
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of secondary organic aerosol generated from the oxidation of laboratory precursors by hydroxyl radicals, chlorine atoms, and bromine atoms in an oxidation flow reactor†
Andrew T.
Lambe
 *a,
Anita M.
Avery
a,
Nirvan
Bhattacharyya
b,
Dongyu S.
Wang‡
b,
Mrinali
Modi
b,
Catherine G.
Masoud
b,
Lea Hildebrandt
Ruiz
b and
William H.
Brune
c
*a,
Anita M.
Avery
a,
Nirvan
Bhattacharyya
b,
Dongyu S.
Wang‡
b,
Mrinali
Modi
b,
Catherine G.
Masoud
b,
Lea Hildebrandt
Ruiz
b and
William H.
Brune
c
aAerodyne Research Inc., Billerica, Massachusetts, USA. E-mail: lambe@aerodyne.com
bThe University of Texas at Austin, USA
cPennsylvania State University, USA
First published on 6th May 2022
Abstract
The role of hydroxyl radicals (OH) as a daytime oxidant is well established on a global scale. In specific source regions, such as the marine boundary layer and polluted coastal cities, other daytime oxidants, such as chlorine atoms (Cl) and even bromine atoms (Br), may compete with OH for the oxidation of volatile organic compounds (VOCs) and/or enhance the overall oxidation capacity of the atmosphere. However, the number of studies investigating halogen-initiated secondary organic aerosol (SOA) formation is extremely limited, resulting in large uncertainties in these oxidative aging processes. Here, we characterized the chemical composition and yield of laboratory SOA generated in an oxidation flow reactor (OFR) from the OH and Cl oxidation of n-dodecane (n-C12) and toluene, and the OH, Cl, and Br oxidation of isoprene and α-pinene. In the OFR, precursors were oxidized using integrated OH, Cl, and Br exposures ranging from 3.1 × 1010 to 2.3 × 1012, 6.1 × 109 to 1.3× 1012 and 3.2 × 1010 to 9.7 × 1012 molecules cm−3 s−1, respectively. Like OH, Cl facilitated multistep SOA oxidative aging over the range of OFR conditions that were studied. In contrast, the extent of Br-initiated SOA oxidative aging was limited. SOA elemental ratios and mass yields obtained in the OFR studies were comparable to those obtained from OH and Cl oxidation of the same precursors in environmental chamber studies. Overall, our results suggest that alkane, aromatic, and terpenoid SOA precursors are characterized by distinct OH- and halogen-initiated SOA yields, and that while Cl may enhance the SOA formation potential in regions influenced by biogenic and anthropogenic emissions, Br may have the opposite effect.
1 Introduction
The atmosphere is an oxidizing environment. Gas-phase oxidants, including ozone (O3), hydroxyl radicals (OH), nitrate radicals (NO3), chlorine atoms (Cl), and bromine atoms (Br), can react with organic and inorganic pollutants to generate a myriad of gas- and condensed-phase oxidation products. The importance of each oxidant in different parts of the atmosphere depends on the local meteorology, emissions, and photochemistry. Globally, OH is the most important oxidant: there are many ways to generate it during the daytime from precursors that are widely distributed throughout the atmosphere, and, unlike O3, it reacts with most inorganic and organic compounds.1 With regards to atmospheric aerosols, OH is particularly important in initiating the oxidation of sulfur dioxide (SO2) to generate sulfuric acid and initiating the oxidation of volatile organic compounds (VOCs) to generate low volatility organic compounds (LVOC) that condense to form secondary organic aerosol (SOA). NO3 is an important oxidant at nighttime2 and in some cases during the daytime,3,4 particularly in source regions influenced by emissions from unsaturated VOCs emitted from plants and wildfires.5Significant Cl production occurs in regions such as the marine boundary layer,6 polluted coastal cities,7 and the Arctic atmosphere.8,9 Most VOCs react with Cl approximately 10 to 100 times faster than their corresponding rate of reaction with OH. Thus, when atmospheric Cl mixing ratios are high enough, Cl may compete with OH in the oxidation of VOCs and/or otherwise enhance the overall oxidation capacity of the atmosphere. Additionally, significant inland Cl production has been observed,10–12 bleach washing has been shown to initiate significant indoor chlorine chemistry,13–15 and both Cl and Br have been linked to enhanced secondary aerosol formation in China.16 Br contributes to springtime polar mercury and O3 depletion9,17,18 reacts with dimethyl sulfide, alkenes, and aldehydes at rates that are similar to their reaction rates with OH, and, like Cl, induces significant multiphase chemistry in organic aerosols.19–21
Large environmental chambers have been used for decades to study complex SOA formation chemistry. Multi-instrument, multi-investigator chamber studies have provided comprehensive data sets that transform existing concepts of SOA formation and aging and SOA models.22 The recent emergence of oxidation flow reactors (OFRs) complements chambers through their lower operation/maintenance costs, portability for in situ oxidative aging of ambient and source emissions, and ability to access photochemical aging timescales of up to several days.23–28 To date, the vast majority of SOA formation studies in chambers and OFRs have used O3, OH, and to a lesser extent NO3, to mimic daytime and nighttime oxidation of hydrocarbons. The handful of studies that have measured yields of SOA obtained from Cl oxidation of VOCs have shown that Cl exposure generates SOA in yields that are comparable to, or exceed, OH oxidation of the same precursors.29–36 SOA formed from Br oxidation of VOCs has not been studied; models including halogen chemistry assume the same yield of SOA is obtained regardless of whether Cl and Br is the initiating oxidant.16
To investigate these knowledge gaps, we characterized the chemical composition and yield of laboratory SOA generated in an OFR from the OH, Cl and Br oxidation of a set of anthropogenic and biogenic VOCs. OFRs use residence times that are on the order of minutes and oxidant concentrations that are typically 100–1000 times higher than ambient levels; these factors may make the chemistry and microphysics in the OFR somewhat different from the chemistry and microphysics in the atmosphere.37,38 Thus, we also compared the chemical composition and mass yields of SOA obtained from OH and Cl oxidation of the same precursors in the OFR with previous chamber studies.
2 Experimental
Experiments were conducted inside a Potential Aerosol Mass (PAM) OFR (Aerodyne Research, Inc.), which is a horizontal 13 L aluminum cylindrical chamber (46 cm long × 22 cm ID) operated in continuous flow mode, with 6.0–6.8 L min−1 flow through the reactor.39 The corresponding calculated mean residence time in the OFR, τOFR, ranged from 114 to 130 s. An electroconductive Teflon coating was applied to the OFR to improve chemical compatibility with halogen precursors while maintaining high transmission of gases and particles.40 Two low-pressure mercury (Hg) lamps that were isolated from the sample flow using type 214 quartz sleeves were used to photolyze oxidant precursors. As discussed in Section 2.1 and shown in Fig. S1,† different lamps were used for different OFR methods to maximize the overlap between the absorption cross section of the oxidant precursor and the range of achievable oxidant exposure. A fluorescent dimming ballast (IZT-2S28-D, Advance Transformer Co.) was used to regulate current applied to the lamps. The UV irradiance was measured using a photodetector (TOCON-GaP6, sglux GmbH) and was varied by changing the control voltage applied to the ballast between 1.5 and 10 VDC. The corresponding actinic flux ranged from approximately 1 × 1014 to 3 × 1015 photons cm−2 s−1.39,412.1 Oxidant generation
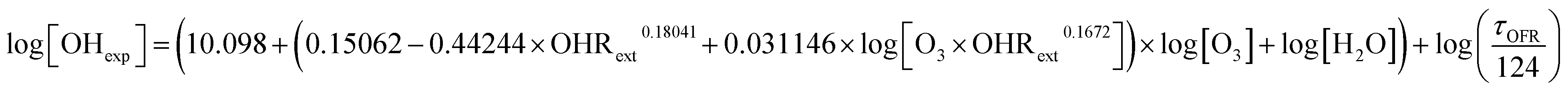 | (1) |
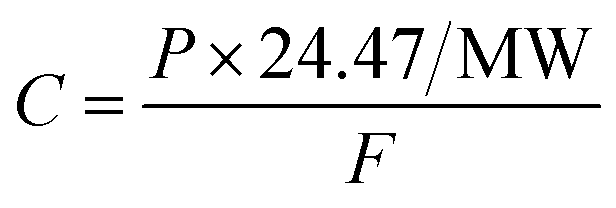 | (2) |
Integrated Cl and Br exposures (Clexp, Brexp) were characterized in offline calibration experiments by measuring the decay of O3 injected into the OFR and measured using an O3 analyzer (2B Technologies) as a function of lamp voltage. O3 concentrations were allowed to stabilize before initiating Clexp and Brexp measurements, during which steady-state levels of O3 were obtained with the lamps turned off (O3,i). Then, the lamps were turned on, and O3 concentrations were allowed to stabilize before being measured at illuminated steady-state conditions (O3,f) following reaction with Cl or Br. The Cl or Br exposure (Clexp, Brexp) at each condition was calculated using eqn (3) and (4):
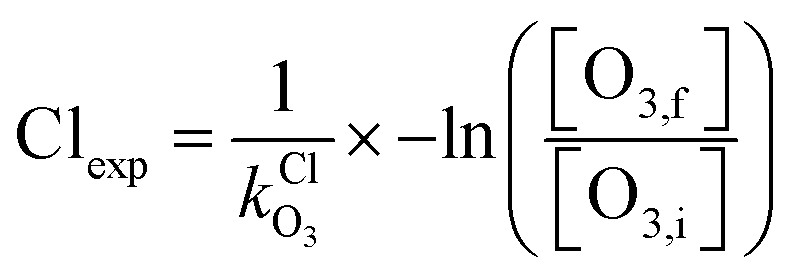 | (3) |
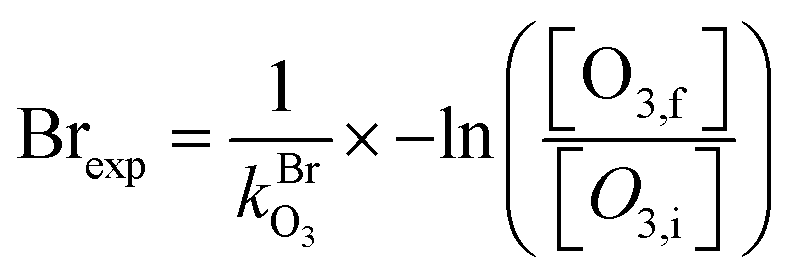 | (4) |
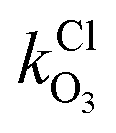 and
and 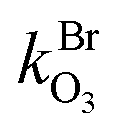 are the bimolecular Cl + O3 and Br + O3 reaction rate coefficients. Here, we used
are the bimolecular Cl + O3 and Br + O3 reaction rate coefficients. Here, we used 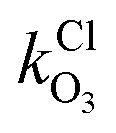 = 1.21 × 10−11 and
= 1.21 × 10−11 and 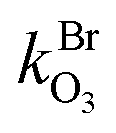 = 1.16 × 10−12 cm3 s−1.53
= 1.16 × 10−12 cm3 s−1.53
Because [Cl2] was varied in experiments that used OFR313-iCl2 and OFR369-iCl2 to generate Cl, separate calibration experiments were performed to measure Clexp as a function of [Cl2]. To correct for Cl or Br suppression that occurs in the presence of external Cl or Br reactivity (ClRext, BrRext), which is the product of the O3 mixing ratio and its bimolecular Cl or Br rate coefficient, [O3,i] was varied from 242 to 3360 ppbv in Cl-OFR calibration experiments and 369 to 7191 ppbv in Br-OFR calibration experiments. These calibration conditions achieved ClRext values ranging from 72 to 1000 s−1 and BrRext values ranging from 12 to 226 s−1, which approximately span the range of ClRext and BrRext values in the OFR conditions listed in Table 1. Example OFR313-iCl2, OFR254-iC2Cl2O2, OFR369-iBr2, OFR254-iC2Br2O2 calibration data are shown in Fig. S2–S5.† Calibration results indicate that Clexp decreased by a factor of 4 to 12 at each lamp setting over the range of ClRext values shown in Fig. S2,† with the largest Cl suppression occurring at lower lamp voltage, as expected. Similarly, Brexp decreased by a factor of 2 to 60 using OFR369-iBr2 (Fig. S4†) and by a factor of 2 to 28 using OFR254-iC2Br2O2 (Fig. S5†). Here, we assumed that a specific ClRext or BrRext value suppressed Clexp or Brexp by the same amount regardless of the source of ClRext or BrRext (e.g. O3 in calibrations, or VOCs in SOA studies). This assumption may have introduced uncertainty in some cases, such as OFR conditions where VOCs were short-lived and their Cl or Br oxidation products had significantly different Cl or Br reaction rates. Because ClOx and BrOx chemistry in Cl-OFR and Br-OFR calibration experiments was more complex than the analogous HOx chemistry in OH-OFR calibration experiments, we assumed ±70% uncertainty in Clexp and Brexp values.
| VOC/oxidant | [VOC]0 (ppb) | [C2Cl2O2] (ppm) | [Cl2] (ppm) | [C2Br2O2] (ppm) | [Br2] (ppm) | RH (%) | T (°C) | Oxidant exposure (cm−3 s) |
|---|---|---|---|---|---|---|---|---|
| n-C12/OH | 21 | — | — | — | — | 30.9 | 26.2 | 2.2 × 1011 to 2.1 × 1012 |
| n-C12/Cl | 21 | 4.2 | — | — | — | 1.2 | 26.1 | 3.3 × 1010 to 2.3 × 1011 |
| Toluene/OH | 45 | — | — | — | — | 31.7 | 26.2 | 2.6 × 1011 to 2.3 × 1012 |
| Toluene/Cl | 45 | 4.2 | — | — | — | 1.1 | 26.9 | 2.7 × 1010 to 4.5 × 1011 |
| Toluene/Cl | 45 | — | 4.9–24.7 | — | — | 1.2 | 24.2 | 2.3 × 1010 to 1.3 × 1012 |
| Isoprene/OH | 48 | — | — | — | — | 43.3 | 27.2 | 3.1 × 1010 to 9.6 × 1011 |
| Isoprene/Cl | 48 | — | 1.9–24.4 | — | — | 4.0 | 26.5 | 6.1 × 109 to 2.9 × 1011 |
| Isoprene/Br | 144 | — | — | 1.8 | — | 4.0 | 27.8 | 3.2 × 1010 to 2.6 × 1012 |
| α-Pinene/OH | 30 | — | — | — | — | 28.2 | 28.8 | 6.7 × 1010 to 1.2 × 1012 |
| α-Pinene/Cl | 30 | 4.2 | — | — | — | 1.4 | 28.0 | 3.9 × 1010 to 1.2 × 1011 |
| α-Pinene/Br | 90 | — | — | 1.8 | — | 4.1 | 25.6 | 9.8 × 1010 to 2.9 × 1012 |
| α-Pinene/Br | 90 | — | — | — | 1.9 | 3.9 | 26.0 | 9.6 × 1011 to 9.7 × 1012 |
A subset of OFR421-iBr2 calibration conditions that were applicable to α-pinene/Br experiments (lamp voltages higher than 4 V and/or BrRext < 90 s−1) depleted all the O3 that was injected into the OFR. To constrain Brexp at these conditions, we compared Brexp obtained using OFR421-iBr2 and OFR369-iBr2 at otherwise identical OFR conditions when Brexp < 3 × 1012 cm−3 s using both methods. Brexp obtained using OFR421-iBr2 was approximately 2.16 times higher than Brexp obtained using OFR369-iBr2 (Fig. S6†), which is consistent with a higher Br2 absorption cross section at λ = 421 nm than at λ = 369 nm.50 Brexp values obtained at the other OFR369-iBr2 conditions were then multiplied by 2.16 to obtain Brexp values at OFR421-iBr2 conditions with equivalent lamp settings and BrRext values.
Calculated Clexp values in SOA experiments ranged from 6.1 × 109 to 1.3 × 1012 cm−3 s, or approximately 1.2 d to 8.2 months of atmospheric oxidation at [Cl] = 6 × 104 cm−3.18 Similarly, calculated Brexp values ranged from 3.2 × 1010 to 9.7 × 1012 cm−3 s, or approximately 1 h to 16 d at [Br] = 7 × 106 cm−3.18 These simple calculations should be interpreted as a rough estimate of the photochemical age in a representative source region with active Cl or Br photochemistry (e.g., Arctic spring), and may vary by orders of magnitude elsewhere.
2.2 Photochemical model
To investigate the fate of O3 in our Clexp and Brexp calibration experiments, and to build a foundation for characterizing the concentrations of inorganic halogens generated in the OFR, we developed a photochemical box model that was implemented in the KinSim chemical kinetic solver.54 The KinSim mechanism shown in Table S1† contains 66 reactions to model HOx concentrations in OFRs39,55,56 plus 139 reactions that were added to model concentrations of Cl2, C2Cl2O2, Br2, C2Br2O2, Cl, ClO, ClO2, ClO3, OClOO, ClOO, Cl2O, Cl2O2, HCl, HOCl, Cl2O3, Br, BrO, BrO2, HBr, HOBr, and BrCl. Inputs to the KinSim model were [O3] (242 to 7191 ppb), UV flux (3.5 × 1013 to 3.5 × 1015 photons cm−2 s−1), RH = 1%, T = 25 °C, and τOFR = 130 s (modeled as plug flow). Over this range of OFR conditions, the model suggests that >97% of reactive O3 loss was due to reaction with Cl across all OFR313-iCl2, OFR369-iCl2, OFR254-iC2Cl2O2, and OFR313-iC2Cl2O2 calibration conditions, with the remaining O3 lost to reaction with ClO. Similarly, we estimate that >99% of reactive O3 loss was due to reaction with Br across all OFR369-iBr2, OFR421-iBr2, OFR254-and iC2Br2O2 calibration conditions, with the remaining O3 lost to reaction with BrO. We estimate that Cl or Br regeneration via photolysis of ClOx and BrOx biased calibrated Clexp or Brexp values by <2% or <14% respectively. Because we already applied ±70% uncertainty estimates to Clexp and Brexp values, we did not apply additional correction factors to the calibration data.2.3 Particle generation
SOA particles were generated via gas-phase OH or Cl oxidation of n-dodecane (n-C12) or toluene, or OH, Cl, or Br oxidation of isoprene or α-pinene, followed by homogeneous nucleation; we hereafter refer to SOA formation initiated by OH, Cl, and Br as “OH-SOA”, “Cl-SOA” or “Br-SOA” respectively. These precursors were chosen to cover a range of surrogate anthropogenic and biogenic compounds that enabled comparison with results from Cl-initiated chamber SOA formation studies.29–33,35,36 Particle number concentrations and mobility size distributions were measured with a TSI scanning mobility particle sizer (SMPS), and ensemble aerosol mass spectra were measured with an Aerodyne long high-resolution time-of-flight aerosol mass spectrometer (L-ToF-AMS). Liquid solutions containing the precursor diluted to 10% (v/v) in carbon tetrachloride were injected into the OFR carrier gas flow at liquid flow rates (QVOC,l) ranging from 0.94 to 2.8 μL h−1 using a syringe pump. The VOC mixing ratio entering the OFR, rVOC,g, was calculated using the ideal gas law as applied by Liu et al.57 in eqn (5):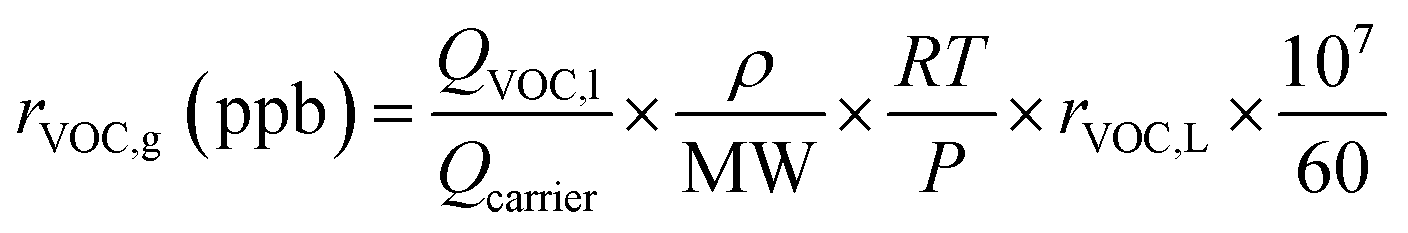 | (5) |
Over the course of our studies, we learned that conditioning the OFR with humidified carrier gas containing O3 and OH while transitioning from Cl-SOA to OH-SOA studies generated >100 μg m−3 and >107 cm−3 of homogenously nucleated particles that gradually subsided over hours to days. As shown in Fig. S7,† L-ToF-AMS spectra of these particles contained chlorinated ion signals (and their primary Cl isotope at m/z +2), at m/z = 35 (Cl+), 36 (HCl+), 51 (ClO+), 67 (ClO2+), 70 (Cl2+), 83 (ClO3+), and 100 (HClO4+), suggesting that these particles contained perchloric acid (HClO4) and/or perchlorate salts.58,59 Significant signal at m/z = 44 (CO2+) was also observed and is a known artifact from the interaction of inorganic salts on surfaces inside the AMS.60 During these transient conditioning periods, we hypothesize that perchlorate was generated from the reaction of O3 with aqueous HCl on the walls of the OFR61 and/or in the gas phase from the reactions:62
| HCl + OH → Cl + H2O | (R1) |
| Cl + O3 → ClO + O2 | (R2) |
| ClO + O3 → ClO2 + O2 | (R3) |
| ClO2 + O3 → ClO3 + O2 | (R4) |
| ClO3 + OH → HClO4 | (R5) |
To minimize the influence of perchlorate on ensuing OH-SOA measurements, the OFR was conditioned with O3 and OH until ClOx+ signals in the AMS returned to background levels.
2.4 Analysis
To compare our measurements with chamber Cl-SOA mass spectra obtained with an Aerodyne quadrupole aerosol chemical speciation monitor (Q-ACSM) with unit mass resolution,32,33,35,36 corresponding O/C ratios for those data were calculated using the equation O/C = 3.82 × f44 + 0.0794,64 where f44 was the fraction of Cl-SOA signal at m/z = 44. We used L-ToF-AMS data to confirm the literature O/C-f44 parameterization was accurate for Cl-SOA generated in the OFR in this study. However, H/C ratios of Cl-SOA calculated using the equation H/C = 1.01 + 6.07 × f43–16.01 × f432,68 where f43 was the fraction of Cl-SOA signal at m/z = 43, were systematically ≈20–40% higher than H/C ratios calculated from the L-ToF-AMS spectra (Fig. S8†). Additionally, significant contributions to m/z = 43 from both C3H7+ and C2H3O+ ions in n-C12 Cl-SOA spectra complicated application of the H/C-f43 correlation proposed by Ng et al.68 Thus, we developed a different parameterization to calculate the H/C ratio of Cl-SOA from the fraction of L-ToF-AMS signal at m/z = 41, which contained one alkyl ion (C3H5+). We then applied the equation H/C = 5.032 × f410.1485 − 1.737 (Fig. S9†) to calculate the H/C ratio of chamber-generated n-C12, isoprene, and α-pinene Cl-SOA obtained with the Q-ACSM. For chamber-generated toluene Cl-SOA data published in Dhulipala et al.,35 because only f43 was available, we calculated f41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :f43 = 0.17 in our toluene Cl-SOA spectra, used this result to estimate f41 for their toluene chamber Cl-SOA spectra, and then calculated H/C ratio using the equation described above.
:f43 = 0.17 in our toluene Cl-SOA spectra, used this result to estimate f41 for their toluene chamber Cl-SOA spectra, and then calculated H/C ratio using the equation described above.
2.5 SOA yields
SOA mass yields were calculated from the ratio of SOA mass formed to precursor gas reacted. The SOA mass was calculated from the integrated SMPS particle volume and the SOA particle density, ρSOA, which was calculated using eqn (6):69 | (6) |
We estimated the fraction of precursor gas reacted from the product of the OHexp, Clexp, or Brexp and the bimolecular rate coefficients of n-C12 + OH/Cl, toluene + OH/Cl, isoprene + OH/Cl/Br, and α-pinene + OH/Cl/Br.70–76 These calculations suggested that 95–100% of n-C12, 75–100% of toluene, 91–100% of isoprene, and 89–100% of α-pinene reacted across the OFR conditions summarized in Table 1.
SOA yields were corrected for size-dependent particle wall losses in the OFR by applying the particle transmission efficiency measurements of Bhattarai et al.77 to the SMPS volume-weighted mobility size distributions. Here, the particle wall loss correction factors ranged from 1.07 to 1.38 for mean volume-weighted particle mobility diameters ranging from 180 nm to 36 nm. To investigate the influence of vapor wall losses on SOA yields, we used the LVOC fate correction model,78,79 which calculates the timescales of diffusional wall losses, gas-phase oxidative loss, and condensation onto aerosols at a user-specified condensation loss rate, OH/Cl/Br exposure and reaction rate coefficient, and OFR residence time values. We assumed that LVOCs had the same OH, Cl, or Br reaction rate coefficients as their VOC precursors. The condensation sink was calculated using the integrated SMPS number-weighted mobility size distributions and assuming a LVOC diffusion coefficient of 0.07 cm2 s−1, mean molecular speed of 2 × 104 cm s−1, and mass accommodation coefficient of unity.78,80,81 In these experiments, mean fractional LVOC wall losses for each precursor/oxidant combination ranged from 0.003 ± 0.001 (n-C12/Cl) to 0.079 ± 0.076 (α-pinene/Br). Thus, we assumed LVOC wall losses were negligible compared to gas-phase oxidative loss and condensation onto aerosols,82,83 and did not modify SOA yield values to account for them.
3 Results & discussion
3.1 Sample anthropogenic OH-SOA and Cl-SOA mass spectra
Fig. 1 shows L-ToF-AMS spectra of SOA generated from the OH and Cl oxidation of n-C12 and toluene. To compare results obtained at lower oxidant exposures that were most applicable to urban atmospheres, the spectra shown were obtained at the lowest OHexp and Clexp values at which particle formation was observed: OHexp = 2.2 × 1011 and Clexp = 3.3 × 1010 cm−3 s for n-C12 OH- and Cl-SOA, and OHexp = 2.6 × 1011 and Clexp = 2.7 × 1010 cm−3 s for toluene OH- and Cl-SOA. For both n-C12 OH-SOA and Cl-SOA, the spectra were dominated by CxHy+ (green) and CxHyO1+ (purple) ion groups, which contributed ∼60% and 31% of the total OH- and Cl-SOA signal, respectively (Fig. 1e and f). Some of the most abundant ions within these groups included signals at m/z = 27 (C2H3+), 29 (CHO+), 41 (C3H5+), 43 (C2H3O+ + C3H7+), and 55 (C3H5O+ + C4H7+). Additional signals were present at m/z = 44 (CO2+), a marker for organic acids in the AMS,84 and at multiple ion clusters above m/z = 60 that contained CxHy+, CxHyO1+, and CxHyO>1+ (pink) ions.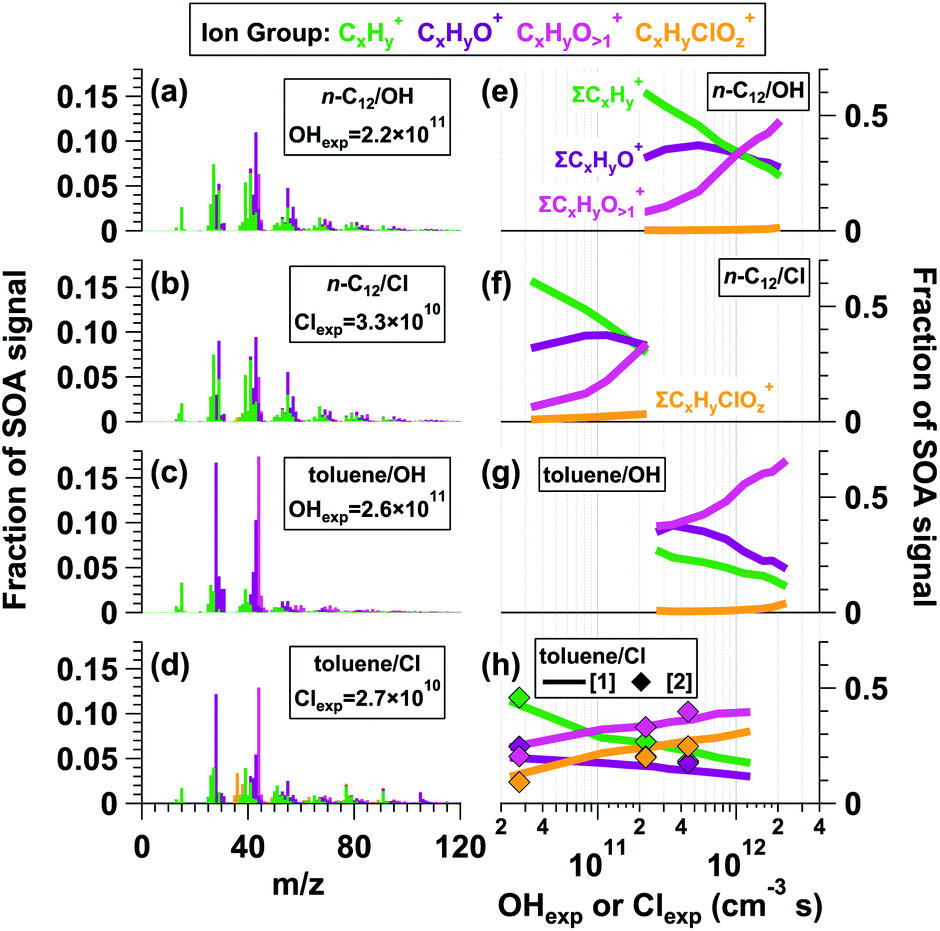 | ||
| Fig. 1 L-ToF-AMS spectra of SOA generated from the (a and e) OH oxidation of n-C12, (b and f) Cl oxidation of n-C12, (c and g) OH oxidation of toluene, and (d and h) Cl oxidation of toluene. OHexp and Clexp values listed in (a)–(d) are in units of cm−3 s. The toluene Cl-SOA spectrum presented in (d) was generated using OFR254-iC2Cl2O2. Additional notes regarding (h): toluene Cl-SOA was generated using (1) OFR313/369-iCl2 or (2) OFR254-iC2Cl2O2. | ||
To characterize the similarity between SOA mass spectra obtained from OH and Cl oxidation of the same precursor, using simple linear regression, we calculated the square of the Pearson correlation coefficient (r2) between the n-C12 OH- and Cl-SOA mass spectra shown in Fig. 1a and b, and between toluene OH- and Cl-SOA mass spectra shown in Fig. 1c and d. OH and Cl oxidation of n-C12 generated SOA with similar AMS spectra (r2 = 0.94) because both OH and Cl oxidation of n-C12 proceeded via hydrogen atom abstraction. In contrast, toluene OH-SOA and Cl-SOA spectra were more distinct from each other: while an r2 value of 0.87 was obtained between the two spectra, r2 decreased to 0.57 when contributions from signals at m/z = 44 and m/z = 28 (CO+, set equal to CO2+ by default) were removed from the regression analysis. Notably, toluene Cl-SOA contained enhanced signals at m/z = 77 (C6H5+), 91 (C7H7+), and 105 (C7H5O+) relative to toluene OH-SOA. While the Clexp used to generate Fig. 1d was 10 times lower than the OHexp used to generate Fig. 1c, toluene reacts with Cl ten times faster than OH;75 thus, to first order, the extent of OH and Cl oxidation was similar in both cases. The most likely explanation is that Cl oxidation of toluene generated a higher yield of ring-retaining C6 and C7 oxidation products because Cl preferentially abstracts H-atoms from the methyl group.30
To compare the similarity of n-C12 and toluene OH-/Cl-SOA spectra across the range of OHexp or Clexp values shown in Fig. 1e–h, Fig. S10† plots r2 values between L-ToF-AMS spectra shown in Fig. 1a or c and n-C12 or toluene OH-/Cl-SOA spectra obtained at other OHexp and Clexp. For example, the r2 value between Fig. 1a and more-oxidized n-C12 OH-SOA spectra decreased from 0.98 to 0.31 with increasing OHexp. For n-C12 Cl-SOA, r2 decreased from 0.94 to 0.43 with increasing Clexp. Likewise, r2 between Fig. 1c and more-oxidized toluene OH-SOA spectra decreased from 0.98 to 0.85 with increasing OHexp, and r2 between Fig. 1c and toluene Cl-SOA spectra decreased from 0.87 to 0.71 with increasing Clexp.
Fig. 1e, f and g, h plot the fractional contributions of the CxHy+, CxHyO1+, CxHyO>1+, and CxHyClOz+ ion groups (fCxHy+, fCxHyO1+, fCxHyO>1+, and fCxHyClOz+) (pale orange) present in n-C12 and toluene OH-/Cl-SOA as a function of OHexp and Clexp. Minimum and maximum fractions of each ion group are provided in Table 2, and the corresponding range of OHexp and Clexp values are listed in Table 1. Minor contributions from the CxHyClO+z ion group (orange) to the toluene OH-SOA spectra in Fig. 1g were observed, primarily from signals at Cl+ and HCl+. These signals may be associated with NH4Cl generated from the reaction of trace NH3 in the system with residual HCl from Cl-OFR studies. While fCxHy+ and fCxHyO1+ values spanned similar ranges, the maximum fCxHyO>1+ value was lower in n-C12 Cl-SOA (0.34) than in n-C12 OH-SOA (0.48). For both n-C12 OH-SOA and Cl-SOA, fCxHy+ decreased monotonically, fCxHyO1+ increased and then decreased, and fCxHyO>1+ and fCxHyClOz+ increased monotonically as a function of OHexp and/or Clexp. Because n-C12 has no double bonds for direct Cl addition, one possible source of particulate organic chlorides (ROCl) may have been Cl oxidation of unsaturated dihydrofuran intermediates.33 Another possible ROCl source involves the reaction RO2 + Cl → RO + ClO followed by the reaction RO2 + ClO → ROCl + O2,85,86 where RO2 represents organic peroxy radicals derived from Cl oxidation of n-C12 and/or its oxidation products and RO represents alkoxy radicals.
| VOC/oxidant | f CxHy+ | f CxHyO+ | f CxHyO>1+ | f CxHyClOz+ | f CxHyBrOz+ | O/C | H/C | C SOA (μg m−3) | Y SOA |
|---|---|---|---|---|---|---|---|---|---|
| n-C12/OH | 0.24–0.60 | 0.30–0.37 | 0.08–0.48 | — | — | 0.27–1.05 | 0.89–1.24 | 25–132 | 0.18–0.91 |
| n-C12/Cl | 0.30–0.61 | 0.32–0.37 | 0.06–0.34 | 0.009–0.033 | — | 0.22–0.82 | 1.20–1.63 | 160–357 | 1.1–2.5 |
| Toluene/OH | 0.11–0.27 | 0.19–0.38 | 0.37–0.66 | — | — | 0.83–1.51 | 0.89–1.24 | 16–93 | 0.098–0.56 |
| Toluene/Cl | 0.18–0.46 | 0.17–0.25 | 0.20–0.40 | 0.09–0.25 | — | 0.46–1.08 | 0.86–1.22 | 24–106 | 0.15–0.64 |
| Toluene/Cl | 0.18–0.44 | 0.12–0.20 | 0.24–0.40 | 0.12–0.31 | — | 0.53–1.02 | 0.82–1.07 | 14–99 | 0.083–0.58 |
| Isoprene/OH | 0.24–0.43 | 0.37–0.50 | 0.08–0.38 | — | — | 0.36–0.91 | 1.17–1.50 | 4–53 | 0.031–0.40 |
| Isoprene/Cl | 0.36–0.60 | 0.30–0.34 | 0.06–0.17 | 0.042–0.065 | — | 0.24–0.55 | 1.22–1.38 | 1–29 | 0.011–0.21 |
| Isoprene/Br | 0.46 | 0.34–0.35 | 0.05–0.07 | — | 0.12–0.13 | 0.29–0.31 | 1.40–1.44 | 2–7 | 0.007–0.018 |
| α-Pinene/OH | 0.28–0.46 | 0.26–0.40 | 0.13–0.42 | — | — | 0.38–0.93 | 1.16–1.48 | 18–52 | 0.11–0.31 |
| α-Pinene/Cl | 0.29–0.45 | 0.28–0.34 | 0.16–0.33 | 0.058–0.095 | — | 0.42–0.80 | 1.18–1.39 | 37–77 | 0.22–0.47 |
| α-Pinene/Br | 0.49–0.56 | 0.31–0.32 | 0.08–0.12 | — | 0.042–0.063 | 0.26–0.34 | 1.36–1.39 | 0.5–18 | 0.0006–0.037 |
| α-Pinene/Br | 0.55–0.59 | 0.25–0.26 | 0.09–0.12 | — | 0.056–0.063 | 0.24–0.30 | 1.33–1.38 | 1–9 | 0.003–0.018 |
Qualitatively similar changes in toluene OH-SOA and Cl-SOA spectra were observed as a function of OHexp and Clexp. Because toluene is more volatile than n-C12, addition of more oxygen-containing functional groups was required to generate condensable oxidation products, resulting in lower initial fCxHy+ and fCxHyO1+ values, with a monotonic decrease in fCxHyO1+ instead of an increase and then decrease as was observed in n-C12 SOA. Additionally, higher fCxHyO>1+ values were measured in toluene OH-/Cl-SOA, and higher fCxHyClOz+ values were observed in toluene Cl-SOA than in n-C12 SOA. Notably, for Cl-SOA generated from toluene (and, to a lesser extent, n-C12), fCxHyClOz+ followed the same trend as fCxHyO>1+. This suggests that this ion group was also associated with later-generation oxidation products. Because direct Cl addition to the aromatic ring is a minor pathway (e.g. Cai et al.30), ROCl in toluene Cl-SOA may have been generated from RO2 + ClO reactions, as was hypothesized earlier for ROCl observed in n-C12 Cl-SOA. Overall, these observations were consistent with multigenerational oxidative aging of n-C12 and toluene OH- and Cl-SOA, where early-generation oxidation products that contributed to the less-oxidized CxHy+ and CxHyO1+ ion groups were converted to later-generation oxidation products that contributed to the CxHyO>1+ ion groups.
3.2 Sample biogenic OH-SOA, Cl-SOA, and Br-SOA mass spectra
Fig. 2a–f shows L-ToF-AMS spectra of SOA generated from the OH, Cl, and Br oxidation of isoprene and α-pinene. The isoprene OH-SOA, Cl-SOA and Br-SOA mass spectra were obtained at OHexp, Clexp, and Brexp values of 3.1 × 1010, 6.1 × 109, and 3.2 × 1010 cm−3 s, and the α-pinene OH-SOA, Cl-SOA, and Br-SOA spectra were obtained at OHexp, Clexp, and Brexp values of 6.8 × 1010, 3.9 × 1010, and 5.9 × 1011 cm−3 s. All SOA spectra were dominated by signals at m/z = 29 (CHO+), 39 (C3H3+), 43 (C2H3O+), 44 (CO2+), and 55 (C3H5O+). Thus, at the lower OH, Cl, and Br exposures used to generate Fig. 2 spectra, the main differences were associated with CxHyClOz+ and CxHyBrOz+ ions present in Cl-SOA and Br-SOA.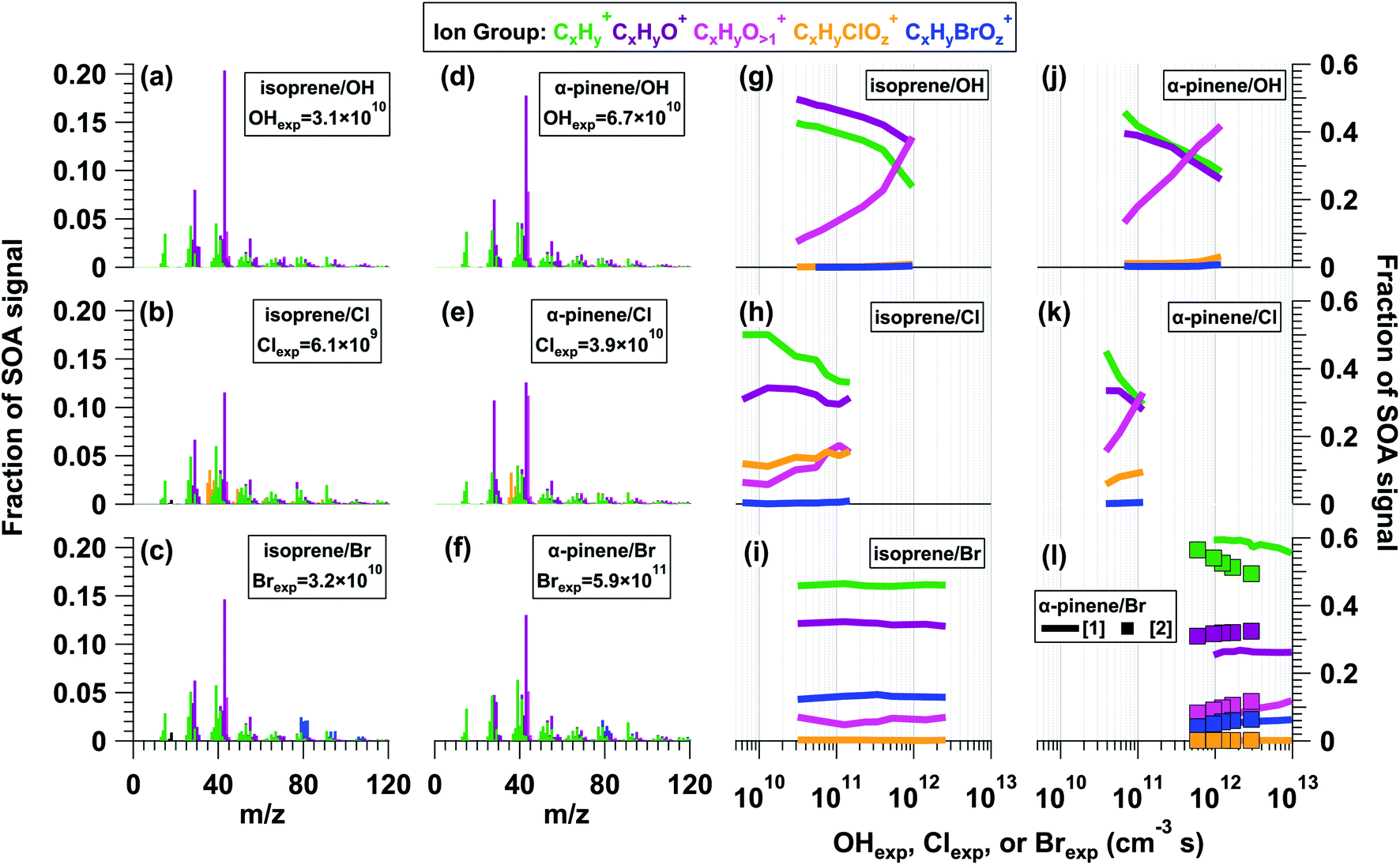 | ||
| Fig. 2 L-ToF-AMS spectra of SOA generated from the (a and g) OH oxidation of isoprene, (b and h) Cl oxidation of isoprene, (c and i) Br oxidation of isoprene, (d and j) OH oxidation of α-pinene, (e and k) Cl oxidation of α-pinene, and (f and l) Br oxidation of α-pinene. OHexp, Clexp, and Brexp values listed in (a)–(f) are in units of cm−3 s. The α-pinene Br-SOA spectrum presented in (f) was generated using OFR254-iC2Br2O2. Additional figure notes regarding (l): α-pinene Br-SOA was generated using (1) OFR369/421-iBr2 or (2) OFR254-iC2Br2O2. | ||
As was done in Section 3.1, to characterize the similarity of SOA mass spectra obtained from OH, Cl, and Br oxidation of the same precursor, we calculated r2 values between the isoprene OH-/Cl-/Br-SOA spectra shown in Fig. 2a–c, and between the α-pinene OH-/Cl-/Br-SOA shown in Fig. 2d–f. The r2 values between isoprene OH-/Cl-SOA and OH-/Br-SOA were 0.83 and 0.93 respectively. Likewise, r2 values between α-pinene OH-/Cl-SOA and OH-/Br-SOA were 0.86 and 0.92. In each of these cases, removing contributions from m/z = 43 or m/z = 44 from the regression analysis resulted in a minimal change in r2. To compare the similarity of isoprene and α-pinene OH-SOA, Cl-SOA and Br-SOA spectra across the full range of experimental conditions that were used, Fig. S11† plots r2 values between the L-ToF-AMS spectra shown in Fig. 2a and d and corresponding isoprene or α-pinene OH-/Cl-/Br-SOA spectra obtained at other OHexp, Clexp, and Brexp. The r2 value between Fig. 2a and more-oxidized isoprene OH-SOA spectra decreased from 0.99 to 0.32 with increasing OHexp. For isoprene Cl-SOA, r2 increased from 0.83 to 0.87 before decreasing to 0.63 at higher Clexp, whereas r2 = 0.91–0.92 as a function of Brexp for isoprene Br-SOA. The r2 value between Fig. 2d and more-oxidized α-pinene OH-SOA spectra decreased from 0.96 to 0.46 with increasing OHexp. For α-pinene Cl-SOA, r2 decreased from 0.86 to 0.57, and for α-pinene Br-SOA, r2 ranged from 0.92–0.93 (OFR254-iC2Br2O2) or 0.83–0.86 (OFR369/421-iBr2) as a function of Brexp.
Fig. 2g–l plot fCxHy+, fCxHyO1+, fCxHyO>1+, fCxHyClOz+, and fCxHyBrOz+ (blue) present in isoprene and α-pinene OH-/Cl-/Br-SOA as a function of oxidant exposure. Here, as was observed in Fig. 1g, minor contributions from the CxHyClOz+ ion group (orange) to the α-pinene OH-SOA spectra in Fig. 2g were mostly Cl+ and HCl+, and may be associated with NH4Cl generated from incidental NH3 + HCl reactions. For isoprene and α-pinene OH-/Cl-SOA, as with toluene OH-/Cl-SOA, fCxHy+ and fCxHyO1+ decreased, and fCxHyO>1+ and fCxHyClOz+ increased with increasing OHexp or Clexp (Table 2). Despite a similar decrease in fCxHy+ of isoprene OH-SOA and Cl-SOA, the decrease in fCxHyO1+ and increase in fCxHyO>1+ were smaller in isoprene Cl-SOA than in isoprene OH-SOA. This was probably because a higher oxidation state was achieved for isoprene OH-SOA than for isoprene Cl-SOA: the maximum OHexp (9.6 × 1011 cm−3 s) was 6.5 times higher than the maximum Clexp (1.5 × 1011 cm−3 s) shown in Fig. 2h, but the isoprene + Cl reaction rate is only 4.3 times faster than the isoprene + OH reaction rate. Due to Cl-induced fragmentation at high Clexp, the yield and size of isoprene SOA particles generated at Clexp = 2.9 × 1011 cm−3 s were too small for efficient transmission through the L-ToF-AMS inlet.
On the other hand, changes in isoprene and α-pinene Br-SOA composition were minor by comparison. For isoprene Br-SOA, fCxHy+ was approximately constant, fCxHyO+ decreased by less than 0.01, and fCxHyO>1+ and fCxHyBrOz+ increased slightly. For α-pinene Br-SOA, fCxHy+ decreased slightly, while fCxHyO1+, fCxHyO>1+, and fCxHyBrOz+ increased slightly. Following direct Br addition to α-pinene, additional organic bromide (ROBr) formation may have occurred via RO2 + Br and/or RO2 + BrO reactions.87,88 At a specific Brexp, α-pinene Br-SOA had lower fCxHyO+ and higher fCxHyO1+ when generated via OFR254-iC2Br2O2 (lines) compared to OFR369/421-iBr2 (symbols). We do not think that α-pinene Br-SOA photolysis at λ = 254 nm was important when using OFR254-iC2Br2O2 (Section 3.5), but other potential reasons for these Br-SOA compositional differences are unclear. Overall, based on these results, we hypothesize that multigenerational oxidative aging of Br-SOA was less extensive than in OH-SOA and Cl-SOA because Br is a more selective oxidant. While OH and Cl are likely reactive towards the majority of early-generation isoprene and α-pinene oxidation products, Br is only known to react efficiently with alkenes and aldehydes, with reactivity towards alcohols, ketones, and peroxides that is orders of magnitude slower than OH or Cl (e.g. Manion et al.89). This hypothesis and its implications will be explored further in Sections 3.4 and 3.5.
3.3 Cl-SOA and Br-SOA mass spectral markers
Fig. 3 shows L-ToF-AMS spectra of CxHyClO+z ions present in the n-C12, toluene, isoprene and α-pinene Cl-SOA spectra plotted in Fig. 1b, d and 2b, e. The y-axis in Fig. 3a (n-C12 Cl-SOA) was multiplied by a factor of 10 to put it on the same scale as Fig. 3b–d. The fCxHyClOz+ was lowest for n-C12 Cl-SOA (0.0083) because Cl oxidation proceeded practically exclusively via H-abstraction, and was highest for isoprene Cl-SOA (0.114) because Cl oxidation proceeded mostly via addition to double bonds. Intermediate fCxHyOzCl+ values of 0.077 and 0.063 were observed for toluene and α-pinene SOA, respectively. Overall, signals at m/z = 35 (Cl+) and m/z = 36 (HCl+) and their isotopes contributed 66 to 81% of the CxHyClOz+ signal across all Cl-SOA types. Other ions that were detected in multiple Cl-SOA spectra included m/z = 49 (CH2Cl+), 61–63 (C2H2−4Cl+), 76 (C2HClO+) and 77 (C2H2ClO+), albeit usually at much lower levels than Cl+ and HCl+.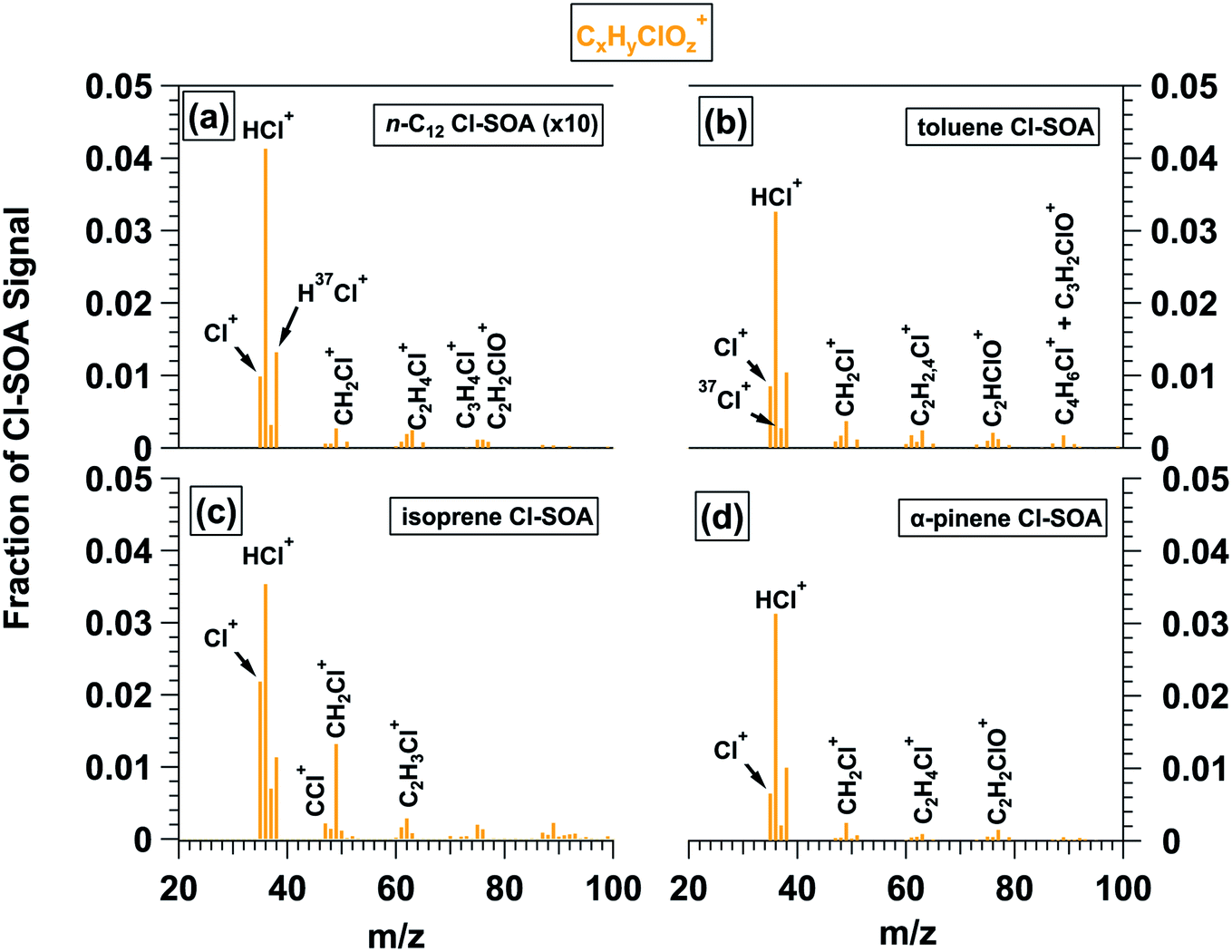 | ||
| Fig. 3 CxHyClOz+ ions present in L-ToF-AMS spectra of (a) n-C12, (b) toluene, (c) isoprene, and (d) α-pinene Cl-SOA displayed in Fig. 1b, d and 2b, e. The y-axis scale in (a) is multipled by 10 to put it on the same scale as (b)–(d). | ||
To compare these results with chamber Cl-SOA studies that reported fHCl+ values, Table 3 lists minimum and maximum fHCl+ values measured in OFR- and chamber-generated Cl-SOA at their corresponding Clexp values. In OFR-generated SOA, as expected, fHCl+ increased as a function of Clexp in a similar manner to the CxHyClOz+ ion signals shown in Fig. 1f, h and 2h, k. The most significant absolute change in fHCl+ was observed for OFR-generated toluene Cl-SOA, where fHCl+ increased from 0.050 to 0.16 as Clexp increased from 1.5 × 1010 to 1.3 × 1012 cm−3 s. By comparison, fHCl+ measured in chamber-generated toluene Cl-SOA ranged from 0.051 to 0.069.35 For the other Cl-SOA types, fHCl+ increased from 0.0059 to 0.020 (n-C12), 0.046 to 0.071 (isoprene), and 0.041 to 0.065 (α-pinene), respectively. Chamber-generated Cl-SOA had fHCl+ values ranging from 0.008 to 0.014 (n-C12), 0.034 to 0.067 (isoprene), and 0.013 to 0.056 (α-pinene).32,33,36
| VOC | Clexp (cm−3 s) | O/C | H/C | f HCl+ | C SOA (μg m−3) | Y SOA | Reference |
|---|---|---|---|---|---|---|---|
| n-C12 | 3.3 × 1010 to 2.3 × 1011 | 0.22–0.82 | 1.63–1.20 | 0.006–0.020 | 160–357 | 1.1–2.5 | This work |
| n-C12 | 9.8 × 1010 to 1.3 × 1011 | 0.32–0.88 | 1.50–1.26 | 0.006–0.014 | 99–149 | 1.10–1.65 | Wang and Hildebrandt Ruiz33 |
| Toluene | 2.3 × 1010 to 1.3 × 1012 | 0.53–1.02 | 1.07–0.82 | 0.067–0.159 | 14–99 | 0.083–0.58 | This work |
| Toluene | 3.3 × 1010 to 2.3 × 1011 | 0.46–1.08 | 1.22–0.86 | 0.050–0.127 | 24–106 | 0.15–0.64 | This work |
| Toluene | N/S | 0.69–0.81 | 0.94–0.89 | 0.050–0.069 | 53–136 | 0.33–0.67 | Dhulipala et al.35 |
| Toluene | N/S | 0.65 | 1.31 | 0.071 | 3–12 | 0.030–0.079 | Cai et al.30 |
| Isoprene | 6.1 × 109 to 2.9 × 1011 | 0.24–0.55 | 1.38–1.22 | 0.042–0.065 | 1–29 | 0.011–0.21 | This work |
| Isoprene | N/S | 0.45–0.72 | 1.32–1.11 | 0.033–0.067 | 9–80 | 0.08–0.29 | Wang and Hildebrandt Ruiz32 |
| α-Pinene | 3.9 × 1010 to 1.2 × 1011 | 0.42–0.80 | 1.39–1.18 | 0.040–0.065 | 37–77 | 0.22–0.47 | This work |
| α-Pinene | 1.9 × 1011 | 0.46–0.65 | 1.25 | 0.013–0.056 | 14–247 | 0.44–0.96 | Masoud and Hildebrandt Ruiz36 |
| α-Pinene | N/S | N/S | N/S | N/S | 0.0013–176 | 3.7 × 10−5 to 0.62 | Ofner et al.31 |
| α-Pinene | N/S | N/S | N/S | N/S | 8–33 | 0.079–0.22 | Cai and Griffin29 |
L-ToF-AMS spectra of Br-SOA contained a series of CxHyBrOz+ ions (Fig. 4), with fCxHyBrOz+ = 0.115 and 0.039 for isoprene and α-pinene Br-SOA, respectively. Because Br oxidation proceeded via double-bond addition, fCxHyBrOz+ was higher in isoprene Br-SOA. Signals at m/z = 79 (Br+) and 80 (HBr+) and their isotopes contributed 58 and 68% of the CxHyBrOz+ signal for isoprene and α-pinene Br-SOA; fHBr+ ranged from 0.035 to 0.042 and from 0.016 to 0.022 for isoprene and α-pinene Br-SOA (Table 3). Analogous to the usage of HCl+ as a marker ion for ROCl in the AMS,32 we hypothesize that H79Br+ and/or H81Br+ may be used as simple markers for ROBr in the absence of inorganic halides such as NH4Br. Depending on the source region, H81Br+ may be easier to resolve than H79Br+ due to sulfate (SO3+) interference at m/z = 79. Other signals that were detected in both isoprene and α-pinene Br-SOA spectra included m/z = 93 and 95 (CH2Br+), 106 and 108 (C2H3Br+), 121 and 123 (C2H2BrO+) and 133 and 135 through 138 (C3H5−6BrO+). Larger CxHyBrOz+-containing ions up to m/z = 245 and 247 were identified in α-pinene Br-SOA (Fig. S12–S13;† both m/z values shown to indicate similar concentrations of C10H14BrO2+ and its 81Br-containing isotope).
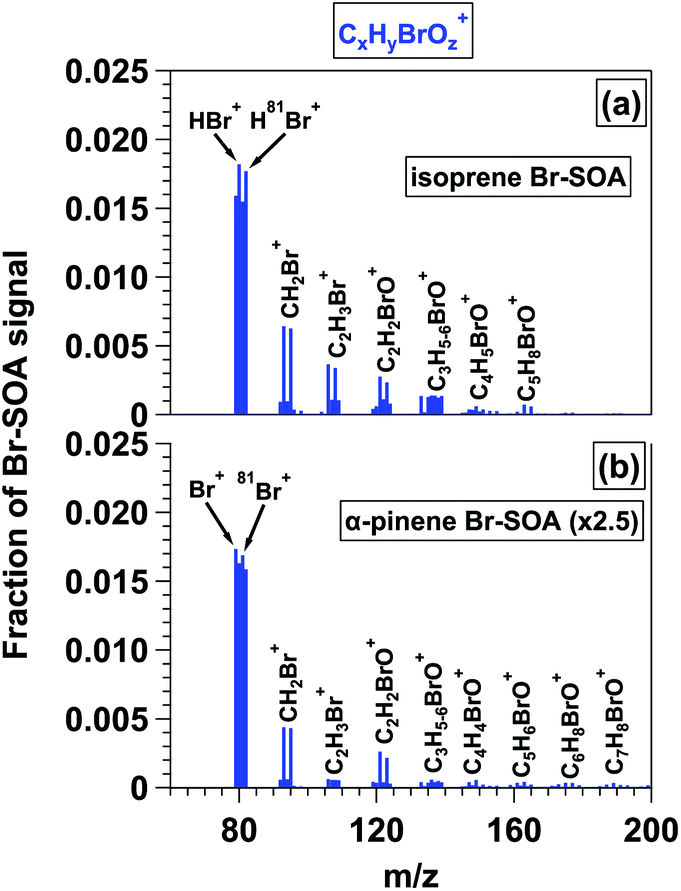 | ||
| Fig. 4 CxHyBrOz+ ions present in L-ToF-AMS spectra of (a) isoprene and (b) α-pinene Br-SOA displayed in Fig. 2c and f. The y-axis scale in (b) is multipled by 2.5 to put it on the same scale as (a). | ||
3.4 H/C and O/C ratios of OH-SOA, Cl-SOA and Br-SOA
Van Krevelen diagrams that show H/C ratio as a function of O/C ratio have been used to provide information about the nature of SOA formation and oxidative aging.90 Typically, with oxidative aging the O/C ratio increases and H/C ratio of SOA decreases as oxygen-containing functional groups are added to a carbon backbone. Here, we use Van Krevelen diagrams to compare the elemental ratios of OH-SOA, Cl-SOA, and Br-SOA discussed in the previous sections. Where applicable, data from chamber SOA studies are included for comparison.Fig. 5a–d show that chamber- and OFR-generated OH-SOA generally have similar Van Krevelen plots within the limited range of overlap of O/C and H/C values. Chamber-generated n-C12 OH-SOA had O/C and H/C values ranging from 0.18 to 0.30 and 1.83 to 1.67, respectively,66 and OFR-generated n-C12 OH-SOA had O/C and H/C values ranging from 0.27 to 1.05 and 1.59 to 1.08. Thus, SOA elemental ratios agree within approximately 5–10% for the most-oxidized chamber-generated n-C12 OH-SOA and the least-oxidized OFR-generated n-C12 OH-SOA. Similar trends were observed with the other OH-SOA types studied in this work (Fig. 5b–d). Whereas chamber studies have limited ability to generate highly oxidized OH-SOA, here, it was possible to compare H/C and O/C ratios of chamber- and OFR-generated Cl-SOA over a wide range of SOA oxidation state. Chamber-generated n-C12 Cl-SOA had O/C and H/C ratios ranging from 0.30 to 0.88 and 1.50 to 1.26 respectively33 compared to O/C and H/C ratios ranging from 0.22 to 0.82 and 1.63 to 1.20 for OFR-generated n-C12 Cl-SOA. At O/C = 0.36, n-C12 Cl-SOA H/C ratios agreed within ∼1% for chamber and OFR-generated Cl-SOA; likewise, at O/C = 0.82, the H/C ratios agreed within ∼5%. These results suggest that Cl-SOA elemental composition is the same regardless of whether it is generated at lower oxidant concentrations over longer exposure times in chambers, or higher oxidant concentrations over shorter exposures times in OFRs, similar to OH-SOA.91,92
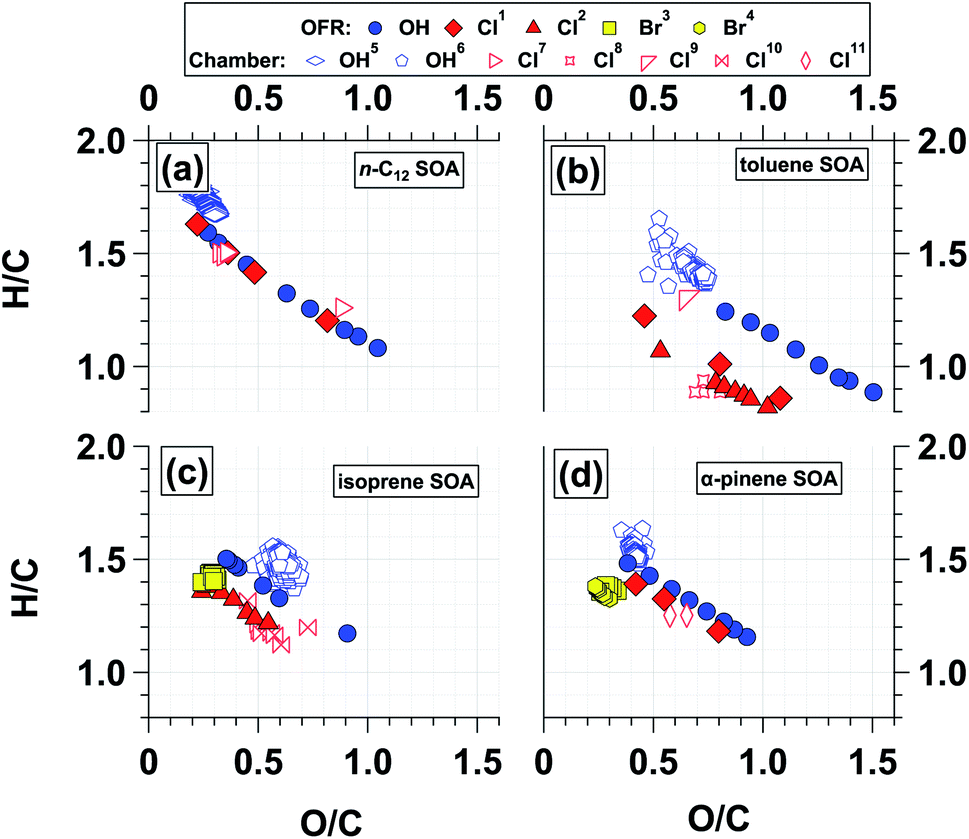 | ||
| Fig. 5 Van Krevelen diagrams showing H/C ratio as a function of O/C ratio for SOA generated from (a) OH and Cl oxidation of n-C12, (b) OH and Cl oxidation of toluene, (c) OH, Cl, and Br oxidation of isoprene, and (d) OH, Cl, and Br oxidation of α-pinene. Additional figure notes for superscripts: 1Cl generated using OFR254/313-iC2Cl2O2; 2Cl generated using OFR313/369-iCl2; 3Br generated using OFR254-iC2Br2O2; 4Br generated using OFR369/421-iBr2; 5Yee et al.;666Chhabra et al.;657Wang and Hildebrandt Ruiz;338Dhulipala et al.;359Cai et al.;3010Wang and Hildebrandt Ruiz;3211Masoud and Hildebrandt Ruiz.36 | ||
Fig. 5 also provides insight into differences between OH-, Cl-, and Br-SOA oxidative aging pathways. For systems where H-atom abstraction was the dominant reaction pathway (e.g., n-C12), the OH- and Cl-SOA Van Krevelen plots were essentially identical within the range of overlapping H/C and O/C ratios. On the other hand, the H/C ratio of toluene Cl-SOA was consistently ∼25–30% lower than the H/C ratio of toluene OH-SOA at a specific O/C ratio. For example, at O/C ≈ 0.8, H/C = 1.24 for toluene OH-SOA and 1.01 for toluene Cl-SOA. Because the Van Krevelen slopes (Δ(H/C)/Δ(O/C)) ranged from −0.50 to −0.55 for toluene OH-/Cl-SOA, the higher H/C ratio observed for toluene OH-SOA was probably a consequence of OH addition to the aromatic ring for (at least) the first toluene + OH reaction step, given that the H/C ratio of toluene is 1.14. This pathway would initially increase the H/C ratio. Thereafter, addition of similar oxygen-containing functional groups to toluene OH-SOA and Cl-SOA over multiple oxidation steps resulted in similar reductions in H/C as a function of O/C. Similar trends were observed for isoprene and α-pinene OH-/Cl-SOA, with H/C offsets of approximately 0.15 (isoprene) and 0.07 (α-pinene) at a specific O/C ratio. Corresponding Van Krevelen slopes ranged from −0.55 to −0.60 for each OH- and Cl-SOA type. As with OH oxidation of toluene via direct addition, OH addition to double bond(s) present in isoprene and α-pinene would have also initially increased the H/C ratio relative to Cl-SOA generated from the same precursor. Here, the lower increase in H/C of isoprene OH-SOA relative to toluene OH-SOA suggests multiple OH additions to the toluene backbone occurred via ring-opening reactions. Subsequent addition of similar oxygen-containing functional groups by both OH and Cl oxidation would have then generated similar Van Krevelen slopes.
Whereas the O/C and H/C ratios of OH-SOA and Cl-SOA changed significantly as a function of OHexp or Clexp, changes in the elemental ratios of isoprene and α-pinene Br-SOA were relatively minor. The O/C and H/C ratios of isoprene Br-SOA ranged from 0.24 to 0.31 and 1.40 to 1.41, and O/C and H/C ratios of α-pinene Br-SOA ranged from 0.24 to 0.32 and 1.33 to 1.39. Along with Fig. 2f and l, Fig. 5 provides additional evidence that multistep oxidative aging of Br-SOA is less extensive than in OH-SOA and Cl-SOA.
3.5 OH-SOA, Cl-SOA, and Br-SOA mass yields
Fig. 6a–d shows mass yields of SOA as a function of OHexp and Clexp for n-C12, toluene, isoprene, and α-pinene. SOA yields obtained from Br oxidation of isoprene and α-pinene are also shown in Fig. 6c and d. Fig. S14† shows the same data that is plotted in Fig. 6, plus the corresponding SOA yield values without applying particle wall loss correction (pWLC) factors (Section 2.3.2). Results obtained from Cl oxidation of the same precursors in environmental chamber studies is provided in Table 3. Some of the environmental chamber α-pinene Cl-SOA yields, and all the n-C12 Cl-SOA yields, were measured in the presence of added NOx, whereas no NOx was added in OFR experiments. However, yields of n-C12 OH-SOA do not display a systematic NOx dependence,93 and it is not yet clear to what extent NOx affects α-pinene Cl-SOA yields.29,31,36 Thus, to first order we assume NOx has less influence than other experimental variables such as oxidant type or Clexp on yields of n-C12 or α-pinene Cl-SOA.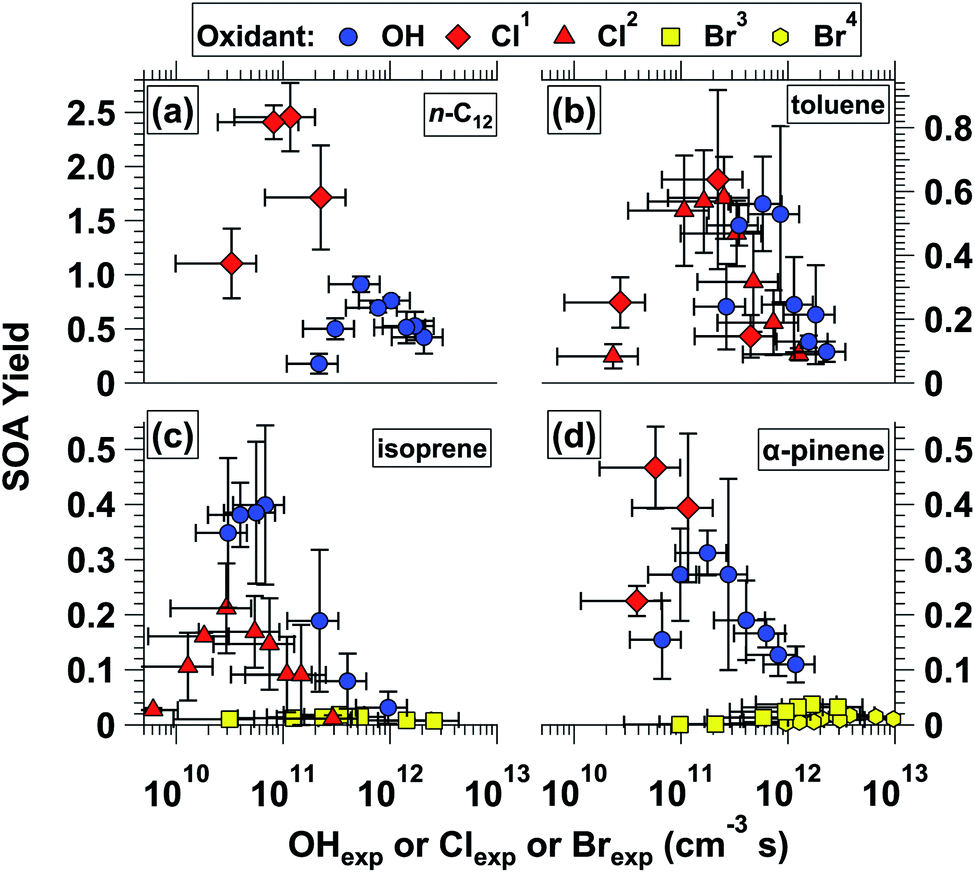 | ||
| Fig. 6 Mass yields of SOA generated from (a) OH and Cl oxidation of n-C12, (b) OH and Cl oxidation of toluene, (c) OH, Cl, and Br oxidation of isoprene, and (d) OH, Cl, and Br oxidation of α-pinene as a function of OHexp, Clexp, or Brexp. Different y-axis scales are used in each subpanel. Error bars indicate ±1σ uncertainty in binned SOA yield values, ±50% uncertainty in OH exposure values, and ±70% uncertainty in Cl and Br exposure values. Additional figure notes: 1Cl generated using OFR254/313-iC2Cl2O2; 2Cl generated using OFR313/369-iCl2; 3Br generated using OFR254-iC2Br2O2; 4Br generated using OFR369/421-iBr2. | ||
Fig. 6a shows that yields of n-C12 Cl-SOA and OH-SOA initially increased at lower Clexp and OHexp following functionalization reactions that produced condensable LVOCs. At higher Clexp and OHexp, yields decreased due to fragmentation reactions that generated higher-volatility oxidation products. Here, the observation that Cl-SOA yields exhibit similar trends as OH-SOA yields with increasing oxidation therefore builds on results obtained in previous OH-OFR laboratory and field studies.23–28,78,94,95 For n-C12 Cl-SOA, the yield increased from 1.1 to 2.5 (0.92 to 1.9 without pWLC; Fig. S14†) as Clexp increased from 3.3 × 1010 to 8.2 × 1010 cm−3 s, then decreased to 1.7 (1.3 without pWLC) at Clexp = 2.3 × 1011 cm−3 s. For n-C12 OH-SOA, the yield increased from 0.18 to 0.91 (0.14 to 0.74 without pWLC) as OHexp increased from 2.3 × 1011 to 5.4 × 1011 cm−3 s, then decreased to 0.42 (0.31 without pWLC) at OHexp = 2.1 × 1012 cm−3 s. Thus, over the range of conditions shown in Fig. 6, the maximum n-C12 Cl-SOA yield was approximately 2.7 times higher than the maximum n-C12 OH-SOA yield. In chamber studies, n-C12 Cl-SOA yields ranged from 1.10 to 1.65 (ref. 33) (Clexp = 9.8 × 1010 to 1.3 × 1011 cm−3 s) and n-C12 OH-SOA yields ranged from 0.15 to 0.28 (OHexp = 2.2 × 1011 to 4.3 × 1011 cm−3 s).93
Fig. 6b shows yields of toluene Cl-SOA and OH-SOA as a function of OHexp and Clexp. Toluene Cl-SOA yields obtained via OFR254-iC2Cl2O2 and OFR313/369-iCl2 are represented by different symbols. At the lowest and highest Clexp values that were used (Table 1), Cl-SOA yields were 0.083 and 0.090, respectively. Maximum toluene Cl-SOA yields were 0.58 ± 0.13 at Clexp = 2.5 × 1011 cm−3 s via OFR313/369-iCl2 and 0.64 ± 0.28 at Clexp = 2.2 × 1011 cm−3via OFR254-iC2Cl2O2. Toluene Cl-SOA yield values obtained in chambers ranged from 0.030 to 0.079 (ref. 30) and 0.33 to 0.67 (ref. 35) (Table 3). For toluene OH-SOA, the yield increased from 0.24 to 0.56 as OHexp increased from 2.6 × 1011 to 5.8 × 1011 cm−3 s, then decreased to 0.079 at OHexp = 2.3 × 1012 cm−3 s. At the lowest OHexp used here, our toluene OH-SOA yield value of 0.24 agrees within 14% of the toluene OH-SOA mass yield of 0.21 obtained at OHexp = 2.3 × 1011 cm−3 by Hildebrandt Ruiz et al.96 Overall, over the range of OFR conditions shown in Fig. 6b, Cl and OH oxidation of toluene generated maximum Cl-SOA and OH-SOA yield values that were within 14% of each other.
Fig. 6c shows yields of isoprene Cl-SOA, Br-SOA, and OH-SOA as a function of OHexp, Clexp, and Brexp. At the lowest and highest Clexp values shown in Fig. 6c, Cl-SOA yields were 0.027 and 0.011, respectively. The maximum isoprene Cl-SOA yield was 0.21 at Clexp = 3.0 × 1010 cm−3 s. By comparison, chamber isoprene Cl-SOA yield values ranged from 0.08 to 0.29.32 The maximum isoprene Br-SOA yield was only 0.018 at Brexp = 3.8 × 1011 cm−3 s, even when using a significantly higher isoprene mixing ratio than what was used in OH and Cl experiments (Table 1). The maximum isoprene OH-SOA yield measured here was 0.40 at OHexp = 6.8 × 1010 cm−3 s, which was a higher yield than expected based on recent isoprene OH-SOA yield values measured in the absence of NOx in both OFRs (0.032 at OHexp = 7.8 × 1011 cm−3 (ref. 92)) and chambers (≤0.15 at OHexp ≈ 5.1 × 1010 cm−3 s (ref. 97)). Our results agree with those obtained by Lambe et al.92 at comparable OHexp because we observed an isoprene OH-SOA yield of 0.031 at OHexp = 9.6 × 1011 cm−3 s. Thus, the lower OHexp achieved in this study was a contributing factor to the higher OFR isoprene OH-SOA yield, presumably due to less fragmentation of the SOA. While OHexp = 6.8 × 1010 cm−3 s is within ≈ 30% of the OHexp we estimate was used by Liu et al.,97 photochemical box modeling calculations suggest that mixing ratios of hydroperoxyl (HO2) radicals were approximately 6 times higher in the OFR (≈3 ppb vs. 0.55 ppb). Because isoprene OH-SOA yields are sensitive to the rate of reaction between HO2 and RO2, we hypothesize that our results may have been obtained under conditions that favored HO2 + RO2 reactions to a greater extent, thereby leading to ≈2 times higher SOA yield values. Overall, over the range of OFR conditions shown in Fig. 6, Cl and Br oxidation of isoprene generated SOA with maximum yields that are 52% and 5% of those obtained via OH oxidation.
Fig. 6d shows yields of α-pinene Cl-SOA, Br-SOA and OH-SOA. As was done in isoprene Br-SOA studies, a higher α-pinene mixing ratio was used to generate enough mass of low-volatility oxidation products to promote homogenous nucleation of Br-SOA (Table 1). The maximum α-pinene Cl-SOA yield was 0.47 at Clexp = 5.8 × 1010 cm−3 s. Maximum α-pinene Br-SOA yields were 0.037 at Brexp = 1.7 × 1012 cm−3 s (OFR254-iC2Br2O2) and 0.018 at Brexp = (2.8–3.8) × 1012 cm−3 s (OFR421-iBr2). As summarized in Table 3, our α-pinene Cl-SOA yield values were within the range of chamber α-pinene Cl-SOA yield values between 0.079 to 0.22,29 0.11 to 0.62,31 and 0.44 to 0.96.36 For α-pinene OH-SOA, the maximum yield was 0.31 at OHexp = 1.8 × 1011 cm−3 s, which is generally consistent with chamber α-pinene OH-SOA yield values obtained at comparable OHexp.98 Over the range of conditions shown in Fig. 6d, Cl and Br oxidation of α-pinene generates SOA at yields that are approximately 150% and 6–12% of those obtained via OH oxidation.
SOA photolysis at λ = 254 nm is a concern under certain OH-OFR conditions,46 but direct experimental evaluation is difficult due to the lack of operable OH sources at longer photolysis wavelengths. Here, our measurements enable an investigation into the potential role of Cl-SOA and Br-SOA photolysis at λ = 254 nm through a comparison of the yields of toluene Cl-SOA generated via OFR254-iC2Cl2O2versus OFR313/369-iCl2, and of α-pinene Br-SOA generated via OFR254-iC2Br2O2versus OFR369/421-iBr2. If SOA photolysis at λ = 254 nm was significant, yields of toluene Cl-SOA and α-pinene Br-SOA obtained using OFR254-iC2Cl2O2 and OFR254-iC2Br2O2 would have been lower than those obtained via OFR313/369-iCl2 and OFR369/421-iBr2. This was not the case. For toluene Cl-SOA, in the three regions of approximate Clexp overlap – (2.3–2.7) × 1010, (2.2–2.5) × 1011, and (4.5–4.7) × 1011 cm−3 s – toluene Cl-SOA yields obtained using OFR254-iC2Cl2O2 were either higher than, or in agreement with, yields obtained using OFR313/369-iCl2 within measurement uncertainties: 0.25 ± 0.08 versus 0.083 ± 0.038, 0.64 ± 0.28 versus 0.58 ± 0.13, and 0.15 ± 0.07 versus 0.32 ± 0.16, respectively. Similarly, α-pinene Br-SOA yields, obtained at Brexp ranging from 9.5 × 1011 to 2.9 × 1012 cm−3 s, were between 0.024 and 0.032 using OFR254-iC2Br2O2, compared to 0.0028 to 0.017 when using OFR369/421-iBr2. Thus, because halocarbons are typically photolabile, our results suggest that photolysis of Cl-SOA or Br-SOA was too slow to compete with multigenerational Cl- or Br-induced oxidative aging in the OFR. Because OH-SOA was also generated under conditions using λ = 254 nm radiation, and because OH-induced oxidative aging of SOA occurs to a similar or greater extent than Cl or Br, our results support previous modeling studies suggesting that OH-SOA measurements obtained using OFR185 are not significantly impacted by SOA photolysis at λ = 254 nm.46
4 Conclusions
In this study we characterized mass spectra, elemental ratios, and yields of SOA generated from the OH and Cl oxidation of representative anthropogenic precursors (n-C12 and toluene) and the OH, Cl and Br oxidation of representative biogenic (isoprene and α-pinene) precursors. Overall, r2-values between L-ToF-AMS spectra of Cl-SOA (and, where applicable, Br-SOA) and OH-SOA generated from the same precursor ranged from 0.57 to 0.94 at low oxidant exposures (Fig. 1a–d and 2a–f). The highest r2-value was observed between n-C12 OH- and Cl-SOA spectra, which was expected because OH- and Cl-induced oxidative aging occurred primarily via hydrogen atom abstraction. Van Krevelen diagrams of n-C12 OH- and Cl-SOA also had the highest degree of similarity (Fig. 5a). In cases where OH, Cl, and/or Br addition to unsaturated precursors (toluene, isoprene, α-pinene) was possible, the r2 values between Cl-/Br-SOA and OH-SOA were lower, and the H/C ratios of Cl-/Br-SOA were systematically lower than H/C ratios of OH-SOA (Fig. 5b–d). Additionally, the presence of CxHyClO+z and CxHyBrO+z ions in Cl-SOA and Br-SOA were clear indicators of halogen-initiated oxidative aging that may be used to investigate Cl- and Br-induced oxidative aging signatures in ambient AMS datasets. Across the full range of oxidant exposures that were studied, fundamental differences between Br- and OH-/Cl-initiated oxidative aging pathways were evident; namely, that multistep SOA oxidative aging is significant when initiated by OH and Cl, but not by Br.Our SOA yield measurements indicate that specific precursors generated SOA at yields that were strongly dependent on the oxidant and exposure time. Across all OFR conditions that were used, OH oxidation of n-C12, toluene, isoprene and α-pinene resulted in maximum OH-SOA yields ranging from 0.31 to 0.91; Cl oxidation of the same precursors, at the same precursor mixing ratios, generated Cl-SOA at maximum yields ranging from 0.21 to 2.5. Br oxidation of isoprene and α-pinene generated Br-SOA at maximum yields ranging from 0.018 to 0.037, suggesting that multigenerational oxidative aging may be required to achieve the range of yield values obtained across the OH-SOA and Cl-SOA systems examined here.
Notably, even though n-C12 OH-SOA and Cl-SOA had the highest degree of similarity in chemical composition, maximum n-C12 Cl-SOA and OH-SOA yields were the most different: the yield of n-C12 Cl-SOA was 2.7 times higher than the corresponding maximum n-C12 OH-SOA yield. This may be due to Cl preferentially reacting with terminal carbons on the n-C12 backbone,99 which could provide a longer effective carbon chain length for peroxy radicals to undergo intramolecular hydrogen shift reactions leading to SOA formation via autooxidation.100 Because OH reacts with both terminal and non-terminal carbons with similar probability, this may have resulted in more n-C12 OH-SOA fragmentation relative to n-C12 Cl-SOA.99 On the other hand, while toluene OH-SOA and Cl-SOA had the lowest degree of chemical similarity, maximum toluene OH-SOA and Cl-SOA mass yields were the closest in value. Overall, our results suggest that alkane, aromatic, and terpenoid SOA precursors are characterized by distinct OH- and halogen-initiated SOA yields, and that while Cl may enhance the SOA formation potential in regions influenced by biogenic and anthropogenic emissions, Br may have the opposite effect. Characterizing the molecular composition of specific oxidation products that contribute to OH-, Cl- and Br-SOA formation, and expanding the photochemical model introduced in Section 2.1.3 to include VOC + Cl/Br oxidation chemistry, may provide further insight into the trends observed here and will be the focus of future work.
Author contributions
AL conceived and planned the experiments. AL and AA carried out the OFR experiments and performed data analysis. DW and CM provided unpublished data from previous chamber Cl-SOA experiments, and MM calculated Cl exposure values in those chamber Cl-SOA experiments. AL, AA, NB, LHR, and WHB contributed to the interpretation of the results. AL took the lead in writing the manuscript. All authors provided feedback on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Atmospheric Chemistry Program of the National Science Foundation: grants AGS-1934352 to Aerodyne Research, Inc.; AGS-1934369 to the University of Texas at Austin; and AGS-1934345 to Pennsylvania State University. AL thanks Lindsay Yee (University of California at Berkeley) for providing published H/C and O/C ratios for n-C12 OH-SOA, and Benjamin Nault, Leah Williams, Donna Sueper, (Aerodyne), Pedro Campuzano-Jost (University of Colorado at Boulder), and Sergey Nizkorodov (University of California at Irvine) for helpful discussions.References
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, 2000 Search PubMed.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. Le Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, Atmos. Environ., Part A, 1991, 25, 1–203 CrossRef.
- S. S. Brown, H. D. Osthoff, H. Stark, W. P. Dubé, T. B. Ryerson, C. Warneke, J. A. de Gouw, A. G. Wollny, D. D. Parrish, F. C. Fehsenfeld and A. Ravishankara, J. Photochem. Photobiol., A, 2005, 176, 270–278 CrossRef CAS.
- Z. C. J. Decker, M. A. Robinson, K. C. Barsanti, I. Bourgeois, M. M. Coggon, J. P. DiGangi, G. S. Diskin, F. M. Flocke, A. Franchin, C. D. Fredrickson, G. I. Gkatzelis, S. R. Hall, H. Halliday, C. D. Holmes, L. G. Huey, Y. R. Lee, J. Lindaas, A. M. Middlebrook, D. D. Montzka, R. Moore, J. A. Neuman, J. B. Nowak, B. B. Palm, J. Peischl, F. Piel, P. S. Rickly, A. W. Rollins, T. B. Ryerson, R. H. Schwantes, K. Sekimoto, L. Thornhill, J. A. Thornton, G. S. Tyndall, K. Ullmann, P. Van Rooy, P. R. Veres, C. Warneke, R. A. Washenfelder, A. J. Weinheimer, E. Wiggins, E. Winstead, A. Wisthaler, C. Womack and S. S. Brown, Atmos. Chem. Phys., 2021, 21, 16293–16317 CrossRef CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS.
- O. W. Wingenter, M. K. Kubo, N. J. Blake, T. W. Smith Jr, D. R. Blake and F. S. Rowland, J. Geophys. Res., 1996, 101, 4331–4340 CrossRef CAS.
- A. K. Baker, C. Sauvage, U. R. Thorenz, P. van Velthoven, D. E. Oram, A. Zahn, C. A. M. Brenninkmeijer and J. Williams, Sci. Rep., 2016, 6, 36821 CrossRef CAS PubMed.
- J. Liao, L. G. Huey, Z. Liu, D. J. Tanner, C. A. Cantrell, J. J. Orlando, F. M. Flocke, P. B. Shepson, A. J. Weinheimer, S. R. Hall, K. Ullmann, H. J. Beine, Y. Wang, E. D. Ingall, C. R. Stephens, R. S. Hornbrook, E. C. Apel, D. Riemer, A. Fried, R. L. Mauldin III, J. N. Smith, R. M. Staebler, J. A. Neuman and J. B. Nowak, Nat. Geosci., 2014, 7, 91 CrossRef CAS.
- K. A. Pratt, Trends Chem., 2019, 1, 545–548 CrossRef CAS.
- T. P. Riedel, N. L. Wagner, W. P. Dubé, A. M. Middlebrook, C. J. Young, F. Öztürk, R. Bahreini, T. C. VandenBoer, D. E. Wolfe, E. J. Williams, J. M. Roberts, S. S. Brown and J. A. Thornton, J. Geophys. Res.: Atmos., 2013, 118, 8702–8715 CrossRef CAS.
- C. B. Faxon, J. K. Bean and L. Hildebrandt Ruiz, Atmosphere, 2015, 6, 1487–1506 CrossRef CAS.
- M. Priestley, M. le Breton, T. J. Bannan, S. D. Worrall, A. Bacak, A. R. D. Smedley, E. Reyes-Villegas, A. Mehra, J. Allan, A. R. Webb, D. E. Shallcross, H. Coe and C. J. Percival, Atmos. Chem. Phys., 2018, 18, 13481–13493 CrossRef CAS.
- J. P. S. Wong, N. Carslaw, R. Zhao, S. Zhou and J. P. D. Abbatt, Indoor Air, 2017, 27, 1082–1090 CrossRef CAS PubMed.
- H. Schwartz-Narbonne, C. Wang, S. Zhou, J. P. Abbatt and J. Faust, Environ. Sci. Technol., 2019, 53, 1217–1224 CrossRef CAS.
- J. M. Mattila, P. S. J. Lakey, M. Shiraiwa, C. Wang, J. P. D. Abbatt, C. Arata, A. H. Goldstein, L. Ampollini, E. F. Katz, P. F. DeCarlo, S. Zhou, T. F. Kahan, F. J. Cardoso-Saldaña, L. H. Ruiz, A. Abeleira, E. K. Boedicker, M. E. Vance and D. K. Farmer, Environ. Sci. Technol., 2020, 54, 1730–1739 CrossRef CAS.
- Q. Li, X. Fu, X. Peng, W. Wang, A. Badia, R. P. Fernandez, C. A. Cuevas, Y. Mu, J. Chen, J. L. Jimenez, T. Wang and A. Saiz-Lopez, Environ. Sci. Technol., 2021, 55, 13625–13637 CrossRef CAS.
- L. A. Barrie, J. W. Bottenheim, R. C. Schnell, P. J. Crutzen and R. A. Rasmussen, Nature, 1988, 334, 138–141 CrossRef CAS.
- C. R. Stephens, P. B. Shepson, A. Steffen, J. W. Bottenheim, J. Liao, L. G. Huey, E. Apel, A. Weinheimer, S. R. Hall, C. Cantrell, B. C. Sive, D. J. Knapp, D. D. Montzka and R. S. Hornbrook, J. Geophys. Res., 2012, 117, D00R11 CrossRef.
- T. Moise and Y. Rudich, Geophys. Res. Lett., 2001, 28, 4083–4086 CrossRef CAS.
- J. Ofner, N. Balzer, J. Buxmann, H. Grothe, P. Schmitt-Kopplin, U. Platt and C. Zetzsch, Atmos. Chem. Phys., 2012, 12, 5787–5806 CrossRef CAS.
- J. Buxmann, S. Bleicher, U. Platt, R. von Glasow, R. Sommariva, A. Held, C. Zetzsch and J. Ofner, Environ. Chem., 2015, 12, 476–488 CrossRef CAS.
- T. B. Nguyen, J. D. Crounse, R. H. Schwantes, A. P. Teng, K. H. Bates, X. Zhang, J. M. St. Clair, W. H. Brune, G. S. Tyndall, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2014, 14, 13531–13549 CrossRef.
- A. T. Lambe, T. B. Onasch, D. R. Croasdale, J. P. Wright, A. T. Martin, J. P. Franklin, P. Massoli, J. H. Kroll, M. R. Canagaratna, W. H. Brune, D. R. Worsnop and P. Davidovits, Environ. Sci. Technol., 2012, 46, 5430–5437 CrossRef CAS.
- D. S. Tkacik, A. T. Lambe, S. Jathar, X. Li, A. A. Presto, Y. Zhao, D. Blake, S. Meinardi, J. T. Jayne, P. L. Croteau and A. L. Robinson, Environ. Sci. Technol., 2014, 48, 11235–11242 CrossRef CAS.
- A. M. Ortega, P. L. Hayes, Z. Peng, B. B. Palm, W. Hu, D. A. Day, R. Li, M. J. Cubison, W. H. Brune, M. Graus, C. Warneke, J. B. Gilman, W. C. Kuster, J. De Gouw, C. Gutiérrez-Montes and J. L. Jimenez, Atmos. Chem. Phys., 2016, 16, 7411–7433 CrossRef CAS.
- B. A. Nault, P. Campuzano-Jost, D. A. Day, J. C. Schroder, B. Anderson, A. J. Beyersdorf, D. R. Blake, W. H. Brune, Y. Choi, C. A. Corr, J. A. de Gouw, J. Dibb, J. P. DiGangi, G. S. Diskin, A. Fried, L. G. Huey, M. J. Kim, C. J. Knote, K. D. Lamb, T. Lee, T. Park, S. E. Pusede, E. Scheuer, K. L. Thornhill, J.-H. Woo and J. L. Jimenez, Atmos. Chem. Phys., 2018, 18, 17769–17800 CrossRef CAS.
- W. Hu, H. Zhou, W. Chen, Y. Ye, T. Pan, Y. Wang, W. Song, H. Zhang, W. Deng, M. Zhu, C. Wang, C. Wu, C. Ye, Z. Wang, B. Yuan, S. Huang, M. Shao, Z. Peng, D. A. Day, P. Campuzano-Jost, A. T. Lambe, D. R. Worsnop, J. L. Jimenez and X. Wang, Environ. Sci. Technol., 2021 DOI:10.1021/acs.est.1c03155.
- K. Liao, Q. Chen, Y. Liu, Y. J. Li, A. T. Lambe, T. Zhu, R.-J. Huang, Y. Zheng, X. Cheng, R. Miao, G. Huang, R. B. Khuzestani and T. Jia, Environ. Sci. Technol., 2021, 55(11), 7276–7286 CrossRef CAS.
- X. Cai and R. J. Griffin, J. Geophys. Res., 2006, 111, D14206 CrossRef.
- X. Cai, L. D. Ziemba and R. J. Griffin, Atmos. Environ., 2008, 42, 7348–7359 CrossRef CAS.
- J. Ofner, K. A. Kamilli, A. Held, B. Lendl and C. Zetzsch, Faraday Discuss., 2013, 165, 135–149 RSC.
- D. S. Wang and L. Hildebrandt Ruiz, Atmos. Chem. Phys., 2017, 17, 13491–13508 CrossRef CAS.
- D. S. Wang and L. Hildebrandt Ruiz, Atmos. Chem. Phys., 2018, 18, 15535–15553 CrossRef CAS.
- Y. Wang, M. Riva, H. Xie, L. Heikkinen, S. Schallhart, Q. Zha, C. Yan, X.-C. He, O. Peräkylä and M. Ehn, Atmos. Chem. Phys., 2020, 20, 5145–5155 CrossRef CAS.
- S. V. Dhulipala, S. Bhandari and L. Hildebrandt Ruiz, Atmos. Environ., 2019, 199, 265–273 CrossRef CAS.
- C. G. Masoud and L. Hildebrandt Ruiz, ACS Earth Space Chem., 2021, 5, 2307–2319 CrossRef CAS.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- H. Zhang, D. R. Worton, S. Shen, T. Nah, G. Isaacman-VanWertz, K. R. Wilson and A. H. Goldstein, Environ. Sci. Technol., 2015, 49, 9768–9777 CrossRef CAS PubMed.
- J. P. Rowe, A. T. Lambe and W. H. Brune, Atmos. Chem. Phys., 2020, 20, 13417–13424 CrossRef CAS.
- B. L. Deming, D. Pagonis, X. Liu, D. A. Day, R. Talukdar, J. E. Krechmer, J. A. de Gouw, J. L. Jimenez and P. J. Ziemann, Atmos. Meas. Tech., 2019, 12, 3453–3461 CrossRef CAS.
- A. T. Lambe, J. E. Krechmer, Z. Peng, J. R. Casar, A. J. Carrasquillo, J. D. Raff, J. L. Jimenez and D. R. Worsnop, Atmos. Meas. Tech., 2019, 12, 299–311 CrossRef CAS.
- J. Mao, X. Ren, W. Brune, J. Olson, J. Crawford, A. Fried, L. Huey, R. Cohen, B. Heikes and H. Singh, Atmos. Chem. Phys., 2009, 9, 163–173 CrossRef CAS.
- A. V. Baklanov and L. N. Krasnoperov, J. Phys. Chem. A, 2001, 105, 97–103 CrossRef CAS.
- B. Ghosh, D. K. Papanastasiou and J. B. Burkholder, J. Chem. Phys., 2012, 137, 164315 CrossRef PubMed.
- M. Riva, R. M. Healy, P.-M. Flaud, E. Perraudin, J. C. Wenger and E. Villenave, J. Phys. Chem. A, 2015, 119, 11170–11181 CrossRef CAS PubMed.
- Z. Peng, D. A. Day, A. M. Ortega, B. B. Palm, W. Hu, H. Stark, R. Li, K. Tsigaridis, W. H. Brune and J. L. Jimenez, Atmos. Chem. Phys., 2016, 16, 4283–4305 CrossRef CAS.
- C.-C. Wu, H.-C. Lin, Y.-B. Chang, P.-Y. Tsai, Y.-Y. Yeh, H. Fan, K.-C. Lin and J. S. Francisco, J. Chem. Phys., 2011, 135, 234308 CrossRef PubMed.
- D. Paul, H. K. Kim, M. M. Rahman and T. K. Kim, J. Appl. Spectrosc., 2021, 88, 737–743 CrossRef CAS.
- D. Maric, J. Burrows, R. Meller and G. Moortgat, J. Photochem. Photobiol., A, 1993, 70, 205–214 CrossRef CAS.
- D. Maric, J. Burrows and G. Moortgat, J. Photochem. Photobiol., A, 1994, 83, 179–192 CrossRef CAS.
- J. E. Tuttle and G. K. Rollefson, J. Am. Chem. Soc., 1941, 63, 1525–1530 CrossRef CAS.
- H. Shimada, R. Shimada and Y. Kanda, Bull. Chem. Soc. Jpn., 1968, 41, 1289–1295 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2007, 7, 981–1191 CrossRef CAS.
- Z. Peng and J. L. Jimenez, J. Chem. Educ., 2019, 96, 806–811 CrossRef CAS.
- R. Li, B. B. Palm, A. M. Ortega, J. Hlywiak, W. Hu, Z. Peng, D. A. Day, C. Knote, W. H. Brune, J. A. De Gouw and J. L. Jimenez, J. Phys. Chem. A, 2015, 119, 150406123535006 Search PubMed.
- Z. Peng and J. L. Jimenez, Chem. Soc. Rev., 2020, 49, 2570–2616 RSC.
- P. F. Liu, N. Abdelmalki, H.-M. Hung, Y. Wang, W. H. Brune and S. T. Martin, Atmos. Chem. Phys., 2015, 15, 1435–1446 CrossRef.
- P. Campuzano-Jost, B. Nault, T. Koenig, H. Guo, J. Schroder, D. Day, J. Jimenez, R. Volkamer, K. Froyd, D. Murphy, A. Kupc, C. Williamson and C. Brock, in 18th AMS Users Meeting, 2018 Search PubMed.
- L. Jaegle, Y. C. Chan, D. Kim, P. Campuzano-Jost and J. L. Jimenez, AGU Fall Meeting, 2021 Search PubMed.
- S. M. Pieber, I. El Haddad, J. G. Slowik, M. R. Canagaratna, J. T. Jayne, S. M. Platt, C. Bozzetti, K. R. Daellenbach, R. Fröhlich, A. Vlachou, F. Klein, J. Dommen, B. Miljevic, J. L. Jiménez, D. R. Worsnop, U. Baltensperger and A. S. H. Prévôt, Environ. Sci. Technol., 2016, 50, 10494–10503 CrossRef CAS PubMed.
- B. Rao, T. A. Anderson, A. Redder and W. A. Jackson, Environ. Sci. Technol., 2010, 44, 2961–2967 CrossRef CAS PubMed.
- L. Jaeglé, Y. L. Yung, G. C. Toon, B. Sen and J.-F. Blavier, Geophys. Res. Lett., 1996, 23, 1749–1752 CrossRef PubMed.
- D. Sueper, 2022, https://cires.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware.
- A. C. Aiken, P. F. DeCarlo, J. H. Kroll, D. R. Worsnop, J. A. Huffman, K. S. Docherty, I. M. Ulbrich, C. Mohr, J. R. Kimmel and D. Sueper, Environ. Sci. Technol., 2008, 42, 4478–4485 CrossRef CAS PubMed.
- P. S. Chhabra, N. L. Ng, M. R. Canagaratna, A. L. Corrigan, L. M. Russell, D. R. Worsnop, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2011, 11, 8827–8845 CrossRef CAS.
- L. D. Yee, J. S. Craven, C. L. Loza, K. A. Schilling, N. L. Ng, M. R. Canagaratna, P. J. Ziemann, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem. A, 2012, 116, 6211–6230 CrossRef CAS PubMed.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams and K. R. Wilson, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- N. L. Ng, M. R. Canagaratna, J. L. Jimenez, P. S. Chhabra, J. H. Seinfeld and D. R. Worsnop, Atmos. Chem. Phys., 2011, 11, 6465–6474 CrossRef CAS.
- M. Kuwata, W. Shao, R. Lebouteiller and S. T. Martin, Atmos. Chem. Phys., 2013, 13, 5309–5324 CrossRef.
- R. Atkinson, Chem. Rev., 1986, 86, 69–201 CrossRef CAS.
- R. Atkinson, Atmos. Chem. Phys., 2003, 3, 2233–2307 CrossRef CAS.
- A. Bierbach, I. Barnes and K. H. Becker, Int. J. Chem. Kinet., 1996, 28, 565–577 CrossRef CAS.
- J. J. Orlando, G. S. Tyndall, E. C. Apel, D. D. Riemer and S. E. Paulson, Int. J. Chem. Kinet., 2003, 35, 334–353 CrossRef CAS.
- L. Renbaum-Wolff and G. D. Smith, J. Phys. Chem. A, 2012, 116, 6664–6674 CrossRef CAS PubMed.
- J. Shi and M. J. Bernhard, Int. J. Chem. Kinet., 1997, 29, 349–358 CrossRef CAS.
- B. Shi, W. Wang, L. Zhou, Z. Sun, C. Fan, Y. Chen, W. Zhang, Y. Qiao, Y. Qiao and M. Ge, Atmos. Environ., 2020, 222, 117166 CrossRef CAS.
- C. Bhattarai, V. Samburova, D. Sengupta, M. Iaukea-Lum, A. C. Watts, H. Moosmüller and A. Y. Khlystov, Aerosol Sci. Technol., 2018, 52, 1266–1282 CrossRef CAS.
- B. B. Palm, P. Campuzano-Jost, A. M. Ortega, D. A. Day, L. Kaser, W. Jud, T. Karl, A. Hansel, J. F. Hunter, E. S. Cross, J. H. Kroll, Z. Peng, W. H. Brune and J. L. Jimenez, Atmos. Chem. Phys., 2016, 16, 2943–2970 CrossRef CAS.
- A. T. Lambe and J. L. Jimenez, PAM Wiki, https://sites.google.com/site/pamwiki/estimation-equations?authuser=0 Search PubMed.
- M. Dal Maso, M. Kulmala, K. E. J. Lehtinen, J. M. Mäkelä, P. Aalto and C. D. O'Dowd, J. Geophys. Res.: Atmos., 2002, 107, PAR 2 CrossRef.
- A. T. Lambe, M. A. Miracolo, C. J. Hennigan, A. L. Robinson and N. M. Donahue, Environ. Sci. Technol., 2009, 43, 8794–8800 CrossRef CAS PubMed.
- W. H. Brune, Environ. Sci. Technol., 2019, 53, 3645–3652 CrossRef CAS PubMed.
- Y. He, A. T. Lambe, J. H. Seinfeld, C. D. Cappa, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2022 DOI:10.1021/acs.est.1c08520 , Article ASAP.
- N. Takegawa, T. Miyakawa, K. Kawamura and Y. Kondo, Aerosol Sci. Technol., 2007, 41, 418–437 CrossRef CAS.
- M. M. Maricq, J. J. Szente, E. W. Kaiser and J. Shi, J. Phys. Chem., 1994, 98, 2083–2089 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson Jr, J. A. Kerr, M. J. Rossi, J. Troe, IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry - Web Version, 2001 Search PubMed.
- J. S. Francisco and J. N. Crowley, J. Phys. Chem. A, 2006, 110, 3778–3784 CrossRef CAS PubMed.
- S. Enami, T. Yamanaka, T. Nakayama, S. Hashimoto, M. Kawasaki, D. E. Shallcross, Y. Nakano and T. Ishiwata, J. Phys. Chem. A, 2007, 111, 3342–3348 CrossRef CAS PubMed.
- J. A. Manion, R. E. Huie, R. D. Levin, D. R. B. Burgess Jr, V. L. Orkin, W. Tsang, W. S. McGivern, J. W. Hudgens, V. D. Knyazev, D. B. Atkinson, E. Chai, A. M. Tereza, C.-Y. Lin, T. C. Allison, W. G. Mallard, F. Westley, J. T. Herron, R. F. Hampson and D. H. Frizzell, NIST Chemical Kinetics Database, NIST Standard Reference Database 17, Version 7.0 (Web Version), Release 1.6.8, Data version 2015.09, National Institute of Standards and Technology technical report, 2015 Search PubMed.
- C. L. Heald, J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. Decarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer and P. Artaxo, Geophys. Res. Lett., 2010, 37, L08803 Search PubMed.
- E. A. Bruns, I. El Haddad, -A. Keller, F. Klein, N. K. Kumar, S. M. Pieber, J. C. Corbin, J. G. Slowik, W. H. Brune, U. Baltensperger and A. S. H. Prevot, Atmos. Meas. Tech., 2015, 8, 2315–2332 CrossRef CAS.
- A. T. Lambe, P. S. Chhabra, T. B. Onasch, W. H. Brune, J. F. Hunter, J. H. Kroll, M. J. Cummings, J. F. Brogan, Y. Parmar, D. R. Worsnop, C. E. Kolb and P. Davidovits, Atmos. Chem. Phys., 2015, 15, 3063–3075 CrossRef CAS.
- C. L. Loza, J. S. Craven, L. D. Yee, M. M. Coggon, R. H. Schwantes, M. Shiraiwa, X. Zhang, K. A. Schilling, N. L. Ng, M. R. Canagaratna, P. J. Ziemann, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2014, 14, 1423–1439 CrossRef.
- K. Li, J. Liggio, P. Lee, C. Han, Q. Liu and S.-M. Li, Atmos. Chem. Phys., 2019, 19, 9715–9731 CrossRef CAS.
- W. Xu, Z. Li, A. T. Lambe, J. Li, T. Liu, A. Du, Z. Zhang, W. Zhou and Y. Sun, Environ. Res., 2022, 209, 112751 CrossRef CAS PubMed.
- L. Hildebrandt Ruiz, A. L. Paciga, K. M. Cerully, A. Nenes, N. M. Donahue and S. N. Pandis, Atmos. Chem. Phys., 2015, 15, 8301–8313 CrossRef CAS.
- J. Liu, E. L. D'Ambro, B. H. Lee, F. D. Lopez-Hilfiker, R. A. Zaveri, J. C. Rivera-Rios, F. N. Keutsch, S. Iyer, T. Kurten, Z. Zhang, A. Gold, J. D. Surratt, J. E. Shilling and J. A. Thornton, Environ. Sci. Technol., 2016, 50, 9872–9880 CrossRef CAS PubMed.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, M. Chan, K. A. Schilling, P. S. Chhabra, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2012, 12, 7413–7427 CrossRef CAS.
- L. G. Jahn, D. S. Wang, S. V. Dhulipala and L. Hildebrandt Ruiz, J. Phys. Chem. A, 2021, 125, 7303–7317 CrossRef CAS PubMed.
- T. F. Mentel, M. Springer, M. Ehn, E. Kleist, I. Pullinen, T. Kurtén, M. Rissanen, A. Wahner and J. Wildt, Atmos. Chem. Phys., 2015, 15, 6745–6765 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: mercury lamp emission spectra, Cl and Br exposure calibration data, supplemental high-resolution aerosol mass spectra, KinSim mechanism. See https://doi.org/10.1039/d2ea00018k |
| ‡ Present address: Paul Scherrerr Institute, Villagen, Switzerland. |
| This journal is © The Royal Society of Chemistry 2022 |