
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical transformation of a long-chain alkyl organosulfate via heterogeneous OH oxidation: a case study of sodium dodecyl sulfate†
Sze In
Madeleine Ng
 a,
Kwan Hung
Ng
b,
Pui Wo
Felix Yeung
b,
Rongshuang
Xu
a,
Pui-Kin
So
c,
Yuanlong
Huang
d,
Jian Zhen
Yu
e,
Chun Kit K.
Choi
f,
Ying-Lung
Steve Tse
*b and
Man Nin
Chan
*ag
a,
Kwan Hung
Ng
b,
Pui Wo
Felix Yeung
b,
Rongshuang
Xu
a,
Pui-Kin
So
c,
Yuanlong
Huang
d,
Jian Zhen
Yu
e,
Chun Kit K.
Choi
f,
Ying-Lung
Steve Tse
*b and
Man Nin
Chan
*ag
aEarth System Science Programme, Faculty of Science, The Chinese University of Hong Kong, Hong Kong, China. E-mail: mnchan@cuhk.edu.hk
bDepartment of Chemistry, The Chinese University of Hong Kong, Hong Kong, China. E-mail: stevetse@cuhk.edu.hk
cThe University Research Facility in Life Sciences, The Hong Kong Polytechnic University, Hong Kong, China
dDivision of Geological and Planetary Sciences, California Institute of Technology, Pasadena, California 91125, USA
eDepartment of Chemistry, The Hong Kong University of Science and Technology, Hong Kong, China
fDepartment of Chemical Pathology, The Chinese University of Hong Kong, Hong Kong, China
gThe Institute of Environment, Energy and Sustainability, The Chinese University of Hong Kong, Hong Kong, China
First published on 1st June 2022
Abstract
Organosulfates (OSs) are regarded as stable tracers of secondary organic aerosols. However, recent studies have reported their potential chemical instability, which is dependent on their structures. In this study, we aim to investigate the transformation and kinetics of a long-chain alkyl OS upon heterogeneous hydroxyl radical (OH) oxidation. We selected sodium dodecyl sulfate (SDS, C12H25O4SNa) as a model compound due to its atmospheric relevance. We conducted experiments using an oxidation flow reactor at 80% RH and room temperature. We analyzed the reaction kinetics and products by liquid chromatography-mass spectrometry and ultrahigh-resolution mass spectrometry. We quantified inorganic sulfate formation by ion chromatography. We have proposed reaction pathways based on aerosol composition data. Our results reveal that dodecyl sulfate decays at (4.09 ± 0.09) × 10−13 cm3 per molecule per s with an atmospheric lifetime of ∼19 days upon heterogeneous OH oxidation. Compared with the literature results, we observe a significant kinetics enhancement when ammonium sulfate is present in aerosols. Our molecular dynamics simulations suggest that ammonium ions tend to displace sodium ions from the air–water interface and attract OH more strongly, which promotes collisions between dodecyl OS and OH. Therefore, the effects of counterions on surface-active organics should be considered during interpretation of experimental kinetics data. We detected sequential oxygenation of dodecyl sulfate, which dominated over fragmentation and inorganic sulfate formation. Our identified products indicate a potential source of some oxygenated aliphatic C6- to C10- and C12-OS detected in the atmosphere. Collectively, our work highlights the need for more comprehensive investigations of structural factors governing OS chemistry.
Environmental significanceOrganosulfates (OSs), occupying an important organic mass fraction in atmospheric aerosols, are frequently considered chemically stable. However, studies have shown that OSs undergoing oxidative aging in the troposphere can produce highly oxidized species, and inorganic sulfates which can heavily alter radiative forcing. Here, we demonstrate that an alkyl OS can readily react with the hydroxyl radical (OH), leading to the formation of oxygenated derivatives. Our results indicate that inorganic sulfate formation is less significant as the carbon chain of the OS lengthens, but the significance increases with OH exposure. Furthermore, the reaction kinetics depend on the presence of ions in aerosols. Therefore, we stress the importance of a thorough understanding of structural factors governing OS oxidative chemistry for a better assessment of OS lifetimes and budget in the atmosphere. |
1. Introduction
Organosulfur compounds are important contributors to secondary organic aerosol formation.1–5 By observing the discrepancy between the total aerosol sulfur measured by X-ray fluorescence spectrometry and the sulfur present as inorganic sulfate measured by ion chromatography (IC) in fine atmospheric aerosols, organosulfur compounds were shown to occupy up to 30% of the total organic aerosol mass at a forest site in K-puszta, Hungary.6 Using the same method, organosulfur compounds were found to contribute as high as 5–10% of the organic aerosol mass at 12 sites in the US.1 More recently, emerging field measurements have suggested that a large variety of organosulfur compounds originate from different precursors in clean and polluted atmospheric environments.6–15 For instance, organosulfur compounds found in cleaner cities such as Mainz, Germany, are mainly derived from biogenic organic compounds, whereas those in polluted atmospheres in China are attributed to anthropogenic organic precursors.16–20 These studies convey that organosulfur compounds contribute to an important fraction of aerosols worldwide, particularly in regions where photochemical activities are strong.5,17,21,22 Among all organosulfur species (including organosulfates (OSs), sulfoxides, sulfonates, and sulfones), OSs with a sulfate ester functional group (R–O–SO3−) are quantified as the most abundant class.6–13,23 The formation pathways of some OSs (e.g. isoprene derived OSs) have been well-studied and incorporated into photochemical box models such as GAMMA and UNIPAR and chemical transport models such as CMAQ, to examine the abundances of OSs in the atmosphere.7,24–29 Numerous studies have also reported the role of OSs in governing the physicochemical properties of aerosols such as surface tension, light absorption, water uptake, and cloud formation potential, which exert direct climatic impacts.30–33Several laboratory studies complemented by field studies identified and quantified a wide variety of OSs in atmospheric aerosols, including hydroxy OSs,6,34 nitroxy and carboxy OSs,19,35–40 aromatic OSs,12,41–43 and aliphatic OSs (i.e., alkyl OSs, alkenyl OSs, and their oxygenated derivatives).16–18,44–46 OSs primarily exist in the aerosol phase due to low volatility,30,47 and are assumed to be relatively stable compounds. Several OSs have frequently been used as tracers of secondary organic aerosols (SOAs).13,48–50 However, recent laboratory investigations have revealed the potential of OSs to transform significantly into other unidentified OSs (OSs which have been detected in the atmosphere but the precursors are unknown) or inorganic sulfates, via hydrolysis and heterogeneous oxidation on a short atmospheric timescale. For instance, Elrod and his co-workers reported their potential to form polyols and sulfuric acid via hydrolysis, with the shortest chemical lifetime being less than half a day.7,9 These findings further suggested that the transformation rate of OSs depends on the aerosol acidity and their chemical structures, e.g., tertiary OSs are prone to faster transformation than their primary and secondary counterparts.
Heterogeneous oxidation, different from aqueous or gas-phase oxidation, is triggered by gas-phase oxidants that can react with aerosol components confined to the aerosol surface.51–53 Heterogeneous oxidation by the hydroxyl radical (OH) is an efficient way to chemically transform OSs.54–57 Literature studies utilized oxidation flow reactors to investigate OS species such as short-chain alkyl OSs (methyl sulfate (methyl OS, CH3SO4−) and ethyl sulfate (ethyl OS, C2H5SO4−)),55,56 monoterpene-derived OSs (α-pinene OS, C10H17O5S−),57 and isoprene-epoxydiol-derived OSs (2-methyltetrol sulfates and 3-methyltetrol sulfates (2-/3-methyltetrol OS, C5H11SO7−)).58 Their results demonstrated that all these OSs can be oxidized by OH efficiently with a chemical lifetime ranging from 10 to 15 days. Importantly, several unidentified OSs can be explained, with respect to formation mechanisms, by heterogeneous OH oxidation of OSs.54,57 Regarding the formation of other products, Xu et al.56 reported a significant molar yield of inorganic sulfates from methyl OS, defined as the number of moles of bisulfate (HSO4−) and sulfate (SO4−) ions generated per mole of parent OS, which increased up to 62 ± 18% upon heterogeneous OH oxidation. From these findings, it is reasonable to hypothesize that a portion of OS may not be as atmospherically stable as previously presumed. Potentially, they can further transform to produce some of the currently unidentified OSs in the atmosphere. However, because the stability of OSs depends heavily on their chemical structures, the potential of structurally different OSs to transform remains unclear.
In addition to short-chain alkyl OSs, their long-chain counterparts, whose precursors are mainly anthropogenic alkanes, have also been detected in atmospheric aerosols.15–18,39,44–46,50,59 Long-chain OSs are commonly present as surfactant coatings on marine and continental aerosols.60–62 Dodecyl sulfate (hereafter referred to as dodecyl OS), with the chemical formula C12H25O4S−, is a relevant example of an anionic surfactant in atmospheric aerosols.15,59,63,64 Because dodecyl OS is widely used as a manufactory detergent, it has been detected to be omnipresent in aerosols generated from waste water.65 Through waste water drainage and river runoff into the seawater, dodecyl OS is also abundant in coastal sea spray aerosols.60 The sodium salt of dodecyl OS, namely SDS, is therefore atmospherically abundant and relevant. Due to the surface-active nature of SDS, heterogeneous oxidation can be one of its potential sinks in the atmosphere. In fact, previous laboratory investigations have studied heterogeneous oxidation of aerosols involving SDS. However, in these studies, the role of dodecyl OS was presented as a surfactant assisting in the probing of the properties of other aerosol constituents.66,67 Faust and Abbatt66 investigated the shielding effect of dodecyl OS on dissolved tricarballylic acid against heterogeneous OH oxidation and suggested that the presence of surfactants in aerosols can prolong the atmospheric lifetime of inner constituents. Huang et al.67 deduced the heterogeneous OH oxidation kinetics of surface-active pinonic acid by using competitive kinetics with dodecyl OS. Their results implied that the air–water interfacial reaction chemistry is crucial in determining the fate of surface-active organic species in aerosols.67 In their study, sequential oxygenation of dodecyl OS was observed.
To the best of our knowledge, direct investigation of the transformation and kinetics of dodecyl OS upon heterogeneous OH oxidation is scarce. Particularly, the potential of dodecyl OS undergoing oxidation to fragment into short-chain OSs and inorganic sulfates is largely unknown. A fundamental study to address these subjects can serve to better characterize the reactivity of long-chain alkyl OSs in atmospheric aerosols. In our study, we conducted experiments via controlled laboratory oxidative aging of SDS aerosols at 80% relative humidity (RH) and at room temperature (Table 1). Heterogeneous OH oxidation of SDS aerosols was performed using an oxidation flow reactor (OFR) (a.k.a. potential aerosol mass reactor, PAM). After oxidation, we collected the aerosols onto Teflon filters for offline chemical analysis using various mass spectrometry techniques. Collectively, the overall study was designed to achieve the following objectives: (1) to determine the heterogeneous reactivity of SDS (pure organic) aerosols, (2) to supplement our findings with molecular dynamics (MD) simulations, in order to account for the difference between our experimental kinetics data and those in the literature utilizing a mixture of SDS and an inorganic salt (ammonium sulfate), (3) to examine the significance of smaller OS (C < 12) formation and inorganic sulfate yield upon heterogeneous OH oxidation of a long-chain alkyl OS, and (4) to propose reaction mechanisms for dodecyl OS. Our results offer new insight into the heterogeneous OH reactivity of a long-chain alkyl OS and the chemical conversions of sulfur between its inorganic and organic forms, initiating a re-evaluation of the sources and budget of atmospheric OSs and inorganic sulfates.
| Compound | Sodium dodecyl OS (SDS) |
|---|---|
| Molecular structure |
|
| Molecular formula | C12H25O4SNa |
| Molecular weight (g mol−1) | 288.37 |
| Experiment | |
|---|---|
| Relative humidity (RH%) | 80 |
| Maximum OH exposure (× 1011 molecules per cm3 s) | 16.3 ± 0.3 |
| Mean surface-weighted particle diameter before/after oxidation (nm) | 279.5/278.0 |
| Effective heterogeneous OH oxidation rate constant, k (×10−13 cm3 per molecule per s) | 4.09 ± 0.09 |
| Atmospheric lifetime against OH oxidation (days) | 18.9 ± 0.4 |
| Inorganic sulfur yield at maximum OH exposure (%) | 1.82 ± 6.57 |
2. Experimental methods
Heterogeneous OH oxidation of SDS aerosols was investigated using an OFR at 80% RH and room temperature. The experimental setup and procedures have been described elsewhere.57,68 In brief, SDS was dissolved in ultra-pure deionized water. Aqueous aerosols were then generated by atomizing the solution through a constant output atomizer (without passing through a diffusion dryer) and subsequently mixing with nitrogen (N2), oxygen (O2), and ozone (O3) before being introduced into the OFR. The RH was controlled by varying the mixing ratios of dry and wet gas flows. A total flow rate of ∼5 L min−1 was fed into the reactor corresponding to ∼156 s of residence time. The aerosols were oxidized inside the reactor by gas-phase OH radicals generated by the photolysis of O3 under ultraviolet (UV) light (λ = 254 nm) illumination in the presence of water vapor. O3 was generated by passing O2 through an ozone generator. The OH concentration was regulated by changing the O3 concentration and was determined by measuring the decay of sulfur dioxide (SO2) in independent experiments in the presence or absence of SDS aerosols. We ascertained that the presence of aerosols did not significantly affect the generation of gas-phase OH radicals and the determination of OH exposure, for which the uncertainty was less than ∼10%. OH exposure, defined as the product of gas-phase OH concentration and residence time, ranging from 0 to (16.3 ± 0.3) × 1011 molecules per cm3 s was utilized. This number corresponds to a maximum of 12.6 ± 0.3 days of exposure time assuming a 24 h averaged ambient OH concentration of 1.5 × 106 molecules per cm3.69After oxidation, the aerosol stream exiting the reactor then passed through an annular Carulite catalyst denuder and an activated charcoal denuder to remove residual O3 and the gas-phase species remaining in the stream, respectively. As such, only aerosol-phase reaction products were collected for offline analysis. The size distribution of the aerosols was measured with a scanning mobility particle sizer (SMPS, TSI). Before oxidation, the mean surface weighted diameter for aerosol distribution was about 279.5 ± 0.5 nm with a geometric standard deviation of 1.3, and the aerosol mass loading was ∼2200 μg m−3. Aerosols were collected onto Teflon filters with a flow of 3 L min−1 for 30 min, allowing for a total gas sampling volume of ∼90 L. The filters were immediately stored at −20 °C in the dark prior to chemical analysis, which was conducted within three weeks after experiments. An overview of the chemical analysis for product identification and reaction kinetics determination is shown in Scheme S1.† Ultrahigh-performance liquid chromatography-electrospray ionization-high resolution mass spectrometry (UHPLC-ESI-HRMS) was employed to quantify the decay of dodecyl OS for derivation of reaction kinetics. The composition of reaction products was characterized using an ultrahigh-resolution Orbitrap Eclipse Tribrid mass spectrometer. The formation of inorganic sulfates was quantified by ion chromatography. The details of the analytical procedures and measurement uncertainties are outlined in the ESI (Sections S1 and S2†).
To decouple the effects of O3 and UV light on the composition of SDS aerosols, we ran control experiments. No significant change in the amount of dodecyl OS was found in the presence of O3 without UV light, suggesting a negligible reaction between SDS and O3. The quantified amount was also similar to that obtained in the absence of O3 with UV light, suggesting that the photolysis of SDS is not likely to occur. These results are consistent with previous observations by Faust and Abbatt.66
3. Results and discussion
3.1 Reaction products
To examine the heterogeneous oxidation of dodecyl OS, we conducted aerosol speciation by ultrahigh-resolution mass spectrometry. Fig. 1 depicts the aerosol mass spectra before and after oxidation. Before oxidation, the peak at m/z 265 with the chemical formula C12H25O4S− was predominantly detected as the major component, corresponding to the parent dodecyl OS. After oxidation, the absolute intensity of this peak substantially decreased by 49.1% while several new peaks appeared, possibly due to the formation of various products. Notably, detectable peaks of bisulfate ions (HSO4−, m/z 97) and other short-chain OSs (C < 12) emerged. Products with m/z values larger than 265 were the oxygenated C12-OS, which were likely formed by functionalization processes without cleavage of the carbon chain. To generalize, the addition of m hydroxyl groups (–OH) and n carbonyl groups (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) to dodecyl OS will produce C12H25−2nO4+m+nS−, in the (m + n)th generation of oxygenation. Specifically, the products from the 1st generation of oxygenation have chemical formulae of C12H23O5S− (m/z 279) and C12H25O5S− (m/z 281), which, respectively, correspond to the addition of a carbonyl and a hydroxyl group to dodecyl OS. The products from the 2nd generation include m/z 293, 295, and 297, with chemical formulae of C12H21O6S−, C12H23O6S−, and C12H25O6S−, respectively. These OSs represent the addition of 2 carbonyl groups, 1 carbonyl and 1 hydroxyl groups, and 2 hydroxyl groups to dodecyl OS, respectively. Similarly, the 3rd generation products include C12H19O7S− (m/z 307), C12H21O7S− (m/z 309), and C12H23O7S− (m/z 311), respectively, formed by the addition of 3 carbonyl groups, 2 carbonyl and 1 hydroxyl groups, and 2 hydroxyl and 1 carbonyl groups. A summary of the OSs detected is shown in Table S1.†
O) to dodecyl OS will produce C12H25−2nO4+m+nS−, in the (m + n)th generation of oxygenation. Specifically, the products from the 1st generation of oxygenation have chemical formulae of C12H23O5S− (m/z 279) and C12H25O5S− (m/z 281), which, respectively, correspond to the addition of a carbonyl and a hydroxyl group to dodecyl OS. The products from the 2nd generation include m/z 293, 295, and 297, with chemical formulae of C12H21O6S−, C12H23O6S−, and C12H25O6S−, respectively. These OSs represent the addition of 2 carbonyl groups, 1 carbonyl and 1 hydroxyl groups, and 2 hydroxyl groups to dodecyl OS, respectively. Similarly, the 3rd generation products include C12H19O7S− (m/z 307), C12H21O7S− (m/z 309), and C12H23O7S− (m/z 311), respectively, formed by the addition of 3 carbonyl groups, 2 carbonyl and 1 hydroxyl groups, and 2 hydroxyl and 1 carbonyl groups. A summary of the OSs detected is shown in Table S1.†
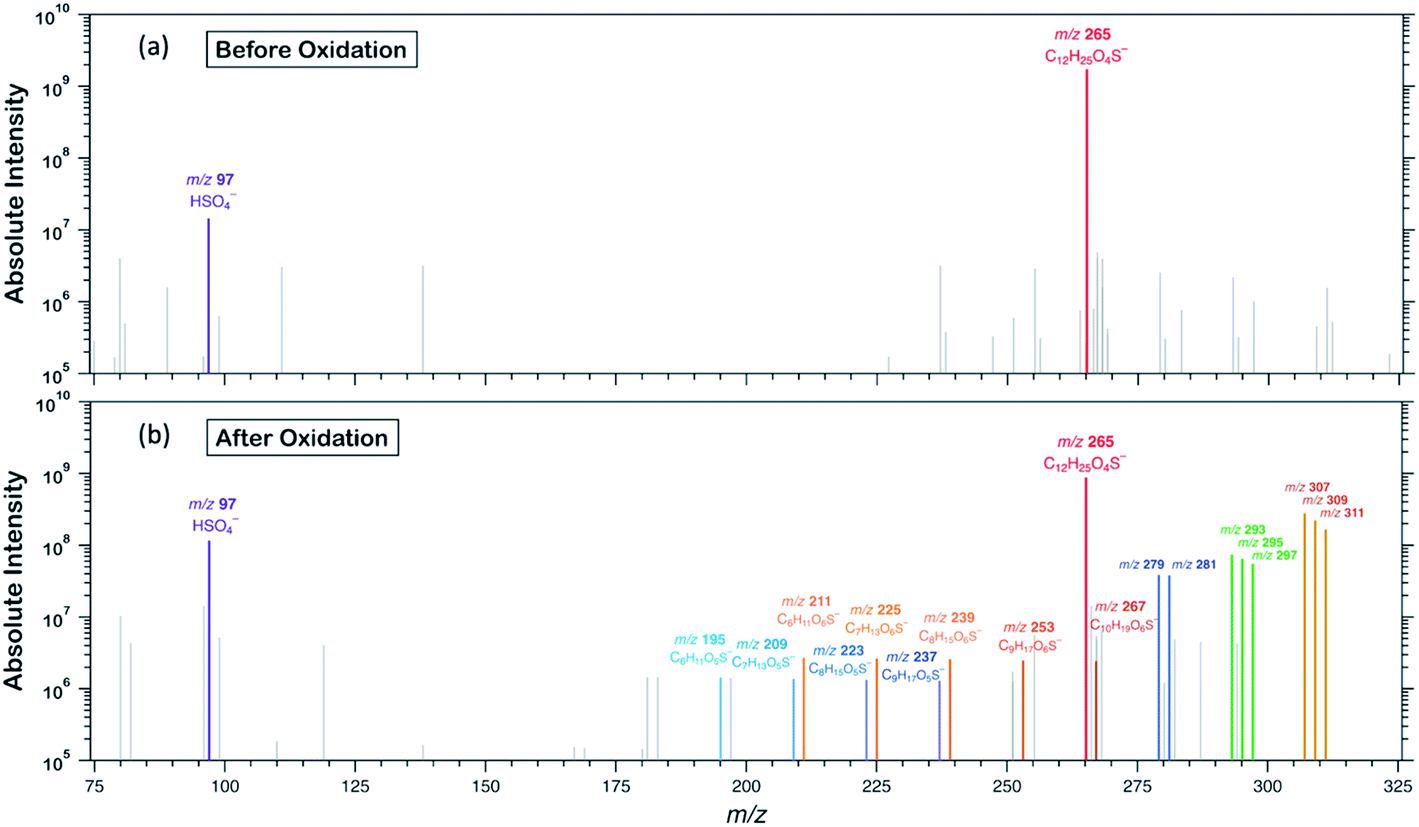 | ||
| Fig. 1 Aerosol mass spectra (y-axis on the logarithmic scale) acquired using an ultrahigh-resolution mass spectrometer (a) before and (b) after heterogeneous OH oxidation of SDS aerosols. The m/z values of 279 and 281 (lst generation products, in blue) represent C12H23O5S− and C12H25O5S−; m/z of 293, 295, and 297 (2nd generation products, in green) represent C12H21O6S−, C12H23O6S−, and C12H25O6S−; m/z of 307, 309, and 311 (3rd generation products, in yellowish brown) represent C12H19O7S−, C12H21O7S−, and C12H23O7S−, respectively. The peaks in light grey are background ions. | ||
Fig. 2a displays the evolution profiles of oxygenated C12-OS from different generations of oxygenation together with the signal decay of dodecyl OS. The intensities of the products from the 1st generation peaked at a lower OH exposure of about 2.5 × 1011 molecules per cm3 s, accompanied by a gradual increase in those from the 2nd generation which peaked afterward at an OH exposure of about 5.5 × 1011 molecules per cm3 s. The signal drop of the 2nd generation products was followed by the signal increase of the 3rd generation products which culminated at about 13.0 × 1011 molecules per cm3 s. This pattern of intensity evolution coupling between consecutive generations is characterized as multi-generation sequential oxygenation, in which the abundancy of carbon atoms within saturated organic molecules allows for continuous OH-initiated hydrogen abstraction from the available C–H to generate highly oxygenated species.70 We did not detect products beyond the 3rd generation, potentially due to the limited range of OH exposure. Nonetheless, more extensive sequential oxygenation of dodecyl OS against higher OH exposure was revealed in a study by Huang et al.,67 who utilized field-induced droplet ionization mass spectrometry to detect chemical species at the interface of a hanging droplet via a surface-selective sampling mechanism. They used a maximum OH exposure of 5.15 × 1014 molecules per cm3 s to oxidize SDS aerosols, which was about 300 times higher than what we used in our study. Their detection of oxygenated C12-OS of 8 generations67 implies that functionalization likely dominates over fragmentation of dodecyl OS for a wide range of OH exposure values.
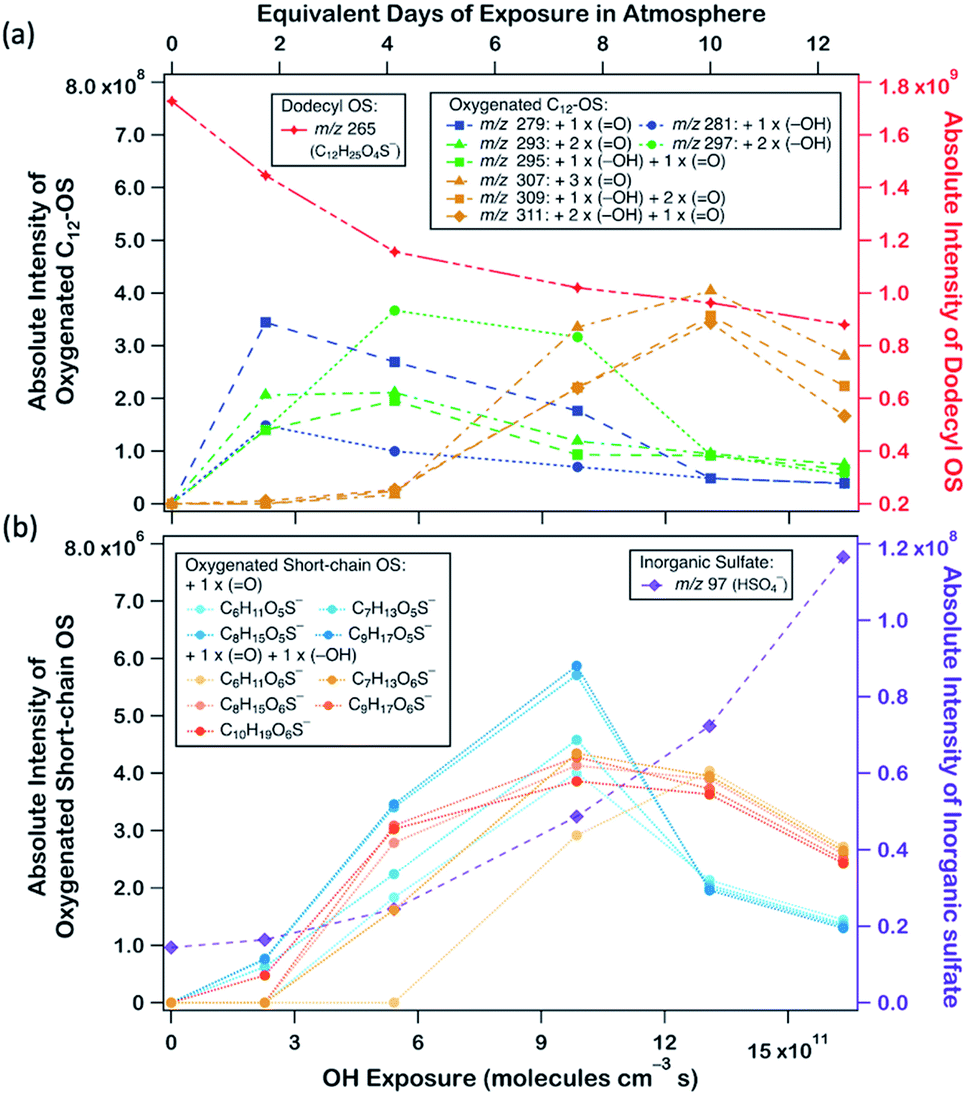 | ||
| Fig. 2 (a) Intensity evolution of oxygenated C12-OS derived from different generations of oxygenation of dodecyl OS formed upon heterogeneous OH oxidation of SDS aerosols (left axis) and dodecyl OS (right axis); (b) intensity evolution of oxygenated short-chain OSs (C6 to C10-OS) (left axis) and bisulfate ions (right axis) formed upon heterogeneous OH oxidation of SDS aerosols. | ||
Formation of oxygenated short-chain OSs and inorganic sulfates was also detected upon a closer examination of the mass spectra, but their intensities were relatively low (∼10−2 times those of oxygenated C12-OS), as evinced in Fig. 1b, 2b and Table S1.† We expect that some short-chain OS products would be present in the aerosol phase, because we experimentally detected the two smallest alkyl OSs (C1- and C2-OS) as aerosols in our previous study.55 In the present study, short-chain OSs with carbon numbers ranging from 6 to 10 (C6- to C10-OS) have been detected and divided into two groups: (1) C6H11O5S−, C7H13O5S−, C8H15O5S−, and C9H17O5S−, all of which are characterized by the addition of one oxygen atom; (2) C6H11O6S−, C7H13O6S−, C8H15O6S−, C9H17O6S−, and C10H19O6S−, which are characterized by the addition of two oxygen atoms. Compared to the products of the same number of oxygen atoms, the peaks of short-chain OSs (Fig. 2b) had a lagged appearance with respect to those of C12-OS products (Fig. 2a). For instance, both short-chain OSs with the addition of 1 and 2 oxygen atoms peaked after the fall of C12-OS from the 1st and 2nd generation. It is plausible that the majority of short-chain OSs were not 1st generation products but were likely fragmented from OSs produced in higher generations, although formation through direct fragmentation of dodecyl OS was also achievable. These results might supplement the finding that functionalization processes in general dominate over fragmentation processes upon oxidation, such that fragmentation to produce short-chain OSs is not favourable until OH exposure further increased.
Other fragmentation products (non-OS organics) are likely formed upon oxidation and partition to the gas phase based on their volatilities. However, measurement of gas-phase species is not the scope of this work, because volatile products will be rapidly further oxidized inside the OFR before detection.71 Detection of volatile species can only provide limited information on the formation and volatilization of fragmentation products formed upon oxidation. Future measurements of gas-phase species formed upon oxidation are warranted, for providing greater insights into the heterogeneous reaction mechanisms, in particular the formation and volatilization of fragmentation products. Based on the observed aerosol-phase products, the proposed formation mechanisms of oxygenated short-chain OSs will be discussed in Section 3.3.2.
3.2 Oxidation kinetics
To obtain an effective second-order heterogeneous OH rate constant (k), the decay of dodecyl OS is fitted into an exponential function72 (see Fig. 3):
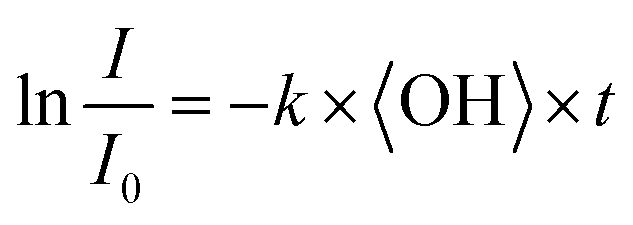 | (1) |
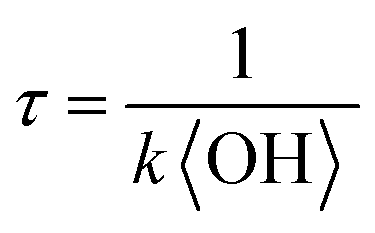 . Assuming the 24 h averaged gas-phase OH concentration (〈OH〉) to be 1.5 × 106 molecules per cm3,69 the chemical lifetime of dodecyl OS against heterogeneous OH oxidation was estimated to be 18.9 ± 0.4 days. Given that the diameters of aerosols were 279.5 ± 0.5 nm (Table 1), the decay via heterogeneous OH oxidation is moderately slower than the removal processes of aerosols with similar sizes (∼200 nm) via wet or dry deposition methods, which have typical timescales of 10–14 days.75,76 In addition, the removal process of dodecyl OS via heterogeneous OH oxidation can possibly contribute to one of the potential sources of some oxygenated alkyl-derived C6- to C10-OS and C12-OS, which have been detected in atmospheric aerosols (Table S1, ESI†).
. Assuming the 24 h averaged gas-phase OH concentration (〈OH〉) to be 1.5 × 106 molecules per cm3,69 the chemical lifetime of dodecyl OS against heterogeneous OH oxidation was estimated to be 18.9 ± 0.4 days. Given that the diameters of aerosols were 279.5 ± 0.5 nm (Table 1), the decay via heterogeneous OH oxidation is moderately slower than the removal processes of aerosols with similar sizes (∼200 nm) via wet or dry deposition methods, which have typical timescales of 10–14 days.75,76 In addition, the removal process of dodecyl OS via heterogeneous OH oxidation can possibly contribute to one of the potential sources of some oxygenated alkyl-derived C6- to C10-OS and C12-OS, which have been detected in atmospheric aerosols (Table S1, ESI†).
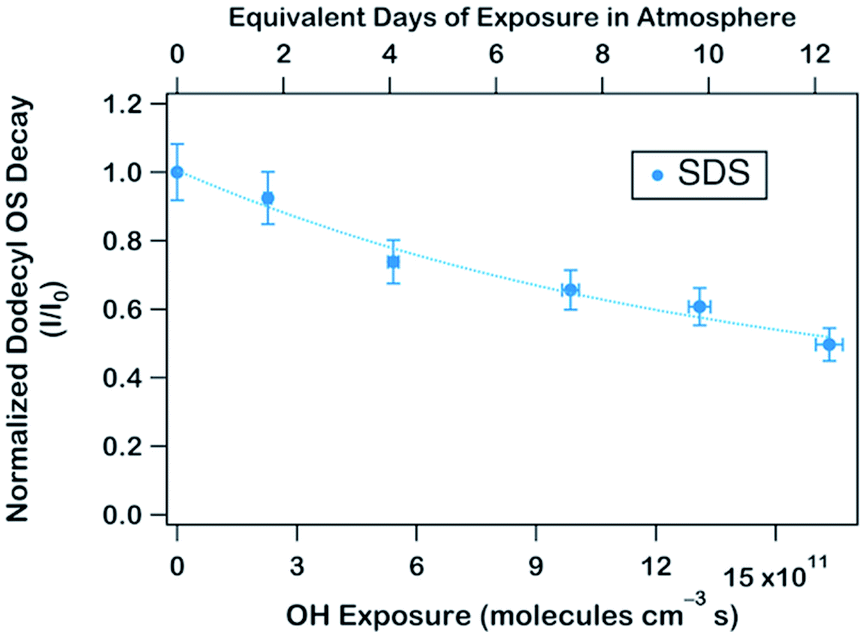 | ||
| Fig. 3 Normalized decay of dodecyl OS upon oxidation as a function of OH exposure. Dodecyl OS was quantified to decay at (4.09 ± 0.09) × 10−13 cm3 per molecule per s. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. During heterogeneous OH oxidation of SDS/AS aerosols under similar atmospheric conditions (∼80% RH and 300 K), dodecyl OS decayed at (4.4 ± 0.4) × 10−11 cm3 per molecule per s, markedly faster than our experimental rate constant by about 100 times. To verify the heterogeneous reactivity enhancement of dodecyl OS in the presence of AS, we performed heterogeneous OH oxidation of SDS/AS aerosols with different mass ratios. Fig. S1c–f† show the TIC before and after heterogeneous OH oxidation of SDS/AS aerosols. The mass spectra of SDS/AS (with a mass ratio of 1.3:1) aerosols before and after oxidation are shown in Fig. S2,† in which the identities of OS products are similar to those of SDS aerosols after oxidation (Fig. 1). Nonetheless, we observed that the intensities of the OS products are enhanced in SDS/AS aerosols. Fig. S3† displays the normalized decay of dodecyl OS with different mass ratios. We found that when the SDS/AS mass ratio decreased from 6:1 to 1.3:1, the rate constant (k) increased from (8.77 ± 0.50) to (11.97 ± 0.96) × 10−13 cm3 per molecule per s, both of which are higher than the rate constant of (4.09 ± 0.09) × 10−13 cm3 per molecule per s when AS was absent. The data suggest that the presence of AS accelerates the reactions between dodecyl OS and OH.
:1. During heterogeneous OH oxidation of SDS/AS aerosols under similar atmospheric conditions (∼80% RH and 300 K), dodecyl OS decayed at (4.4 ± 0.4) × 10−11 cm3 per molecule per s, markedly faster than our experimental rate constant by about 100 times. To verify the heterogeneous reactivity enhancement of dodecyl OS in the presence of AS, we performed heterogeneous OH oxidation of SDS/AS aerosols with different mass ratios. Fig. S1c–f† show the TIC before and after heterogeneous OH oxidation of SDS/AS aerosols. The mass spectra of SDS/AS (with a mass ratio of 1.3:1) aerosols before and after oxidation are shown in Fig. S2,† in which the identities of OS products are similar to those of SDS aerosols after oxidation (Fig. 1). Nonetheless, we observed that the intensities of the OS products are enhanced in SDS/AS aerosols. Fig. S3† displays the normalized decay of dodecyl OS with different mass ratios. We found that when the SDS/AS mass ratio decreased from 6:1 to 1.3:1, the rate constant (k) increased from (8.77 ± 0.50) to (11.97 ± 0.96) × 10−13 cm3 per molecule per s, both of which are higher than the rate constant of (4.09 ± 0.09) × 10−13 cm3 per molecule per s when AS was absent. The data suggest that the presence of AS accelerates the reactions between dodecyl OS and OH.
We postulate that AS may exhibit an ion-specific effect on modifying interactions between dodecyl OS and OH, which subsequently alters their reactivities. To gain fundamental insight, we performed non-reactive all-atom molecular dynamics (MD) simulations of interactions between species in aerosols of different mass fractions. A detailed description of MD simulations is provided in Section S3 (ESI†). As implied by previous MD studies,77 the occurrence of OH reaction with dodecyl OS requires OH to be first absorbed into the aqueous aerosol, followed by diffusion to collide with dodecyl OS at the right orientation with sufficient energy. As the reaction is triggered by a successful collision between dodecyl OS and OH, the average distance between them can serve as an indicative parameter of reaction rate,77,78 that is, when the distance decreases, the collision frequency will increase, and vice versa. To delineate the role of AS in modifying reactivities, we first observed how its presence can affect the spatial distributions of other ions and species, illustrated by the density profiles as a function of the distance away from the water slab center in the direction normal to the interface as shown in Fig. 4.
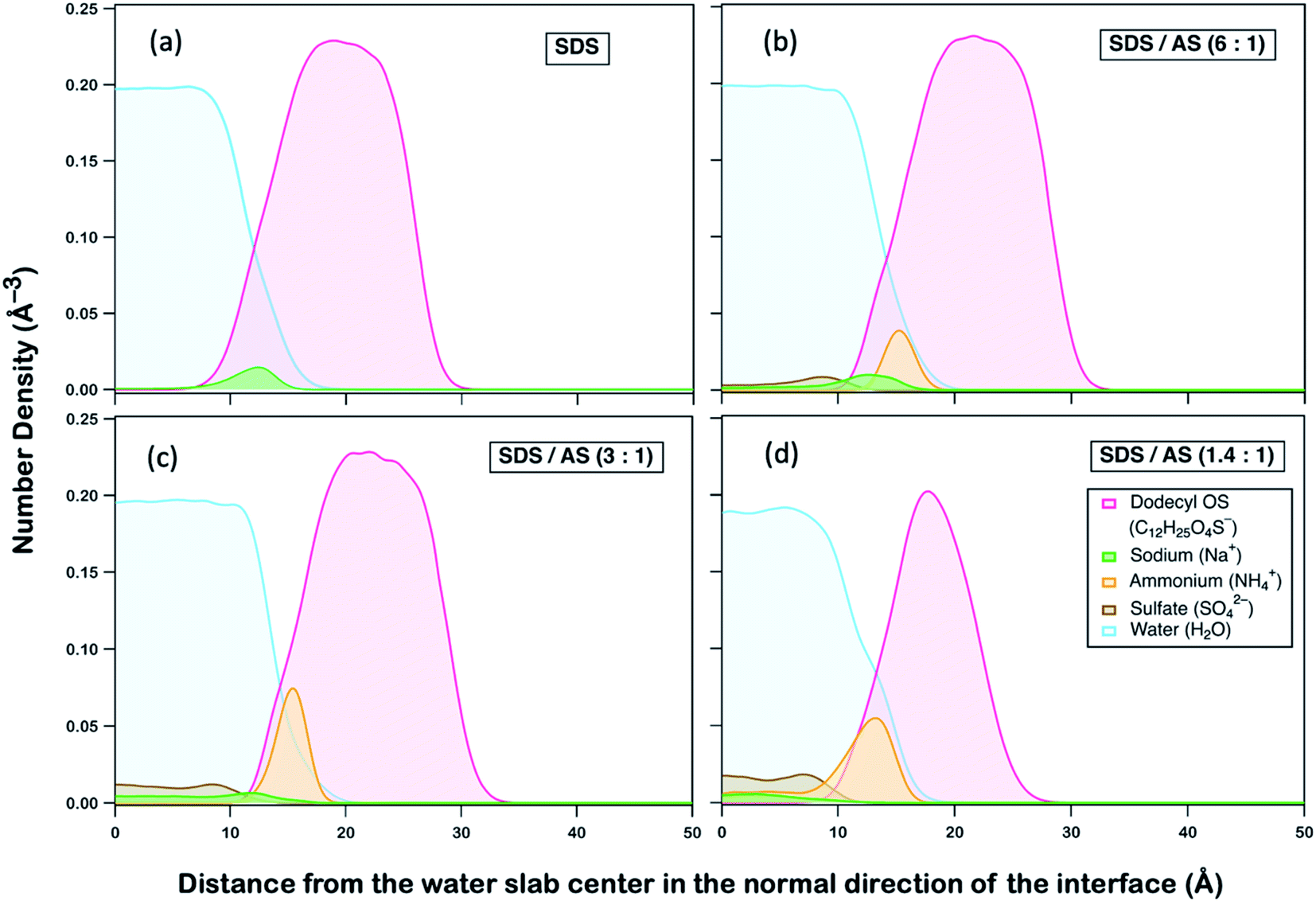 | ||
| Fig. 4 Number density profiles of the species NH4+, C12H25O4S−, SO42−, Na+, and H2O as a function of the distance from the water slab center in the direction normal to the interface for four SDS/AS mass ratios. | ||
We compared four aqueous slab systems, including SDS and SDS/AS with mass ratios of 6:1, 3:1, and 1.4:1. In each scenario, equilibrium distributions of ammonium (NH4+), dodecyl OS (C12H25O4S−), sulfate (SO42−), sodium (Na+), and water (H2O) were analyzed, as illustrated in Fig. 4. Generally, as represented by our simulation (Fig. 4a), dodecyl OS tends to stay at the air–water interface, which is expected as it is a surfactant in SDS aerosol. Na+ being the counterion prefers to stay near dodecyl OS while being dissolved in water away from the air–water interface. Upon addition of AS, it is remarkable that NH4+ tends to replace Na+ from the air–water interface, while SO42− is dispersed within the water body (Fig. 4b–d). Competition between NH4+ and Na+ for the air–water interface is observed, and there is a stronger tendency of NH4+ to go to the interface. In fact, NH4+ preference for the interface was reported in a study by Gopalakrishnan et al.,79 in which they examined the air–water interfacial behaviour of important atmospheric aerosol species, including NH4+, Na+, and SO42− ions. Combining vibrational sum frequency generation spectroscopy with polarizable MD simulations, they also reported that NH4+ has a stronger propensity for the air–water interface than Na+.79,80 When the AS mass fraction increases, there is substitution of Na+ by NH4+ near the interface, accompanied by the displacement towards the water interior, as depicted in Fig. S4.†
The molecular interactions between OH and ions have been rarely studied. Since these interactions are determined using the electronic structures which in turn can be determined using quantum mechanics, we utilized first-principles quantum mechanical (ab initio) calculations to better understand the interactions between OH and NH4+/SO42−. We observed that there is a reasonably strong attraction between OH and NH4+, while there is a reasonably strong repulsion between OH and SO42−. Our classical MD simulations were parametrized to reproduce these ion–OH interactions as much as possible, and the results are shown in Section S3 (ESI†).
Because dodecyl OS is a surfactant and NH4+ has a propensity for the air–water interface, the attraction between dissolved OH and NH4+ brings the radical species closer to dodecyl OS from the aerosol center. To demonstrate this, we calculated the average distance between OH and the center of mass of dodecyl OS as a function of SDS/AS ratio as shown in Fig. 5 (alternatively, we also calculated the average smallest possible distances between OH and various carbon atoms of dodecyl OS, as shown in Fig. S5†). As more AS is added, the average distance tends to decrease. It implies that the presence of NH4+ facilitates the approach of dissolved OH to the air–water interface where dodecyl OS is located, which subsequently promotes the collision frequency, and thus the oxidation reaction rate. This enhancement of the reaction rate may be specific to the propensity of the ion (NH4+ in this case) for the surface and its attraction to OH. In general, we expect that a similar enhancement can be observed if there is another ion sharing a similar surface propensity with OS and exhibiting strong attraction to OH.
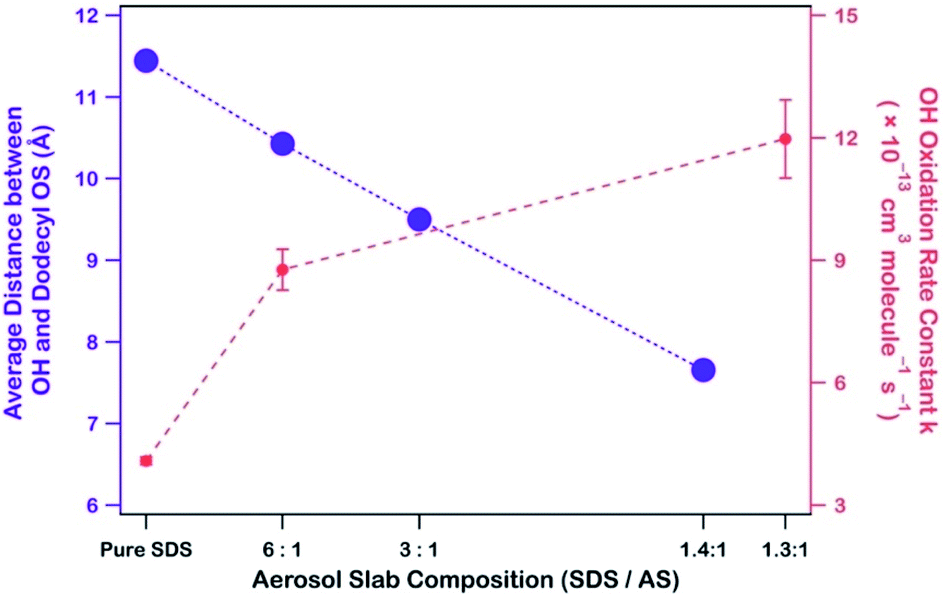 | ||
| Fig. 5 Average distance between OH and the center of mass of dodecyl OS as a function of SDS/AS mass ratios (left axis) and the corresponding rate constant k (right axis). When more AS is added, OH comes closer to dodecyl OS on average, in line with the increase in k. | ||
It is noteworthy that the rate constant (i.e. k) depends on the aerosol size because of the variation in surface-to-volume ratio for oxidation.81,82 Since polydisperse aerosols were generated in the OFR, the effect of aerosol size and the spread of aerosol size on reaction kinetics cannot be determined. We suggest future investigations to measure reaction kinetics for both monodisperse (size-selected) aerosols and polydisperse aerosols in the same reaction system upon heterogeneous oxidation. The effect of aerosol size on the reaction kinetics can be examined using different monodisperse aerosol populations. In addition, the kinetics assembled from different monodisperse aerosol populations can be compared with those obtained from polydisperse aerosol populations using the mean surface-weighted diameter in order to assess how the spread in size distribution of polydisperse aerosols affects the determination of reaction kinetics.
3.3 Reaction mechanisms
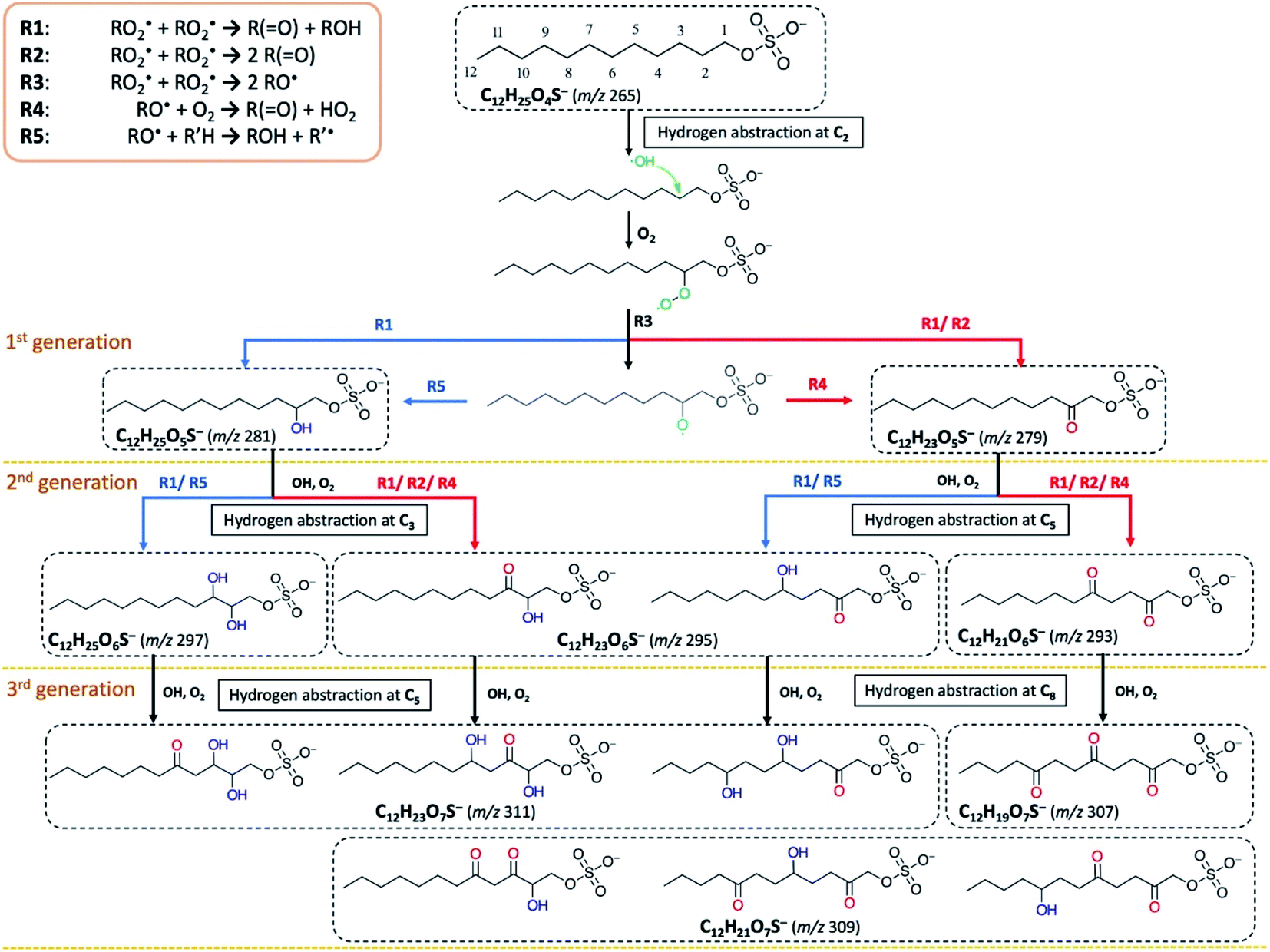 | ||
| Scheme 1 Proposed formation mechanisms of oxygenated C12-OS detected upon heterogeneous OH oxidation of dodecyl OS via R1–R5. Certain hydrogen abstraction sites are selected for demonstration. Cn refers to the n-th carbon atom bonded next to the sulfate (–OSO3−) group in an OS molecule. The formation pathways of products in the 3rd generation are simplified to illustrate OSs of all chemical formulae detected. Structural isomers are not included for simplicity. | ||
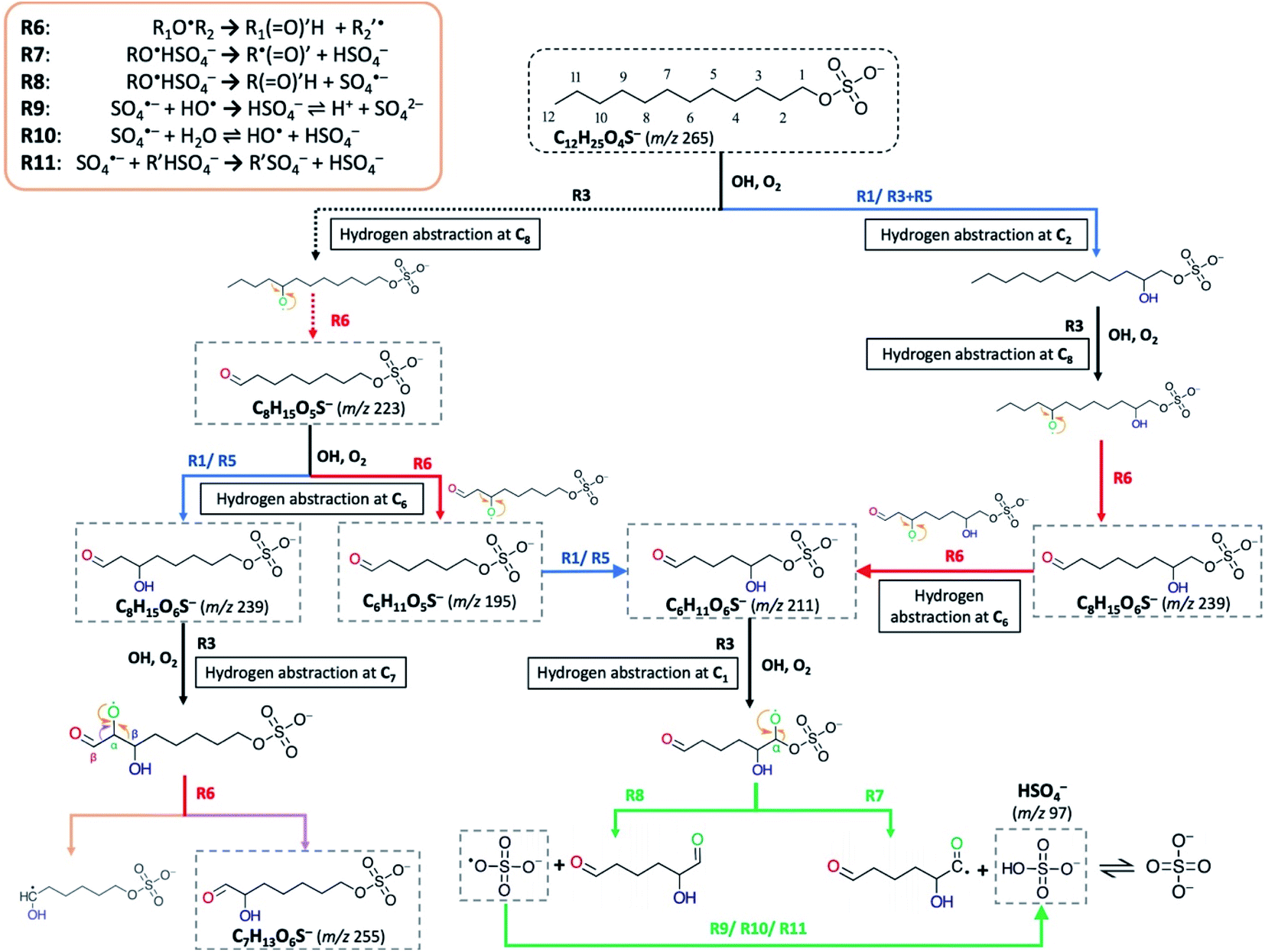 | ||
| Scheme 2 Proposed formation mechanisms of oxygenated short-chain OS and inorganic sulfate ions detected upon heterogeneous OH oxidation of dodecyl OS. C6H11O5S−, C6H11O6S−, C7H13O6S−, C8H15O5S−, and C8H15O6S− are used as examples for the demonstration of fragmentation mechanisms via R6–R8. Cn refers to the n-th carbon atom bonded next to the –OSO3− group in an OS molecule. | ||
Functionalization of dodecyl OS refers to the process of forming oxygenated C12-OS (Scheme 1). Initiated by the hydrogen abstraction from one of the carbon sites, an alkyl radical (R˙) is formed, which quickly reacts with O2 to form a peroxy radical (RO2˙). As well-known aerosol-phase reactions,51 the self-reaction of two RO2˙ can produce products via carbonylation by the Russell mechanism (R1)83 or Bennett–Summers reactions (R2),84 characterized by the addition of one ketone group to form C12H23SO5− (m/z 279). The Russell mechanism (R1) is also responsible for the formation of hydroxyl products.83 Alternatively, the bimolecular reaction of RO2˙ can produce two alkoxy radicals (RO˙) (R3), which abstract another O2 (R4)83 to form C12H23SO5− (m/z 279), or undergo intermolecular hydrogen abstraction to form products from hydroxylation (R5), such that C12H25SO5− (m/z 281) is also a 1st generation product. The products from higher generations of oxygenation can be formed by sequential hydrogen abstraction by OH. The observations of products from sequential functionalization processes are consistent with those in the literature.66,67
Following the formation of RO˙ in (R3), it may undergo either hydrogen abstraction (R5) or decomposition at the β-carbon (R6) (Scheme 2), in which the latter process is known as β-scission that breaks the RO˙ into an alkyl radical (R′˙) and a formyl alkyl molecule (R(O)′H) (R6).85–89 The phenomenon of functionalization dominating over fragmentation at early oxidation stages for long-chain organics (such as squalene (C30H62) and bis(2-ethylhexyl) sebacate (CH2)8(COOC8H17)2) in aerosols has previously been observed in the literature.72,90,91 These studies observed that as more oxygen atoms are added to the organic molecules upon OH exposure, the molecules undergo more fragmentation instead of functionalization. This is consistent with a stochastic simulation study by Wiegel et al.,89 which provided mechanistic insights into the factors (e.g., O/C, H/C, and degree of carbon) governing the fragmentation processes of squalene. It was suggested that when none or few oxygen functional groups have been attached to the parent molecule, the fragmentation of alkyl RO˙ (via R6) will encounter a higher activation barrier than functionalization, resulting in a slow reaction contributing to only ∼2% of the total OH-induced hydrogen abstraction rate.89 This instantly accounts for the insignificance of fragmentation at early stages of oxidation. Nonetheless, as oxidation proceeds, functional groups (i.e., carbonyl and hydroxyl) are continuously added (as reflected by an increase in O/C), stepwise decreasing the number of abstractable hydrogen atoms; in the process, the probability of RO˙ forming adjacent to these functional groups gradually increases89 (i.e. the chance of β-carbon bonding to functional group(s) increases). Meanwhile, carbonyl and hydroxyl group substituents at β-carbon can substantially lower the activation energy for decomposition, the effects of which are statistically more intense than the barrier reduction caused by an alkyl substituent.87,88 This leads to the essential involvement of fragmentation as an emerging reaction pathway at later stages of oxygenation. The products derived from R6 can promote further radical chemistry and decompose into volatile molecules.89
We acknowledge that multiple formation pathways are responsible for the detected short-chain OSs. Scheme 2 illustrates a few of the possible pathways, using C6-OS, C7-OS, and C8-OS as examples. For the sake of simplicity, structural isomers were not completely included in the proposed pathways. Briefly, we postulate that fragmentation can occur for both dodecyl OS and oxygenated C12-OS. Based on the above discussion and comparison of evolution profiles (Fig. 2b), we expect that short-chain OSs (C6- to C10-OS) are more prone to be fragmented from the oxygenated C12-OS than from dodecyl OS. As shown in Scheme 2, C8H15O6S− can be generated in two steps, by direct fragmentation of dodecyl OS, followed by the addition of a hydroxyl group, or by reversing the steps. C8H15O5S− generated by the forward process could further fragment into C6H11O5S−, prior to hydroxylation to result in C6H11O6S−; otherwise, C6H11O6S− could be produced by fragmentation of C8H15O6S−. For demonstration of functional group driven fragmentation, after RO˙ formation at C7 in C8H15O6S−, decomposition can be activated at the β-carbon by its bonded carbonyl group and hence, C7H13O6S− is formed.
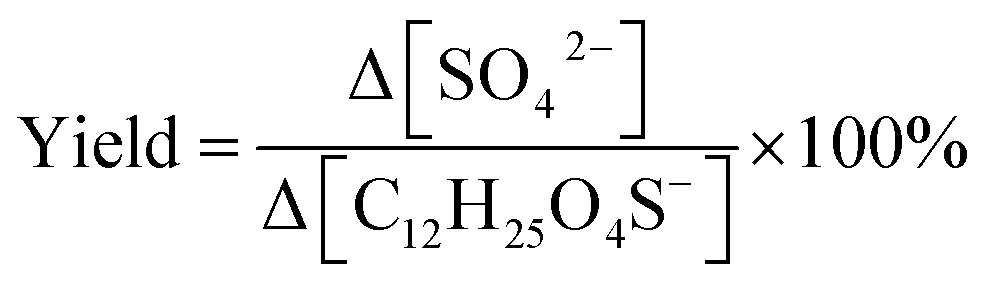 | (2) |
R7–R11 provide the possible pathways for inorganic sulfate production from fragmentation of OSs. Scheme 2 further depicts a formation pathway of inorganic sulfate from a short-chain OS (C6H11O6S−). Similar to the β-scission mechanism to generate short-chain OSs (Section 3.3.2), the proposed mechanisms for OSs to dissociate at C–OSO3− requires RO˙ to form at C1 (R7–R8). The presence of an unpaired electron at the oxygen atom bonded to C1 would reduce the C–O bond strength leading to ready dissociation. This is known as the inductive cleavage of bonds, where preferential bond dissociation would occur between the electronegative group and its bonded carbon, and the negative charge would be preferably transferred to the fragmented electronegative group (R7) to restore thermodynamic stability.92,93 On the other hand, fragmentation can also subsequently generate the sulfate radical anions (SO4˙−) (R8), which can further participate in radical chemistry or react to generate HSO4− (R9–R11).55,58 It is acknowledged that R7–R8 do not explicitly represent all reactions between C1 and OH, but only account for the major pathways of inorganic sulfate production.
As indicated by Fig. 2b and IC results, inorganic sulfate ions tend to originate from products from the 2nd or higher generations. A possible reason to account for this phenomenon is that at early oxidation stages, hydrogen abstraction at C1 may not be favorable given that there is a larger number of hydrogen atoms available for OH abstraction. Also, the electron-rich sulfate group possibly exhibits an uncharacterized effect on the neighbouring carbon sites; for instance, it may have a mesomeric withdrawing effect on the bonded carbon atoms, similar to the effect of a nitrate group in gas-phase organonitrates, which was found to deactivate the neighbouring sites against hydrogen abstraction by OH.94 However, as oxidation proceeds, the probability of hydrogen abstraction occurring at C1 increases when more functional groups are added to the carbon backbone, which can also alter the site selectivity for OH-induced hydrogen abstraction.95,96 In short, we expect that the fragmentation of the sulfate group occurs at a lower frequency in the initial stages of oxidation, which is contrarily more favored as oxidation proceeds, consistent with the formation pathways of oxygenated short-chain OSs.
Although SO42− and/or HSO4− formation was not significantly detected for heterogeneous OH oxidation of SDS, an appreciable inorganic sulfate yield (∼60%) was observed for short-chain OSs (C1-, C2-, and C5-OS).54–56,58 This implies that the phenomenon of fragmentation domination in methyl and ethyl OSs might be special cases of alkyl OSs, in which the probability of hydrogen abstraction at C1 is enhanced by the smaller number of carbon atoms. Similarly, 3-methyltetrol OS undergoing fragmentation with negligible functionalization can be explained by the presence of vicinal hydroxyl groups, which substantially lower the activation energy required for the decomposition of RO˙.58 The presence of vicinal hydroxyl groups, however, is also structurally unique to this C5-OS among other long-chain OSs.
4. Conclusion and atmospheric implications
The findings of this study imply that dodecyl OS, an atmospherically relevant long-chain alkyl OS, can undergo heterogeneous OH oxidation at a relatively slow rate with an atmospheric lifetime of ∼19 days. Nonetheless, our calculation shows that about 52% of dodecyl OS in aerosols can chemically transform within 14 days, prior to removal via wet and dry deposition. In addition, when AS is present, the atmospheric lifetime of dodecyl OS can reduce to 6.45 ± 0.52 days (for SDS/AS (1.3:1) aerosols). Yet, fragmentation to form inorganic sulfate was found to be rather insignificant in the present study. Several atmospheric implications are drawn from the results:
(1) Dodecyl OS reacts predominantly by attaching multiple carbonyl and hydroxyl groups. Although the formation of short-chain OSs is a minor process, fragmentation from their long-chain precursors can represent one of their potential formation mechanisms. These oxygenated short-chain OSs and C12-OS would exhibit different propensities for the air–water interface compared to dodecyl OS, for instance, by formation of hydrogen bonds with water molecules. Meanwhile, oxygenation of long-chain OSs with minimal fragmentation, responsible for the O/C ratio enhancement in SOAs, can potentially alter the physicochemical properties of aerosols such as viscosity and hygroscopicity. Our results point out the need to revisit our conventional assumption that OSs, particularly surfactants such as dodecyl OS, can reside stably at the interface to prolong the lifetimes of inner compounds in aerosols. We argue that surface activities will decrease as surfactants become oxidized, due to the modification in their polarities and hydrophilicity. It is therefore worthwhile to investigate the changes in protective effects of these oxygenated products and physicochemical properties of aerosols, which may consequently alter cloud nucleation, growth, and albedo effects.97,98
(2) Since the formation of inorganic sulfate requires the occurrence of OH-induced hydrogen abstraction at C1 prior to the breakage of the C–OSO3− bond, it is plausible that the inorganic sulfate yielded from long-chain OSs will be lower in amount than that from short-chain OSs in general, as expected due to the decreased probability of OH reactions with C1. The number of carbon atoms in alkyl OSs might therefore represent a factor limiting the inorganic sulfate yield upon heterogeneous OH oxidation, which is also in agreement with the detected low sulfate yield from α-pinene OS.57 Our results might imply that our fundamental understanding of the chemical evolution principles of structurally different OSs is incomplete; therefore, further research in the related area is required.
(3) OH oxidation kinetics of dodecyl OS are accelerated by the presence of NH4+. We conjecture that this is caused by the co-occurrence of their propensity for the air–water interface and the strong NH4+–OH interaction, which brings OH closer to dodecyl OS. Na+ and NH4+, both being atmospherically abundant cations, respectively, originating from marine and anthropogenic sources,99–101 exhibit different air–water interface propensities and interactions with OH, eventually resulting in distinct effects on reactivity alteration of dodecyl OS. Despite the phenomenon being unique to the investigated combination of species, future studies on atmospheric fates of organic compounds, especially surface-active compounds, may require careful consideration of the effects of their counterions and other unexplored factors, which altogether potentially affect the interpretation of experimental kinetics outcomes.
Overall, the study provides fundamental insight into the heterogeneous OH oxidation of long-chain alkyl OSs, complementary to the previous studies on short-chain OSs. The aqueous-phase structure–activity relationship (SAR) of OH with aliphatic organic molecules suggests that OH has no significant preference to any carbon sites in long-chain alkyl OSs when no carbonyl or hydroxyl group is bonded;95,96 however, the partial site reactivity influenced by the sulfate (–OSO3−) group has not been included in the SAR, neither in the aqueous phase nor in the gas phase. As many OSs are surface-active, development of a multiphase SAR is advantageous for quantitative prediction of the effect of –OSO3− on its neighbouring hydrogen-abstractable sites. Besides, heterogeneous reactivity was investigated at a single high RH in this work. Recent studies have demonstrated that aerosol composition and environmental conditions such as RH and temperature can determine aerosol viscosity, which subsequently alters heterogeneous reactivity.102–104 In general, high viscosity limits bulk diffusion within aerosols, resulting in slower heterogeneous reactions.105,106 Although both SDS and SDS/AS aerosols are likely not viscous at high RH, future explorations into the effects of environmental conditions and aerosol viscosity on heterogeneous OH reactivity are highly desired. These would greatly assist in the estimation of atmospheric lifetimes of aliphatic OSs and their potential transformational products under tropospheric conditions. To sum up, future investigation of the fates of structurally different OSs upon heterogeneous OH oxidation is necessary, through coupling laboratory studies with computational approaches to obtain a thorough mechanistic insight.
Data availability
Data are available upon request from the corresponding authors.Author contributions
Sze In Madeleine Ng, Ying-Lung Steve Tse, and Man Nin Chan designed the experiments. Sze In Madeleine Ng and Rongshuang Xu ran the experiments. Rongshuang Xu, Chun Kit K. Choi, Pui-Kin So, and Jian Zhen Yu helped with the chemical analysis. Kwan Hung Ng, Pui Wo Felix Yeung, and Ying-Lung Steve Tse performed the MD simulations. Yuanlong Huang helped provide scientific explanations of the chemistry involved. Sze In Madeleine Ng, Ying-Lung Steve Tse, and Man Nin Chan prepared the manuscript. All authors provided comments and suggestions for improving the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by the Hong Kong Research Grants Council (HKRGC): Project ID 2130626 (Ref 14300118) and Project ID 2130705 (Ref 14300920). The authors are thankful for the support from the Li Ka Shing Foundation for ultrahigh-resolution Orbitrap Eclipse Tribrid mass spectrometry in the Li Ka Shing Translational Omics Platform.References
- M. P. Tolocka and B. Turpin, Contribution of Organosulfur Compounds to Organic Aerosol Mass, Environ. Sci. Technol., 2012, 46, 7978–7983 CrossRef CAS PubMed.
- K. Shakya and R. Peltier, Non-sulfate sulfur in fine aerosols across the United States: insight for organosulfate prevalence, Atmos. Environ., 2014, 100, 159–166 CrossRef PubMed.
- K. M. Shakya and R. E. Peltier, Investigating Missing Sources of Sulfur at Fairbanks, Alaska, Environ. Sci. Technol., 2013, 47, 9332–9338 CrossRef CAS PubMed.
- K. Gao and T. Zhu, Analytical methods for organosulfate detection in aerosol particles: current status and future perspectives, Sci. Total Environ., 2021, 784, 147244 CrossRef CAS PubMed.
- W. Fan, T. Chen, Z. Zhu, H. Zhang, Y. Qiu and D. Yin, A review of secondary organic aerosols formation focusing on organosulfates and organic nitrates, J. Hazard. Mater., 2022, 430, 128406 CrossRef CAS PubMed.
- J. D. Surratt, Y. Gómez-González, A. W. H. Chan, R. Vermeylen, M. Shahgholi, T. E. Kleindienst, E. O. Edney, J. H. Offenberg, M. Lewandowski, M. Jaoui, W. Maenhaut, M. Claeys, R. C. Flagan and J. H. Seinfeld, Organosulfate Formation in Biogenic Secondary Organic Aerosol, Sci. Total Environ., 2008, 112, 8345–8378 CAS.
- A. I. Darer, N. C. Cole-Filipiak, A. E. O'Connor and M. J. Elrod, Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates, Environ. Sci. Technol., 2011, 45, 1895–1902 CrossRef CAS PubMed.
- J. Liggio and S.-M. Li, Organosulfate formation during the uptake of pinonaldehyde on acidic sulfate aerosols, Geophys. Res. Lett., 2006, 33, L13808 CrossRef.
- E. C. Minerath, M. T. Casale and M. J. Elrod, Kinetics Feasibility Study of Alcohol Sulfate Esterification Reactions in Tropospheric Aerosols, Environ. Sci. Technol., 2008, 42, 4410–4415 CrossRef CAS PubMed.
- B. Nozière, S. Ekström, T. Alsberg and S. Holmström, Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols, Geophys. Res. Lett., 2010, 37, L05806 CrossRef.
- M. J. Perri, Y. B. Lim, S. P. Seitzinger and B. J. Turpin, Organosulfates from glycolaldehyde in aqueous aerosols and clouds: Laboratory studies, Atmos. Environ., 2010, 44, 2658–2664 CrossRef CAS.
- M. Riva, S. Tomaz, T. Cui, Y.-H. Lin, E. Perraudin, A. Gold, E. A. Stone, E. Villenave and J. D. Surratt, Evidence for an Unrecognized Secondary Anthropogenic Source of Organosulfates and Sulfonates: Gas-Phase Oxidation of Polycyclic Aromatic Hydrocarbons in the Presence of Sulfate Aerosol, Environ. Sci. Technol., 2015, 49, 6654–6664 CrossRef CAS PubMed.
- H. Zhang, D. R. Worton, M. Lewandowski, J. Ortega, C. L. Rubitschun, J.-H. Park, K. Kristensen, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, M. Jaoui, J. H. Offenberg, T. E. Kleindienst, J. Gilman, W. C. Kuster, J. de Gouw, C. Park, G. W. Schade, A. A. Frossard, L. Russell, L. Kaser, W. Jud, A. Hansel, L. Cappellin, T. Karl, M. Glasius, A. Guenther, A. H. Goldstein, J. H. Seinfeld, A. Gold, R. M. Kamens and J. D. Surratt, Organosulfates as Tracers for Secondary Organic Aerosol (SOA) Formation from 2-Methyl-3-Buten-2-ol (MBO) in the Atmosphere, Environ. Sci. Technol., 2012, 46, 9437–9446 CrossRef CAS PubMed.
- L. Xu, Z. Yang, N. T. Tsona, X. Wang, C. George and L. Du, Anthropogenic–Biogenic Interactions at Night: Enhanced Formation of Secondary Aerosols and Particulate Nitrogen- and Sulfur-Containing Organics from β-Pinene Oxidation, Environ. Sci. Technol., 2021, 55, 7794–7807 CrossRef CAS PubMed.
- P. G. Kanellopoulos, S. P. Kotsaki, E. Chrysochou, K. Koukoulakis, N. Zacharopoulos, A. Philippopoulos and E. Bakeas, PM2.5-bound organosulfates in two Eastern Mediterranean cities: the dominance of isoprene organosulfates, Chemosphere, 2022, 297, 134103 CrossRef CAS PubMed.
- S. Tao, X. Lu, N. Levac, A. P. Bateman, T. B. Nguyen, D. L. Bones, S. A. Nizkorodov, J. Laskin, A. Laskin and X. Yang, Molecular Characterization of Organosulfates in Organic Aerosols from Shanghai and Los Angeles Urban Areas by Nanospray-Desorption Electrospray Ionization High-Resolution Mass Spectrometry, Environ. Sci. Technol., 2014, 48, 10993–11001 CrossRef CAS PubMed.
- K. Wang, Y. Zhang, R.-J. Huang, M. Wang, H. Ni, C. J. Kampf, Y. Cheng, M. Bilde, M. Glasius and T. Hoffmann, Molecular Characterization and Source Identification of Atmospheric Particulate Organosulfates Using Ultrahigh Resolution Mass Spectrometry, Environ. Sci. Technol., 2019, 53, 6192–6202 CrossRef CAS PubMed.
- X. K. Wang, S. Rossignol, Y. Ma, L. Yao, M. Y. Wang, J. M. Chen, C. George and L. Wang, Molecular characterization of atmospheric particulate organosulfates in three megacities at the middle and lower reaches of the Yangtze River, Atmos. Chem. Phys., 2016, 16, 2285–2298 CrossRef CAS.
- Y. Wang, Y. Zhao, Y. Wang, J. Z. Yu, J. Shao, P. Liu, W. Zhu, Z. Cheng, Z. Li, N. Yan and H. Xiao, Organosulfates in atmospheric aerosols in Shanghai, China: seasonal and interannual variability, origin, and formation mechanisms, Atmos. Chem. Phys., 2021, 21, 2959–2980 CrossRef CAS.
- H. Jiang, J. Li, J. Tang, M. Cui, S. Zhao, Y. Mo, C. Tian, X. Zhang, B. Jiang, Y. Liao, Y. Chen and G. Zhang, Molecular Characteristics of Organosulfur Compounds in Guangzhou, South China: Heterogeneous Secondary Reactions Drivers the Molecular Distribution, Atmos. Chem. Phys. Discuss., 2022, 2022, 1–21 Search PubMed.
- H. Jiang, Y. He, Y. Wang, S. Li, B. Jiang, L. Carena, X. Li, L. Yang, T. Luan, D. Vione and S. Gligorovski, Formation of organic sulfur compounds through SO2-initiated photochemistry of PAHs and dimethylsulfoxide at the air-water interface, Atmos. Chem. Phys., 2022, 22, 4237–4252 CrossRef CAS.
- H. Ren, J. A. Sedlak and M. J. Elrod, General Mechanism for Sulfate Radical Addition to Olefinic Volatile Organic Compounds in Secondary Organic Aerosol, Environ. Sci. Technol., 2021, 55, 1456–1465 CrossRef CAS PubMed.
- M. Brüggemann, R. Xu, A. Tilgner, K. C. Kwong, A. Mutzel, H. Y. Poon, T. Otto, T. Schaefer, L. Poulain, M. N. Chan and H. Herrmann, Organosulfates in Ambient Aerosol: State of Knowledge and Future Research Directions on Formation, Abundance, Fate, and Importance, Environ. Sci. Technol., 2020, 54, 3767–3782 CrossRef PubMed.
- R. L. Beardsley and M. Jang, Simulating the SOA formation of isoprene from partitioning and aerosol phase reactions in the presence of inorganics, Atmos. Chem. Phys., 2016, 16, 5993–6009 CrossRef CAS.
- S. H. Budisulistiorini, A. Nenes, A. G. Carlton, J. D. Surratt, V. F. McNeill and H. O. T. Pye, Simulating Aqueous-Phase Isoprene-Epoxydiol (IEPOX) Secondary Organic Aerosol Production During the 2013 Southern Oxidant and Aerosol Study (SOAS), Environ. Sci. Technol., 2017, 51, 5026–5034 CrossRef CAS PubMed.
- V. F. McNeill, J. L. Woo, D. D. Kim, A. N. Schwier, N. J. Wannell, A. J. Sumner and J. M. Barakat, Aqueous-Phase Secondary Organic Aerosol and Organosulfate Formation in Atmospheric Aerosols: A Modeling Study, Environ. Sci. Technol., 2012, 46, 8075–8081 CrossRef CAS PubMed.
- H. O. T. Pye, B. N. Murphy, L. Xu, N. L. Ng, A. G. Carlton, H. Guo, R. Weber, P. Vasilakos, K. W. Appel, S. H. Budisulistiorini, J. D. Surratt, A. Nenes, W. Hu, J. L. Jimenez, G. Isaacman-VanWertz, P. K. Misztal and A. H. Goldstein, On the implications of aerosol liquid water and phase separation for organic aerosol mass, Atmos. Chem. Phys., 2017, 17, 343–369 CrossRef CAS PubMed.
- H. O. T. Pye, R. W. Pinder, I. R. Piletic, Y. Xie, S. L. Capps, Y.-H. Lin, J. D. Surratt, Z. Zhang, A. Gold, D. J. Luecken, W. T. Hutzell, M. Jaoui, J. H. Offenberg, T. E. Kleindienst, M. Lewandowski and E. O. Edney, Epoxide Pathways Improve Model Predictions of Isoprene Markers and Reveal Key Role of Acidity in Aerosol Formation, Environ. Sci. Technol., 2013, 47, 11056–11064 CrossRef CAS PubMed.
- J. L. Woo and V. F. McNeill, simpleGAMMA v1.0 – a reduced model of secondary organic aerosol formation in the aqueous aerosol phase (aaSOA), Geosci. Model Dev., 2015, 8, 1821–1829 CrossRef CAS.
- A. D. Estillore, A. P. S. Hettiyadura, Z. Qin, E. Leckrone, B. Wombacher, T. Humphry, E. A. Stone and V. H. Grassian, Water Uptake and Hygroscopic Growth of Organosulfate Aerosol, Environ. Sci. Technol., 2016, 50, 4259–4268 CrossRef CAS PubMed.
- L. T. Fleming, N. N. Ali, S. L. Blair, M. Roveretto, C. George and S. A. Nizkorodov, Formation of Light-Absorbing Organosulfates during Evaporation of Secondary Organic Material Extracts in the Presence of Sulfuric Acid, ACS Earth Space Chem., 2019, 3, 947–957 CrossRef CAS.
- A. M. K. Hansen, J. Hong, T. Raatikainen, K. Kristensen, A. Ylisirniö, A. Virtanen, T. Petäjä, M. Glasius and N. L. Prisle, Hygroscopic properties and cloud condensation nuclei activation of limonene-derived organosulfates and their mixtures with ammonium sulfate, Atmos. Chem. Phys., 2015, 15, 14071–14089 CrossRef CAS.
- M. Riva, Y. Chen, Y. Zhang, Z. Lei, N. E. Olson, H. C. Boyer, S. Narayan, L. D. Yee, H. S. Green, T. Cui, Z. Zhang, K. Baumann, M. Fort, E. Edgerton, S. H. Budisulistiorini, C. A. Rose, I. O. Ribeiro, R. L. e Oliveira, E. O. dos Santos, C. M. D. Machado, S. Szopa, Y. Zhao, E. G. Alves, S. S. de Sá, W. Hu, E. M. Knipping, S. L. Shaw, S. Duvoisin Junior, R. A. F. de Souza, B. B. Palm, J.-L. Jimenez, M. Glasius, A. H. Goldstein, H. O. T. Pye, A. Gold, B. J. Turpin, W. Vizuete, S. T. Martin, J. A. Thornton, C. S. Dutcher, A. P. Ault and J. D. Surratt, Increasing Isoprene Epoxydiol-to-Inorganic Sulfate Aerosol Ratio Results in Extensive Conversion of Inorganic Sulfate to Organosulfur Forms: Implications for Aerosol Physicochemical Properties, Environ. Sci. Technol., 2019, 53, 8682–8694 CrossRef CAS PubMed.
- A. P. S. Hettiyadura, E. A. Stone, S. Kundu, Z. Baker, E. Geddes, K. Richards and T. Humphry, Determination of atmospheric organosulfates using HILIC chromatography with MS detection, Atmos. Meas. Tech., 2015, 8, 2347–2358 CrossRef CAS.
- K. E. Altieri, B. J. Turpin and S. P. Seitzinger, Oligomers, organosulfates, and nitrooxy organosulfates in rainwater identified by ultra-high resolution electrospray ionization FT-ICR mass spectrometry, Atmos. Chem. Phys., 2009, 9, 2533–2542 CrossRef CAS.
- Q.-F. He, X. Ding, X.-M. Wang, J.-Z. Yu, X.-X. Fu, T.-Y. Liu, Z. Zhang, J. Xue, D.-H. Chen, L.-J. Zhong and N. M. Donahue, Organosulfates from Pinene and Isoprene over the Pearl River Delta, South China: Seasonal Variation and Implication in Formation Mechanisms, Environ. Sci. Technol., 2014, 48, 9236–9245 CrossRef CAS PubMed.
- A. Kahnt, S. Behrouzi, R. Vermeylen, M. Safi, J. Vercauteren, E. Roekens, M. Claeys and W. Maenhaut, One-year study of nitro-organic compounds and their relation to wood burning in PM10 aerosol from a rural site in Belgium, Atmos. Environ., 2013, 81, 561–568 CrossRef CAS.
- M. Le Breton, Y. Wang, Å. M. Hallquist, R. K. Pathak, J. Zheng, Y. Yang, D. Shang, M. Glasius, T. J. Bannan, Q. Liu, C. K. Chan, C. J. Percival, W. Zhu, S. Lou, D. Topping, Y. Wang, J. Yu, K. Lu, S. Guo, M. Hu and M. Hallquist, Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS, Atmos. Chem. Phys., 2018, 18, 10355–10371 CrossRef CAS.
- P. Lin, J. Z. Yu, G. Engling and M. Kalberer, Organosulfates in Humic-like Substance Fraction Isolated from Aerosols at Seven Locations in East Asia: A Study by Ultra-High-Resolution Mass Spectrometry, Environ. Sci. Technol., 2012, 46, 13118–13127 CrossRef CAS PubMed.
- Y. Wang, Y. Ma, X. Li, B. Y. Kuang, C. Huang, R. Tong and J. Z. Yu, Monoterpene and Sesquiterpene α-Hydroxy Organosulfates: Synthesis, MS/MS Characteristics, and Ambient Presence, Environ. Sci. Technol., 2019, 53, 12278–12290 CrossRef CAS PubMed.
- S. Kundu, T. A. Quraishi, G. Yu, C. Suarez, F. N. Keutsch and E. A. Stone, Evidence and quantitation of aromatic organosulfates in ambient aerosols in Lahore, Pakistan, Atmos. Chem. Phys., 2013, 13, 4865–4875 CrossRef.
- Y. Ma, X. Xu, W. Song, F. Geng and L. Wang, Seasonal and diurnal variations of particulate organosulfates in urban Shanghai, China, Atmos. Environ., 2014, 85, 152–160 CrossRef CAS.
- S. Staudt, S. Kundu, H.-J. Lehmler, X. He, T. Cui, Y.-H. Lin, K. Kristensen, M. Glasius, X. Zhang, R. J. Weber, J. D. Surratt and E. A. Stone, Aromatic organosulfates in atmospheric aerosols: synthesis, characterization, and abundance, Atmos. Environ., 2014, 94, 366–373 CrossRef CAS PubMed.
- B. Y. Kuang, P. Lin, M. Hu and J. Z. Yu, Aerosol size distribution characteristics of organosulfates in the Pearl River Delta region, China, Atmos. Environ., 2016, 130, 23–35 CrossRef CAS.
- S. L. Blair, A. C. MacMillan, G. T. Drozd, A. H. Goldstein, R. K. Chu, L. Paša-Tolić, J. B. Shaw, N. Tolić, P. Lin, J. Laskin, A. Laskin and S. A. Nizkorodov, Molecular Characterization of Organosulfur Compounds in Biodiesel and Diesel Fuel Secondary Organic Aerosol, Environ. Sci. Technol., 2017, 51, 119–127 CrossRef CAS PubMed.
- L. Qi, Z. Zhang, X. Wang, F. Deng, J. Zhao and H. Liu, Molecular characterization of atmospheric particulate organosulfates in a port environment using ultrahigh resolution mass spectrometry: identification of traffic emissions, J. Hazard. Mater., 2021, 419, 126431 CrossRef CAS PubMed.
- X. H. H. Huang, Q. Bian, W. M. Ng, P. K. K. Louie and J. Z. Yu, Characterization of PM2.5 Major Components and Source Investigation in Suburban Hong Kong: A One Year Monitoring Study, Aerosol Air Qual. Res., 2014, 14, 237–250 CrossRef CAS.
- M. N. Chan, J. D. Surratt, M. Claeys, E. S. Edgerton, R. L. Tanner, S. L. Shaw, M. Zheng, E. M. Knipping, N. C. Eddingsaas, P. O. Wennberg and J. H. Seinfeld, Characterization and Quantification of Isoprene-Derived Epoxydiols in Ambient Aerosol in the Southeastern United States, Environ. Sci. Technol., 2010, 44, 4590–4596 CrossRef CAS PubMed.
- A. P. S. Hettiyadura, L. Xu, T. Jayarathne, K. Skog, H. Guo, R. J. Weber, A. Nenes, F. N. Keutsch, N. L. Ng and E. A. Stone, Source apportionment of organic carbon in Centreville, AL using organosulfates in organic tracer-based positive matrix factorization, Atmos. Environ., 2018, 186, 74–88 CrossRef CAS.
- M. Riva, T. Da Silva Barbosa, Y. H. Lin, E. A. Stone, A. Gold and J. D. Surratt, Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 2016, 16, 11001–11018 CrossRef CAS.
- I. J. George and J. P. D. Abbatt, Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- J. H. Kroll, C. Y. Lim, S. H. Kessler and K. R. Wilson, Heterogeneous Oxidation of Atmospheric Organic Aerosol: Kinetics of Changes to the Amount and Oxidation State of Particle-Phase Organic Carbon, Sci. Total Environ., 2015, 119, 10767–10783 CAS.
- Y. Rudich, N. M. Donahue and T. F. Mentel, Aging of Organic Aerosol: Bridging the Gap Between Laboratory and Field Studies, Annu. Rev. Phys. Chem., 2007, 58, 321–352 CrossRef CAS PubMed.
- Y. Chen, Y. Zhang, A. T. Lambe, R. Xu, Z. Lei, N. E. Olson, Z. Zhang, T. Szalkowski, T. Cui, W. Vizuete, A. Gold, B. J. Turpin, A. P. Ault, M. N. Chan and J. D. Surratt, Heterogeneous Hydroxyl Radical Oxidation of Isoprene-Epoxydiol-Derived Methyltetrol Sulfates: Plausible Formation Mechanisms of Previously Unexplained Organosulfates in Ambient Fine Aerosols, Environ. Sci. Technol. Lett., 2020, 7, 460–468 CrossRef CAS.
- K. C. Kwong, M. M. Chim, J. F. Davies, K. R. Wilson and M. N. Chan, Importance of sulfate radical anion formation and chemistry in heterogeneous OH oxidation of sodium methyl sulfate, the smallest organosulfate, Atmos. Chem. Phys., 2018, 18, 2809–2820 CrossRef CAS.
- R. Xu, Y. Ge, K. C. Kwong, H. Y. Poon, K. R. Wilson, J. Z. Yu and M. N. Chan, Inorganic Sulfur Species Formed upon Heterogeneous OH Oxidation of Organosulfates: A Case Study of Methyl Sulfate, ACS Earth Space Chem., 2020, 4, 2041–2049 CrossRef CAS.
- R. Xu, S. I. M. Ng, W. S. Chow, Y. K. Wong, Y. Wang, D. Lai, Z. Yao, P. K. So, J. Z. Yu and M. N. Chan, Chemical Transformation of α-Pinene derived Organosulfate via Heterogeneous OH Oxidation: Implications for Sources and Environmental Fates of Atmospheric Organosulfates, Atmos. Chem. Phys. Discuss., 2021, 2021, 1–24 Search PubMed.
- H. K. Lam, K. C. Kwong, H. Y. Poon, J. F. Davies, Z. Zhang, A. Gold, J. D. Surratt and M. N. Chan, Heterogeneous OH oxidation of isoprene-epoxydiol-derived organosulfates: kinetics, chemistry and formation of inorganic sulfate, Atmos. Chem. Phys., 2019, 19, 2433–2440 CrossRef CAS.
- A. P. S. Hettiyadura, T. Jayarathne, K. Baumann, A. H. Goldstein, J. A. de Gouw, A. Koss, F. N. Keutsch, K. Skog and E. A. Stone, Qualitative and quantitative analysis of atmospheric organosulfates in Centreville, Alabama, Atmos. Chem. Phys., 2017, 17, 1343–1359 CrossRef CAS.
- R. E. Cochran, O. Laskina, T. Jayarathne, A. Laskin, J. Laskin, P. Lin, C. Sultana, C. Lee, K. A. Moore, C. D. Cappa, T. H. Bertram, K. A. Prather, V. H. Grassian and E. A. Stone, Analysis of Organic Anionic Surfactants in Fine and Coarse Fractions of Freshly Emitted Sea Spray Aerosol, Environ. Sci. Technol., 2016, 50, 2477–2486 CrossRef CAS PubMed.
- M. T. Latif and P. Brimblecombe, Surfactants in Atmospheric Aerosols, Environ. Sci. Technol., 2004, 38, 6501–6506 CrossRef CAS PubMed.
- H. Tervahattu, J. Juhanoja and K. Kupiainen, Identification of an organic coating on marine aerosol particles by TOF-SIMS, J. Geophys. Res., 2002, 107, D16 Search PubMed.
- V. F. McNeill, J. Patterson, G. M. Wolfe and J. A. Thornton, The effect of varying levels of surfactant on the reactive uptake of N2O5 to aqueous aerosol, Atmos. Chem. Phys., 2006, 6, 1635–1644 CrossRef CAS.
- N. L. Prisle, M. Dal Maso and H. Kokkola, A simple representation of surface active organic aerosol in cloud droplet formation, Atmos. Chem. Phys., 2011, 11, 4073–4083 CrossRef CAS.
- M. Radke, Sterols and Anionic Surfactants in Urban Aerosol: Emissions from Wastewater Treatment Plants in Relation to Background Concentrations, Environ. Sci. Technol., 2005, 39, 4391–4397 CrossRef CAS PubMed.
- J. A. Faust and J. P. D. Abbatt, Organic Surfactants Protect Dissolved Aerosol Components against Heterogeneous Oxidation, Sci. Total Environ., 2019, 123, 2114–2124 CAS.
- Y. Huang, K. M. Barraza, C. M. Kenseth, R. Zhao, C. Wang, J. L. Beauchamp and J. H. Seinfeld, Probing the OH Oxidation of Pinonic Acid at the Air–Water Interface Using Field-Induced Droplet Ionization Mass Spectrometry (FIDI-MS), Sci. Total Environ., 2018, 122, 6445–6456 CAS.
- E. Kang, M. J. Root, D. W. Toohey and W. H. Brune, Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 2007, 7, 5727–5744 CrossRef CAS.
- J. Mao, X. Ren, W. H. Brune, J. R. Olson, J. H. Crawford, A. Fried, L. G. Huey, R. C. Cohen, B. Heikes, H. B. Singh, D. R. Blake, G. W. Sachse, G. S. Diskin, S. R. Hall and R. E. Shetter, Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 2009, 9, 163–173 CrossRef CAS.
- Z. Wang, M. Popolan-Vaida Denisia, B. Chen, K. Moshammer, Y. Mohamed Samah, H. Wang, S. Sioud, A. Raji Misjudeen, K. Kohse-Höinghaus, N. Hansen, P. Dagaut, R. Leone Stephen and S. M. Sarathy, Unraveling the structure and chemical mechanisms of highly oxygenated intermediates in oxidation of organic compounds, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 13102–13107 CrossRef CAS PubMed.
- Z. Peng and J. L. Jimenez, Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 2020, 49, 2570–2616 RSC.
- J. D. Smith, J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop and K. R. Wilson, The heterogeneous reaction of hydroxyl radicals with sub-micron squalane particles: a model system for understanding the oxidative aging of ambient aerosols, Atmos. Chem. Phys., 2009, 9, 3209–3222 CrossRef CAS.
- J. F. Davies and K. R. Wilson, Nanoscale interfacial gradients formed by the reactive uptake of OH radicals onto viscous aerosol surfaces, Chem. Sci., 2015, 6, 7020–7027 RSC.
- E. Woods III, H. S. Kim, C. N. Wivagg, S. J. Dotson, K. E. Broekhuizen and E. F. Frohardt, Phase Transitions and Surface Morphology of Surfactant-Coated Aerosol Particles, Sci. Total Environ., 2007, 111, 11013–11020 Search PubMed.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- J. H. P. S. N. Seinfeld, Atmospheric Chemistry and Physics : from Air Pollution to Climate Change, 2016 Search PubMed.
- R. Xu, H. K. Lam, K. R. Wilson, J. F. Davies, M. Song, W. Li, Y. L. S. Tse and M. N. Chan, Effect of inorganic-to-organic mass ratio on the heterogeneous OH reaction rates of erythritol: implications for atmospheric chemical stability of 2-methyltetrols, Atmos. Chem. Phys., 2020, 20, 3879–3893 CrossRef CAS.
- H. K. Lam, R. Xu, J. Choczynski, J. F. Davies, D. Ham, M. Song, A. Zuend, W. Li, Y. L. S. Tse and M. N. Chan, Effects of liquid–liquid phase separation and relative humidity on the heterogeneous OH oxidation of inorganic–organic aerosols: insights from methylglutaric acid and ammonium sulfate particles, Atmos. Chem. Phys., 2021, 21, 2053–2066 CrossRef CAS.
- S. Gopalakrishnan, P. Jungwirth, D. J. Tobias and H. C. Allen, Air–Liquid Interfaces of Aqueous Solutions Containing Ammonium and Sulfate: Spectroscopic and Molecular Dynamics Studies, J. Phys. Chem. B, 2005, 109, 8861–8872 CrossRef CAS PubMed.
- P. Jungwirth and D. J. Tobias, Specific Ion Effects at the Air/Water Interface, Chem. Rev., 2006, 106, 1259–1281 CrossRef CAS PubMed.
- F. A. Houle, W. D. Hinsberg and K. R. Wilson, Oxidation of a model alkane aerosol by OH radical: the emergent nature of reactive uptake, Phys. Chem. Chem. Phys., 2015, 17, 4412–4423 RSC.
- C. Y. Lim, E. C. Browne, R. A. Sugrue and J. H. Kroll, Rapid heterogeneous oxidation of organic coatings on submicron aerosols, Geophys. Res. Lett., 2017, 44, 2949–2957 CrossRef CAS.
- G. A. Russell, Deuterium-isotope Effects in the Autoxidation of Aralkyl Hydrocarbons. Mechanism of the Interaction of PEroxy Radicals1, J. Am. Chem. Soc., 1957, 79, 3871–3877 CrossRef CAS.
- J. E. Bennett and R. Summers, Product Studies of the Mutual Termination Reactions of sec-Alkylperoxy Radicals: Evidence for Non-Cyclic Termination, Can. J. Chem., 1974, 52, 1377–1379 CrossRef CAS.
- C. T. Cheng, M. N. Chan and K. R. Wilson, The role of alkoxy radicals in the heterogeneous reaction of two structural isomers of dimethylsuccinic acid, Phys. Chem. Chem. Phys., 2015, 17, 25309–25321 RSC.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, n. null, J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- J. Peeters, G. Fantechi and L. Vereecken, A Generalized Structure-Activity Relationship for the Decomposition of (Substituted) Alkoxy Radicals, J. Atmos. Chem., 2004, 48, 59–80 CrossRef CAS.
- L. Vereecken and J. Peeters, Decomposition of substituted alkoxy radicals—part I: a generalized structure–activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 2009, 11, 9062–9074 RSC.
- A. A. Wiegel, K. R. Wilson, W. D. Hinsberg and F. A. Houle, Stochastic methods for aerosol chemistry: a compact molecular description of functionalization and fragmentation in the heterogeneous oxidation of squalane aerosol by OH radicals, Phys. Chem. Chem. Phys., 2015, 17, 4398–4411 RSC.
- C. W. Harmon, C. R. Ruehl, C. D. Cappa and K. R. Wilson, A statistical description of the evolution of cloud condensation nuclei activity during the heterogeneous oxidation of squalane and bis(2-ethylhexyl) sebacate aerosol by hydroxyl radicals, Phys. Chem. Chem. Phys., 2013, 15, 9679–9693 RSC.
- K. R. Wilson, J. D. Smith, S. H. Kessler and J. H. Kroll, The statistical evolution of multiple generations of oxidation products in the photochemical aging of chemically reduced organic aerosol, Phys. Chem. Chem. Phys., 2012, 14, 1468–1479 RSC.
- J. A. Dean and N. A. Lange, Lange's Handbook of Chemistry, McGraw-Hill, New York, 15th edn, 1999 Search PubMed.
- S. Oae and J. T. Doi, Organic Sulfur Chemistry: Structure and Mechanism, CRC Press, Boca Raton, 2007 edn, 1991 Search PubMed.
- R. Atkinson, A structure-activity relationship for the estimation of rate constants for the gas-phase reactions of OH radicals with organic compounds, Int. J. Chem. Kinet., 1987, 19, 799–828 CrossRef CAS.
- J. F. Doussin and A. Monod, Structure–activity relationship for the estimation of OH-oxidation rate constants of carbonyl compounds in the aqueous phase, Atmos. Chem. Phys., 2013, 13, 11625–11641 CrossRef.
- A. Monod and J. F. Doussin, Structure-activity relationship for the estimation of OH-oxidation rate constants of aliphatic organic compounds in the aqueous phase: alkanes, alcohols, organic acids and bases, Atmos. Environ., 2008, 42, 7611–7622 CrossRef CAS.
- M. C. Facchini, M. Mircea, S. Fuzzi and R. J. Charlson, Cloud albedo enhancement by surface-active organic solutes in growing droplets, Nature, 1999, 401, 257–259 CrossRef CAS.
- S. D. Forestieri, S. M. Staudt, T. M. Kuborn, K. Faber, C. R. Ruehl, T. H. Bertram and C. D. Cappa, Establishing the impact of model surfactants on cloud condensation nuclei activity of sea spray aerosol mimics, Atmos. Chem. Phys., 2018, 18, 10985–11005 CrossRef CAS.
- T. Cheng, L. Luo, L. Yang, H. Fan and H. Wu, Formation and Emission Characteristics of Ammonium Sulfate Aerosols in Flue Gas Downstream of Selective Catalytic Reduction, Energy Fuels, 2019, 33, 7861–7868 CrossRef CAS.
- R. M. Harrison and C. A. Pio, Size-differentiated composition of inorganic atmospheric aerosols of both marine and polluted continental origin, Atmos. Environ., 1983, 17, 1733–1738 CrossRef CAS.
- A. Ooki, M. Uematsu, K. Miura and S. Nakae, Sources of sodium in atmospheric fine particles, Atmos. Environ., 2002, 36, 4367–4374 CrossRef CAS.
- M. Shiraiwa, A. Zuend, A. K. Bertram and J. H. Seinfeld, Gas–particle partitioning of atmospheric aerosols: interplay of physical state, non-ideal mixing and morphology, Phys. Chem. Chem. Phys., 2013, 15, 11441–11453 RSC.
- J. H. Slade and D. A. Knopf, Multiphase OH oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity, Geophys. Res. Lett., 2014, 41, 5297–5306 CrossRef CAS.
- M. Song, P. Liu, S. Hanna, S. Martin and A. Bertram, Relative humidity-dependent viscosities of isoprene-derived secondary organic material and atmospheric implications for isoprene-dominant forests, Atmos. Chem. Phys. Discuss., 2015, 15, 1131–1169 Search PubMed.
- M. M. Chim, C. Lim, J. Kroll and M. Chan, Evolution in the Reactivity of Citric Acid toward Heterogeneous Oxidation by Gas-Phase OH Radicals, ACS Earth Space Chem., 2018, 2, 1323–1329 CrossRef CAS.
- F. H. Marshall, T. Berkemeier, M. Shiraiwa, L. Nandy, P. B. Ohm, C. S. Dutcher and J. P. Reid, Influence of particle viscosity on mass transfer and heterogeneous ozonolysis kinetics in aqueous–sucrose–maleic acid aerosol, Phys. Chem. Chem. Phys., 2018, 20, 15560–15573 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ea00026a |
| This journal is © The Royal Society of Chemistry 2022 |