
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigator series: deposited particles and human lung lining fluid are dynamic, chemically-complex reservoirs leading to thirdhand smoke emissions and exposure†
Roger
Sheu
 a,
Tori
Hass-Mitchell
a,
Akima
Ringsdorf
b,
Thomas
Berkemeier
b,
Jo
Machesky
a,
Achim
Edtbauer
b,
Thomas
Klüpfel
b,
Alexander
Filippi
b,
Benjamin A.
Musa Bandowe
b,
Marco
Wietzoreck
b,
Petr
Kukučka
c,
Haijie
Tong‡
b,
Gerhard
Lammel
bc,
Ulrich
Pöschl
b,
Jonathan
Williams
b and
Drew R.
Gentner
*ab
a,
Tori
Hass-Mitchell
a,
Akima
Ringsdorf
b,
Thomas
Berkemeier
b,
Jo
Machesky
a,
Achim
Edtbauer
b,
Thomas
Klüpfel
b,
Alexander
Filippi
b,
Benjamin A.
Musa Bandowe
b,
Marco
Wietzoreck
b,
Petr
Kukučka
c,
Haijie
Tong‡
b,
Gerhard
Lammel
bc,
Ulrich
Pöschl
b,
Jonathan
Williams
b and
Drew R.
Gentner
*ab
aDepartment of Chemical and Environmental Engineering, Yale University, 17 Hillhouse Ave., New Haven, CT 06511, USA. E-mail: drew.gentner@yale.edu
bMax Planck Institute for Chemistry, Mainz, Germany
cRECETOX, Masaryk University, Brno, Czech Republic
First published on 27th June 2022
Abstract
Thirdhand smoke (THS) persists in locations where smoking previously occurred and can be transported into non-smoking environments, leading to non-smoker exposure. Laboratory experiments using high-resolution mass spectrometry demonstrate that deposited particulate matter (PM) and smoke-exposed surrogate lung lining fluid (LLF) are substantial, chemically-complex reservoirs of gas-phase THS emissions, including hazardous air pollutants, polycyclic aromatic compounds, and nitrogen/oxygen-containing species. Both PM and LLF are persistent real-world THS reservoirs that chemically evolve over time, and can act as vehicles for the transport and emission of reactive pollutants and their reaction byproducts (e.g., acrolein). Deposited PM on clothes, furnishings, bodies, and/or airways will emit volatile to semi-volatile gases over long lifetimes, which can re-partition to other indoor materials and increase their overall persistence. On the other hand, LLF off-gassing consists predominantly of volatile organic compounds in amounts influenced by their aqueous solubilities, and their persistence in breath will be prolonged by re-distribution across internal aqueous reservoirs, as corroborated by multicompartment modeling in this study.
Environmental significanceThirdhand smoke (THS) is a known health concern, but understudied source of hazardous, reactive gases. Reservoirs of THS, including deposited particulate matter and smoke-exposed lung lining fluid, can expose non-smokers to elevated concentrations of these toxic or carcinogenic tobacco smoke-derived compounds over long periods of time and sometimes at similar gas-phase concentrations to fresh secondhand smoke. Detailed chemical speciation of these chemically-complex reservoirs shows their off-gassing composition and behavior is influenced by their chemical properties (i.e., volatility for particulate matter and aqueous solubility for lung lining fluid) and chemical transformations of the THS reservoirs. These findings can inform detailed THS risk assessments, elevate ongoing tobacco research, and exemplify the importance of multiphase partitioning processes involved in indoor air quality. |
1 Introduction
Tobacco smoke contains a combination of organic and inorganic particulate matter (primarily PM2.5) and gas-phase organic compounds that have been shown to be hazardous to human health, with no “safe” level of exposure.1–3 Mainstream and secondhand smoke (SHS) exposure are known to be harmful to humans2,4 with many comorbidities5,6 and are thus a focus of considerable tobacco smoke research along with extensive public health campaigns. However, non-smoker exposure to thirdhand smoke (THS) in locations where smoking has previously occurred also comes with major health implications1,4,7–10 and can exist in non-smoking environments via either THS transport by humans or through building air recirculation systems.11–13 THS contains an understudied mixture of compounds with varying reactivities, which can undergo chemical transformations that impact its toxicity, including surface-phase reactions converting nicotine to highly carcinogenic nitrosamines.14–16SHS can contaminate surfaces (e.g., clothing, walls, furnishings, human bodies) or other building materials with the accumulation of volatile organic compounds (VOCs), as well as intermediate-volatility and semi-volatile organic compounds (IVOCs, SVOCs).11,17–19 Over time, condensed organic compounds originating from any source can gradually re-partition to the gas-phase from organic films and other surfaces/materials,18,20,21 where they can either condense onto aerosols11,12,22 or re-deposit onto other surfaces (incl. dust).8,23–25 Inhalation of these chemicals, either in the gas-phase or adsorbed to aerosols/dust, represents a major pathway for exposure, along with ingestion and dermal uptake,1,26 which are especially important pathways for young children.1 Previous studies involving the exposure of cells in vitro to THS and some of its most prominent components have demonstrated increased incidence of DNA strand breaks, oxidative damage, and cell death.1 Furthermore, laboratory studies on animal subjects (e.g., mice) found damage to various organs upon continuous THS exposure.1
THS off-gassing has been directly studied for only a limited number of targeted gas-phase organic compounds, either in laboratory studies or in real-world smoking (and non-smoking) environments.4,11,17,19 THS chemical speciation remains limited compared to mainstream smoke and SHS,27–32 which inhibits fundamental knowledge on THS contamination processes across the gas, aerosol, and aqueous phases. Specifically, THS off-gassing has been observed for a subset of compounds in controlled laboratory studies with simulated rooms or fabrics17,19 and breath analysis from smokers and non-smokers,19 though largely to identify efficacious markers of tobacco exposure.33–36 However, the underlying mechanisms of THS contamination and transport via humans remain unclear, especially for non-smoking environments and for many lighter VOCs.11
Using recent analytical advances in offline high-resolution mass spectrometry37,38 and improvements in real-time, online mass spectrometry39,40 to enable the detailed chemical speciation of the complex mixtures present in THS over a wide range of volatilities and chemical functionalities, we seek to elucidate the composition, dynamics, and underlying physical mechanisms of THS contamination and off-gassing from particle- and aqueous-phase reservoirs. Our specific objectives include: (a) examine chemically speciated THS emissions from both deposited tobacco PM (i.e., SHS) and surrogate lung lining fluid (LLF) exposed to SHS gases; (b) investigate the factors governing THS multi-phase partitioning dynamics and their timescales by comparing observed THS to SHS and by evaluating the chemically speciated evolution of both PM and LLF off-gassing emissions with age; and (c) contextualize our findings to real-world THS contamination, persistence, and exposure related to these THS reservoirs.
2 Methods
2.1 Overall experimental design
SHS was generated within a 1 m3 chamber with online and offline gas-phase measurements of the SHS (Fig. S1†). In each trial, two different common cigarettes from several popular brands purchased in Germany (Table S1†) were lit simultaneously to generate sidestream SHS representative of typical SHS and allowed to smolder until completely combusted.During the 7–14 minute cigarette combustion periods, the PM from fresh SHS emissions was collected onto PTFE filters. After PM collection, these filters were transferred to a clean filter holder and purified air was passed through the collected PM to measure THS off-gassing from deposited PM in real time using an Aerodyne proton transfer reaction, high-resolution (M/ΔM ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) time-of-flight mass spectrometer (Vocus PTR-TOF (2R model); Aerodyne/TOFWERK).
000) time-of-flight mass spectrometer (Vocus PTR-TOF (2R model); Aerodyne/TOFWERK).
As the PM was being collected, the filtered SHS gases were also bubbled through 25 mL of a surrogate lung lining fluid (LLF) solution, comprised of high-purity (HPLC grade) water that was phosphate-buffered to a pH of 7.2–7.4 containing four added antioxidants (ascorbic acid, citric acid, glutathione, and uric acid), to simulate gas-phase SHS uptake into lung lining fluid.41 Thereafter, the LLF was connected to the PTR-TOF and purified air was bubbled through the LLF to quantify off-gassing THS-related compounds.
Both PM and LLF off-gassing were monitored in real-time for initial 0.5–2 hour periods using the PTR-TOF, with additional gas-phase samples collected for subsequent offline analysis (see below). Additionally, to simulate the continued off-gassing of PM in real-world environments, purified air was continuously flowed through the filters for 6, 18, and up to 72 hours with periodic measurements of off-gassing emissions from the aged PM.
A detailed description of all methods, equations, extensive supplemental data, and a list of experimental trials with the duration and age of PTR-TOF measurements and a list of associated offline adsorbent tube samples of SHS and THS at various ages can be found in the ESI† for each of the five separate experiments conducted (Table S1†).
2.2 Real-time gas-phase measurements of SHS and THS via Vocus PTR-TOF
PTR-TOF data were first mass-calibrated, baseline subtracted, and duty-cycle-corrected; formulas were then assigned using a list of formulas of the form CiHjN0–2O0–4 and further manual processing. Compound sensitivities (counts per second per parts per billion, cps ppb−1) were calculated using calibrations from a 79 component standard cylinder (Table S2†) and the parameterizations from Sekimoto et al.40 High-time resolution off-gassing dynamics and representative concentrations of SHS, PM off-gassing, and LLF off-gassing were then determined for 417 priority compound formulas (see Sections S2.1–S2.3 in the ESI†).Replicate trials for both fresh PM off-gassing and LLF off-gassing with varied cigarette brands generally found agreement in terms of chemical composition and compound concentrations across the volatility range of compounds measured by PTR-TOF (Fig. S2–S4†). SHS, PM off-gassing, and LLF off-gassing data were combined and averaged across experimental trials to examine the overall chemical composition where appropriate. When comparing within and across these experiments, data from individual trials or across trials were used as specified and compared as a function of calculated saturation concentrations (e.g., Fig. S3 and S4†) or published Henry's law constants.42
2.3 Offline gas-phase and aerosol-phase sampling and analysis
While online real-time sampling was conducted using the aforementioned PTR-TOF, offline gas-phase samples of SHS and THS (from PM and LLF off-gassing) were collected on custom-packed adsorbent tubes and analyzed via thermal desorption and gas chromatography with atmospheric pressure chemical ionization time-of-flight mass spectrometry (GC-TOF; Agilent 7890B, 6550) with high mass resolution (M/ΔM > 25000).38,43 For comparison, PM samples were collected on both quartz filters and PTFE filters and analyzed via GC-TOF and liquid chromatography with electrospray time-of-flight mass spectrometry (LC-TOF; Agilent, 1260, 6550), respectively. Both utilized chromatographic separation to aid accurate chemical formula determinations within the complex mixture across a wide volatility (VOC–LVOC) and chemical functionality range, spanning hydrocarbons to highly-functionalized organic compounds. For more information and details on adsorbent tube and filter preparation, handling, sampling, and analysis, see Sections S1.5 and S1.6 in the ESI† and prior work.38,43
Supplemental polyurethane foam (PUF) samples were collected and analyzed to quantify levels of oxygen/nitrogen-containing polycyclic aromatic compounds (PACs), polychlorinated biphenyls (PCBs), and polychlorinated dibenzo-dioxins/furans (PCDD/Fs). They were extracted and run using gas chromatography, triple quadrupole mass spectrometry (GC-QQQ; Thermo Scientific TSQ8000 Evo), following detailed procedures provided in Section S1.7 of the ESI.†
2.4 Multi-compartment body modeling to examine THS in exhaled breath following smoking
For comparison to laboratory measurements of LLF off-gassing, the partitioning of smoke-associated, gaseous organic compounds into lung lining fluid, blood, and tissues was simulated using a kinetic multi-compartment model similar to the KM-SUB-ELF model presented in Lakey et al. and further described in Section S5 of the ESI.†44 The model consists of four compartments—lung gas phase, lung lining fluid, blood, and moderately-perfused tissues (Fig. S5†)—with aqueous-phase partitioning between the reservoirs according to Henry's law constants.3 Results
3.1 Tobacco PM and smoke-exposed LLF are substantial off-gassing reservoirs of THS
Real-time observations of THS off-gassing emissions from both PM and LLF detected an extensive array of off-gassing compounds, including key compounds related to tobacco smoke (Fig. 1). Out of the 3362 molecular masses observed using online high-resolution mass spectrometry during PM and LLF off-gassing (Fig. S6†), we focused on detailed time-resolved data for 417 prominent, unique chemical formulas important to tobacco smoke (Table S3†), each of which may include multiple isomers. These high-time resolution (i.e., 1 Hz) measurements ranged from mass-to-charge ratios (m/z) of 31.0163 (i.e., formaldehyde) to 391.283 (i.e., C24H38O4) (see mass spectra in Fig. S6†). Prominent formulas included those for many light VOCs (e.g., acetone, acetaldehyde, acetonitrile), hazardous air pollutants (e.g., acrolein, furan, acrylonitrile, 1,3-butadiene, formaldehyde, benzene, 2,5-dimethylfuran, phenol),4,19,45 and reactive organic compounds (e.g., terpenes, single-ring aromatics, alkenes, alkaloids) (Table S3†).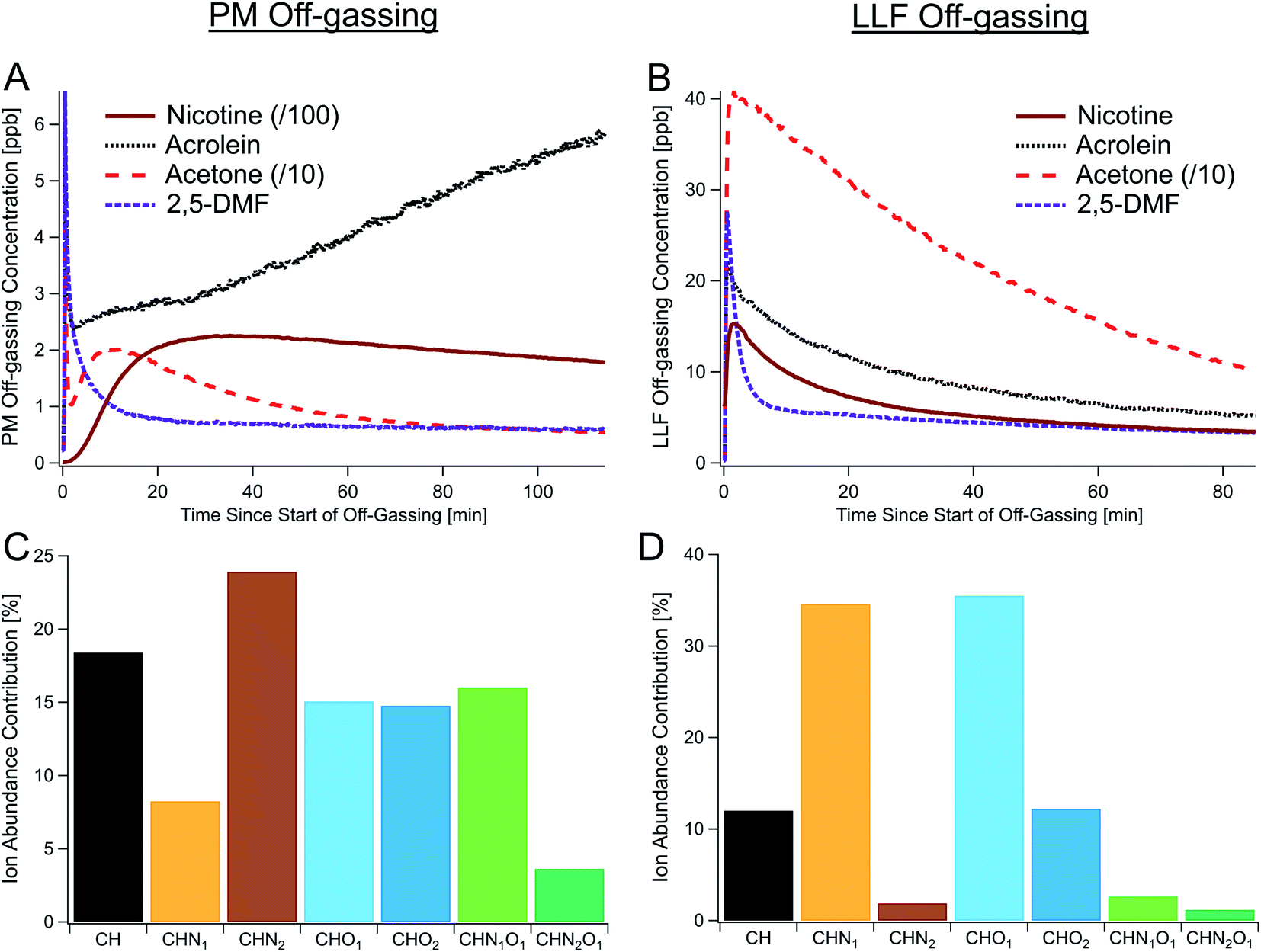 | ||
| Fig. 1 Observations of THS off-gassing from deposited PM and SHS-exposed LLF. (A and B) Time series of major THS compounds off-gassing from (A) PM and (B) surrogate LLF in individual experiments measured via online PTR-TOF. (C and D) Relative ion abundances by elemental composition (PM, (C); LLF, (D)) from offline high-resolution GC-TOF data, each shown as the average of three samples collected during the first 1–1.5 hours of off-gassing (see Table S1†). To show all the traces on one axis, concentrations of acetone and nicotine in (A and B) were scaled down as indicated. CH, CHN1, CHN2, CHO1, CHO2, CHN1O1, and CHN2O1 represent different classes (types) of compounds as defined in Table 1, with each class encompassing the full range of formulas shown in Fig. 2. | ||
A wide range of gas-phase IVOCs and SVOCs were also observed in real-time data, including an array of heterocyclic nitrogen- and oxygen-containing compounds; single-ring and polycyclic aromatic compounds, such as polycyclic aromatic hydrocarbons (PAHs); and chemical formulas corresponding to a broad suite of other aromatic and aliphatic oxygenated compounds (e.g., phenols, phthalates, and carboxylic acids) (Table S3†).
In fresh PM off-gassing, nicotine (C10H14N2) was the most abundant compound, accounting for approximately 30% of the total gas-phase concentration among the 417 most important compounds quantified by PTR-TOF, followed by acetic acid, myosmine (C9H10N2), triacetin (C9H14O6), and acetone. In addition to nicotine and acetone, Fig. 1 shows time series for acrolein, given its toxicity,4 and 2,5-dimethylfuran, which has been used as an indoor tobacco smoke tracer.46 Other studies have found highly hazardous nitrosamines, such as nicotine-derived nitrosamine ketone/aldehyde (NNK/NNA) and N-nitrosonornicotine (NNN), in SHS, on surfaces, and in dust.14,47 In this study, we observed real-time data on C10H13N3O2 (e.g., NNK) in SHS and PM off-gassing via PTR-TOF, though NNN could not be differentiated from other compounds with similar m/z.
3.2 Speciating the extensive chemical complexity of THS
While detailed speciation has been carried out for direct emissions from cigarettes,29,32,48–50 measurements of THS have been relatively limited to a subset of prominent individual compounds.4,11,12,18,19 Here, we applied a powerful combination of online and offline high-resolution mass spectrometers with soft ionization to chemically speciate complex THS mixtures, along with the gases and PM that comprise SHS, from which THS is derived.Offline data analysis employed a hybrid targeted/non-targeted approach similar to prior work.11,38,43,51 An initial non-targeted survey of molecular features in gas-phase GC-TOF data found major contributions from various compound classes, which are summarized here as a function of their heteroatom counts. These features included CH (i.e., hydrocarbons, equivalent to CxHy), CHO1, CHO2, CHN1, CHN2, CHN1O1, and CHN2O1 compounds (Fig. 1C, D, S7 and S8†), with smaller, but notable contributions from CHO3, CHO4, and CHN1O2 compounds, among others. Subsequent targeted analysis of this complex mixture via GC-TOF spanned C6–30 hydrocarbons and C4–25 for other compound classes, with molecular formulas designated by carbon number and decreasing hydrogen atom counts (i.e., double bond equivalents; DBEs), such as CnH2n+2−(2×DBE) for CH compounds.
THS off-gassing from PM and LLF contained a diverse range of hydrocarbons and nitrogen- and/or oxygen-containing compounds (Fig. 1C and D), with their breakdown within each compound type enumerated in Fig. 2. Observed hydrocarbons included major contributions from single-ring aromatics and PAHs, as well as C10, C15, and C20 terpenoids (incl. monoterpenes, sesquiterpenes, and diterpenes). The underlying chemical functionalities of prominent nitrogen- and oxygen-containing features in Fig. 2 likely include similar structures as those found from prior observations of tobacco smoke and biomass burning.11,52 For instance, prominent features in the CHN data likely include alkylpyridines (CnH2n−5N), quinolines (CnH2n−11N), and pyrazines (CnH2n−4N2). Similarly, CnH2n−4O and CnH2n−6O2 compounds likely had major contributions from furans and furanaldehydes, respectively, and examples of common CHNO compounds included pyrrolidones (CnH2n−5NO), oxazoles (CnH2n−3NO), and alkoxypyrazines (CnH2n−4N2O).
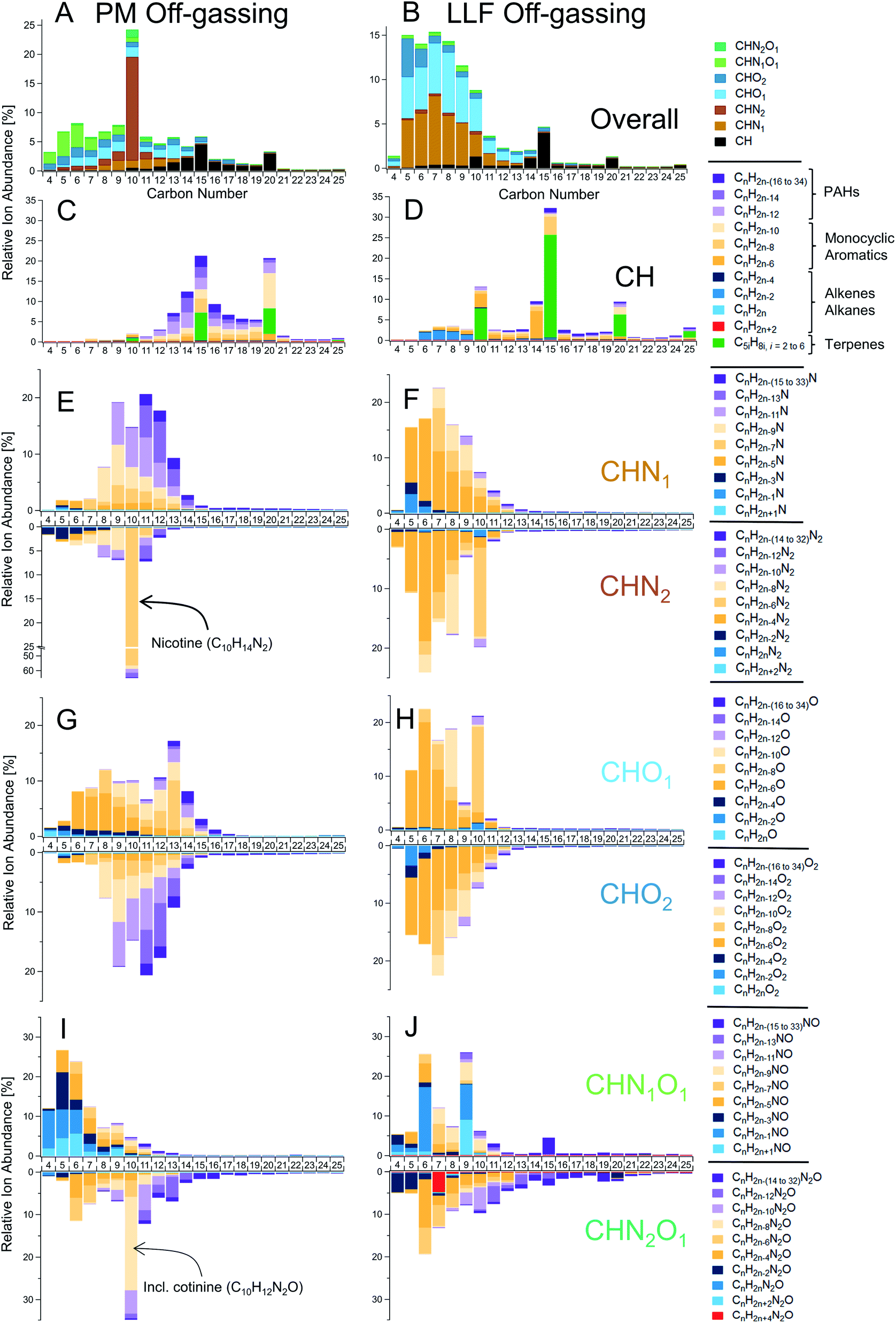 | ||
| Fig. 2 Detailed speciation of chemically-complex THS off-gassing from PM and LLF reservoirs. Measurements via GC-TOF (with soft ionization to reduce fragmentation and preserve molecular structure) show the relative chemical composition of gas-phase compounds emitted from slightly-aged PM (left column) and LLF (right column) samples following initial off-gassing (i.e., 10–80 minutes) into the PTR-TOF. (A and B) A summary of ion abundances by carbon number and compound class, followed by breakdowns of (C and D) hydrocarbons, (E and F) CHO1–2 compounds, (G and H) CHN1–2 compounds, and (I and J) CHN1–2O1 compounds with specific chemical formulas indicated in each row's legend and generalized compound types for hydrocarbons. Note: initial concentrations from PM and LLF off-gassing were analyzed using PTR-TOF to capture high time-resolution dynamics and very volatile (C2–C5) compounds (e.g., Fig. 1, 3, 4, 5 and Table S3†), so the abundances of some compounds shown here may be relatively lower than their peak in initial off-gassing (e.g., furans, pyrrole, pyrroline) or some small compounds may be underestimated via GC-TOF (e.g., C4–C6 hydrocarbons) due to reduced ionization efficiencies relative to larger compounds. See the ESI† for additional details on samples and relative abundance calculations. | ||
Given the complexity of the THS emissions we observed here, the objective of the GC-TOF analysis was focused on grouping isomers by molecular formula as opposed to identifying each individual isomer. Confirmation analyses were conducted on offline adsorbent tubes using GC with electron ionization mass spectrometry (i.e., GC-EIMS; Agilent 5977A) to provide mass spectral identifications for some major compounds observed in GC-TOF and PTR-TOF data. A survey of the GC-EIMS data (e.g., Fig. S9†) similarly reveals a broad range of VOCs to SVOCs present in PM off-gassing, LLF off-gassing, and SHS across a combination of resolvable peaks and unresolved complex mixtures of compounds. Along with compounds frequently reported in the tobacco smoke literature (e.g., nicotine, 3-ethenylpyridine, phenol, single-ring aromatics),11,29,46,53,54 a wide array of other compounds were observed in SHS, including hydrocarbons (e.g., alkanes, alkenes, PAHs) and functionalized compounds such as furanoids (e.g., 2-methylfuran, 2,5-dimethylfuran, furfural, ethylmethylfuran). Notable examples of compounds found in PM off-gassing include triacetin, glycerol, nicotine-related alkaloids, nitrogen-containing heterocycles in the CHN1–2 category (e.g., pyridines, quinolines, pyrroles, indoles), aromatic nitriles (e.g., benzonitriles, naphthalenecarbonitrile), CHO1–2 compounds (e.g., cresols, other alkyl phenols, guaiacols, benzofurans, acids), PAHs (e.g., C11–C15 alkyl naphthalenes, biphenyls, acenaphthylene, phenalene, fluorene, alkyl-fluorenes, phenanthrene, anthracene, fluoranthene, pyrene), and CHN1O1 compounds similar to those seen in Fig. 2I (e.g., pyrrolidones/pyrrolidinones, pyridinols).
| Compound class | Gas-phase secondhand smoke (SHS) | Deposited PM in SHS | PM off-gassing | LLF off-gassing | |
|---|---|---|---|---|---|
| a See ESI Section S3.1 for discussion of potential underestimates in acyclic alkane abundances as a result of their lower response factors relative to other compounds. Cumulative sums may exceed 100% due to rounding. | |||||
| CH | 16 (14–23) | 20 (20–23) | 18 (15–20) | 13 (7–18) | |
| Acyclic alkanes | 0 (0–0) | 10 (9–11) | 0 (0–0) | 0 (0–1) | |
| Alkenes & cycloalkanes | 22 (18–26) | 4 (3–4) | 2 (1–3) | 11 (8–13) | |
| Terpenes | 13 (12–14) | 10 (10–10) | 15 (9–22) | 41 (36–45) | |
| Single-ring aromatics | 52 (48–53) | 32 (30–35) | 41 (40–42) | 37 (33–39) | |
| PAHs | 13 (8–16) | 44 (42–45) | 43 (34–48) | 12 (7–15) | |
| CHN1 | 19 (17–24) | 6 (5–7) | 8 (8–10) | 39 (26–52) | |
| CHN2 | 16 (7–13) | 22 (21–23) | 24 (18–25) | 2 (1–3) | |
| CHO1 | 23 (16–23) | 7 (7–8) | 15 (12–16) | 28 (5–52) | |
| CHO2 | 19 (13–23) | 21 (18–22) | 15 (14–15) | 14 (8–18) | |
| CHN1O1 | 5 (3–7) | 9 (5–10) | 16 (13–25) | 3 (0–6) | |
| CHN2O1 | 1 (1–2) | 14 (14–17) | 4 (3–5) | 1 (0–3) | |
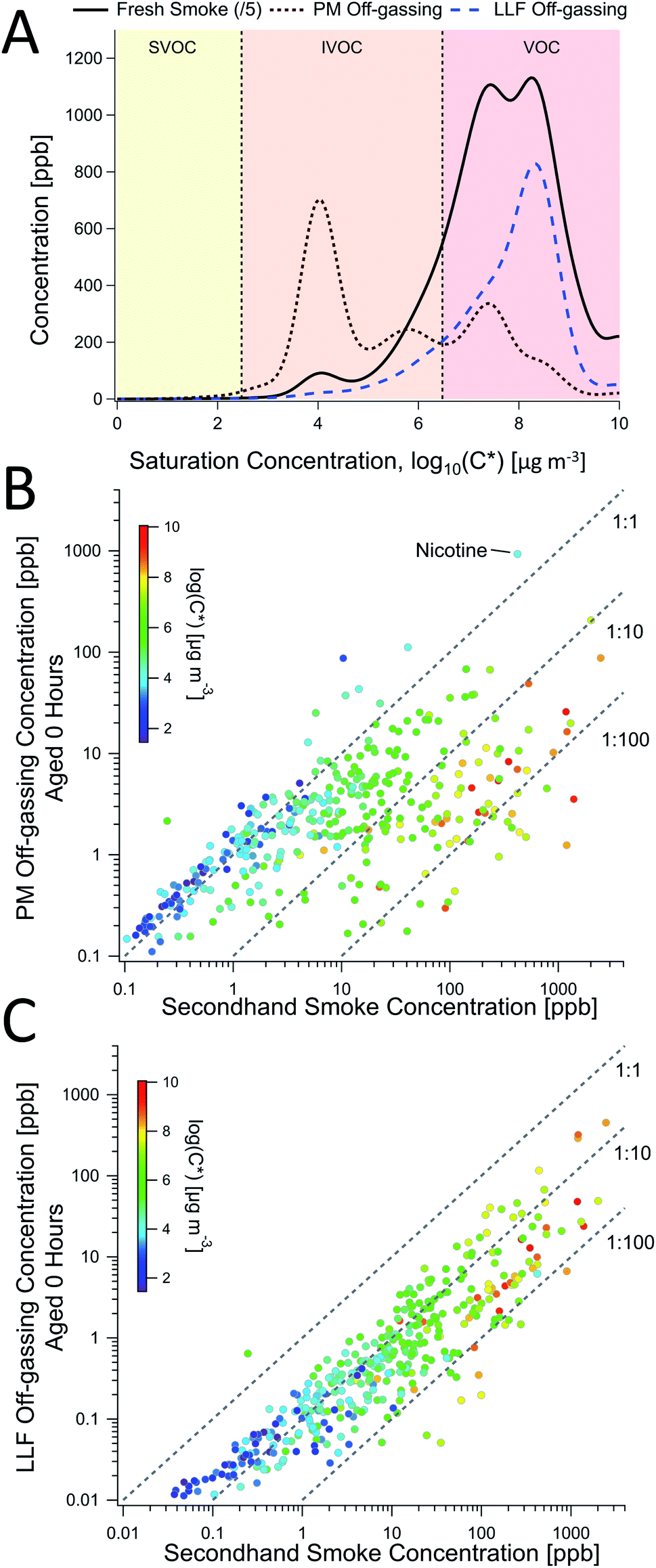 | ||
| Fig. 3 Comparisons of PM and LLF off-gassing to secondhand smoke. (A) The volatility distribution of gas-phase compound concentrations found in secondhand smoke, PM off-gassing, and LLF off-gassing, each averaged across 4 experiments from online PTR-TOF data. Scatterplots of (B) PM and (C) LLF off-gassing concentrations for individual compounds were compared to secondhand smoke and colored as a function of volatility (i.e., saturation concentration; C*), shown here for a single experiment (Trial 4; see ESI for other examples, Fig. S12, S14, S15, S18–S20 and S22†). The three dashed lines in (B and C) represent concentration ratios of 1:1, 1:10, and 1:100. | ||
The overall chemical composition of PM off-gassing peaked at C10 compounds, given the prominence of nicotine (C10H14N2) and cotinine (C10H12N2O) (Fig. 2). However, the distribution of PM off-gassing included large contributions of other nitrogenous and oxygenated VOCs–IVOCs from C1 to C15 and marked emissions of aromatics across the VOC (e.g., benzene) to SVOC (e.g., C20) range (Fig. 2, Tables S3 and S4A†). The multicyclic compounds and PACs were not just limited to hydrocarbons (e.g., PAHs), but included an extensive range of multicyclic nitrogen-containing compounds (e.g., CHN1–2) and oxygen-containing PACs, especially for CHO2 compounds. A supplemental targeted analysis for select oxygen- and nitrogen-containing PACs via GC with triple quadrupole MS/MS confirmed that PM off-gassing contains notable levels of multiple key oxygenated PACs (e.g., 1,4-naphthoquinone, 2-methyl-1,4-naphthoquinone, 9-fluorenone, 9,10-anthraquinone, 9,10-phenanthrenequinone, benzo([a]/[b])fluorenone, benzanthrone) and less significant levels of nitro-PACs (Table S5†). Along with online PTR-TOF measurements of benzoquinone at 6.8 ± 2.1 ppb in off-gassing PM (Table S3†), this targeted analysis demonstrated the presence and emissions of individual carbonyl- and quinone-containing functionalities in THS, some with high oxidative potential leading to ROS formation and potential cytotoxicity (e.g., 1,4-naphthoquinone, 9,10-phenanthrenequinone, 2-methyl-1,4-naphthoquinone).55,56
In contrast with off-gassing from PM, LLF off-gassing emissions tended towards lighter and more reduced compounds, with outsized contributions from terpenoids. Aromatic CH and CHO1–2 compounds were prominent but with relatively less PAC content than PM off-gassing (Fig. 2, S13† and Table 1). A majority of the abundance appeared on compounds with ≤10 carbon atoms (Fig. 2) including CHN1, CHO1 and CHO2 compounds, such as CnH2n−5N, CnH2n−6O and CnH2n−6O2, likely corresponding to pyridines, phenols, and catechols, respectively.
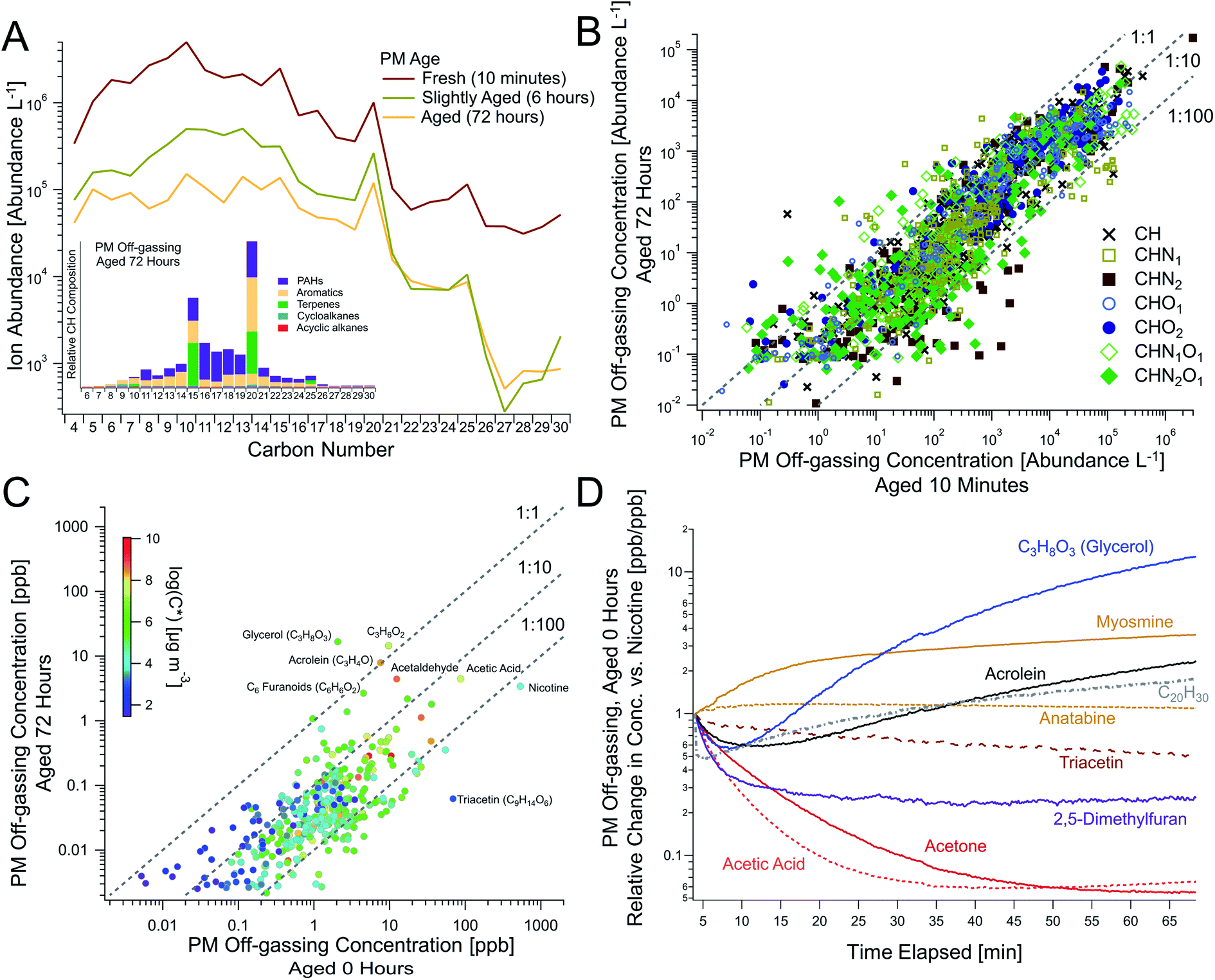 | ||
| Fig. 4 Persistent and dynamic THS off-gassing from aging PM. (A) The temporal evolution of PM over time as a function of carbon number, shown with the chemical speciation of hydrocarbons from PM aged for 72 hours (inset, analogous to Fig. 2C), measured via offline GC-TOF. This comparison between “fresh” vs. 72 hour-aged PM demonstrates persistent off-gassing for (B) major compound classes across the complex mixture (as ion abundances measured via GC-TOF) and for (C) the highest concentration compounds (where each point represents a chemical formula measured via online PTR-TOF), including elevated concentrations of acrolein and C3H8O3 (e.g., glycerol), among others. (D) The temporal evolution of notable off-gassing compounds from fresh PM relative to nicotine demonstrates dynamic changes in major species with time (Trial 5, see Table S3† for average absolute ratios to nicotine). Triacetin in (C and D) was detected as the fragment C7H11O4+via PTR-TOF and its prominence in minimally-aged PM off-gassing was confirmed via GC-EIMS (Fig. S9†). See Fig. S10† for scatterplots of panel B data broken up elemental composition. | ||
In addition to being enhanced in lower volatility compounds, PM off-gassing included considerable aromatic content (Fig. 2) with a general shift towards more aromatic compounds (vs. alkanes/alkenes) for both CH and CHO1–2 compounds compared to the chemical profile of SHS (Fig. S7 and S13†). While this observation also holds for CHN1, which had more highly unsaturated compounds, the distribution of CHN1O1 and CHN2O1 compounds demonstrated greater similarity between gas-phase SHS and PM off-gassing (Fig. 2 and S7†). Furthermore, aromatic compounds made up a greater fraction of hydrocarbons (i.e., CH) than alkanes/alkenes in PM off-gassing (Fig. 2, S7, S9, S13, S16, S17† and Table 1) as well as in SHS, which was similarly noted in prior work on SHS with a smaller range of compounds.46 Despite their high volatility, emissions of lighter CH and CHO1-2 aromatics were also observed in PM off-gassing (e.g., Fig. 2 and Table S3†), where C6–C9 aromatic hydrocarbons comprised 0.64% ± 0.03% of the observed PTR-TOF abundance during initial PM off-gassing.
Despite considerable aromatic contributions to THS, non-aromatic hydrocarbons, including alkanes, terpenes, and non-terpenoid alkenes, were observed in THS off-gassing from PM (and LLF), especially in the IVOC-SVOC range for PM off-gassing (Fig. S7 and Table S3†). While quantitative differentiation of cycloalkanes and alkenes was not feasible with GC-TOF in Fig. 2, observations of PM off-gassing via PTR-TOF (for alkenes) and GC-EIMS (for alkanes/alkenes) included alkenes (e.g., C5–18), dienes (e.g., isoprene, terpenes, neophytadiene (C20H38)) benzyl-alkenes (e.g., styrene, indenes), linear alkanes (e.g., n-C11–21), and sparse evidence for prominent branched/cyclo-alkanes. Similar to the general volatility-dependent trends in Fig. 3, concentrations of C16–20 acyclic alkenes in PM off-gassing are similar to those in SHS with smaller alkenes having progressively lower concentrations relative to SHS (Fig. S16†). Similarly, oxygenated compounds with aliphatic formulas are prominent in SHS at lower molecular sizes (i.e., ≤C10) in GC-TOF (Fig. 2) and PTR-TOF data (Table S3†) and are observed in THS, but oxygenated aromatic compounds are also generally enhanced in PM off-gassing (Fig. 2, S7 and S13†).
While there are some differences with chemical properties, our results establish a general volatility-dependent relationship between THS from PM off-gassing and SHS composition (Fig. S3†), where the THS to SHS ratio for a given compound is approximated by:
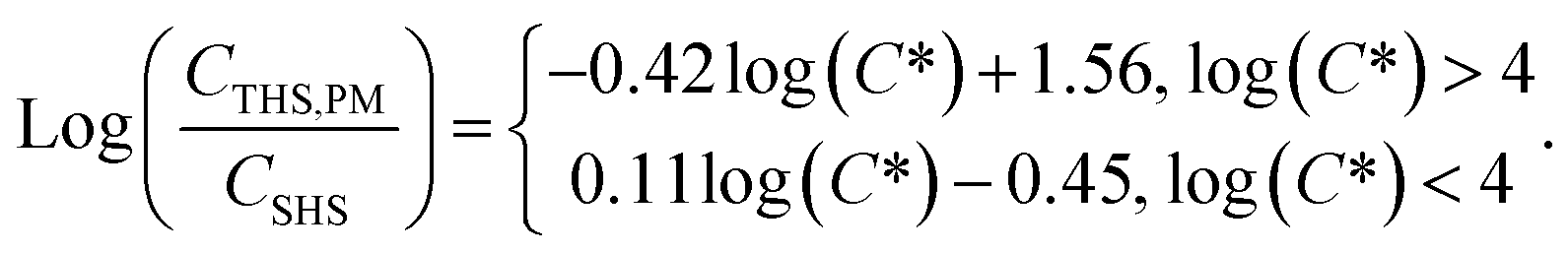 | (1) |
The general chemical composition and volatility distribution of emissions from LLF over the initial off-gassing period was more similar to SHS (Fig. 3A, S4 and S18†), though individual compound concentrations were all lower than in SHS (Fig. 3C), which may have been influenced by the LLF exposure and off-gassing setup used. The composition was not dependent on volatility (Fig. 3C) but was instead sensitive to aqueous solubility, which led to notable differences between the evolution of SHS and LLF composition over the off-gassing period (see Section 3.5). These differences included reduced contributions from CHN2 (esp. nicotine) and CHN1–2O1 compounds, relative enhancements in CHN1 and CHO1 compounds, and some variations in the overall CH compound distribution shown in Fig. 2 after the initial off-gassing period. Similar to the PTR-TOF observations of alkenes in LLF off-gassing (Table S3†), a greater prevalence of ion fragments associated with alkanes/alkenes in the GC-EIMS data (e.g., m/z 57, 55) was observed in the VOC range (<C12), which is consistent with the lack of major volatility-dependent differences (Fig. 3).
3.3 THS off-gassing from PM reservoirs is persistent and chemically-evolves over time
We simulated long-term PM off-gassing to demonstrate deposited PM's ability to act as a persistent THS source due to the large reservoir of VOCs, IVOCs, and SVOCs present within the PM. While the “ages” presented here do not directly equate to real-world THS ages given the dependence on ventilation conditions, they provide key information on the evolution of chemical composition during prolonged PM off-gassing, and will likely extend past the multi-day timescales examined here.Online and offline measurements after continued PM off-gassing were compared to SHS and unaged (“fresh”) PM off-gassing for all aging experiments (Fig. S12, S19 and S20†). Over shorter time periods in the various experiments (e.g., 1–6 hours), some individual compounds showed minor changes and others decreased by up to an order of magnitude depending on volatility, and slightly larger decreases were observed after 18 hours (Fig. S12 and S14†). For one experiment shown in Fig. 4 with sampling from the same PM filter at 0, 6, and 72 hours, we observed persistent off-gassing, with concentrations decreasing slightly over an order of magnitude on average (Fig. S10 and S14†), and with larger decreases for some compounds (e.g., nicotine) (Fig. 4C). Some shifts in the volatility distribution were observed, including larger decreases in many light VOCs as well as larger compounds that were relatively more persistent (Fig. 4A–C). A greater fraction of PAHs was also observed at 72 hours (Fig. 4A inset) compared to fresh PM off-gassing (Fig. 2C).
We observed three distinct trends in the temporal dynamics of PM off-gassing that are important over short and long timescales.
Type 1 (transient): most small to moderately sized hydrocarbons and relatively non-polar compounds, including 2,5-dimethylfuran and acetone, exhibited an early concentration spike in fresh samples and slowly decreased from their maxima while remaining substantially elevated over the course of the initial hour-long sampling period (Fig. 1A) and after 6 hours (Fig. S10†). This early concentration spike was likely indicative of the initial off-gassing from the surface layer of the predominantly organic and highly-viscous particles, prior to the off-gassing rate becoming limited by internal diffusion (Fig. S21†), which can result in sustained emissions of even light VOCs.
Type 2 (persistent): off-gassing concentrations of alkaloids (e.g., nicotine, nornicotine, myosmine, cotinine), strongly-associated with tobacco combustion, along with many larger and/or polar compounds, often appeared to rise slowly and decayed more slowly. While persistent, compounds in this category still followed general trends with substantial decreases from their peak concentrations after 18 and 72 hours of simulated off-gassing. For example, nicotine was observed to be a large fraction of alkaloids present in SHS and appeared to take approximately 15 minutes to reach maximum off-gassing concentrations (Fig. 1A). Myosmine and anatabine, though observed at much lower concentrations than nicotine, showed similar temporal trends (Fig. 4D), suggesting that their particle-phase concentrations do not drive these timescales. For the alkaloids, their equilibrium between free base form (i.e., protonated) and deprotonated form may both dampen the rate of initial off-gassing and limit the amount of available alkaloid for partitioning to the gas-phase. Based on prior instrument characterization, a lag in response times on the PTR-TOF is not expected to be responsible for this delayed increase either,57 and the effect was not observed in LLF off-gassing for nicotine (Fig. 1B) nor for all C14 hydrocarbons with similar volatilities (Fig. S17†).
A notable non-alkaloid compound that also appears to also follow this trend is triacetin (C9H11O6, observed in PTR-TOF as its fragment ion C7H11O4+), which is the second most abundant compound in PM off-gassing after nicotine (Table S3†). It is commonly used as a plasticizer in cigarette filters, though it has also been used as a humectant additive to preserve moisture or as a solvent during production, and previous work has observed large triacetin emissions from cigarette butts.48 Its magnitude and persistence, similar to nicotine, make it an important, dynamic THS component.
C20 compounds also showed noteworthy persistence (Fig. 4A). Of these, C20H30 has been reported in prior literature on tobacco smoke, albeit in the particle phase,58 and could serve as an important marker for persistent PM-sourced THS, since it is relatively less prevalent in gas-phase SHS (Fig. S7†). As measured via PTR-TOF (e.g., Table S3†), the [C20H30]/[nicotine] ratio sampled from the same filter for fresh, 6 hour old, and 72 hour old PM increased over time with values of 0.0011, 0.0042, and 0.045, respectively.
Type 3 (degradation products): a select few compounds increased or stayed constant throughout long-term PM off-gassing (Fig. 1A and 4C), including C3H4O (e.g., acrolein), C3H8O3 (e.g., glycerol), C3H6O2 (e.g., hydroxyacetone, glycidol), and C2H4O (e.g., acetaldehyde). Since many of them are also prevalent SHS components from combustion or as cigarette additives (e.g., glycerol), they initially dropped in concentration, exhibiting a trend similar to compounds like acetone and 2,5-dimethylfuran, but then began rising after 10–15 minutes (Fig. 1 and 4D). This rise in concentrations during off-gassing was observed across multiple experiments and ages (Fig. S11 and S14†). Their concentrations after 18 hours were similar to those during initial (fresh) PM off-gassing and even larger after 72 hours—making them some of the most abundant compounds observed (Fig. 4C).
These results suggest that they may be formed as degradation products from other larger species, and that the consideration of processes aside from volatility-dependent dynamics are necessary. The most prominent candidate parent compound is the aforementioned triacetin, which was prevalent in SHS and the second most abundant compound observed in PM off-gassing, with its identification confirmed via GC-EIMS. Triacetin degradation has been reported in the literature, including under thermal conditions with acetic acid or acetic anhydride loss, to yield acrolein, acetaldehyde, formaldehyde, hydroxyacetone, glycidol, and others.59,60 These papers do not show glycerol as a degradation product of triacetin (the glycerol triester), though ester hydrolysis of triacetin would be expected to yield glycerol. However, glycerol formation from triacetin pyrolysis was suggested by Laino et al.,60 who also predict that triacetin has a relatively high tendency to (thermally) decompose. Glycerol, derived from either its use as a tobacco additive or from triacetin degradation, may also produce acrolein via acid-catalyzed dehydration (e.g., via acetic acid),59 and acrolein and glycidol formation via thermal degradation of glycerol has been previously mentioned in literature.61 While most studies of triacetin/glycerol degradation focus on pyrolysis, glycerol mechanism modeling reported acid-catalyzed dehydration of protonated glycerol at lower temperatures with activation barriers of only 21–25 kcal mol−1,62 and cigarette smoke's measured pH between 5.3 and 6.5 makes it acidic in nature.63
While not ascribed to triacetin degradation here, the abundance of several other compounds similarly remained elevated over time, including C6H6O2 (e.g., 5-methylfurfural, 2-acetylfuran, catechols), C5H5NO (e.g., pyridinols), C3H6O3 (e.g., lactic acid), and C5H6N2O (e.g., methoxypyrazine). We note that the decomposition of other species may be occurring concurrently, such as those involving propylene glycol, which was also reported to yield hydroxyacetone (C3H6O2) during pyrolysis.61 Additionally, while C3H6O2 isomers were prevalent in PTR-TOF data, the breakdown between glycidol and hydroxyacetone could not be determined by GC analysis given limitations with measuring the epoxide glycidol in PM off-gassing. Regardless of the parent species and decomposition pathways, increasing levels of acrolein to 10+ ppb throughout THS aging, along with potential contributions from glycidol and formaldehyde, imply major health consequences, since acrolein is one of the most hazardous known components of THS4 and glycidol, if present, is a probable carcinogen (Group 2A agent per the International Agency for Research on Cancer).61
3.4 Speciating the underlying PM reservoir of THS emissions
Offline speciation of collected PM samples (without off-gassing) demonstrated extensive reservoirs of VOC–SVOC THS compounds, which were likely abundant in PM due to their formation in the combustion plume, where elevated concentrations of these compounds (e.g., 30–60 ppm benzene in-plume) drive partitioning to the condensed phase.64 PM samples thermally desorbed from quartz filters showed a greater fraction of CH, CHN2, CHO2, and CHN2O1 compounds (Fig. S8†). A broad array of these compounds had a high degree of unsaturation across GC-TOF observations (i.e., C4–C30), which was especially true for functionalized compounds, including substantial amounts of C5–C15 oxygenates similar to those in PM off-gassing; an array of nitrogenous species including nicotine and other alkaloids; and a range of formulas for single-ring aromatics and PAHs that were also observed in PM off-gassing, including C20H30 and C20H32 (Fig. S7†). As expected, there were greater proportions of larger compounds (i.e., I/SVOCs) when compared to the gas-phase compound distributions in SHS and PM off-gassing, which illustrates their role as reservoirs for long-term PM off-gassing (Fig. 2 and S7†).Following PM solvent extraction from PTFE filters, LC-TOF was used for confirmation and to examine more LC-amenable, highly-functionalized compounds. Non-targeted analysis in both positive and negative mode identified large quantities of CHN2, CHO2, and CHN2O1 compounds similar to GC-TOF results as well as notable contributions from CHN2O2, CHN1O1, CHO2S1, CHN1O1S1, CHO4, CHO3, CHO3S1, and CHN1O4 (possibly nitrocatechols) compounds (Fig. S2†). While CHON compounds span the IVOC–SVOC volatility range in positive mode, CHN compounds were primarily in the IVOC range, largely between 3 < log(C*) < 5 (Fig. S2†), including known alkaloids from cigarette smoke. CHO compounds were observed in negative mode ionization across the VOC–SVOC range with a large CHO signal between 1 < log(C*) < 2 that was also observed in GC-TOF PM data, and a large fraction of the observed CHOS compounds also fall in the IVOC range (Fig. S2†).
3.5 LLF off-gassing dynamics and dependence on Henry's law constants
The concentrations of some compounds in LLF off-gassing decayed relatively quickly over 5–60 minutes in the experimental setup, albeit with different first-order rates of decay (Fig. 1B). Some compounds, like acetone and acetic acid, decreased relatively slowly before leveling out at elevated concentrations (Fig. 1B and 5A), while more non-polar compounds (e.g., benzene, methylnaphthalenes), decreased rapidly with an e-folding time of less than 15 minutes.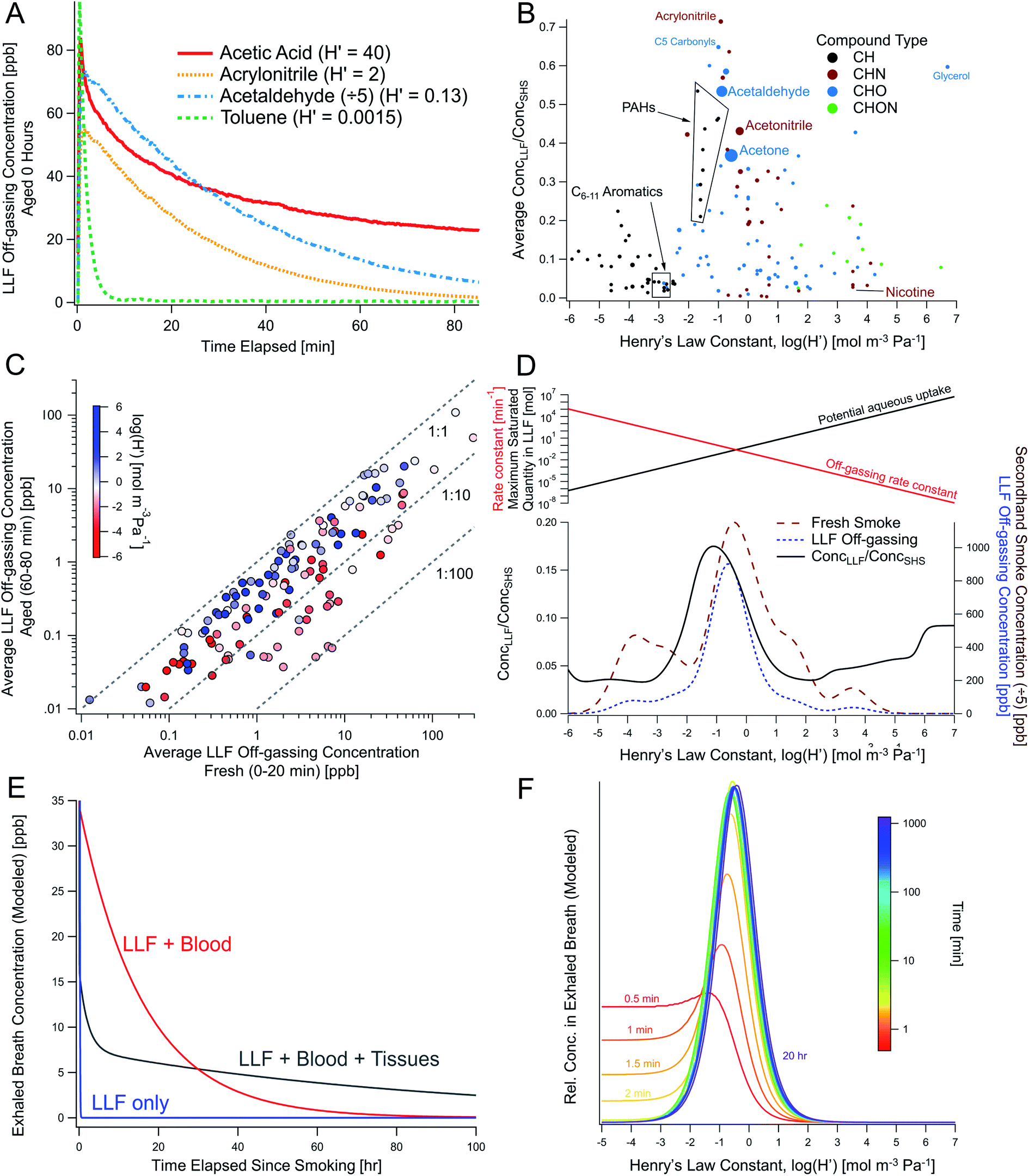 | ||
Fig. 5 THS off-gassing from LLF and the effect of compound-specific Henry's law constants in lab experiments and multi-compartment body models. (A) Concentrations of selected THS compounds from freshly-exposed surrogate LLF (measured via online PTR-TOF), shown with their Henry's law constants (H′). (B) Average peak concentrations observed from LLF off-gassing in the first 80 min compared to SHS 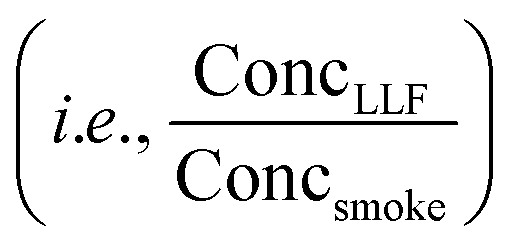 averaged across multiple trials, showing enhanced near-term off-gassing around H′ of 10−3–100 mol m−3 Pa−1. (C) Comparison of LLF off-gassing concentrations after 1 h to initial concentration in the first 20 min, with compounds colored by H′. (D) Average concentration-weighted distribution of near-term concentration enhancements from LLF shown with the distribution of SHS and THS from LLF (within the same experiment). The top panel in (D) depicts the competing roles of aqueous uptake (i.e., maximum possible uptake in LLF exposed to smoke) and off-gassing rates, which are both a function of Henry's law and drive enhanced initial off-gassing of compounds with lower H′ and lead to longer residence times for compounds with a greater affinity for the aqueous phase. (E) Modeled long-term breath concentrations of a compound with H′ = 0.5 mol m−3 Pa−1 over 100 hours considering LLF only compared to multi-compartment body models with blood and moderately-perfused tissues. (F) The temporal evolution of the relative chemical composition of breath as a function of H′ including LLF, blood, and tissues (given an equivalent initial concentration in mainstream smoke). See Fig. S25 and S26† for relevant experimental and modeling results, including additional depictions of (E) and the absolute changes of data in (F). averaged across multiple trials, showing enhanced near-term off-gassing around H′ of 10−3–100 mol m−3 Pa−1. (C) Comparison of LLF off-gassing concentrations after 1 h to initial concentration in the first 20 min, with compounds colored by H′. (D) Average concentration-weighted distribution of near-term concentration enhancements from LLF shown with the distribution of SHS and THS from LLF (within the same experiment). The top panel in (D) depicts the competing roles of aqueous uptake (i.e., maximum possible uptake in LLF exposed to smoke) and off-gassing rates, which are both a function of Henry's law and drive enhanced initial off-gassing of compounds with lower H′ and lead to longer residence times for compounds with a greater affinity for the aqueous phase. (E) Modeled long-term breath concentrations of a compound with H′ = 0.5 mol m−3 Pa−1 over 100 hours considering LLF only compared to multi-compartment body models with blood and moderately-perfused tissues. (F) The temporal evolution of the relative chemical composition of breath as a function of H′ including LLF, blood, and tissues (given an equivalent initial concentration in mainstream smoke). See Fig. S25 and S26† for relevant experimental and modeling results, including additional depictions of (E) and the absolute changes of data in (F). | ||
These differences were explored using Henry's law constants to represent variations in gas–aqueous-phase interactions. Enhancements in short-term LLF off-gassing were examined by plotting the ratio of LLF off-gassing to gas-phase SHS concentrations for each compound against the Henry's law constants (Fig. 5B) available for 139 compounds.42 The gases in SHS have Henry's law constants (Hcp, i.e., H′) that are largely distributed in the 10−4–104 mol m−3 Pa−1 range (Fig. S22†). The resulting distribution of the LLF to SHS concentration ratios peaked in the range of H′ = 10−3–100 mol m−3 Pa−1 at a ratio of approximately 0.7, which indicates relatively enhanced near-term emissions (e.g., during the first hour) that is influenced by a combination of their aqueous uptake and off-gassing (Fig. 5B–D). Compounds with Henry's law constants of 100 mol m−3 Pa−1 and greater generally remained substantially elevated over an hour of off-gassing (e.g., Fig. 5C, S23 and S24†). We also note that compounds with a H′ below 10−3 mol m−3 Pa−1 were consistently found at concentrations approximately an order of magnitude lower in the LLF off-gassing compared to SHS.
These observations suggest that this peak is the result of the competing processes of aqueous uptake (i.e., maximum possible uptake in LLF exposed to smoke) and off-gassing rates, which are both a function of Henry's law (Fig. 5D). If the Henry's law constant of a given compound is very low, then the quantity of uptake to the LLF is greatly limited by the Henry's law constant value. The off-gassing will be rapid, but the low overall quantity able to be sequestered by the LLF means that the ratio of LLF off-gassing to SHS concentration will be low. On the other hand, compounds with a higher Henry's law constant will be taken up in greater quantities but will off-gas more slowly when exposed to cleaner air. Therefore, compounds with a higher H′ will be more persistent in the aqueous phase and off-gas over long timescales as they return to equilibrium (in the absence of biological processing). Compounds in the middle with an H′ around 10−2 enter the LLF in substantial amounts, but do not have a prohibitively high aqueous-phase affinity, allowing them to off-gas in greater quantities over shorter timescales.
4 Discussion
4.1 Real-world implications and considerations
These highly volatile compounds would likely not have been able to achieve sufficient sorption solely from the more diluted gas-phase (e.g., outside the concentrated plume) to clothing (and external bodily surfaces) to produce the immediate, large concentration enhancements observed in the theater with the entry of new audiences.11 While gas-phase adsorption and desorption of these components to fabrics and materials have been studied in prior work,18,19 the particle-phase and aqueous-phase reservoirs explored in this work represent highly-concentrated and viably persistent THS reservoirs that are dosed in very-high concentration gradient conditions, specifically in the cigarette plume or respiratory tract after mainstream smoke inhalation, respectively.
The underlying source strength of off-gassing PM originates during tobacco combustion, which produces high concentrations of tobacco smoke-related gases and particles, resulting in a conducive environment for aerosol growth. Condensed-phase particles are generated and grow in the highly-concentrated tobacco smoke plume during combustion; they thus incorporate a wide volatility range of gas-phase organic compounds present at extremely elevated gas-phase concentrations, which, along with high organic aerosol loadings, drive their condensation to the particle phase in spite of high vapor pressures. These extremely-elevated concentrations can even induce some of the most volatile species to be present in the particle phase in substantial quantities (e.g., acetone) (Table S3†), though the extent of light VOC content in PM-related THS is likely sensitive to the duration and extent of airborne dilution prior to PM deposition. One prior study on two VOCs supports this conclusion, estimating that 20–70% of acrolein and formaldehyde in SHS emissions were in the condensed phase.65 Once in the particle matrix, viscosity and diffusion limitations can then limit their rate of off-gassing,66,67 effectively “trapping” them in a viscous and/or phase-separated THS reservoir. The viscosity of these particles may evolve over time, similar to observations of biomass burning organic aerosol outdoors.68
As a result, in the experiments, the high viscosity of these particles extended measured off-gassing lifetimes, such that the PM still emitted a considerable amount of THS contaminants even at 18 and 72 hours of aging with continuous air flow (Fig. 4A–C). These results support PM as a major, highly-concentrated THS reservoir in real-world indoor spaces with off-gassing concentrations of VOCs–SVOCs that remain elevated over extended timescales (Fig. 1, 4, S10 and S14†). Since gas-phase concentrations of many off-gassing compounds (e.g., I/SVOCs) remained similar to their SHS gas-phase concentrations (Fig. 4C and S12†), both initially and over several hours in the controlled PM experiments, gas-phase concentrations in real-world indoor environments are likely dependent on the saturation concentrations (C*) of many compounds, especially in spaces with limited ventilation.
First, the flow velocity through the filter was ∼10 m h−1, which is slightly higher than the 1–5 m h−1 range of vertical transport velocities expected in indoor environments (e.g., 2.5–3.9 m h−1)20 and likely resulted in more rapid air exchange per time than in real indoor spaces. Second, the use of purified air in the PM experiments may have accelerated aging in this study, as real-world diffusion will often have smaller concentration gradients between the PM surface and in-room air. With low to moderate ventilation for indoor spaces contaminated with deposited PM from SHS, off-gassing may approach saturation concentrations for higher-MW compounds, similar to their gas-phase (in-room) concentrations during smoking, resulting in elongated emission timescales and exposure to gas-phase THS over longer periods, though the impact of ventilation variations (after deposition) may not dramatically lower gas-phase concentrations of many species based on prior work.51
Third, the underlying surface that the viscous PM deposits on could impact THS timescales via several mechanisms. THS from PM (or from the surrounding gas-phase) can penetrate deep into a sufficiently thick surface (e.g., paint) or other building materials and re-partition between the particle and the underlying sorptive surface/material—increasing the overall persistence of THS off-gassing.21 Fourth, while the intention of our experimental design was to isolate and focus on off-gassing from deposited PM (or LLF) to the gas-phase, in real-world indoor environments, off-gassing VOCs, IVOCs, and SVOCs will partly re-partition to indoor surfaces, other bulk material reservoirs (e.g., building materials, furnishings, fabrics), and other (non-tobacco related) aerosols.12 This sorptive partitioning will contribute to extended persistence of THS indoors and could be impacted by humidity levels. Similarly, off-gassed THS may not be isolated to a single room, as other aerosols can act as condensation sites for PM off-gassing emissions, and this aerosol-phase transport can allow them to move to other parts of buildings via either connected rooms or through forced air (HVAC) systems12 in addition to gas-phase THS transport.
In summary, the timescales for THS PM off-gassing in real-world conditions will vary as a function of several factors: initial particle size, particle viscosity (i.e., internal diffusion coefficients), compound volatility and polarity, THS layer thickness resulting from deposited PM, ventilation rates, and possible effects from phase state, viscosity, and pH.70–72 For comparison, single particles have modeled equilibrium timescales ranging from less than a second to weeks once settled on surfaces, with a strong dependence on viscosity and diffusive length scales (Fig. S21†).
In past observations of smokers' breath over time, the concentration of exhaled particles returned to baseline one minute after cessation of smoking,74 while time-resolved observations of exhaled VOCs were limited to only a few compounds. Ueta et al. observed fairly rapid initial decay rates (i.e., e-folding time (τ) < 2 min) for four VOCs (benzene, 2,5-dimethylfuran, toluene, and limonene) from peak concentrations in smokers' breath after the last cigarette puff.19 Others reported similar initial decay timescales for benzene, 2,5-dimethylfuran, and 1,3-butadiene, though concentrations sometimes persisted at elevated levels during the 1–2 hours tested.33,34 In contrast, exhaled acetonitrile was found to decay much more slowly over multiple days.34,75
Our observed decay trends in LLF off-gassing are consistent with these prior observations19,33,34,75 and the slower decay in many compounds can be explained by their Henry's law solubility constants (Fig. 5). Specifically, benzene (H′ = 0.0018 mol m−3 Pa−1), 2,5-dimethylfuran (0.0015), toluene (0.0015), and limonene (0.0004) are all largely non-polar, undergo relatively less aqueous uptake, and exhibit faster off-gassing (Fig. 5D top). By contrast, acetonitrile has a higher Henry's law constant (0.52 mol m−3 Pa−1), and its persistence relative to benzene has been attributed to differences in solubility.75 A greater tendency towards the aqueous phase means that aqueous fluids/tissues not only have a greater capacity to take up the extensive array of more soluble compounds (Fig. 5B, D and S23†), but also that the gas-phase equilibrium concentration over the liquid surface is low. Thus, at a constant exchange rate of the gas phase, off-gassing for compounds with higher solubility is slow and aqueous fluids/tissues may act as more persistent THS reservoirs over longer periods of time.
Our LLF off-gassing data showed an elevated abundance of compounds with H′ = 10−2–101 mol m−3 Pa−1 (relative to SHS), including acetonitrile (Fig. 5B). Therefore, real-world THS off-gassing from LLF can be expected to include a prevalent set of VOCs (e.g., Fig. 2) that will likely behave similarly to acetonitrile in exhaled breath. Other compounds with greater solubility coefficients may be even more readily retained in the aqueous phase and thus persist over longer timescales. This assertion is supported by our observations of LLF off-gassing after an hour of off-gassing compared to fresh LLF (Fig. 5C), where concentrations of some compounds decreased considerably, but compounds with Henry's law constants of 100 mol m−3 Pa−1 and greater generally remained similar to, or within an order of magnitude of their initial values at steady elevated levels (e.g., Fig. 5A and C).
Though the total volume of LLF used in this study was based on LLF in an average human respiratory system (25 mL), the bubbler containing surrogate LLF was not intended to mimic the human respiratory system.76 In order to examine expected concentrations of THS from thin films of LLF in the human body and the role of uptake to other bodily reservoirs, we used a multi-compartment model that included aqueous uptake to LLF, blood, and moderately-perfused tissues (Fig. S5†), with timescales based on prior work.33,34 The model was validated against the limited available data on acetonitrile breath concentrations,34 which is prevalent in LLF THS and represents a compound with a median H′ value in THS (Fig. 5D and S22†).
Model simulations were performed in three stages, first only including LLF and then successively adding the blood and tissue compartments. When including only LLF in the model, peak exhaled compound concentrations were generally lower than mainstream smoke by an order of magnitude (e.g., Fig. S25 and S26†), similar to the experimental results (Fig. 3C). When partitioning from LLF to blood/tissues was included, greater uptake of smoke components was observed (Fig. S25†), but the initial off-gassing concentrations were lower (Fig. S25†). However, incorporating the blood and then tissue reservoirs increased the overall persistence, extending over days for some compounds (Fig. 5E), where the incorporation of tissues provided an additional reservoir that dropped compound loadings in the blood and thus led to lower breath concentrations over the first day (Fig. 5E and S26†). The multi-day profile, shown for a compound with H′ = 0.5 mol m−3 Pa−1 (Fig. 5E and S25A†), reveals information about distinct processes in off-gassing via breath. In the first minutes, breath concentrations are driven by the off-gassing of the THS-rich LLF layer, along with further uptake into blood. Thereafter, breath concentrations are determined by partitioning between blood and tissues, which moderate the re-partitioning to LLF and subsequent exhalation.
Concentrations of compounds with smaller H′ (e.g., ≤10−2) started elevated, but decreased faster than other compounds (Fig. 5F and S26†), similar to the lab observations (Fig. 5A, C, S25 and S26†). Normalized concentration plots as a function of H′ (Fig. 5F) show that compounds with H′ of 10−1–101 mol m−3 Pa−1 comprise a greater fraction of emissions over time, which aligns with the observed peak in LLF off-gassing (Fig. 5D).
The extent of recent prior smoking in the hours preceding to the last cigarette affected the initial loadings of compounds in blood and tissue reservoirs, which thus impacted the breath concentrations and is an important consideration for THS transport. In the absence of this prior exposure, the tissues act as a much stronger sink. Prior smoking (e.g., several cigarettes in the preceding hours) dramatically increases the in-body loadings (e.g., 8× in Fig. S25†) and thus the breath concentrations from a moderate to heavy smoker, which is consistent with a study focused on acetonitrile where general breath concentrations increased with the number of cigarettes consumed per day.75,77
A few differences remain that may distinguish our experimental and modeling results from real-world THS off-gassing via LLF, with a more detailed discussion in Section S5.2 of the ESI.† (1) Differences between the properties of surrogate LLF and real LLF can include possible variations in pH, the inclusion of lipids and proteins, chemical interactions of THS compounds with human LLF components not included here, and increased viscosity that may affect relevant physical processes in the respiratory tract, and potentially in blood.77 (2) Similarly, other tissues in the respiratory tract or throughout the body will engage in non-aqueous partitioning, especially for compounds with low Henry's law constants. The slightly slower decay and persistence of lower H′ compounds in breath studies following smoking (e.g., at 10's of ppb for benzene, acetylene, 2,5-dimethylfuran, 2-methylfuran33,35,78) compared to our multicompartment model may suggest non-aqueous uptake to tissues (along with particle deposition) playing a role in extending the off-gassing timescales of lower H′ THS components. (3) Additionally, within these multi-compartment bodily reservoirs, the metabolism of tobacco smoke contaminants (e.g., the conversion of nicotine to cotinine) also needs to be considered as an in-body loss process for the complex mixture of THS compounds and their metabolites. Other possible loss processes should also be considered, such as the cycling rates of the various aqueous reservoirs, including LLF and blood, and how exactly these aqueous reservoirs are processed or eliminated. (4) Lastly, with real-world smoking, large concentrations of particles from mainstream smoke will deposit on airway surfaces or dissolve into LLF.79 While particles were intentionally filtered in this study to independently focus on gas-phase uptake into LLF (with the intentional exception of one experiment, Fig. S4 and S22†), this particle deposition would provide an additional in-lung THS reservoir and may affect the composition and persistence of THS in smokers' breath.
The multi-faceted health effects of THS have been explored and reported across a number of publications for both exposure to real THS (e.g., animal trials with THS exposure, human cohort studies) and by estimating effects from exposure to a subset of previously-known THS components (e.g., acrolein) and select transformation products (e.g., TSNAs).1,4,14,80–85 The results of this study expand the scope of identified THS components whose direct effects and secondary transformation products will help to elucidate observed THS health effects and evaluate chemically-speciated contributions to health effects estimates.
While many of the observed compound formulas may not have sufficient health effects data, PAHs, oxygenated PACs (e.g., quinones), quinolines (e.g., quinoline/isoquinoline and methyl-quinolines at 23 ppb and 9.2 ppb in off-gassing PM, respectively), and possible contributions from nitro-PACs (e.g., 1-nitropyrene, 1/2-nitronaphthalene; Table S5†) are newly-identified THS compounds of particular interest, given their mutagenicity, genotoxicity, and their ability to cause oxidative stress.86–91 We also observed immediate off-gassing of nitrosamines (e.g., NNK) from fresh PM and persistent enhanced off-gassing of acrolein from PM. Among these potential health effects, THS represents a source of reactive oxygen species (ROS) generation in non-smokers' respiratory systems due to compounds such as quinones and nitrated-PACs.86,92,93 ROS, which includes a range of radicals that lead to oxidative stress,94 has also been similarly observed with SHS95–97 and biomass burning98via dithiothreitol assays or electron paramagnetic resonance spectroscopy.
The oxidation of deposited PM or other THS-contaminated surfaces will lead to further functionalization of THS organic compounds (Fig. 2 and S7†), potentially increasing their toxicity99,100 or carcinogenicity, such as that observed with nitrosamines that are formed on surfaces via nicotine + nitrous acid (HONO) reactions.14 Additionally, the diverse range of off-gassing compounds in THS includes a number of highly-reactive species important for both airborne and surface chemistry indoors, including the formation of secondary organic aerosols and gas-phase oxidation products.
5 Conclusions and future work
5.1 Conclusions
The laboratory experiments conducted here with multiple state-of-the-art measurement methods demonstrate that both deposited PM from tobacco smoke and aqueous bodily fluids/tissues (e.g., LLF) previously exposed to tobacco smoke gases can be abundant reservoirs of THS emissions that off-gas substantial quantities of THS over immediate and extended timescales. These observations of THS are more chemically complex than previously reported, spanning a broad volatility spectrum with a range of chemical functionalities and varying physical/chemical properties that drive the dynamics of both uptake and emission processes. Emissions of compounds ranging from light VOCs to SVOCs included hazardous and highly-reactive compounds that were persistent over the timescales considered here, whose health impacts could be amplified by known real-world transformations, such as tobacco smoke nitrosamine (TSNA) formation from nicotine and similar alkaloids following exposure to HONO.14The observed THS off-gassing from surrogate LLF and PM, which can deposit on clothes, furnishings, bodies, and/or airways, are complementary to previously observed gas-phase sorption of THS to fabrics and other materials18,19 by representing key underlying processes of efficient THS off-gassing that need to be considered for THS contamination in locations where smoking has previously occurred and for transport into non-smoking environments.11 For instance, these processes elucidate how elevated levels of nicotine have been observed on surfaces in a multitude of non-smoking environments1 and how PAHs, which have been linked to an increase in lung cancer incidence,101 can be found in the homes of smokers who did not smoke indoors.7 This re-partitioning of THS initially from PM and LLF to other indoor surfaces/materials and internal aqueous bodily reservoirs, respectively, will extend the lifetimes of THS contamination, as discussed above.
5.2 Opportunities for future work
While this study elucidates underlying THS reservoirs, including those that facilitate both persistent contamination and prompt off-gassing following transport for a wide range of THS contaminants, continued research on the magnitude and dynamics of THS reservoirs will further our understanding of the driving factors and impacts of THS off-gassing. This knowledge on their dynamics will also inform the ongoing assessment of mitigation strategies (e.g., dust removal, surface cleaning, ventilation, ozonation). While our detailed examination of chemically-dependent processes focused on THS, these results are also relevant to other combustion emissions related to indoor air quality (e.g., biomass burning or cooking), outdoor air quality (as seen in past wildfire plume dilution studies102,103), and aqueous-phase emissions/uptake, such as with vaping,104,105 waterpipe tobacco smoking,106 or humidifiers.107The role of PM deposition into LLF also requires further examination to determine its impact relative to gas-phase uptake in a thin film surrogate LLF system. To inform this future work, in one experiment (Fig. S18†), LLF was exposed to SHS without a particle filter to allow for some fraction of the particle mass to enter the LLF in the bubbler. The overall concentration was slightly elevated with minor increases for some compounds and generally similar composition (Fig. S19†), but future work is warranted with deposition in a thin film with quantified PM uptake to examine the interactive effect of PM and LLF.
One area of potential variance between tobacco products is the incorporation of cigarette contaminants into SHS and thus THS. Notable levels of phthalates (Table S3†) and cyclic siloxanes (i.e., D3, D4, D5, D6, D7) were observed in this study, and THS may also contain pesticides used in tobacco production and their combustion by-products. Additionally, polychlorinated biphenyls (PCBs) and polychlorinated dibenzo-dioxins/furans (PCDD/Fs) have been previously observed in tobacco smoke.111 Hence, they are likely present in deposited PM and may also be a component of off-gassing THS. Exploratory off-gassing measurements as part of this work were inconclusive for PCDD/Fs, but dioxin-like PCBs were observed at varying levels across samples (Table S5B†) and further research should be done to quantify their off-gassing rates relative to typical indoor levels.
Given chemical-dependent interactions with sorptive reservoirs, future work should employ online and offline high-resolution mass spectrometry techniques to examine uptake and off-gassing of organic compounds from these and other potential reservoirs. Furthermore, the distinct physical–chemical processes impacting LLF uptake and off-gassing observed here warrant investigation into the role of surface humidity on the uptake of SHS gases to common indoor materials and surfaces.
Lastly, this expanded chemical characterization of gas-phase THS and condensed-phase THS (from deposited PM) provides opportunities to expand potential toxicology assessments and provides a detailed speciation of gas- and particle-phase SHS (Fig. 2 and S7†) for similar purposes. Past bottom-up assessments of THS health effects have been limited to a finite number of speciated compounds1,4 that may lead to underestimates, and toxicity may also be enhanced by chemical transformations of the broader set of reactive precursors shown here, extending beyond TSNAs.4 Future work can leverage the data collected here and similar advanced techniques to examine contributions from functional group classes and individual high-toxicity compounds to elucidate the multifaceted health effects of THS and the underlying mechanisms of human exposure.
Funding
R. S. and D. R. G. would like to acknowledge financial support from NSF CBET (CBET-2011362) and NSF GRFP (DGE1122492 and DGE1752134), and also from NSF AGS (AGS-1764126), which enabled the development of the offline analytical capabilities applied here. D. R. G. is also grateful for support from the Alexander von Humboldt Fellowship, which facilitated the collaboration with MPIC. Furthermore, the work was supported by the Max Planck Society. Additionally, D. R. G. acknowledges support from the U.S. EPA in this area of research. Laboratory resources at the Masaryk University were supported by the Ministry of Education, Youth, and Sports of the Czech Republic (RECETOX Research Infrastructure LM2018121). This publication was developed under assistance agreement no. RD835871 awarded by the U.S. EPA to Yale University. It has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the EPA. EPA does not endorse any products or commercial services mentioned in this publication.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Additional data related to this paper may be requested from the authors.Author contributions
R. S., J. W., and D. R. G. conceived the study. A. F. and H. T. prepared surrogate LLF samples and assisted with PTFE filter preparations. T. K., A. R., A. E., and D. R. G. ran the Vocus PTR-TOF, and A. R. pre-processed of the data output. D. R. G. sampled tubes and filters, and R. S. and J. E. M. analyzed these offline samples at Yale. B. A. M. B., M. W., P. K., and G. L. conducted analysis of the PUF samples. R. S., T. H.-M., J. E. M., and D. R. G. analyzed and interpreted the experimental data. T. B., D. R. G., and R. S. modeled timescales for THS off-gassing from PM and LLF. R. S. and D. R. G. wrote the paper. All authors reviewed the manuscript.Conflicts of interest
Authors declare they have no competing interests.Acknowledgements
The authors would like to acknowledge Kanako Sekimoto and especially Jordan Krechmer (Aerodyne) for their assistance with analyzing the Vocus PTR-TOF data. The authors also recognize Hugo Destaillats (LBNL) for feedback on an early version of the manuscript. The authors thank GERSTEL for their collaboration with the TD 3.5 used in this study.References
- P. Jacob, N. L. Benowitz, H. Destaillats, L. Gundel, B. Hang, M. Martins-Green, G. E. Matt, P. J. E. Quintana, J. M. Samet, S. F. Schick, P. Talbot, N. J. Aquilina, M. F. Hovell, J. H. Mao and T. P. Whitehead, Thirdhand smoke: new evidence, challenges, and future directions, Chem. Res. Toxicol., 2017, 30, 270–294 Search PubMed.
- W. W. Nazaroff and B. C. Singer, Inhalation of hazardous air pollutants from environmental tobacco smoke in US residences, J. Expo. Sci. Environ. Epidemiol., 2004, 14, S71–S77 CrossRef CAS PubMed.
- G. E. Matt, P. J. E. Quintana, H. Destaillats, L. A. Gundel, M. Sleiman, B. C. Singer, P. Jacob, N. Benowitz, J. P. Winickoff, V. Rehan, P. Talbot, S. Schick, J. Samet, Y. Wang, B. Hang, M. Martins-Green, J. F. Pankow and M. F. Hovell, Thirdhand Tobacco Smoke: Emerging Evidence and Arguments for a Multidisciplinary Research Agenda, Environ. Health Perspect., 2011, 119, 1218–1226 CrossRef CAS PubMed.
- M. Sleiman, J. M. Logue, W. Luo, J. F. Pankow, L. A. Gundel and H. Destaillats, Inhalable constituents of thirdhand tobacco smoke: chemical characterization and health impact considerations, Environ. Sci. Technol., 2014, 48, 13093–13101 CrossRef CAS PubMed.
- J. Barnoya and S. A. Glantz, Cardiovascular Effects of Secondhand Smoke, Circulation, 2005, 111, 2684–2698 CrossRef.
- Centers for Disease Control and Prevention (US) and U.S. Department of Health and Human Services, The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General, 2006 Search PubMed.
- G. E. Matt, P. J. E. Quintana, J. M. Zakarian, A. L. Fortmann, D. A. Chatfield, E. Hoh, A. M. Uribe and M. F. Hovell, When smokers move out and non-smokers move in: Residential thirdhand smoke pollution and exposure, Tob. Control, 2011, 20, 1–8 CrossRef PubMed.
- E. Hoh, R. N. Hunt, P. J. E. Quintana, J. M. Zakarian, D. A. Chatfield, B. C. Wittry, E. Rodriguez and G. E. Matt, Environmental tobacco smoke as a source of polycyclic aromatic hydrocarbons in settled household dust, Environ. Sci. Technol., 2012, 46, 4174–4183 CrossRef CAS PubMed.
- G. E. Matt, P. J. E. Quintana, J. M. Zakarian, E. Hoh, M. F. Hovell, M. Mahabee-Gittens, K. Watanabe, K. Datuin, C. Vue and D. A. Chatfield, When smokers quit: exposure to nicotine and carcinogens persists from thirdhand smoke pollution, Tob. Control, 2017, 26, 548–556 CrossRef PubMed.
- M. Martins-Green, N. Adhami, M. Frankos, M. Valdez, B. Goodwin, J. Lyubovitsky, S. Dhall, M. Garcia, I. Egiebor, B. Martinez, H. W. Green, C. Havel, L. Yu, S. Liles, G. Matt, H. Destaillats, M. Sleiman, L. A. Gundel, N. Benowitz, P. Jacob, M. Hovell, J. P. Winickoff and M. Curras-Collazo, Cigarette smoke toxins deposited on surfaces: implications for human health, PLoS One, 2014, 9, 86391 CrossRef PubMed.
- R. Sheu, C. Stönner, J. C. Ditto, T. Klüpfel, J. Williams and D. R. Gentner, Human transport of thirdhand tobacco smoke: a prominent source of hazardous air pollutants into indoor nonsmoking environments, Sci. Adv., 2020, 6, eaay4109 CrossRef CAS PubMed.
- P. F. DeCarlo, A. M. Avery and M. S. Waring, Thirdhand smoke uptake to aerosol particles in the indoor environment, Sci. Adv., 2018, 4, eaap8368 CrossRef.
- G. E. Matt, P. J. E. Quintana, A. L. Fortmann, J. M. Zakarian, V. E. Galaviz, D. A. Chatfield, E. Hoh, M. F. Hovell and C. Winston, Thirdhand smoke and exposure in California hotels: non-smoking rooms fail to protect non-smoking hotel guests from tobacco smoke exposure, Tob. Control, 2014, 23, 264–272 CrossRef PubMed.
- M. Sleiman, L. A. Gundel, J. F. Pankow, P. Jacob, B. C. Singer and H. Destaillats, Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6576–6581 CrossRef CAS PubMed.
- N. Ramírez, M. Z. Özel, A. C. Lewis, R. M. Marcé, F. Borrull and J. F. Hamilton, Exposure to nitrosamines in thirdhand tobacco smoke increases cancer risk in non-smokers, Environ. Int., 2014, 71, 139–147 CrossRef PubMed.
- K. Yeh, L. Li, F. Wania and J. P. D. Abbatt, Environ. Int., 2022, 160, 107063 CrossRef CAS PubMed.
- B. C. Singer, A. T. Hodgson, K. S. Guevarra, E. L. Hawley and W. W. Nazaroff, Gas-phase organics in environmental tobacco smoke. 1. Effects of smoking rate, ventilation, and furnishing level on emission factors, Environ. Sci. Technol., 2002, 36, 846–853 CrossRef CAS PubMed.
- B. C. Singer, K. L. Revzan, T. Hotchi, A. T. Hodgson and N. J. Brown, Sorption of organic gases in a furnished room, Atmos. Environ., 2004, 38, 2483–2494 CrossRef CAS.
- I. Ueta, Y. Saito, K. Teraoka, T. Miura and K. Jinno, Determination of Volatile Organic Compounds for a Systematic Evaluation of Third-Hand Smoking, Anal. Sci., 2010, 26, 569–574 CrossRef CAS PubMed.
- C. J. Weschler and W. W. Nazaroff, Semivolatile organic compounds in indoor environments, Atmos. Environ., 2008, 42, 9018–9040 CrossRef CAS.
- L. B. Algrim, D. Pagonis, J. A. de Gouw, J. L. Jimenez and P. J. Ziemann, Measurements and modeling of absorptive partitioning of volatile organic compounds to painted surfaces, Indoor Air, 2020, 30, 745–756 CrossRef CAS.
- D. B. Collins, C. Wang and J. P. D. Abbatt, Selective Uptake of Third-Hand Tobacco Smoke Components to Inorganic and Organic Aerosol Particles, Environ. Sci. Technol., 2018, 52, 13195–13201 CrossRef CAS.
- T. P. Whitehead, C. Havel, C. Metayer, N. L. Benowitz and P. Jacob, Tobacco Alkaloids and Tobacco-Specific Nitrosamines in Dust from Homes of Smokeless Tobacco Users, Active Smokers, and Nontobacco Users, Chem. Res. Toxicol., 2015, 28, 1007–1014 Search PubMed.
- P. J. E. Quintana, G. E. Matt, D. Chatfield, J. M. Zakarian, A. L. Fortmann and E. Hoh, Wipe sampling for nicotine as a marker of thirdhand tobacco smoke contamination on surfaces in homes, cars, and hotels, Nicotine Tob. Res., 2013, 15, 1555–1563 Search PubMed.
- K. Min, P. Guo, D. Chen, S. Huang, W. Luo, M. Ma, B. Chen, S. Yao and H. Zuilhof, Direct and quantitative in situ analysis of third-hand smoke in and on various matrices by ambient desorption corona beam ionization mass spectrometry, Talanta, 2020, 219, 121330 CrossRef CAS PubMed.
- G. Bekö, G. Morrison, C. J. Weschler, H. M. Koch, C. Pälmke, T. Salthammer, T. Schripp, J. Toftum and G. Clausen, Measurements of dermal uptake of nicotine directly from air and clothing, Indoor Air, 2017, 27, 427–433 CrossRef PubMed.
- M. Borgerding and H. Klus, Analysis of complex mixtures – cigarette smoke, Exp. Toxicol. Pathol., 2005, 57, 43–73 CrossRef CAS PubMed.
- W. F. Rogge, L. M. Hildemann, M. A. Mazurek, G. R. Cass and B. R. T. Simonelt, Sources of Fine Organic Aerosol. 6. Cigarette Smoke in the Urban Atmosphere, Environ. Sci. Technol., 1994, 28, 1375–1388 CrossRef CAS PubMed.
- A. Rodgman and T. Perfetti, The Chemical Components of Tobacco and Tobacco Smoke, 2nd edn, 2013 Search PubMed.
- R. Talhout, T. Schulz, E. Florek, J. van Benthem, P. Wester and A. Opperhuizen, Hazardous compounds in tobacco smoke, Int. J. Environ. Res. Public Health, 2011, 8, 613–628 CrossRef PubMed.
- R. L. Stedman, The chemical composition of tobacco and tobacco smoke, Chem. Rev., 1968, 68, 153–207 CrossRef CAS PubMed.
- J. Dallüge, L. L. P. van Stee, X. Xu, J. Williams, J. Beens, R. J. Vreuls and U. A. T. Brinkman, Unravelling the composition of very complex samples by comprehensive gas chromatography coupled to time-of-flight mass spectrometry: cigarette smoke, J. Chromatogr. A, 2002, 974, 169–184 CrossRef.
- S. M. Gordon, L. A. Wallace, M. C. Brinkman, P. J. Callahan and D. V. Kenny, Volatile organic compounds as breath biomarkers for active and passive smoking, Environ. Health Perspect., 2002, 110, 689–698 CrossRef CAS PubMed.
- A. Jordan, A. Hansel, R. Holzinger and W. Lindinger, Acetonitrile and benzene in the breath of smokers and non-smokers investigated by proton transfer reaction mass spectrometry (PTR-MS), Int. J. Mass Spectrom. Ion Processes, 1995, 148, L1–L3 CrossRef CAS.
- J. M. M. Sanchez and R. D. D. Sacks, Development of a multibed sorption trap, comprehensive two-dimensional gas chromatography, and time-of-flight mass spectrometry system for the analysis of volatile organic compounds in human breath, Anal. Chem., 2006, 78, 3046–3054 CrossRef CAS PubMed.
- S. M. Gordon, Identification of exposure markers in smokers' breath, J. Chromatogr. A, 1990, 511, 291–302 CrossRef CAS PubMed.
- J. C. Ditto, E. B. Barnes, P. Khare, M. Takeuchi, T. Joo, A. A. T. Bui, J. Lee-Taylor, G. Eris, Y. Chen, B. Aumont, J. L. Jimenez, N. L. Ng, R. J. Griffin and D. R. Gentner, An omnipresent diversity and variability in the chemical composition of atmospheric functionalized organic aerosol, Commun. Chem., 2018, 1, 1–13 CrossRef CAS.
- P. Khare, A. Marcotte, R. Sheu, A. N. Walsh, J. C. Ditto and D. R. Gentner, Advances in offline approaches for trace measurements of complex organic compound mixtures via soft ionization and high-resolution tandem mass spectrometry, J. Chromatogr. A, 2019, 1598, 163–174 CrossRef CAS PubMed.
- J. Krechmer, F. Lopez-Hilfiker, A. Koss, M. Hutterli, C. Stoermer, B. Deming, J. Kimmel, C. Warneke, R. Holzinger, J. Jayne, D. Worsnop, K. Fuhrer, M. Gonin and J. de Gouw, Evaluation of a New Reagent-Ion Source and Focusing Ion-Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 2018, 90, 12011–12018 CrossRef CAS PubMed.
- K. Sekimoto, S. M. Li, B. Yuan, A. Koss, M. Coggon, C. Warneke and J. de Gouw, Calculation of the sensitivity of proton-transfer-reaction mass spectrometry (PTR-MS) for organic trace gases using molecular properties, Int. J. Mass Spectrom., 2017, 421, 71–94 CrossRef CAS.
- J. G. Charrier, A. S. McFall, N. K. Richards-Henderson and C. Anastasio, Hydrogen peroxide formation in a surrogate lung fluid by transition metals and quinones present in particulate matter, Environ. Sci. Technol., 2014, 48, 7010–7017 CrossRef CAS PubMed.
- R. Sander, Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- R. Sheu, A. Marcotte, P. Khare, S. Charan, J. C. Ditto and D. R. Gentner, Advances in offline approaches for chemically speciated measurements of trace gas-phase organic compounds via adsorbent tubes in an integrated sampling-to-analysis system, J. Chromatogr. A, 2018, 1575, 80–90 CrossRef CAS PubMed.
- P. S. J. Lakey, T. Berkemeier, H. Tong, A. M. Arangio, K. Lucas, U. Pöschl and M. Shiraiwa, Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract, Sci. Rep., 2016, 6(1), 1–6 CrossRef.
- V. Bahl, N. J. H. Weng, S. F. Schick, M. Sleiman, J. Whitehead, A. Ibarra and P. Talbot, Cytotoxicity of Thirdhand Smoke and Identification of Acrolein as a Volatile Thirdhand Smoke Chemical That Inhibits Cell Proliferation, Toxicol. Sci., 2016, 150, 234–246 CrossRef CAS.
- S. M. Charles, C. Jia, S. A. Batterman and C. Godwin, VOC and particulate emissions from commercial cigarettes: analysis of 2,5-DMF as an ETS tracer, Environ. Sci. Technol., 2008, 42, 1324–1331 CrossRef CAS PubMed.
- S. F. Schick, K. F. Farraro, C. Perrino, M. Sleiman, G. van de Vossenberg, M. P. Trinh, S. K. Hammond, B. M. Jenkins and J. Balmes, Thirdhand cigarette smoke in an experimental chamber: evidence of surface deposition of nicotine, nitrosamines and polycyclic aromatic hydrocarbons and de novo formation of NNK, Tob. Control, 2014, 23, 152–159 CrossRef.
- M. Gong, N. Daniels and D. Poppendieck, Measurement of chemical emission rates from cigarette butts into air, Indoor Air, 2020, 30, 711–724 CrossRef CAS.
- X. Lu, J. Cai, H. Kong, M. Wu, R. Hua, M. Zhao, J. Liu and G. Xu, Analysis of cigarette smoke condensates by comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry I acidic fraction, Anal. Chem., 2003, 75, 4441–4451 CrossRef CAS PubMed.
- T. Gröger, W. Welthagen, S. Mitschke, M. Schäffer and R. Zimmermann, Application of comprehensive two-dimensional gas chromatography mass spectrometry and different types of data analysis for the investigation of cigarette particulate matter, J. Sep. Sci., 2008, 31, 3366–3374 CrossRef PubMed.
- R. Sheu, C. F. Fortenberry, M. J. Walker, A. Eftekhari, C. Stönner, A. Bakker, J. Peccia, J. Williams, G. C. Morrison, B. J. Williams and D. R. Gentner, Evaluating Indoor Air Chemical Diversity, Indoor-to-Outdoor Emissions, and Surface Reservoirs Using High-Resolution Mass Spectrometry, Environ. Sci. Technol., 2021, 55, 10255–10267 CrossRef CAS PubMed.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. de Gouw, Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 2018, 18, 3299–3319 CrossRef CAS.
- B. C. Singer, A. T. Hodgson and W. W. Nazaroff, Gas-phase organics in environmental tobacco smoke: 2. Exposure-relevant emission factors and indirect exposures from habitual smoking, Atmos. Environ., 2003, 37, 5551–5561 CrossRef CAS.
- M.-S. Lee, R. F. LeBouf, Y.-S. Son, P. Koutrakis and D. C. Christiani, Nicotine, aerosol particles, carbonyls and volatile organic compounds in tobacco- and menthol-flavored e-cigarettes, Environ. Health, 2017, 16, 42 CrossRef PubMed.
- V. Klaus, T. Hartmann, J. Gambini, P. Graf, W. Stahl, A. Hartwig and L. O. Klotz, 1,4-Naphthoquinones as inducers of oxidative damage and stress signaling in HaCaT human keratinocytes, Arch. Biochem. Biophys., 2010, 496, 93–100 CrossRef CAS PubMed.
- C. E. Rodriguez, M. Shinyashiki, J. Froines, R. C. Yu, J. M. Fukuto and A. K. Cho, An examination of quinone toxicity using the yeast Saccharomyces cerevisiae model system, Toxicology, 2004, 201, 185–196 CrossRef CAS PubMed.
- X. Liu, B. Deming, D. Pagonis, D. A. Day, B. B. Palm, R. Talukdar, J. M. Roberts, P. R. Veres, J. E. Krechmer, J. A. Thornton, J. A. De Gouw, P. J. Ziemann and J. L. Jimenez, Effects of gas-wall interactions on measurements of semivolatile compounds and small polar molecules, Atmos. Meas. Tech., 2019, 12, 3137–3149 CrossRef CAS.
- D. Arndt, C. Wachsmuth, C. Buchholz and M. Bentley, A complex matrix characterization approach, applied to cigarette smoke, that integrates multiple analytical methods and compound identification strategies for non-targeted liquid chromatography with high-resolution mass spectrometry, Rapid Commun. Mass Spectrom., 2020, 34(2), 1–13 CrossRef PubMed.
- S. Vreeke, D. H. Peyton and R. M. Strongin, Triacetin Enhances Levels of Acrolein, Formaldehyde Hemiacetals, and Acetaldehyde in Electronic Cigarette Aerosols, ACS Omega, 2018, 3, 7165–7170 CrossRef CAS PubMed.
- T. Laino, C. Tuma, P. Moor, E. Martin, S. Stolz and A. Curioni, Mechanisms of Propylene Glycol and Triacetin Pyrolysis, J. Phys. Chem. A, 2012, 116, 4602–4609 CrossRef CAS PubMed.
- M. Sleiman, J. M. Logue, V. N. Montesinos, M. L. Russell, M. I. Litter, L. A. Gundel and H. Destaillats, Emissions from electronic cigarettes: key parameters affecting the release of harmful chemicals, Environ. Sci. Technol., 2016, 50, 9644–9651 CrossRef CAS PubMed.
- M. R. R. Nimlos, S. J. J. Blanksby, X. Qian, M. E. E. Himmel and D. K. K. Johnson, Mechanisms of Glycerol Dehydration, J. Phys. Chem. A, 2006, 110, 6145–6156 CrossRef CAS PubMed.
- A. K. Armitage and D. M. Turner, Absorption of nicotine in cigarette and cigar smoke through the oral mucosa, Nature, 1970, 226, 1231–1232 CrossRef CAS PubMed.
- B. R. Appel, G. Guirguis, I. S. Kim, O. Garbin, M. Fracchia, C. P. Flessel, K. W. Kizer, S. A. Book and T. E. Warriner, Benzene, benzo(a)pyrene, and lead in smoke from tobacco products other than cigarettes, Am. J. Public Health, 1990, 80, 560–564 CrossRef CAS PubMed.
- H. E. Ayer and D. W. Yeager, Irritants in cigarette smoke plumes, Am. J. Public Health, 1982, 72, 1283–1285 CrossRef CAS PubMed.
- E. John, S. Coburn, C. Liu, J. McAughey, D. Mariner, K. G. McAdam, I. Bakos and S. Dóbé, Gas-Particle Partitioning of Formaldehyde in Mainstream Cigarette Smoke, Beitr. Tab. Int./ Contrib. Tob. Res., 2020, 29, 2–20 CAS.
- J. P. Reid, A. K. Bertram, D. O. Topping, A. Laskin, S. T. Martin, M. D. Petters, F. D. Pope and G. Rovelli, The viscosity of atmospherically relevant organic particles, Nat. Commun., 2018, 9, 1–14 CrossRef CAS PubMed.
- K. Adachi, A. J. Sedlacek, L. Kleinman, S. R. Springston, J. Wang, D. Chand, J. M. Hubbe, J. E. Shilling, T. B. Onasch, T. Kinase, K. Sakata, Y. Takahashi and P. R. Buseck, Spherical tarball particles form through rapid chemical and physical changes of organic matter in biomass-burning smoke, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19336–19341 CrossRef CAS PubMed.
- H. Mai, M. Shiraiwa, R. C. Flagan and J. H. Seinfeld, Under What Conditions Can Equilibrium Gas–Particle Partitioning Be Expected to Hold in the Atmosphere?, Environ. Sci. Technol., 2015, 49, 11485–11491 CrossRef CAS PubMed.
- J. C. Ditto, T. Joo, P. Khare, R. Sheu, M. Takeuchi, Y. Chen, W. Xu, A. A. T. Bui, Y. Sun, N. L. Ng and D. R. Gentner, Effects of Molecular-Level Compositional Variability in Organic Aerosol on Phase State and Thermodynamic Mixing Behavior, Environ. Sci. Technol., 2019, 53, 13009–13018 CrossRef CAS PubMed.
- W.-S. W. DeRieux, Y. Li, P. Lin, J. Laskin, A. Laskin, A. K. Bertram, S. A. Nizkorodov and M. Shiraiwa, Predicting the glass transition temperature and viscosity of secondary organic material using molecular composition, Atmos. Chem. Phys., 2018, 18, 6331–6351 CrossRef.
- M. Shiraiwa, Y. Li, A. P. Tsimpidi, V. A. Karydis, T. Berkemeier, S. N. Pandis, J. Lelieveld, T. Koop and U. Pöschl, Global distribution of particle phase state in atmospheric secondary organic aerosols, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- E. Fröhlich, A. Mercuri, S. Wu and S. Salar-Behzadi, Measurements of Deposition, Lung Surface Area and Lung Fluid for Simulation of Inhaled Compounds, Front. Pharmacol., 2016, 7, 1–10 CrossRef PubMed.
- G. Invernizzi, A. Ruprecht, C. De Marco, P. Paredi and R. Boffi, Residual tobacco smoke: measurement of its washout time in the lung and of its contribution to environmental tobacco smoke, Tob. Control, 2007, 16, 29–33 CrossRef PubMed.
- P. Prazeller, T. Karl, A. Jordan, R. Holzinger, A. Hansel and W. Lindinger, Quantification of passive smoking using proton-transfer-reaction mass spectrometry, Int. J. Mass Spectrom., 1998, 178, L1–L4 CrossRef CAS.
- S. I. Rennard, G. Basset, D. Lecossier, K. M. O’Donnell, P. Pinkston, P. G. Martin and R. G. Crystal, Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution, J. Appl. Physiol., 1986, 60, 532–538 CrossRef CAS PubMed.
- M. Li, J. Ding, H. Gu, Y. Zhang, S. Pan, N. Xu, H. Chen and H. Li, Facilitated Diffusion of Acetonitrile Revealed by Quantitative Breath Analysis Using Extractive Electrospray Ionization Mass Spectrometry, Sci. Rep., 2013, 3(1), 1–6 Search PubMed.
- M. Metsälä, F. M. Schmidt, M. Skyttä, O. Vaittinen and L. Halonen, Acetylene in breath: background levels and real-time elimination kinetics after smoking, J. Breath Res., 2010, 4, 046003 CrossRef PubMed.
- R. R. Baker and M. Dixon, The retention of tobacco smoke constituents in the human respiratory tract, Inhal. Toxicol., 2006, 18, 255–294 CrossRef CAS PubMed.
- C. Sarkar, V. Sinha, V. Kumar, M. Rupakheti, A. Panday, K. S. Mahata, D. Rupakheti, B. Kathayat and M. G. Lawrence, Overview of VOC emissions and chemistry from PTR-TOF-MS measurements during the SusKat-ABC campaign: High acetaldehyde, isoprene and isocyanic acid in wintertime air of the Kathmandu Valley, Atmos. Chem. Phys., 2016, 16, 3979–4003 CrossRef CAS.
- B. Hang, P. Wang, Y. Zhao, H. Chang, J.-H. Mao and A. M. Snijders, Thirdhand smoke: genotoxicity and carcinogenic potential, Chronic Dis. Transl. Med., 2020, 6, 27–34 Search PubMed.
- B. Hang, A. M. Snijders, Y. Huang, S. F. Schick, P. Wang, Y. Xia, C. Havel, P. Jacob, N. Benowitz, H. Destaillats, L. A. Gundel and J. H. Mao, Early exposure to thirdhand cigarette smoke affects body mass and the development of immunity in mice, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- B. Hang, P. Wang, Y. Zhao, A. Sarker, A. Chenna, Y. Xia, A. Snijders and J.-H. Mao, Adverse Health Effects of Thirdhand Smoke: From Cell to Animal Models, Int. J. Mol. Sci., 2017, 18, 932 CrossRef PubMed.
- A. M. Snijders, M. Zhou, T. P. Whitehead, B. Fitch, P. Pandey, A. Hechmer, A. Huang, S. F. Schick, A. J. De Smith, A. B. Olshen, C. Metayer, J. H. Mao, J. L. Wiemels and S. C. Kogan, In utero and early-life exposure to thirdhand smoke causes profound changes to the immune system, Clin. Sci., 2021, 135, 1053–1063 CrossRef CAS PubMed.
- T. F. Northrup, A. L. Stotts, R. Suchting, G. E. Matt, P. J. E. Quintana, A. M. Khan, C. Green, M. R. Klawans, M. Johnson, N. Benowitz, P. Jacob, E. Hoh, M. F. Hovell and C. J. Stewart, Thirdhand smoke associations with the gut microbiomes of infants admitted to a neonatal intensive care unit: an observational study, Environ. Res., 2021, 197, 111180 CrossRef CAS PubMed.
- D. Gao, S. Ripley, S. Weichenthal and K. J. G. Pollitt, Ambient particulate matter oxidative potential: chemical determinants, associated health effects, and strategies for risk management, Free Radic. Biol. Med., 2020, 151, 7–25 CrossRef CAS PubMed.
- A. Clergé, J. Le Goff, C. Lopez, J. Ledauphin and R. Delépée, Crit. Rev. Toxicol., 2019, 49, 302–328 CrossRef PubMed.
- M. C. Geier, A. C. Chlebowski, L. Truong, S. L. M. Simonich, K. A. Anderson and R. L. Tanguay, Comparative developmental toxicity of a comprehensive suite of polycyclic aromatic hydrocarbons, Arch. Toxicol., 2018, 92, 571–586 CrossRef CAS PubMed.
- K. Misaki, T. Takamura-Enya, H. Ogawa, K. Takamori and M. Yanagida, Tumour-promoting activity of polycyclic aromatic hydrocarbons and their oxygenated or nitrated derivatives, Mutagenesis, 2016, 31, 205–213 CrossRef CAS PubMed.
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Chem. Res. Toxicol., 2000, 13, 135–160 Search PubMed.
- S. Lundstedt, P. A. White, C. L. Lemieux, K. D. Lynes, I. B. Lambert, L. Öberg, P. Haglund and M. Tysklind, Sources, fate, and toxic hazards of oxygenated Polycyclic Aromatic Hydrocarbons (PAHs) at PAH-contaminated sites, Ambio, 2007, 36, 475–485 CrossRef CAS PubMed.
- H. Andersson, E. Piras, J. Demma, B. Hellman and E. Brittebo, Low levels of the air pollutant 1-nitropyrene induce DNA damage, increased levels of reactive oxygen species and endoplasmic reticulum stress in human endothelial cells, Toxicology, 2009, 262, 57–64 CrossRef CAS PubMed.
- Y. Motoyama, K. Bekki, W. C. Sang, N. Tang, T. Kameda, A. Toriba, K. Taguchi and K. Hayakawa, Oxidative stress more strongly induced by ortho- than para-quinoid polycyclic aromatic hydrocarbons in A549 cells, J. Health Sci., 2009, 55, 845–850 Search PubMed.
- W. Y. Tuet, F. Liu, N. De Oliveira Alves, S. Fok, P. Artaxo, P. Vasconcellos, J. A. Champion and N. L. Ng, Chemical Oxidative Potential and Cellular Oxidative Stress from Open Biomass Burning Aerosol, Environ. Sci. Technol. Lett., 2019, 6, 126–132 Search PubMed.
- R. Goel, Z. Bitzer, S. M. Reilly, N. Trushin, J. Foulds, J. Muscat, J. Liao, R. J. Elias and J. P. Richie, Variation in Free Radical Yields from U.S. Marketed Cigarettes, Chem. Res. Toxicol., 2017, 30, 1038–1045 Search PubMed.
- J. Zhao and P. K. Hopke, Concentration of reactive oxygen species (ROS) in mainstream and sidestream cigarette smoke, Aerosol Sci. Technol., 2012, 46, 191–197 CrossRef CAS.
- F. Hasan, L. Khachatryan and S. Lomnicki, Comparative Studies of Environmentally Persistent Free Radicals on Total Particulate Matter Collected from Electronic and Tobacco Cigarettes, Environ. Sci. Technol., 2020, 54, 5710–5718 CrossRef CAS PubMed.
- J. T. Bates, R. J. Weber, J. Abrams, V. Verma, T. Fang, M. Klein, M. J. Strickland, S. E. Sarnat, H. H. Chang, J. A. Mulholland, P. E. Tolbert and A. G. Russell, Reactive Oxygen Species Generation Linked to Sources of Atmospheric Particulate Matter and Cardiorespiratory Effects, Environ. Sci. Technol., 2015, 49, 13605–13612 CrossRef CAS PubMed.
- P. H. Chowdhury, Q. He, T. L. Male, W. H. Brune, Y. Rudich and M. Pardo, Exposure of Lung Epithelial Cells to Photochemically Aged Secondary Organic Aerosol Shows Increased Toxic Effects, Environ. Sci. Technol. Lett., 2018, 5, 424–430 CrossRef CAS.
- C. A. Weitekamp, T. Stevens, M. J. Stewart, P. Bhave and M. I. Gilmour, Health effects from freshly emitted versus oxidatively or photochemically aged air pollutants, Sci. Total Environ., 2020, 704, 135772 CrossRef CAS PubMed.
- M. Shrivastava, S. Lou, A. Zelenyuk, R. C. Easter, R. A. Corley, B. D. Thrall, P. J. Rasch, J. D. Fast, S. L. M. Simonich, H. Shen and S. Tao, Global long-range transport and lung cancer risk from polycyclic aromatic hydrocarbons shielded by coatings of organic aerosol, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1246–1251 CrossRef CAS PubMed.
- H. M. Rogers, J. C. Ditto and D. R. Gentner, Evidence for impacts on surface-level air quality in the northeastern US from long-distance transport of smoke from North American fires during the Long Island Sound Tropospheric Ozone Study (LISTOS) 2018, Atmos. Chem. Phys., 2020, 20, 671–682 CrossRef CAS.
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 2011, 11, 4039–4072 CrossRef CAS.
- T. Schripp, D. Markewitz, E. Uhde and T. Salthammer, Does e-cigarette consumption cause passive vaping?, Indoor Air, 2013, 23, 25–31 CrossRef CAS PubMed.
- M. L. Goniewicz, J. Knysak, M. Gawron, L. Kosmider, A. Sobczak, J. Kurek, A. Prokopowicz, M. Jablonska-Czapla, C. Rosik-Dulewska, C. Havel, P. Jacob and N. Benowitz, Levels of selected carcinogens and toxicants in vapour from electronic cigarettes, Tob. Control, 2014, 23, 133–139 CrossRef PubMed.
- M. El Hourani, R. Salman, S. Talih, N. A. Saliba and A. Shihadeh, Does the Bubbler Scrub Key Toxicants from Waterpipe Tobacco Smoke?: Measurements and Modeling of CO, NO, PAH, Nicotine, and Particulate Matter Uptake, Chem. Res. Toxicol., 2020, 33, 727–730 Search PubMed.
- H. Schwartz-Narbonne, B. Du and J. A. Siegel, Volatile organic compound and particulate matter emissions from an ultrasonic essential oil diffuser, Indoor Air, 2021, 1–11 Search PubMed.
- L. M. Petrick, A. Svidovsky and Y. Dubowski, Thirdhand smoke: heterogeneous oxidation of nicotine and secondary aerosol formation in the indoor environment, Environ. Sci. Technol., 2011, 45, 328–333 CrossRef CAS PubMed.
- X. Tang, N. R. González, M. L. Russell, R. L. Maddalena, L. A. Gundel and H. Destaillats, Chemical changes in thirdhand smoke associated with remediation using an ozone generator, Environ. Res., 2021, 198, 110462 CrossRef CAS PubMed.
- T. Joo, J. C. Rivera-Rios, D. Alvarado-Velez, S. Westgate and N. L. Ng, Formation of Oxidized Gases and Secondary Organic Aerosol from a Commercial Oxidant-Generating Electronic Air Cleaner, Environ. Sci. Technol. Lett., 2021, 8, 691–698 CrossRef CAS.
- G. Löfroth and Y. Zebühr, Polychlorinated dibenzo-p-dioxins (PCDDs) and dibenzofurans (PCDFs) in mainstream and sidestream cigarette smoke, Bull. Environ. Contam. Toxicol., 1992, 48, 789–794 CrossRef PubMed.
- Y. C. Chien, C. P. Chang and Z. Z. Liu, Volatile organics off-gassed among tobacco-exposed clothing fabrics, J. Hazard. Mater., 2011, 193, 139–148 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1ea00107h |
| ‡ Now at: Department of Civil and Environmental Engineering, Hong Kong Polytechnic University, Hong Kong. |
| This journal is © The Royal Society of Chemistry 2022 |