
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-enhanced uptake of SO2 on Icelandic volcanic dusts†
Jerome
Lasne
 *a,
Darya
Urupina
a,
Elena C.
Maters
b,
Pierre
Delmelle
c,
Manolis N.
Romanias
*a and
Frederic
Thevenet
a
*a,
Darya
Urupina
a,
Elena C.
Maters
b,
Pierre
Delmelle
c,
Manolis N.
Romanias
*a and
Frederic
Thevenet
a
aIMT Nord Europe, Institut Mines-Télécom, Univ. Lille, CERI EE, F-59000 Lille, France. E-mail: jerome.lasne@imt-nord-europe.fr; emmanouil.romanias@imt-nord-europe.fr; Tel: +33 3 27 71 26 33 Tel: +33 3 27 71 22 93
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
cEnvironmental Sciences, Earth & Life Institute, UCLouvain, Croix du sud 2, B-1348 Louvain-la-Neuve, Belgium
First published on 8th February 2022
Abstract
Iceland is the largest volcanic dust (v-dust) desert on Earth, with an estimated area of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km2. In addition, Iceland is one of the most active aeolian areas in the world, with frequent high-velocity winds resuspending v-dust from the ground into the atmosphere. Suspended v-dust particles can then be transferred over thousands of kilometers, reaching as far as central Europe. Once released into the atmosphere, v-dust particles can interact or react with atmospheric pollutants. In this study, we investigate the heterogeneous reactivity of sulfur dioxide (SO2), one of the most prominent gases released by volcanic eruptions, with Icelandic v-dust particles, and the influence of relevant atmospheric parameters, such as relative humidity (RH) and ultraviolet (UV) light flux, on this reactivity. The experiments are conducted at atmospheric pressure in a coated-wall flow-tube reactor coupled with an SO2 analyzer. To quantify the heterogeneous processes, we determine the initial number of SO2 molecules taken up by dust, NS, and the steady-state uptake coefficient, γss, of SO2 on different v-dusts. NS increases with RH and with the photon flux characterized by the photolysis rate of NO2 in the setup, JNO2. The photo-enhanced removal of SO2 is also found to depend on the surface elemental composition of v-dust particles, and an empirical parametrization of the photo-induced effect is proposed to account for the most important environmental factors, leading to the general expression: NS,light/NS,dark = 6.1 × (1 + 7.76 × 10−2 × RH) × (1 + 480.5 × JNO2) × (Ti/Si). The steady-state uptake coefficients of SO2 are in the 10−8 range, once normalized to the specific surface area of v-dust. RH and UV light influence the value of γss, but to a lesser extent than they influence NS. Our results suggest that the photo-induced heterogeneous uptake of SO2 on v-dust particles may provide a significant sink of sulfur in volcanic clouds, and should be taken into account in atmospheric modeling.
000 km2. In addition, Iceland is one of the most active aeolian areas in the world, with frequent high-velocity winds resuspending v-dust from the ground into the atmosphere. Suspended v-dust particles can then be transferred over thousands of kilometers, reaching as far as central Europe. Once released into the atmosphere, v-dust particles can interact or react with atmospheric pollutants. In this study, we investigate the heterogeneous reactivity of sulfur dioxide (SO2), one of the most prominent gases released by volcanic eruptions, with Icelandic v-dust particles, and the influence of relevant atmospheric parameters, such as relative humidity (RH) and ultraviolet (UV) light flux, on this reactivity. The experiments are conducted at atmospheric pressure in a coated-wall flow-tube reactor coupled with an SO2 analyzer. To quantify the heterogeneous processes, we determine the initial number of SO2 molecules taken up by dust, NS, and the steady-state uptake coefficient, γss, of SO2 on different v-dusts. NS increases with RH and with the photon flux characterized by the photolysis rate of NO2 in the setup, JNO2. The photo-enhanced removal of SO2 is also found to depend on the surface elemental composition of v-dust particles, and an empirical parametrization of the photo-induced effect is proposed to account for the most important environmental factors, leading to the general expression: NS,light/NS,dark = 6.1 × (1 + 7.76 × 10−2 × RH) × (1 + 480.5 × JNO2) × (Ti/Si). The steady-state uptake coefficients of SO2 are in the 10−8 range, once normalized to the specific surface area of v-dust. RH and UV light influence the value of γss, but to a lesser extent than they influence NS. Our results suggest that the photo-induced heterogeneous uptake of SO2 on v-dust particles may provide a significant sink of sulfur in volcanic clouds, and should be taken into account in atmospheric modeling.
Environmental significanceSO2 is an atmospheric pollutant that plays a key role in the environment. It is well established in the literature that SO2 can interact/react with atmospheric particles. However, there is limited knowledge on SO2 interaction with natural volcanic particles, despite the significance of this heterogeneous process on air quality and climate. In this study, we investigate the heterogeneous uptake of SO2 on natural volcanic particles. Using a well-established experimental approach, the SO2 uptake coefficients and surface coverages are determined under atmospheric levels of relative humidity (RH) and light radiation. A multi-parametric equation is developed and applied to fit SO2 removal on volcanic particles. |
1. Introduction
Volcanic eruptions are a highly variable source of solid particles; annual emissions of fine volcanic ash (<63 μm) are estimated between 176 and 256 Tg on average,1 accounting for 5.1–7.4% of total aerosol emissions.2 Iceland is a very active geological site that features volcanic eruptions typically every 3 to 5 years,3 and thus it is a major source of volcanic particles in the atmosphere. The Eyjafjallajökull eruption in 2010 emitted 8.3 ± 4.2 Tg of fine ash in the 2.8–28 μm diameter range, as determined by inverse modeling of satellite observations.4 Ash subsequently settles on the ground, we refer to this material as volcanic dust (hereafter ‘v-dust’), and can be remobilized by the strong winds that cause frequent dust storm events in Iceland.5 It is estimated that around 30 to 40 Tg of Icelandic v-dust is resuspended annually.5 The dust event observed in september 2013 in Southern Iceland was composed of remobilized ash from the 2010 Eyjafjallajökull and 2011 Grimsvötn eruptions, and accounted for roughly 0.2 Tg of dust over the two days of observations.6 Icelandic v-dust can travel thousands of kilometers before eventually being deposited. For instance, recent studies have observed the transportation of v-dust to the central Balkans.7 Interestingly, v-dust exhibits thermal and optical properties similar to black carbon.8 In general, volcanic particles play an important role in climate and can impact directly or indirectly the energy budget of the planet.1,9–12In addition to particles, explosive volcanic eruptions release sulfur dioxide (SO2) along with a range of other gases, among which water and carbon dioxide dominate.1 Non-eruptive outgassing is a less spectacular, but continuous source of tropospheric volcanic gases. Typical concentrations of SO2 in the plume range from 100 ppb (part per billion) to 3000 ppb.13 Measurements while flying through the eruptive plume of Mount Hekla in 2000 recorded SO2 concentrations up to 1000 ppb, with a large spatial variation at the edge of the cloud.14 Observations of the plume of the Eyjafjallajökull volcano in 2010 showed average SO2 mixing ratios of 40 ppb.15
SO2 is an atmospheric species that plays a key role in the environment. The tropospheric background concentration of SO2 is very low (ppt range),16 and thus volcanic SO2 emissions receive great attention. Once released into the atmosphere, SO2 can either be (i) oxidized in the gas phase by hydroxyl radicals to produce sulfuric acid (H2SO4), a relatively minor atmospheric sink of SO2, or (ii) dissolved in water droplets and oxidized by hydrogen peroxide, ozone or oxygen to form sulfate (SO42−) species, which is a relatively fast atmospheric process. This conversion of SO2 in the troposphere can affect human health and air quality,17–19 as well as the radiative balance of the Earth due to the formation of sulfate aerosols.19 SO2 can also interact/react with airborne solid particles, especially during high particle loading, but this atmospheric process has received less attention than the gas and liquid phase oxidation of SO2. Several literature studies have evidenced the uptake of SO2 on mineral surrogates20 and natural mineral dusts.21,22 Reactions of SO2 on desert and volcanic origin particles during dust storms could potentially impact the sulfur budget in the troposphere, but this heterogeneous transformation of SO2 has not been yet fully assessed in the literature. The interaction of SO2 with volcanic material (i.e., ash or v-dust) is of particular interest due to the coexistence of high atmospheric loadings of particles and SO2. Maters et al. measured the uptake of SO2 on natural volcanic ash and synthetic volcanic glass samples under tropospherically relevant conditions of SO2 concentration and temperature, but under dry conditions.23 Recently, we have investigated for the first time the transformation of SO2 on Icelandic v-dust;24,25 we expand this work here by evaluating the uptake of SO2 on v-dusts in the dark and under UV light irradiation. In particular, the aging of v-dusts by SO2 is investigated under atmospherically relevant conditions, i.e., at atmospheric pressure, an SO2 concentration of 75 ppb, relative humidity (RH) in the 0–72% range, and in the dark and under different UV photon fluxes, performing long laboratory experiments over timescales of typically 24 hours. As a result, we quantify the initial number of SO2 molecules taken up (NS) and the steady-state uptake (γss,BET) of SO2 by Icelandic v-dust.
2. Methods
2.1 Materials
To further characterize the v-dust samples, we determined the surface (topmost 2–10 nm) elemental composition of the v-dusts by X-ray Photoelectron Spectroscopy (XPS) using a Kratos Axis Ultra Instrument. Relative concentrations (in at%) were determined by high-resolution scans at a pass energy of 40 eV for C1s, O1s, Si2p, Al2p, Fe2p, Mg2p, Ca2p, Na1s, K2p, Ti2p, and Mn2p using a monochromatic Al X-ray beam centered at 1486.6 eV. Data treatment was performed using the CasaXPS software. The binding energy scale was calibrated by fixing the C1s peak for adventitious carbon at 284.8 eV.27 The results are presented in Table 1.
| V-dust | Si | Al | Fe | Mg | Ca | Na | K | Ti | Mn |
|---|---|---|---|---|---|---|---|---|---|
| Mýrdalssandur | 53.1 | 17.6 | 11.7 | 4.4 | 6.6 | 3.3 | 0.7 | 2.6 | <0.1 |
| Dyngjusandur | 53.9 | 19.9 | 9.1 | 6.4 | 7.7 | 1.4 | <0.1 | 1.4 | 0.3 |
| Hagavatn | 45.0 | 35.8 | 5.7 | 3.1 | 6.9 | 1.9 | <0.1 | 1.1 | 0.1 |
| Maelifellssandur | 49.8 | 22.3 | 13.0 | 4.1 | 5.2 | 1.9 | 0.4 | 3.0 | 0.4 |
2.2. Experimental setup
The heterogeneous interaction of SO2 with the v-dust materials is studied in a horizontal coated-wall flow-tube (CWFT) reactor at atmospheric pressure and room temperature T = 296 K (Fig. 1).24,26,28 The v-dust sample is deposited on the inner surface of a Pyrex tube. Further details about the sample preparation can be found in recent publications.24,26 The tube is then inserted into the reactor along its main axis, and kept in position by two Viton O-rings sealing the space between the tube and the wall of the reactor. Before each experiment, the sample is left for at least 1 hour under a zero air flow at the RH to be used during the subsequent experiment. This allows equilibration of the surface of the v-dusts with H2O at the desired level of RH, prior to exposure to SO2.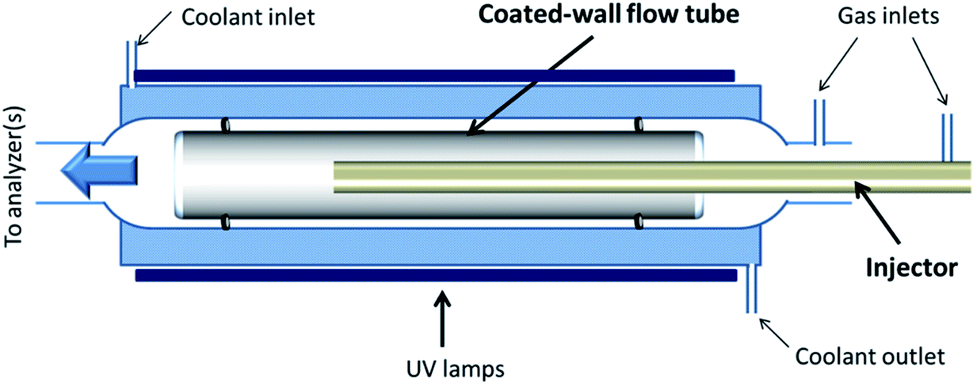 | ||
| Fig. 1 Schematic representation of the coated-wall flow-tube (CWFT) reactor. The space filled with coolant between the two walls is shaded in blue. The dust sample coating the inner surface of the Pyrex tube is shown in gray. | ||
Air and SO2 are flowed through a movable injector with an internal diameter of 0.3 cm. The total flow rate ranges from 250 to 500 sccm, ensuring laminar flow conditions (Reynolds number Re < 150). A thermoregulation unit (Huber, Ministat 230) controls the temperature by circulating water between the double-wall of the flow-tube. In separate experiments, the temperature profile of the gas flow along the reactor was recorded by inserting a K-type thermocouple into the movable injector; the temperature was found to be constant along the flow-tube and consistent with the value displayed by the thermoregulation unit (±1%).
Downstream of the reactor, the outgoing flow is directed to a SO2 analyzer (Model 43C, Thermo Environmental Instruments Inc.), where SO2 is detected via its fluorescence signal after absorption of UV photons. The limit of detection is 2.0 ppb for the average time of 10 seconds set in our experiments. The precision of the measurement given by the supplier of the instrument is 1.0 ppb or 1% of the reading, whichever is greater. Throughout this study, gas concentrations are given in ppb; under usual experimental conditions (T = 296 K, P = 1 atm), 1 ppb (SO2) ≈ 2.5 × 1010 molecules cm−3.
Three UVA lamps (UVA, Philips Lighting PL-L 18 W/10/4P; 315–400 nm with maximum emission at 352 nm) are placed evenly around the flow-tube at a distance of 12 cm in order to investigate possible photo-induced reactions on the surface of the v-dusts. The light source is characterized by measuring the photolysis rate of NO2 in the reactor, JNO2. The JNO2 values measured after switching on one, two or three UV lamps are 1.5 × 10−3 s−1, 3.0 × 10−3 s−1 and 4.5 × 10−3 s−1, respectively, similar to the JNO2 measured in the Earth's atmosphere under cloudy and clear sky conditions.29–31
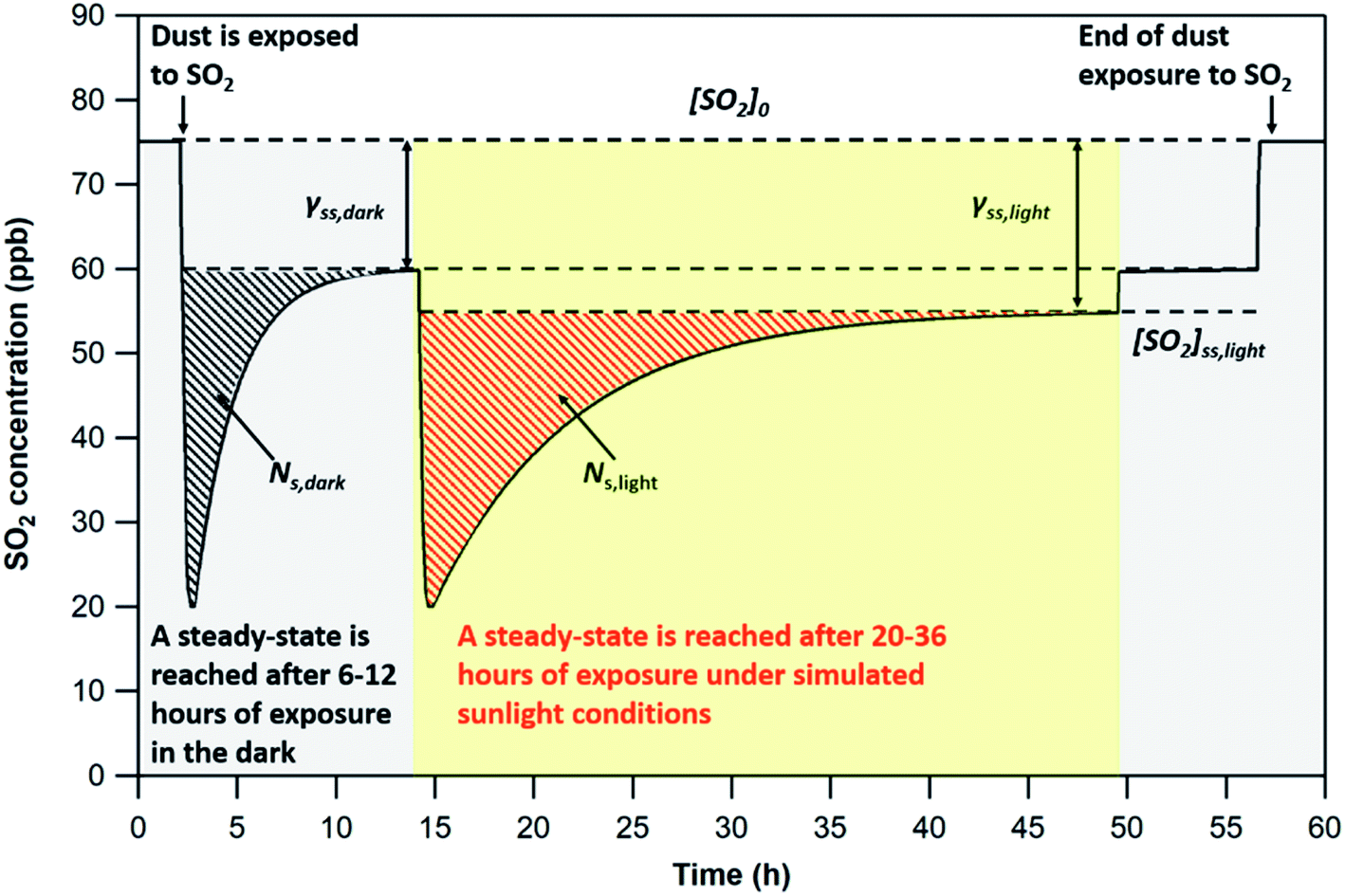 | ||
| Fig. 2 Theoretical SO2 concentration profile during a typical flow-tube experiment with v-dust at 296 K. At the beginning of the experiment, the dust is isolated from the gas mixture, in which an initial SO2 concentration, [SO2]0, is set. The dust is then exposed to SO2 in the dark (gray area), and [SO2] decreases initially strongly, before recovering to a steady-state value [SO2]SS,dark within typically 6–12 hours. Thereafter, the surface is irradiated with UV light (yellow area), and a second large consumption of SO2 is observed, before recovering to a steady-state value [SO2]SS,light within typically 20–36 hours. Lights are turned off (second gray area) and [SO2] returns to its steady-state value in the dark. The SO2 concentration is then returned to [SO2]0 by pushing the injector in, and the dust is no longer exposed to the gas. Black-shaded and red-shaded areas correspond to the initial number of SO2 molecules taken up per surface unit of v-dust (in molecules cm−2), respectively in the dark and under UV, NS,dark and NS,light. The moment at which the steady-state is reached (as discussed earlier, see text) is chosen as integration limit for the determination of NS. [SO2]SS,dark and [SO2]SS,light are used to measure the steady-state uptake coefficients, γss,dark and γss,light. | ||
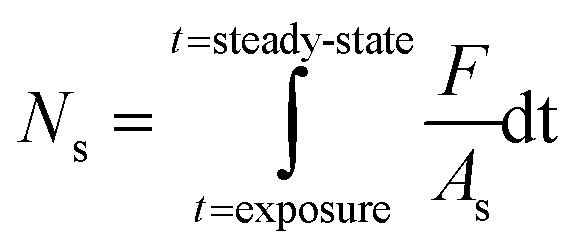 | (1) |
Since the SO2 concentrations recorded at steady-state both in the dark and under UV irradiation are lower than pre-exposure concentrations, only the initial removal of SO2 is considered (dashed areas in Fig. 2) in the determination of NS. The integration limits for the determination of NS correspond to the period of time elapsed between exposure of the dust to SO2 and steady-state (i.e., maximum signal variation of 1.5% for at least three hours). This criterion is adopted to compare the uptake capacities of the four v-dusts at the initial stage of interaction with SO2.
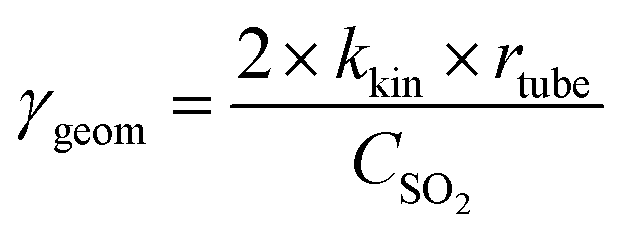 | (2) |
The uptake coefficient determined using the geometric surface area (Sgeom) of dust can be considered an upper limit.20 A more realistic estimate of the surface area probed by gas molecules is made using the Brunauer, Emmett and Teller specific surface area, SBET (m2 g−1), of the solid sample.37 The BET surface areas of the v-dusts studied here are presented in Table S1.† The effective surface area of the v-dust is then given by As = mdust × SBET, where mdust is the mass of dust deposited on the walls of the flow-tube. The corresponding uptake coefficient (γBET) is given by the following expression:
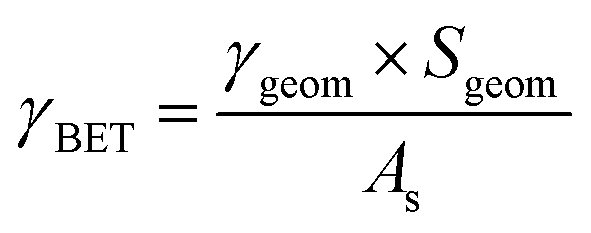 | (3) |
In order to investigate whether the entire v-dust surface is accessible to SO2 or not, kkin was determined under steady-state conditions in a series of experiments where different v-dust masses were exposed to SO2. The results obtained for Mýrdalssandur v-dust are displayed in Fig. 3. For comparison purpose, kkin calculated for a clean tube is also presented. While a negligible adsorption of SO2 occurs on a clean tube, the precision of the SO2 analyser (i.e., 1 ppb) enables determination of the value of kkin in a clean tube. The results shown in Fig. 3 were fitted with a linear function that excludes the datapoint estimated for a clean tube. Note that kkin increases linearly with the mass of dust, which indicates that the entire surface of dust is accessible to SO2 gas and participates in the heterogeneous loss of SO2. The linear fit intersects (within error) the kkin value estimated for a clean tube, confirming the robustness of our measurements. In the following, experiments are performed with v-dust masses in the linear regime and adjusting the flow rate to lower values to increase the precision of the kkin values measured under steady-state conditions.
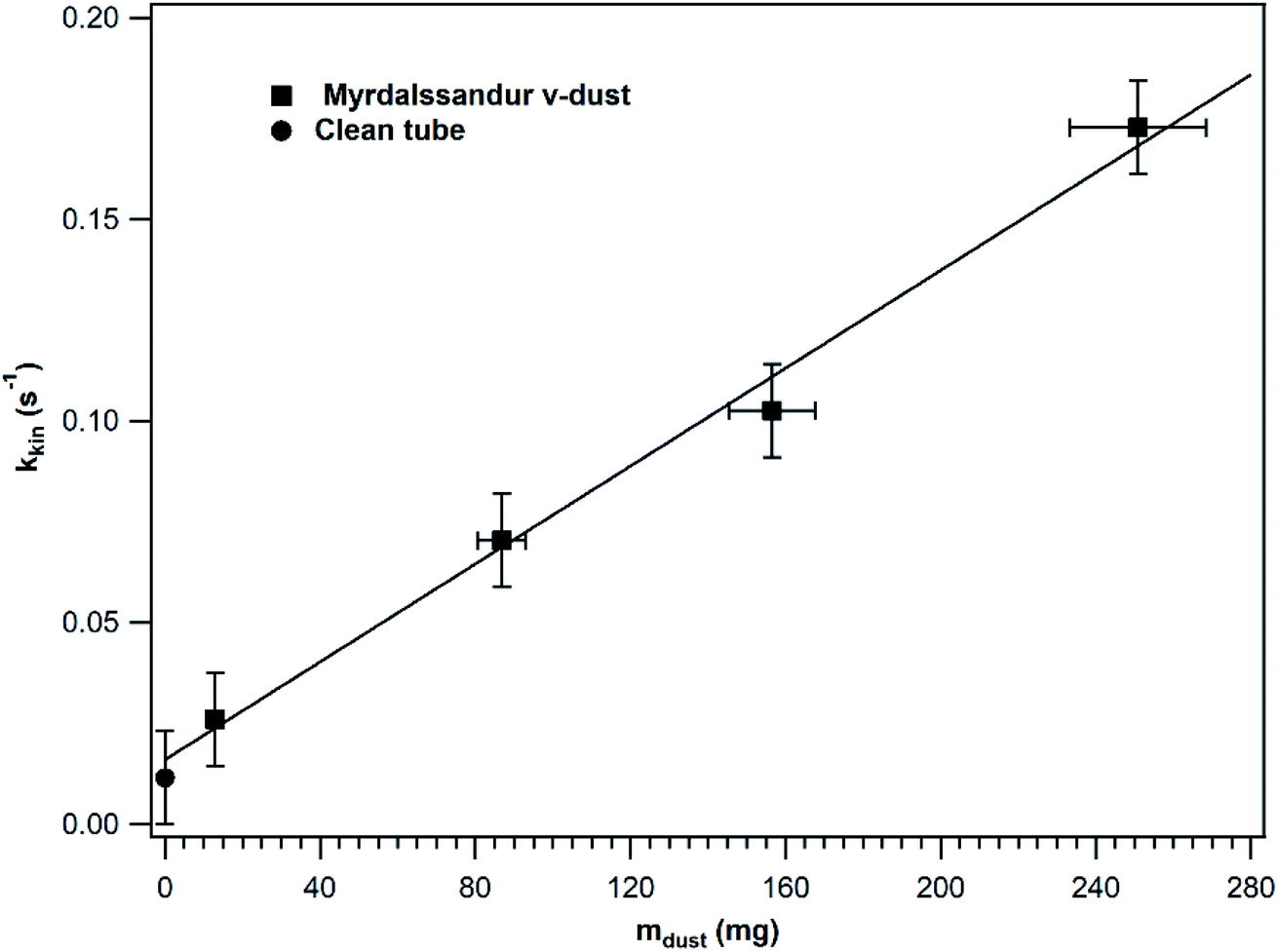 | ||
| Fig. 3 k kin values (in s−1) measured under steady-state conditions as a function of the mass of Mýrdalssandur v-dust deposited (squares). The experiments were performed at 296 K under dry and dark conditions, with [SO2] fixed at 75 ppb. For comparison, the kkin value estimated on a clean tube is also presented (circle). The solid line is the linear fit of the experimental results (excluding the value estimated for a clean tube). The error on the values of kkin corresponds to the precision of the measurement and includes estimated uncertainties. The error bars on mdust reflect the uncertainties on the mass of v-dust deposited, and mainly originate from the mass lost (<7%) during the experiment, as measured before and after the experiment. | ||
Regarding the steady-state uptake coefficient, considering the precision of the signal (1 ppb) and its propagation to kobs, the determination of SBET and all relevant uncertainties on the gas flow measurement, temperature, mass weight, and length of the exposed dust coating, the overall uncertainty is ∼8%. The total error calculated for the γ values is estimated to be ∼35%, and mainly stems from the precision of the SO2 concentration measured at steady-state and after isolation of the surface. In all experiments, a conservative limit of 40% uncertainty in γSS,BET is given.
3. Results and discussion
3.1. Initial number of SO2 molecules taken up on v-dust samples
| V-dust | Initial number of SO2 molecules taken up, NS (in 1013 molecules cm−2) | ||
|---|---|---|---|
| N S,dark | N S,light | N S,light/NS,dark | |
| Mýrdalssandur | 3.0 ± 0.9 | 9.3 ± 3.0 | 3.1 ± 0.31 |
| Dyngjusandur | 2.1 ± 0.6 | 3.6 ± 1.0 | 1.7 ± 0.17 |
| Hagavatn | 1.2 ± 0.4 | 2.0 ± 0.6 | 1.7 ± 0.17 |
| Maelifellssandur | 0.96 ± 0.29 | 3.7 ± 1.1 | 4.0 ± 0.40 |
Under dark conditions, NS values determined for the different v-dust samples range from 0.96 to 2.5 × 1013 molecules cm−2. The variation observed may be attributed to differences in the physical (e.g., morphology, crystallinity, hygroscopicity) and/or chemical (e.g., surface elemental composition) properties of the particles. A schematic representation of the general reaction mechanism of SO2 at the surface of v-dust reported in a previous study24 is given below:
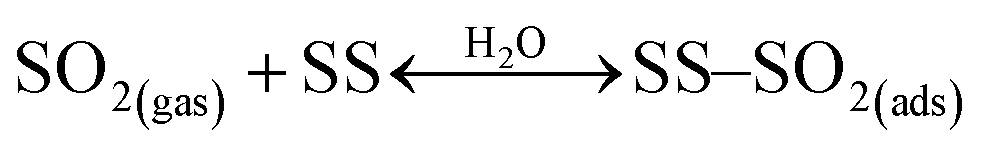 | (R1) |
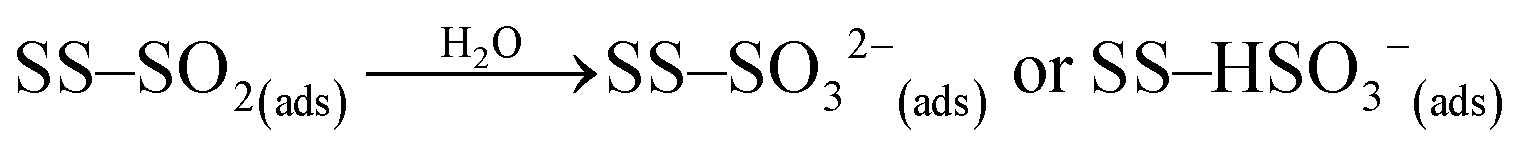 | (R2) |
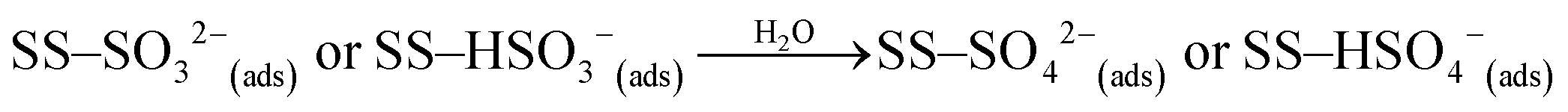 | (R3) |
Under UV irradiation, NS increases by a factor ranging from 1.7 to 3.8 relative to dark conditions, depending on the type of v-dust. The enhanced removal of SO2 in the presence of UV photons may be attributed to photo-induced processes occurring at the v-dust/air interface. In our experiments, we cannot discriminate photon activation of v-dust from photon activation of surface groups or of adsorbed species. Therefore, several points are useful to highlight in considering the nature of this photo-induction:
(i) It is well established that reactive species (i.e., hydroxyl radicals, ˙OH, hydroperoxyl radicals, ˙O2H, or superoxide radicals, O2˙−, etc.) form on the surface of mineral dusts containing Ti and Fe under UV/visible light irradiation in the presence of H2O.38–40 Therefore, these reactive species likely formed on the surface of v-dust, contributing to SO2 uptake.
(ii) Under our experimental conditions, no photolysis of SO2 has been observed, since it requires photons with wavelengths lower than 220 nm,33 which are absent from the emission spectrum of our lamps. Direct photochemistry of SO42− species does not occur either, since SO42− does not absorb photons between 310 and 400 nm.41
(iii) Although the absorption of photons by SO32− in the 310–410 nm range remains very low, it cannot be neglected. Thus, irradiation of v-dust may lead to SO32− surface photochemistry, forming low amounts of ˙SO3−(ads),42 which in turn, can lead to H2SO4 formation in the presence of H2O.43
Thus, irradiation of the surface of v-dust may create new active surface sites that promote SO2 uptake (reaction (R1)) and/or provide new reactive pathways enhancing the oxidation of intermediate species in the mechanism of SO2 transformation (reactions (R2) and (R3)). However, monitoring only the gas phase, as we do in these experiments, does not enable assessment of the contribution of each process to the overall consumption of SO2 gas.
In an attempt to evaluate further the observed trends, we investigate whether the amplification factor, NS,light/NS,dark is correlated with the surface elemental composition of the v-dust samples. It is well established that mineral oxides containing Ti are photoactivated under UV-A light irradiation, and create reactive surface species.38–40,44 Iron-bearing minerals are also photoactive, but mainly under visible light. In this context, we display in Fig. 4 the NS,light/NS,dark ratio plotted as a function of the surface Ti concentration (Fig. 4a), and of the surface Ti/Si ratio (Fig. 4b). We observe a linear increase of NS,light/NS,dark with both surface Ti and Ti/Si. Trends observed with elemental Fe are not straightforward, and are therefore not discussed. We propose that the photo-enhanced uptake of SO2 is related to the presence of Ti at the surface of v-dust. To parametrize these results (see also Section 3.1.4.), we choose the surface Ti content relative to the most abundant element in v-dust (i.e. the surface Ti/Si ratio) to facilitate comparison across samples of different composition. Therefore, based on Fig. 4b, we propose an empirical parametrization of the amplification factor (4) relevant under our experimental conditions and for the v-dust samples studied:
| NS,light/NS,dark = 65.1 × (Ti/Si) | (4) |
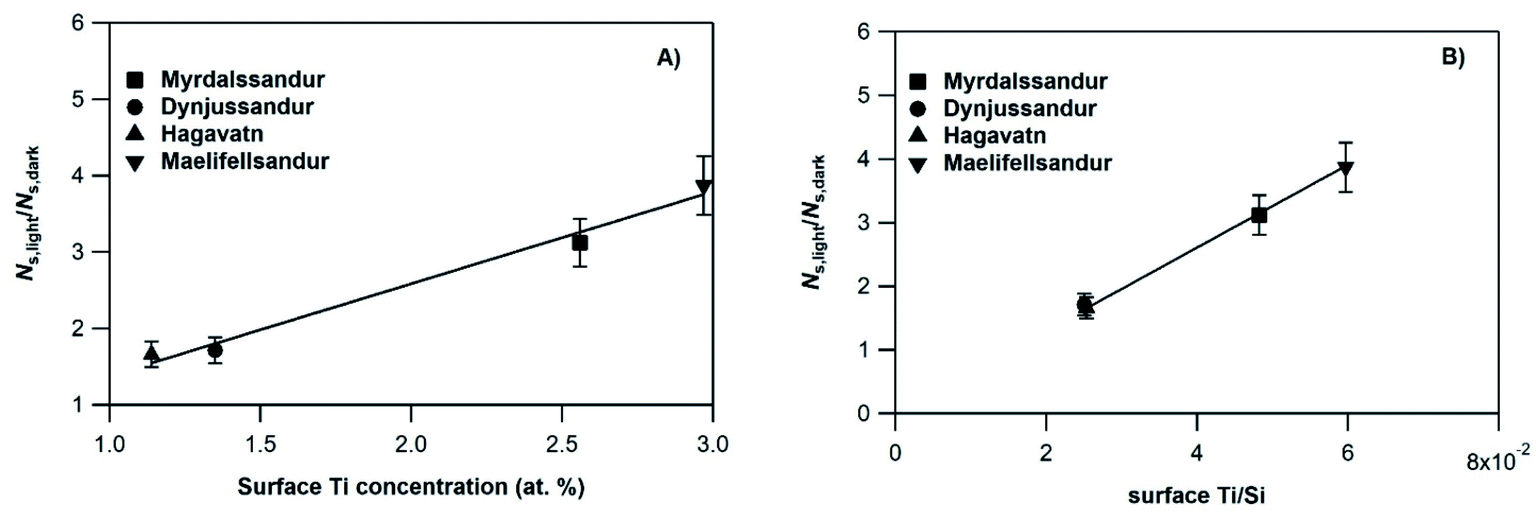 | ||
| Fig. 4 Amplification by light of the initial number of SO2 molecules taken up on the v-dust samples, NS,light/NS,dark, as a function of: (A) surface Ti concentration, and (B) surface Ti/Si concentration ratio, as measured by XPS. The experimental conditions were: [SO2]0 = 75 ppb, RH = 30% and T = 296 K, in the dark, and JNO2 = 4.5 × 10−3 s−1 during UV irradiation experiments. The solid lines show linear fits of the experimental results, with a correlation coefficient R2 > 0.97 for Ti surface concentration and R2 > 0.99 for the Ti/Si ratio. | ||
The dependence of the amplification factor on surface Ti/Si content suggests that active surface species (e.g. ˙OH, ˙O2H, O2˙−) may be formed on the surface of v-dust (point (i) above), enhancing SO2 consumption. Nevertheless, considering the uptake profiles in Fig. S1† (and illustrated in Fig. 2), and the gradual saturation/deactivation of the surface with time, the process cannot be characterized as catalytic. In addition, other photo-induced processes, such as photolysis of surface intermediate species (e.g. SO32−; point (iii) above), should not be discarded.
In subsequent experiments to evaluate the impact of other relevant atmospheric parameters (photon flux and RH) on the photo-promoted removal of SO2, we choose Mýrdalssandur v-dust as a model sample. The major criterion for this selection is the high uptake capacity (high NS values) of the sample under both dark and UV light conditions, coupled with a large amplification factor under light with respect to dark conditions (see Table 2).
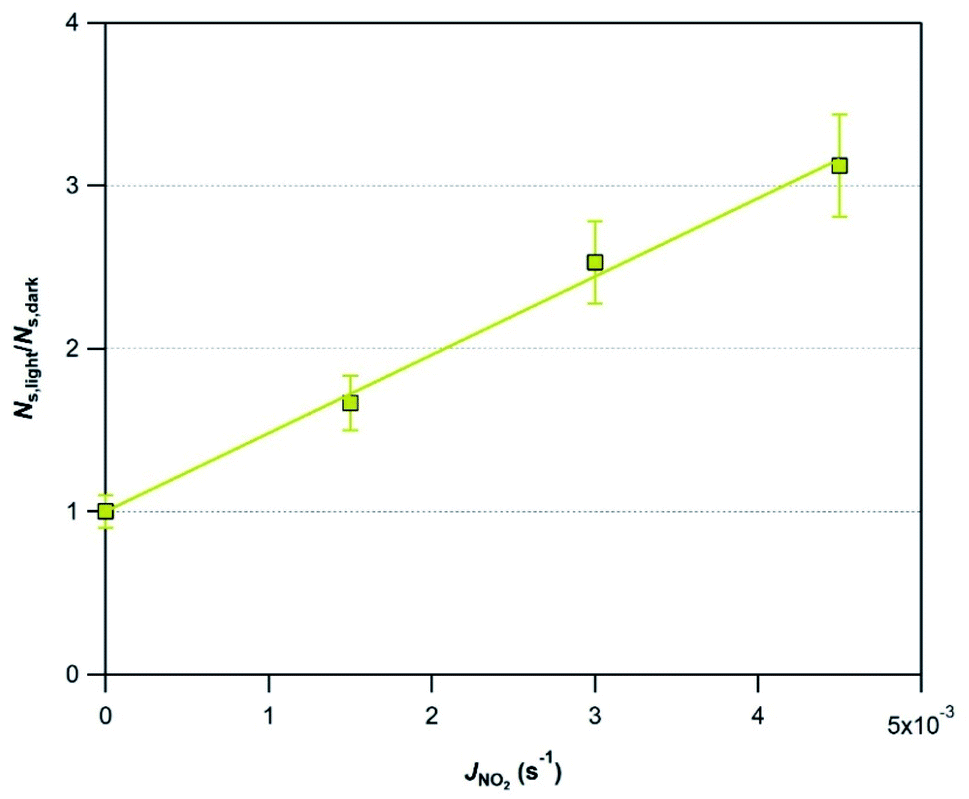 | ||
| Fig. 5 Amplification by light of the initial number of SO2 molecules taken up on Mýrdalssandur v-dust, NS,light/NS,dark, as a function of the photon flux (JNO2 = 0–4.5 × 10−3 s−1). Mýrdalssandur v-dust was exposed to 75 ppb of SO2 at RH = 30% and T = 296 K. | ||
Photo-induced removal of atmospheric species has been highlighted in previous studies dealing with the degradation of pollutants on the surface of mineral dust proxies,34,45,46 and on v-dusts,26 but an accurate description of the heterogeneous physicochemical pathway involved is still lacking. The nature of the photo-induced surface processes can involve complementary surface sites activated by light, or alternatively photo-enhanced surface reactions promoting the transformation of SO2 into surface species such as SO32−/HSO3− or SO42−/HSO4− (reactions (R1)–(R3)).
In all cases, based on the breakthrough profiles recorded under different irradiation intensities, a gradual recovery of SO2 concentration with time is observed (see Fig. 2). In spite of the enhancement brought by UV light, SO2 uptake remains a transient process, attesting to the aging of the surface and its deactivation irrespective of the magnitude of the photon enhancement observed here.
Eqn (5) is proposed as an empirical description of the photo-enhanced consumption of SO2 under the current experimental conditions:
| Ns,light/Ns,dark = 1 + 480.5 × JNO2 | (5) |
To evaluate the impact of RH on the uptake of SO2, we exposed Mýrdalssandur v-dust to a fixed concentration of SO2 under both dark conditions and UV irradiation (JNO2 = 4.5 × 10−3 s−1) at 296 K, and varying RH in the 0.1–72% range. The results are presented in Table 3. NS,dark increases from 0.1 to 30% RH before reaching a saturation regime and remaining almost constant. This observation points to a two-fold role played by water molecules in the removal of SO2. As RH increases from 0.1 to 30%, the consumption of SO2 is enhanced by the formation of OH groups on the surface of v-dust. Surface OH groups assist the transformation of SO2 to surface SO3− and SO42− species, as evidenced by Urupina et al.24 Above 30% RH, i.e., beyond the water monolayer formation on dust,52,53 we suggest a competition between two processes: the formation of OH groups promoting the removal of SO2, and the blocking of active surface sites by molecularly adsorbed water. The antagonistic nature of the two processes may account for the saturation of NS,dark.
| RH (%) | Initial number of SO2 molecules taken up, NS (in 1013 molecules cm−2) | ||
|---|---|---|---|
| N S,dark | N S,light | N S,light/NS,dark | |
| 0.1 | 0.39 ± 0.12 | — | — |
| 10 | 2.3 ± 0.7 | 4.0 ± 1.2 | 1.7 ± 0.2 |
| 30 | 3.0 ± 0.9 | 9.5 ± 2.8 | 3.2 ± 0.3 |
| 50 | 3.3 ± 1.0 | 17 ± 5.0 | 5.2 ± 0.5 |
| 72 | 3.6 ± 1.1 | 23 ± 7.0 | 6.4 ± 0.6 |
In contrast, when the surface is irradiated, NS,light increases over the whole range of RH values and saturation does not occur. The blank experiments (in the absence of dust), carried out under UV light, evidence a negligible removal of SO2 at RH = 30%. During these experiments, multilayers of water molecules are formed on the walls of the flow-tube reactor. The absence of any contribution from water-covered surfaces to SO2 uptake indicates that solvation reactions at the adsorbed water/air interface are negligible. The linear increase of NS,light with RH cannot be directly attributed to RH, but to photo-induced processes. These processes may be related to Ti-surface sites activation, leading to the formation of reactive surface species,38,39 or to surface photochemistry involving adsorbed species and promoting the oxidation mechanism of SO2.40,54,55 These observations support the hypothesis made in Section 3.1.1.; the increase of SO2 removal observed under light for various values of RH is due to the generation of new surface sites and/or to the activation of new reaction pathways promoting the transformation of SO2 or of intermediate species to final products (reactions (R1)–(R3)).
To quantify the enhancement of NS in the presence of light, in Fig. 6 the amplification factor is plotted as a function of RH. The results are linearly fitted with eqn (6):
| NS,light/NS,dark = 1 + (7.76 × 10−2) × RH | (6) |
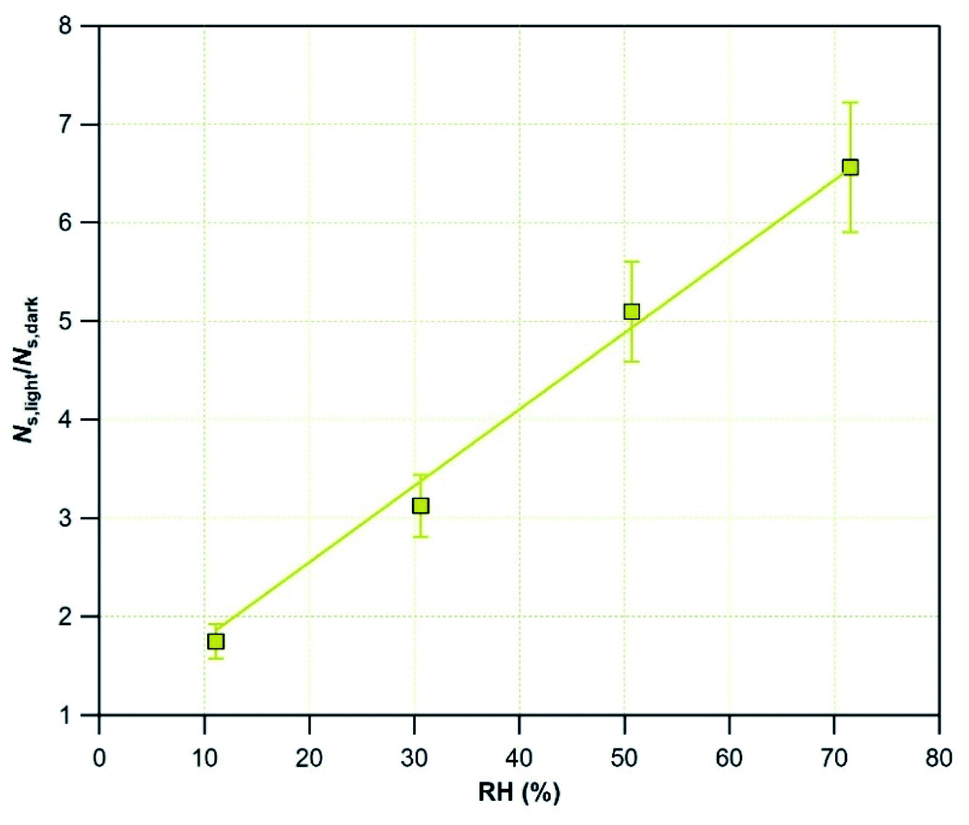 | ||
| Fig. 6 The amplification factor, NS,light/NS,dark, plotted as a function of RH for Mýrdalssandur v-dust exposed to 75 ppb of SO2 at T = 296 K. The yellow line is the linear fit of the results. | ||
Based on the experimental approach followed, including (i) screening the v-dusts to assess their capacity for SO2 uptake and (ii) tuning individual atmospheric parameters (UV light irradiation and RH) and evaluating their impacts on SO2 removal, the experimental results for Mýrdalssandur v-dust are parametrized with eqn (4)–(6). We propose a general eqn (7) describing the correlation between the amplification factor with the chemical composition of the sample, the photon flux, and RH:
| NS,light/NS,dark = 6.1 × (1 + 7.76 × 10−2 × RH) × (1 + 480.5 × JNO2) × (Ti/Si) | (7) |
Despite the fact that we did not perform a detailed RH dependence for all types of v-dusts, eqn (7) succeeds in describing the expected amplification factors for the other samples within 10% uncertainty. This type of empirical equation is useful in modeling because relevant atmospheric parameters are gathered to give a global description of the photo-enhanced removal of SO2. It should be stressed that the proposed parametrization concerns the results of the current study performed under specific experimental conditions of RH and photon flux, using a limited range of v-dust compositions, and requires validation with a more experimental conditions and samples before a general application can be recommended.
The values of NS,dark measured in our study range between 1 and 3 × 1013 molecules cm−2 at RH = 30%. They are in good agreement with Maters et al.,23 who report values of NS,dark in the 1011–1013 molecules cm−2 range for SO2 uptake by volcanic ash and glass powders. Our values are also close to those measured on Saharan dusts at 27% RH, but on a much shorter timescale and under different experimental conditions (pressure, absence of oxygen, etc.).56
Sulfur dioxide removed from the gas phase is expected to be converted into SO32− and SO42− species. Under dark conditions, several authors have reported different pathways of SO2 transformation (see Urupina et al.24 and references therein). Under UV light, the transformation of SO2 has been studied in a simulation chamber on airborne Arizona Test Dust (ATD) particles. Nevertheless, these experiments were carried out in the presence of NOx and O3, generating ˙OH radicals in the gas phase, which further oxidize SO2 to SO42−. Even in the set of experiments carried out in the absence of NOx, ˙OH radicals were always present (due to outgassing of NOx and NOy from the wall of the reactor).57,58 Therefore, no direct comparison to our data can be made.
3.2. Steady-state uptake of SO2 on v-dust samples
| V-dust | Steady-state uptake coefficients, γSS,BET (10−8) | ||
|---|---|---|---|
| γ SS,BET,dark | γ SS,BET,light | γ SS,BET,light/γSS,BET,dark | |
| Mýrdalssandur | 6.2 ± 2.5 | 7.1 ± 2.9 | 1.1 |
| Dyngjusandur | 1.6 ± 0.6 | 7.3 ± 2.9 | 4.6 |
| Hagavatn | 0.66 ± 0.26 | 2.6 ± 1.0 | 3.9 |
| Maelifellssandur | 3.2 ± 1.3 | 6.6 ± 2.7 | 2.1 |
All v-dust samples exhibit a photo-enhanced uptake of SO2 under initial and steady-state regimes; they are nonetheless characterized by contrasted amplification factors. Interestingly, Mýrdalssandur and Maelifellssandur samples have high NS,light/NS,dark ratios, but exhibit the lowest γSS,BET,light/γSS,BET,dark ratios, whereas Hagavatn and Dyngjusandur samples exhibit lower NS,light/NS,dark values, but higher γSS,BET,light/γSS,BET,dark values. For Mýrdalssandur and Maelifellssandur samples, the number and/or the reactivity of the surface sites at the beginning of UV irradiation is significantly higher than for Dyngjusandur and Hagavatn, leading to an important light-induced consumption of gas-phase SO2, as reflected by the values of NS,light/NS,dark (Table 2). This is consistent with Mýrdalssandur and Maelifellssandur samples having the highest surface Ti/Si ratios (Table 1). However, the surface sites of Mýrdalssandur and Maelifellssandur dusts appear to be deactivated/exhausted faster than on the less reactive Dyngjusandur and Hagavatn samples, probably due to higher concentrations of surface products on the former materials, leading to lower γSS,BET,light/γSS,BET,dark values. Considering the inverse correlation between initial and steady-state amplification factors, we suggest that the larger the number of surface sites involved in initial uptake, the lower the magnitude of the steady-state uptake coefficient. The initial uptake, which corresponds to NS, seems to act as an ageing process on surface reactivity, hence lowering the uptake process at steady-state, characterized by γSS,BET.
Mýrdalssandur v-dust was used as a model sample to further evaluate the impact of atmospheric conditions on γSS,BET (see Section 3.1.1.). Considering the relatively low increase in γSS,BET after irradiation of the v-dust surface with the highest photon flux (i.e., JNO2 = 4.5 × 10−3 s−1), experiments were not performed with lower JNO2. However, additional experiments were conducted to determine the uptake coefficients as a function of RH under dark and light irradiation (JNO2 = 4.5 × 10−3 s−1).
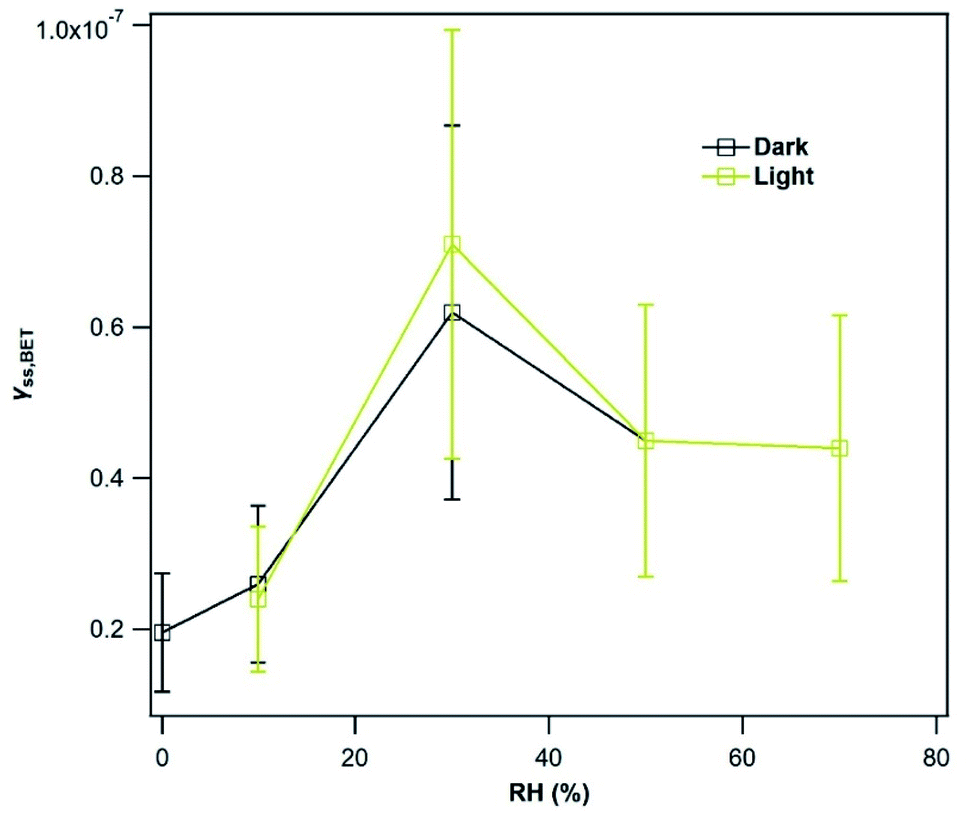 | ||
| Fig. 7 Steady-state BET uptake coefficient, γSS,BET, as a function of RH during exposure of Mýrdalssandur v-dust to 75 ppb of SO2 in the dark (black squares) and in the presence of UV light (JNO2 = 4.5 × 10−3 s−1; yellow squares) at T = 296 K. Datapoints in the dark at 50% and 70% RH overlap with those under light. | ||
An increase of γSS,BET by Mýrdalssandur v-dust is observed as RH increases from 0.1 to 30%. We suggest that this variation is related to the formation of a water monolayer at the surface of Mýrdalssandur v-dust. As reported by Urupina et al.,24 the increase of the surface water content promotes the transformation of SO2 into intermediate and final products (reactions (R1)–(R3)), and enhances the uptake of SO2. The small reduction of the uptake coefficient as RH increases from 30 to 50% could be due to changes in the equilibrium by two effects: the enhanced removal/transformation of SO2 on hydroxylated surface sites, and the competition of SO2 with H2O molecules for the same adsorption sites. Above 50%, the uptake coefficient seems to stabilize and become independent of RH. Nevertheless, we do not know what would be the behavior of SO2 uptake for RH greater than 72%, where an exponential increase in the surface water concentration is anticipated based on the adsorption isotherms reported by Joshi et al.52 Under UV light, despite the increasing initial consumption of SO2 discussed previously, the formation of low-volatility final surface products deactivates the surface, and after 36 hours of exposure to light, γSS,BET,light values are similar to those recorded in the dark.
The variations in the SO2 uptake coefficient on the v-dusts with RH are qualitatively similar to those reported for two types of Chinese mineral dust, although the authors of these works observe a change in uptake above RH = 40%.21 The difference in behavior with respect to RH may be due to the differing chemical and mineralogical composition of the dusts under investigation. Huang et al.22 have studied the RH-dependence of SO2 uptake on Asian Mineral Dust (AMD), ATD and Tengger Desert Dust (TDD); they observe a decrease in SO2 uptake with increasing RH on AMD, but the opposite on ATD and TDD. Moreover, the data presented by Huang et al. show that on each type of dust investigated, a change in the behavior of SO2 uptake occurs around 30% RH, in agreement with our observations for the v-dust samples. Consistently, the formation of a water monolayer is observed at 28 ± 2% RH on the 40–80 μm fraction of ATD.60 Li et al.61 investigated the uptake of SO2 by CaCO3 in the presence of O3, and did not observe a dependence of the initial SO2 uptake coefficient on RH. However, they report an increase of γSS,BET from 2 × 10−9 at 5% RH to 2 × 10−8 at 60% RH, although the variation in SO2 uptake between these RH values is not reported. The latter value of 2 × 10−8 at 60% RH is consistent with our value of roughly 4.5 × 10−8 between 50% and 72% RH.
4. Concluding remarks
We investigated the heterogeneous interaction of SO2 with four Icelandic v-dust samples under atmospherically relevant conditions, with and without UV light. The uptake of SO2 on the v-dusts is time-dependent both in the dark and under UV irradiation. In particular, a substantial quantity of SO2 is taken up initially in a transient process, with NS values measured in the dark on the order of 1013 molecules cm−2 using the SBET surface. After several hours of exposure to SO2, the v-dust surface saturates/deactivates, probably due to the formation of low volatility surface products such as SO32− and SO42−. Following SO2 uptake in the dark, UV irradiation of the v-dust surfaces promotes an additional consumption of SO2, with NS values measured under UV light on the order of 1014 molecules cm−2 using SBET. The photo-enhanced removal of SO2, characterized by NS,light/NS,dark, is correlated with the surface Ti/Si composition, the UV irradiance intensity and RH. Based on our experimental observations, a general equation is proposed to extrapolate the amplification of SO2 removal by a photo-induced heterogeneous process as a function of the atmospheric parameters studied. The proposed parametrization concerns the results of the current study, and requires validation with more experiments and samples before general application. We attribute the photo-enhanced heterogeneous uptake of SO2 to the formation of active surface species on Ti-containing surface sites and/or to photochemical reactions of intermediate surface groups. Both processes promote the uptake and transformation of SO2 into final surface products such as SO42− and HSO4− species. However, the clarification of the reaction mechanism requires more studies, preferably through real-time monitoring of surface species both in the dark and under UV light.This work has implications for atmospheric models aiming to simulate the removal of SO2 by resuspended volcanic material, and to elucidate the importance of heterogeneous processes both under nighttime and daytime tropospheric conditions. Based on our results, we anticipate that the impact of SO2 heterogeneous chemistry is more important on irradiated volcanic particles. Indeed, under tropospheric conditions, the photo-enhanced removal of SO2 evidenced in the present study is up to seven times higher than under dark conditions. Climate or chemical transport models should therefore include diurnal profiles of SO2 uptake by volcanic particles to improve representation of the sulfur budget, and of its evolution in the atmosphere following an eruption. However, to the best of our knowledge there are no modeling studies incorporating the heterogeneous removal of SO2 on solid particles of volcanic origin in the troposphere.
In a recent study, Zhu et al.62 performed aerosol-climate model simulations to estimate the impact of the heterogeneous removal of SO2 on volcanic ash particles in the stratosphere. Specifically, they compared the results of model simulations which included the uptake of SO2 on ash with satellite and aircraft observations following the 2014 Kelud eruption. The best match of the model results to the observational data was achieved using γ and NS values (3 × 10−3 and 3 × 1016 molecules cm−2, respectively), normalized to geometric surface area of the ash particles, of the same order of magnitude as those measured in laboratory experiments in the absence of UV light and at room temperature. However, note that SO2 uptake normalized to geometric surface area represents an upper limit and may exceed by more than one order of magnitude the SO2 uptake normalized to the true surface area calculated from SBET.20 The resulting model simulations of Zhu et al.62 reveal for the first time that heterogeneous reaction of SO2 with volcanic ash could remove up to 43% more sulfur from the stratosphere within two months of the eruption. However, it should be mentioned that these authors did not include any daytime photo-induced loss of SO2 on volcanic ash, a process that could be important under stratospheric conditions where short wavelength radiation is present (UV-A and UV-B), as highlighted by the present study. The absence of these photo-processes in their model might contribute to the relatively high γ and NS values needed by Zhu et al.62 to match simulations with observations.
Building on recent advances in laboratory and model studies, there is still much to learn regarding controls on SO2 uptake by volcanic ash/dust, the underlying reaction mechanisms, and the potential atmospheric and climate impacts. Laboratory temperature-dependent studies are necessary to provide uptake coefficient and surface coverage values under relevant tropospheric/stratospheric conditions. The inclusion of diurnal heterogeneous removal of SO2 in both tropospheric and stratospheric model simulations is also recommended to better capture the photo-induced variation in heterogeneous SO2 loss, evidenced and characterized in this work.
Funding sources
This work was achieved in the frame of Labex CaPPA, funded by ANR through the PIA under contract ANR-11-LABX-0005-01, and CPER CLIMIBIO project, both funded by the Hauts-de-France Regional Council and the European Regional Development Fund (ERDF).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Mr V. Gaudion and Dr M. Zeineddine (CERI EE, IMT Nord Europe) for their assistance in the lab. Mr E. Tison (CERI EE, IMT Nord Europe) is gratefully acknowledged for providing the SO2 analyzer, and Dr Pavla Dagsson-Waldhauserova (University of Iceland) for providing the volcanic dust samples. We thank P. Eloy for performing the XPS analysis. JL acknowledges invaluable discussions with Dr D. Marchione (Università degli Studi di Perugia, Italy) and Dr A. Rosu-Finsen (University College London, UK). This work was achieved in the frame of Labex CaPPA, funded by ANR through the PIA under contract ANR-11-LABX-0005-01, and CPER CLIMIBIO project, both funded by the Hauts-de-France Regional Council and the European Regional Development Fund (ERDF). JL acknowledges support from the Labex CaPPA, CPER CLIMIBIO, the Hauts-de-France Regional Council, and IMT Nord Europe (project IRAPAQ) for post-doctoral fellowships. EM acknowledges the Leverhulme Trust and Isaac Newton Trust for an Early Career Fellowship.References
- A. J. Durant, C. Bonadonna and C. J. Horwell, Atmospheric and Environmental Impacts of Volcanic Particulates, Elements, 2010, 6, 235–240 CrossRef CAS.
- M. O. Andreae, in World Survey of Climatology, ed. A. Henderson-Sellers, Elsevier, 1995, vol. 16, pp. 347–398 Search PubMed.
- T. Thordarson and Á. Höskuldsson, Postglacial volcanism in Iceland, Jokull, 2008, 58, 197–228 Search PubMed.
- A. Stohl, A. J. Prata, S. Eckhardt, L. Clarisse, A. Durant, S. Henne, N. I. Kristiansen, A. Minikin, U. Schumann, P. Seibert, K. Stebel, H. E. Thomas, T. Thorsteinsson, K. Tørseth and B. Weinzierl, Determination of time- and height-resolved volcanic ash emissions and their use for quantitative ash dispersion modeling: the 2010 Eyjafjallajökull eruption, Atmos. Chem. Phys., 2011, 11, 4333–4351 CrossRef CAS.
- O. Arnalds, P. Dagsson-Waldhauserova and H. Olafsson, The Icelandic volcanic aeolian environment: Processes and impacts — A review, Aeolian Res., 2016, 20, 176–195 CrossRef.
- F. Beckett, A. Kylling, G. Sigurðardóttir, S. von Löwis and C. Witham, Quantifying the mass loading of particles in an ash cloud remobilized from tephra deposits on Iceland, Atmos. Chem. Phys., 2017, 17, 4401–4418 CrossRef CAS.
- D. Đorđević, I. Tošić, S. Sakan, S. Petrović, J. Đuričić-Milanković, D. C. Finger and P. Dagsson-Waldhauserová, Can Volcanic Dust Suspended From Surface Soil and Deserts of Iceland Be Transferred to Central Balkan Similarly to African Dust (Sahara)?, Front. Earth Sci., 2019, 7, 142 CrossRef.
- A. Yoshida, N. Moteki, S. Ohata, T. Mori, R. Tada, P. Dagsson-Waldhauserová and Y. Kondo, Detection of light-absorbing iron oxide particles using a modified single-particle soot photometer, Aerosol Sci. Technol., 2016, 50, 1–4 CrossRef CAS.
- E. J. Highwood and D. S. Stevenson, Atmospheric impact of the 1783–1784 Laki Eruption: Part II Climatic effect of sulphate aerosol, Atmos. Chem. Phys., 2003, 3, 1177–1189 CrossRef CAS.
- D. S. Stevenson, C. E. Johnson, E. J. Highwood, V. Gauci, W. J. Collins and R. G. Derwent, Atmospheric impact of the 1783–1784 Laki eruption: Part I Chemistry modelling, Atmos. Chem. Phys., 2003, 3, 487–507 CrossRef CAS.
- J.-P. Vernier, T. D. Fairlie, T. Deshler, M. Natarajan, T. Knepp, K. Foster, F. G. Wienhold, K. M. Bedka, L. Thomason and C. Trepte, In situ and space-based observations of the Kelud volcanic plume: The persistence of ash in the lower stratosphere, J. Geophys. Res.: Atmos., 2016, 121, 11104–111118 Search PubMed.
- A. Vogel, S. Diplas, A. J. Durant, A. S. Azar, M. F. Sunding, W. I. Rose, A. Sytchkova, C. Bonadonna, K. Krüger and A. Stohl, Reference data set of volcanic ash physicochemical and optical properties, J. Geophys. Res.: Atmos., 2017, 122, 9485–9514 CrossRef CAS.
- C. Oppenheimer, P. Francis, M. Burton, A. J. H. Maciejewski and L. Boardman, Remote measurement of volcanic gases by Fourier transform infrared spectroscopy, Appl. Phys. B, 1998, 67, 505–515 CrossRef CAS.
- D. E. Hunton, A. A. Viggiano, T. M. Miller, J. O. Ballenthin, J. M. Reeves, J. C. Wilson, S.-H. Lee, B. E. Anderson, W. H. Brune, H. Harder, J. B. Simpas and N. Oskarsson, In-situ aircraft observations of the 2000 Mt. Hekla volcanic cloud: Composition and chemical evolution in the Arctic lower stratosphere, J. Volcanol. Geotherm. Res., 2005, 145, 23–34 CrossRef CAS.
- K. P. Heue, C. A. M. Brenninkmeijer, A. K. Baker, A. Rauthe-Schöch, D. Walter, T. Wagner, C. Hörmann, H. Sihler, B. Dix, U. Frieß, U. Platt, B. G. Martinsson, P. F. J. van Velthoven, A. Zahn and R. Ebinghaus, SO2 and BrO observation in the plume of the Eyjafjallajökull volcano 2010: CARIBIC and GOME-2 retrievals, Atmos. Chem. Phys., 2011, 11, 2973–2989 CrossRef CAS.
- M. Höpfner, C. D. Boone, B. Funke, N. Glatthor, U. Grabowski, A. Günther, S. Kellmann, M. Kiefer, A. Linden, S. Lossow, H. C. Pumphrey, W. G. Read, A. Roiger, G. Stiller, H. Schlager, T. von Clarmann and K. Wissmüller, Sulfur dioxide (SO2) from MIPAS in the upper troposphere and lower stratosphere 2002–2012, Atmos. Chem. Phys., 2015, 15, 7017–7037 CrossRef.
- H. K. Carlsen, U. Valdimarsdóttir, H. Briem, F. Dominici, R. G. Finnbjornsdottir, T. Jóhannsson, T. Aspelund, T. Gislason and T. Gudnason, Severe volcanic SO(2) exposure and respiratory morbidity in the Icelandic population - a register study, Environ. Health: Global Access Sci. Source, 2021, 20, 23 CrossRef CAS PubMed.
- H. K. Carlsen, E. Ilyinskaya, P. J. Baxter, A. Schmidt, T. Thorsteinsson, M. A. Pfeffer, S. Barsotti, F. Dominici, R. G. Finnbjornsdottir, T. Jóhannsson, T. Aspelund, T. Gislason, U. Valdimarsdóttir, H. Briem and T. Gudnason, Increased respiratory morbidity associated with exposure to a mature volcanic plume from a large Icelandic fissure eruption, Nat. Commun., 2021, 12, 2161 CrossRef CAS PubMed.
- J. Penner, Three ways through the soot, sulfates and dust, Nature, 2019, 570, 158–159 CrossRef CAS PubMed.
- J. N. Crowley, M. Ammann, R. A. Cox, R. G. Hynes, M. E. Jenkin, A. Mellouki, M. J. Rossi, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V – heterogeneous reactions on solid substrates, Atmos. Chem. Phys., 2010, 10, 9059–9223 CrossRef CAS.
- L. Zhou, W. Wang, Y. Gai and M. Ge, Knudsen cell and smog chamber study of the heterogeneous uptake of sulfur dioxide on Chinese mineral dust, J. Environ. Sci., 2014, 26, 2423–2433 CrossRef PubMed.
- L. Huang, Y. Zhao, H. Li and Z. Chen, Kinetics of Heterogeneous Reaction of Sulfur Dioxide on Authentic Mineral Dust: Effects of Relative Humidity and Hydrogen Peroxide, Environ. Sci. Technol., 2015, 49, 10797–10805 CrossRef CAS PubMed.
- E. C. Maters, P. Delmelle, M. J. Rossi and P. M. Ayris, Reactive Uptake of Sulfur Dioxide and Ozone on Volcanic Glass and Ash at Ambient Temperature, J. Geophys. Res.: Atmos., 2017, 122, 10077–10088 CrossRef CAS.
- D. Urupina, J. Lasne, M. N. Romanias, V. Thiery, P. Dagsson-Waldhauserova and F. Thevenet, Uptake and surface chemistry of SO2 on natural volcanic dusts, Atmos. Environ., 2019, 217, 116942 CrossRef CAS.
- D. Urupina, M. N. Romanias and F. Thevenet, How Relevant Is It to Use Mineral Proxies to Mimic the Atmospheric Reactivity of Natural Dust Samples? A Reactivity Study Using SO2 as Probe Molecule, Minerals, 2021, 11, 282 CrossRef CAS.
- M. N. Romanias, Y. Ren, B. Grosselin, V. Daële, A. Mellouki, P. Dagsson-Waldhauserova and F. Thevenet, Reactive uptake of NO2 on volcanic particles: A possible source of HONO in the atmosphere, J. Environ. Sci., 2020, 95, 155–164 CrossRef PubMed.
- M. J. Genet, C. C. Dupont-Gillain and P. G. Rouxhet, XPS analysis of biosystems and biomaterials, J. Environ. Sci., 2008, 177–309 Search PubMed.
- J. Lasne, M. N. Romanias and F. Thevenet, Ozone Uptake by Clay Dusts under Environmental Conditions, ACS Earth Space Chem., 2018, 2, 904–914 CrossRef CAS.
- J. C. Barnard, E. G. Chapman, J. D. Fast, J. R. Schmelzer, J. R. Slusser and R. E. Shetter, An evaluation of the FAST-J photolysis algorithm for predicting nitrogen dioxide photolysis rates under clear and cloudy sky conditions, Atmos. Environ., 2004, 38, 3393–3403 CrossRef CAS.
- B. Bohn, F. Rohrer, T. Brauers and A. Wahner, Actinometric measurements of NO2 photolysis frequencies in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 2005, 5, 493–503 CrossRef CAS.
- C. Topaloglou, S. Kazadzis, A. F. Bais, M. Blumthaler, B. Schallhart and D. Balis, NO2 and HCHO photolysis frequencies from irradiance measurements in Thessaloniki, Greece, Atmos. Chem. Phys., 2005, 5, 1645–1653 CrossRef CAS.
- T. Huthwelker, M. Ammann and T. Peter, The Uptake of Acidic Gases on Ice, Chem. Rev., 2006, 106, 1375–1444 CrossRef CAS PubMed.
- M. T. C. Martins-Costa, J. M. Anglada, J. S. Francisco and M. F. Ruiz-López, Photochemistry of SO2 at the Air–Water Interface: A Source of OH and HOSO Radicals, J. Am. Chem. Soc., 2018, 140, 12341–12344 CrossRef CAS PubMed.
- M. N. Romanias, A. El Zein and Y. Bedjanian, Heterogeneous Interaction of H2O2 with TiO2 Surface Under Dark and UV Light Irradiation Conditions, J. Phys. Chem. A, 2012, 116, 8191–8200 CrossRef CAS PubMed.
- M. N. Romanias, Y. Bedjanian, A. M. Zaras, A. Andrade-Eiroa, R. Shahla, P. Dagaut and A. Philippidis, Mineral Oxides Change the Atmospheric Reactivity of Soot: NO2 Uptake Under Dark and UV Irradiation Conditions, J. Phys. Chem. A, 2013, 117, 12897–12911 CrossRef CAS PubMed.
- M. J. Tang, R. A. Cox and M. Kalberer, Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: volume 1. Inorganic compounds, Atmos. Chem. Phys., 2014, 14, 9233–9247 CrossRef.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. M. Herrmann, Heterogeneous photocatalysis: state of the art and present applications, Top. Catal., 2005, 34, 49–65 CrossRef CAS.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Titanium Dioxide Photocatalysis in Atmospheric Chemistry, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Heterogeneous Photochemistry in the Atmosphere, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS PubMed.
- C. Anastasio, M. Hoffmann, P. Klán and J. Sodeau, in The Science of Solar System Ices, ed. M. S. Gudipati and J. Castillo-Rogez, Springer, New York, NY, 2013, pp. 583–644, DOI: DOI:10.1007/978-1-4614-3076-6_18.
- L. Dogliotti and E. Hayon, Flash photolysis study of sulfite, thiocyanate, and thiosulfate ions in solution, J. Phys. Chem., 1968, 72, 1800–1807 CrossRef CAS.
- P. Neta and R. E. Huie, Free-Radical Chemistry of Sulfite, Environ. Health Perspect., 1985, 64, 209–217 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Understanding TiO2 Photocatalysis: Mechanisms and Materials, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- A. E. Zein, M. N. Romanias and Y. Bedjanian, Heterogeneous Interaction of H2O2 with Arizona Test Dust, J. Phys. Chem. A, 2014, 118, 441–448 CrossRef CAS PubMed.
- M. N. Romanias, A. El Zein and Y. Bedjanian, Reactive uptake of HONO on aluminium oxide surface, J. Photochem. Photobiol., A, 2012, 250, 50–57 CrossRef CAS.
- M. N. Zeineddine, M. N. Romanias, V. Gaudion, V. Riffault and F. Thevenet, Heterogeneous Interaction of Isoprene with Natural Gobi Dust, ACS Earth Space Chem., 2017, 1, 236–243 CrossRef CAS.
- M. N. Zeineddine, M. N. Romanias, V. Riffault and F. Thévenet, Heterogeneous Interaction of Various Natural Dust Samples with Isopropyl Alcohol as a Probe VOC, J. Phys. Chem. A, 2018, 122, 4911–4919 CrossRef CAS PubMed.
- J. Shang, J. Li and T. Zhu, Heterogeneous reaction of SO2 on TiO2 particles, Sci. China: Chem., 2010, 53, 2637–2643 CrossRef CAS.
- M. Tang, X. Huang, K. Lu, M. Ge, Y. Li, P. Cheng, T. Zhu, A. Ding, Y. Zhang, S. Gligorovski, W. Song, X. Ding, X. Bi and X. Wang, Heterogeneous reactions of mineral dust aerosol: implications for tropospheric oxidation capacity, Atmos. Chem. Phys., 2017, 17, 11727–11777 CrossRef CAS.
- M. N. Romanias, M. N. Zeineddine, V. Riffault and F. Thevenet, Isoprene Heterogeneous Uptake and Reactivity on TiO2: A Kinetic and Product Study, Int. J. Chem. Kinet., 2017, 49, 773–788 CrossRef CAS.
- N. Joshi, M. N. Romanias, V. Riffault and F. Thevenet, Investigating water adsorption onto natural mineral dust particles: Linking DRIFTS experiments and BET theory, Aeolian Res., 2017, 27, 35–45 CrossRef.
- M. Tang, D. J. Cziczo and V. H. Grassian, Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation, Chem. Rev., 2016, 116, 4205–4259 CrossRef CAS PubMed.
- J. Park, M. Jang and Z. Yu, Heterogeneous Photo-oxidation of SO2 in the Presence of Two Different Mineral Dust Particles: Gobi and Arizona Dust, Environ. Sci. Technol., 2017, 51, 9605–9613 CrossRef CAS PubMed.
- Y. Dupart, S. M. King, B. Nekat, A. Nowak, A. Wiedensohler, H. Herrmann, G. David, B. Thomas, A. Miffre, P. Rairoux, B. D'Anna and C. George, Mineral dust photochemistry induces nucleation events in the presence of SO2, Proc. Natl. Acad. Sci., 2012, 12297, DOI:10.1073/pnas.1212297109%J.
- J. W. Adams, D. Rodriguez and R. A. Cox, The uptake of SO2 on Saharan dust: a flow tube study, Atmos. Chem. Phys., 2005, 5, 2679–2689 CrossRef CAS.
- J. Y. Park and M. Jang, Heterogeneous photooxidation of sulfur dioxide in the presence of airborne mineral dust particles, RSC Adv., 2016, 6, 58617–58627 RSC.
- Z. Yu, M. Jang and J. Park, Modeling atmospheric mineral aerosol chemistry to predict heterogeneous photooxidation of SO2, Atmos. Chem. Phys., 2017, 17, 10001–10017 CrossRef CAS.
- Y. Dupart, Impact de la chimie des poussières minérales sur la photochimie atmosphérique, PhD thesis, Université de Lyon 1, p. 2012 Search PubMed.
- S. Ibrahim, M. N. Romanias, L. Y. Alleman, M. N. Zeineddine, G. K. Angeli, P. N. Trikalitis and F. Thevenet, Water Interaction with Mineral Dust Aerosol: Particle Size and Hygroscopic Properties of Dust, ACS Earth Space Chem., 2018, 2, 376–386 CrossRef CAS.
- L. Li, Z. M. Chen, Y. H. Zhang, T. Zhu, J. L. Li and J. Ding, Kinetics and mechanism of heterogeneous oxidation of sulfur dioxide by ozone on surface of calcium carbonate, Atmos. Chem. Phys., 2006, 6, 2453–2464 CrossRef CAS.
- Y. Zhu, O. B. Toon, E. J. Jensen, C. G. Bardeen, M. J. Mills, M. A. Tolbert, P. Yu and S. Woods, Persisting volcanic ash particles impact stratospheric SO2 lifetime and aerosol optical properties, Nat. Commun., 2020, 11, 4526 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00094b |
| This journal is © The Royal Society of Chemistry 2022 |