., 2020, 720, 137470 CrossRef CAS PubMed.
- L. E. Hatch, R. J. Yokelson, C. E. Stockwell, P. R. Veres, I. J. Simpson, D. R. Blake, J. J. Orlando and K. C. Barsanti, Multi-instrument comparison and compilation of non-methane organic gas emissions from biomass burning and implications for smoke-derived secondary organic aerosol precursors, Atmos. Chem. Phys., 2017, 17, 1471–1489 CrossRef CAS.
- Y. Zhao, C. J. Hennigan, A. A. May, D. S. Tkacik, J. A. de Gouw, J. B. Gilman, W. C. Kuster, A. Borbon and A. L. Robinson, Intermediate-volatility organic compounds: a large source of secondary organic aerosol, Environ. Sci. Technol., 2014, 48, 13743–13750 CrossRef CAS PubMed.
- E. S. Cross, A. G. Sappok, V. W. Wong and J. H. Kroll, Load-Dependent Emission Factors and Chemical Characteristics of IVOCs from a Medium-Duty Diesel Engine, Environ. Sci. Technol., 2015, 49, 13483–13491 CrossRef CAS PubMed.
- D. S. Tkacik, A. A. Presto, N. M. Donahue and A. L. Robinson, Secondary organic aerosol formation from intermediate-volatility organic compounds: cyclic, linear, and branched alkanes, Environ. Sci. Technol., 2012, 46, 8773–8781 CrossRef CAS PubMed.
- A. W. H. Chan, K. E. Kautzman, P. S. Chhabra, J. D. Surratt, M. N. Chan, J. D. Crounse, A. Kurten, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: implications for oxidation of intermediate volatility organic compounds (IVOCs), Atmos. Chem. Phys., 2009, 9, 3049–3060 CrossRef CAS.
- Q. Lu, B. N. Murphy, M. Qin, P. J. Adams, Y. Zhao, H. O. T. Pye, C. Efstathiou, C. Allen and A. L. Robinson, Simulation of organic aerosol formation during the CalNex study: updated mobile emissions and secondary organic aerosol parameterization for intermediate-volatility organic compounds, Atmos. Chem. Phys., 2020, 20, 4313–4332 CrossRef CAS.
- A. P. Grieshop, J. M. Logue, N. M. Donahue and A. L. Robinson, Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution, Atmos. Chem. Phys., 2009, 9, 1263–1277 CrossRef CAS.
- N. M. Donahue, A. L. Robinson, C. O. Stanier and S. N. Pandis, Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 2006, 40, 02635–02643 CrossRef CAS PubMed.
- R. Ots, D. E. Young, M. Vieno, L. Xu, R. E. Dunmore, J. D. Allan, H. Coe, L. R. Williams, S. C. Herndon, N. L. Ng, J. F. Hamilton, R. Bergström, C. Di Marco, E. Nemitz, I. A. Mackenzie, J. J. P. Kuenen, D. C. Green, S. Reis and M. R. Heal, Simulating secondary organic aerosol from missing diesel-related intermediate-volatility organic compound emissions during the Clean Air for London (ClearfLo) campaign, Atmos. Chem. Phys., 2016, 16, 6453–6473 CrossRef CAS.
- S. Eluri, C. D. Cappa, B. Friedman, D. K. Farmer and S. H. Jathar, Modeling the formation and composition of secondary organic aerosol from diesel exhaust using parameterized and semi-explicit chemistry and thermodynamic models, Atmos. Chem. Phys., 2018, 18, 13813–13838 CrossRef CAS.
- S. Samy and B. Zielinska, Secondary organic aerosol production from modern diesel engine emissions, Atmos. Chem. Phys., 2010, 10, 609–625 CrossRef CAS.
- T. T. Win-Shwe, C. Kyi-Tha-Thu, Y. Moe, Y. Fujitani, S. Tsukahara and S. Hirano, Exposure of BALB/c Mice to Diesel Engine Exhaust Origin Secondary Organic Aerosol (DE-SOA) during the Developmental Stages Impairs the Social Behavior in Adult Life of the Males, Front. Neurosci., 2016, 9, 524 CrossRef PubMed.
- R. D. McWhinney, S. S. Gao, S. Zhou and J. P. Abbatt, Evaluation of the effects of ozone oxidation on redox-cycling activity of two-stroke engine exhaust particles, Environ. Sci. Technol., 2011, 45, 2131–2136 CrossRef CAS PubMed.
- S. Okayama, Exhaust emission regulation and exhaust reduction technology for automobiles, J. Jpn. Soc. Atmos. Environ., 2019, 54, A43–A55 Search PubMed.
- Y. Nakashima, N. Kamei, S. Kobayashi and Y. Kajii, Total OH reactivity and VOC analyses for gasoline vehicular exhaust with a chassis dynamometer, Atmos. Environ., 2010, 44, 468–475 CrossRef CAS.
- G. T. Drozd, Y. Zhao, G. Saliba, B. Frodin, C. Maddox, R. J. Weber, M. O. Chang, H. Maldonado, S. Sardar, A. L. Robinson and A. H. Goldstein, Time Resolved Measurements of Speciated Tailpipe Emissions from Motor Vehicles: Trends with Emission Control Technology, Cold Start Effects, and Speciation, Environ. Sci. Technol., 2016, 50, 13592–13599 CrossRef CAS PubMed.
- S. Inomata, H. Tanimoto, Y. Fujitani, K. Sekimoto, K. Sato, A. Fushimi, H. Yamada, S. Hori, Y. Kumazawa, A. Shimono and T. Hikida, On-line measurements of gaseous nitro-organic compounds in diesel vehicle exhaust by proton-transfer-reaction mass spectrometry, Atmos. Environ., 2013, 73, 195–203 CrossRef CAS.
- S. Inomata, H. Yamada and H. Tanimoto, Investigation on VOC Emissions from Automobile Sources by Means of Online Mass Spectrometry, Curr. Pollut. Rep., 2016, 2, 188–199 CrossRef CAS.
- A. C. Aiken, P. F. Decarlo, J. H. Kroll, D. R. Worsnop, J. A. Huffman, K. S. Docherty, I. M. Ulbrich, C. Mohr, J. R. Kimmel, D. Sueper, Y. Sun, Q. Zhang, A. Trimborn, M. Northway, P. J. Ziemann, M. R. Canagaratna, T. B. Onasch, M. R. Alfarra, A. S. H. Prevot, J. Dommen, J. Duplissy, A. Metzger, U. Baltensperger and J. L. Jimenez, O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 2008, 42, 4478–4485 CrossRef CAS PubMed.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- R. Bahreini, B. Ervens, A. M. Middlebrook, C. Warneke, J. A. de Gouw, P. F. DeCarlo, J. L. Jimenez, C. A. Brock, J. A. Neuman, T. B. Ryerson, H. Stark, E. Atlas, J. Brioude, A. Fried, J. S. Holloway, J. Peischl, D. Richter, J. Walega, P. Weibring, A. G. Wollny and F. C. Fehsenfeld, Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas, J. Geophys. Res.: Atmos., 2009, 114, D00F16 CrossRef.
- P. F. DeCarlo, E. J. Dunlea, J. R. Kimmel, A. C. Aiken, D. Sueper, J. Crounse, P. O. Wennberg, L. Emmons, Y. Shinozuka, A. Clarke, J. Zhou, J. Tomlinson, D. R. Collins, D. Knapp, A. J. Weinheimer, D. D. Montzka, T. Campos and J. L. Jimenez, Fast airborne aerosol size and chemistry measurements above Mexico City and Central Mexico during the MILAGRO campaign, Atmos. Chem. Phys., 2008, 8, 4027–4048 CrossRef CAS.
- R. Atkinson and J. Arey, Atmospheric degradation of volatile organic compounds, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- B. Warren, R. L. Austin and D. R. Cocker, Temperature dependence of secondary organic aerosol, Atmos. Environ., 2009, 43, 3548–3555 CrossRef CAS.
- P. J. Ziemann and R. Atkinson, Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC.
- K. S. Docherty, W. Wu, Y. B. Lim and P. J. Ziemann, Contributions of organic peroxides to secondary aerosol formed from reactions of monoterpenes with O-3, Environ. Sci. Technol., 2005, 39, 4049–4059 CrossRef CAS PubMed.
- A. A. May, A. A. Presto, C. J. Hennigan, N. T. Nguyen, T. D. Gordon and A. L. Robinson, Gas-particle partitioning of primary organic aerosol emissions: (1) Gasoline vehicle exhaust, Atmos. Environ., 2013, 77, 128–139 CrossRef CAS.
- W. P. L. Carter, Documentation of the SAPRC-99 Chemical Mechanism for VOC Reactivity Assessment, 2000 Search PubMed.
- B. N. Murphy and S. N. Pandis, Simulating the Formation of Semivolatile Primary and Secondary Organic Aerosol in a Regional Chemical Transport Model, Environ. Sci. Technol., 2009, 43, 4722–4728 CrossRef CAS PubMed.
- A. G. Carlton and B. J. Turpin, Particle partitioning potential of organic compounds is highest in the Eastern US and driven by anthropogenic water, Atmos. Chem. Phys., 2013, 13, 10203–10214 CrossRef.
- H. O. T. Pye, B. N. Murphy, L. Xu, N. L. Ng, A. G. Carlton, H. Guo, R. Weber, P. Vasilakos, K. W. Appel, S. H. Budisulistiorini, J. D. Surratt, A. Nenes, W. Hu, J. L. Jimenez, G. Isaacman-VanWertz, P. K. Misztal and A. H. Goldstein, On the implications of aerosol liquid water and phase separation for organic aerosol mass, Atmos. Chem. Phys., 2017, 17, 343–369 CrossRef CAS PubMed.
- Ramboll Environment and Health, CAMx User's Guide Version 7.00, p. 2020 Search PubMed.
- C. L. Loza, J. S. Craven, L. D. Yee, M. M. Coggon, R. H. Schwantes, M. Shiraiwa, X. Zhang, K. A. Schilling, N. L. Ng, M. R. Canagaratna, P. J. Ziemann, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol yields of 12-carbon alkanes, Atmos. Chem. Phys., 2014, 14, 1423–1439 CrossRef.
- J. F. Pankow, An Absorption-Model of the Gas Aerosol Partitioning Involved in the Formation of Secondary Organic Aerosol, Atmos. Environ., 1994, 28, 189–193 CrossRef CAS.
- A. P. Tsimpidi, V. A. Karydis, M. Zavala, W. Lei, L. Molina, I. M. Ulbrich, J. L. Jimenez and S. N. Pandis, Evaluation of the volatility basis-set approach for the simulation of organic aerosol formation in the Mexico City metropolitan area, Atmos. Chem. Phys., 2010, 10, 525–546 CrossRef CAS.
- B. N. Murphy, N. M. Donahue, C. Fountoukis and S. N. Pandis, Simulating the oxygen content of ambient organic aerosol with the 2D volatility basis set, Atmos. Chem. Phys., 2011, 11, 7859–7873 CrossRef CAS.
- P. S. Chhabra, A. T. Lambe, M. R. Canagaratna, H. Stark, J. T. Jayne, T. B. Onasch, P. Davidovits, J. R. Kimmel and D. R. Worsnop, Application of high-resolution time-of-flight chemical ionization mass spectrometry measurements to estimate volatility distributions of alpha-pinene and naphthalene oxidation products, Atmos. Meas. Tech., 2015, 8, 1–18 CrossRef CAS.
- C. D. Cappa and K. R. Wilson, Multi-generation gas-phase oxidation, equilibrium partitioning, and the formation and evolution of secondary organic aerosol, Atmos. Chem. Phys., 2012, 12, 9505–9528 CrossRef CAS.
- J. E. Krechmer, D. Pagonis, P. J. Ziemann and J. L. Jimenez, Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 2016, 50, 5757–5765 CrossRef CAS PubMed.
- Y. Morino, K. Sato, K. Tanabe, S. Inomata, Y. Fujitani, S. H. Jathar, S. Ramasamy and C. D. Cappa, Modeling the Effects of Dimerization and Bulk Diffusion on the Evaporative Behavior of Secondary Organic Aerosol Formed from α-Pinene and 1,3,5-Trimethylbenzene, ACS Earth Space Chem., 2020, 4, 1931–1946 CrossRef CAS.
- P. Roth, J. Yang, E. Fofie, D. R. Cocker III, T. D. Durbin, R. Brezny, M. Geller, A. Asa-Awuku and G. Karavalakis, Catalyzed Gasoline Particulate Filters Reduce Secondary Organic Aerosol Production from Gasoline Direct Injection Vehicles, Environ. Sci. Technol., 2019, 53, 3037–3047 CrossRef CAS PubMed.
- E. Z. Nordin, A. C. Eriksson, P. Roldin, P. T. Nilsson, J. E. Carlsson, M. K. Kajos, H. Hellén, C. Wittbom, J. Rissler, J. Löndahl, E. Swietlicki, B. Svenningsson, M. Bohgard, M. Kulmala, M. Hallquist and J. H. Pagels, Secondary organic aerosol formation from idling gasoline passenger vehicle emissions investigated in a smog chamber, Atmos. Chem. Phys., 2013, 13, 6101–6116 CrossRef.
- S. Dusanter, D. Vimal, P. S. Stevens, R. Volkamer, L. T. Molina, A. Baker, S. Meinardi, D. Blake, P. Sheehy, A. Merten, R. Zhang, J. Zheng, E. C. Fortner, W. Junkermann, M. Dubey, T. Rahn, B. Eichinger, P. Lewandowski, J. Prueger and H. Holder, Measurements of OH and HO2 concentrations during the MCMA-2006 field campaign - Part 2: Model comparison and radical budget, Atmos. Chem. Phys., 2009, 9, 6655–6675 CrossRef CAS.
- Y. Kanaya, R. Q. Cao, H. Akimoto, M. Fukuda, Y. Komazaki, Y. Yokouchi, M. Koike, H. Tanimoto, N. Takegawa and Y. Kondo, Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004, J. Geophys. Res.: Atmos., 2007, 112, D21312 CrossRef.
- P. M. Edwards, S. S. Brown, J. M. Roberts, R. Ahmadov, R. M. Banta, J. A. deGouw, W. P. Dube, R. A. Field, J. H. Flynn, J. B. Gilman, M. Graus, D. Helmig, A. Koss, A. O. Langford, B. L. Lefer, B. M. Lerner, R. Li, S. M. Li, S. A. McKeen, S. M. Murphy, D. D. Parrish, C. J. Senff, J. Soltis, J. Stutz, C. Sweeney, C. R. Thompson, M. K. Trainer, C. Tsai, P. R. Veres, R. A. Washenfelder, C. Warneke, R. J. Wild, C. J. Young, B. Yuan and R. Zamora, High winter ozone pollution from carbonyl photolysis in an oil and gas basin, Nature, 2014, 514, 351–354 CrossRef CAS PubMed.
- A. A. Presto, K. E. H. Hartz and N. M. Donahue, Secondary organic aerosol production from terpene ozonolysis. 2. Effect of NOx concentration, Environ. Sci. Technol., 2005, 39, 7046–7054 CrossRef CAS PubMed.
- N. L. Ng, P. S. Chhabra, A. W. H. Chan, J. D. Surratt, J. H. Kroll, A. J. Kwan, D. C. McCabe, P. O. Wennberg, A. Sorooshian, S. M. Murphy, N. F. Dalleska, R. C. Flagan and J. H. Seinfeld, Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 2007, 7, 5159–5174 CrossRef CAS.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 2007, 7, 3909–3922 CrossRef CAS.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- Y. Morino, T. Ohara, Y. Yokouchi and A. Ooki, Comprehensive source apportionment of volatile organic compounds using observational data, two receptor models, and an emission inventory in Tokyo metropolitan area, J. Geophys. Res.: Atmos., 2011, 116, D02311 Search PubMed.
- S. H. Jathar, M. A. Miracolo, D. S. Tkacik, N. M. Donahue, P. J. Adams and A. L. Robinson, Secondary organic aerosol formation from photo-oxidation of unburned fuel: experimental results and implications for aerosol formation from combustion emissions, Environ. Sci. Technol., 2013, 47, 12886–12893 CrossRef CAS PubMed.
- A. Hodzic, S. Madronich, P. S. Kasibhatla, G. Tyndall, B. Aumont, J. L. Jimenez, J. Lee-Taylor and J. Orlando, Organic photolysis reactions in tropospheric aerosols: effect on secondary organic aerosol formation and lifetime, Atmos. Chem. Phys., 2015, 15, 9253–9269 CrossRef CAS.
- Y. Zhao, R. Saleh, G. Saliba, A. A. Presto, T. D. Gordon, G. T. Drozd, A. H. Goldstein, N. M. Donahue and A. L. Robinson, Reducing secondary organic aerosol formation from gasoline vehicle exhaust, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6984–6989 CrossRef CAS PubMed.
- T. Liu, X. Wang, W. Deng, Q. Hu, X. Ding, Y. Zhang, Q. He, Z. Zhang, S. Lü, X. Bi, J. Chen and J. Yu, Secondary organic aerosol formation from photochemical aging of light-duty gasoline vehicle exhausts in a smog chamber, Atmos. Chem. Phys., 2015, 15, 9049–9062 CrossRef CAS.
- G. T. Drozd, Y. Zhao, G. Saliba, B. Frodin, C. Maddox, M. C. Oliver Chang, H. Maldonado, S. Sardar, R. J. Weber, A. L. Robinson and A. H. Goldstein, Detailed Speciation of Intermediate Volatility and Semivolatile Organic Compound Emissions from Gasoline Vehicles: Effects of Cold-Starts and Implications for Secondary Organic Aerosol Formation, Environ. Sci. Technol., 2019, 53, 1706–1714 CrossRef CAS PubMed.
- K. Sato, S. Hatakeyama and T. Imamura, Secondary organic aerosol formation during the photooxidation of toluene: NOx dependence of chemical composition, J. Phys. Chem. A, 2007, 111, 9796–9808 CrossRef CAS PubMed.
- S. M. Pieber, N. K. Kumar, F. Klein, P. Comte, D. Bhattu, J. Dommen, E. A. Bruns, D. Kılıç, I. El Haddad, A. Keller, J. Czerwinski, N. Heeb, U. Baltensperger, J. G. Slowik and A. S. H. Prévôt, Gas-phase composition and secondary organic aerosol formation from standard and particle filter-retrofitted gasoline direct injection vehicles investigated in a batch and flow reactor, Atmos. Chem. Phys., 2018, 18, 9929–9954 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb and D. R. Worsnop, Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 2011, 3, 133–139 CrossRef CAS PubMed.
- B. N. Murphy, N. M. Donahue, C. Fountoukis, M. Dall'Osto, C. O'Dowd, A. Kiendler-Scharr and S. N. Pandis, Functionalization and fragmentation during ambient organic aerosol aging: application of the 2-D volatility basis set to field studies, Atmos. Chem. Phys., 2012, 12, 10797–10816 CrossRef CAS.
- C. D. Cappa, X. Zhang, C. L. Loza, J. S. Craven, L. D. Yee and J. H. Seinfeld, Application of the Statistical Oxidation Model (SOM) to Secondary Organic Aerosol formation from photooxidation of C-12 alkanes, Atmos. Chem. Phys., 2013, 13, 1591–1606 CrossRef.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- R. A. Zaveri, J. E. Shilling, J. D. Fast and S. R. Springston, Efficient Nighttime Biogenic SOA Formation in a Polluted Residual Layer, J. Geophys. Res.: Atmos., 2020, 125, e2019JD031583 Search PubMed.
- H. O. T. Pye, D. J. Luecken, L. Xu, C. M. Boyd, N. L. Ng, K. R. Baker, B. R. Ayres, J. O. Bash, K. Baumann, W. P. L. Carter, E. Edgerton, J. L. Fry, W. T. Hutzell, D. B. Schwede and P. B. Shepson, Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 2015, 49, 14195–14203 CrossRef CAS PubMed.
- D. K. Farmer, A. Matsunaga, K. S. Docherty, J. D. Surratt, J. H. Seinfeld, P. J. Ziemann and J. L. Jimenez, Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6670–6675 CrossRef CAS PubMed.
- M. R. Alfarra, D. Paulsen, M. Gysel, A. A. Garforth, J. Dommen, A. S. H. Prevot, D. R. Worsnop, U. Baltensperger and H. Coe, A mass spectrometric study of secondary organic aerosols formed from the photooxidation of anthropogenic and biogenic precursors in a reaction chamber, Atmos. Chem. Phys., 2006, 6, 5279–5293 CrossRef CAS.
- J. L. Fry, A. Kiendler-Scharr, A. W. Rollins, P. J. Wooldridge, S. S. Brown, H. Fuchs, W. Dube, A. Mensah, M. dal Maso, R. Tillmann, H. P. Dorn, T. Brauers and R. C. Cohen, Organic nitrate and secondary organic aerosol yield from NO3 oxidation of beta-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 2009, 9, 1431–1449 CrossRef CAS.
- C. M. Boyd, T. Nah, L. Xu, T. Berkemeier and N. L. Ng, Secondary Organic Aerosol (SOA) from Nitrate Radical Oxidation of Monoterpenes: Effects of Temperature, Dilution, and Humidity on Aerosol Formation, Mixing, and Evaporation, Environ. Sci. Technol., 2017, 51, 7831–7841 CrossRef CAS PubMed.
- C. M. Boyd, J. Sanchez, L. Xu, A. J. Eugene, T. Nah, W. Y. Tuet, M. I. Guzman and N. L. Ng, Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 2015, 15, 7497–7522 CrossRef CAS.
- D. Zhao, S. H. Schmitt, M. Wang, I.-H. Acir, R. Tillmann, Z. Tan, A. Novelli, H. Fuchs, I. Pullinen, R. Wegener, F. Rohrer, J. Wildt, A. Kiendler-Scharr, A. Wahner and T. F. Mentel, Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of alpha-pinene and limonene, Atmos. Chem. Phys., 2018, 18, 1611–1628 CrossRef CAS.
- S. Ramasamy, T. Nakayama, T. Imamura, Y. Morino, Y. Kajii and K. Sato, Investigation of dark condition nitrate radical- and ozone-initiated aging of toluene secondary organic aerosol: Importance of nitrate radical reactions with phenolic products, Atmos. Environ., 2019, 219, 117049 CrossRef CAS.
- K. Sato, A. Takami, T. Isozaki, T. Hikida, A. Shimono and T. Imamura, Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons, Atmos. Environ., 2010, 44, 1080–1087 CrossRef CAS.
- K. Sato, A. Takami, Y. Kato, T. Seta, Y. Fujitani, T. Hikida, A. Shimono and T. Imamura, AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1,3,5-trimethylbenzene in the presence of NOx: effects of chemical structure on SOA aging, Atmos. Chem. Phys., 2012, 12, 4667–4682 CrossRef CAS.
- A. Kiendler-Scharr, A. A. Mensah, E. Friese, D. Topping, E. Nemitz, A. S. H. Prevot, M. Äijälä, J. Allan, F. Canonaco, M. Canagaratna, S. Carbone, M. Crippa, M. Dall Osto, D. A. Day, P. De Carlo, C. F. Di Marco, H. Elbern, A. Eriksson, E. Freney, L. Hao, H. Herrmann, L. Hildebrandt, R. Hillamo, J. L. Jimenez, A. Laaksonen, G. McFiggans, C. Mohr, C. O'Dowd, R. Otjes, J. Ovadnevaite, S. N. Pandis, L. Poulain, P. Schlag, K. Sellegri, E. Swietlicki, P. Tiitta, A. Vermeulen, A. Wahner, D. Worsnop and H. C. Wu, Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 2016, 43, 7735–7744 CrossRef CAS.
- K. R. Daellenbach, G. Uzu, J. H. Jiang, L. E. Cassagnes, Z. Leni, A. Vlachou, G. Stefenelli, F. Canonaco, S. Weber, A. Segers, J. J. P. Kuenen, M. Schaap, O. Favez, A. Albinet, S. Aksoyoglu, J. Dommen, U. Baltensperger, M. Geiser, I. El Haddad, J. L. Jaffrezo and A. S. H. Prevot, Sources of particulate-matter air pollution and its oxidative potential in Europe, Nature, 2020, 587, 414–419 CrossRef CAS PubMed.
- W. Y. Tuet, Y. Chen, L. Xu, S. Fok, D. Gao, R. J. Weber and N. L. Ng, Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds, Atmos. Chem. Phys., 2017, 17, 839–853 CrossRef CAS.
- A. Fushimi, D. Nakajima, A. Furuyama, G. Suzuki, T. Ito, K. Sato, Y. Fujitani, Y. Kondo, A. Yoshino, S. Ramasamy, J. Schauer, J. P. Fu, Y. Takahashi, K. Saitoh, S. Saito and A. Takami, Source contributions to multiple toxic potentials of atmospheric organic aerosols, Sci. Total Environ., 2021, 773, 145614 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sections S1 and S2, Tables S1–S13 and Fig. S1–S8. See DOI: 10.1039/d1ea00045d |
| This journal is © The Royal Society of Chemistry 2022 |