
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEssential data for industrially relevant development of bifunctional cathodes and biopolymer electrolytes in solid-state zinc–air secondary batteries
Domenico
Frattini
 a,
Estíbaliz
García Gaitán
abc,
Ainhoa
Bustinza Murguialday
a,
Michel
Armand
*a and
Nagore
Ortiz-Vitoriano
*ad
a,
Estíbaliz
García Gaitán
abc,
Ainhoa
Bustinza Murguialday
a,
Michel
Armand
*a and
Nagore
Ortiz-Vitoriano
*ad
aCentre for Cooperative Research on Alternative Energies (CIC energiGUNE), Basque Research and Technology Alliance (BRTA), Alava Technology Park, Albert Einstein 48, Vitoria-Gasteiz 01510, Spain. E-mail: marmand@cicenergigune.com; nortiz@cicenergigune.com
bCEGASA Energía SLU, Marie Curie, 1, Parque Tecnológico de Álava, 01510 Miñano, Spain
cUniversity of the Basque Country (UPV/EHU), Barrio Sarriena, s/n, 48940 Leioa, Spain
dIkerbasque, Basque Foundation for Science, María Díaz de Haro 3, 48013 Bilbao, Spain
First published on 13th October 2022
Abstract
Presently, primary zinc–air batteries (ZABs), with a KOH-based liquid electrolyte, represent the first applied metal–air batteries and the most prevalent non-Li technology. The ZABs’ barrier to challenge Li–ion batteries is represented by rechargeability (needing a bifunctional cathode) and durability of the liquid electrolyte (due to leakage and/or evaporation). The liquid electrolyte should be replaced by a solid or gelled one but should not involve fossil-derived polymers or critical ceramic materials. Many naturally occurring biopolymers can be considered to prepare gelled electrolytes for ZABs, but focused literature about synthesis, properties, and applications in ZABs is still needed. Moreover, there is extensive literature about bifunctional cathodes for electrically rechargeable ZABs, but their assessment and performance for further industrialization are insufficient. The bottlenecks of sustainable gel electrolytes, extended cyclability, and relevant depth-of-discharge (DoD) per cycle should be met. In fact, industry seeks rechargeable materials, components, and assemblies capable of providing high current densities (e.g., >5–10 mA cm−2) in long cycles (e.g., >6–12 h) for as large as possible DoD (e.g., >5–10% per cycle, >100% total). The integration in the cells of gelled electrolytes and bifunctional cathode materials could overcome these problems if the correct calculations and testing are performed when carrying out experimental research. In this work, the actual state-of-the-art, key information, limitations, and calculations needed to assess a real promising cell integration between a biopolymer gel electrolyte and a cathode material in, at least, lab scale devices for rechargeability are reported. Finally, a wealth of experimental data spanning cyclability performance at low/medium drain rates (i.e., from 1–2 to 5–10 mA cm−2) at very short cycles (e.g., minutes) and long cycles (e.g., hours), enclosing a DoD analysis, are also shown, serving as a template for future studies.
Broader contextElectrochemical energy storage technologies can help mitigate the dependency on fossil sources and greenhouse gas emissions, enabling energy storage from renewable sources and their integration into the grid, thus driving energy transition worldwide. Presently, the most commercialized technology is lithium-ion batteries, from small portable devices to electric vehicles, and up to stationary applications at the grid level. Lithium-ion batteries are characterized by high power and energy density, a high voltage range, and very good rechargeability, thus dominating the field of secondary batteries. However, they are plagued by low environmental compatibility and strong dependency on critical raw materials (CRMs) for which supply chain and/or abundancy are not compatible with sustainable development. Therefore, it is strategically not convenient to rely only on this technology to implement electrochemical energy storage, but few valid alternative candidates can be foreseen considering the electrochemical window and energy density. Zinc–air secondary batteries are one of these; however, affordable development based on industrially and quantitatively relevant breakthroughs is required. In this comprehensive perspective, suitable guidelines are proposed to drive future research in this area, and the most important aspects, advantages, techniques, and testing protocols are critically discussed to make these batteries an attractive reality. |
1. Introduction
The environmental crisis society is facing has driven us to study high-performance energy generation, conversion, and storage systems.1 Though lithium-ion technologies continue to top the landscape of rechargeable batteries,2 primary and secondary zinc–air batteries (ZABs) have made considerable progress in the last decade,3 advancing at all component levels. Zn-based batteries, and ZABs as well, are not new technologies, but they are being revisited nowadays in the energy storage field as an alternative to lithium ion batteries (LIBs) due to a series of advantages like zinc being an abundant non-critical raw material (CRM), cheap (Li, as battery grade LiCO3, is rated at 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 $ ton−1 in 2022,4 whereas Zn, as a commodity metal, is approx. 3500 $ ton−1 in 20225), and easy to recover/recycle; ZABs use a safe, non-toxic, non-flammable aqueous electrolyte and have interesting specific capacity (theoretically, Li metal is 3860 mA h g−1, Zn metal is ≈820 mA h g−1, but practical LIBs are limited to 200–250 W h kg−1, while ZABs can approach 400–450 W h kg−1).2 However, secondary ZABs suffer from limited voltage, lifetime, cyclability, and rate capability, compared to LIBs. Reliable rechargeability, in ZABs, would be ideal and highly profitable for a valid, economic, and sustainable alternative to LIBs, for portable as well as stationary applications. The main chemical reactions involved in ZABs are:6
000 $ ton−1 in 2022,4 whereas Zn, as a commodity metal, is approx. 3500 $ ton−1 in 20225), and easy to recover/recycle; ZABs use a safe, non-toxic, non-flammable aqueous electrolyte and have interesting specific capacity (theoretically, Li metal is 3860 mA h g−1, Zn metal is ≈820 mA h g−1, but practical LIBs are limited to 200–250 W h kg−1, while ZABs can approach 400–450 W h kg−1).2 However, secondary ZABs suffer from limited voltage, lifetime, cyclability, and rate capability, compared to LIBs. Reliable rechargeability, in ZABs, would be ideal and highly profitable for a valid, economic, and sustainable alternative to LIBs, for portable as well as stationary applications. The main chemical reactions involved in ZABs are:6| Negative: Zn + 2OH− ↔ ZnO + H2O + 2e−, E = −1.26 V vs. SHE | (1) |
| Positive: O2 + 2H2O + 4e− ↔ 4OH−, E = +0.40 V vs. SHE | (2) |
| Overall: 2Zn + O2 ↔ 2ZnO, E = +1.66 V vs. SHE | (3) |
In the actual ZABs’ state-of-the-art, the negative electrode (anode during discharge) is based on pure Zn (foil or powder) or Zn alloys (corrosion resistant),7 the liquid electrolyte is a KOH aqueous solution in the 4–8 M range with dissolved Zn-based salts (acetates, chlorides, or oxides) and additives, and the cathode is composed of a hydrophobic gas diffusion layer (GDL) supported on a soft carbon material (paper, cloth, felt, etc.) upon which the active catalyst layer is dispersed. The benchmark positive electrode (cathode during discharge) catalysts are based on platinum group metals (PGMs), especially Pt, Ru, Ir, and their oxides, as well as Co and non-precious transition metal (TM) oxides.8,9 These oxides, or just the metal atoms, are also variously dispersed on nanostructured carbon materials.10
The Zn negative electrode has a complex physico-chemical behaviour, with the main issues being corrosion, hydrogen evolution, dendrite growth, and Zn modifications (i.e., passivation and shape change), which will not be covered here. Alleviating strategies and design concepts are discussed in numerous outstanding studies to which the reader is warmly referred to, for example, ref. 11–14. Liquid electrolytes have always played an essential role in electrochemical energy storage, mainly due to their high ionic conductivities and good contact with electrodes; however, the use of liquid electrolytes entails risks such as leakages and even combustion of organic electrolytes which render them risky for this kind of device. There are other drawbacks of liquid electrolytes such as dendrite growth in liquid solution, which is caused by inhomogeneities that produce preferential nucleation and uneven currents when charging, evaporation and handling issues among others.15 Therefore, the use of gel polymer electrolytes (GPEs) would be more suitable in electrochemical storage devices. A GPE contains a liquid electrolyte entrapped in a polymer matrix, to retain the ionic conductivity. GPEs eliminate the need for a separator between the two electrodes as the polymer matrix adsorbs the liquid while acting as a physical barrier preventing the internal short-circuit. GPEs have several advantages including safety and resistance against corrosion, excellent flexibility, and processability.16
The other pivotal component, that determines the difference between primary and secondary ZABs and between ZABs that are mechanically or electrically rechargeable, is the bifunctional cathode.17,18 The use of such a cathode enables a simple one-cathode electrode battery design, the absence of a switching system (design with two specialized cathodes), and the absence of complex cases to replace exhausted elements (mechanically rechargeable designs) which makes secondary ZABs compact, easy to connect and operate, and potentially require minimum maintenance.19 The bifunctional cathode is devoted to the oxygen reduction reaction (ORR) during discharge and the oxygen evolution reaction (OER) during charge. One of the bottlenecks limiting the development of secondary ZABs is the overpotential of the bifunctional catalyst, at discharge as well as at charge. This is a critical aspect of aqueous electrolyte-based batteries because when high currents are applied during cycling, failures occur due to hydrogen evolution or carbon oxidation.20–23 The same applies to solid and semi-solid gelled electrolytes, where high overpotentials may cause severe deterioration of the gel. The most documented techniques to evaluate the catalyst activity are linear sweep voltammetry (LSV) and cyclic voltammetry (CV) with the rotating disk electrode (RDE). The methodologies, procedures, and selection of the activity metrics in fundamental electrochemical characterization are sources of discussion, hence straightforward comparisons in the literature are often not possible. In early studies, different procedures were adopted, hence some metrics were not defined or available. More comparisons can be found in the supplementary information of a recent work,24 showing that conditions and RDE loadings may vary, but this has improved recently in new studies. Additionally, projecting encouraging fundamental findings to full cells at the lab scale is not straightforward. The operating conditions of a lab-scale cell might be very different from those of an electrochemical three-electrode cell. This is surely the case of catalysts characterized in a liquid electrolyte, e.g., by RDE in KOH 0.1 M, and then tested in a full cell using a solid GPE. There is an unsaid, non-trivial contribution due to the engineering from the catalyst to the cathode. Generally, these aspects are pertinent also to the anode electrode design (beyond the scope of this work).11–14
In this perspective, GPEs derived from synthetic or natural biopolymers are critically analysed, and their limitations are overviewed; bifunctional cathodes, from catalyst formulation and characterization to electrode design, are discussed and evaluated to guide future work directions. A focused part is devoted to demonstrating, quantitatively, how reliable and usable rechargeability should be assessed to fill the gap between fundamental characterization and testing at the device level, for a market breakthrough, and possible application field expansions envisaged.
2. Biopolymers for gel electrolytes in primary/secondary ZABs
2.1 Naturally occurring vs. synthetic biopolymers
In the past decade, research has focused on the design of GPEs for battery applications to replace commonly used liquid electrolytes. The term biopolymers refers to polymers, natural or synthetic, that are naturally degraded and decomposed within a reasonable period of time under environmental conditions by the effect of atmospheric agents, like solar radiation and oxygen, or living agents, like bacteria and enzymes.25Synthetic polymers can be biodegradable, but derive from non-renewable sources totally or partially, depending on the production process. Synthetic polymer gels have found more flexible and diverse biomedical applications26,27 as well as in several fields28–30 due to their versatility.31
Natural polymers are those derived from natural biomass, animals, or plants and are mainly composed of a few types of repeating carbon-containing chemical building blocks, originating from the cells of living organisms. Naturally occurring biopolymers (e.g., polysaccharides and proteins) have attracted the interest of the scientific community to integrate as GPEs, due to their good biocompatibility, biodegradability, flexibility, mechanical properties, natural abundance, and sustainability. Moreover, they present several reactive sites that can be chemically modified or crosslinked.32–34
If the solvent used is aqueous based, the term hydrogel is used to refer to the GPE. Hydrogels are composed of three-dimensional networks that absorb and retain large amounts of water within their solid polymeric matrices.35 Among GPEs, hydrogels from cellulose, starch, chitosan, and agars are being extensively studied for electrochemical devices.
Naturally derived polymers, however, have a complex structural composition and are therefore difficult to handle and control.36 As a result, research has mainly focused on developing synthetic biodegradable polymers due to their lower cost and affordability37 for electrochemical energy storage devices.38 For example, alkaline GPEs such as polyvinyl alcohol (PVA),39 polyacrylic acid (PAA),40 polyacrylamide (PAM), and polyethylene oxide (PEO)15,41 can be listed.15
Much of the early, but also recent, work in metal air batteries (especially in Zn and Na air batteries) has been devoted to the study of synthetic biopolymer-based membranes that act as separators and electrolytes to substitute liquid electrolytes. Recent studies have demonstrated that many biopolymers could be used to obtain semi-solid electrolytes, having a significant impact on the performance of ZABs. The challenge is to develop a gel electrolyte for aqueous systems with stability, conductivity, and physical properties similar to those of organic electrolytes. In the following subsections, synthesis methods, physicochemical properties, and electrochemical performance are summarized to provide at the end a critical outlook of the possible developments.
2.2 Gel biopolymer electrolytes: syntheses and characteristics
GPEs can be synthesized from monomers where the network structure is formed simultaneously during the synthesis and from polymers where the network is obtained by crosslinking the already existing polymer chains.42In this section, current gelation methods and resulting properties are briefly discussed.
Selecting the adequate gelation method, to prepare a GPE, is a matter of the specific biopolymer to be used and the degree of bonding to be obtained. It is important to understand these methods to tailor the GPE formulation, fabrication, and properties, but also to exploit diverse and alternative sources of biopolymers.
Bonding nature. In physical gels, the nature of the bonding is due to physical interaction which is normally achieved via phenomena such as hydrophobic association, chain aggregation, crystallization, polymer chain complexation, and above all, hydrogen bonding. Physical hydrogels are easy to produce, do not need crosslinking agents during their synthesis and could be reversible due to conformational changes.34 The methods for producing physically crosslinked hydrogels are ionic interactions, freeze-thawing, stereocomplex formation, non-covalent interaction, and thermoreversible gels.34 On the other hand, simultaneous gelation/polymerization or post-polymerization gelation is utilized to prepare a chemical hydrogel. In chemical hydrogels, phenomena like covalent crosslinking are utilized for preparation; they are permanent and irreversible because conformational changes are inhibited. They are basically constructed by crosslinking networks; hence, covalent bonds between polymer chains can be established by the reaction of functional groups. Typical crosslinking agents are glutaraldehyde,43,47 epichlorohydrin, adipic acid, dihydrazides, etc. To chemically crosslink a polymer, grafting, radical polymerization, and condensation reactions can be used.34,45,48
Response triggered. Polymers can have a physical (e.g., temperature, pressure, and light), chemical (e.g., pH and chemical initiators), or biochemical response to a trigger. The type of response determines the strength, structure, and properties of the gel. For temperature response, gelation of a polymer by freeze-drying and freeze-thawing techniques is very well known45 and used for example in PVA hydrogels, providing more porous, spongier, and highly elastic structures.34,49 Some polymers can be responsive to light, leading to photopolymerization.50,51 Photopolymerization is an effective method to obtain in situ crosslinked (chemical) hydrogels with various architectures in a fairly short time, under solvent-free conditions, at room temperature, or below. The process is typically done in the presence of a photo-initiator upon exposure to a light source (UV or visible), where UV light is the most common one, widely used in industrial processes. There are several photopolymerizable biopolymers that can give hydrogels, such as gelatine, alginate, PEG derivatives, PVA, hyaluronic acid (HA), dextran, cellulose, and chitosan.50
pH responsive (ionic charges). Hydrogels with ionic pendant groups can accept or donate protons due to a pH change according to their own degree of ionization (pKa or pKb), giving anionic or cationic hydrogels.45,52 Cationic hydrogels contain pendant groups, such as amine groups, where ionization takes place below the pKb, which increases swelling due to the increased electrostatic repulsions. These are also known as cation exchange membranes (CEMs), and Nafion and poly(styrene sulfonic acid) are the most common ones which are chemically stable at high acidities; however, the membranes afford low voltage efficiency, even at low working current densities30 which makes their use in batteries using alkaline electrolytes not suitable. Anionic hydrogels consist of polymer chains functionalized with several groups such as polysulfonium,53 where deprotonation occurs when the environmental pH is above the pKa, leading to the ionization of the pendant groups similarly to CEMs, but triggered by a different pH. These are known as anion exchange membranes (AEMs) and most are made from hydrocarbon polymer backbones with covalently attached quaternary ammonium (QA) groups.44,47,52
The ionic conductivity of a solid electrolyte can be enhanced by dissolving in situ an alkaline salt into the polymer formulation. Adding ex situ KOH solutions (by soaking) will also increase the conductivity, depending on water uptake: GPEs with higher water uptake could benefit from adsorbing lower KOH concentration solutions, while GPEs with limited water uptake would need concentrated KOH solutions to achieve the highest possible ionic conductivity. However, it is reported that at KOH concentrations higher than 6 M, ionic conductivity decreases.35 The transport easiness of OH− is associated with its size and its transport via the Grotthuss mechanism from one water molecule to the next via hydrogen bonding. An increase in temperature causes a faster movement of the anions due to thermal agitation and expansion of ion transport channels, i.e., pores. Plasticizers (e.g., PEG, glycerol, etc.56,57) can penetrate and alter the cohesive forces between the biopolymer chains, increasing segmental movement, and creating more free volume for ion transport. For liquid electrolytes, ionic conductivity is generally higher compared to that of GPEs.55 Solid electrolytes lack good ionic conductivity but are easier to manage and reduce leakage and evaporation. The ionic conductivity of some reported GPEs from synthetic and natural biopolymers is shown in Fig. 1.
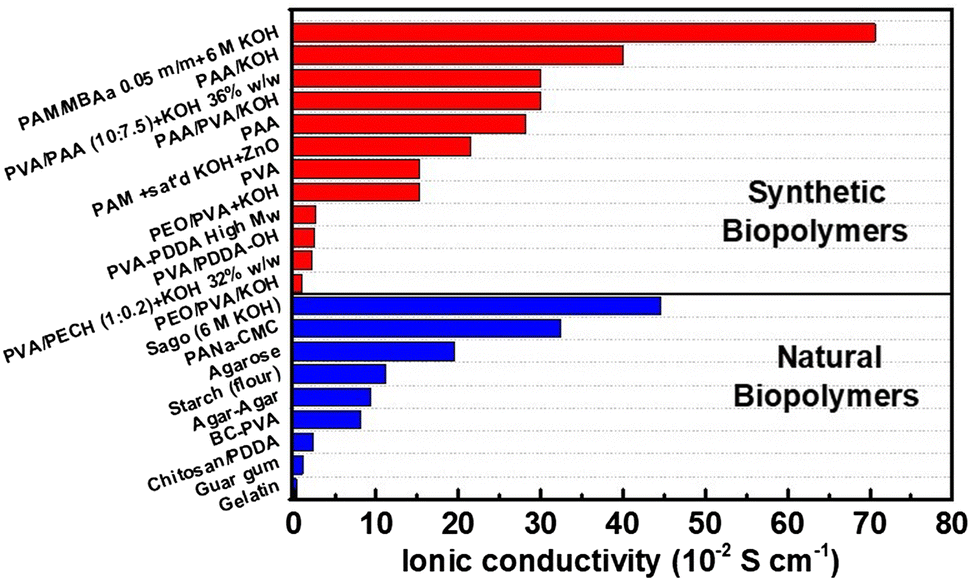 | ||
| Fig. 1 Bar chart of ionic conductivity for GPEs used in ZABs. Synthetic biopolymers: PAM/MBAa + 6 M KOH;59 PAA/KOH;60 PVA/PAA + KOH 36% w/w;46 PAA/PVA/KOH;60 PAA;61 PAM + sat’d KOH + ZnO;62 PVA;61 PEO/PVA + KOH;41 PVA-PDDA High Mw;63 PVA/PDDA-OH;64 PVA/PECH + KOH 32% w/w;65 and PEO/PVA/KOH.60 Natural biopolymers: sago (6 M KOH);66 PANa-CMC;67 agarose;68 starch (flour);62 agar–Agar;69 BC-PVA;49 chitosan/PDDA;43 guar gum;70 and gelatin.71 PAM: poly(acryl amide); MBAa: N,N′-methylenebis-(acryl amide); PVA: poly(vinyl alcohol); PAA: poly(acrylic acid); PEO: poly(ethylene oxide); PDDA: poly(diallyl dimethyl ammonium chloride); PECH: poly(epichlorohydrin); PANa: sodium poly(acrylate); CMC: carboxymethyl cellulose; and BC: bacterial cellulose. | ||
Water uptake/content is connected to ionic conductivity also due to swelling. The formation of swelling nanochannels in the polymer matrix is significant; the higher the number of pores in the GPE, the easier the transport of OH−.54 GPEs with high electrolyte absorption/retention capability act as electrolyte reservoirs also. Low anisotropic swelling boosted the specific capacity and improved the cycling stability of the battery.54 Good water retention capacity can greatly extend the service life of ZABs that are semi-open systems in which evaporation has a high influence.58 Thanks to a crosslinked network, hydrogels can absorb and retain a large amount of water.
On the other hand, the higher the crosslinking degree is, the lower the water absorption is.58 A limited water absorption deteriorates the ionic conductivity, thus leading to early cell failure.72
Gel strength has been usually related to the ability of the polymer to form sufficient helical structures for network formation and mechanical stability. Indeed, a linear correlation has been reported between the gel strength and the number of triple helices present in gel-based materials.73 The gel strength of GPEs from natural biopolymers strongly depends on the specific animal/plant source, the amino acid composition if present, and molecular weight distribution, which differ according to species and the processing conditions, respectively.74,75 The gel strength also depends on the isoelectric point and can be controlled by adjusting the pH.55 From pH 4 to pH 9, the strength is not affected to a significant extent. Gels obtained at high alkaline pH (in ZABs the pH is 13–14) generally show higher strengths. The gel strength can be enhanced by increasing the density of the internal and surface-crosslinking. The aim of surface-crosslinking technology is to enhance the strength of a gel when it absorbs large amounts of liquid, and various parameters can be adjusted to improve its performance.73
Therefore, when designing a GPE, these three properties should be tuned carefully to obtain a good compromise between conductivity, electrolyte retention, and mechanical resistance. The concurrent optimization of these properties is very challenging and is a bottleneck for GPE industrialization because industries and end-users look for ready-to-use performing solutions.
2.3 Current performance of gel electrolytes in ZaBs
GPEs have been earlier tested in primary ZABs and recently they have been proposed also for secondary ZABs, showing interesting performance, in terms of specific capacity, Zn utilization (referred to primary ZABs, and intended as the percentage of zinc utilized and converted to ZnO), and current density. Cyclability needs further effort to be confidently demonstrated (see subsection 3.4 and Section 4), and is more focused on flexible, small devices, showing encouraging performance as power sources for wearable electronics. Although the overall performance strongly depends on anode/cathode materials, cell design, and assembly, GPEs lead to different performances when integrated with ZABs. In primary ZABs:• Synthetic biopolymer gels obtained from PAA, PVA, and their crosslinked blends give high specific capacities; Zn extraction (calculated as the specific capacity delivered by a fully discharged cell divided by the theoretical capacity of pure zinc, i.e., 819.73 mA h g−1) within 80–90% at current densities from 1–5 mA cm−2 and up to 10–50 mA cm−2, in few cases, has been demonstrated.
• Gels from naturally occurring cellulose, nanocellulose, and derivatives usually give good specific capacities, but lower Zn extraction, approx. 70–80%, hardly approaching 85% in few cases.
This was ascribed to the high ionic conductivity of synthetic biopolymer gels, superior water electrolyte uptake, and mechanical strength with small anisotropic swelling.
An overview of the performance of various GPEs in primary/secondary ZABs is given in Table 1. In secondary ZABs, more examples are available for synthetic GPEs whereas a few natural biopolymers (agar-based, cellulose, starches, and gelatines) have been reported. For example, the use of jellified aqueous-based agar–KOH electrolytes has been reported, preventing the water loss due to overcharging and improving the cycle-life and high-charge acceptance. These gels are stable (at near-neutral and alkaline pH) and mechanically flexible, and their ionic conductivity (see Fig. 1) is high enough. Cellulose derivatives have gained immense popularity as gels due to their versatile pH responsive behaviour. Carboxymethyl cellulose (CMC) is an important ionic ether derivative of cellulose and can improve the mechanical integrity of the final film without affecting the ionic mobility and the electrode/electrolyte interfacial characteristics. Moreover, it shows good solubility in water, low cost, biodegradability, biocompatibility, and lack of toxicity.76 Another commonly used derivative is bacterial cellulose (BC), whose hydrogels have long chains rich in hydroxyl groups, conferring high hydrophilicity. BC hydrogels contain more than 98% of water and exhibit outstanding liquid retention ability.77
| GPE | Anode/cathode type | ZAB type (primary/secondary) | Performances | Ref. |
|---|---|---|---|---|
| Primary: current density, specific capacity, discharge time | ||||
| Secondary: current density, cycle time (areal capacity), n. of cycles | ||||
| PAA/6 M KOH gel | Electrodeposited Zn/Pt–Ir/C | Primary | 1 mA cm−2, ≈700 mA h g−1, — | 40 |
| 5 mA cm−2, ≈684 mA h g−1, — | ||||
| PVA/PAA + 32 wt% KOH | Zn powder, carbopol 940, 32 wt% KOH/KMnO4/PTFE/Vulcan C/Ni foam | Primary | PVA:PAA (10:7.5): 150 mA (C/10) 1347 mA h, 8.98 h |
46 |
| PVA:PAA (10:7.5): 300 mA (C/5), 1077 mA h, 3.59 h |
||||
| (theoretical capacity 1476 mA h) | ||||
| Quaternary ammonium functionalized cellulose film + 1 M KOH | Free-standing Zn film/Co3O4/carbon cloth | Primary | 25 mA g−1, —, 1100 min | 54 |
| Starch gel saturated with KOH + ZnO | Zn foil/Mn–Co–Fe@CNT | Primary | 2 mA cm−2, 752 mA h g−1, — | 62 |
| Sago + 6 M KOH | Zn powder/— | Primary | 100 mA, 505 mA h g−1, — | 66 |
| PVA/poly(epichlorohydrin) (PECH) + 32 wt% KOH | Zn powder, carbopol 940, 32 wt% KOH/carbon slurry for gas-diffusion, acetylene black and 30 wt% PTFE as a binder | Primary | PVA 150 mA (C/10), 1338 mA h, 8.92 h | 65 |
| PVA:PECH (1:0.2) 150 mA (C/10), 1290 mA h, 8.60 h |
||||
| (theoretical capacity is 1476 mA h) | ||||
| Quaternary ammonium functionalized cellulose film gelled with 6 M KOH | Free-standing Zn film/Co3O4-NCNT | Primary | 5 mA cm−2, 652.6 mA h g−1, — | 78 |
| 50 mA cm−2, 632.3 mA h g−1, — | ||||
| PVA + KOH | Zn plate in 6 M KOH/Co4N/CNW/CC | Primary | 10 mA cm−2, 774 mA h g−1, ≈15 h | 79 |
| P-Co4N/CNW/CC | 50 mA cm−2, 701 mA h g−1, ≈10 h | |||
| PAA/6 M KOH gel | Electrodeposited Zn/Pt–Ir/C | Secondary | 1 mA cm−2, 40 min per cycle (0.33 mA h cm−2), 100 cycles | 40 |
| Chitosan and poly(diallyl dimethyl ammonium chloride) (PDDA) + 2 M KOH | Zn foil/IrO2/Pt/C/carbon paper | Secondary | 3 mA cm−2, 10 min per cycle (0.25 mA h cm−2), 16 cycles | 43 |
| PVA/guar hydroxy propyl trimonium chloride in 2 M KOH (PGG-GP membrane) | Zn foil/IrO2/Pt/C/carbon cloth | Secondary | 2 mA cm−2, 10 min per cycle (0.167 mA h cm−2), 57 cycles | 47 |
| BC-PVA in 6 M KOH + 0.2 M Zn(Ac) | Zn foil/Co3O4@Ni foam | Secondary | 0.5 mA cm−2, 40 min per cycle (0.167 mA h cm−2), 650 cycles | 49 |
| Quaternary ammonium functionalized cellulose + 1 M KOH | Free-standing Zn film/Co3O4/carbon cloth | Secondary | 250 mA g−1, 1 h per cycle (125 mA h g−1), 35 cycles | 54 |
| Starch gel saturated with KOH + ZnO | Zn foil/Mn–Co–Fe@CNT | Secondary | 2 mA cm−2, 10 min per cycle (0.167 mA h cm−2), 216 cycles | 62 |
| PVA-KOH (8.3 wt%) PAA-KOH | Zn foil/Co3O4/Ag@N-rGO/carbon cloth | Secondary | 2 mA cm−2, 10 min per cycle (0.167 mA h cm−2), 100 cycles | 61 |
| PAM crosslinked with MBAa in 6 M KOH | Zn foil/C-CoPAN900/carbon paper | Secondary | 2 mA, 1 h per cycle, 55 cycles | 59 |
| Crosslinked PVA/PAA in 6 M KOH | Zn powder/PEI/ | Secondary | 0.5 mA cm−2, 10 min per cycle (0.042 mA h cm−2), 150 cycles | 80 |
| Co3O4@MWCNT | ||||
| PVA-PEO-KOH | Zn foil/CaMnO3-S/carbon cloth | Secondary | 5 mA cm−2, 400 s per cycle (0.278 mA h cm−2), 120 cycles | 81 |
| PAA + 6 M KOH | Zn foil/Pt/Ru/C | Secondary | 0.5 mA cm−2, 10 min per cycle (0.042 mA h cm−2), 25 cycles | 82 |
| Quaternary ammonium functionalized cellulose in 6 M KOH | Zinc plate/S-C2N aerogel/stainless steel mesh | Secondary | 25 mA cm−2, 2 h per cycle (25 mA h cm−2), 230 cycles | 83 |
Although very cheap, the conductivities of these biopolymers are low, and more cell data, as well as fundamental characterization, are needed to assess their potential.84 For these reasons, the choice by industry of a specific type of GPE is not fixed yet. Going from liquid electrolytes to gels involves making investments in coating technologies that are capital cost intensive. Resorting to chemical crosslinking can be the production rate determining step, and this may become the bottleneck for industrialisation.
2.4 Cell integration: advantages and current limitations
For ZAB technology, strong alkaline GPEs are necessary to provide high ionic conductivity and fast reaction kinetics, while naturally occurring biopolymers can push sustainability.85 The use of jellified electrolytes for ZABs can improve the performance of these batteries. However, it is still needed to design electrolytes with higher mechanical strengths, temperature tolerance, and high stability during charging to enhance the lifetime of ZABs.It is also well-known that adding suitable additives to the electrolytes is a straightforward method to reduce the growth of dendrites during cycling.86 GPEs usually display a lower conductivity compared to liquid electrolytes, but in zinc–ion based technology they are highly desirable because of the protection they offer against dendrite growth due to the superior regulation of Zn cation transport to minimize gradient, and the better current distribution thanks to the nanochannels in the polymer matrix. These effects are supported also by a planar, flat, and homogeneous contact between the GPE and the electrodes.52 GPEs have good adhesive properties, which provide well-bonded interfaces for the charge and the mass transfer, limiting the dendrite growth and decreasing the corrosion on Zn foils when used as anodes.16 On the other hand, the extension of the triple-phase boundary zone is generally reduced because of the semi-solid nature of the GPEs.
By cross-linking, it is possible to almost eliminate leakage, retain larger amounts of liquid electrolyte, and increase mechanical resistance, hence the thicknesses of GPEs can be reduced to compensate for the lower ionic conductivity. Comparing the values in Fig. 1, the ionic conductivity of GPEs from synthetic/naturally occurring biopolymers varies within 2 orders of magnitude, depending on the KOH concentration and retention, with higher average values for synthetic GPEs. Mechanical properties, water uptake, biopolymer content, and synthesis determine the ionic conductivity, as explained previously. Oscillations in ionic conductivity are due also to the measurement methodology used to estimate values, because this is not always reported and detailed.
However, reducing the thickness too much, i.e., the distance between the anode and the cathode, without proper physical protection, inherently increases the risk of short circuit and oxygen crossover. For stable and durable ZABs, a uniform and minimized GPE thickness is necessary, considering all the issues mentioned above (i.e., balancing ionic conductivity, internal resistance, mechanical integrity, ion/gas crossover, and dendrite growth). Practicable thicknesses are also dependent on the coating technology available, representing a major investment for companies. Typical thicknesses for advanced fabrication of polymeric membranes and GPEs are in the range of 25–200 μm52 whereas, by using inexpensive and simple processes, gels with >0.5 mm thickness are prepared. Another drawback in GPEs is the possible dissolution of some metals, e.g., Ni or Mn, from the cathode into the GPEs, or the oxidation of the organic carbon of the biopolymers during the charge phase. For this, less positive voltages at moderate currents for limited charging times should be applied, i.e., lower OER overpotentials. Therefore, the development and modification of hydrogel electrolytes for ZABs, in the aspects of ion conductivity, mechanical properties, environmental adaptability and interfacial compatibility of electrolytes and cathodes still need to be further studied.87 Several other critical issues await impending solutions, e.g., the CO2 neutralization toward alkaline electrolytes and temperature adaptability. Blocking CO2 from the outside environment is challenging, and a possible mitigation strategy is the functionalization of the polymer matrix with specific –OCO2− groups.88 Temperature tolerant ZABs are even more challenging because many polymers give hydrogels non-adaptable to temperature due to the poor interaction between the GPE and the air cathode.89 The major limit is the aqueous nature of the electrolyte entrapped in the GPE due to freezing below 0 °C and evaporation at high temperature. This can be tackled, for example, by grafting or crosslinking highly polarized functional groups to the polymer matrix thus decreasing the activity of free water molecules in the GPE,90 or by blending with ionic liquids (ILs).91
Positive factors to be considered are the simple preparation process of the electrolytes and their cost. Despite great progress in developing alkaline GPEs, the raw material prices of PVA, PAA, and PAM, for example, are higher compared to those of some biopolymers obtained from natural resources that can be used, like starch-based or agar–agar. Therefore, future directions in this field should make more effort in developing low-cost, affordable, and reliable GPEs62 by optimizing the conductivity-water uptake-gel strength triad, avoiding carbonate formation by either changing KOH to another salt or implementing selective sieves to reject CO2 or using non-alkaline pH.
3. Bifunctional cathode materials and extended cyclability
3.1 Trends and development in cathode materials for secondary ZABs
From a rational point of view, it is possible to contemplate as bifunctional catalysts the following options:• A single compound (multi metallic) capable of catalysing both reactions;
• A mix of two or more compounds/phases, each capable of catalysing the ORR or OER;
• A single support with specific active sites, each capable of catalysing the ORR or OER.
The first option is the straightforward meaning of “bifunctional”, but it is slightly tricky because the double activity is often more pronounced for one reaction than the other (see subsection 3.3). It can be obtained via compositional engineering of materials, such as alloys or ceramic oxides. The second option represents a simple approach based on several combined steps like physical mixing, sonication, mechanochemistry, hydrothermal treatments, in situ growth, etc., to prepare composites, from the macro to the nano level, seeking synergies between compounds/phases. The third option is more complex and specifically regards carbon-based catalysts,92,93 doped with metal atoms and/or non-metal elements like boron, nitrogen, sulphur, and phosphorus. These catalysts can be obtained via defect/doping engineering, even from waste biomass as raw carbonaceous materials,94 showing variety in feedstock, syntheses, and procedures. Active sites can be carbon defects, dispersed single atoms, or small clusters of nanoparticles. An overview of the catalysts recently proposed in the literature for ZABs is shown in Table 2.
| Type | Cathode materials | Synthesis procedurea | Year | Ref. |
|---|---|---|---|---|
| a Common operations like mixing, washing, filtering, centrifugation, etc. are not mentioned, for details please refer to the cited references. | ||||
| Composites, nanocomposites | MnO2/N-CNT | Hydrothermal + CVD + physical mixing | 2012 | 100 |
| CoO/N-CNT | Various steps (Hummer's, precipitation, freeze-drying, and annealing) | 2013 | 101 | |
| NiFe LDH/CNT | ||||
| Co3O4/MnO2NT | Two-step hydrothermal + calcination | 2013 | 102 | |
| MnCo2O4/CNT | Various steps (precipitation, hydrothermal, freeze-drying, calcination, physical mixture, and annealing) | 2014 | 103 | |
| CoMn2O4/N-rGO | Hummer's + two-step hydrothermal | 2014 | 104 | |
| NCNT/CoO–NiO–NiCo | Various steps (coprecipitation, hydrothermal, freeze-drying, and multiple annealing) | 2015 | 105 | |
| Co3O4/NPGC | Multistep hydrothermal + annealing | 2016 | 106 | |
| Co-doped TiO2 | Sol–gel | 2016 | 107 | |
| α-MnO2@XC-72 + Fe0.1Ni0.9Co2O4/Ti | Coprecipitation | 2016 | 108 | |
| Co4N/CNW/CC | Various steps (electrodeposition, impregnation, and pyrolysis) | 2016 | 79 | |
| NiCo2S4/N-CNT | Various steps (Hummer's, precipitation, solvothermal, and freeze-drying) | 2017 | 109 | |
| NiFe-LDH/Co,N-CNF | Various steps (from ZIFs, sol–gel, pyrolysis, etching, and coprecipitation) | 2017 | 110 | |
| Fe@C–NG/NCNTs | Various steps (Hummer's, freeze drying, and pyrolysis) | 2018 | 111 | |
| Mn/Fe–HIBMOFs | Various steps (amination, protonation, oxidation, hydrothermal, and annealing) | 2019 | 112 | |
| FeP–Fe2O3/N, P-doped carbon | Coprecipitation, freeze drying, and annealing | 2020 | 113 | |
| FeCo–N–C hollow nanospheres | Pyrolysis + acid leaching of ZIF derived complexes | 2021 | 114 | |
| Heterodoped carbons | Fe–N–graphene | Various steps (pseudo-Hummer's, freeze-drying, annealing, physical mixing, and pyrolysis) | 2013 | 115 |
| N-doped carbon from phenylenediamine | Hard template polymerization + pyrolysis/etching + activation | 2014 | 116 | |
| N-P-C from PANi pyrolysis | Polymerization + freeze drying + pyrolysis | 2015 | 117 | |
| Defective graphene | Multistep pyrolysis | 2016 | 118 | |
| N-doped graphene | Polymerization + pyrolysis | 2016 | 119 | |
| Co-Nx-C/graphene | Various steps (gelification, pyrolysis, leaching, and freeze-drying) | 2017 | 120 | |
| S, N-Fe/N/C-CNT | Precipitation + pyrolysis/leaching | 2017 | 121 | |
| FeCo–N–C | Direct pyrolysis | 2017 | 122 | |
| P, S-CNS | Various steps (precipitation, freeze drying, pyrolysis, etc.) | 2017 | 123 | |
| S-doped C2N aerogels | Various steps (sol–gel, freeze drying, and annealing) | 2018 | 83 | |
| N-doped HPC from raw wood | Enzymatic hydrolysis + pyrolysis | 2019 | 124 | |
| N, S, F-doped carbon | Triazine polymer self-templated carbonization | 2020 | 125 | |
| N, S, P-doped graphene | Ball milling + freeze drying + pyrolysis | 2021 | 126 | |
| Perovskite based | La0.6Ca0.4CoO3 | Sol–gel | 2011 | 127 |
| LaNiO3/N-CNT | Hydrothermal + CVD | 2012 | 128 | |
| La1.7Sr0.3NiO4 | Sol–gel | 2013 | 129 | |
| LaNiO3/N-CNT | Hydrothermal + CVD | 2015 | 130 | |
| LaCoO3 | Sol–gel | 2015 | 131 | |
| LaTi0.65Fe0.35O3−δ/N-doped carbon nanorods | Electrospinning | 2015 | 132 | |
| La(Co0.71Ni0.25)0.96O3−δ | Electrospinning | 2016 | 133 | |
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ nanofiber | Electrospinning | 2017 | 134 | |
| La0.9Y0.1MnO | Sol–gel | 2018 | 135 | |
| PrBa0.5Sr0.5Co1.9Ni0.1O5+δ | Sol–gel | 2019 | 136 | |
| SrMnO3 | Sol–gel | 2020 | 137 | |
| CoSx/Pr0.5Ba0.5Mn0.25Fe0.75O3−δ | Sol–gel + photochemical deposition | 2021 | 138 | |
The table reports examples of the three options discussed above, and a rough classification by the type of material employed, i.e., metal-free carbon-based,95 ceramic perovskites,96 metal/metal oxides over carbons, doped carbons, and nanostructured carbons.97,98 These catalysts are based on a recurrent group of “traditional” elements for this purpose, like Ni, Co, Mn, Ag, Fe, Cu, and rare earths.9,99 Carbon-based catalysts, doped with N, S, P, or B, and metal atoms (mainly Fe or Co), derived from metal–organic frameworks (MOFs), like zeolitic imidazolate frameworks (ZIFs), are also very popular. Another “evergreen” class of catalysts, recently revisited for secondary ZABs, is that of the long-known perovskites and/or ceramic oxides.100
In perovskites, recurrent elements are Ni, Co, Mn, Fe, and Cu, with an important use of rare earth elements, especially La, and minor use of alkaline earths like Ca and Sr.
Despite the promising performance and progress in this field, some crucial challenges, at the material and device level, must be undertaken. These can be summarised as follows:
• Cathodes enabling extended cyclability and stability.
• Catalysts with low ORR/OER overpotentials.
• Electrocatalysts based on no/less critical raw materials (CRMs).
Extended cyclability means reliable operations for long cycles (>6 h, up to 24 h per cycle) at 2–5 mA cm−2 that can compete with Li-ion technology (see subsection 3.4). Cobalt-containing catalysts have very high and reliable performance, especially for the OER, but Co is listed by the European Union as a CRM, so its availability, supply, and abundance are not compatible with sustainable development.139 Unfortunately, the same applies to the PGM elements, other elements like Ti, P, and W (relatively abundant but considered CRMs), and light rare earth elements (LREE), hence including La, Ce, and Sm. Catalysts based on these elements provide the lowest known overpotentials at the fundamental characterization level, but non-CRM based catalysts could be competitive in real devices. Reducing/eliminating the use of CRMs in cathodes for ZABs will be a major obstacle soon for the development of the technology. In view of the shortage/supply of these raw materials, it is necessary to improve the formulation, atomic structure, and intrinsic/practical activity of catalysts based on no/less CRMs. The future development of cathode materials for secondary ZABs should be focused more to overcome these aspects.
3.2 Essential electrochemical characterization of ORR/OER catalysts
When investigating a new catalyst, it is of primary importance to correctly characterize its activity. This means basically observing the electrochemical behaviour in the form of a current vs. potential plot, then correcting potentials and normalizing currents to extract specific curves/values related to independent parameters that characterize the system. Raw data must be always corrected for the deviation of capacitive current effects (a sort of “background”) and ohmic drops (the so called “iR” correction) due to the resistance of aqueous solution, and then potentials referred to the reversible hydrogen electrode (vs. RHE) to consider the effect of the pH (and temperature) in alkaline media. The CV and/or LSV techniques, the type of working electrode, RDE and/or the rotating ring disk electrode (RRDE), to perform the basic characterization are consolidated tools.140Considering some of the entries in Table 2, the most popular metrics, together with the RDE mass loading, the electrolyte, and normalization parameters used, are reported in Table 3. The most diffused normalization is based on the geometric area of the RDE working electrode used, and overpotential metrics are the ORR half-wave potential (E1/2), the OER potential at a current density j = 10 mA cm−2 (E10), both reported versus RHE, and the voltage gap between these two points (ΔE = E10 − E1/2).
| Type | Cathode material | E 1/2 vs. RHE (V) | E 10 vs. RHE (V) | ΔE (V) | RDE mass loading (mg cm−2) | Normalizations and electrolytes | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| a n.r. = not reported; the values are not available vs. RHE, not defined, not measured, not explicitly mentioned, or cannot be reliably calculated from the reference. For details, please refer to the cited reference. | ||||||||
| Composites, nanocomposites | MnO2/N-CNT | n.r.a | n.r. | n.r. | n.r. | Geometric area, 0.1 M KOH | 2012 | 100 |
| Co3O4/MnO2NT | n.r. | n.r. | n.r. | 0.100 | Geometric area, 0.1 M KOH, 2000 rpm | 2013 | 102 | |
| MnCo2O4/CNT | n.r. | n.r. | n.r. | 0.100 | Geometric area, 0.1 M KOH | 2014 | 103 | |
| CoMn2O4/N-rGO | 0.80 | 1.66 | 0.86 | 0.900 | Geometric area, 0.1 M KOH | 2014 | 104 | |
| NCNT/CoO–NiO–NiCo | 0.83 | 1.5 | 0.67 | 0.21 | Geometric area, 0.1 M KOH | 2015 | 105 | |
| Co3O4/NPGC | 0.84 | 1.68 | 0.84 | 0.204 | Geometric area, 0.1 M KOH | 2016 | 106 | |
| Co-doped TiO2 | n.r. | n.r. | n.r. | 0.108 | Geometric area, 0.1 M KOH | 2016 | 107 | |
| α-MnO2@XC-72 + Fe0.1Ni0.9Co2O4/Ti | n.r. | n.r. | n.r. | n.r. | Geometric area, 0.1 M KOH | 2016 | 108 | |
| Co4N/CNW/CC | 0.80 | 1.54 | 0.74 | n.r. | Geometric area, 0.1 M KOH | 2016 | 79 | |
| NiCo2S4/N-CNT | 0.80 | 1.6 | 0.80 | 0.248 | Geometric area, 0.1 M KOH | 2017 | 109 | |
| NiFe-LDH/Co,N-CNF | 0.79 | 1.54 | 0.75 | 0.120 | Geometric area, 0.1 M KOH | 2017 | 110 | |
| Fe@C–NG/NCNTs | 0.84 | 1.68 | 0.84 | 0.240 | Geometric area, 0.1 M KOH | 2018 | 111 | |
| Mn/Fe–HIBMOFs | 0.883 | 1.51 | 0.627 | 0.15 | Geometric area, 0.1 M KOH | 2019 | 112 | |
| FeP-Fe2O3/N, P-doped carbon | 0.838 | 1.632 | 0.794 | 0.255 | Geometric area, 0.1 M KOH | 2020 | 113 | |
| FeCo–N–C hollow nanospheres | 0.86 | 1.59 | 0.73 | 0.200 | Geometric area, 0.1 M KOH | 2021 | 114 | |
| Heterodoped carbons | Defective graphene | 0.76 | 1.60 | 0.84 | 0.283 | Geometric area, 0.1 M KOH | 2016 | 118 |
| N-doped graphene | 0.84 | 1.66 | 0.82 | 0.6 (ORR) | Geometric area, 0.1 M KOH | 2016 | 119 | |
| 0.3 (OER) | ||||||||
| Co-Nx-C/graphene | 0.79 | 1.74 | 0.95 | 0.250 | Geometric area, 0.1 M KOH | 2017 | 120 | |
| S, N-Fe/N/C-CNT | 0.85 | 1.60 | 0.75 | 0.600 | Geometric area, 0.1 M KOH | 2017 | 121 | |
| FeCo–N–C | 0.92 | 1.73 | 0.81 | 0.200 | Geometric area, 0.1 M KOH | 2017 | 122 | |
| P, S-CNS | 0.87 | 1.56 | 0.69 | 0.150 | Geometric area, 0.1 M KOH | 2017 | 123 | |
| S-doped C2N aerogels | 0.88 | 1.53 | 0.65 | 0.150 | Geometric area, 0.1 M KOH | 2018 | 83 | |
| N-doped HPC from raw wood | 0.85 | 1.68 | 0.83 | 0.255 | Geometric area, 0.1 M KOH | 2019 | 124 | |
| N, S, F-doped carbon | 0.81 | 1.57 | 0.76 | 0.285 | Geometric area, 0.1 M KOH | 2020 | 125 | |
| N, S, P-doped graphene | 0.82 | 1.76 | 0.94 | 0.200 | Geometric area, 0.1 M KOH | 2021 | 126 | |
| Perovskite based | La0.6Ca0.4CoO3 | n.r. | n.r. | n.r. | 0.17–0.24 | Geometric area, 1–4–6 M KOH | 2011 | 127 |
| LaNiO3/N-CNT | n.r. | n.r. | n.r. | 1.22 | Geometric area, 0.1 M KOH | 2012 | 128 | |
| La1.7Sr0.3NiO4 | 0.83 | n.r. | n.r. | 0.141 | Absolute current, 0.1 M KOH | 2013 | 129 | |
| LaNiO3/N-CNT | n.r. | n.r. | n.r. | 1.22 | Geometric area, 0.1 M KOH | 2015 | 130 | |
| La(Co0.71Ni0.25)0.96O3−δ | n.r. | 1.554 | n.r. | 0.700 | Geometric area, mass, 0.1 M KOH | 2016 | 133 | |
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ nanofiber | 0.73 | 1.53 | 0.80 | 0.796 | Geometric area, 0.1 M KOH | 2017 | 134 | |
| La0.9Y0.1MnO | 0.75 | n.r. | n.r. | 0.236 | Geometric area, 0.1 M KOH | 2018 | 135 | |
| PrBa0.5Sr0.5Co1.9Ni0.1O5+δ | n.r. | n.r. | n.r. | 0.796 | Geometric area, 0.1 M KOH | 2019 | 136 | |
| SrMnO3 | 0.81 | n.r. | n.r. | 0.25 | Geometric area, mass, 0.1 M KOH | 2020 | 137 | |
| CoSx/Pr0.5Ba0.5Mn0.25Fe0.75O3−δ | 0.70 | 1.56 | 0.86 | 0.226 | Geometric area, 0.1 M KOH | 2021 | 138 | |
Geometric current density, although very useful at the practical cathode level, is challenging for defining intrinsic activity because bulk and porous catalysts would be normalized to the same value leading to overestimations.141 An excessive RDE mass loading would lead to a thick layer over the RDE, and electrolyte or oxygen could be accumulated, trapped, and not readily consumed, so an effect of the thickness is observed instead of surface activity.140,141E1/2 and E10 are strongly dependent on a correct RHE conversion, so pH, temperature, and experimental calibration are very important because they determine the exact offset on the potential axis; E10 is quite arbitrary, based on geometric current density, and strongly dependent upon RDE mass loading, because of the catalyst's thickness effect due to porosity; ΔE is the difference, so virtually independent of the RHE offset, but it is dependent on the iR correction, and cross comparison between different catalysts is biased by the different surface areas. Therefore, if these popular metrics are being used, all suitable corrections must be done, and the observed currents normalized by the specific surface area obtained, e.g., from BET analysis.141
These and further procedural details about RDE experiments, just summarised here, together with benchmarking examples on how to process and report intrinsic activity data, have been excellently discussed in a series of studies141–144 to which readers are warmly referred to. In general, a golden rule is to seek a mass loading capable of covering completely the working electrode and giving a homogeneous thin film. Of extreme interest is to always report in published studies the rotation speeds, volume and concentration of the electrolyte, the RDE mass loading, catalysts’ BET surface areas, pH and temperature used for experiments, the evidence of iR correction (EIS or the value of R) or experimental calibration (CV with a Pt working electrode in pure H2 and the same cell and electrolyte).
For the ORR and the OER, Tafel plots (semi-log plot of j vs. E (vs. RHE)) of well-corrected-and-normalized CV/LSV data are warmly recommended as the essential, principal, info required to determine Tafel slopes, a valuable metric to compare activity. When reporting Tafel plots based on specific surface area and catalyst mass, overpotential cross comparisons could be done between different material classes. When comparing current densities, whatever is the normalization used, E1/2 is acceptable for the ORR, whereas comparing current densities at a fixed (over)potential vs. RHE would be more appropriate for the OER. In the ORR it is possible to have insights by a complete Koutecky–Levich analysis, but the description is beyond the scope of this work. This is necessary to seek/validate enhanced catalysts for the ORR/OER by essential electrochemical characterization.
3.3 Specific activity-driven bifunctional catalyst design: the case of spinel and perovskite-type oxides
To demonstrate how a rational approach and precise electrochemical characterization are fundamental to progress in the field and to provide research directions, the case of spinel (e.g., NiCo2O4, MnCo2O4, etc.) and perovskite oxide activity determination is critically explained. By correctly applying the corrections and normalizations described in Section 3.2, it is possible to obtain, for each oxide, the specific activity (i.e., current density per specific surface area of the catalyst) as a function of voltage vs. RHE. This fundamental information, however, is not able alone to describe, explain, and predict the activity of different oxides. Therefore, the activity was linked to a “descriptor” factor, related to the catalyst structure. Many descriptors have been proposed, like the B site-OH bond strength, covalency of metals, and the number of d orbital electrons, giving as a result a characteristic volcano-shaped plot. Finally, the electron occupancy in the antibonding orbitals, eg, was found to be a key descriptor, applicable to spinel as well as perovskite oxides.145–147 From these exemplary studies, together with others,148,149 a critical analysis of the fine volcano plots reported leads to fascinating conclusions on the data available so far:• Oxides with the same value of eg can present small or even relevant different activities. A reason could be due to particle size, morphology, and shape, other than the stoichiometry and chemical formula, hence could be optimized. It is also known that, depending on the overlap degree of the O–2p orbitals with the orbitals of the atom in the active site, the spin state and eg can change;
• Considering the reported oxides, the same oxide, although presenting a relevant bifunctionality, could show different activities for the ORR and the OER, hence it is not the “best in class” for both reactions;
• Doping the basic ABO3 structure of a specific perovskite by introducing another element in the A and/or B site can alleviate this activity discrepancy between the ORR and the OER, reducing the relative difference between the overpotentials;
• Although doping could balance and leverage the bifunctionality of the oxide, it does not always reduce the absolute overpotentials; hence, there are combinations of elements that work better than others and oxygen-defective oxides seem to display improved activity;
• The theoretical vertexes of the volcano plots for the ORR and the OER seem to fall at different values of eg (in perovskites, for the ORR, the max activity should be around eg 0.71,145 for the OER at around 1.25146 or 1150), depending on current density, hence a single oxide cannot be the best for both reactions, but just as close as possible to both values (the mathematical averages being 0.98 or 0.86);
• However, the exact position of the top of the volcano could depend on the number of data points, i.e., the number of perovskite oxides, the precision in the measurement of eg, especially for solid solutions, i.e., doped systems, and the current density at which potentials or overpotentials are taken, i.e., the Tafel slope somehow.
Therefore, although being a powerful descriptor tool, it is not faultless; it is not the only one, and should not be used alone, but for sure can guide and unify future research on specific directions, like the development of new syntheses, active materials with no/less CRMs, and their correct activity classification within the field. By accepting a descriptor as robust and reliable, and considering these observations, future research trends, with a solid basis, can be proposed to achieve fundamental progress in electrocatalysis as well as materials science.
3.4 From catalysts to cathodes: electrode design and performance evaluation for scale-up
Upscaling electrocatalysis findings is not straightforward because a cathode, at the full cell level, may have different properties (thickness, porosity, formulation, etc.), far from the catalyst powders used for the RDE. The cathode is a structured full-scale component, of which the bifunctional catalyst is a part, and it is composed of the following elements (with relative functions):• Conductive support/filler (e.g., carbon paper, metal mesh, and/or carbon powder): serving as the main structural part to convey/distribute charges, in the form of ions and/or electrons, from the electrolyte/external circuit. It is characterized by electrical conductivity, porosity, and wettability (i.e., hydrophobic/hydrophilic behaviour).
• Current collector (e.g., Ni wires): serving as the component to collect electrons from/to the cathode. It can be a physically separated piece, hence just in contact with the cathode, or integrated, welded directly to the supporting metal mesh, if any, thus effectively becoming the collector and support of the cathode.
• Gas diffusion layer (GDL): serving as a hydrophobic barrier, obtained by depositing/painting/mixing, for example, poly(tetrafluoroethylene) (PTFE) with the conductive support. It also has a binder function when used in the form of powder/solution and mixed with the catalyst. It can prevent liquid electrolyte leakage/evaporation and can facilitate oxygen transport from air to the catalyst, and vice versa.
• Catalyst layer: serving as the active material where the cathodic discharge/charge half reaction takes place. As in the air electrodes of other technologies (e.g., fuel cells151), around the particles of the catalyst, the critical three-phase boundary (TPB) zone is formed.
The formation of an extended and favourable TPB zone is the microscopic result of the electrode design, i.e., the integration of the different parts listed above, because all of them contribute to the contact with the electrolyte. At the engineering design level, some aspects to be carefully considered are:151
• Selection of materials (for the conductive support, current collector, GDL, catalysts).
• Formulation (e.g., catalyst loading, binder amount, and other additives).
• Wettability and porosity (e.g., to control the flooding of electrolytes, leakage, evaporation, oxygen crossover, etc.).
• Processing (final thickness, surface finishing, geometry, and homogeneity).
In cathode design, selection and formulation are related to the intrinsic electrochemical behaviour, while the other two determine the physical behaviour, especially the balance between the hydrophobicity and hydrophilicity of the surface in contact with the electrolyte, and oxygen diffusion vs. oxygen barrier of the surface exposed to air.152 In alkaline media, it is believed that the hydrophobic portion of the cathode, with catalytic sites exposed, is involved in the ORR, whereas the hydrophilic portion, with relevant catalytic sites, participates in the OER (Fig. 2).
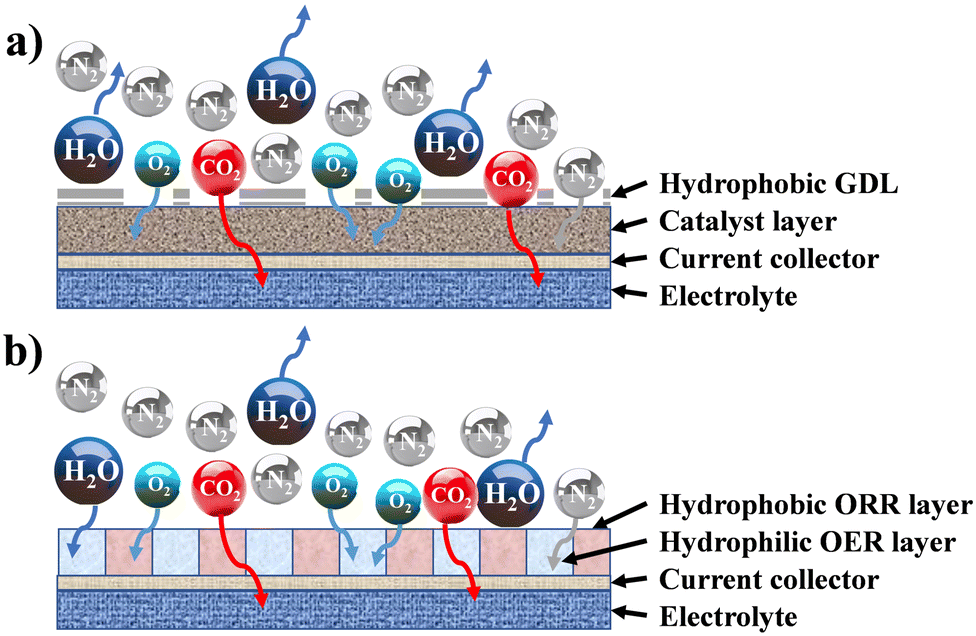 | ||
| Fig. 2 Architecture of a gas diffusion electrode: (a) standard structure (coupled ORR/OER activity); (b) decoupled ORR/OER hydrophobic/philic structure. | ||
This concept led to the separation (decoupling) of cathode functions, i.e., discharge/charge, together with the formation and control of separated hydrophobic and hydrophilic portions within the same cathode.152,153 The consequence is an asymmetric design in terms of material composition and wettability152,154 that is capable of extending the reactive interface from a 2D to a 3D multiphase interface.154,155 This is further complicated in ZABs with solid or gelled electrolytes because the physical surface in contact with the cathode is smaller, hence the porous structure, channel networks for mass transport and the number of available active sites for the reactions must be strictly controlled, together with flexibility and mechanical strength in the case of flexible ZABs.156 Another example is the interference of non-catalyst components on the OER during the charging process. It is true that, during the charging process, some current collectors and electrical contacts (e.g., stainless steel, Ni-based, etc.) are not inert due to the applied oxidizing potentials, possibly interfering with the OER process.157 Resistance to the corrosion and oxidation of the electrocatalyst, as well as of the conductive support or current collectors, is critical for stability and rechargeability. Apart from materials selection, i.e., carbon conductive additives, the addition of a protective layer, corrosion resistant current collectors, etc., the issue is mitigated, and electrocatalysts’ stability is improved, if the overpotential at charging is reduced (as measured from LSV by the RDE). Therefore, electrocatalyst stability should be tested alone, without interfering agents, and cathode corrosion resistance, during charging, could be checked considering different designs and formulations of the electrode. This is necessary for the correct estimation of stability in the long term, and to distinguish the intrinsic behaviour of the catalyst from the other components of the electrode. These are some of the challenges when scaling up catalysts to cathodes, creating discrepancies with the results from fundamental electrochemical characterization.
Considering a potentially interesting novel catalyst, with encouraging intrinsic activity determined by suitable descriptors, a poor electrode design, in cathodes bearing the novel catalyst, could invalidate the encouraging fundamental results. On the other hand, a poor performance evaluation of full cells with novel cathodes can lead to hyped claims, not reflecting device-relevant performance for lab-scale cells as well as scaled-up batteries. When considering performance evaluation, cells must be studied as a whole, and any information about their components (dimensions, weights, formulations, etc.) should be provided and mentioned to conduct a proper evaluation.158 All the components contribute to the overall performance of the cells, even if the only component varied in a study is the cathode. Testing relevant cycling conditions is also necessary to validate the real benefits and improvements due to the catalyst as well as the electrode and cell design. In the work of Parker et al.,2 this issue is addressed, and guiding equations are provided to formulate convincing and solid protocols for performance evaluation. Here, the most important equations are analysed, further elaborated, and critically commented. The main descriptor of performance for a ZAB is the depth of discharge (DoD) of zinc, the only active material stored in the battery (O2 is not stored inside the battery and is virtually “infinite”). The DoD is defined as the ratio between the capacity extracted from the battery and the capacity theoretically available from the active material, and is given by:2
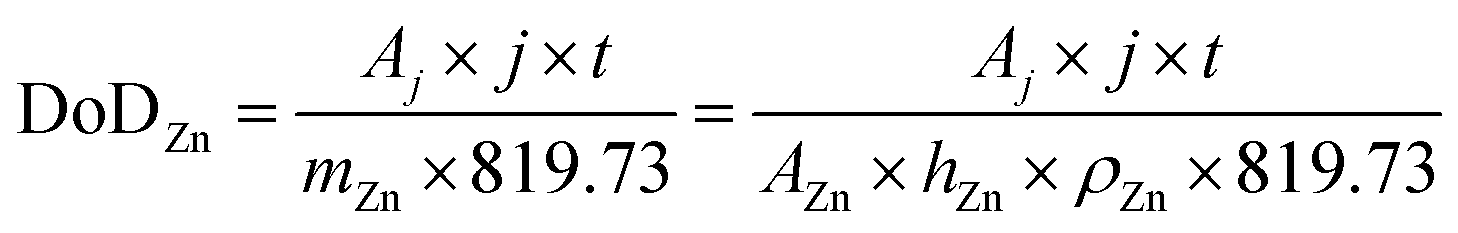 | (4) |
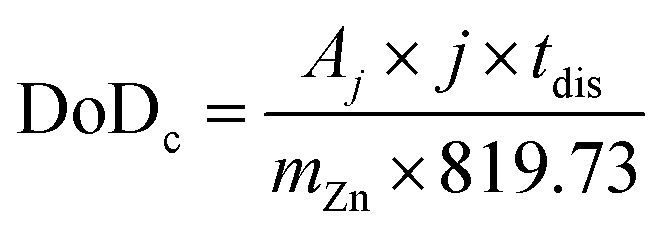 | (5) |
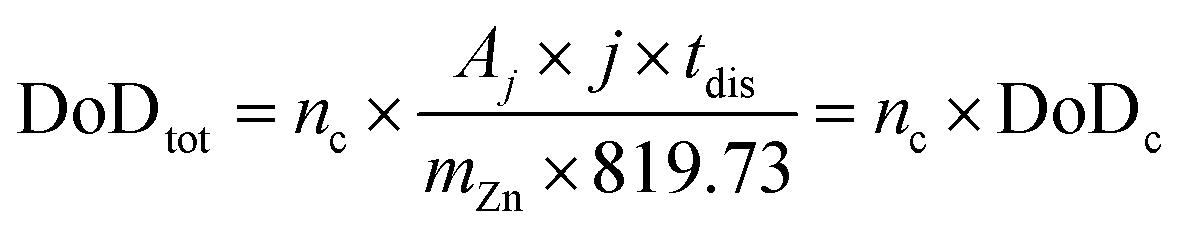 | (6) |
Another notable example of performance is given by the calculation of the total specific energy2 and specific energy per cycle, both in W h kg−1:
| Specific energyc = 819.73 × DoDc × fZn × Vavg | (7) |
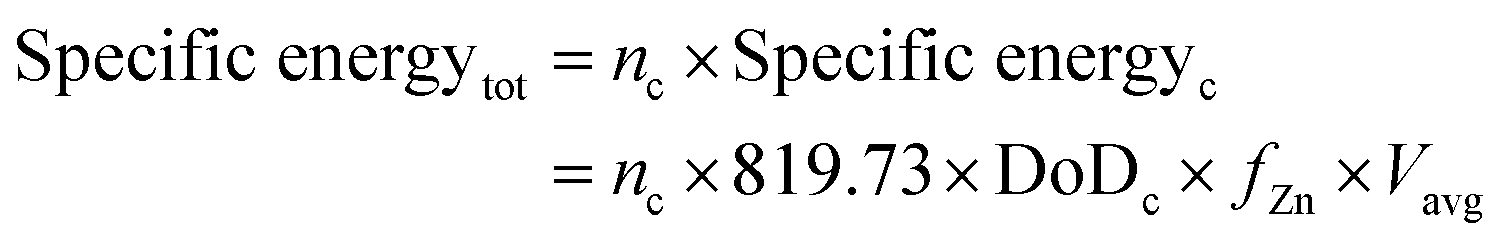 | (8) |
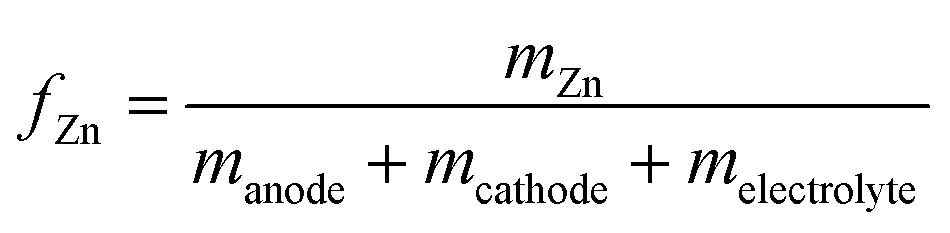 | (9) |
Although the equations presented here, and the others in the mentioned work, do not offer a relationship between the macroscopic and microscopic behaviours of each component, they can describe, at least, the effect of improved cell and cathode electrode designs. Viable directions are the mixing of two specialized catalysts, one for the ORR and one for the OER, with the lowest overpotentials, using a corrosion resistant additive and a structured GDL, capable of balancing hydrophobic and hydrophilic zones with the active sites, into a robust thin electrode to optimize weight and thickness for a compact design. This will increase fZn, Vavg, round-trip efficiency, and therefore energy density.
4. Cell integration and perspective performance for extended cyclability
In this section, experimental cyclability data for electrically rechargeable ZABs are shown and briefly discussed. The aim is to compare the behaviour under very short/long cycles (i.e., small/large areal capacity) to understand the current limitations. The anode is obtained by mixing commercial zinc powder (Zn > 99%, Cegasa, Spain) and a 45% w/w KOH solution (KOH pellets, >99%, Scharlab, Spain) to form a paste with 65% w/w of active zinc. The electrolyte is a semi solid GPE patent pending, containing KOH and a naturally occurring biopolymer. The bifunctional cathode is a mixture of a MnO2/carbon/PTFE primary catalyst (Cegasa, Spain), nickel cobalt oxide (NCO, NiCo2O4, >99%, Sigma-Aldrich, Germany), and ethanol in a 1:1:1 mass ratio to obtain a paste that is then pressed on a Ni mesh and dried at 60 °C overnight. A home-made design, consisting of a Teflon case with circular compartments for the components, was used. Anode paste fills a 13 mm in diameter compartment; upon it a GPE disk of 24 mm in diameter is placed, and then a 20 mm diameter MnO2/NCO on Ni mesh disk cathode (area 3.14 cm2) completes the stack. Ni-based current collectors/wires are placed as electrical contacts. The cell is closed by a Teflon cap with passing-through aeration holes, held in place by an O-ring and two bolts. The current density is normalized by the anode geometric area (1.33 cm2) and the specific capacity is normalized by the precise mass of active zinc used. The cells are tested in a constant current mode (CC) in a BCS 815 potentiostat (BioLogic, Seyssinet-Pariset, France), and cycles are defined as symmetric discharge/charge. The cutoffs are set at 0.8 and 2.0 V vs. Zn/Zn2+, for discharge and charge, respectively. Once the exact amount of active zinc is known, together with the weights of the electrolyte and the cathode, the information given above is sufficient to apply all the equations reported in subsection 3.4 to perform a consistent evaluation of the results.
4.1 Cyclability analysis and DoD for short cycles
A very commonly used cycling condition, especially for aqueous ZABs, is 10 min per cycle, i.e., 5 min discharge and 5 min charge, at a current density of 5 mA cm−2. This is equal to an areal capacity of 0.416 mA h cm−2. By assembling a cell as described above, and cycling it under these conditions, a voltage profile is shown in Fig. 3.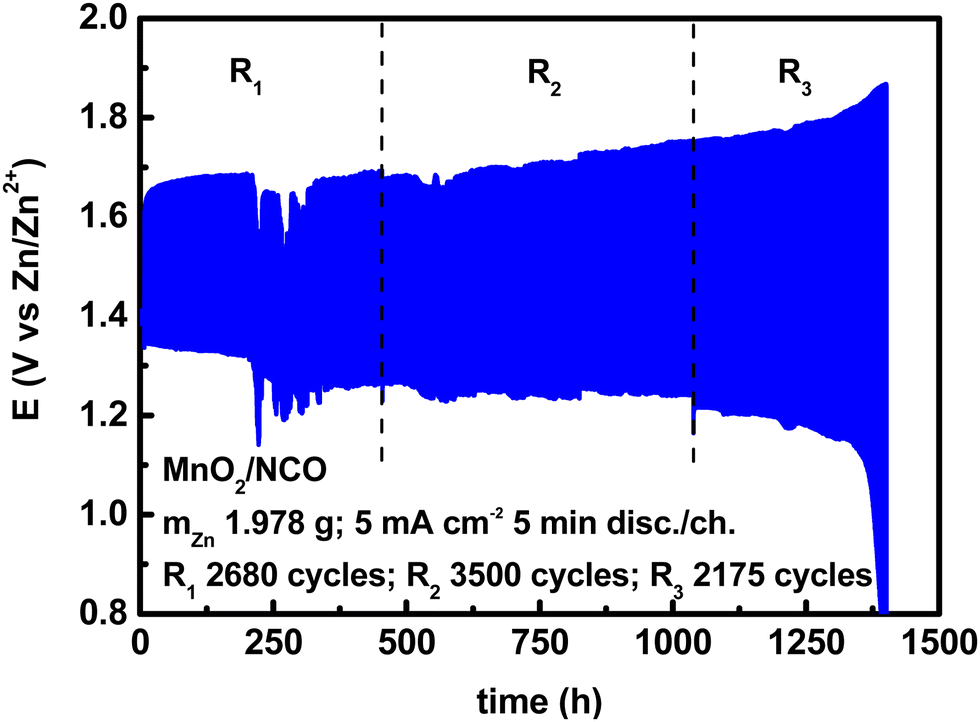 | ||
| Fig. 3 Electrically rechargeable ZABs (anode paste, GPEs from natural biopolymers, bifunctional cathodes) operated under “short cycles”. | ||
In Fig. 3, the voltage profile was divided into three regions to consider the change in the discharge voltage, due to a large number of cycles, and almost stable discharge/charge cycles. Between each region, there was a resting time in open circuit voltage mode overnight, and it was possible to relaunch the same cell twice. Under the same conditions (with no resting time), and using the same materials, duplicated cells gave an average number of cycles of 2500–3000 (see Fig. 5 in the next subsection). The cell above is used as an example to explain the cumulative effect of the number of cycles on metrics, the effect of resting time will be studied in future works. According to eqn (4)–(9), using a specific capacity of 819.73 mA h gZn−1, and the voltage profile in Fig. 3, the following figures can be calculated.
Although there are no formal definitions of short and long cycles, considering a threshold of ≈12 mA h cm−2 suggested by Parker et al.,2 and current densities suitable with ZABs, 0.416 mA h cm−2 is a very small value due to the small discharge/charge time, hence is referred to as a (very) short cycle. The DoD per cycle is therefore very small, but it depends also on the active zinc mass. With a small areal capacity, high DoD can be achieved by using less zinc, but anyway this discharge time has no practical application at the device level. Both parameters should be taken into consideration when setting cycling conditions. For energy, by using the weights, DoDc, and nc in Table 4, the specific capacity (819.73 mA h gZn−1), and the voltage profiles in Fig. 3, the figures in Table 5 are calculated.
| Region | m anode (g) | m Zn (g) | m electrolyte (g) | m cathode (g) | n c (−) | DoDc (%) | DoDtot (%) |
|---|---|---|---|---|---|---|---|
| R1 | 2.931 | 1.978 | 1.626 | 0.495 | 2680 | 0.034 | 91.120 |
| R2 | 2.931 | 1.978 | 1.626 | 0.495 | 3500 | 0.034 | 119.000 |
| R3 | 2.931 | 1.978 | 1.626 | 0.495 | 2175 | 0.034 | 73.950 |
| Overall | 2.931 | 1.978 | 1.626 | 0.495 | 8355 | 0.034 | 284.070 |
| Region | V avg (V) | f Zn (−) | Specific energyc (W h kg−1) | Specific energytot (W h kg−1) |
|---|---|---|---|---|
| R1 | 1.300 | 0.392 | 0.142 | 380.560 |
| R2 | 1.250 | 0.392 | 0.137 | 479.500 |
| R3 | 1.200 | 0.392 | 0.131 | 284.925 |
| Overall | 1.250 | 0.392 | 0.137 | 1144.985 |
The cumulated figures are really promising, showing interesting values for the total DoD, nc, and specific energytot. However, this is an effect of the number of cycles, because the figures per cycle, DoDc and specific energyc, are low. The DoD per cycle can be improved by optimizing/minimizing the mass of zinc, but to a limited extent. In Table 4 a large mass of zinc paste is used, whereas thin zinc foils are widely used in the literature. Considering a zinc density of 7.13 g cm−3, Acathode = Aanode = 1 cm2, and the same areal capacity of 0.416 mA h cm−2, prospective DoDc (eqn (5)) by changing foil thickness is given in Table 6.
| Foil thickness (cm) | Area (cm2) | Volume (cm3) | Mass (g) | Theoretical capacity (mA h) | Areal capacity (mA h cm−2) | DoDc (%) |
|---|---|---|---|---|---|---|
| 0.030 (300 μm) | 1 | 0.030 | 0.214 | 175.422 | 0.416 | 0.237 |
| 0.020 (200 μm) | 1 | 0.020 | 0.143 | 117.221 | 0.416 | 0.355 |
| 0.010 (100 μm) | 1 | 0.010 | 0.071 | 58.200 | 0.416 | 0.715 |
| 0.001 (10 μm) | 1 | 0.001 | 0.007 | 5.738 | 0.416 | 7.250 |
As expected, by reducing the foil thickness, for constant areal capacity and cycling conditions, zinc mass decreases, the available theoretical capacity decreases, and DoDc increases. However, for full cells, the real cycling feasibility of very thin zinc foil anodes must be proven. The last example of a 10 μm thick zinc foil, giving 7.25% of DoD each cycle of 5 min discharge per 5 min charge at 5 mA cm−2 is just a calculation, showing that even by reducing thickness, an interesting DoDc is not met anyway. The only other way to increase the DoDc is to increase the areal capacity or at least to increase the discharging/charging time. Something similar was also claimed by Parker et al.,2 together with the doubt that these “short cycles” are more useful as fundamental electrodeposition study rather than device-relevant testing. In this regard it must be also added that in liquid electrolyte secondary ZAB tests in the literature, the KOH-based electrolyte also contains zinc acetate or zinc chloride, very frequently at 0.2 M. This means that, other than zinc foil, Zn is also present as a cation or as Zn(OH)42− derived from the action of KOH, at an equivalent zinc concentration of approx. 13 mg mL−1 in the liquid, giving an equivalent extra capacity of 10.6 mA h mL−1. In the GPE used in the cell shown in Fig. 3 there is no extra zinc. Depending on the amount of zinc, foil thickness, and the volume of liquid electrolyte, a non-negligible source of extra capacity (approx. 6–18% per mL, with Zn foil of 0.1–0.3 mm thickness and active area of 1 cm2) coming from the liquid could participate in the Zn deposition during charge, especially if the areal capacity, discharging/charging times, and DoDc are small (hence, the zinc foil could be under-used or misused). Therefore, it is extremely important to clearly report the geometry/area of electrodes, which one is used to calculate current density, and the total weight of the anode and active zinc, at least.
4.2 Long cycles at higher DoD for battery-relevant performance
When the areal capacity is increased at cycling, the performance starts fading, and the figures from cyclability and energy analyses are less encouraging. In this work some larger cycles, at higher areal capacities, have been tested using the same materials and assembly. Voltage profiles for three different areal capacities are given in Fig. 4.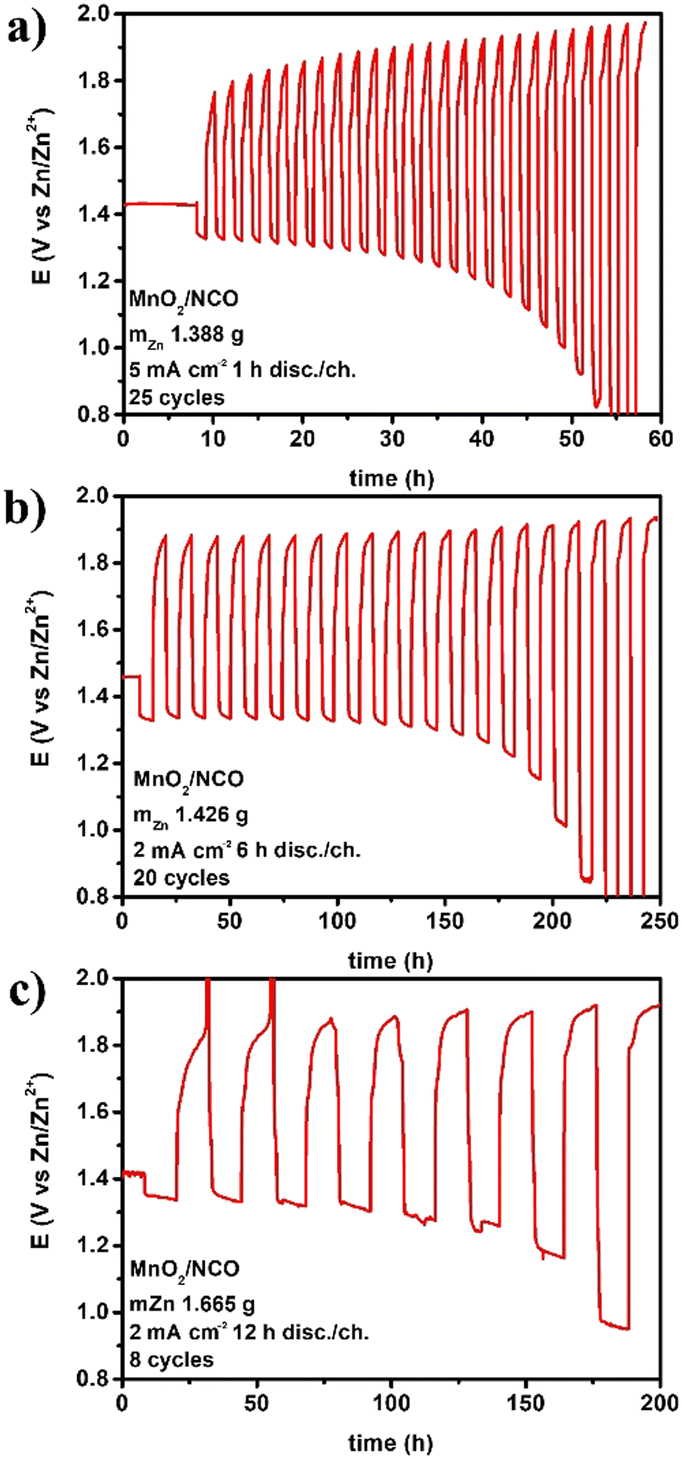 | ||
| Fig. 4 Electrically rechargeable ZABs (anode paste, GPEs from natural biopolymers, bifunctional cathodes) operated under “long cycles” at (a) 5 mA h cm−2, (b) 12 mA h cm−2, and (c) 24 mA h cm−2. | ||
As expected, by increasing the areal capacity, especially discharge/charge time, the number of cycles drastically decreases from thousands to a few cycles. On the other hand, DoDc and specific energyc increase, and the discharge times are compatible with portable or stationary applications (i.e., hours). Cyclability and energy calculation for the three examples in Fig. 4 are reported in Table 7.
| Areal capacity (mA h cm−2) | m anode (g) | m Zn (g) | m electrolyte (g) | m cathode (g) | n c (−) | DoDc (%) | DoDtot (%) | V avg (V) | f Zn (−) | Specific energyc (W h kg−1) | Specific energytot (W h kg−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 2.056 | 1.388 | 1.889 | 0.457 | 25 | 0.585 | 14.625 | 1.200 | 0.315 | 1.813 | 45.325 |
| 12 | 2.112 | 1.426 | 1.507 | 0.487 | 20 | 1.366 | 27.320 | 1.250 | 0.347 | 4.857 | 97.140 |
| 24 | 2.467 | 1.665 | 1.646 | 0.563 | 8 | 2.338 | 18.704 | 1.260 | 0.356 | 8.597 | 68.776 |
The cumulated figures in these cases are lower than those in Tables 4 and 5 mainly due to the small number of cycles, while the amount of active zinc is reduced, areal capacity is increased, and hence DoDc and specific energyc increased substantially. These conditions are more attractive for practical applications at the device level; the challenge is thus to increase under these cycling conditions the number of cycles, the total DoD, and hence the total specific energy.
Another aspect emerging from these data, to be highly considered, is that the electrical rechargeability of a lab-scale device can be improved by optimizing the cell design. In fact, in all the examples, the materials used are the same but components' weights change (affecting directly fZn, DoDc, and indirectly the others), and the electrode area of the home-made cell used are different (anode 1.33 cm2vs. cathode 3.14 cm2) not directly affecting any calculation (the area used here for current density is always the anodic one), but operations (i.e., real current density experienced by the anode during discharge and by the cathode during charge may differ). Materials science (for anodes, electrolytes, and bifunctional catalysts), engineering design (for each component, single cell geometry, and assembly), and electrochemistry (for cycling conditions, operating strategy during discharge/charge) are highly connected, all affecting the figures reported. Other areal capacities have been explored in this work, and the trend ncvs. areal capacity is shown in Fig. 5.
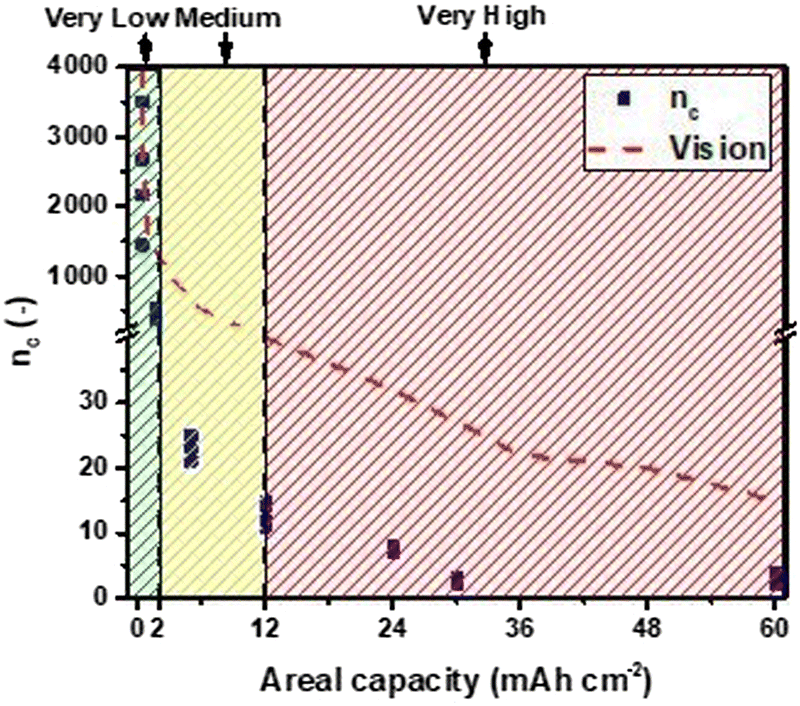 | ||
| Fig. 5 Number of cycles, nc, vs. areal capacity trend with prospective development for electrically rechargeable ZABs. | ||
Future development of electrically rechargeable ZABs should aim therefore to improve and assess enhancement in materials, design, and testing of devices considering all these parameters to achieve more cycles at higher areal capacities (hence DoDtot > 100%, meaning real recharge).
According to the literature and the threshold recommended,2 three areal capacity regions are suggested here: very low (from 0 to 2 mA h cm−2), medium (from 2 to 12 mA h cm−2) and very high (>12 mA h cm−2) regions, based on areal capacity, and supposing that current densities are in the range of 1–5 mA cm−2 and the discharge/charge time is in the range of a few hours. This is because high current densities, especially at charge, are associated with various failure mechanisms,1 as well as long cycling time could lead to dendrite formation, if not suppressed.159 The DoD per cycle is also connected to this map but, apart from areal capacity, it depends on the structure, geometry and active zinc mass in the single cell,159 shaping the voltage profile when it is the limiting electrode in the device.107,160
5. Current and potential applications for primary/secondary ZABs
Following the discussion in previous sections about the research and development of ZABs, it is critical to reflect on the progress made in fundamental studies into field applications and relevant environments. Considering the technology readiness level (TRL) scale, this means performing the “hard jump” from TRL 1–3 to TRL 4–6 and beyond. For electrically rechargeable ZABs this is actually the most limiting aspect for prospective development toward commercialization, delayed also by the unreconciled validations of integrated cells. The existing applications for primary and secondary ZABs are overviewed below, together with the possible expansion of the field if the challenges discussed for GPEs and bifunctional cathodes are resolved.5.1 Primary ZABs in portable applications
Today, primary ZABs are a commercial reality, with a TRL 9, but are limited to their initial market niche of power sources for hearing aid devices. The field of portable applications, including secondary cells, is today an unconquerable domain of LIBs and other intercalation chemistries.161 The small market share, compared to other technologies, is also limited due to the small number of manufacturers (Varta, Rayovac, Sony, etc.)162 offering the same type of standard product. For other portable applications, low power uses, and remote-but-yet-portable installations, commercial primary ZAB stacks, with higher nominal capacity, are available from very few manufacturers. For example, Cegasa company (Spain) offers primary ZAB stacks with high nominal capacity (from 100 to 4800 A h packs), used for railway signalling, electric fencing, portable LED lights, mobile warning systems, mobile LED devices, and other remote applications,163 showing innovative technology applied to many fields outside of the hearing aids. However, the use in larger applications, requiring more power, is limited by the low-rate capability, short lifetime, and leakage of liquid electrolytes of ZABs.Although focused on electrically rechargeable ZABs, primary batteries are considered here because they can also benefit from the potential progress in GPEs and cathodes, as described in previous sections. For example, natural biopolymer-based GPEs could improve zinc utilization and capacity extraction, replace the separator, and shift from liquid to semi-solid electrolyte to increase durability, apart from being eco-friendly, biodegradable and/or recyclable; the advanced cathode, bifunctional or not, could increase the rate capability, i.e., the maximum current density allowable, reducing the thickness and amount of catalyst required, hence increasing energy and power density.
Nowadays, an interesting and increasing academic field is that of flexible ZABs,6,164,165 characterized by a multidisciplinary approach where miniaturization, electrochemistry, engineering design, physics, and materials science need to cooperate. Flexible ZABs, primary the most, but also electrically rechargeable, using gelled electrolytes and advanced zinc anodes (powder, paste, and 3D-printed structured foils) could break the market of flexible/wearable electronics or the disposable/wearable non-invasive devices for sensing/detection/biomedical applications.166–169 Another reason for extending the application of advanced primary ZABs in portable power sources is about future restrictions, environmental concerns, and supply issues of raw materials for LIBs, which could turn the table in favour of aqueous chemistries with no critical raw materials in a post-lithium battery era. Virtually, apart from current catalysts for the OER and carbon-derived materials (coking coal and natural graphite are listed as CRMs), primary ZABs are already CRM-free.
5.2 Secondary ZABs in stationary applications
Available secondary ZABs are nowadays based on mechanical recharge or flowing anode/electrolyte concepts (TRL 6).161 They present high recharging, recycling, operational and maintenance costs, compared to other technologies for stationary applications, nevertheless they increase the environmental impact of a technology that is virtually eco-friendly by principle. Early developments of high TRL secondary ZABs were historically dedicated to electric vehicles and small stationary applications.161 Unfortunately, the history of companies in this field (e.g., ReVolt, NantEnergy, and Zinium) includes uncertain success, unclear performances in the field, enthusiastic hyped claims, true liquidations and running outs of the business. A notable, still-running experience is Zinc8170 among all, proposing a flowing-anode regenerative solution capable of storing energy from 8 to 24 h. The main bottleneck of such configurations is that flowing needs energy, and solid slurries tend to dry and harden quickly, limiting the storage duration, and hence cycling.Nowadays, electrically rechargeable ZABs are believed to be at TRL 3–4, with a lower rate capability than primary ZABs, and the need for additional components for air management (filters). The niche of small and flexible secondary ZABs for portable devices appears to be more advanced than stationary applications. There is voluminous literature on the topic;164–167,169 however, higher TRL values seem hard to be achieved in real applications, and reliable commercial products, at any scale, size, or price, are missing, to the best of authors’ knowledge.
Electrically rechargeable ZABs would be profitable and desirable in portable as well as stationary, small as well as large (in terms of capacity) applications. Recently, the focus has been on stationary applications because of the low volumetric energy density of zinc.161 Prospective uses of this technology include renewable energy storage (i.e., solar and wind) in large storage elements for grid support and peak-demand attenuation, small storage elements for residential/single house applications, or hybrid systems together with LIBs to increase the rate capability and response time.161 Considering recent and future electrocatalyst development for the OER, the advantages of these concepts are clear: they would facilitate the storage of intermittent renewable sources for a short (hours to a few days) to the mid (weeks) term in a cheap, maintenance free, non-CRM-based (or at least with a very small incidence of CRMs) device. To accomplish the goal, electrically rechargeable ZABs would need long cycles (from 6 to 24 h for a discharge/charge cycle), and hence high areal capacities, and higher cycle numbers to approach high total DoD (see Section 4). For stationary applications, more fundamental and solid research, with reliable results, to consolidate a TRL 4, is required to prospectively develop this electrochemical storage direction.
6. Conclusions and final remarks
Electrically rechargeable ZABs are at the border between lab-scale infancy and technological maturity; they need concrete device-level assessment to jump to higher TRLs, getting reliable confidence and a solid basis for sustainable mass production, field application, and market penetration. This would be possible only if extended cyclability and rate capability are unlocked. Overall development of materials, components, and cell design is needed because all of them equally participate in setting the device performance. In this perspective, GPEs, bifunctional catalysts/cathodes, and device testing protocols have been discussed. Developing bio-based GPEs, using naturally occurring biopolymers, is ongoing and highly attractive to push the eco-friendliness and sustainable character of ZABs. There is a wide choice of biopolymers and versatile approaches to enhance the physical, chemical, and mechanical properties of hydrogels. Moreover, catalysts with no/less CRMs to design bifunctional cathodes must be further developed and accurately characterized, as well as lab-scale full cells by adopting the correct protocols. The most important aspect is to report suitable metrics and quantitative analysis to assess performances and concrete breakthroughs. Characterization techniques and tools for these purposes are already available and should be systematically applied in future studies. This is fundamental to win the market of portable as well as stationary electrochemical energy storage devices to prevent future crisis due to key materials scarcity, supply chain problems, or environmental issues. The dominance of LIBs hides an unsaid risk of not having a valid alternative technology for energy storage to respond to global concerns about decarbonization, energy cost, and the large use of renewables. ZABs have the theoretical potential to be an alternative to LIBs, reducing the environmental impact, costs, and dependence on non-abundant non-globally distributed sources. A bright future is predicted for this technology if the current gaps and bottlenecks can be solved.Author contributions
Conceptualization: N. O. V., D. F.; data curation: D. F.; formal analysis: D. F., E. G. G., N. O. V.; investigation: D. F., A. B. M., E. G. G.; funding acquisition: N. O. V.; methodology: N. O. V., D. F.; visualization: D. F., N. O. V.; supervision: N. O. V., M. A.; writing – original draft preparation: all authors; writing – review and editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the R&D&I project PID2020-117626RA-I00 funded by MCIN/AEI/10.13039/501100011033. E. García Gaitán and N. Ortiz-Vitoriano thank the Basque Government for the Beca Bikaintek 01-AF-W2-2019-00003 and Ramon y Cajal grant (RYC-2020-030104-I) funded by MCIN/AEI/10.13039/501100011033 and by FSE invest in your future, respectively.References
- Z. Song, J. Ding, B. Liu, Y. Shen, J. Liu, X. Han, Y. Deng, C. Zhong and W. Hu, Chem. Eng. J., 2022, 429, 132331 CrossRef CAS.
- J. F. Parker, J. S. Ko, D. R. Rolison and J. W. Long, Joule, 2018, 2, 2519–2527 CrossRef CAS.
- N. Radenahmad, R. Khezri, A. A. Mohamad, M. T. Nguyen, T. Yonezawa, A. Somwangthanaroj and S. Kheawhom, J. Alloys Compd., 2021, 883, 160935 CrossRef CAS.
- J. Spector and M. V. Olano, https://www.canarymedia.com/articles/batteries/chart-lithium-prices-are-through-the-roof-this-year.
- https://markets.businessinsider.com/commodities/zinc-price .
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef.
- N. Borchers, S. Clark, B. Horstmann, K. Jayasayee, M. Juel and P. Stevens, J. Power Sources, 2021, 484, 229309 CrossRef CAS.
- H. Osgood, S. v Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- V. Neburchilov, H. Wang, J. J. Martin and W. Qu, J. Power Sources, 2010, 195, 1271–1291 CrossRef CAS.
- D. Yang, D. Chen, Y. Jiang, E. H. Ang, Y. Feng, X. Rui and Y. Yu, Carbon Energy, 2021, 3, 50–65 CrossRef CAS.
- X. G. Zhang, J. Power Sources, 2006, 163, 591–597 CrossRef CAS.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 RSC.
- D. Stock, S. Dongmo, J. Janek and D. Schröder, ACS Energy Lett., 2019, 4, 1287–1300 CrossRef CAS.
- J. Zheng and L. A. Archer, Sci. Adv., 2021, 7(2), eabe0219 CrossRef CAS.
- S. Lorca, F. Santos and A. J. Fernández Romero, Polymers, 2020, 12, 2812 CrossRef CAS.
- E. Lizundia and D. Kundu, Adv. Funct. Mater., 2021, 31, 2005646 CrossRef CAS.
- F. Mahlendorf, A. Heinzel, C. Mueller and D. Fuchs, Electrochemical Power Sources: Fundamentals, Systems, and Applications, Elsevier, 2021, pp. 99–123 Search PubMed.
- L. Jörissen, Electrochemical Power Sources: Fundamentals, Systems, and Applications, Elsevier, 2021, pp. 81–97 Search PubMed.
- M. Krebs, H.-U. Reichardt, R. Sojka and P. Stevens, Electrochemical Power Sources: Fundamentals, Systems, and Applications, Elsevier, 2021, pp. 217–245 Search PubMed.
- Y. He, W. Shang, M. Ni, Y. Huang, H. Zhao and P. Tan, Chem. Eng. J., 2022, 427, 130862 CrossRef CAS.
- S. Velraj, A. K. Estes, B. L. Bates and J. H. Zhu, Electrochim. Acta, 2018, 292, 446–457 CrossRef CAS.
- S. Möller, S. Barwe, S. Dieckhöfer, J. Masa, C. Andronescu and W. Schuhmann, ChemElectroChem, 2020, 7, 2680–2686 CrossRef.
- Y. J. Min, S. J. Oh, M. S. Kim, J. H. Choi and S. Eom, J. Appl. Electrochem., 2018, 48, 405–413 CrossRef CAS.
- S. S. Shinde, C. H. Lee, J.-Y. Jung, N. K. Wagh, S.-H. Kim, D.-H. Kim, C. Lin, S. U. Lee and J.-H. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC.
- A. B. Balaji, H. Pakalapati, M. Khalid, R. Walvekar and H. Siddiqui, Biodegradable and Biocompatible Polymer Composites, Elsevier, 2018, pp. 3–32 Search PubMed.
- R. Rebelo, M. Fernandes and R. Fangueiro, Procedia Eng., 2017, 200, 236–243 CrossRef CAS.
- Q. Wu, Y. Wang and G.-Q. Chen, Artif. Cells, Blood Substitutes, Biotechnol., 2009, 37, 1–12 CrossRef CAS PubMed.
- J. Pandey, Biopolymers and Their Application in Wastewater Treatment, in Emerging Eco-friendly Green Technologies for Wastewater Treatment, ed. R. Bharagava, 2020, pp. 245–266 Search PubMed.
- G. Robertson, Environmentally Compatible Food Packaging, Elsevier, 2008, pp. 3–28 Search PubMed.
- J. N. de Freitas, J. E. Benedetti, F. S. Freitas, A. F. Nogueira and M. A. de Paoli, Polymer Electrolytes, Elsevier, 2010, pp. 381–430 Search PubMed.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS.
- K. H. Hong, Polym. Bull., 2017, 74, 2861–2872 CrossRef CAS.
- C. Wang, T. Yokota and T. Someya, Chem. Rev., 2021, 121, 2109–2146 CrossRef CAS PubMed.
- K. Varaprasad, G. M. Raghavendra, T. Jayaramudu, M. M. Yallapu and R. Sadiku, Mater. Sci. Eng., C, 2017, 79, 958–971 CrossRef CAS.
- T. N. T. Tran, D. Aasen, D. Zhalmuratova, M. Labbe, H. Chung and D. G. Ivey, Batteries Supercaps, 2020, 3, 917–927 CrossRef CAS.
- B. Guo and P. X. Ma, Sci. China: Chem., 2014, 57, 490–500 CrossRef CAS PubMed.
- J. Kundu, F. Pati, Y. Hun Jeong and D.-W. Cho, Biofabrication, Elsevier, 2013, pp. 23–46 Search PubMed.
- X. Fu and W. Zhong, Adv. Energy Mater., 2019, 9, 1901774 CrossRef CAS.
- C.-C. Yang, J. M. Yang and C.-Y. Wu, J. Power Sources, 2009, 191, 669–677 CrossRef CAS.
- O. Kwon, H. J. Hwang, Y. Ji, O. S. Jeon, J. P. Kim, C. Lee and Y. G. Shul, Sci. Rep., 2019, 9, 3175 CrossRef.
- Y. Zhang, C. Li, X. Cai, J. Yao, M. Li, X. Zhang and Q. Liu, Electrochim. Acta, 2016, 220, 635–642 CrossRef CAS.
- O. E. Philippova and A. R. Khokhlov, Polymer Science: A Comprehensive Reference, Elsevier, 2012, pp. 339–366 Search PubMed.
- Y. Wei, M. Wang, N. Xu, L. Peng, J. Mao, Q. Gong and J. Qiao, ACS Appl. Mater. Interfaces, 2018, 10, 29593–29598 CrossRef CAS.
- J. Zhang, J. Fu, X. Song, G. Jiang, H. Zarrin, P. Xu, K. Li, A. Yu and Z. Chen, Adv. Energy Mater., 2016, 6, 1600476 CrossRef.
- F. Ullah, M. B. H. Othman, F. Javed, Z. Ahmad and H. Md Akil, Mater. Sci. Eng., C, 2015, 57, 414–433 CrossRef CAS.
- G. M. Wu, S. J. Lin and C. C. Yang, J. Membr. Sci., 2006, 280, 802–808 CrossRef CAS.
- M. Wang, N. Xu, J. Fu, Y. Liu and J. Qiao, J. Mater. Chem. A, 2019, 7, 11257–11264 RSC.
- J. Fu, D. U. Lee, F. M. Hassan, L. Yang, Z. Bai, M. G. Park and Z. Chen, Adv. Mater., 2015, 27, 5617–5622 CrossRef CAS.
- N. Zhao, F. Wu, Y. Xing, W. Qu, N. Chen, Y. Shang, M. Yan, Y. Li, L. Li and R. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 15537–15542 CrossRef CAS.
- I. Chiulan, E. B. Heggset, Ş. I. Voicu and G. Chinga-Carrasco, Biomacromolecules, 2021, 22, 1795–1814 CrossRef CAS PubMed.
- S. Bastani and M. Mohseni, Handbook of Nanoceramic and Nanocomposite Coatings and Materials, Elsevier, 2015, pp. 155–182 Search PubMed.
- M. T. Tsehaye, F. Alloin, C. Iojoiu, R. A. Tufa, D. Aili, P. Fischer and S. Velizarov, J. Power Sources, 2020, 475, 228689 CrossRef CAS.
- E. L. Dewi, K. Oyaizu, H. Nishide and E. Tsuchida, J. Power Sources, 2003, 115, 149–152 CrossRef CAS.
- J. Fu, J. Zhang, X. Song, H. Zarrin, X. Tian, J. Qiao, L. Rasen, K. Li and Z. Chen, Energy Environ. Sci., 2016, 9, 663–670 RSC.
- Biopolymer Membranes and Films, ed. M. Agostini de Moraes, C. Ferreira da Silva and R. Silveira Vieira, Elsevier, 1st edn, 2020 Search PubMed.
- X. Zhu, K. Wang, Y. Xu, G. Zhang, S. Li, C. Li, X. Zhang, X. Sun, X. Ge and Y. Ma, Energy Storage Mater., 2021, 36, 291–308 CrossRef.
- E. Raphael, C. O. Avellaneda, M. A. Aegerter, M. M. Silva and A. Pawlicka, Mol. Cryst. Liq. Cryst., 2012, 554, 264–272 CrossRef CAS.
- K. Lu, T. Jiang, H. Hu and M. Wu, Front. Chem., 2020, 8, 546728 CrossRef CAS.
- M. J. Tan, B. Li, P. Chee, X. Ge, Z. Liu, Y. Zong and X. J. Loh, J. Power Sources, 2018, 400, 566–571 CrossRef CAS.
- A. Poosapati, E. Jang, D. Madan, N. Jang, L. Hu and Y. Lan, MRS Commun., 2019, 9, 122–128 CrossRef CAS.
- Q. Wang, H. Miao, S. Sun, Y. Xue and Z. Liu, Chem. – Eur. J., 2018, 24, 14816–14823 CrossRef CAS PubMed.
- Y. Zuo, K. Wang, M. Wei, S. Zhao, P. Zhang and P. Pei, Cell Rep. Phys. Sci., 2022, 3, 100687 CrossRef CAS.
- J. Zhang, T. Zhou, J. Qiao, Y. Liu and J. Zhang, Electrochim. Acta, 2013, 111, 351–358 CrossRef CAS.
- J. Qiao, J. Zhang and J. Zhang, J. Power Sources, 2013, 237, 1–4 CrossRef CAS.
- C.-C. Yang, S.-J. Lin and S.-T. Hsu, J. Power Sources, 2003, 122, 210–218 CrossRef CAS.
- M. N. Masri, M. F. M. Nazeri and A. A. Mohamad, Sago Gel Polymer Electrolyte for Zinc-Air Battery, 5th FORUM ON NEW MATERIALS PART A, 2010, vol. 72, pp. 305–308, DOI:10.4028/www.scientific.net/AST.72.305.
- J. Lu, P. Jaumaux, T. Wang, C. Wang and G. Wang, J. Mater. Chem. A, 2021, 9, 24175–24194 RSC.
- J. Hur, K. Im, S. W. Kim, J. Kim, D.-Y. Chung, T.-H. Kim, K. H. Jo, J. H. Hahn, Z. Bao, S. Hwang and N. Park, ACS Nano, 2014, 8, 10066–10076 CrossRef CAS.
- S. Y. Liew, J. C. Juan, C. W. Lai, G.-T. Pan, T. C.-K. Yang and T. K. Lee, Ionics, 2019, 25, 1291–1301 CrossRef CAS.
- U. Bhardwaj, A. Sharma, A. Mathur, A. Halder and H. S. Kushwaha, Electrochem. Sci. Adv., 2022, 2(3), e202100056 Search PubMed.
- J. Park, M. Park, G. Nam, J. Lee and J. Cho, Adv. Mater., 2015, 27, 1396–1401 CrossRef CAS.
- Z. Song, J. Ding, B. Liu, X. Liu, X. Han, Y. Deng, W. Hu and C. Zhong, Adv. Mater., 2020, 32, 1908127 CrossRef CAS PubMed.
- J. Enrione, C. Char, M. Pepczynska, C. Padilla, A. González-Muñoz, Y. Olguín, C. Quinzio, L. Iturriaga and P. Díaz-Calderón, Polymers, 2020, 12, 1587 CrossRef CAS.
- F. A. Johnston-Banks, Food Gels, Springer Netherlands, Dordrecht, 1990, pp. 233–289 Search PubMed.
- R. A. S. N. Ranasinghe, W. L. I. Wijesekara, P. R. D. Perera, S. A. Senanayake, M. M. Pathmalal and R. A. U. J. Marapana, Food Rev. Int., 2022, 38, 812–855 CrossRef CAS.
- F. Colò, F. Bella, J. R. Nair, M. Destro and C. Gerbaldi, Electrochim. Acta, 2015, 174, 185–190 CrossRef.
- Y. Zhang, Y. Chen, X. Li, M. Alfred, D. Li, F. Huang and Q. Wei, J. Power Sources, 2021, 482, 228963 CrossRef CAS.
- J. Fu, F. M. Hassan, J. Li, D. U. Lee, A. R. Ghannoum, G. Lui, Md. A. Hoque and Z. Chen, Adv. Mater., 2016, 28, 6421–6428 CrossRef CAS PubMed.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- D. Lee, H.-W. Kim, J.-M. Kim, K.-H. Kim and S.-Y. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 22210–22217 CrossRef CAS.
- S. Peng, X. Han, L. Li, S. Chou, D. Ji, H. Huang, Y. Du, J. Liu and S. Ramakrishna, Adv. Energy Mater., 2018, 8, 1800612 CrossRef.
- T. N. T. Tran, H.-J. Chung and D. G. Ivey, Electrochim. Acta, 2019, 327, 135021 CrossRef CAS.
- S. S. Shinde, C. H. Lee, J.-Y. Yu, D.-H. Kim, S. U. Lee and J.-H. Lee, ACS Nano, 2018, 12, 596–608 CrossRef CAS.
- A. R. M. Zahid, M. N. Masri, M. H. Hussin and M. B. A. Bakar, AIP Conf. Proc., 2018, 2030, 020278 CrossRef.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127 RSC.
- S. Hosseini, S. Masoudi Soltani and Y.-Y. Li, Chem. Eng. J., 2021, 408, 127241 CrossRef CAS.
- P. Zhang, K. Wang, P. Pei, Y. Zuo, M. Wei, X. Liu, Y. Xiao and J. Xiong, Mater. Today Chem., 2021, 21, 100538 CrossRef CAS.
- Y. Zhou, J. Pan, X. Ou, Q. Liu, Y. Hu, W. Li, R. Wu, J. Wen and F. Yan, Adv. Energy Mater., 2021, 11, 2102047 CrossRef CAS.
- Z. Pei, L. Ding, C. Wang, Q. Meng, Z. Yuan, Z. Zhou, S. Zhao and Y. Chen, Energy Environ. Sci., 2021, 14, 4926–4935 RSC.
- Z. Pei, Z. Yuan, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 4793–4799 CrossRef CAS.
- X. Liu, X. Fan, B. Liu, J. Ding, Y. Deng, X. Han, C. Zhong and W. Hu, Adv. Mater., 2021, 33, 2006461 CrossRef CAS.
- J. Pang, R. G. Mendes, A. Bachmatiuk, L. Zhao, H. Q. Ta, T. Gemming, H. Liu, Z. Liu and M. H. Rummeli, Chem. Soc. Rev., 2019, 48, 72–133 RSC.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- E. Antolini, Renewable Sustainable Energy Rev., 2016, 58, 34–51 CrossRef CAS.
- D. Liu, Y. Tong, X. Yan, J. Liang and S. X. Dou, Batteries Supercaps, 2019, 2, 743–765 CrossRef.
- Y. Dai, J. Yu, C. Cheng, P. Tan and M. Ni, Chem. Eng. J., 2020, 397, 125516 CrossRef CAS.
- R. Cao, J.-S. Lee, M. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816–829 CrossRef CAS.
- Z. Chen, J.-Y. Choi, H. Wang, H. Li and Z. Chen, J. Power Sources, 2011, 196, 3673–3677 CrossRef CAS.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- Z. Chen, A. Yu, R. Ahmed, H. Wang, H. Li and Z. Chen, Electrochim. Acta, 2012, 69, 295–300 CrossRef CAS.
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef.
- G. Du, X. Liu, Y. Zong, T. S. A. Hor, A. Yu and Z. Liu, Nanoscale, 2013, 5, 4657 RSC.
- X. Ge, Y. Liu, F. W. T. Goh, T. S. A. Hor, Y. Zong, P. Xiao, Z. Zhang, S. H. Lim, B. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12684–12691 CrossRef CAS.
- M. Prabu, P. Ramakrishnan and S. Shanmugam, Electrochem. Commun., 2014, 41, 59–63 CrossRef CAS.
- X. Liu, M. Park, M. G. Kim, S. Gupta, G. Wu and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 9654–9658 CrossRef CAS PubMed.
- G. Li, X. Wang, J. Fu, J. Li, M. G. Park, Y. Zhang, G. Lui and Z. Chen, Angew. Chem., Int. Ed., 2016, 55, 4977–4982 CrossRef CAS.
- L.-N. Han, L.-B. Lv, Q.-C. Zhu, X. Wei, X.-H. Li and J.-S. Chen, J. Mater. Chem. A, 2016, 4, 7841–7847 RSC.
- P.-C. Li, Y.-J. Chien and C.-C. Hu, J. Power Sources, 2016, 313, 37–45 CrossRef CAS.
- X. Han, X. Wu, C. Zhong, Y. Deng, N. Zhao and W. Hu, Nano Energy, 2017, 31, 541–550 CrossRef CAS.
- Q. Wang, L. Shang, R. Shi, X. Zhang, Y. Zhao, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2017, 7, 1700467 CrossRef.
- Q. Wang, Y. Lei, Z. Chen, N. Wu, Y. Wang, B. Wang and Y. Wang, J. Mater. Chem. A, 2018, 6, 516–526 RSC.
- S. S. Shinde, C. H. Lee, J.-Y. Jung, N. K. Wagh, S.-H. Kim, D.-H. Kim, C. Lin, S. U. Lee and J.-H. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC.
- K. Wu, L. Zhang, Y. Yuan, L. Zhong, Z. Chen, X. Chi, H. Lu, Z. Chen, R. Zou, T. Li, C. Jiang, Y. Chen, X. Peng and J. Lu, Adv. Mater., 2020, 32, 2002292 CrossRef CAS.
- V. Jose, H. Hu, E. Edison, W. Manalastas, H. Ren, P. Kidkhunthod, S. Sreejith, A. Jayakumar, J. M. V. Nsanzimana, M. Srinivasan, J. Choi and J. Lee, Small Methods, 2021, 5, 2000751 CrossRef CAS.
- B. J. Kim, D. U. Lee, J. Wu, D. Higgins, A. Yu and Z. Chen, J. Phys. Chem. C, 2013, 117, 26501–26508 CrossRef CAS.
- H.-W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- H. bin Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2(4), e1501122 CrossRef PubMed.
- C. Tang, B. Wang, H. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185 CrossRef.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- C. Su, H. Cheng, W. Li, Z. Liu, N. Li, Z. Hou, F. Bai, H. Zhang and T. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef.
- S. S. Shinde, C.-H. Lee, A. Sami, D.-H. Kim, S.-U. Lee and J.-H. Lee, ACS Nano, 2017, 11, 347–357 CrossRef CAS.
- X. Peng, L. Zhang, Z. Chen, L. Zhong, D. Zhao, X. Chi, X. Zhao, L. Li, X. Lu, K. Leng, C. Liu, W. Liu, W. Tang and K. P. Loh, Adv. Mater., 2019, 31, 1900341 CrossRef.
- Y. Zheng, H. Song, S. Chen, X. Yu, J. Zhu, J. Xu, K. A. I. Zhang, C. Zhang and T. Liu, Small, 2020, 16, 2004342 CrossRef CAS PubMed.
- Y. Wang, N. Xu, R. He, L. Peng, D. Cai and J. Qiao, Appl. Catal., B, 2021, 285, 119811 CrossRef CAS.
- X. Li, W. Qu, J. Zhang and H. Wang, J. Electrochem. Soc., 2011, 158, A597 CrossRef CAS.
- Z. Chen, A. Yu, D. Higgins, H. Li, H. Wang and Z. Chen, Nano Lett., 2012, 12, 1946–1952 CrossRef CAS.
- K.-N. Jung, J.-H. Jung, W. bin Im, S. Yoon, K.-H. Shin and J.-W. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 9902–9907 CrossRef CAS PubMed.
- D. U. Lee, H. W. Park, M. G. Park, V. Ismayilov and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 902–910 CrossRef CAS PubMed.
- K. Lopez, G. Park, H.-J. Sun, J.-C. An, S. Eom and J. Shim, J. Appl. Electrochem., 2015, 45, 313–323 CrossRef CAS.
- M. Prabu, P. Ramakrishnan, P. Ganesan, A. Manthiram and S. Shanmugam, Nano Energy, 2015, 15, 92–103 CrossRef CAS.
- A. Vignesh, M. Prabu and S. Shanmugam, ACS Appl. Mater. Interfaces, 2016, 8, 6019–6031 CrossRef CAS PubMed.
- Y. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, ACS Nano, 2017, 11, 11594–11601 CrossRef CAS.
- H. Miao, Z. Wang, Q. Wang, S. Sun, Y. Xue, F. Wang, J. Zhao, Z. Liu and J. Yuan, Energy, 2018, 154, 561–570 CrossRef CAS.
- H. Lee, O. Gwon, C. Lim, J. Kim, O. Galindev and G. Kim, ChemElectroChem, 2019, 6, 3154–3159 CrossRef CAS.
- C. Zhao, X. Zhang, M. Yu, A. Wang, L. Wang, L. Xue, J. Liu, Z. Yang and W. Wang, Adv. Mater., 2020, 32, 2006145 CrossRef CAS.
- X. Shi, Y. Deng, L. Zhao, Y. Gong, R. Wang, H. Wang and B. He, Electrochim. Acta, 2021, 391, 138951 CrossRef CAS.
- European Commission, Critical raw materials resilience: charting a path towards greater security and sustainability, https://ec.europa.eu/docsroom/documents/42849/attachments/2/translations/en/renditions/native, (accessed 18 February 2022).
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc, New York, 2 edn, 2001 Search PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- C. A. Campos-Roldán, R. G. González-Huerta and N. Alonso-Vante, J. Electrochem. Soc., 2018, 165, J3001–J3007 CrossRef.
- Y. Wu and Y. Nabae, Curr. Opin. Electrochem., 2021, 25, 100633 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 1979, 2011(334), 1383–1385 Search PubMed.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef.
- I. Yamada, A. Takamatsu, K. Asai, T. Shirakawa, H. Ohzuku, A. Seno, T. Uchimura, H. Fujii, S. Kawaguchi, K. Wada, H. Ikeno and S. Yagi, J. Phys. Chem. C, 2018, 122, 27885–27892 CrossRef CAS.
- J. A. Dias, M. A. S. Andrade, H. L. S. Santos, M. R. Morelli and L. H. Mascaro, ChemElectroChem, 2020, 7, 3173–3192 CrossRef CAS.
- I. Yamada, A. Takamatsu, K. Asai, T. Shirakawa, H. Ohzuku, A. Seno, T. Uchimura, H. Fujii, S. Kawaguchi, K. Wada, H. Ikeno and S. Yagi, J. Phys. Chem. C, 2018, 122, 27885–27892 CrossRef CAS.
- J. J. Martin, V. Neburchilov, H. Wang and W. Qu, 2009 IEEE Electrical Power & Energy Conference (EPEC), IEEE, 2009, pp. 1–6.
- Y. Zhong, X. Xu, P. Liu, R. Ran, S. P. Jiang, H. Wu and Z. Shao, Adv. Energy Mater., 2020, 10, 2002992 CrossRef CAS.
- Y. Dai, J. Yu, P. Tan, C. Cheng, T. Liu, S. Zhao, Z. Shao, T. Zhao and M. Ni, J. Power Sources, 2022, 525, 231108 CrossRef CAS.
- J. Yu, B. Li, C. Zhao, J. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS PubMed.
- S. Askari, D. Mariotti, R. McGlynn and J. Benedikt, Energy Technol., 2021, 9, 2000999 CrossRef CAS.
- X. X. Wang, X. Yang, H. Liu, T. Han, J. Hu, H. Li and G. Wu, Small Struct., 2022, 3, 2100103 CrossRef CAS.
- X. Hu, X. Tian, Y.-W. Lin and Z. Wang, RSC Adv., 2019, 9, 31563–31571 RSC.
- S. Dongmo, J. J. A. Kreissl, K. Miyazaki, T. Abe, T.-H. You, C.-C. Hu and D. Schröder, Sci. Data, 2020, 7, 395 CrossRef CAS PubMed.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 RSC.
- Y. Huang, Z. Li, Z. Pei, Z. Liu, H. Li, M. Zhu, J. Fan, Q. Dai, M. Zhang, L. Dai and C. Zhi, Adv. Energy Mater., 2018, 8, 1802288 CrossRef.
- M. Krebs, H.-U. Reichardt, R. Sojka and P. Stevens, Electrochemical Power Sources: Fundamentals, Systems, and Applications, Elsevier, 2021, pp. 217–245 Search PubMed.
- Zinc air battery technical specifications (by brand), https://www.microbattery.com/zinc-air-battery-specs, (accessed 4 May 2022).
- Cegasa Zinc air alkaline, https://www.cegasa.com/en/ez8, (accessed 4 May 2022).
- P. Tan, B. Chen, H. Xu, H. Zhang, W. Cai, M. Ni, M. Liu and Z. Shao, Energy Environ. Sci., 2017, 10, 2056–2080 RSC.
- Y. Xu, X. Xu, M. Guo, G. Zhang and Y. Wang, Front. Chem., 2022, 10, 827563 CrossRef CAS PubMed.
- J. Zhou, J. Cheng, B. Wang, H. Peng and J. Lu, Energy Environ. Sci., 2020, 13, 1933–1970 RSC.
- S. Lorca, F. Santos and A. J. Fernández Romero, Polymers, 2020, 12, 2812 CrossRef CAS.
- P. Bocchetta, D. Frattini, S. Ghosh, A. M. V. Mohan, Y. Kumar and Y. Kwon, Materials, 2020, 13, 2733 CrossRef CAS PubMed.
- P. Zhang, K. Wang, P. Pei, Y. Zuo, M. Wei, X. Liu, Y. Xiao and J. Xiong, Mater. Today Chem., 2021, 21, 100538 CrossRef CAS.
- Zinc8 storage solutions, https://www.zinc8energy.com/, (accessed 5 May 2022).
| This journal is © The Royal Society of Chemistry 2022 |