
An electrochemical method for deborylative selenylation of arylboronic acids under metal- and oxidant-free conditions†
Zhengjiang
Fu
 *ab,
Jian
Yin
a,
Dongdong
He
a,
Xuezheng
Yi
a,
Shengmei
Guo
a and
Hu
Cai
*a
*ab,
Jian
Yin
a,
Dongdong
He
a,
Xuezheng
Yi
a,
Shengmei
Guo
a and
Hu
Cai
*a
aCollege of Chemistry, Nanchang University, Nanchang, Jiangxi 330031, China. E-mail: fuzhengjiang@ncu.edu.cn; caihu@ncu.edu.cn
bState Key Laboratory of Structural Chemistry, Fuzhou, Fujian 350002, China
First published on 30th November 2021
Abstract
An efficient protocol to synthesize aryl selenoethers through deborylative selenylation of widely available arylboronic acids has been established under electrochemical conditions in the absence of metal catalyst and external oxidant. The synthesis of bioactive molecules and gram-scale transformation have been performed to highlight the synthetic utility of the protocol. CV (cyclic voltammetry) experiment indicates that anodic oxidation of diselenide occurs prior to that of the arylboronic acid substrate under the standard conditions.
Organic selenium compounds are not only of great interest as versatile synthetic intermediates for synthetic chemists to create more complex molecules,1 but as essential structural constituents in a wealth of functional materials and potential drug candidates.2 For example, several aryl selenide moieties with N- or O-containing substituents on the aromatic ring have been proven to possess anti-Alzheimer,2c antitubulin and cytotoxic activities,2d as well as the effect of 5-lipoxygenase inhibition2e (Fig. 1). Therefore, the fundamental importance of organoselenides has fascinated synthetic chemists. In order to efficiently synthesize organoselenides, reported methods usually involved selenylation of aryl halides3 or C–H selenation of arenes2c,4 with selenol(ate)s, diselenides or selenium powder as selenation agents. Bench-stable organoboronic acids are common chemicals, which have significant advantages of simple operation and distinct reaction site. In this regard, Taniguchi5 and Ranu6 described the methods for Cu-catalyzed preparation of monoselenides from organoboronic acids with diselenides, Wu and coworkers7 developed Ag-catalyzed cyclization of arylboronic acids with elemental selenium for the synthesis of selenaheterocycles. However, the aforementioned protocols often suffered from certain issues for the requirement of transition metal catalysts, external oxidants and harsh conditions with high temperature, which restricts their practical applications and further improvement, especially the residual trace metals may result in a contamination to final product. From the perspective of sustainability and green synthesis, it is desirable to develop an environmentally friendly, efficient and simple selenization process.
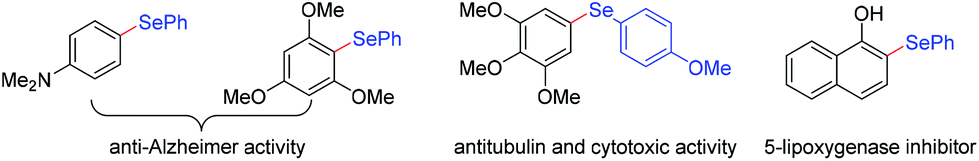 | ||
| Fig. 1 The examples of aryl selenide moieties with N- or O-containing substituents. | ||
As an efficient and environmentally benign synthetic strategy, electro-organic synthesis has attracted widespread attention in recent years, which can minimize waste dramatically and improve functional group tolerance significantly by using electrons as the “reagent” instead of stoichiometric amounts of oxidants or reductants in conventional synthetic methods.8 Employing easily available arylboronic acids as starting material, in this context, our group has recently gained considerable knowledge on both metal- and oxidant-free electrochemical methods for deborylative halogenation and seleno/thiocyanation of various arylboronic acids.9a,b Toward the synthesis of selenylated compounds under electrochemical conditions, the groups of Sun,10 Mendes,11 Xu,12 and Cao13 reported iodide ion-mediated electrochemical oxidative C(sp2)–H selenylation of arenes with diselenides. Lei revealed electro-induced aminoselenation and oxyselenation of styrenes to prepare vicinal difunctionalized organoselenium compounds,14 while Braga,15 Pan16 and Sarkar17 developed electrochemical oxidative selenation-cyclization conversion between diselenides and various coupling partners including allylnaphthols, allylphenols, acrylamides, β,γ-unsaturated amides and oximes. Nevertheless, to the best of our knowledge, metal-free electrocatalytic selenylation of cheap arylboronic acids to streamline the synthesis of unsymmetrical (hetero)aryl selenides is important and remains underexplored. As a part of our efforts to continuously develop cheap and eco-friendly catalytic methods under electrochemical conditions,9 to this end, we herein firstly described an electrochemical method for deborylative selenylation of arylboronic acids under metal-free and oxidant-free conditions.
At the outset, electrochemical deborylative selenylation between p-methoxyphenylboronic acid (1a) and dimethyl diselenide (2a) in an undivided cell was chosen as the model reaction to probe various reaction parameters under electrolytic conditions (Table 1). Employing a two-electrode system with a platinum plate anode and cathode, when deborylative selenylation of p-methoxyphenylboronic acid (1a) was performed with 1.2 equivalents of MeSeSeMe under constant-current electrolysis (6 mA) in CH3CN (10 mL) at 50 °C for 3 h, to our delight, the transformation afforded desired p-methoxyselenoanisole (3aa) in an isolated yield of 86% (Table 1, entry 1). Notably, replacing platinum plate with graphite felt as anode or cathode furnished reduced efficiency, thus, electrode material had a significant effect on the transformation (entries 2 and 3). As for the choice of electrolyte, decreased yield was obtained when nBu4NBr was used as electrolyte under otherwise identified conditions (entry 4), moreover, the introduction of nBu4NOAc instead of nBu4NBF4 led to a shut-off for the desired transformation, presumably because of the decomposition of nBu4NOAc via Kolbe electrolysis under this electrochemical condition (entry 5). Elaborate solvent screening showed that the effect of CH3CN was better than CH2Cl2 or CH3OH (entries 6 and 7), which might be due to higher conductivity and better solubility of acetonitrile for the reaction mixtures. Unfortunately, either reducing or elevating reaction temperature led to a diminished outcome, therefore, the reaction temperature (50 °C) was the best for the transformation (entries 8 and 9). On the other hand, decreasing the operating current or shortening reaction time did not improve the conversion efficiency (entries 10 and 11). Finally, control experiment demonstrated that no reaction took place in the absence of electric current under otherwise equal conditions (entry 12), implying that the conversion could be unambiguously controlled by switching the electric current on or off.
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Conditions: 1a (0.2 mmol), MeSeSeMe (1.2 equiv., 0.24 mmol), nBu4NBF4 (1 equiv., 0.2 mmol), CH3CN (10 mL) in an undivided cell with two platinum plates (each 1.0 × 1.0 cm2), air, 50 °C, 6 mA, 3 h (3.36 F mol−1). b Yield of isolated product. | ||
| 1 | None | 86 |
| 2 | C (+) instead of Pt (+) | 46 |
| 3 | C (−) instead of Pt (−) | 62 |
| 4 | nBu4NBr instead of nBu4NBF4 | 54 |
| 5 | nBu4NOAc instead of nBu4NBF4 | 0 |
| 6 | CH2Cl2 instead of CH3CN | 35 |
| 7 | CH2OH instead of CH3CN | 43 |
| 8 | 25 °C instead of 50 °C | 54 |
| 9 | 60 °C instead of 50 °C | 72 |
| 10 | 4 mA instead of 6 mA | 63 |
| 11 | 2 h instead of 3 h | 62 |
| 12 | No electric current | 0 |
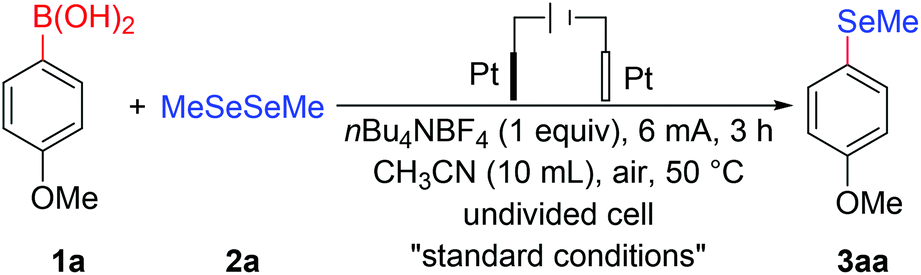
With the optimized reaction conditions in hand, we investigated the scope of arylboronic acid substrates to evaluate generality of the protocol. As depicted in Table 2, electrochemical deborylative selenylation transformations between various arylboronic acids (1a–n) and dimethyl diselenide (2a) readily provided desired aryl methyl selenoethers (3aa-na) with good functional group tolerance in moderate to satisfactory yields. In general, arylboronic acids bearing electron-donating groups showed good reaction efficiency, whereas the substrates with halide substituents afforded low yields as a result of low conversion. Under optimized reaction conditions, although methoxy-substituted phenylboronic acid (1a–c) gave deborylative selenylation products in satisfactory yields, para-substituted phenylboronic acid (1a) showed better efficiency than that of meta-and ortho-substituted counterparts (1b–c), thus the para-substituent had a noticeable effect on the reaction efficiency. Compared to 4-methoxyphenylboronic acid (1a), a slightly decreased yield was obtained for electrochemical deborylative selenylation of 2,4-dimethoxyphenylboronic acid (1d), presumably due to steric encumbrance of ortho-substituted group. Although inert atmosphere was employed to prevent aldehyde group from further oxidation, 3-formyl-4-methoxyphenylboronic acid (1e) furnished a decreased yield probably owing to inductive effect of aldehyde group. Besides 4-methoxyphenylboronic acid (1a), the substrates bearing electron-rich substituents at para-position, such as 4-hydroxybenzeneboronic acid (1f), p-cumeneboronic acid (1g) and p-biphenylboronic acid (1h), produced corresponding organoselenium compounds with satisfactory yields. Fortunately, both ring-fused α-naphthylboronic acid (1i) and β-naphthylboric acid (1j) were all compatible to form designated products at synthetically useful levels. Albeit in moderate yields, it's found that p-halophenylboronic acids (1k–l) were amenable to giving halo-substituted products with intactness of halo-moieties at room temperature, which offered late-stage modification through classical activation of C-Halo bond to provide further potential application of this methodology. Since the inherently good activity of C3-substituted fused five-membered heterocyclics, and oxygen is more electronegative than sulfur, the desired product yield for benzo[b]thien-3-ylboronic acid (1m) involved in the reaction was higher than that of 2-benzofuranboronic acid (1n) participating in the transformation.
| a Arylboronic acid (0.2 mmol), MeSeSeMe (1.2 equiv., 0.24 mmol), nBu4NBF4 (1 equiv., 0.2 mmol), CH3CN (10 mL) in an undivided cell with two platinum plates (each 1.0 × 1.0 cm2), air, 50 °C, 6 mA, 3 h (3.36 F mol−1). b N2, 2 h (2.24 F mol−1). c CH3CN/HOAc (9: 1, 10 mL), 60 °C, 3 h (3.36 F mol−1). d 60 °C, 2 h (2.24 F mol−1). e CH2Cl2 (10 mL), rt. f 2 h (2.24 F mol−1). g 1.5 h (1.68 F mol−1). |
|---|
|
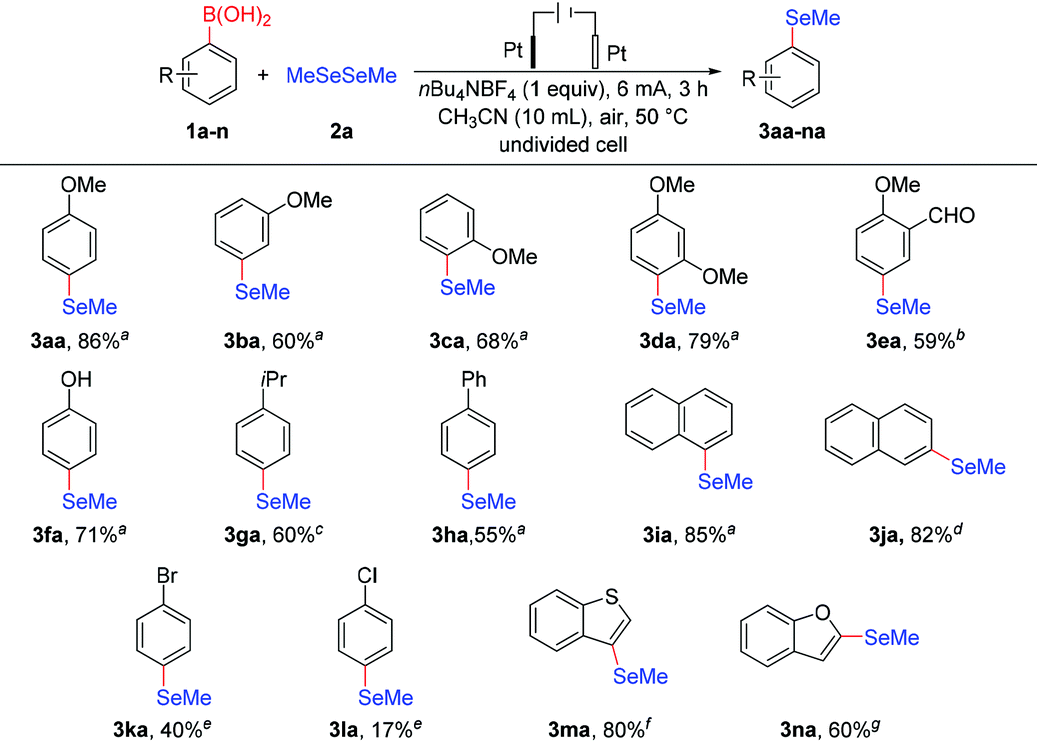
Encouraged by the feasibility of variously substituted phenylboronic acids as coupling partners in electrochemical deborylative selenylation, we subsequently proceeded to explore the scope with respect to a series of diselenides. As summarized in Table 3, we were delighted to find the protocol was quite general and could be extended to both dialkyl (2b–e) and diaryl (2f–r) diselenides, which underwent the transformation smoothly to form corresponding products in moderate to good yields. Aside from dimethyl diselenide (2a), other dialkyl diselenides including primary (2b–d) and secondary (2e) alkyl groups were competent substrates to furnish desired alkyl aryl selenides under the current conditions. For the case of diaryl diselenides substrates, this electrochemical deborylative selenylation conversion was shown to be effective for a broad range of diphenyl diselenides (2f–o) as well as N- and S-containing diheteroaryl diselenides (2p–s). It's noteworthy that diphenyl diselenide (2k) worked well to afford the desired product in 85% yield, which was also the divide between electron-donating (methyl, isopropyl and methoxy) and electron-withdrawing (chloro, bromo and fluoro) substituents at different positions on the aromatic ring. Albeit with moderate yield in the case of bis(2-bromophenyl) diselenide (2l), the substrates with ortho- (2f), para- (2h) and 2,6-dimethyl (2g) gave the similar yields, thus increased steric encumbrance had a negative influence on the transformation. Likewise, the coupling partners (2i–j) of diselenide with isopropyl and methoxy substituents at the para-position delivered target products at synthetically useful levels respectively. Although strong electronegativity of fluorine made substrate (2o) more electron-deficient than bromo or chloro-substituted substrates (2m–n), bis(4-fluorophenyl) diselenide (2o) still achieved a yield as good as the substrates (2m–n) under standard electrochemi-cal conditions, which also provided the possibility for further de-rivatization of the products. In particular, 2,2′-dipyridyl diselenide (2p) afforded hoped-for product with almost complete consumption of coupling partner 4-methoxyphenylboronic acid, while 3,3′-dipyridyl diselenide (2q) gave nearly as excellent yield as its analogue 2,2′-dipyridyl diselenide under slightly modified conditions. In addition to six-membered heteroaromatic diselenides, dithienyl diselenides (2r–s) underwent the transformations smoothly to provide anticipated products in 65% and 75% yields respectively, either at α or β position of thiophene.
| a Arylboronic acid (0.2 mmol), RSeSeR (1.2 equiv., 0.24 mmol), nBu4NBF4 (1 equiv., 0.2 mmol), CH3CN (10 mL) in an undivided cell with two platinum plates (each 1.0 × 1.0 cm2), air, 50 °C, 6 mA, 3 h (3.36 F mol−1). b nBu4NBr (1 equiv., 0.2 mmol) with carbon cloth (1.5 × 1.5 cm2) as anode and platinum plate (1.0 × 1.0 cm2) as cathode. c nBu4NBr (1 equiv., 0.2 mmol) with graphite felt (1.5 × 1.5 cm2) as anode and platinum plate (1.0 × 1.0 cm2) as cathode. |
|---|
|
As described above, some aryl selenides tolerated N-containing substituent exhibit anti-Alzheimer activity,2c while bearing O-containing functional group showcase the effect of 5-lipoxygenase inhibition (see Fig. 1).2e As indicated in Scheme 1A, interestingly, electrochemical deborylative selenylation of p-(dimethylamino)phenylboronic acid (1o) or 3,4,5-trimethoxyphenylboronic acid (1p) with diaryl diselenide to furnish biologically relevant compounds N,N-dimethyl-4-(phenylseleno)benzenamine (3ok) or 1,2,3-trimethoxy-5-((4-methoxyphenyl)seleno)benzene (3pa) at synthetically useful levels under standard electrochemical conditions in the absence of metal catalyst and exogenous oxidants. Furthermore, a 25-fold scale-up electrochemical deborylative selenylation between 4-methoxyphenylboronic acid (1a) and diphenyl diselenide (2k) was performed on gram scale under the standard conditions with 500 mL undivided cell setup, as outlined in Scheme 1B, the desired product 4-(phenylseleno)an-isole (3ak) was obtained in 75% isolated yield (0.99 g) under constant-current electrolysis for 24 h. Consequently, the robustness of scale-up transformation underscored the synthetic utility of this electrochemical approach for practical organic synthesis.
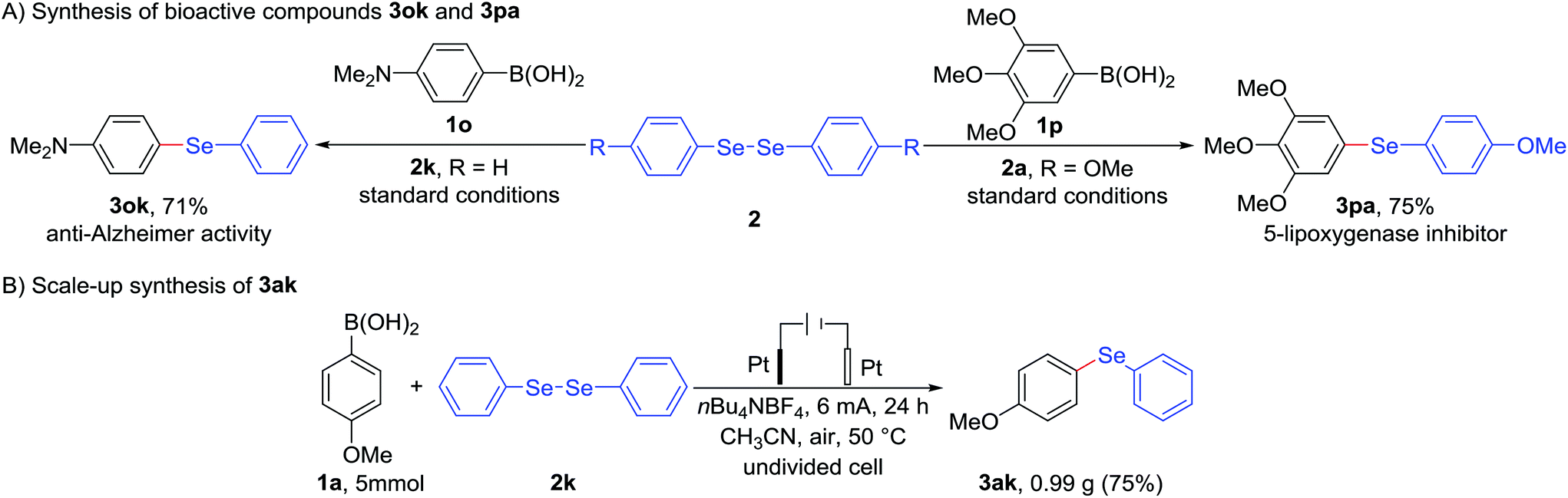 | ||
| Scheme 1 Preparation of biologically relevant compounds 3ok and 3pa, and gram-scale synthesis of 4-(phenylseleno)anisole 3ak. | ||
In order to gain insight into reaction mechanism, several experiments were carried out. Firstly, the key role of electrical current was confirmed by control experiment, which was necessary for this deborylative selenylation transformation (Table 1, entry 12). Secondly, electrochemical deborylative selenylation between p-methoxyphenylboronic acid (1a) and dimethyl diselenide (2a) was performed in the presence of 2 equiv. of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) or BHT (2,4-di-tert-butyl-4-methylphenol) under otherwise identified conditions, the drastically diminished yields suggested the involvement of a radical pathway in the process (Scheme 2). Furthermore, the coupling partners of 4-methoxyphenylboronic acid (1a) and diphenyl diselenide (2k) were subjected to CV experiments. For the case of PhSe-SePh (2k), an oxidation peak was observed at 1.85 V (Fig. 2A, curve b), while a reduction peak was obtained at – 1.40 V under the reaction solvent system (Fig. 2B, curve b). Therefore, PhSe-SePh might involve both oxidation and reduction processes in electrochemical deborylative selenylation transformation. In contrast, the CV of 4-methoxyphenylboronic acid (1a) presented a higher oxidation peak (2.12 V, Fig. 2A, curve c) than that of PhSe-SePh (Fig. 2A, curve b). As a result, the comparison highlighted that the oxidation of diselenide should be prior to that of arylboronic acid substrate under this galvanostatic mode.
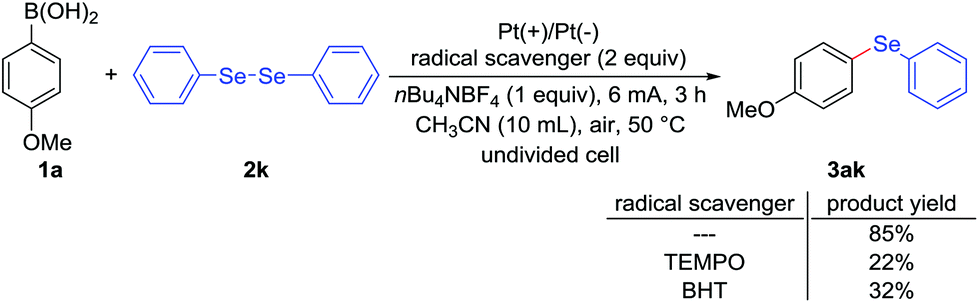 | ||
| Scheme 2 Experiments with radical scavengers. | ||
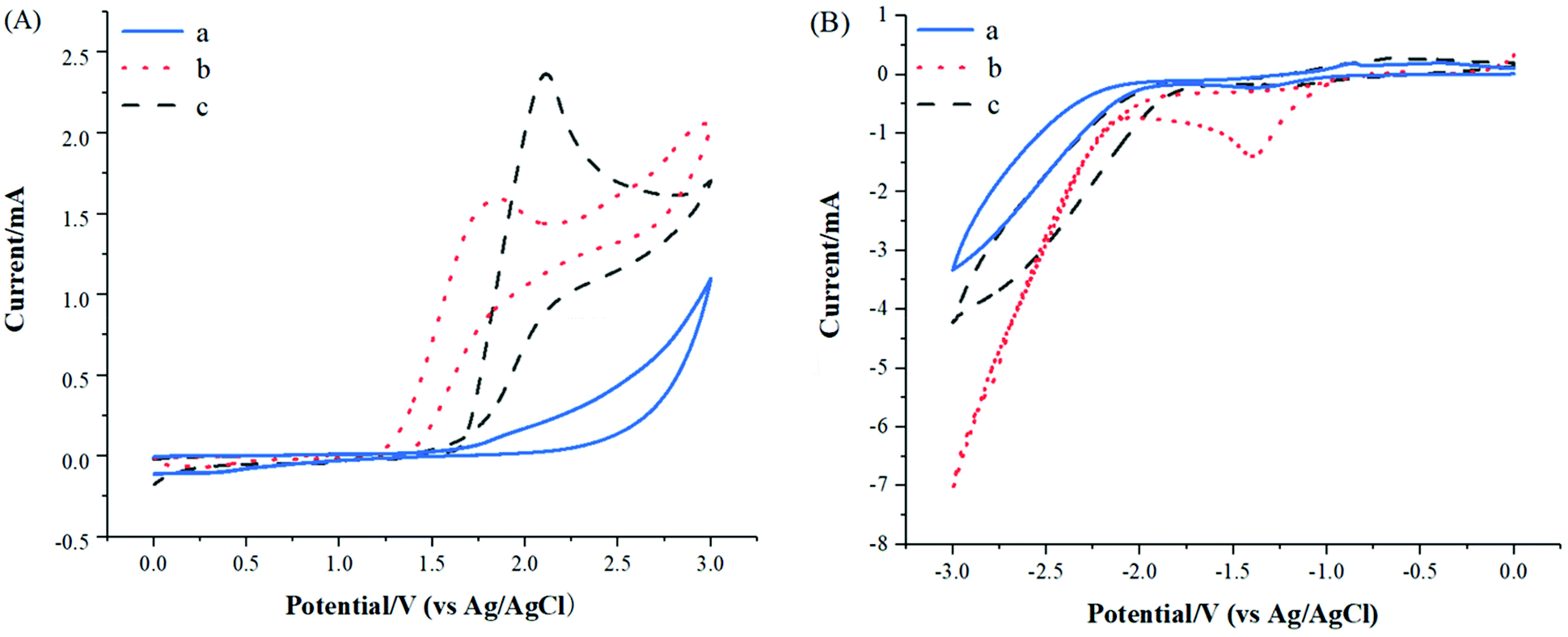 | ||
| Fig. 2 Cyclic voltammograms of substrates: (a) background (nBu4NBF4 0.02 mol L−1 in CH3CN); (b) diphenyl diselenide (0.02 mol L−1); (c) 4-methoxyphenylboronic acid (0.02 mol L−1). | ||
Although a detailed mechanism should await further investi-gation, on the basis of preliminary mechanistic studies and literature reports,9,14–18 a plausible mechanism of electrochemical deborylative selenylation was proposed in Scheme 3. Initially, anodic oxidation of diselenide RSeSeR led to the generation of selenium cation RSe+ and seleno radical RSe˙. After that, the addition of the seleno radical RSe˙ to arylboronic acid I and then the following oxidation to furnish aryl cation III through intermediate II at the anode, and rearomatization of the intermediate III by losing +B(OH)2 afforded the desired aryl selenide product. At the cathode, electro-reduction of +B(OH)2 generated tetrahydroxydiboron B2(OH)4 detected by LC-MS, concomitant regeneration of starting material RSeSeR via reduction of generated selenium cation RSe+ intermediate.
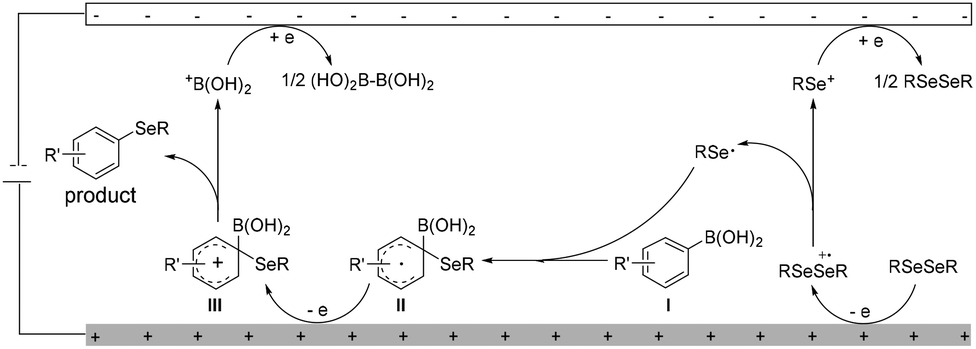 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, an effective electrochemical deborylative selenylation of readily accessible arylboronic acids with various diselenides was realized in an undivided cell under metal-free and external oxidant-free conditions, which provided a simple and green way to synthesize value-added aryl selenoethers. The synthesis of bioactive molecules and gram-scale reaction were conducted to evaluate the practicability of the method. Further study of the application of this approach is underway, and the results will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (NSFC) (21761021 and 21861026) is gratefully acknowledged.Notes and references
- (a) D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409–8411 CrossRef CAS PubMed; (b) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed; (c) J. Luo, Z. Zhu, Y. Liu and X. D. Zhao, Org. Lett., 2015, 17, 3620–3623 CrossRef CAS PubMed.
- (a) R. L. Brutchey, Acc. Chem. Res., 2015, 48, 2918–2926 CrossRef CAS; (b) A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Chem. Mater., 2011, 23, 896–906 CrossRef CAS; (c) J. Rodrigues, S. Saba, A. C. Joussef, J. Rafique and A. Braga, Asian J. Org. Chem., 2018, 7, 1819–1824 CrossRef CAS; (d) E. A. Santos, E. Hamel, R. Bai, J. C. Brunett, C. S. S. Tozatti, D. Bogo, R. T. Perdomo, A. M. M. Antunes, M. M. Marques, M. F. C. Matos and D. P. Llima, Bioorg. Med. Chem. Lett., 2013, 23, 4669–4673 CrossRef; (e) L. Engman, D. Stern, H. Frisell, K. Vessman, B. Ek and C. M. Andersson, Bioorg. Med. Chem. Lett., 1995, 3, 1255–1262 CrossRef CAS.
- (a) E. Senol, T. Scattolin and F. Schoenebeck, Chem. – Eur. J., 2019, 25, 9419–9422 CrossRef CAS PubMed; (b) C. Cremer, M. A. Eltester, H. Bourakhouadar, I. L. Atodiresei and F. W. Patureau, Org. Lett., 2021, 23, 3243–3247 CrossRef CAS PubMed; (c) F. Li, D. Wang, H. Chen, Z. He, L. Zhou and Q. Zeng, Chem. Commun., 2020, 56, 13029–13032 RSC.
- (a) X. Zhou, J. Hao, Y. Kong and R. Xu, Chem. Commun., 2021, 57, 5426–5429 RSC; (b) L. Gu, X. Fang, Z. Weng, Y. Song and W. Ma, Eur. J. Org. Chem., 2019, 1825–1829 CrossRef CAS; (c) L. Gu, X. Fang, Z. Weng, J. Lin, M. He and W. Ma, Adv. Synth. Catal., 2019, 361, 4998–5004 CrossRef CAS; (d) M. Iwasaki, N. Miki, Y. Tsuchiya, K. Nakajima and Y. Nishihara, Org. Lett., 2017, 19, 1092–1095 CrossRef CAS; (e) X.-H. Jiang, L.-S. Zhang, H.-Y. Liu, D.-S. Wu, F.-Y. Wu, L. Tian, L.-L. Liu, J.-P. Zou, S.-L. Luo and B.-B. Chen, Angew. Chem., Int. Ed., 2020, 59, 23112–23116 CrossRef CAS; (f) W. Ma, Z. Weng, T. Rogge, L. Gu, J. Lin, A. Peng, X. Luo, X. Gou and L. Ackermann, Adv. Synth. Catal., 2018, 360, 704–710 CrossRef CAS; (g) W. Song, J. Shi, X. Chen and G. Song, J. Org. Chem., 2020, 85, 11104–11115 CrossRef CAS; (h) P. Ghosh, G. Chhetri, E. Perl and S. Das, Adv. Synth. Catal., 2021, 363, 2148–2156 CrossRef CAS.
- N. Taniguchi, J. Org. Chem., 2007, 72, 1241–1245 CrossRef CAS.
- D. Kundu, N. Mukherjee and B. C. Ranu, RSC Adv., 2013, 3, 117–125 RSC.
- X. Zhang, X. Huang, W. Gao, Y. Zhou, M. Liu and H. Wu, Adv. Synth. Catal., 2020, 362, 5639–5644 CrossRef CAS.
- (a) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS; (b) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef; (c) L. Lu, H. Li, Y. Zheng, F. Bu and A. Lei, CCS Chem., 2020, 2, 2669–2675 Search PubMed; (d) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- (a) Z. Fu, G. Hao, Y. Fu, D. He, X. Tuo, S. Guo and H. Cai, Org. Chem. Front., 2020, 7, 590–595 RSC; (b) D. He, J. Yao, B. Ma, J. Wei, G. Hao, X. Tuo, S. Guo, Z. Fu and H. Cai, Green Chem., 2020, 22, 1559–1564 RSC; (c) S. Guo, S. Li, W. Yan, Z. Liang, Z. Fu and H. Cai, Green Chem., 2020, 22, 7343–7347 RSC; (d) L. Sun, L. Wang, H. Alhumade, H. Yi, H. Cai and A. Lei, Org. Lett., 2021, 23, 7724–7729 CrossRef CAS.
- X. Zhang, C. Wang, H. Jiang and L. Sun, Chem. Commun., 2018, 54, 8781–8784 RSC.
- A. G. Meirinho, V. F. Pereira, G. M. Martins, S. Saba, J. Rafique, A. L. Braga and S. R. Mendes, Eur. J. Org. Chem., 2019, 6465–6469 CrossRef CAS.
- Q. Wang, X.-L. Ma, Y.-Y. Chen, C.-N. Jiang and Y.-L. Xu, Eur. J. Org. Chem., 2020, 4384–4388 CrossRef CAS.
- X. Liu, Y. Wang, D. Song, Y. Wang and H. Cao, Chem. Commun., 2020, 56, 15325–15328 RSC.
- L. Sun, Y. Yuan, M. Yao, H. Wang, D. Wang, M. Gao, Y.-H. Chen and A. Lei, Org. Lett., 2019, 21, 1297–1300 CrossRef CAS.
- M. R. Scheide, A. R. Schneider, G. A. M. Jardim, G. M. Martins, D. C. Durigon, S. Saba, J. Rafique and A. L. Braga, Org. Biomol. Chem., 2020, 18, 4916–4921 RSC.
- X.-Y. Wang, Y.-F. Zhong, Z.-Y. Mo, S.-H. Wu, Y.-L. Xu, H.-T. Tang and Y.-M. Pan, Adv. Synth. Catal., 2021, 363, 208–214 CrossRef CAS.
- S. Mallick, M. Baidya, K. Mahanty, D. Maiti and S. D. Sarkar, Adv. Synth. Catal., 2020, 362, 1046–1052 CrossRef CAS.
- J. Hua, Z. Fang, J. Xu, M. Bian, C. K. Liu, W. He, N. Zhu, Z. Yang and K. Guo, Green Chem., 2019, 21, 4706–4711 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02962b |
| This journal is © The Royal Society of Chemistry 2022 |