
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suppression of colonic oxidative stress caused by chronic ethanol administration and attenuation of ethanol-induced colitis and gut leakiness by oral administration of sesaminol in mice†
Hideo
Ohira
 a,
Daiki
Oikawa
b,
Yoichi
Kurokawa
c,
Yuichi
Aoki
d,
Ayano
Omura
e,
Kunio
Kiyomoto
e,
Wao
Nakagawa
a,
Rie
Mamoto
a,
Yoshio
Fujioka
a and
Toru
Nakayama
b
a,
Daiki
Oikawa
b,
Yoichi
Kurokawa
c,
Yuichi
Aoki
d,
Ayano
Omura
e,
Kunio
Kiyomoto
e,
Wao
Nakagawa
a,
Rie
Mamoto
a,
Yoshio
Fujioka
a and
Toru
Nakayama
b
aDivision of Clinical Nutrition, Faculty of Nutrition, Kobe Gakuin University, Kobe 651-2180, Japan
bDepartment of Biomolecular Engineering, Graduate School of Engineering, Tohoku University, Sendai, Miyagi 980-8579, Japan
cFaculty of Bioscience, Fukui Prefectural University, Fukui 910-1195, Japan
dTohoku University Tohoku Medical Megabank Organization, Seiryo 2-1, Sendai, Miyagi 980-8573, Japan
eKiyomoto Co., Ltd., 6-1633 Totoro-cho, Nobeoka, Miyazaki 889-0595, Japan
First published on 8th August 2022
Abstract
Chronic consumption of excess ethanol is one of the major risk factors for colorectal cancer (CRC), and the pathogenesis of ethanol-related CRC (ER-CRC) involves ethanol-induced oxidative-stress and inflammation in the colon and rectum, as well as gut leakiness. In this study, we hypothesised that oral administration of sesaminol, a sesame lignan, lowers the risk of ER-CRC because we found that it is a strong antioxidant with very low prooxidant activity. This hypothesis was examined using a mouse model, in which 2.0% v/v ethanol was administered ad libitum for 2 weeks with or without oral gavage with sesaminol (2.5 mg per day). Oral sesaminol administration suppressed the ethanol-induced colonic lesions and the ethanol-induced elevation of the colonic levels of oxidative stress markers (8-hydroxy-2′-deoxyguanosine, malondialdehyde, and 4-hydroxyalkenals). It consistently suppressed the chronic ethanol-induced expressions of cytochrome P450-2E1 and inducible nitric oxide synthase and upregulated heme oxygenase-1 expression, probably via the nuclear factor erythroid-derived 2-like 2 pathway in the mouse colon. Oral sesaminol administration also suppressed the chronic ethanol-induced elevation of colonic inflammation marker levels, such as those of tumour necrosis factor-α, interleukin-6, and monocyte chemoattractant protein-1, probably via the nuclear factor-kappa B pathway. Moreover, it prevented the chronic ethanol-induced gut leakiness by restoring tight junction proteins, giving rise to lower plasma endotoxin levels compared with those of ethanol-administered mice. All of these results suggest that dietary supplementation of sesaminol may lower the risk of ER-CRC by suppressing each of the above-mentioned steps in ER-CRC pathogenesis.
1. Introduction
Chronic consumption of excess ethanol has been shown to be one of the major risk factors for colorectal cancer (CRC).1,2 Although the mechanism by which chronic ethanol consumption increases CRC risk is yet to be fully clarified, one possible scenario for ethanol-related CRC (ER-CRC) pathogenesis is as follows. Chronic ethanol consumers retain high blood concentrations of ethanol for prolonged periods, which equilibrates among tissues, including the colon and rectum.3 Ethanol oxidation, which takes place not only in liver, but also in other tissues and organs including the colon and rectum, is coupled to the mitochondrial electron transport system, which coincidentally produces reactive oxygen species (ROS).2 In chronic ethanol consumers, CYP2E1 is also involved in ethanol oxidation in the colon and rectum and potentially produces ROS.4,5 ROS thus produces enhanced oxidative stress and promotes inflammation via multiple pathways in the colorectal tissues.2,6,7 Moreover, it has been shown that ethanol can also directly or indirectly cause impaired gut barrier function, such as dysfunction of the tight junction (TJ), giving rise to leaky gut.8,9 The gut leakiness may result in translocation of intestinal bacteria via the cardiovascular system to result in appreciable plasma levels of lipopolysaccharides (LPS), which induce inflammation.10 The oxidative stress and inflammation thus generated cause DNA damage, which initiates malignant transformation of colorectal cells,2,6,7,11–13 leading to ER-CRC.14The relative CRC risk of heavy drinkers has been estimated to be 1.52-times higher than that of non-drinkers/occasional drinkers, with a stronger association (relative CRC risk, 1.81) in Asian people.15 We previously compared gut microbiota structures of alcoholics and non-alcoholics to identify the ecophysiological consequences of alcoholism on human gut microbiota.16 It was found that gut microbiota of alcoholics were in a dysbiotic state, in which obligate anaerobes (such as Bacteroides and Ruminococcus) decreased, and facultative anaerobes (such as Streptococcus and bacterial species belonging to family Enterobacteriaceae) were enriched. Because obligate anaerobes are generally more susceptible to ROS than facultative anaerobes, these observations were consistent with the above-described ER-CRC pathogenesis in chronic ethanol consumers.16 The induction of colonic oxidative stress by ethanol was further corroborated by our recent study using a mouse model.17
Sesame oil extracted from sesame (Sesamum indicum L.) seeds is known to display exceptionally high resistance to lipid peroxidation among other edible oils, and this unique nature of sesame oil arises from the presence of a variety of sesame lignans in it, such as sesaminol and sesamol, which display strong antioxidant activities.18 A wide variety of bioactivities that are beneficial to human health have been reported for these lignans, and these useful bioactivities likely arise from their antioxidant activities.
Of these sesame lignans, sesaminol (Fig. 1) has particularly strong antioxidant activity and serves as a potential inhibitor of the oxidation of low-density lipoproteins.19,20 It also inhibits 6-hydroxydopamine-induced ROS production in human neuroblastoma cells21 and protects against β-amyloid peptide-induced cognitive deficits in mice.22
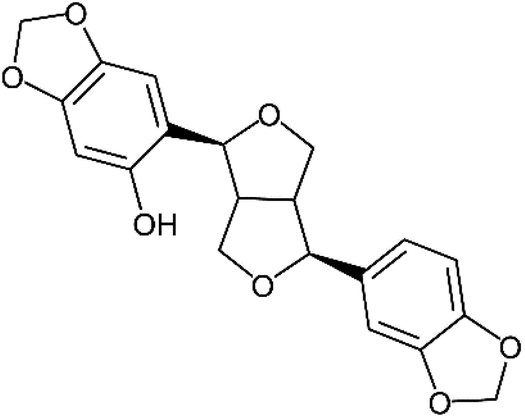 | ||
| Fig. 1 Structure of sesaminol. | ||
Thus, we hypothesised that oral administration of sesaminol lowers the risk of ER-CRC based on its strong antioxidant activity. As the first step to examine this hypothesis, we re-characterised the antioxidant activity of sesaminol to show that it is a strong antioxidant with very low prooxidant activity, unlike quercetin (a well-known phytochemical antioxidant).23 We then examined the hypothesis by analysing its effects on the proposed steps of ER-CRC pathogenesis involving ethanol-induced oxidative-stress and inflammation in the colon and rectum, as well as gut leakiness, using a mouse model.
2. Materials and methods
2.1. Chemicals
Sesaminol was prepared as described previously24 and was of 95% or higher purity as judged by HPLC analysis (section 2.16). Luminol was purchased from Tokyo Chemical Industry (Tokyo, Japan). Hydrogen peroxide and copper chloride were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan). The plasmid pBR322 was from New England Biolab (Ipswich, MA, USA). Other chemicals were of analytical grade and purchased from FUJIFILM Wako Pure Chemical or Nacalai Tesque, Kyoto, Japan.2.2. Antioxidant and prooxidant assays
Antioxidant activity was evaluated by the method of Parejo et al.25 Briefly, luminol (10 μM) and H2O2 (1000 μM) were mixed with or without test compounds in 50 mM boric acid–sodium hydroxide buffer (pH 9.0). To initiate the Fenton reaction, CuCl2 (100 μ M) was added, and after 5 min incubation at room temperature, chemiluminescence at 450 nm was recorded on a microplate reader (SpectraMax M2, MolecularDevices, CA, USA). The experiments were performed in triplicate, and the potency of antioxidant activity was expressed as the mean ratio of light extinction.Prooxidant activity was evaluated by the method of Aruoma et al.26 Briefly, plasmid DNA pBR322 (10 ng) and CuCl2 (200 μM) were mixed with or without test compounds in 50 mM sodium phosphate–150 mM sodium chloride buffer (pH 7.4) in a microtube. After incubation at 37 °C for 1 h, an aliquot of the reaction mixture (10 μL) was mixed with loading buffer (FUJIFILM Wako Pure Chemical) and loaded onto 1% agarose gel. Electrophoresis was performed at 100 volts for 40 min in TAE buffer (0.04 M tris-acetate and 1 mM EDTA, pH 7.4) using a DNA subcell (Cosmo-bio, Tokyo, Japan). The agarose gel was stained with ethidium bromide for 10 min, and DNA bands were visualised with a gel-imaging system FAS-IV (Nippon Genetics, Tokyo, Japan).
2.3. Overall animal experimental design
Prior to the start of this work, all animal experiments were approved by the Ethics Committee for Animal Experimentation of Kobe Gakuin University, Kobe, Japan (approval number, A19-55). As required by the Guidelines for Proper Conduct of Animal Experiments by the Science Council of Japan, methods and procedures were used to minimize the pain and/or distress of all animals used in this study.Acclimated, eight-week-old mice (n = 32) were randomly divided into 4 groups (groups C, E, S, and ES; n = 8 per group) and bred in parallel for 14 days with different feeding programs in terms of added ethanol and sesaminol (Fig. 2; see also section 2.4). The dose and duration of sesaminol administration used in this study were comparable with those of previous reports using mice.22,27 On day 15, all subjects were sacrificed painlessly, and the colon, liver, and plasma of each mouse were obtained by necropsy. The colonic tissue samples of these mice were examined for lesions by histochemical examinations (section 2.5) and microscopic evaluation (section 2.6), and for oxidative stress-related and inflammation-related markers by immunohistochemical staining (section 2.7), enzyme-linked immunosorbent assays (ELISA, sections 2.8 and 2.13), western blotting (WB) analysis (section 2.9), quantitative reverse transcription PCR (qRT-PCR) (section 2.10), and lipid peroxide analysis (section 2.11) (n = 5 per group for each analysis). Colonic tissue and plasma samples of these mice were also used to examine ethanol-induced gut tight-junction leakiness by WB analysis of colonic epithelial proteins (section 2.9) and endotoxin assays (section 2.12) (n = 5 per group for each analysis). In addition, liver functions of these mice were examined by blood biochemical analyses (section 2.14) and liver lipid analyses (section 2.15) (n = 8 per group for each analysis). Faeces collected on day 14 and plasma (see above) from each mouse were used for determination of faecal and plasma sesaminol levels by HPLC (section 2.16).
 | ||
| Fig. 2 Schematic of the dietary intervention procedure for mice. | ||
2.4. Mouse groups and their feeding programs
C57BL/6NCr male mice (n = 32, 8 weeks old, with an average weight of 23.0 ± 1.0 g) were obtained from SLC (Hamamatsu, Japan). All mice were housed, one mouse per cage, in an animal breeding room of SPF level, Animal Experimentation Facility, Kobe Gakuin University, under standard conditions [23–24 °C, 12 h light/dark cycle (lights on from 8 a.m. to 8 p.m.)]. The Animal Experimentation Facility undertakes a twice-a-year health screening, covering various bacterial, viral, and parasitic organisms, and the obtained colony screened negative for all of them. The mice, which were 8 weeks old, were randomly assigned to four groups (groups C, S, E, and ES; n = 8 mice per group), which were fed in parallel as follows. For acclimation, all 8-week-old mice (n = 32) were fed Rodent Chow diet (AIN93M, SLC) and water ad libitum during the first week, then Lieber-DeCarli control (dextrose) liquid diet (termed LDCLD, ESI Table 1†) (Research Diets; New Brunswick, NJ, USA)28 and water ad libitum during the next week using a liquid diet feeder (model KN-676, Natsume Seisakusho Co., Ltd, Tokyo Japan).Group C (the control group)—mice (n = 8) were fed LDCLD ad libitum with a daily oral gavage of 1 mL of LDCLD using a disposable feeding needle FG5202K (silicon 20-gauge; AS ONE, Osaka, Japan) for 14 days, in which water was available ad libitum.
Group S (the sesaminol group)—mice (n = 8) were fed LDCLD ad libitum with a daily oral gavage of 1 mL of LDCLD containing 2.5 mg sesaminol per mouse (corresponding to a dosage of 100 mg sesaminol per kg body weight per day) using a disposable feeding needle for 14 days. Water was available ad libitum.
Group E (the ethanol group)—mice (n = 8) were fed LDCLD containing 2.0% v/v ethanol (ESI Table 1†) ad libitum with a daily oral gavage of 1 mL of LDCLD using a disposable feeding needle for 14 days. Water was available ad libitum.
Group ES (the ethanol-plus-sesaminol group)—mice (n = 8) were fed LDCLD containing 2.0% v/v ethanol (ESI Table 1†) ad libitum with a daily oral gavage of 1 mL of LDCLD containing 2.5 mg sesaminol per mouse using a disposable feeding needle for 14 days. Water was available ad libitum.
It must be noted that LDCLD and LDCLD containing 2% v/v ethanol were isocaloric, adjusted by both maltodextrin and 100% ethanol formulation ratio (see ESI Table 1†). Gavage was conducted in the laboratory room of SPF level in the above-mentioned facility.
On day 15, which was the experimental endpoint, all mice were anesthetised 12 h after gavage by initial exposure to 4% isoflurane (FUJIFILM Wako Pure Chemical) using an anaesthesia vaporizer (Muromachi Kikai; Tokyo, Japan), followed by sustained exposure to 3% isoflurane. The mice were then sacrificed painlessly and subjected to necropsy.
The liver, colon, and plasma were collected from each mouse. The colon was dissected and immediately frozen at −80 °C or stored in RNAprotect Tissue Reagent (QIAGEN; Venlo, Netherlands) for further analysis. Blood samples were treated with 10 IU mL−1 heparin sodium (Nacalai Tesque). Plasma was obtained by centrifuging the blood at 1000g for 15 min at 4 °C. The rest of the tissues and the plasma were quickly frozen and stored at −80 °C until analysis.
2.5. Histology
Colonic tissue samples were fixed overnight in 10% v/v neutral formaldehyde solution (Nacalai Tesque) at room temperature and then embedded in paraffin. The colonic tissue slices (4 μm in thickness) were subjected to staining with haematoxylin/eosin (HE) and toluidine blue (TB). Hepatic tissue slices were also prepared essentially as described above and subjected to staining with HE and Oil red O. These procedures were outsourced to Kyodo Byori (Kobe, Japan). Finally, these histology specimens were observed using a model VH-8000 microscope (KEYENCE; Osaka, Japan).2.6. Microscopic evaluation of colonic lesions
For each animal, five parts were randomly taken from colonic tissue section samples and subjected to microscopic evaluation. Semi-quantitative degrees of inflammation of the colonic lumen were graded from 0 (the absence of inflammation) to 11 (the most severe inflammation) according to the sum of the scores based on the following observations: (a) loss of mucosal architecture (score, 0–3); (b) cellular infiltration (score, 0–3); (c) muscle thickening (score, 0–3); (d) crypt abscess formation (score, 0–1); and (e) goblet cell depletion (score, 0–1).17,29,302.7. Immunohistochemical staining of MUC2 and 8-OHdG
Colonic tissue slices (4 μm in thickness; see above) were placed on clean, positively charged microscope slide glasses and dried by heating for 2 h at 60 °C in a tissue-drying oven. The section slices were deparaffinised by washing in xylene for 5 min, 3 times, and dehydrated in a series of graded concentrations of ethanol. For heat-induced antigen retrieval, the slide glasses were heated at 100 °C for 20 min in 10 mM sodium citrate buffer, pH 6.0, and then cooled to room temperature in the same buffer for 20 min, followed by washing with 1× Tris-buffered saline containing 0.1% Tween 20 at room temperature for 1 min.For immunohistochemical staining, endogenous peroxidase activity was blocked by incubation of the slices with 3% H2O2 for 5 min. The slices were then incubated with 2% bovine serum albumin in phosphate-buffered saline at room temperature for 30 min, followed by incubation at 4 °C overnight with rabbit anti-intestinal mucin 2 (MUC2) polyclonal antibody (Abcam; Cambridge, UK) or rabbit anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) monoclonal antibody (Abcam). The slices were subsequently incubated with the biotinylated goat anti-rabbit IgG antibody (Abcam) at room temperature for 20 min. Immune complexes were visualised by peroxidase-catalysed oxidation of diaminobenzidine (Dojindo Laboratories; Kumamoto, Japan), and the sections were counterstained with haematoxylin. These procedures were outsourced to Kyodo Byori. Immuno-positive cells were observed by microscopic examination in randomly selected high-power fields (400×) of these histology specimens using a model VH-8000 microscope (KEYENCE).
2.8. Enzyme-linked immunosorbent assay (ELISA)
For ELISA of colonic 8-OHdG levels, colonic tissue samples (50 mg) were homogenised with 1 mL of DNAzol Reagent (Thermo Fisher Scientific; Waltham, MA, USA) using a Polytron homogenizer (model 1300D; Central Scientific Commerce; Tokyo, Japan), in which tissues were homogenised for 10 s, 5 times, using a loose pestle, followed by homogenization for 10 s, 5 times, using a tight pestle. The resulting homogenate was centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 3 h. DNA was isolated from the supernatant by ethanol precipitation and subsequently treated with the 8-OHdG assay preparation reagent set (FUJIFILM Wako Pure Chemical) according to the manufacturer's guidance. Equal amounts of the extracted DNAs were used for the analysis of the 8-OHdG levels using the OxiSelect Oxidative DNA Damage ELISA Kit (Cell Biolabs; San Diego, CA, USA).
000g at 4 °C for 3 h. DNA was isolated from the supernatant by ethanol precipitation and subsequently treated with the 8-OHdG assay preparation reagent set (FUJIFILM Wako Pure Chemical) according to the manufacturer's guidance. Equal amounts of the extracted DNAs were used for the analysis of the 8-OHdG levels using the OxiSelect Oxidative DNA Damage ELISA Kit (Cell Biolabs; San Diego, CA, USA).
For ELISA of colonic levels of tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1), colonic tissue samples (100 mg) were homogenised with 1 mL of M-PER Mammalian protein extraction reagent (Pierce; Rockford, IL, USA) supplemented with a recommended dilution of a protease inhibitor cocktail (Nacalai Tesque) using a homogenizer, essentially as described above, followed by centrifugation at 20000g for 15 min at 4 °C. The protein concentration of the supernatant was determined using a protein assay reagent (Bio-Rad Laboratories; Hercules, CA, USA). Equal amounts of protein were used for the analysis of the levels of TNF-α, IL-6, and MCP-1 using ELISA kits (commercial sources of the kits for TNF-α and MCP-1 assays were RayBiotech, Peachtree Corners, GA, USA, and Thermo Fisher Scientific, for the IL-6 assay) following the manufacturers’ protocols.
For ELISA of hepatic 8-OHdG levels, liver tissue was homogenised using a homogenizer for 10 s, 5 times, using a loose pestle at 4 °C with phosphate-buffered saline, pH 7.4, containing 1 mM phenylmethyl sulfonyl fluoride (Nacalai Tesque). Hepatic cells were collected from the resulting homogenate by centrifugation at 600g for 5 min at 4 °C. Mitochondrial DNA (mtDNA) was isolated from the collected cells using the Mitochondrial DNA isolation kit (BioVision; Milpitas, CA, USA), according to the manufacturer's protocols. Equal amounts of the mtDNAs thus obtained were used for the analysis of the 8-OHdG contents using the OxiSelect oxidative DNA damage ELISA kit (Cell Biolabs).
2.9. Western blotting (WB) analysis
Colonic tissue samples (100 mg) were homogenised with 1 mL of M-PER Mammalian protein extraction reagent (Pierce) supplemented with a recommended dilution of a protease inhibitor cocktail (Nacalai Tesque) using a homogenizer, essentially as described in section 2.8, followed by centrifugation at 20000g for 15 min at 4 °C. The protein concentration of the supernatant was determined using a protein assay reagent (Bio-Rad Laboratories). Equal amounts of protein were subjected to SDS-PAGE, and the electrophoresed proteins in the gels were transferred to PVDF membranes, which were then blocked with a recommended dilution of a blocking reagent (Bullet Blocking One for Western Blotting, Nacalai Tesque) at room temperature overnight. The membranes were then incubated with a recommended dilution of one of the following primary rabbit polyclonal antibodies: anti-mouse CYP2E1, anti-mouse inducible nitric oxide synthase (iNOS), anti-mouse zonula occludens-1 protein (ZO-1), anti-mouse occludin, anti-mouse claudin-1 (each from Abcam), anti-mouse heme oxygenase-1 (HO-1) (Proteintech Group), and anti-mouse β-actin (Genetex; Irvine, CA, USA). Subsequently, the PVDF membranes were washed with 1× Tris-buffered saline containing 0.1% Tween 20 for 10 min, 3 times. The immune complexes were probed with secondary horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Cytiva; Marlborough, MA, USA) and visualised by means of a peroxidase-catalysed chemiluminescence reaction using the ECL western blotting kit (Cytiva). The intensities of the immuno-reactive bands were quantified by densitometry using Image J software (National Institutes of Health; Bethesda, MD, USA).
2.10. Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted from colonic tissue samples using TRIzol reagent (for RNA extraction; Invitrogen). First strand cDNA was synthesised at 37 °C for 60 min from 1 μg of total RNA using the ReverTra Ace-α first strand cDNA synthesis kit (Toyobo; Osaka, Japan) using a thermal cycler (Gene Amp PCR System model 9700; Applied Biosystems; Carlsbad, CA, USA). The mixture was then incubated at 94 °C for 5 min to inactivate the enzyme, followed by incubation at 4 °C. To quantify TNF-α, IL-6, MCP-1, and β-actin mRNAs, qRT-PCR was performed using a LightCycler System (Roche Diagnostics; Mannheim, Germany) and a kit (LightCycler Fast Start DNA Master Plus SYBR Green 1, Roche Diagnostics). Thermal cycling conditions were as follows: 45 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 10–20 s. Nucleotide sequences of the qRT-PCR primers used are listed in ESI Table 2.†2.11. Lipid peroxidation levels in colonic tissues
Colonic tissue samples (100 mg) were homogenised with 1 mL of 20 mM phosphate-buffered saline, pH 7.4, using a homogenizer, essentially as described in section 2.8, followed by centrifugation at 3000g for 10 min at 4 °C. The precipitate was assayed for malondialdehyde (MDA) and 4-hydroxyalkenals (HAE) using the lipid peroxidation assay kit (Oxford Biomedical Research; Oxford, MI, USA) following the manufacturer's instructions.2.12. Endotoxin assays
Plasma endotoxin levels were determined by the Limulus Amebocyte Lysate assay method,31 using the ToxinSensor™ Chromogenic LAL endotoxin assay kit with a concentration range of 0.01–0.10 EU mL−1 (GenScript Biotech; Piscataway, NJ, USA).2.13. Nuclear factor-kappa B (NF-κB) and nuclear factor erythroid-derived 2-like 2 (Nrf2) assays in colonic tissues
Nuclear proteins of colonic tissues were extracted by the nuclear extraction kit (Cayman Chemical; Ann Arbor, MI, USA) following the manufacturer's protocols. Protein concentration was determined using a protein assay reagent (Bio-Rad Laboratories). Known amounts of the extracted nuclear proteins were used for ELISA for activation of NF-κB and Nrf2 by using the TransAM® NFκB p65 transcription factor assay kit (Active Motif; Carlsbad, CA, USA) and Nrf2 transcription factor assay kit (Cayman Chemical), respectively, according to the manufacturers’ guidelines.2.14. Blood biochemical analyses
Activities of plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST), as well as plasma triacylglycerol (TG), were measured using kits (L-Type Wako ALT, L-Type Wako AST, and L-Type Wako TG M, respectively), each obtained from FUJIFILM Wako Pure Chemical.2.15. Liver tissue lipid levels
Liver homogenate was prepared at 4 °C with a kit (lipid droplet isolation kit; Cell Biolabs) using a homogenizer, essentially as described above in section 2.8. The resulting homogenate was centrifuged at 20000g at 4 °C for 3 h. TG and proteins in the supernatant were determined using kits [L-type Wako TG M and protein assay reagents (Bio-Rad Laboratories), respectively] according to the manufacturers’ guidelines.
2.16. Plasma and faecal sesaminol contents
Plasma was filtered through a 0.45 μm filter, and the filtrate was subjected to HPLC. Faeces (20–100 mg) were suspended in 4 volumes of 80% v/v ethanol and dispersed by shaking at 1500 rpm for 10 min with glass beads (0.5 mm in diameter, 100 μL) in a plastic tube using Multi-BeadsShocker (Yasui Kikai; Osaka, Japan), followed by centrifugation at 20000g at 4 °C for 10 min. The supernatant was filtered through a 0.45 μm filter and mixed with an equal volume of 80% v/v ethanol, followed by HPLC analysis. Analytical HPLC conditions were as follows: HPLC apparatus, Shimadzu Prominence HPLC system (Shimadzu; Kyoto, Japan); column, J′ Sphere ODS-H80 (4.6 × 150 mm, YMC, Kyoto, Japan); column temperature, ambient (25 °C); solvent A, 0.1% v/v trifluoroacetic acid in water; solvent B, 0.1% v/v trifluoroacetic acid in a 9:1 v/v mixture of acetonitrile and H2O; and flow rate, 0.7 mL min−1. The column was equilibrated with 55% B. After injection, the column was isocratically developed with 55% B for 3 min, followed by linear gradients from 55% B to 65% B in 8 min and then 65% B to 100% B in 1 min. The column was then washed with 100% B for 3 min, followed by a linear gradient from 100% B to 55% B in 1 min. The column was then developed with 55% B for 5 min for equilibration. The chromatograms were obtained with detection at 280 nm using a photodiode array model SPD M-20A UV-visible detector (Shimadzu). The retention time of the sesaminol was 8.5 min under these HPLC conditions. The detection limits of sesaminol in plasma and faeces were 0.04 μg mL−1-serum and 0.4 μg g−1-faeces, respectively.
2.17. Statistical analysis
Data were assessed using ANOVA with the Tukey–Kramer post hoc comparison test and are expressed as means ± SD. Statistical analysis was performed using GraphPad Prism ver 6.0 (GraphPad Software; San Diego, CA, USA). Significance was set at p < 0.05. Mouse sample size was calculated using a power analysis, which indicated that 8 animals per group were required to detect a 25% difference among means, considering a standard deviation of 20% for basic in vivo data,32 an α error of 0.05, and power of 0.8 (80%). The other sample size was established considering the probability (p = 1/2) of an increase or decrease occurring in the variables of interest. Thus, considering the significance level of p = 0.03, the minimum number of samples used in the statistical analysis was p = (1/2)events; so, if n = 4 (1/2)5 or p = 0.03 (falls within the range of p < 0.05).333. Results
3.1. Evaluation of antioxidant and prooxidant activities of sesaminol
The antioxidant and prooxidant activities of sesaminol were evaluated and compared with those of sesamin (a structural analogue of sesaminol without a hydroxy group) and quercetin (a representative antioxidant phytochemical), using the luminol chemiluminescence extinction assay25 and plasmid DNA nicking activity assay,26 respectively. The latter assay measures generation of the most detrimental ROS, hydroxyl radical,34,35 as the change of the electrophoresis pattern of a plasmid DNA, which is indicative of prooxidant activity.When the luminol chemiluminescence extinction assay was carried out with sesamin, the intensity of luminescence hardly decreased at 1 and 100 μM (Fig. 3A). In contrast, either sesaminol or quercetin decreased the intensity of luminescence in a concentration-dependent manner. The intensity of luminescence decreased to about 50% of the control reaction with 10 μM sesaminol or quercetin, and even to the basal level (control without oxidant) with 100 μM (Fig. 3A). This was comparable to the results with a positive control, ethylenediaminetetraacetic acid (EDTA), showing extinction to 99% at the same concentration (Fig. 3A). These results suggest that sesaminol has a comparable antioxidant activity to that of quercetin or EDTA, whereas sesamin lacks such activity.
 | ||
| Fig. 3 Antioxidant and prooxidant activities of sesamin, sesaminol, and quercetin. (A) Evaluation of antioxidant activity. Luminol was incubated with the compounds indicated in the figure, and luminol chemiluminescence was recorded according to the experimental procedures. The data are expressed as means ± SD (n = 3). Chem., chemical. (B) Evaluation of prooxidant activity. Plasmid DNA pBR322 was incubated with the compounds indicated in the figure and applied to gel electrophoresis, followed by visualization according to the experimental procedures. The ccc form represents non-oxidised plasmid DNA, both the nicked and linear forms represent oxidised DNA. | ||
When plasmid DNA (pBR322) was incubated in the presence of copper chloride and sesamin, followed by agarose gel electrophoresis and staining, plasmid DNA was detected as the covalently closed circular (ccc) form (Fig. 3B), suggesting that the plasmid DNA was not converted to the nicked form, which is caused by hydroxyl radical generation. When plasmid DNA was incubated with sesaminol and copper chloride, less than 10% of the plasmid was detected as linear form, whereas the rest (more than 90%) was detected as the ccc form (Fig. 3B). In sharp contrast, when plasmid DNA was incubated with quercetin and copper chloride, the plasmid was detected as the nicked or linear form, whereas the ccc form was not detected (Fig. 3B). These results strongly suggest that while both sesamin and sesaminol display little pro-oxidant activity, quercetin has detectable prooxidant activity in the presence of Fenton-like reaction reagents. Taken together, sesaminol showed high antioxidant activity, like quercetin, but only very low prooxidant activity, like sesamin.
3.2. Body weight and general observations
Acclimated, 8-week-old mice were randomly divided into 4 groups (groups C, S, E, and ES; n = 8 per group) and bred in parallel for 14 days with different feeding programs in terms of ethanol and sesaminol content. There was no sudden death, no loss of appetite, and no immobility among the mice used, and thus no mice dropped out. There were no significant differences between group C and the other groups in terms of initial and final body weights and the courses of body weight changes (Fig. 4).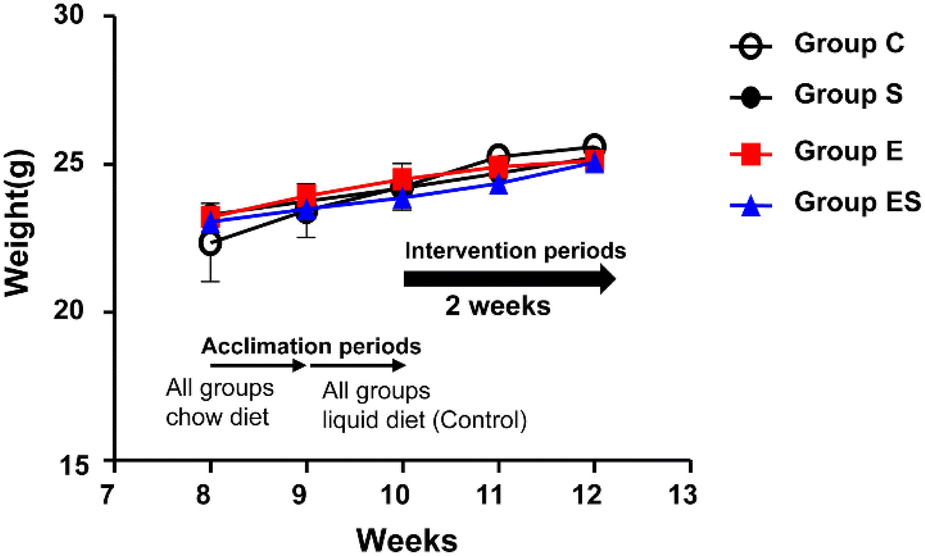 | ||
| Fig. 4 Body weight changes during the experiment. Group C, open circles; group S, closed circles; group E, red squares, and group ES, blue triangles. Body weights are measured every week. The data are expressed as means ± SD (n = 8). Significant differences between group C compared with other groups in terms of initial, every week, and final body weights, were assessed using ANOVA with the Tukey–Kramer test. See the text for the experimental details. | ||
When plasma and faecal sesaminol contents of the mice of these four groups (n = 3 for each group) were measured at the experimental endpoint (on day 15), no detectable level of sesaminol could be found in the plasma of any mouse examined, whereas a very low level of sesaminol (2.67 ± 1.44 μg g−1 faeces) was found in the mice of group ES (ESI Table 3†).
3.3. Effects of oral sesaminol administration on ethanol-induced lesions of mouse colon
The colonic epithelia of the mice of groups C, S, E, and ES were stained with HE (Fig. 5A) and TB (Fig. 5B), and immuno-stained using anti-MUC2 antibody (Fig. 5C), followed by microscopic examination. The colonic epithelia of the mice of group E showed an architectural alteration with a thinner mucosal layer when compared with those of group C (Fig. 5A and B). Consistent with this, the colonic tissues of the mice of group E showed a reduced level of mucus secretion due to a decreased number of goblet cells (Fig. 5B and C). The colonic epithelia of the mice of group ES showed a decreased degree of disorder, whereas no appreciable disorder could be observed in those of groups C and S (Fig. 5A–C). When microscopic lesion scores of the gut tissues (colonic epithelium, colonic crypts, submucosa, and mucosa) were compared among these four groups, the highest scores were obtained with group E (Fig. 5D). The score of group ES was significantly lower than that of group E, but slightly higher, though not significantly, than those of groups C and S, which showed no lesion (Fig. 5D).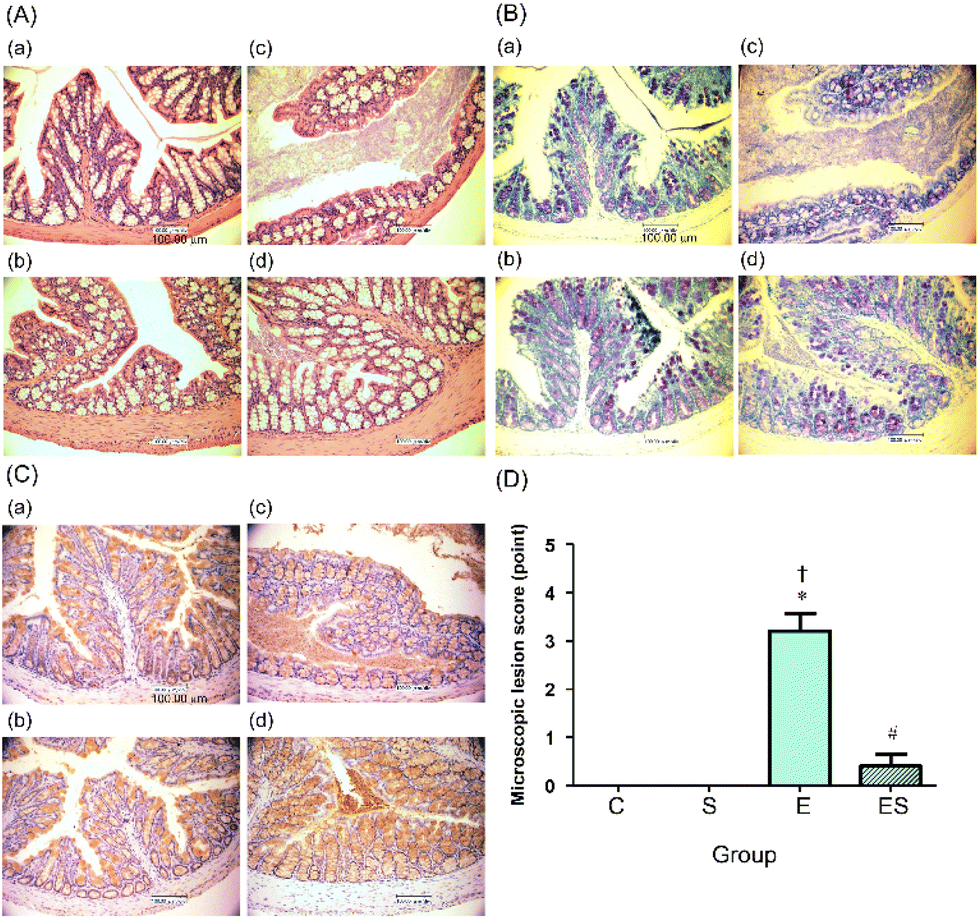 | ||
| Fig. 5 Effects of oral sesaminol administration on ethanol-induced colonic lesions on microscopic evaluation. Colonic tissue sections of the mice on day 15 were observed microscopically. (A) HE-stained images at 400× magnification, (B) TB-stained images at 400× magnification, and (C) MUC2 immunohistochemically stained images at 400× magnification; (a) group C, (b) group S, (c) group E, and (d) group ES. (D) The microscopic lesion scores were evaluated as described in the Materials and methods. The scores are expressed as means ± SD (n = 5). *p < 0.05 versus group C, †p < 0.05 versus group S, #p < 0.05 versus group E as assessed by ANOVA with the Tukey–Kramer test. | ||
3.4. Effects of oral sesaminol administration on ethanol-induced oxidative stress in mouse colon
We previously showed that chronic oral administration of ethanol in mice increases oxidative stress in colonic tissues.17 Thus, to examine the effects of oral administration of sesaminol on the chronic ethanol-induced colonic oxidative stress, colonic oxidative stress marker levels were measured in the four groups.Immunohistochemical staining results showed that the highest colonic 8-OHdG level was obtained in the mice of group E, followed by those of group ES, whereas no appreciable levels of 8-OHdG were detected in the mice of groups C and S (Fig. 6A). These results were further quantitatively confirmed by ELISA results (Fig. 6B). The colonic levels of MDA + HAE as markers for lipid peroxidation were also analysed. The highest value was obtained in group E, followed by group ES, which showed a slightly higher value than groups C and S (Fig. 6C).
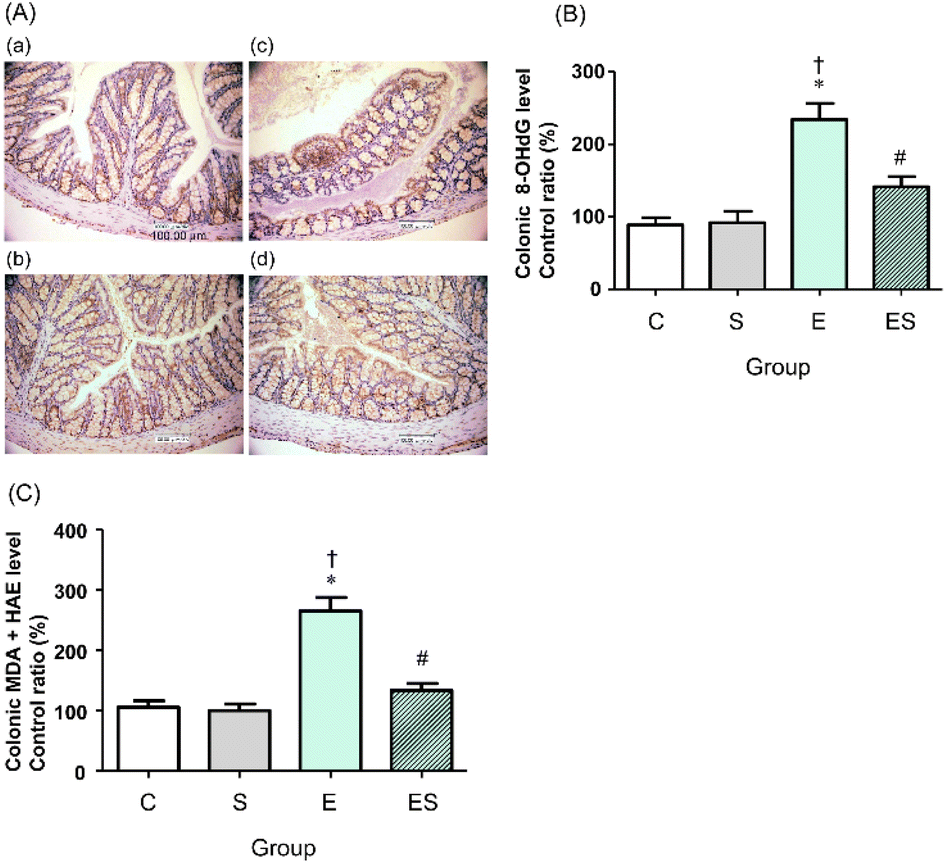 | ||
| Fig. 6 Effects of oral sesaminol administration on ethanol-induced oxidative stress markers (8-OHdG and MDA + HAE) in colonic tissues. (A) The 8-OHdG immunohistochemical staining of colon section images at 400× magnification. (a) Group C, (b) group S, (c) group E, and (d) group ES. (B) The 8-OHdG levels in colonic tissue extracts were determined by ELISA, and the relative percentages of the 8-OHdG levels are shown with the level obtained with a group C subject taken to be 100%. (C) The MDA + HAE levels in colonic tissue extracts were determined, and the relative percentages of the MDA + HAE levels are shown with the level obtained in a group C subject taken to be 100%. The data are expressed as means ± SD (n = 5). *p < 0.05 versus group C, †p < 0.05 versus group S, #p < 0.05 versus group E as assessed by ANOVA with the Tukey–Kramer test. | ||
Chronic administration of excess ethanol has been reported to elevate levels of CYP2E14,5,12,13,36,37 and iNOS38 causing ethanol-induced oxidative stress in the liver and other tissues in experimental animals. Moreover, the nuclear factor erythroid 2-related factor 2/heme oxygenase-1 (Nrf2/HO-1) system is widely involved in the protection of cells from oxidative injury.39,40 Thus, the levels of these proteins (CYP2E1, iNOS, and HO-1) in colonic tissues of the mice of the four groups were examined by means of WB analysis. The results showed that the colonic level of CYP2E1 in group E was significantly higher than the levels observed in group C and other groups, with no significant difference in CYP2E1 levels among groups C, S, and ES (Fig. 7A and B). These observations were also the case for the colonic iNOS levels (Fig. 7A and B). In contrast, the colonic level of HO-1 in group E was significantly lower than in group C and other groups, with no significant difference in HO-1 levels among groups C, S, and ES (Fig. 7A and B).
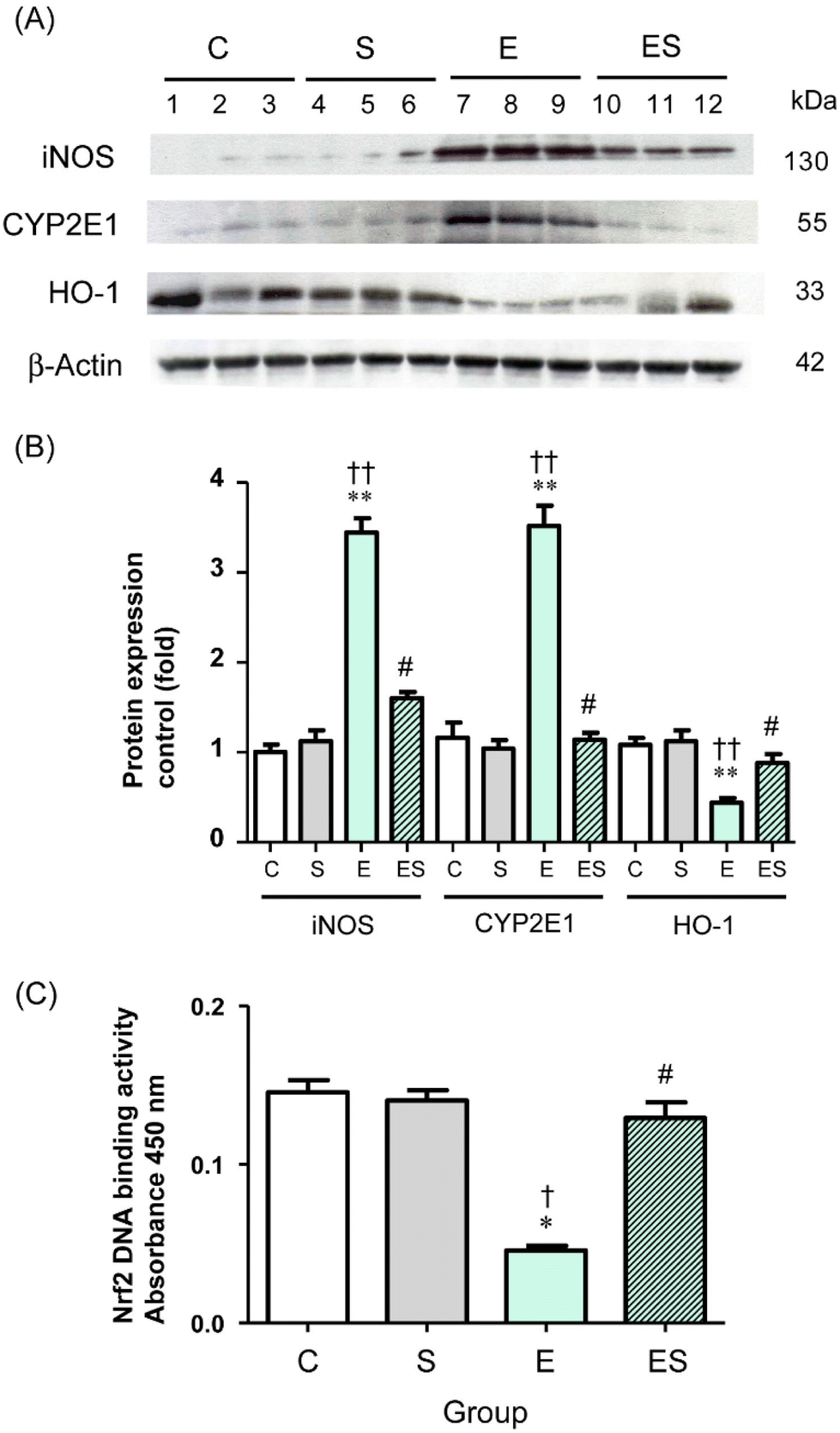 | ||
| Fig. 7 Effects of oral sesaminol administration on the levels of ethanol-induced oxidative-stress proteins (iNOS, CYP2E1, HO-1) and transcription factor protein (Nrf2) in colonic tissues. (A) WB analyses of iNOS, CYP2E1, HO-1, and β-actin (from top to bottom) in colonic tissue extracts of the mice of groups C, S, E, and ES. Lanes 1–3, group C; lanes 4–6, group S; lanes 7–9, group E; and lanes 10–12, group ES. (B) Protein expression levels of iNOS, CYP2E1, and HO-1 were analysed by WB. The band intensities were measured by densitometry and normalised to β-actin. The relative band intensities (-fold) are shown with the intensity obtained with a subject of group C taken to be 1.0. The data are expressed as means ± SD (n = 5). (C) Nrf2 activation levels were determined by ELISA in colonic tissue extracts, whose protein concentrations were identical (1 μg μL−1) among the four groups. The data are expressed as means ± SD (n = 5). *p < 0.05, **p < 0.01 versus group C, †p < 0.05, ††p < 0.01 versus group S, #p < 0.05 versus group E as assessed by ANOVA with the Tukey–Kramer test. | ||
The observation that the HO-1 level was decreased in group E prompted further analysis of the activation levels of Nrf2 transcription factor, which is the major regulator for cytoprotective programs which activate HO-1 gene expression during cellular responses to oxidative stress.40 The results showed that the activation level of Nrf2 was significantly decreased in group E compared with group C and restored to the control level in group ES, whereas the activation level in group S was comparable with the control (Fig. 7C).
3.5. Effects of oral sesaminol administration on ethanol-induced colonic inflammation in mouse colon
It has been reported that chronic consumption of excess ethanol may accelerate inflammation of colonic tissues to potentially promote tumourigenesis.41 Thus, whether oral sesaminol administration prevents ethanol-induced colitis was examined by determining the expression levels of inflammatory cytokines (TNF-α and IL-6) and a chemokine (MCP-1)41 in colonic lamina propria of the mice of the four groups by means of qRT-PCR and ELISA. NF-κB activation analysis was also carried out in these mice because this transcription factor is known to be involved in the expression of genes that regulate inflammation and is activated by exposure of immune cells to inflammatory cytokines.41The results showed that the expressions of TNF-α, IL-6, and MCP-1 in colonic tissues of the mice of group E were all significantly higher than those of groups C and S, as analysed by qRT-PCR (Fig. 8A–C) and ELISA (Fig. 8D–F). These colonic inflammation marker levels in group ES showed higher (but not significant) tendencies than in group C, but were significantly lower than the levels in group E, whereas those in group S were all comparable with the control levels based on both methods (Fig. 8A–F). Consistently, the NF-κB p65 activation level was significantly higher in group E than in groups C and S, whereas the level was significantly lower in group ES than in group E (Fig. 8G). The activation level in group S was comparable with that in group C.
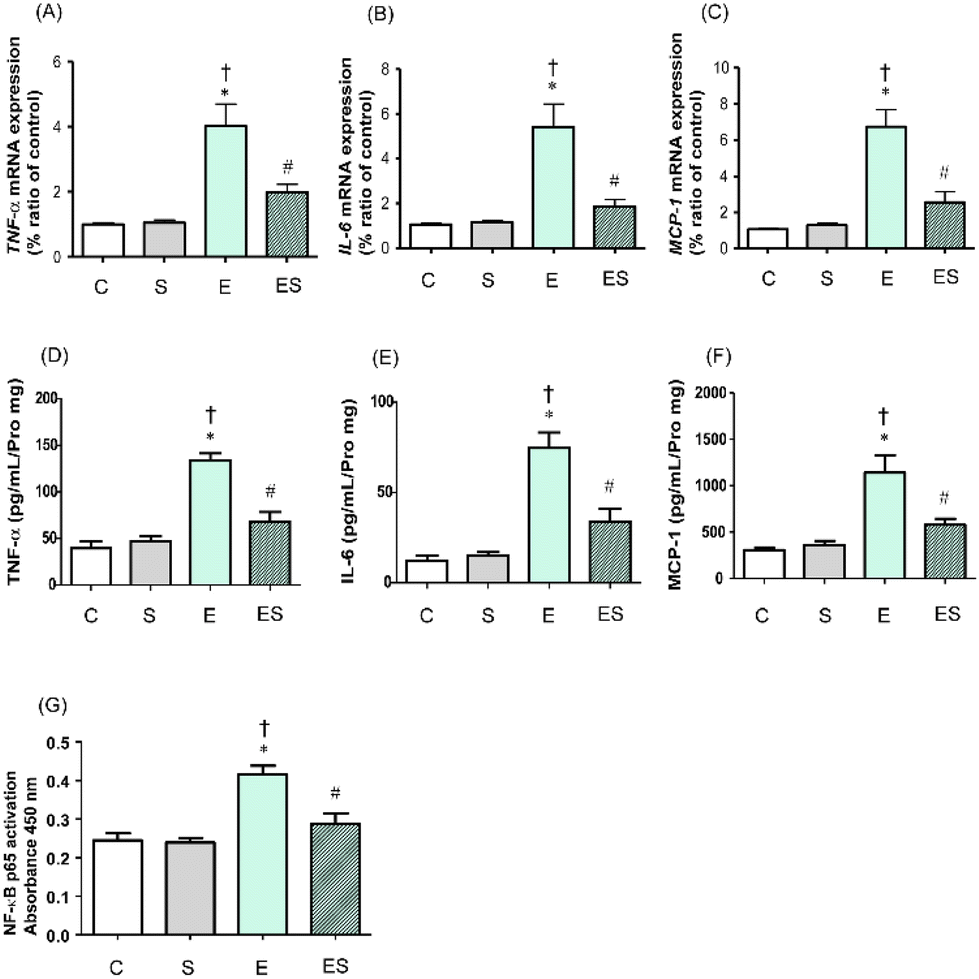 | ||
| Fig. 8 Effects of oral sesaminol administration on ethanol-induced colonic inflammatory marker levels and NF-κB transcriptional activity in mice. The mRNA levels of (A) TNF-α, (B) IL-6, and (C) MCP-1 were analysed by qRT-PCR and are shown as relative values with the respective mRNA level of a subject of group C taken to be 1.0. The protein levels of (D) TNF-α, (E) IL-6, and (F) MCP-1 were measured by ELISA. (G) NF-κB p65 activation levels in colonic tissue extracts were determined by ELISA, and the relative NF-κB p65 activation levels are shown with absorbance at 450 nm. The data are expressed as means ± SD (n = 5). *p < 0.05 versus group C, †p < 0.05 versus group S, #p < 0.05 versus group E as assessed by ANOVA with the Tukey–Kramer test. | ||
3.6. Effects of oral sesaminol administration on ethanol-induced gut tight-junction dysfunction and leakiness
Acetaldehyde, which accumulates during oxidative ethanol metabolism, is known to disrupt intestinal TJs via dissociation of colonic epithelial TJ proteins (ZO-1, occludin, and claudin-1) from the cytoskeleton.8,42,43 This results in the loss of cell–cell adhesion and an increased permeability to bacterial toxins (lipopolysaccharides, LPS) from the colonic lumen into the systemic circulation and potentially causes endotoxemia.8 Therefore, whether oral sesaminol administration affects the ethanol-induced gut barrier dysfunction was examined by analysing the levels of colonic epithelial TJ proteins and plasma LPS levels in the mice of the four groups.The results indicated that the colonic tissues of the mice of group E showed significantly decreased levels of the three TJ protein species compared with those of groups C and S (Fig. 9A and B). In group ES, the levels of these protein species were slightly lower, but not significantly, than in group C, but significantly higher than in group E, whereas the levels in group S were comparable with the control. Consistently, the plasma LPS level of the mice of group E was significantly higher than the levels of group C and other groups, with no significant difference in plasma LPS levels among groups C, S, and ES (Fig. 9C).
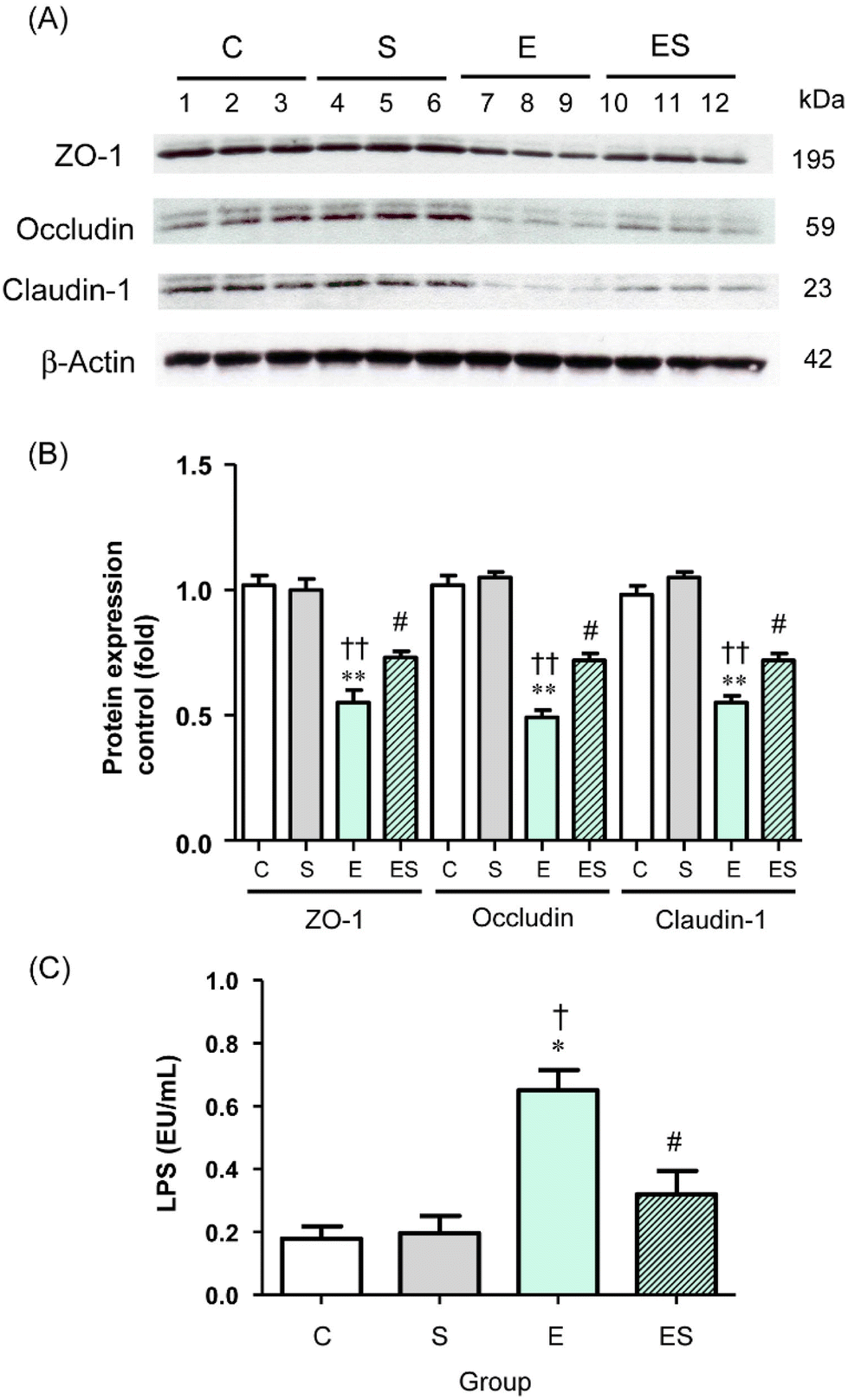 | ||
| Fig. 9 Effects of oral sesaminol administration on the expression levels of gut TJ proteins (ZO-1, occludin, and claudin-1) and the plasma LPS level. (A) WB images of ZO-1, occludin, claudin-1, and β-actin (from top to bottom) in the colonic tissue extracts of the mice (n = 3 in each group) of group C (lanes 1–3), group S (lanes 4–6), group E (lanes 7–9), and group ES (lanes 10–12). (B) Intensities of protein bands in WB images (A) were normalised to that of β-actin in each subject, and the relative band intensities are shown with a subject in group C taken to be 1.0; the data are expressed as means ± SD (n = 5). (C) The plasma LPS concentrations were measured by means of the limulus amebocyte lysate assay, and the data are expressed as means ± SD (n = 5). *p < 0.05, **p < 0.01 versus group C, †p < 0.05, ††p < 0.01 versus group S, #p < 0.05 versus group E as assessed by ANOVA with the Tukey–Kramer test. | ||
3.7. Effects of oral sesaminol administration on ethanol-induced liver dysfunction and oxidative stress
The effects of sesaminol administration on ethanol-induced liver dysfunction in the four groups of mice were also examined. The liver to body weight ratio of the mice on day 15 was significantly higher in group E than in group C, and this increase was significantly suppressed in group ES (Table 1), whereas the ratio in group S was comparable to that in group C.| Valuea | Group (n = 8 per each group) | |||
|---|---|---|---|---|
| C | S | E | ES | |
| a Values of eight independent biological replicates are expressed as the mean ± SD. Data were assessed using ANOVA with Tukey–Kramer test. *p < 0.05, **p < 0.01, and ***p < 0.001: versus group C, †p < 0.05, ††p < 0.01, and †††p < 0.001: versus group S, #p < 0.05, ##p < 0.01: versus group E. TG, triglyceride; AST, aspartate aminotransferase; ALT, alanine aminotransferase. | ||||
| Body weight (g) | 25.7 ± 0.6 | 25.3 ± 0.4 | 25.0 ± 1.1 | 25.1 ± 0.6 |
| Liver/body weight ratio (g/g, %) | 3.23 ± 0.29 | 3.52 ± 0.25 | 4.38 ± 0.39**, †† | 3.77 ± 0.29 *,# |
| Liver TG (mg g−1 of protein) | 54.9 ± 10.9 | 58.9 ± 5.0 | 104.5 ± 11.5**, †† | 66.0 ± 8.5## |
| Plasma AST (U L−1) | 38.3 ± 5.3 | 40.5 ± 5.7 | 51.5 ± 7.0**, †† | 44.1 ± 4.1 |
| Plasma ALT (U L−1) | 30.3 ± 5.9 | 31.6 ± 4.8 | 63.8 ± 7.0***, ††† | 45.3 ± 5.6**,†, ## |
| Plasma TG (mmol L−1) | 0.49 ± 0.11 | 0.47 ± 0.07 | 0.70 ± 0.10**, †† | 0.57 ± 0.07# |
On HE and oil red O staining, a slightly irregular arrangement of hepatocytes (indicative of histological liver injury) and elevated fat accumulation in liver were observed in group E and, to a lesser extent, in group ES, but they were not observed in group S when compared with group C (ESI Fig. 1A and B†).
Plasma ALT and AST levels, as well as plasma and hepatic TG levels, were significantly higher in group E than the control values. In group ES, these values were only slightly higher (for plasma AST and plasma, hepatic TG levels) or significantly higher (for plasma ALT and liver/body weight ratio (g/g, %)) than the control values, but comparable to the control values in group S (Table 1).
The content of 8-OHdG in the liver was higher in the mice of group E than in the other three groups, as analysed by ELISA using hepatic mtDNA (ESI Fig. 2A and B†).
4. Discussion
The primary objective of this study was to examine the potentials of dietary phytochemicals44 to lower the risk of ER-CRC, the pathogenesis of which has been proposed to involve ethanol-induced oxidative-stress and inflammation in the colon and rectum, as well as gut leakiness.2 We hypothesised that oral administration of sesaminol lowers the risk of ER-CRC because this lignan is known to have strong antioxidant activity by itself.18,19 In the present study, the antioxidant activity of sesaminol, which was comparable with that of quercetin, a well-known phytochemical antioxidant, was confirmed by the luminol chemiluminescence extinction assays (Fig. 3A). The plasmid DNA nicking activity assay results showed that quercetin displays an appreciable level of prooxidant activity, consistent with previous observations.45,46 Remarkably, however, sesaminol was distinguished from quercetin by the fact that it showed only very low prooxidant activity under the same assay conditions (Fig. 3B). Subsequently, it was shown that oral administration of sesaminol suppresses colonic oxidative stress caused by chronic ethanol administration, attenuating ethanol-induced colitis and gut leakiness in mice, and these observations strongly supported our hypothesis.It would be important to note, however, that only trace amounts of sesaminol could be detected in the faeces collected from the mice of groups S and ES during the experiments, suggesting that most of the ingested sesaminol is converted to other metabolite(s) after absorption in the small intestine, followed by xenobiotic metabolism in the liver and/or by the action of colonic microbiota. In relation to this, in rats, ingested sesamin (i.e., 2′-deoxysesaminol) was reported to be excreted in urine and faeces within 24 h after ingestion, and the excreted amounts corresponded to 0.02% and 9.33% of the amount of the ingested sesaminol, respectively.47 It has also been reported that sesaminol was converted to 5′′-methylated sesamin-6-catechol and sesaminol-6-catechol in the rat liver, and very interestingly, the antioxidant activities of these metabolites were 1.6-fold higher than the original lignans.48 Sesaminol was also reported to be metabolised by rat colonic microbiota to produce tetrahydrofuranoid metabolites,47 which were shown to weakly inhibit ROS-producing P450 cytochromes such as CYP1A2 and CYP2C9.49 Thus, the effects of oral sesaminol administration on mice observed in the present study could arise not only from sesaminol itself, but also from metabolites that were generated from sesaminol as a result of mouse and microbial metabolism.
To examine the potential of oral sesaminol administration to lower the ER-CRC risk, the levels of colonic oxidative stress, colonic inflammation, colonic lesions, and gut leakiness were analysed in the mice of groups C, S, E, and ES. In the first step, the effects of chronic oral administration of excess ethanol were confirmed by comparing the results with the mice of groups E and C, and the results obtained were all consistent with our previous findings.16,17 Namely, higher colonic levels of 8-OHdG (Fig. 6A and B) and lipid peroxidation (Fig. 6C) were found in group E than in group C. Consistently, the mice of group E displayed higher levels of CYP2E1 and iNOS in the colonic tissues than in group C (Fig. 7A and B), which potentially gave rise to higher ROS levels.4,5,12,13,36,37,39 Thus, chronic oral administration of excess ethanol induced oxidative stress in the mouse colon. In the present analysis, the colonic HO-1 level in group E was found to be rather lower than that in group C (Fig. 7A and B), consistent with a decreased level of the colonic Nrf2 level in group E (Fig. 7C), implying that the oxidative-stress response in the mouse gut might be undermined after 2-week chronic administration of excess ethanol. The mice of group E showed higher levels of inflammation in the colonic tissues than those of group C, as shown by the elevated colonic levels of an inflammation-related transcription factor (NF-κB), cytokines (TNF-α and IL-6), and a chemokine (MCP-1) (see Fig. 8), which most likely arose from the ethanol-induced oxidative stress mentioned above.2,6,7,17 In addition, the mice of group E showed more severe colonic lesions (i.e., higher colonic lesion scores; Fig. 5D), consistent with the elevated levels of inflammation mentioned above, with a lower number of goblet cells to cause a thinner mucosal layer than those of group C (Fig. 5C). Moreover, elevated plasma LPS levels of the mice of group E, consistent with the increased gut leakiness in this group of mice, were found.8–10 It was observed that chronic oral administration of excess ethanol resulted in decreased levels of multiple TJ proteins (ZO-1, occludin, and claudin-1) in the mouse colonic epithelia (Fig. 9A and B). Although the mechanism by which the decrease of these proteins took place remains to be examined, it is likely related to the suggested gut leakiness in group E.
The effects of oral administration of sesaminol alone were then examined by comparing the results with the mice of groups S and C. It was found that the mice of group S were indistinguishable from those of group C in terms of colonic levels of oxidative stress markers (8-OHdG and MDA + HAE) and related proteins (CYP2E1, iNOS, HO-1, and Nrf2), as well as inflammation-related markers (TNF-α, IL-6, MCP-1, and NF-κB), colonic lesion scores, goblet cell numbers, mucosal layer thickness, and TJ protein contents in colonic epithelia, and plasma LPS levels.
The effects of oral sesaminol administration on the above-mentioned ethanol-induced alterations in the mouse colon were subsequently examined by comparing the results with the mice of group ES, group E, and other groups. The results showed that the ethanol-induced generation of colonic oxidative stress, colonic inflammation, colonic lesions, and gut leakiness were all partly, though not fully, suppressed in the mice of group ES, indicating that oral administration of sesaminol could prevent the above-described ethanol-induced alterations in the mouse colon, as summarised in Fig. 10.
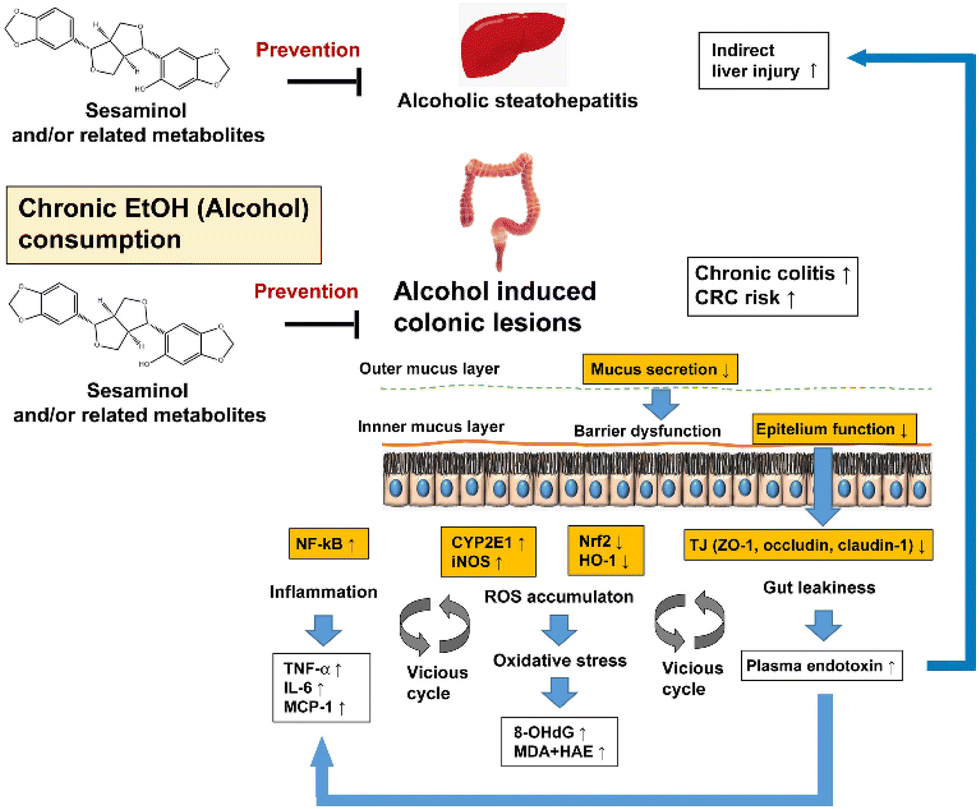 | ||
| Fig. 10 Hypothesis of the preventive effects of sesaminol and/or related metabolites against ethanol-oxi induced dative stress in the colon. | ||
More specifically, the colonic levels of 8-OHdG and lipid peroxidation (MDA + HAE) in group ES were significantly lower than those in group E, but slightly higher than those in groups C and S (Fig. 6), indicating that oral sesaminol administration partly suppressed the ethanol-induced oxidative stress in the mouse colon. This might, at least partly, arise from the antioxidant activities of sesaminol and/or related metabolites (see above) that were delivered to the colonic tissues (Fig. 10). This might also be explained in terms of lower levels of CYP2E1 and iNOS, as well as higher levels of Nrf2 and HO-1, in the colonic tissues of the mice of group ES than in those of group E (see also Fig. 7), although the mechanistic aspects of the effects of oral sesaminol administration on the expressions of these oxidative stress-related proteins remain to be analysed. The mice of group ES showed lower levels of inflammation in the colonic tissues, as shown by the lower levels of an inflammation-related transcription factor (NF-κB), cytokines (TNF-α and IL-6), and a chemokine (MCP-1) (Fig. 8), which most likely arose from the suppression of ethanol-induced oxidative stress (see Fig. 10).
The colonic tissues of the mice of group ES showed lower lesion scores (Fig. 5D), consistent with the lower levels of inflammation. The colonic tissues of the mice of group ES had larger numbers of goblet cells and showed a thicker mucosal layer than those of the mice of group E (Fig. 5A–C; see also Fig. 10). Plasma LPS levels of the mice of group ES were lower than those of group E and slightly higher than the levels of groups C and S (Fig. 9C), suggesting that gut leakiness induced by acetaldehyde in the in vitro model42,43,50 could be partly circumvented by oral sesaminol administration (Fig. 10). This observation could be partly explained in terms of partial recovery of TJ protein levels in the colonic tissues in the mice of group ES (Fig. 9A and B).
In the present study, it was also observed that oral sesaminol administration dramatically suppressed signs of alcoholic liver disease that arose from chronic administration of excess ethanol. Namely, it prevented ethanol-induced liver enlargement (Table 1), steatosis (ESI Fig. 1B and Table 1†), and liver injury (ESI Fig. 1A†), consistent with the fact that the levels of plasma ALT and AST and 8-OHdG in hepatic mtDNA were significantly lower in the mice of group ES than in those of group E (Table 1 and ESI Fig. 2†). Of note, it was recently suggested that sesaminol induces brown and beige adipocyte formation in high-fat diet-conditioned mice,51 and this might be related to the observed suppression of steatosis by oral sesaminol administration. It might be possible that improvement of liver functions via oral sesaminol administration also plays an indirect role in prevention of ethanol-induced colonic oxidative stress, colonic inflammation, colonic lesions, and gut leakiness (Fig. 10).
Gut leakiness and plasma LPS pathways might also be targets to treat patients with alcoholic liver disease (ALD).52 In hepatic immune cells (involving Kupffer cells), plasma LPS causes excessive inflammatory signalling to be induced by TLR4, with the latter being proposed as a therapeutic target for the treatment of chronic ALD (Fig. 10).53
One of the limitations of the present study is that whether the observed effects of oral sesaminol administration arose from sesaminol itself, its metabolite(s), or both was not clarified. Determination of the chemical structures of sesaminol-related metabolites produced in mice would be an important issue to be addressed in future studies. Moreover, the observed effects of oral sesaminol administration may not only arise from the antioxidant activities of sesaminol and its metabolites, but also from other mechanisms. For example, dietary polyphenols have recently been shown to have the ability to modulate expression of microRNAs, which may be partially relevant to their physiological effects.54 The ability of sesaminol and its metabolites to modulate expression of microRNAs in animal cells, however, remains to be clarified. In addition, the minimal amount of dietary supplementation of sesaminol to prevent and lower ER-CRC risk should be determined in future studies. Finally, the gut microbiota play an important role in maintaining gut functions and have been shown to change with long-term chronic ethanol consumption. However, the effects of oral sesaminol administration on gut microbiota structures remain uncertain and will be addressed in future studies.
5. Conclusions
Sesaminol serves as an antioxidant with very low prooxidant activity. Dietary supplementation of sesaminol may lower the risk of ER-CRC by suppressing ethanol-induced oxidative stress, inflammation, and injury in the colon and rectum, as well as gut leakiness, which have been proposed to be important steps in ER-CRC pathogenesis.Author contributions
H. O., Y. K., Y. F., and T. N. conceived and designed the experiments; H. O., D. O., Y. K., W. N., R. M., and Y. A. performed the experiments; H. O., Y. K., D. O., A. O., K. K., and Y. A. analysed the experimental data; H. O., D. O., Y. A., and T. N. performed data curation; H. O., Y. K., D. O., Y. F., and T. N. wrote the manuscript. All authors have read and approved the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was supported in part by JSPS KAKENHI grants (15K14682, 18K11087 to T. N., and 18K19721 to H. O.). The authors are thankful to each facility to which they belong for providing necessary support.References
- T. Mizoue, M. Inoue, K. Wakai, C. Nagata, T. Shimazu, I. Tsuji, T. Otani, K. Tanaka, K. Matsuo, A. Tamakoshi, S. Sasazuki and S. Tsugane, Research Group for Development and Evaluation of Cancer Prevention Strategies in Japan, Alcohol drinking and colorectal cancer in Japanese: a pooled analysis of results from five cohort studies, Am. J. Epidemiol., 2008, 167, 1397–1406 CrossRef PubMed.
- H. K. Seitz and F. Stickel, Molecular mechanisms of alcohol-mediated carcinogenesis, Nat. Rev. Cancer, 2007, 7, 599–612 CrossRef CAS PubMed.
- C. H. Halsted, E. A. Robles and E. Mezey, Distribution of ethanol in the human gastrointestinal tract, Am. J. Clin. Nutr., 1973, 26, 831–834 CrossRef CAS PubMed.
- A. I. Cederbaum, Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity, Dig. Dis., 2010, 28, 802–811 CrossRef PubMed.
- F. P. Guengerich and N. G. Avadhani, Roles of cytochrome P450 in metabolism of ethanol and carcinogens, Adv. Exp. Med. Biol., 2018, 1032, 15–35 CrossRef CAS PubMed.
- H. K. Na and J. Y. Lee, Molecular basis of alcohol-related gastric and colon cancer, Int. J. Mol. Sci., 2017, 18, 1116 CrossRef PubMed.
- M. Rossi, M. J. Anwar, A. Usman, A. Keshavarzian and F. Bishehsari, Colorectal cancer and alcohol consumption-populations to molecules, Cancers, 2018, 10, 38 CrossRef PubMed.
- S. Patel, R. Behara, G. R. Swanson, C. B. Forsyth, R. M. Voigt and A. Keshavarzian, Alcohol and the intestine, Biomolecules, 2015, 5, 2573–2588 CrossRef CAS PubMed.
- C. B. Forsyth, R. M. Voigt and A. Keshavarzian, Intestinal CYP2E1: a mediator of alcohol-induced gut leakiness, Redox Biol., 2014, 3, 40–46 CrossRef CAS PubMed.
- M. Meroni, M. Longo and P. Dongiovanni, Alcohol or gut microbiota: who Is the guilty?, Int. J. Mol. Sci., 2019, 20, 4568 CrossRef CAS PubMed.
- S. K. Das and D. M. Vasudevan, Alcohol-induced oxidative stress, Life Sci., 2007, 81, 177–187 CrossRef CAS PubMed.
- D. Wu and A. I. Cederbaum, Alcohol, oxidative stress, and free radical damage, Alcohol Res. Health, 2003, 27, 277–284 Search PubMed.
- K. Linhart, H. Bartsch and H. K. Seitz, The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts, Redox Biol., 2014, 3, 56–62 CrossRef CAS PubMed.
- G. Vanella, L. Archibugi, S. Stigliano and G. Capurso, Alcohol and gastrointestinal cancers, Curr. Opin. Gastroenterol., 2019, 35, 107–113 CrossRef CAS PubMed.
- V. Fedirko, I. Tramacere, V. Bagnardi, M. Rota, L. Scotti, F. Islami, E. Negri, K. Straif, I. Romieu, C. L. Vecchia, P. Boffetta and M. Jenab, Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies, Ann. Oncol., 2011, 22, 1958–1972 CrossRef CAS PubMed.
- A. Tsuruya, A. Kuwahara, Y. Saito, H. Yamaguchi, T. Tsubo, S. Suga, M. Inai, Y. Aoki, S. Takahashi, E. Tsutsumi, Y. Suwa, H. Morita, K. Kinoshita, Y. Totsuka, W. Suda, K. Oshima, M. Hattori, T. Mizukami, A. Yokoyama, T. Shimoyama and T. Nakayama, Ecophysiological consequences of alcoholism on human gut microbiota: implications for ethanol-related pathogenesis of colon cancer, Sci. Rep., 2016, 6, 27923 CrossRef CAS PubMed.
- H. Ohira, A. Tsuruya, D. Oikawa, W. Nakagawa, R. Mamoto, M. Hattori, T. Waki, S. Takahashi, Y. Fujioka and T. Nakayama, Alteration of oxidative-stress and related marker levels in mouse colonic tissues and fecal microbiota structures with chronic ethanol administration: implications for the pathogenesis of ethanol-related colorectal cancer, PLoS One, 2021, 16, e0246580 CrossRef CAS PubMed.
- M. Andargie, M. Vinas, A. Rathgeb, E. Möller and P. Karlovsky, Lignans of sesame (sesamum indicum L.): a comprehensive review, Molecules, 2021, 26, 883 CrossRef CAS PubMed.
- M. H. Kang, H. Katsuzaki and T. Osawa, Inhibition of 2,2′-azobis(2,4-dimethylvaleronitrile)-induced lipid peroxidation by sesaminols, Lipids, 1998, 33, 1031–1036 CrossRef CAS PubMed.
- M. H. Kang, M. Naito, K. Sakai, K. Uchida and T. Osawa, Mode of action of sesame lignans in protecting low-density lipoprotein against oxidative damage in vitro, Life Sci., 2000, 66, 161–171 CrossRef CAS PubMed.
- H. Kaji, I. Matsui-Yuasa, K. Matsumoto, A. Omura, K. Kiyomoto and A. Kojima-Yuasa, Sesaminol prevents Parkinson's disease by activating the Nrf2-ARE signaling pathway, Heliyon, 2020, 6, e05342 CrossRef PubMed.
- M. Y. Um, J. Y. Ahn, S. Kim, M. K. Kim and T. Y. Ha, Sesaminol glucosides protect beta-amyloid peptide-induced cognitive deficits in mice, Biol. Pharm. Bull., 2009, 32, 1516–1520 CrossRef CAS PubMed.
- A. W. Boots, G. R. M. M. Haenen and A. Bast, Health effects of quercetin: From antioxidant to nutraceutical, Eur. J. Pharmacol., 2008, 585, 325–337 CrossRef CAS PubMed.
- A. Nair, A. Kuwahara, A. Nagase, H. Yamaguchi, T. Yamazaki, M. Hosoya, A. Omura, K. Kiyomoto, M. Yamaguchi, T. Shimoyama, S. Takahashi and T. Nakayama, Purification, gene cloning, and biochemical characterization of a β-glucosidase capable of hydrolyzing sesaminol triglucoside from Paenibacillus sp. KB0549, PLoS One, 2013, 8, e60538 CrossRef CAS PubMed.
- I. Parejo, C. Petrakis and P. Kefalas, A transition metal enhanced luminol chemiluminescence in the presence of a chelator, J. Pharmacol. Toxicol. Methods, 2000, 43, 183–190 CrossRef CAS PubMed.
- O. I. Aruoma, B. Halliwell, E. Gajewski and M. Dizdaroglu, Copper-ion-dependent damage to the bases in DNA in the presence of hydrogen peroxide, Biochem. J., 1991, 273, 601–604 CrossRef CAS PubMed.
- A. Jahagirdar, D. Usharani, M. Srinivasan and R. Rajasekharan, Sesaminol diglucoside, a water-soluble lignan from sesame seeds induces brown fat thermogenesis in mice, Biochem. Biophys. Res. Commun., 2018, 507, 155–160 CrossRef CAS PubMed.
- F. Guo, K. Zheng, R. Benedé-Ubieto, F. J. Cubero and Y. A. Nevzorova, The Lieber-DeCarli Diet-a flagship model for experimental alcoholic liver disease, Alcohol.: Clin. Exp. Res., 2018, 42, 1828–1840 CrossRef PubMed.
- C. B. Appleyard and J. L. Wallace, Reactivation of hapten-induced colitis and its prevention by anti-inflammatory drugs, Am. J. Physiol., 1995, 269, G119–G125 CAS.
- G. Tahan, R. Gramignoli, F. Marongiu, S. Aktolga, A. Cetinkaya, V. Tahan and K. Dorko, Melatonin expresses powerful anti-inflammatory and antioxidant activities resulting in complete improvement of acetic-acid-induced colitis in rats, Dig. Dis. Sci., 2011, 56, 715–720 CrossRef CAS PubMed.
- J. L. Ding and B. Ho, Endotoxin detection–from limulus amebocyte lysate to recombinant factor C, Subcell. Biochem., 2010, 53, 187–208 CAS.
- L. H. Maurer, C. B. B. Cazarin, A. Quatrin, N. M. Minuzzi, S. M. Nichelle, C. d. A. Lamas, V. H. A. Cagnon, J. Morari, L. A. Velloso, M. R. Maróstica Júnior and T. Emanuelli, Grape peel powder attenuates the inflammatory and oxidative response of experimental colitis in rats by modulating the NF-κB pathway and activity of antioxidant enzymes, Nutr. Res., 2020, 76, 52–70 CrossRef CAS PubMed.
- L. M. Cruz-Orive and E. R. Weibel, Recent stereological methods for cell biology: a brief survey, Am. J. Physiol., 1990, 258, L148–L156 CAS.
- J. A. Imlay, S. M. Chin and S. Linn, Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro, Science, 1988, 240, 640–642 CrossRef CAS PubMed.
- B. Lipinski, Hydroxyl radical and its scavengers in health and disease, Oxid. Med. Cell. Longevity, 2011, 2011, 809696 Search PubMed.
- S. K. Mantena, A. L. King, K. K. Andringa, H. B. Eccleston and S. M. Bailey, Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases, Free Radical Biol. Med., 2008, 44, 1259–1272 CrossRef CAS PubMed.
- M. A. Abdelmegeed, A. Banerjee, S. Jang, S. Yoo, J. Yun, F. J. Gonzalez, A. Keshavarzian and B. Song, CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis, Free Radical Biol. Med., 2013, 65, 1238–1245 CrossRef CAS PubMed.
- J. Lee, T. Oh, Y. Kim, J. Baik, S. So, K. Hahm and Y. Surh, Protective effects of green tea polyphenol extracts against ethanol-induced gastric mucosal damages in rats: stress-responsive transcription factors and MAP kinases as potential targets, Mutat. Res., 2005, 579, 214–224 CrossRef CAS PubMed.
- J. C. Onyiah, S. Z. Sheikh, N. Maharshak, L. E. Otterbein and S. E. Plevy, Heme oxygenase-1 and carbon monoxide regulate intestinal homeostasis and mucosal immune responses to the enteric microbiota, Gut Microbes, 2014, 5, 220–224 CrossRef PubMed.
- A. Loboda, M. Damulewicz, E. Pyza, A. Jozkowicz and J. Dulak, Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism, Cell. Mol. Life Sci., 2016, 73, 3221–3247 CrossRef CAS PubMed.
- H.-K. Na and J. Y. Lee, Molecular basis of alcohol-related gastric and colon cancer, Int. J. Mol. Sci., 2017, 18, 1116 CrossRef PubMed.
- K. J. Atkinson and R. K. Rao, Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions, Am. J. Physiol.: Gastrointest. Liver Physiol., 2001, 280, G1280–G1288 CrossRef CAS PubMed.
- S. Basuroy, P. Sheth, C. M. Mansbach and R. K. Rao, Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine, Am. J. Physiol.: Gastrointest. Liver Physiol., 2005, 289, G367–G375 CrossRef CAS PubMed.
- D. Dragos, M. Gilca, L. Gaman, A. Vlad, L. Iosif, I. Stoian and O. Lupescu, Phytomedicine in Joint Disorders, Nutrients, 2017, 9, 70 CrossRef PubMed.
- N. Yamashita, H. Tanemura and S. Kawanishi, Mechanism of oxidative DNA damage induced by quercetin in the presence of Cu(II), Mutat Res., 1999, 425, 107–115 CrossRef CAS PubMed.
- J. Tan, B. Wang and L. Zhu, DNA binding and oxidative DNA damage induced by a quercetin copper(II) complex: potential mechanism of its antitumor properties, J. Biol. Inorg. Chem., 2009, 14, 727–739 CrossRef CAS PubMed.
- K. Jan, K. Ku, Y. Chu, L. S. Hwang and C. Ho, Intestinal distribution and excretion of sesaminol and its tetrahydrofuranoid metabolites in rats, J. Agric. Food Chem., 2011, 59, 3078–3086 CrossRef CAS PubMed.
- M. Mochizuki, Y. Tsuchie, Y. Nakamura and T. Osawa, Identification and characterization of sesaminol metabolites in the liver, J. Agric. Food Chem., 2009, 57, 10429–10434 CrossRef CAS PubMed.
- K. Jan, Y. Chang, L. S. Hwang and C. Ho, Tissue distribution and cytochrome P450 inhibition of sesaminol and its tetrahydrofuranoid metabolites, J. Agric. Food Chem., 2012, 60, 8616–8623 CrossRef CAS PubMed.
- P. K. Shukla, A. S. Meena, K. Dalal, C. Canelas, G. Samak, J. F. Pierre and R. Rao, Chronic stress and corticosterone exacerbate alcohol-induced tissue injury in the gut-liver-brain axis, Sci. Rep., 2021, 11, 826 CrossRef CAS PubMed.
- S. J. Divakaran, S. Srivastava, A. Jahagirdar, R. Rajendran, S. V. Sukhdeo and S. Rajakumari, Sesaminol induces brown and beige adipocyte formation through suppression of myogenic program, FASEB J., 2020, 34, 6854–6870 CrossRef CAS PubMed.
- B. Gao and R. Bataller, Alcoholic liver disease: pathogenesis and new therapeutic targets, Gastroenterology, 2011, 141, 1572–1585 CrossRef CAS PubMed.
- A. Mencin, J. Kluwe and R. F. Schwabe, Toll-like receptors as targets in chronic liver diseases, Gut, 2009, 58, 704–720 CrossRef CAS PubMed.
- S. Yamada, S. Tsukamoto, Y. Huang, A. Makio, M. Kumazoe, S. Yamashita and H. Tachibana, Epigallocatechin-3-O-gallate up-regulates microRNA-let-7b expression by activating 67 kDa laminin receptor signaling in melanoma cells, Sci. Rep., 2016, 6, 19225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1fo04120g |
| This journal is © The Royal Society of Chemistry 2022 |