
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemi-continuous and continuous processes for enantiomeric separation†
Marina
Ciriani
 *ab,
Rudi
Oliveira
a and
Carlos A. M.
Afonso
b
*ab,
Rudi
Oliveira
a and
Carlos A. M.
Afonso
b
aHovione Farmaciência, S.A. Sete Casas, 2674-506 Loures, Portugal. E-mail: marinaciriani@tecnico.ulisboa.pt; rsoliveira@hovione.com; carlosafonso@ff.ulisboa.pt
bResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Avenida Professor Gama Pinto, 1649-003 Lisbon, Portugal
First published on 20th May 2022
Abstract
Chiral resolution is an operation of great interest and increasing importance for the scientific community and industry. There are two approaches to provide enantiomerically pure compounds: by asymmetric synthesis of just one of the enantiomers or by resolution of racemates consisting of separating a mixture of both enantiomers. In the past years, extraordinary progress has been achieved in continuous enantiomeric separation, implementing more efficient procedures, and contributing to developing greener and more sustainable separation processes. This review article covers the main topics and applications of semi-continuous and continuous chiral separation focusing, in particular, on preferential crystallization, membrane separations, and continuous chromatography. It also offers an authors’ perspective on potential future directions in the field.
 Marina Ciriani | Marina Ciriani is currently a Scale-up Design Scientist at Hovione Farmaciência S.A and, since 2016, she has been working on the process development of sensitive active pharmaceutical compounds using continuous flow technologies and modeling. She obtained a Bachelor of Science in Chemical Engineering and a Master of Science in Pharmaceutical Engineering. She is also pursuing her Ph.D. in the Medicinal Chemistry Department of the Faculty of Pharmacy at the University of Lisbon in partnership with Hovione Farmaciência, S.A. |
 Rudi Oliveira | Rudi Oliveira is currently a Senior Scientist at Hovione FarmaCiência SA and an Invited Auxiliary Lecturer at the University of Lisbon. In 2014, he received his Ph.D. in Medicinal Chemistry under the supervision of Professors Francisca Lopes and Rui Moreira from the University of Lisbon (PT) and Professor Paul M. O'Neill from the University of Liverpool (UK). After a PostDoc in Chemical Biology (2015), he joined Hovione FarmaCiência SA to participate in the build-up of a Continuous Manufacturing group. Since then, he has worked in the development and scale-up of chemical processes both in batch and continuous. His research interests are currently focused on continuous unit operations and modern scale-up tools. |
 Carlos A. M. Afonso | Carlos A. M. Afonso is currently Full Professor at Faculty of Pharmacy, University of Lisbon. He received his PhD in 1990 under supervision of Professor C. D. Maycock, subsequently became Assistant Professor at New University of Lisbon and Associate Professor at Instituto Superior Técnico in Lisbon (2004–2010), and took a postdoctoral fellow (1990) at Imperial College of Science Technology and Medicine (W.B. Motherwell) and sabbatical leave (1997/1998) at University of Bath, UK (Jonathan Williams) and at the University of Toronto (Robert Batey). His current research is mainly focused on synthetic methodologies, more sustainable chemistry, synthetic valorisation of national natural resources and synthesis of bioactive molecules. |
1. Introduction
Molecular chirality was discovered by Louis Pasteur, in 1848.1 He found out that some molecules could exist as two non-superimposable-mirror image forms. Today, we know this type of molecule as enantiomers. Enantiomers are stereoisomers molecules with identical atomic constitution but have a non-superimposable mirror image, in other words, their three-dimensional arrangement of atoms is different. They have identical physical properties except for optical rotatory.2,3 The classical notations for enantiomers are D or L (commonly used for amino acids and sugars), R or S, and/or (+) or (−) which relates the ordering of the ligands to the chiral center.4Nowadays, it is known that chirality influences the biological and physical properties of molecules. The Global Chiral Chemicals Market size is estimated to be USD 57.79 billion in 2019 and is predicted to reach USD 150.64 billion by 2030 with a compound annual growth rate of 9.1% from 2020–2030.5
Increasing government focus on pharmaceutical manufacturing and rising investment by the manufacturers in emerging economies are the major factors propelling the market growth. Today, more than 80% of the drugs that are manufactured contain a chiral center due to the approval by FDA as safe ingredients.5 The growing demand for targeted pharmaceutical offerings and eco-friendly agrochemical products continues to be a key factor driving growth.6
Evidence exists that frequently only one enantiomer of a chiral active pharmaceutical ingredient provides the desired pharmacokinetic, physiological, and toxicological properties.4,5 Also, regulatory authorities require a scientific-based justification for any proposal to market a racemic mixture.7–10 Thus, producing pure enantiomers is key for the pharmaceutical industry.11 Chiral active pharmaceutical ingredients available in the market are for example: (S)-propanolol, (S)-naproxen, levosalbutamol, flavopiridol, etc. And, some racemic drugs also available are ibuprofen, salbutamol (Ventilan™), fluoxetine (Prozac™), etc.12,13
There are two approaches to provide enantiomerically pure compounds: by asymmetric synthesis of just one enantiomer (chemical or chemoenzymatic) or by resolution of racemic mixtures. The attractive option in terms of reagent consumption would be asymmetric synthesis that makes use of fermentation, chiral source synthesis (chiral pools), and asymmetric catalysis.14 Significant progress was achieved in this field in the last years but the procedures are still limited in scope and do not provide directly the required purity.15 Consequently, it is crucial developing new processes to separate enantiomers. The two major operations for chiral resolution are chiral chromatography and crystallization.16 Different types of chromatography exist for enantiomeric resolution namely, high-performance liquid chromatography, simulated moving bed chromatography, supercritical fluid chromatography, ionic-liquid assisted ligand exchange chromatography,17 and others. Most often, crystallization is the chosen operation because it is widely applicable, scalable, and cost-effective.11
An article guideline for an efficient selection of promising process operations for enantiomeric separation can be found in the literature,18 and it is based on experience gained with a large number of industrial case studies. Fig. 1 can be used to identify feasible unit operations for each case of enantiomeric separation based on physicochemical properties available in an early stage of development. It must be known if racemization is possible, if conglomerate is present and which is the eutectic composition of the chiral system. As a result of this knowledge, it can be identified possible strategies to perform the chiral resolution. Although asymmetric synthesis is not approached in this review, if racemization is unachievable, a stereoselective synthesis might be performed if economically attractive. The other options would be chromatography, crystallization, membrane separation, or a combination thereof. If racemization is possible, it should be applied in sequence with a resolution operation feasible for: (1) conglomerate systems that would be preferential crystallization or; (2) the compound-forming system with two eutectics at symmetrical compositions that would be chromatography, crystallization, and membrane separation.
 | ||
| Fig. 1 Decision tree based on qualitative criteria for the selection of feasible unit operations for the obtention of pure enantiomers. Adapted from ref. 11 and 18. | ||
1.1. Continuous processes and Green Chemistry
The field of Green Chemistry demonstrates how process scientists and chemists might develop products and processes that can be profitable and at the same time good for human health and the environment.19 Throughout in this review, we will address the pertinent Green Chemistry principles by the references listed in Table 1.| Reference | Green chemistry principle |
|---|---|
| P1 | Prevention |
| P2 | Atom economy |
| P3 | Less hazardous chemical syntheses |
| P4 | Designing safer chemicals |
| P5 | Safer solvents and auxiliaries |
| P6 | Design for energy efficiency |
| P7 | Use of renewable feedstocks |
| P8 | Reduce derivatives |
| P9 | Catalysis |
| P10 | Design for degradation |
| P11 | Real-time analysis for pollution prevention |
| P12 | Inherently safer chemistry for accident prevention |
Continuous crystallization (chapter 2) can offer considerable advantages such as elimination of batch to batch variability, particle size control, scalability of the process, reduced capital expenditure (CapEx) and operational expenditure (OpEx) along with the more efficient use of energy and materials (P6).20
Membrane separation (chapter 3) is a robust and green unit operation for the purification and separation of desired compounds streams that takes a shift towards a continuous methodology and presents minimal energy requirements (P1, P6).20
The use of supercritical fluids, namely supercritical carbon dioxide (scCO2) is an interesting approach for continuous processes since they are inaccessible without high-pressure environments. The application of supercritical fluids in a flow system offers numerous advantages over batch vessels and has proven to be one valuable alternative to traditional solvents.19 The operations may be performed on a small scale, improving safety and reducing the amount of material required (P1, P5).21 Examples using scCO2 for enantiomeric separation are given in chapters 4 and 5 of this review.
The bibliography of this work was obtained through the SciFinder database of the American Chemical Society and the Web of Knowledge database of the Clarivate Analytics. No restriction to publication date was applied. The search was done in the period between January and December of 2021.
2. Crystallization
Crystallization is a solid–liquid separation and purification process very commonly used in the fine chemical, pharmaceutical, agrochemical, and food industries. More than 90% of active pharmaceutical ingredients (API) are isolated as crystalline products.22,23 During crystallization, parameters such as morphology and size distribution must be strictly controlled due to their great influence on the quality, physical and chemical properties of the resulting product. It can also affect downstream operations such as the time of drying, milling, and filtration.34 A tutorial review on how to rationally conduct a crystallization of a stable or a metastable phase in solution is given by Coquerel.24 Vetter et al.25 described a methodology that allows finding regions of attainable particle sizes in batch and continuous crystallization processes.The obtention of enantiopure chemicals can be achieved by using crystallization procedures such as the classical resolution, preferential crystallization, optically active solvent methods, and attrition-enhanced deracemization (Viedma Ripening).11 In 1853, Pasteur developed the classical and most known method for enantiomers purification, the Pasteurian resolution.26 He performed the resolution of tartaric acid using chiral quinuclidine bases. The method is based on adding a chiral agent to the racemic solution, breaking the thermodynamic symmetry of the enantiomers by forming diastereomeric compounds in the form of complexes or salts.15,27 Diastereomers have different physicochemical properties, for example, solubility, which allow the separation to be achieved. This is a feasible approach used in large-scale processes for enantiomers resolution. A synthetic intermediate of the anticancer drug flavopiridol, and (S)-naproxen an anti-inflammatory drug are produced on an industrial-scale using the Pasteurian resolution.28,29 However, this classical technique presents some disadvantages. The chiral agent must be added in stoichiometric amounts (going against P2, P8 and P9), must be inexpensive, and be available in the market with high chemical and optical purities.15 Moreover, there is no rule to find an optimal chiral resolving agent. Numerous candidates must be experimentally tested to discover the chiral agent that changes considerably the solubility between the forming diastereomers allowing the isolation of the desired one by crystallization. Finally, a noncovalent bond between the chiral agent and the pair of enantiomers is required for an easy recovery of the target enantiomer following crystallization.
In a preferential crystallization process feasible only for conglomerate-forming systems, by seeding of a racemic or enantiomerically enriched solution with crystals of the preferred pure enantiomer, control over the crystallizing enantiomer is obtained.30 In 1969, Sato et al.31 described a preferential crystallization batch procedure to prepare 95% pure (D)-lysine-3,5-dinitrobenzoate from a racemic mixture. Also, kinetically controlled crystallization of a racemic conglomerate is possible in the presence of chiral polymers at a certain temperature. The polymer inhibits the precipitation of the undesired enantiomer, collecting the desired one by filtration (against P2).
In 2005, Viedma described an abrasion-grinding method for total symmetry breaking and obtention of complete chiral purity (P1 and P2).32 The author demonstrated the development of a homochiral solid phase for the inorganic compound NaClO3, initially present as a racemic mixture of two enantiomorphic solid phases in equilibrium with the achiral aqueous phase. A population balance model was described by Iggland et al.33 to explain the experimental behavior observed by Viedma. Later, this new symmetry-breaking technique was used for the enantio-enrichment of racemic mixtures of aminoacids compounds such as aspartic acid.34,35 In 2019, Baglai et al.36 described a procedure to synthesize a precursor of Levetiracetam and Brivaracetam using Viedma Ripening approach. A review covering the asymmetric crystallization process emphasizing Viedma approach is given by Rutjes et al.37
In recent years, improved processes variants involving either preferential crystallization, Viedma ripening, or combinations thereof have been reported in the literature in batch and continuous mode. In this review chapter, we will discuss the remarkable improvements for continuous preferential crystallization to obtain enantiopure compounds.
2.1. Preferential crystallization
Preferential Crystallization (PC) is a productive and cost-effective method to obtain crystals of a single enantiomer from a racemic solution.27 To use this methodology, the chiral system must occur as a conglomerate meaning that the enantiomers crystallize as separate enantiopure crystals. Only 10% of the racemic mixtures belong to the conglomerate forming group.38 Phase diagram screenings of enantiomer mixtures in a solution can give important indications on the possibility of chiral resolution through crystallization.39The process is carried out in the metastable zone determined by phase diagrams analysis. In this specific zone, it is possible to preferentially develop crystals of the desired enantiomer with high yields and productivities through a seeded process. The seeding will avoid the nucleation of counter-enantiomer due to the initial homochiral surface area.16 To obtain high purity and yields, the process must be stopped before the undesired counter enantiomer crystallizes.38 Using preferential crystallization to obtain pure enantiomeric compounds can be labor-intensive and require careful monitoring and control of process parameters.
There are several techniques and operation set-ups that can be used with single or coupled crystallizers of different types. Seeded Isothermal Preferential Crystallization is a preferential crystallization technique where the pure enantiomer is crystallized by seeding a supersaturated solution at a constant temperature. An isothermal batch crystallization process should be stopped before the nucleation of crystals of the undesired enantiomer occurs so that only pure crystals of the desired enantiomer can be collected. Petruševska-Seebach et al.40 described the resolution of (L)-asparagine in water using simple isothermal batch preferential crystallization. A precise stopping of the crystallization and rapid filtration of the product crystals is a very high-risk operation, thus preferential crystallization processes can be carried out in a polythermal method.16
Auto-Seeded Polythermal Programmed Preferential Crystallization (AS3PC) is one approach to perform preferential crystallization developed by Coquerel et al.41 where the pure enantiomer is crystallized by application of a controlled cooling until the obtention of a saturated solution. Some examples using this method are the preferential crystallization of 4-carboxyphenylglycine,42 omeprazole,43 (±)-5-(4-bromophenyl)-5-methylhydantoin,44 baclofenium hydrogenomaleate45 and (D/L)-threonine.46 Stirring may also be an important parameter to monitor and control during preferential crystallization as described for the resolution of 5-ethyl-5-methylhydantoin.47,48
Due to the complexity of crystallization, it is necessary the use of online analytical tools for real-time monitoring and control of the operation and to gather some data for a better process understanding. These tools are known as Process Analytical Technologies (PAT) and can be for example spectroscopy technologies as attenuated total reflectance (ATR) Fourier-transform infrared57 or ATR-UV-Vis, NIR, Raman, or other techniques such as focused beam reflectance measurement46,57–59 and particle vision and measurement.57 In addition to the above widely used crystallization monitoring methods, conductivity, refractive index, turbidity measurements, are also available in PAT.50 They are very important to monitor critical process parameters (CPPs) to control the critical quality attributes (CQAs).
In crystallization, PAT can be used to obtain in situ information about the solution and one important CPP is supersaturation because it can affect the nucleation, particle agglomeration, and crystal growth.22,60 By using PAT, crystallization process development can be faster and reproducibility can be enhanced.61,62 Continuous online monitoring was done for the resolution of (D/L)-threonine using an online polarimetric detector, a refractometer, and a density meter.63,64
The coupled preferential crystallizer (CPC) configuration is similar to coupled MSMPR except that there is no continuous feed and product removal.66 The scale-up can be done by using pilot-scale stirred tanks and traditional equipment but some difficulties can be the lack of heat transfer in specific areas of the vessel and homogeneous mixing.51 To improve yield, productivity, and quality, MSMPR crystallizers can be equipped with a recycle system67 or they can be used in multiple stages where a cascade of several crystallizers are connected.49
Reviews comparing the differences between batch and continuous crystallizers and analysis of the advantages of each type have been reported.22,50,62,69,70
In a traditional procedure, a supersaturated solution is fed at the bottom of the crystallizer and this upward liquid flow agitates the crystals in the crystallizer to form a fluidized bed.70 The flow exit at the top of the crystallizer. Seeds are generated by ultrasonication, at the bottom of the crystallizer. The conical shape of the crystallizer allows larger crystals to drop to the bottom, where they can be removed from the vessel. Moreover, since the fluidized bed crystallizer is conical at the lower part, the fluid flow velocity decreases with the increasing diameter which has a great impact on particle size.74
| Inline methods | Purpose | Equipment | Crystallizer |
|---|---|---|---|
| Mixers | Mixing streams | Static mixers | PFC |
| Ultrasonication | Accelerate nucleation and crystal breakage | Sonicator | MSMPR, PFC; FBC |
| Milling | Crystal breakage | Rotor-stator mill | MSMPR |
| T cycling | Control undesired nucleation | Temperature control units | MSMPR, PFC; FBC |
| Recycling | Increase yield | Filters, columns, membranes | MSMPR, FBC |
This method was first described in 1966 by Ito et al.76 by using either a batch crystallizer or a fluidized bed crystallizer for the resolution of amino acids for example (D/L)-glutamic acid and (D/L)-threonine. Eicke et al.77 showed the importance of fines dissolution on the delay of undesired nucleation of the counter-enantiomer and consequently increase the quality and purity of the desired enantiomer. This is accomplished through the use of mills or ultrasounds in the process.16 In 2012, Qamar et al.78 mathematically described for the first time continuous preferential enantioselective crystallization using a single crystallizer equipped with a fines dissolution unit (Fig. 2 and Table 3).
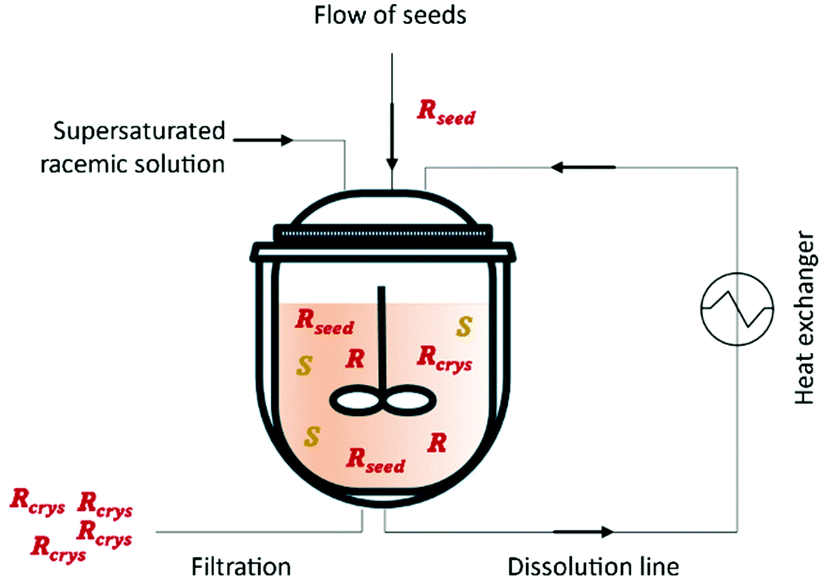 | ||
| Fig. 2 Illustration of a simple continuous procedure for an enantiomeric resolution using a single crystallizer. Adapted from ref. 78. | ||
| Authors | Type of crystallizer | Substance | Studied parameters | Findings | Year | Ref. |
|---|---|---|---|---|---|---|
| Qamar et al. | Single-MSMPR with fines dissolution | D/L-Thr | Continuous seeding without fines dissolution; continuous seeding with fines dissolution; effect of the ratio between solid and liquid phases | This model can be used to find the optimum operating conditions for improving the product quality and for reducing the operational cost of continuous preferential crystallization | 2012 | 78 |
| Qamar et al. | Two-coupled MSMPR with fines dissolution | D/L-Thr | Continuous seeding without fines dissolution; continuous seeding with fines dissolution; periodic seeding without fines dissolution | Significant improvements were achieved by using two coupled MSMPR when compared to a single-MSMPR. This model can be used to find the optimum operating conditions for improving the product quality and for reducing the operational cost of continuous preferential crystallization | 2013 | 65 |
| Chaaban et al. | Two-coupled MSMPR with a feed tank | D/L-Asn | Mean residence time of the liquid phase; fraction of the feed solution inlet; amount, average particle size, and morphological effects of seeds | A mathematical model describing the resolution of (D/L)-asparagine in two-coupled MSMPR and a feed tank under isothermal conditions was developed. It was shown that the increase in the mean residence time of the liquid phase, the increase in the mass of seeds, and the reduction of the size of the seed crystals all contribute positively to the overall process performance | 2014 | 79 |
| Köllges et al. | MSMPRC via Viedma Ripening with mill unit | Generic | Process configuration using different crystallization techniques: (1) resolution via preferential crystallization and (2) deracemization via Viedma Ripening | The configuration (1) achieves the complete separation of enantiomers of a conglomerate forming system using a single-MSMPR. Configuration (2) obtained an enrichment of roughly 90% ee but requires excessively long residence times with correspondingly low productivities to achieve this | 2017 | 80 |
| Mangold M. et al. | FB with a mill unit | Generic | Influence of the fluid and product flow rate; influence of the flow rate to the mill and its properties; influence of the crystallizer geometry; influence of the crystal growth rate | It is found that the inlet flow rate has a strong influence on productivity; the product flow rate hardly influences the productivity; more effective means to change the product size distribution are modifications of the crystallizer geometry; and modifications of the mill affecting the size distribution of the seeds | 2017 | 81 |
| Kollges et al. | MSMPRC with recycling and solvent removal; MSMPRC via Viedma Ripening with a pre-crystallizer | Generic | Process configuration using different crystallization techniques: (1) resolution via preferential crystallization and (2) deracemization via Viedma Ripening | two MSMPRC cascades with equal residence times perform worse than the two MSMPRC cascade with variable residence times unless a very high enantiomeric purity is demanded; adding further crystallizers does not boost the productivity substantially for the variable residence time case; in the case of equal-sized MSMPRCs, the PC process outperforms the processes that rely on ripening stages in terms of productivity | 2018 | 75 |
| Mangold M. et al. | FB with a mill unit | Generic | Particle velocity profiles at different positions in the crystallizer for a constant liquid flow rate crystal phase purity; minimum operation flow rate | The linear model studied can be useful for process optimization or control purposes | 2020 | 82 |
Kollges et al.75 designed a crystallizer unit that is continuously fed with a supersaturated solution of racemic composition. Provision of seed crystals with an enriched composition during the startup phase and by selecting appropriate feed concentration and residence time, a steady state with pure solid phase composition can be obtained from the MSMPR crystallizer. The suspension is continuously transferred into the filtration unit where the solids are separated from the solution. The mother liquor resulting from the filtration is recycled back. The system operates with a distillation system to remove a determined amount of the solvent and maintain the level of the reactor (Fig. 3 and Table 3).
 | ||
| Fig. 3 Illustration of a continuous preferential crystallization with mother liquor recycling and a milling unit for large crystals breakage and fines dissolution. Adapted from ref. 75. | ||
To prevent waste (P1), the distilled solvent might be used for future trials. Moreover, the distillation system could be substituted by a nanofiltration membranes system (P6) where the retentate would be fed back into the crystallizer and the permeate could be recycled for future trials.
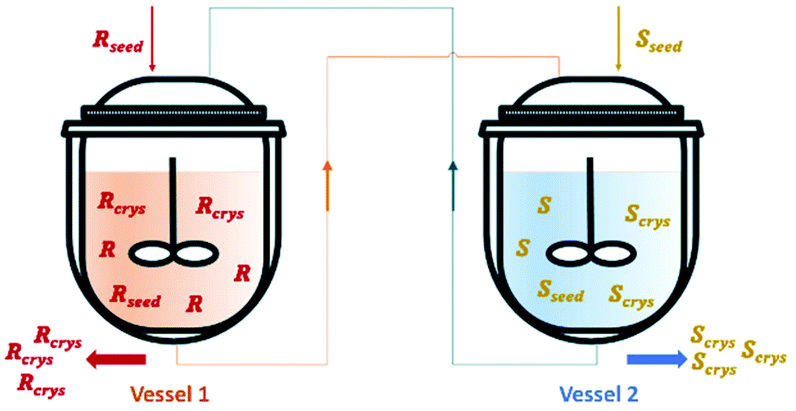 | ||
| Fig. 4 Illustration of a simultaneous preferential crystallization with two vessels coupled via mother liquors exchange. Adapted from ref. 83. | ||
Vetter et al.16 coupled two continuous crystallizers by exchanging their clear liquid phases. Each crystallizer was connected to a grinder responsible for in situ seed generation through large crystals breakage. The authors concluded that, by using this setup, it is possible to continuously obtain the desired enantiomer in one crystallizer and the counter-enantiomer in the other. They determined that an enantiomerically pure steady state can be obtained. Moreover, the process also recovers from the sudden appearance of undesired crystals of the counter-enantiomer, which is a phenomenon encountered from time to time in industrial crystallizers due to scale formation at the crystallizer walls.
Qamar et al.65,78 developed mathematical models exploiting the dynamics of continuously operated single and two-coupled MSMPR crystallizers equipped with a fines dissolution system. The authors used available solubility and kinetic data of (D/L)-threonine in water systems and carried out simulation studies. The impact of different seeding strategies and residence time distributions on the yield, purity, productivity, and crystal size was investigated (Table 3 and Fig. 5).
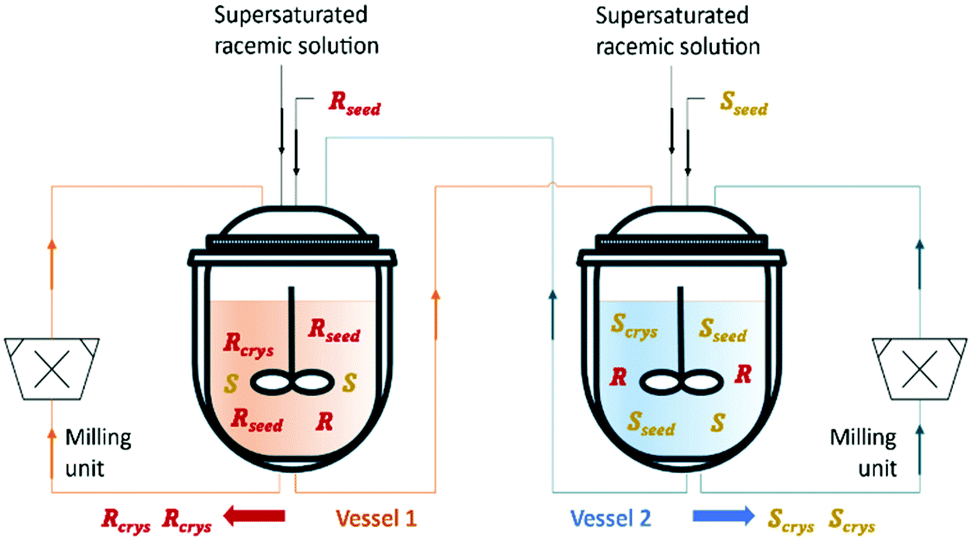 | ||
| Fig. 5 Illustration of a simultaneous preferential crystallization with two vessels coupled via mother liquors exchange and milling units for large crystals breakage and fines dissolution. Adapted from ref. 65. | ||
Later, Galan et al.51 investigated experimentally and compared the separation of (D/L)-threonine using continuous operated single MSMPR crystallizer and two-coupled MSMPR with continuous exchange of mother liquors. The co-workers used Qamar process models to identify suitable operating conditions. Although it was possible to perform continuous preferential crystallization using a single MSMPR crystallizer, under identical conditions almost the double of productivity was achieved by using two-coupled MSMPR vessels, at the same high level for both configurations. Furthermore, the two-coupled MSMPR allowed harvesting both enantiomers simultaneously. Majumder et al.66 also studied the resolution of (D/L)-threonine in the water system. A detailed study on the effect of process parameters (e.g., feed flow rate, seed mass, and liquid phase exchange) on productivity and yield was presented. Both works have achieved enantiomeric excess >99% for the L-enantiomer.
Chaaban et al.53,79 achieved a continuous preferential crystallization of (D/L)-asparagine monohydrate in water using a two-stage MSMPR crystallizer coupled via mother liquors exchange. They showed, by mathematical modeling and validation, how productivities, yields, and purities of solid products were influenced by the morphological differences in the seed crystals. Final purities of 100% and productivity of 0.94 g L−1 h−1 were obtained for each enantiomer (Table 3).
In the described processes, the maximum isolated yield obtained is 50% for the desired enantiomer, leading to a poor atom economy (P2) and the generation of a high amount of waste (P1). A great option would be the racemization of the undesired enantiomer and future preferential crystallization of the obtained racemic mixture to increase the yield and atom economy. Moreover, the Viedma Ripening procedure could be used for the achievement of 100% theoretical yield.
Hein et al.93 proposed a method based on the attrition-enhanced deracemization (Viedma Ripening) approach where preferential crystallization occurs in one vessel and selective dissolution of the counter-enantiomer in the second vessel. The authors successfully obtained S-omeprazole with a productivity of 0.84 g L−1 h−1 and 98% purity.
Additionally, the authors showed how this procedure can be applied for continuous resolution of the (D/L)-threonine system. Obtaining enantiopure L and D enantiomers separately with 76% and 79% yields, respectively (Fig. 6).
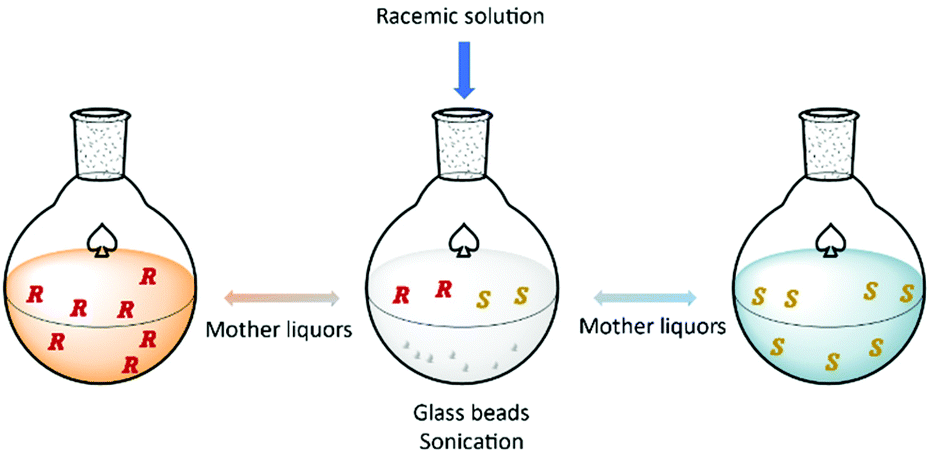 | ||
| Fig. 6 Schematic illustration of coupled preferential crystallization using attrition. Adapted from ref. 93. | ||
Kollges et al.75 designed a continuous Viedma Ripening process where a pre-crystallization step (it generates a racemic mixture of crystals) is followed by one or more ripening stages that successively increase the enantiomeric excess. In the pre-crystallization unit, the mother liquors are filtered, concentrated, and recycled into the vessel. It also has a milling unit to provide attrition. Following the ripening stage, the final solid containing high enantiomeric excess is filtered in a second filtration unit and the mother liquors are recycled back to the first ripening stage (Table 3).
In 2018, Majumder proposed a novel PFC configuration involving two coupled PFCs with liquid phase exchange. Simulation studies were carried out using (D/L)-threonine in a water system as a model. The author used a set of coupled population balance equations to describe the evolution of the crystal phase of both enantiomers in each crystallizer. The results predict that the proposed configuration has higher productivity compared to the currently used continuous crystallization configurations (MSMPR and single-PFC) while maintaining the same level of purity (Table 4 and Fig. 7).91
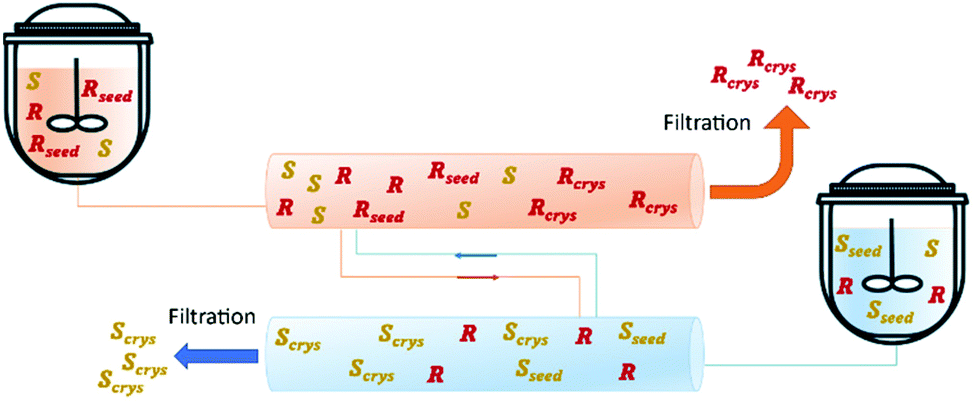 | ||
| Fig. 7 Illustration of coupled plug flow crystallizer via mother liquor exchange. Adapted from ref. 91. | ||
| Product | Type of crystallizer | Technique | Purity (%) | Productivity (g L−1 h−1) | Ref. |
|---|---|---|---|---|---|
| a We always picked the best-case scenario reported in each article. b Calculated by having into consideration the feed flow rate. | |||||
| (D/L)-Asparagine monohydrate | Fluidized bed crystallizers | Two-coupled FBC | 97.0 | 14 | 90 |
| 97.0 | 40 | 52 | |||
| MSMPR | Two-coupled MSMPR | 100 | 0.94 | 53 | |
| 100 | 0.38 | 30 | |||
| (D/L)-Threonine | MSMPR | Single MSMPR | 98.8 | 3.2 | 51 |
| Two-coupled MSMPR | 99.3 | 6.3 | |||
| Two-coupled MSMPR | 99.0 | 7.7b | 66 | ||
| Plug flow crystallizer (PFC) | Single PFC | 99.0 | 9.3b | 91 | |
| Two-coupled PFC | 99.0 | 13.3b | |||
| Omeprazole | MSMPR | Two-coupled MSMPR | 98.0 | 0.84 | 92 |
| Indoline derivatives | Plug flow crystallizer | Two-coupled PFC | 98.0 | 20 | 54 |
Recently, Cameli et al.54 reported the continuous preferential crystallization of isoindoline-1-ones derivatives using a plug flow crystallizer immersed in two thermostatic baths set at different temperatures. This configuration allowed them to obtain productivities of 20 g L−1 h−1 due to the excellent heat transfer and low residence periods (Table 4 and Fig. 8).
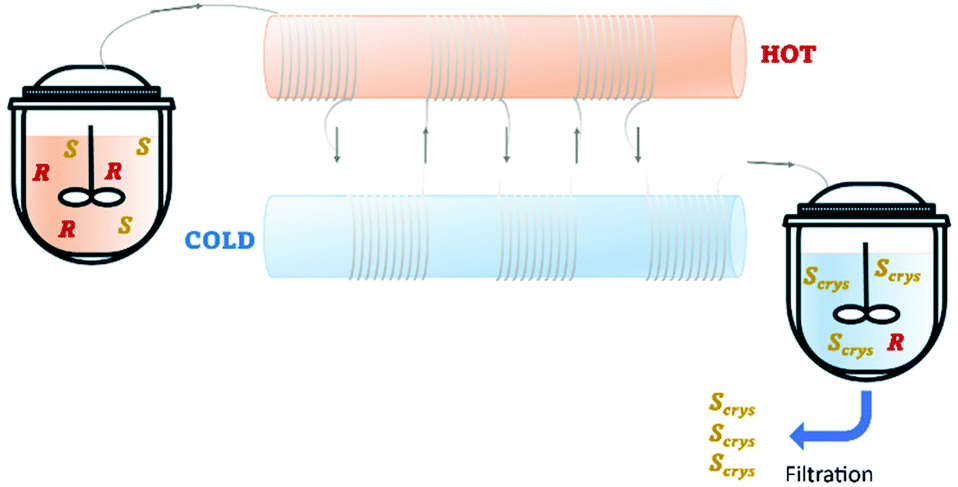 | ||
| Fig. 8 Illustration of a plug flow crystallizer with hot and cold zones for dissolution and re-crystallization, respectively. Adapted from ref. 54. | ||
Binev et al.90 and Temmel et al.52 studied the feasibility of a fluidized bed process with two coupled crystallizers for continuous preferential crystallization of a racemic mixture of (D/L)-asparagine monohydrate (L-asn·H2O) in water. The first authors obtained final purities up to 97% and productivities up to 14 g L−1 h−1 for each enantiomer (Table 4). The second authors also obtained final purities up to 97% but productivities up to 40 g L−1 h−1 for each enantiomer.
In this process, the seed crystals move continuously through the fluidized crystal bed until they are grown to a size at which they settle at the bottom of the fluidized bed. Then, they are removed via peristaltic pumps into a bypass and flow through high-speed dispersers, which were utilized as mills. After that, the milled crystals are feedback as seed material to the process. The authors showed how important it is the effect of the conically shaped tubular crystallizers in the crystal size distribution, low fine contents, high purities, and robust production (Fig. 9).
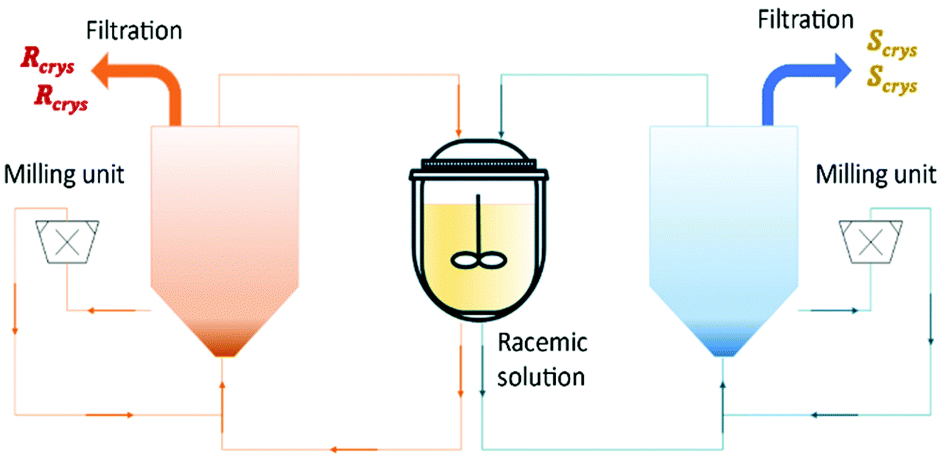 | ||
| Fig. 9 Illustration of coupled tubular fluidized bed crystallizers with a conical lower section and milling units for crystal breakage and fines dissolution. Adapted from ref. 90. | ||
Mangold et al.81 presented a mathematical process model based on population balance equations for a continuous fluidized bed crystallizer, they identified the key operation and design parameters for maximizing the productivity and better control of crystal size distribution (Table 3).
Other approaches are described for example for the resolution of a racemic mixture of menthyl benzoate on a scale of 300 kg h−1 where a new type of crystallization apparatus is described.94 A further variation involves a three-container system of two coupled crystallizers and a feed tank. This setup relies on simultaneous preferential crystallization of both enantiomers in parallel into the two crystallizers, while the racemic mixture is progressively dissolved from the dissolver tank.
Continuous preferential crystallization has gained interest due to its many advantages over discontinuous mode including better reproducibility, yields, and precise control of process parameters. Moreover, it can offer reduced capital expenditure (CapEx) and operational expenditure (OpEx) along with the more efficient use of energy and materials (P1 and P6).20
When comparing both procedures of preferential crystallization and Pasteurian resolution, there is no doubt that the first method is greener. It does not involve additional chemical steps by firstly forming diastereomers salts for the crystallization procedure and secondly, forming the desired enantiomer at the end of the process. When preferential crystallization is used, the atom economy (P2) is higher, and the energy/material efficiency is increased.
A possibility to make the resolution greener and increase the inherently maximum 50% yield would be to racemize the isolated undesired enantiomer after preferential crystallization. Consequently, obtaining a racemic solution that would be fed into the crystallizer.
3. Membrane-based chiral separation
Membrane-based chiral separation is a promising low-cost technique for efficient separation of enantiomers due to its intrinsic advantages such as being operated in continuous mode, low energy consumption (P6), high efficiency of the resolution, and ease of scale-up.95,96 Moreover, it should offer a good transport rate with high selectivity, to be stable in a high range of pH and compatible with different types of solvents.97 The membrane act as a barrier and may selectively transport one enantiomer due to specific interactions (hydrogen bonding, coulombic or van der Waals) between the enantiomer molecule and a chiral recognition site of the membrane, thus producing a permeate solution enriched with this specific enantiomer. Within the last decades, a large variety of synthetic membranes were developed for specific process optimization. They can be classified according to their backbone material, their morphology, preparation method, and shape.98An efficient separation can be achieved by using an enantioselective liquid or solid membrane that contains a chiral recognition site that may be a chiral side chain, a chiral backbone, or an immobilized chiral selector. Also, a non-enantioselective solid membrane with a specific molecular weight cut-off (MWCO) can be used for nano or ultrafiltration procedures where the preferred enantiomer may complex with a specific chiral selector before filtration. Therefore, the resolution method for non-enantioselective membranes is different from enantioselective membranes.95,99
The membrane properties that determine the feasibility and characteristics of the processes are permeability and selectivity. The permeability of a membrane is defined as the normalized flux regarding the concentration change and the thickness of the membrane. The selectivity of a membrane is defined as the permeability ratio of the compounds of interest, in this case, the two enantiomers.98
The selectivity of the process can be calculated in terms of enantiomeric excess (ee) and separation factor (α).
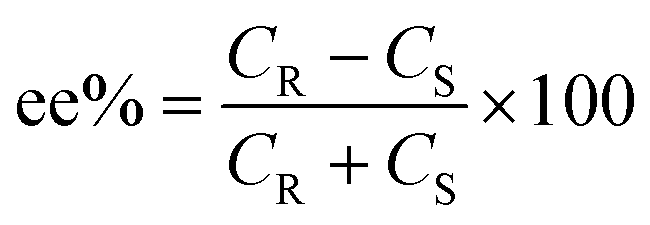
where JR and JS are the normalized flux of the R and S enantiomers, respectively that can be calculated following the formula below.
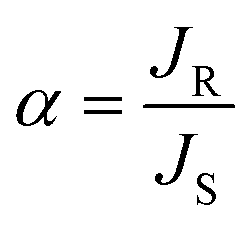
where ΔC is the concentration difference of each enantiomer, Δt is the permeation time, V is the downstream volume, and A is the effective membrane area.

The transport process through the membranes can be categorized as filtration, dialysis, electrodialysis, and pervaporation. Several factors can influence the transport mechanism, it depends on the main driving force of the permeation of compounds through the membranes such as binding affinity forces, pressure gradient, solutions concentration, and electric field or vapor difference.100
The two transport mechanisms can be categorized as facilitated (diffusion-selective membranes) or retarded (sorption-selective membranes). An enantioselective membrane preferentially allows a specific enantiomer to adsorb or to diffuse into the membrane. In diffusion selective membranes, one enantiomer is preferentially adsorbed into the chiral recognition sites, and it is continuously adsorbed and de-adsorbed from one site to the next, and finally it reaches the stripping phase by increasing driving forces such as pressure or pH. The other enantiomer, which has no affinity to the chiral sites, passes through the membrane by diffusion.98 Mostly chiral liquid and solid membranes built from a chiral polymer or coated with an enantioselective polymeric thin-layer employ facilitated transport.99 Sorption selective membranes, which selectively absorb and immobilize one of the enantiomers through incorporated chiral selectors while permitting the other to flow due to lower affinity to the carrier, normally are built from porous membrane material.101 Since the bound with the enantiomer is stronger and the selectivity can be maintained with the convective flow, these types of membranes are more attractive commercially.102
In 1980, the first example of enantiomers separation by host–guest complexation was described by Cram et al.103–105 The authors developed an enantioselective extraction procedure using crown ethers as chiral selectors in chloroform. Examples of chiral selectors can be found in the literature namely different types of cyclodextrins, albumin or other proteins, chiral polysaccharide chains like chitosan or sodium alginate, DNA, crown ether derivatives, and oligopeptides.100,106 In 1990, Pirkle et al.107 patented an invention related to a continuous process to separate enantiomers by using a supported liquid membrane and a chiral carrier that selectively complexes with one of the two enantiomers.
3.1. Liquid membranes for chiral resolution
Liquid membranes consist of a liquid phase that acts as a membrane between two fluid phases.108 Chiral liquid membrane technology has been extensively investigated for the separation of enantiomers due to its high separation factor and increased mass transfer as a result of the higher solubility and diffusivity coefficients of compounds in a liquid medium than in a solid membrane.97 The three basic types of liquid membranes are supported liquid membrane (SLM), emulsion liquid membrane (ELM), and bulk liquid membrane (BLM) – Fig. 10. | ||
| Fig. 10 Illustration of different types of liquid membranes. Adapted from ref. 97. | ||
The set-up is known as the “resolving machine” developed by Cram et al.105 consists of two U-tube containing different selectors for each enantiomer in chlorinated solvents (Fig. 11). The outcome is that the (R)-enantiomer will be in a receiving phase different than the (S)-enantiomer.
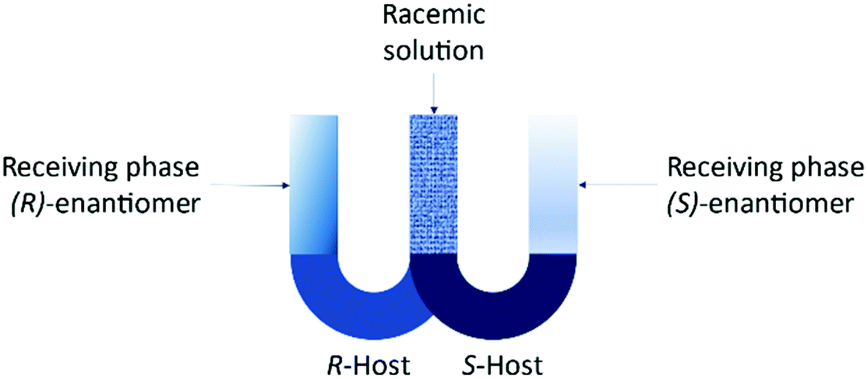 | ||
| Fig. 11 Resolving machine developed by Cram et al.105 | ||
Feringa et al.110 worked on the separation of amino acid enantiomers using Palladium (S)-BINAP complexes as chiral carriers using the “resolving machine”. One advantage of this process is that the host can be prepared in situ from commercially available compounds. The system also showed a great selectivity for Tryptophan.
Krieg et al.111 described the resolution of racemic chlorthalidone using a U-tube multiple membrane cell consisting of three membrane phases and three stripping phases with β-cyclodextrin as chiral selector.
To obtain (D)-phenylalanine from a racemic mixture, Pickering et al.112,115 developed a chiral ELM using copper(II) N′-decyl-(L)-hydroxyproline as chiral selector in a mixture of decane/hexanol. An enantiomeric excess of 40% was obtained. Huang et al.116 reported the resolution of racemic α-cyclohexyl mandelic acid across microemulsion liquid membranes system using tartaric acid benzyl ester as chiral selector, sodium dodecyl sulfate as the surfactant, and mixture n-butyl alcohol and n-heptane as the organic solvents. The effect of several variables such as chiral selector concentration, pH value in the external aqueous phase, and volume ratio of the external aqueous phase to membrane phase was studied. The maximum separation factor obtained was up to 2.0.
In the referred cases, some use metallic chiral selectors. For toxicological, environmental, and economic reasons, it should be avoided and substituted for environmentally friendly hosts (P3).
 | ||
| Fig. 12 Principle of enantiomeric separation by hollow-fiber membranes. The (R)-enantiomers are being transported through the membrane by diffusion. Adapted from ref. 125. | ||
The use of hollow fiber membranes usually results in a relatively compact system because of a high membrane surface area per volume.96 Several case studies are found in the literature demonstrating how the solute exchange between two crystallizers separated by a hollow-fiber membrane module results in both high purity and high yield.126,127 Moreover, a series of membranes can be used for a better efficient mass transfer.128 In 1992, Cussler et al.129 described the separation of Leucine enantiomers with an enantiomeric excess of 99% by fractional extraction across microporous hollow fibers (Fig. 13). The authors considered the two-phase system for enantioselective separation developed by Takeuchi et al.130
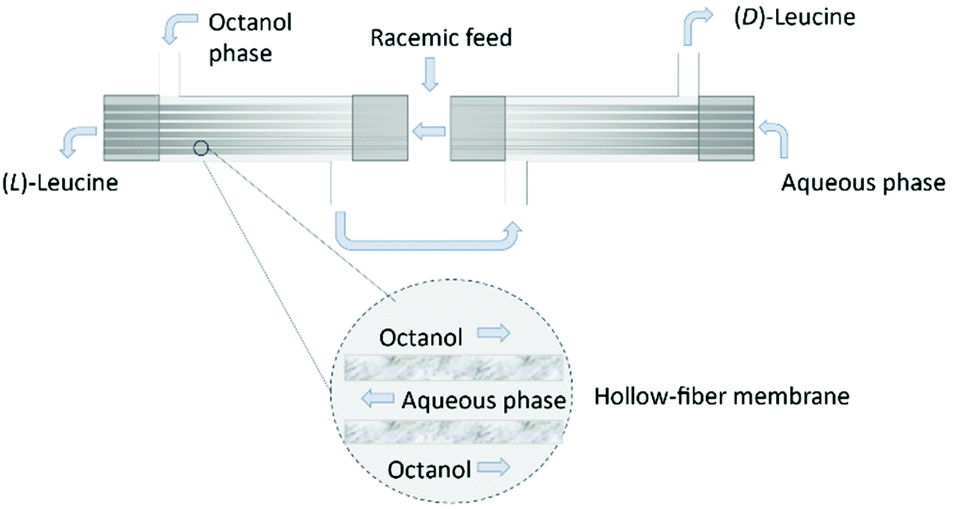 | ||
| Fig. 13 Separation of leucine enantiomers in a hollow fiber module. Shell side: octanol, lumen side: aqueous solution. Leucine enantiomers are collected in the exit streams of the module. Adapted from ref. 129. | ||
Firstly, the water-immiscible solution (N′-decyl-(L)-hydroxyproline in octanol) is fed into the column on the shell side. Then, the aqueous phase is fed into the lumen side in counter current. When a stationary state is reached in both the lumen and shell side, the feed flow consisting of a racemic solution of leucine is feed. The enantiomers are collected in opposite leaving streams of the column and quantified by chromatography.
Four years later, Keurentjes et al.96 determined the enantioselectivity for Ibuprofen, Salbutamol, Propanolol, Norephedrine, Ephedrine, Mirtazapine, Phenylglycine and Terbutaline testing different chiral selectors solutions, namely (R,R)/(S,S)-di-hexyltartrate and dibenzoyl tartaric acid in heptane or chloroform, and different lipophilic anions to form complexes with the racemates to increase drug solubility. The modules were built with regenerated cellulose hollow-fiber membranes. Norephedrine was obtained with an enantiomeric excess of 99% (Table 5).
| Hollow fiber material | Membrane solution phase | Compound | Enantiomeric excess (ee) and/or separation factor (α) | Ref. |
|---|---|---|---|---|
| Polypropylene cross-linked with polyvinyl alcohol gel | Dodecyl-hydroxyproline and octanol | Leucine | 99% | 129 |
| Regenerated cellulose (RC) | 10 wt% di-hexyl tartrate (DHT) in heptane | Norephedrine | >99% | 96 |
| Polyacrylonitrile enzyme activated membrane | Toluene | Diltiazem intermediate | 85% | 137 |
| Polypropylene | N-3,5-Dinitrobenzoyl-alanine-octyl ester in toluene | Lactic acid | 33.50% (α: 2.00) | 131 |
| Alanine | 27.17% (α: 1.75) | |||
| Polypropylene | N-3,5-Dinitrobenzoyl-alanine-octylester in toluene | Lactic acid | 34.60% | 132 |
| Polysulfone | Adamantyl-carbamoyl-11-octadecylthio ether-quinine in 1-decanole/pentadecane | N-Protected amino acid derivatives (DNB-(D/L)-leucine) | 98% | 128 |
| Polyvinylidene fluoride | Isopentyl tartrate, sulfobutylether-cyclodextrin | Ketoconazole | 90% | 133 |
| Polypropylene | DBTA in octanol | Phenylalanine | 55% | 134 |
| Polysulfone | Exchanging mother-liquors | Glutamic acid | 99% | 126, 135 and 136 |
| Polypropylene | DBTA in 1-decanol | Amlodipine | 40.4% | 139 |
| Polypropylene | DBTA in 1-decanol | Cetirizine | 75% | 140 |
| Polypropylene (asymmetric synthesis) | Undecane | 1-Methyl-3-phenylpropyl-amine | 98% | 138 |
| Polyvinylidene fluoride coated with molecularly imprinted polymer | MAA + EGDMA + S-ADB | Amlodipine besylate | 90% (α: 1.98) | 141 |
| Polyvinylidene fluoride | (D)-Tartaric acid and a commercial extractant in heptane | Amlodipine | 90.7% | 127 |
Hadik et al.131,132 reported the resolution of (D/L)-lactic acid and (D/L)-alanine by using N′-3,5-dinitrobenzoy-(L)-alanine-octylester as the chiral selector. The authors compared the enantiomeric resolution capability of a hollow fiber-supported liquid membrane and a chiral solid membrane. On one hand, the maximum enantiomeric excess obtained was 33.5% for (D)-lactic acid and 27.2% for (D)-alanine when SLM was used. On the other hand, when using a chiral solid membrane, the maximum enantiomeric excess obtained was 100% for (D)-lactic acid but no separation of Alanine enantiomers was achieved.
Supported hollow fiber membranes can be used in series for a better process efficiency and productivity. In 2006, Maximini et al.128 developed a continuous SLM system (two in series) to separate enantiomers of racemic N′-protected amino acid derivatives using carbamylated quinine and quinidine derivatives as chiral selectors. Wang et al.133 developed a process to separate ketoconazole enantiomers using a hydrophobic chiral selector – isopentyl tartrate which will recognize (2R,4S)-ketoconazole – in the organic phase (shell side) and a hydrophilic chiral selector – sulfobutylether-β-cyclodextrin in the aqueous phase (lumen side) which will recognize (2S,4R)-ketoconazole. The optical purity for the enantiomers is up to 90% when used two modules in series.
Prapasawat et al.134 modeled and validated the resolution of racemic phenylalanine using O,O-dibenzoy-(L)-(2S,3S)-tartaric acid (DBTA) as chiral selector. Several parameters were investigated, such as concentration of DBTA and feed solution, initial pH, and equal flow rates of feed and stripping solutions. 55% of enantiomeric excess was obtained for (L)-phenylalanine at pH 5.
Svang-Ariyaskul et al.126,135,136 reported the resolution of (D/L)-glutamic acid using a hybrid system where two streams are recirculated in two crystallization vessels and feed in counter-current in a hollow-fiber membrane module, which allowed solute interchange between the vessels but a limited transfer of crystals. Each crystallizer contained a racemic solution of glutamic acid. Seed crystals of a specific enantiomer were fed to one of the crystallizers when the solution was at or near saturation conditions, while seed crystals of the opposite enantiomer were added to the other crystallizer. The feasibility of the process was demonstrated, and key operating variables were identified. The enantiomeric excess of 99% of both enantiomers was obtained (Fig. 14).
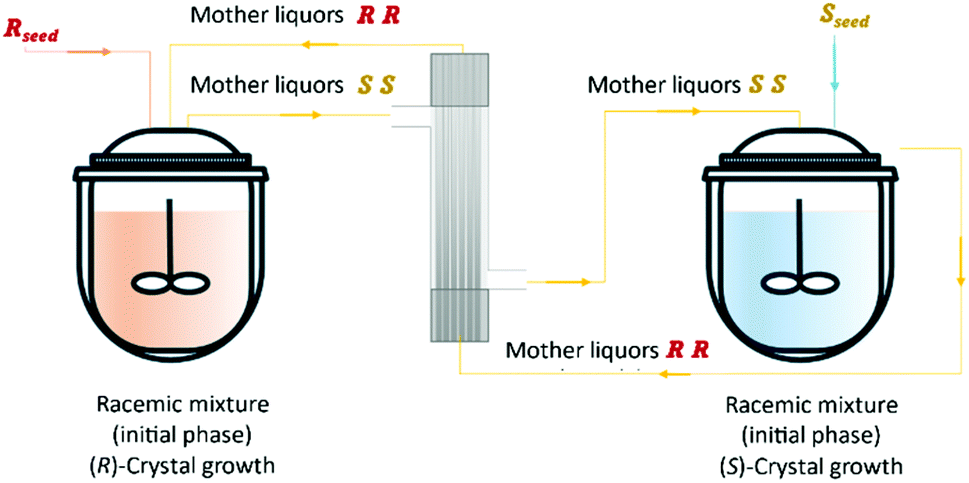 | ||
| Fig. 14 Schematic representation of the hybrid crystallization/filtration process. By seeding the vessels, it will promote the preferential crystallization of a specific enantiomer. The counter enantiomer rich mother liquors will flow through the membrane only transporting the dissolved compounds. | ||
Lopez et al.137 described a hollow-fiber membrane reactor to an enzyme-mediated resolution of a racemic mixture of a Diltiazem intermediate. The authors developed the process from bench-scale to pilot plant. They built a reactor containing hydrophilic enzyme-activated porous located at the interface between organic and aqueous streams. Thus, the hollow fibers were used to ‘immobilize’ the biocatalyst and allow the enzyme phase to be in contact with the substrate organic phase. Börner et al.138 reported the amine transaminase-catalyzed synthesis of chiral amines. The authors investigated the feasibility of using alanine as an amine donor for the reductive amination of a poorly water-soluble ketone in combination with the SLM strategy for in situ product removal. The amine product (1-methyl-3-phenylpropylamine) was obtained with enantiomeric excess above 98% without any additional purification (Fig. 15).
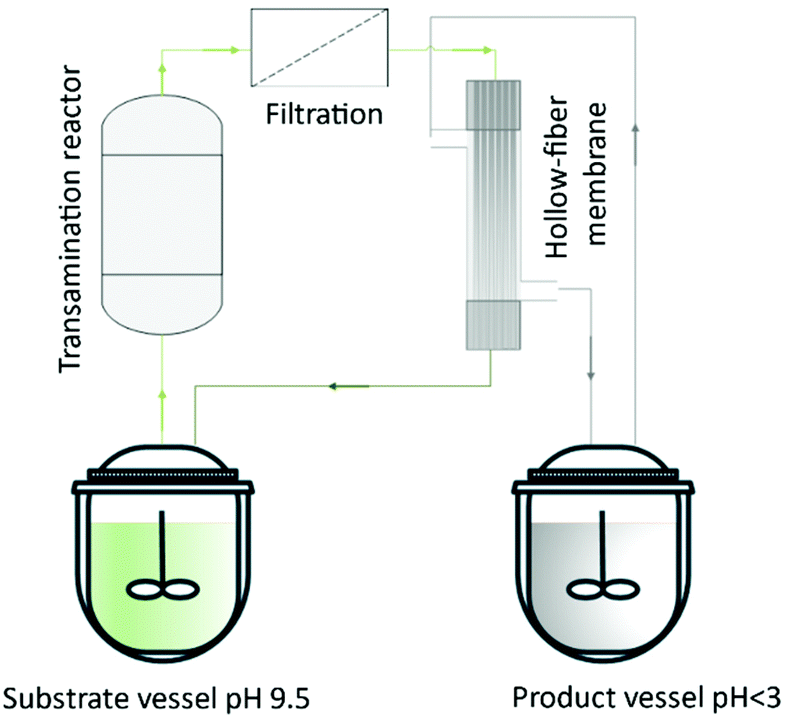 | ||
| Fig. 15 System to produce 1-methyl-3-propylamine (MPPA) from (L)-alanine and benzylacetone. The system consists of a transamination reactor containing an immobilized enzyme, an in-line filter and a hollow-fiber module that separates the product from a co-product by a pH differential of lumen/shell sides. | ||
Sunsandee et al.139,140 studied the enantioselective separation of (S)-amlodipine and (S)-cetirizine via a hollow fiber-supported liquid membrane. In both cases, DBTA was used as a chiral selector in 1-decanol. The maximum enantiomeric excess obtained for (S)-amlodipine was 40.5% and for (S)-cetirizine was 75%.
To promote the selectivity of hollow fiber, molecularly imprinted hollow-fiber membranes can be prepared by immobilizing the molecularly imprinted layer on the cavities of the hollow-fiber via an interfacial polymerization technique. Lai et al.141 prepared molecularly imprinted hollow fiber membranes with methacrylic acid (MAA) as the functional monomer, ethylene glycol dimethacrylate (EGDMA) as the cross-linker, and (S)-amlodipine as an imprinted template. As a result, (S)-amlodipine was obtained with 90% enantiomeric excess and a separation factor of 1.98.
Recently, Zeng et al.127 developed a novel method for the enantioseparation of Amlodipine using a hollow fiber module as an extraction and crystallization system. For the extraction operation, the authors used a commercial extractant P507 (2-ethylhexyl phosphonic acid mono-2-ethylhexyl ester and heptane and for the crystallization operation, (D)-tartaric acid as the chiral selector and DMSO as antisolvent. The obtained enantiomeric excess was 90.7% and a global yield of 48.8% for the isolated product, (S)-amlodipine (Fig. 16).
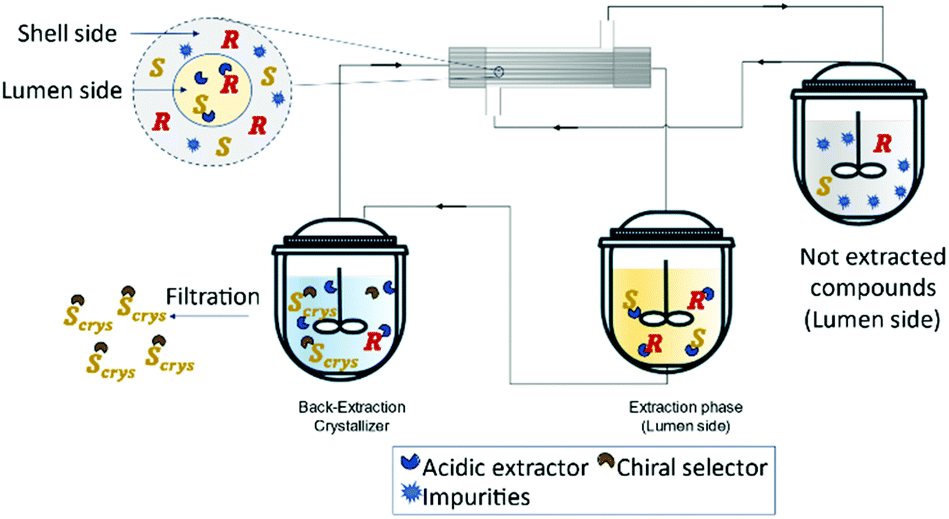 | ||
| Fig. 16 Schematic representation of a solution purification and enantiomeric separation. The lumen side contains an acidic extractor selective to the racemic compounds. The extracted solution is feed into the crystallizer where it contains a chiral selector that will form a salt with the (S)-enantiomer and precipitate as a crystal. Adapted from ref. 127. | ||
From a Green Chemistry perspective, more studies must be performed using supported liquid membranes focusing on more environmentally friendly conditions. The used organic solvents must preferably present low toxic potential (P3). The more concentrated solutions, the less waste is generated (P1) and it is certainly recommended the recycling the used solvents and chiral hosts.
The strategy of Svang-Ariyaskul et al. is very interesting because it uses concepts of crystallization and membrane separation in a very innovative way. A good advantage of this process is that there is no need for the addition of a chiral host but only seeds of the enantiomers. The advantage of not using a chiral host is that the purification procedure is simpler because it is not necessary separating the host of the enantiomer, which would require more solvents, reagents, unit operations and energy expenditure (P1, P6).
The use of an enzyme-mediated resolution process using supported liquid membranes seems a promiscing field and good results were achieved. This approach relies on transformations that make use of natural compounds as reagents under mild conditions. Moreover, enzymes have proven to be more regioselective (P5, P9).19
3.2. Solid membranes for chiral resolution
Taki et al.144 tested the enantioseparation of racemic tryptophan with poly-(L)-glutamic acid (PLGA) crosslinked with polyetherdiisocyanate membranes immersed in aqueous ethanol. (D)-Tryptophan was selectively transported with a maximum separation factor of 2.6. Kim et al.145 also carried on the resolution of tryptophan, the authors prepared sodium alginate crosslinked with glutaraldehyde membranes. The degree of crosslinking and its effect on enantioselectivity was studied and the outcome was that the less crosslinking, a higher swelling index is obtained, the flux increases but the enantiomeric excess decreases. Therefore, a higher membrane swelling was unfavorable for good optical resolution. A maximum enantiomeric excess of 54% was obtained from a membrane with a swelling index of 80%.
Later, the same co-authors prepared an enantioselective membrane of chitosan for the separation of racemic tryptophan having obtained 98% of enantiomeric excess with a swelling index of 79%.146 Aoki et al.149 synthesized nine chiral phenylacetylenes containing pinanyl groups and investigated the introduction of chirality in the main chain and formation of chiral helical backbone during polymerization, and its enantioselectivity in the permeation of the polymeric membranes. Results showed that by using the membrane made of poly-[dimethyl(10-pinanyl) silyl]phenylacetylene, (poly-PSPA), the maximum enantiomeric excess was obtained for phenylalanine, valine, and tryptophan were 53.4%, 28.2%, and 99.9% respectively.
Hazarika et al.150 synthesized a polymeric membrane of 1,2-bis(2-methyl-1-triethylsiloxy-1-propenyloxy) ethane and investigated the optical resolution of racemic trans-sobrerol by pressure-driven permeation through the membrane. The highest enantiomeric excess was obtained as 98.59% for (S)-trans-sobrerol using NMP as process solvent.
Xie et al.147 reported the preparation of a cellulose acetate butyrate membrane for the separation of racemic 2-phenyl-1-propanol and enantiomeric excess of up to 98% was achieved.
Ma et al.151 prepared a cellulose membrane and studied the effect of cellulose concentration, drying time, operating pressure, and feed concentration in the membrane properties and its enantioselectivity for (D)-mandelic acid. The maximum obtained enantiomeric excess was 90%. A cellulose membrane was also used by Yuan et al.152 for the resolution of racemic mandelic acid having obtained the enantiomeric excess of 89.1% for the (D)-enantiomer – Table 6.
| Material | Product | ee and/or α | Ref. |
|---|---|---|---|
| Poly(L)-glutamic acid | (D)-Tryptophan | α: 2.60 | 144 |
| Poly(γ-methyl-(L)-glutamate) derivatives | (D)-Tryptophan | 16% | 142 |
| (D)-Tryptophan | 20% (α: 3.00) | 143 | |
| Sodium alginate | (D)-Tryptophan | 54% | 145 |
| Chitosan | Tryptophan | 98% | 146 |
| Poly[p-(oligopinanyl-siloxanyl)phenylacetylene] | Phenylalanine | 53.4% | 149 |
| Valine | 31.5% | ||
| Tryptophan | 99.9% | ||
| 1,2-Bis(2-methyl-1-triethylsiloxy-1-propenyloxy)ethane | S-trans-Sobrerol | 98.59% | 150 |
| Cellulose acetate | 2-Phenyl-1-propanol | 98% | 147 |
| Cellulose | (D)-Mandelic acid | 90% | 151 |
| (D)-Mandelic acid | 89.1% | 152 |
The end-life management of membrane modules is a great concern. According to the principles of Green Chemistry, the raw material should be renewable rather than depleting (P7), thus the membrane material should be selected accordingly. Chitosan, Cellulose, PLGA and sodium alginate are renewable feedstocks and presented good results as it can be seen in Table 6.
In 2011, Ingole et al.155 reported the chiral separation of racemic lysine and arginine through enantioselective polysulfone membranes containing a superficial layer of chiral metal–Schiff base complexes. The same co-workers described the resolution of racemic arginine using trans-1,4-diaminocyclohexane as the chiral selector (ee: 92%)156 and the resolution of lysine and asparagine using (L)-arginine as the chiral selector (ee: 91.6 and 67.8% respectively).157 Devi et al.158 reported the optical resolution of racemic mixtures of arginine and alanine. The chiral selective layer of the membrane was prepared by interfacial polymerization of metaphenylenediamine, trimesoyl chloride, and (S)-2-acetoxypropionyl chloride in situ on the top of polysulfone membrane (Table 7).
| Support layer | Superficial layer | Chiral selector | Product | ee or α | Ref. |
|---|---|---|---|---|---|
| Polysulfone | TMC in hexane | Metal Schiff complex | Lysine | 94% | 155 |
| Arginine | 84% | ||||
| Polysulfone | TMC in hexane | trans-1,4-Diaminocyclohexane | Arginine | 92% | 156 |
| Polysulfone | TMC in hexane | (L)-Arginine | Lysine | 91.6% | 157 |
| Asparagine | 67.8% | ||||
| Polysulfone | meta-Phenylenediamine, TMC in hexane | (S)-2-Acetoxypropionyl chloride | Arginine | 92% | 158 |
| Alanine | 68% | ||||
| Polysulfone | Polydopamine | Ethylenediamine-β-cyclodextrin | Tryptophan | 3.2% | 159 |
| PE/PP + polyamide nanofiber (PA6) | MPD/DACH in aqueous phase, TMC in hexane | DACH | Tryptophan | 45% | 160 |
| Polysulfone | m-Phenylene-diamine, TMC in hexane | α-Cyclodextrin | Tryptophan | 1.55 | 161 |
| Polysulfone | Carbon nanotubes | (D)-Tryptophan | Tyrosine | 98.86% | 162 |
| Cellulose acetate | TMC | Ethylenediamine-β-cyclodextrin | Warfarin | 9.29% | 163 |
| Ibuprofen | 3.77% | ||||
| Tryptophan | 27.2% |
Miao et al.159 described the chiral resolution of tryptophan by using a modified polysulfone membrane prepared via mussel chemistry. The membranes were modified with dopamine, and β-cyclodextrin as a chiral selector. Gaálová et al.160 prepared a composite membrane from a polyethylene/polypropylene and polyamide nanofiber. The authors prepared a thin, selective layer by interfacial polymerization of two immiscible phases on the porous nanofiber layer: m-phenylene diamine (MPD) and (S,S)-1,2-diaminocyclohexane (DACH) were dissolved in water while 1,3,5-trimesoyl chloride (TMC) was dissolved in n-hexane; the active film was obtained by remittent immersion of the fibrous composite in both phases and subsequent thermal treatment.
Zhou et al.161 reported the synthesis of modified polysulfone membrane with a superficial thin layer containing α-CD as the chiral selector and MPD monomer for interfacial polymerization. (L)-Tryptophan was obtained with a separation factor of 1.55 by a concentration gradient mode.
Gogoi et al.162 demonstrated how single-walled carbon nanotubes can be used to increase the hydrophilicity, mechanical strength, and thermal stability of the membrane. The prepared membrane was applied for the separation of (D/L)-tyrosine having obtained 98.86% of enantiomeric excess for (D)-tyrosine.
Ke et al.163 reported a novel chiral thin-film composite polyamide membrane that was fabricated by using in situ interfacial polymerization. Ethylene-beta-cyclodextrin (EDA-β-CD) was the chosen chiral selector and it was polymerized with TMC to form a thin layer at the surface of a commercial cellulose acetate membrane.
Meng et al.166 integrated a chiral selector (L)-glutamic acid into GO flake via a simple vacuum filtration method. Enantioseparation performances were studied of amino acid modified GO membranes toward 3,4-dihydroxy-(D/L)-phenylalanine with a separation factor of 2.05. The results demonstrated how modified GO membranes might provide enantioseparation by enabling high-flux and high-selectivity.
The same co-authors prepared GO-based composite membranes via tuning the interlayer spacing between the GO-sheets using PLGA. The incorporated PLGA would not only serve as an additional chiral selector, facilitating the transport of (D)-enantiomer of the chiral probe, but also as a kind of filler, reducing the size of pores or channels between GO sheets. The separation factor obtained was 2.80 for 3,4-dihydroxy-(D)-phenylalanine (Table 8).167
| Chiral selector | Product | ee or α | Ref. |
|---|---|---|---|
| (L)-Glutamic acid | 3,4-Dihydroxy-(D)-phenylalanine | 2.05 | 166 |
| PLGA | 3,4-Dihydroxy-(D)-phenylalanine | 2.80 | 167 |
| β-Cyclodextrin (β-CD) | (L)-Asparagine | 99% | 168 |
| (L)-Glutamic acid-functionalized with an ionic liquid | 3,4-Dihydroxy-(D)-phenylalanine | 3.83 | 169 |
| (L)-Phenylalanine | (D)-Phenylalanine | 1.72 | 170 |
| (D)-Methionine | 1.10 | ||
| N′-Acyl-(D)-phenylalanine | 1.80 | ||
| N′-Acyl-(D)-methionine | 1.30 |
Qie et al.168 prepared nanoporous graphene oxides covalently functionalized with β-cyclodextrin as chiral selector. (L)-Asparagine was obtained with great enantioselectivity with more than 99% of enantiomeric excess.
Meng et al.169 proposed a new strategy to improve the enantioseparation performances of GO membranes by controlling the degree of functionalization with an epoxide ring-opening reaction with a carboxyl-terminated ionic liquid as a spacer and an active site for the chiral selectors, followed by an amidation reaction of (L)-glutamic acid as chiral selectors. The authors concluded that these membranes are superior in the enantioselectivity and 1–3 orders of magnitude higher in the flux than traditional chiral separation membranes. The separation factor obtained was 3.89 for 3,4-dihydroxy-(D)-phenylalanine.
Recently, the same co-workers170 prepared an (L)-phenylalanine modified GO-based membranes, and its enantioselectivity was tested for racemic phenylalanine, methionine, N′-acyl-phenylalanine, and N′-acyl-methionine. Results showed that the prepared membrane presented stronger interaction toward (D)-enantiomers essentially caused by the additional non-stereoselective component between GO surfaces and chiral probes.
Carbon nanomaterials play an important role in resolving the increasingly urgent energy and environmental crises due to their intrinsic chemical and mechanical properties. However, the harmful environmental impacts on their preparation might reduce the sustainability (P3). Therefore, new technologies for carbon nanomaterials preparation must be developed to minimize environmental impacts.171
MOFs are a family of porous crystalline materials constructed with coordinate bonds between inorganic secondary building units and organic ligands.179,186 HOFs have some advantages such as easy purification, straightforward regeneration, and reusability by simple recrystallization.187 This type of membrane presents high enantioselectivity because of its porous chiral channels and consequently high permeation flux and low mass transfer resistance.188 Moreover, enantiomer separation can be optimized by tuning the pore size or shape and introducing a chiral functional group into a MOF.176 Also, the use of mixed matrix membranes (MMMs) that combine the advantages of a polymeric matrix and the molecular selectivity of microporous materials can provide a simpler approach to achieve enhanced permeability and selectivity.101,189
A few MOFs and their derivative membranes for chiral separation have been reported recently. Also, coupling chiral ligands to the MOFs has been applied to tune the chirality of achiral MOFs for asymmetric catalysis.190–193
Kepert et al.194 reported that the stereochemistry of alcohol templates spontaneously resolves to form homochiral helical structures featuring helical networks. Therefore, the enantioselective adsorption property was successfully established for the first time by the homochiral MOFs.
In 2011, the resolution of 2,5-hexanediol was performed under continuous flow mode by Liu et al.195 The authors selected surface-mounted metal–organic frameworks to study the adsorption of (R/S)-hexanediol from the gas phase (vapor) in a continuous-flow mode using nitrogen gas as a carrier.
In 2012, a homochiral MOF membrane [Zn2(bdc)((L)-lac)(dmf)] was prepared for the enantioselective separation of (S)-methyl phenyl sulfoxide that can be used as synthetic auxiliary and valuable pharmaceutical.188
In the following year, Kang et al.196,197 developed a method named “Single nickel source” for in situ fabrication of a high thermal stable homochiral MOF. A diol isomer mixture was used to test the separation efficiency of the membrane at different temperatures and pressures obtaining ee of 32.5% (Table 9).
| Framework | Ligand | Enantiomer | ee% | Ref. |
|---|---|---|---|---|
| [Zn2(bdc)((L)-lac)(dmf)] | 1,4-Benzenedicarboxylate (bdc) | (S)-Methyl phenyl sulfoxide | 20 | 188 |
| (L)-Lactic acid ((L)-lac) | ||||
| Ni2((L)-asp)2(bipy) | 4,40-Bipyridine(bipy) | 2-Methyl-2,4-pentanediol | 32.5 | 196 and 197 |
| HOF-2 | (R)-1,10-Bi-2-naphthol scaffold into 2,4-diaminotriazinyl | 1-Phenylethanol | 92 | 187 |
| 1-(4-Chlorophenyl)ethanol 1-(3-chlorophenyl)ethanol | 79 | |||
| 2-Butanol 2-pentanol 2-hexanol | 66 | |||
| 2-Heptanol | 77 | |||
| 48 | ||||
| <10 | ||||
| <4 | ||||
| (S)-[DyNaL(H2O)4] | 3,30-Di-tert-butyl-5,50-di (3,5-carboxyphenyl-1-yl)-6,60-dimethylbipheny(L)-2,20-diol ligand (H4L) | (L)-Methyl mandelate ethyl mandelate i-propyl mandelate | 93.1 | 207 |
| (D)-Methyl mandelate benzyl mandelate | 64.3 | |||
| 90.7 | ||||
| 89.9 | ||||
| 73.5 | ||||
| (D)-His-ZIF-8 | (D)-Histidine | Alanine | 78.52 | 199 |
| Glutamic acid | 79.44 | |||
| [Co2(S-man)2(bpy)3](NO3)2·guest | Mandelate and 4,4′-bipyridine | 1-Phenyl-1-propanol | 60 | 200 |
| (L)-His-ZIF-8 | (L)-Histidine | 1-Phenylethanol | 76 | 204 |
| H-(D)-his-ZIF-8 | (D)-Histidine | Alanine | 90.5 | 205 |
| Glutamic acid | 95.2 | |||
| lysine | 92.6 | |||
| [Cd2(CO2)4(DMF)2(H2O)] | 1,1′-Biphenol | 1-Phenylethanol | 99 | 206 |
| 1-Phenylpropanol | 92 | |||
| 1-(1-Naphthyl)ethanol | 99.4 | |||
| 1-(4-Fluorophenyl)ethanol | 93 | |||
| 1-(4-Chlorophenyl)ethanol | 98 | |||
| 1-(4-Bromophenyl)ethanol | 98 | |||
| Methyl phenyl sulfoxide | 83 | |||
| Ethyl phenyl sulfoxide | 80 | |||
| 3-Methoxy phenyl methyl sulfoxide | 96 | |||
| Lansoprazole | 91 | |||
| MIL-53-NH2/PES MMM | (L)-Histidine | 1-Phenylethanol | 100 | 189 |
| (L)-Glutamic acid | ||||
| γ-CD-MOF/PES MMM | γ-Cyclodextrin | 1-Phenylethanol | 100 | 101 |
In 2014, Li et al.187 prepared a homochiral microporous hydrogen-bonded organic framework (HOF-2) based on a BINOL derivative that has been synthesized and structurally characterized, presenting permanent porosity and highly enantioselective separation of chiral secondary alcohols with ee value up to 92% for 1-phenylethanol. Based on the work of Brunet et al.,198 they showed that 2,4-diaminotriazinyl is a very powerful hydrogen-bonding backbone for the construction of porous robust HOFs, and the use of highly enantioselective separation of small molecules.
In 2015, Zhao et al.199 prepared a microporous MOF structure, with a chiral environment using (D)-histidine as chiral ligand, (D)-his-ZIF-8. The membrane shows selective separation capability for racemic alanine and glutamic acid, with an ee value of 78.5% and 79.4% respectively.
Zhang et al.200 reported a single-step synthesis of MOFs based upon mandelate (man) and 4,4′-bypyridine ligands and enantioselective recognition toward the (R) enantiomer of 1-phenyl-1-propanol was observed with ee values of up to 60%.
In 2017, Liu et al.201,202 demonstrated a superficial chiral etching process to introduce chiral function on achiral MOF surface for enantioselective adsorption toward limonene enantiomers. In the following year, the co-workers203 prepared a MOF–chiral polymer composite, which integrates chirality, porosity, and conductivity into the composite for enantioselective adsorption and sensing for carvone enantiomers.
Chan et al.204 prepared (L)-his-ZIF-8 that exhibited good selectivity for the (R)-enantiomer of 1-phenylethanol over the (S)-enantiomer, showing a high ee value up to 76%. Wang et al.205 prepared chiral ZIF-8 hollow nanospheres with (D)-histidine as ligand. Due to the higher surface area and consequently more accessible chiral groups, the hollow cavity can be more attractive for guest-sorption.
Abbas et al.206 described a chiral porous 3D MOF built from an enantiopure carboxylate ligand of 1,1′-biphenol, which can be utilized as an adsorbent for the separation of aromatic alcohols and sulfoxides with enantioselectivity of up to 99.4%.
In 2019, Wang et al.208 reported a facile and scalable approach: a thermally induced phase separation-hot pressing to fabricate flexible membranes with ultrahigh MOF loading (up to 86 wt%) for separation applications. The membranes presented good mechanical behavior, hierarchical porous structure, and large surface area. High flux, and rejection rate for water treatment with the ability to catch small molecules under crossflow filtration mode.
Lu et al.189 described a new type of enantioselective MMM derived from homochiral MIL-53 nanocrystals with polyethersulfone as a polymeric matrix and aminoacids as chiral ligands. And recently the authors reported γ-CD-MOF-based MMMs with polyethersulfone as a matrix for efficient separation of racemic 1-phenylethanol.101 More information about the preparation and applications of cyclodextrin-MOF can be found in the reviews.209–212
Chen et al.213 described the preparation of a three-dimensional hierarchically porous organic/inorganic composite by incorporation of the hyper-cross-linked resin organic polymer with macroporous silica gel sheet, followed by a chiral selector post-modification. Racemic 1-phenylethanol, ibuprofen, and naproxen could be separated successfully with complete resolution.
In a classical approach, it is needed a template, a functional monomer, a cross-linker, a polymerization initiator, and a solvent.214 The functional monomers will be designed to interact with the template via covalent or non-covalent bonding.215 Furthermore, the monomers are polymerized in the presence of the template that afterward will be removed. The resulting membrane will have specific binding sites to the compound of interest. MIMs may be used on a large scale due to their obvious advantages including ease of preparation, scalability, stability, and low material cost.216 Interesting reviews about the application of molecularly imprinted membranes can be found in the literature.98,214,215,217–220
The first application of molecularly imprinted membrane was developed by Michael's research group in 1962.221 They prepared polyethylene membranes imprinted with p-xylene that were able to selective transport p-xylene over the corresponding isomers, ortho or m-xylene. In 1972, Wulff and co-workers developed a methodology for the introduction of molecular recognition sites into a polymeric matrix in a specific steric arrangement.222–224
Enantioseparation using MIM has been intensively studied by using alternative molecular imprinting, a technique that can convert any polymeric material into an enantioseparation membrane.225–233 In general, it is necessary to employ the optically pure print molecule to obtain MIM for chiral recognition and separation.219 The chiral recognition ability was found to be dependent on the absolute configurations of both the print molecule and the amino acid oligopeptide residues. Specifically, a membrane consisting of oligopeptide residues from a (D)-amino acid and imprinted with a (D)-amino acid derivative recognized the (D)-enantiomer in preference to the corresponding (L)-enantiomer and vice versa.220
Since 1995, Yoshikawa and co-workers published a considerable amount of work based on molecularly imprinted membranes for enantiomers resolution. The authors explored molecularly imprinted nanofiber and polymeric membranes prepared from carboxylated polysulfone, polysulfone-aldehyde, myrtenal-containing polysulfones, cellulose acetate, novel polyurea membranes synthesized from lysinyl residues, polyamide, tri, and tetrapeptide derivatives, and N-α-benzyloxycarbonyl-(D/L)-glutamic acid (Boc-(D/L)-Glu) and N-α-benzyloxycarbonyl-(D/L)-tryptophan (Boc-(D/L)-Trp) as templates. They determined separation factors for tryptophan enantiomers and their derivatives,227,230–237 glutamic acid,225,226,228,238–244,245 and other aminoacids (Table 10).229,246
| Mode | Framework | Imprinted molecule | Enantiomer | ee or α | Ref. |
|---|---|---|---|---|---|
| Electrodialysis | Polysulfone | (L)-Phe | (L)-Phenyl-alanine | 4.10 | 255 |
| Polystyrene resin + tetrapeptide derivative (Boc-DIDE-resin) | (L)-Trp | (D)-Tryptophan | 1.40 | 234 | |
| Nylon-6 | (L)-Glutamine | (L)-Glutamine | 1.0 | 248 | |
| Tetrapeptide derivative | Boc-(L)-Trp | N′-α-Acetyl-(L)-tryptophan | 6 | 236 | |
| Polystyrene resin | 6 | 232 | |||
| Tripeptide derivative | 4.5 | 233 | |||
| Aminomethylated polystyrene resin + oligopeptide tweezers | 2.5 | 235 | |||
| Tetrapeptide derivative | Boc-(D)-Trp | (D)-Tryptophan | 1.0 | 229 | |
| (D)-Phenylalanine | 1.0 | ||||
| (D)-Alanine | 1.0 | ||||
| Tetrapeptide derivative | Boc-(L)-Trp | (L)-Arginine | 1.3 | 246 | |
| (L)-Lysine | 1.3 | ||||
| (L)-Histidine | 1.4 | ||||
| (L)-Asparagine | 1.2 | ||||
| (L)-Glutamic acid | 1.3 | ||||
| Electro-dialysis | Cellulose acetate | Boc-(D)-Glu | (D)-Glutamic acid | 2.30 | 244 |
| Pressure driven | Cellulose acetate | (D)-Glutamic acid | 1.45 | 243 | |
| Electrodialysis | Myrtena(L)-containing polysulfones | Boc-(L)-Glu | (L)-Glutamic acid | 1.59 | 228 |
| Chiral polyurea | 1.14 | 239–242 | |||
| Polysulfone-aldehyde derivatized | 1.25 | 245 | |||
| Conc. driven | Chitosan/GPTMS | (L)-Phenylalanine | (D)-Phenylalanine | 4.5 | 249 |
| Temp. driven | Poly-(methyl methacrylate)-N-isopropyl acrylamide (PMMA-PNIPAm) | (R)-Equol | (S)-Equol | 2.60 | 252 |
| Conc. and pressure | Chloromethylated polysulfone (CMPSF) + dimethylaminoethyl methacrylate (DMAEMA), N,N′-methylene-bisacrylamide (MBA) | (L)-Aspartic acid | (L)-Aspartic acid | 82% (7.52) | 253 |
| Pressure driven | Alumina template with ZrO2 + cellulose acetate | (L)-Mandelic acid | (D)-Mandelic acid | 94.5% (35) | 254 |
The co-workers also studied how the number of constituent amino acid residues influence the molecular recognition ability of the membrane (tri and tetrapeptide derivatives) and it was concluded that the tetrapeptide derivative was the best candidate for forming molecular recognition sites. Moreover, it was interesting to identify those membranes that were imprinted with an amino acid with a certain configuration, recognizing not only the target amino acid but also analogs that had the same absolute configuration.
Electrodialysis was recognized as a suitable driving force for membrane enantioselective transport. In 1999, Dzgoev and Haupt prepared molecularly imprinted polypropylene membranes with Boc-(L)-tyrosine as a template. The results confirmed that the imprinted molecule recognizes the compounds with the same absolute configuration, in this case, Boc-(L)-tyrosine.247
Screenivasulu Reddy et al.248 functionalized Nylon-6 by alternative molecularly imprinted membrane technique with (L)-glutamine. The authors acknowledged that the binding of (L)-glutamine increased with the increase of the amino acid concentration. A maximum separation factor of 1 was obtained. Jiang et al.249 prepared an enantioselective MIM using chitosan as bulk polymer, (L)-phenylalanine as imprinting molecules, and γ-glycidoxypropyltrimethoxysilane as the crosslinking agent. Significantly selectivity with a separation factor of 4.5 for (D)-phenylalanine was achieved.
Molecularly imprinted composite membranes were prepared by Son and Jegal.250 They used polysulfone as a support layer, piperazine and TMC in hexane as a superficial layer, and (D)-serine as a chiral template. The membrane proved to be effective for the selective permeation of (D)-serine having obtained a maximum enantiomeric excess of 80%. Also, Bing et al.251 developed PVDF hollow-fiber membranes modified by a thin layer of a molecularly imprinted polymer prepared with levofloxacin, methacrylic acid, and ethylene glycol dimethacrylate that allowed selective separation of levofloxacin.
Lei et al.252 prepared a molecularly imprinted cation-exchange membrane for the resolution of racemic Equol. The membrane preparation resumed to a one-pot approach to obtain monodispersed methyl methacrylate-N-isopropyl acrylamide polymer by atom transfer radical precipitation polymerization. The maximum obtained separation factor was 2.60.
Gao et al.253 used a chloromethylated polysulfone membrane and applied an advanced surface imprinting technique for the separation of a racemic mixture of aspartic acid. By amine modification, an aminated polysulfone membrane was obtained. The imprinting layer was attained from dimethylaminoethyl methacrylate as functional monomer, N,N′-methylene-bisacrylamide as the crosslinking agent, (NH4)2S2O8 as an initiator, and (L)-aspartic acid as a template. A separation factor of 7.52 was obtained for (L)-aspartic acid with an enantiomeric excess of 82%.
Li et al.254 prepared an alumina template modified with ZrO2 coated with cellulose acetate containing imprinted molecules for a novel enantioselective membrane. (S)-Mandelic acid was used as a chiral template. The co-workers studied the impact of changing the concentration of cellulose acetate, and the procedure of molecular imprinting in the morphology and particle size of the ZrO2-modified Al2O3 membrane. The obtained separation factor was as high as 35 for (S)-mandelic acid with an enantiomeric excess of 94.5%.
Based on the described studies, it seems that complete enantio-separation of a racemic mixture through one cycle process with a molecularly imprinted membrane is not feasible. As far as we know, 100% ee cannot be achieved by a single operation. Multi-cycle operation processes might be the path to achieve better separation.217
Many advances in synthetic approaches have been made in the last years, however, molecular imprinting methodology still uses high quantities of solvents and intensive process steps.256
Computational modeling might be used in the selection of reagents required for the synthesis of MIMs. It can be very useful to avoid unnecessary screening reactions and waste formation (P1). The monomers required for the membrane skeleton are selected computationally based on their ability to interact with the template molecule. Similarly, the used solvent can also be selected using computational tools. This results in a careful selection of the necessary reagents to prepare the desired MIM with no generation of waste.257
Reusability is a great advantage of MIMs as they can be used continually after a regeneration step. Recycling allows the reduction of reagents that would be required for the preparation procedure. In several studies, MIMs have been repeatedly applied many times with minimum loss in their extraction efficiency (P1).257
Commonly, the template removal from the MIMs is performed through exhaustive washing with organic solvents. Subsequently, the organic solvents are discarded into the environment.258 To employ greener methods of template removal, supercritical solvents might be used to replace the organic solvents. Furthermore, the ability to reuse MIMs in many applications reduces the solid waste in the environment.257
Our planet is facing environmental pollution and a serious energy crisis. A green mindset may offer important help to overcome these challenges. The green chemistry principles encourage the use of safer solvents and processes with lower impact on the environment by preventing waste and increasing energy efficiency. Membrane technology is considered a green and sustainable technology because of its relatively low energy consumption.260 Nevertheless, traditional membrane fabrication methods negatively affect sustainability.261 As described in this review, membranes should offer a good transport rate with high selectivity, be stable in a high range of pH, compatible with different types of solvents, superior antifouling properties, and minimal aging or swelling.261
Membrane fabrication frequently employs non-environmentally friendly solvents as NMP, DMF, and DMAC. These traditional procedures must be absolutely substituted by manufacturing processes that make use of green solvents.261
Certainly, green solvents might provide a similar membrane performance when compared to the traditional ones, however, the manufacturing processes are still costly. In fact, due to the inherent advantages compared to the greener alternatives, namely reproducibility in large scales, increased solubility, and lower cost, classical solvents have been preferred. Fortunately, some researches indicate that it is completely possible to achieve these goals with greener solvents, not yet now but very soon.262 Moreover, the treatment and recycling of the used organic solvents may be a viable approach to reduce waste.
The end-life management of membrane modules is another great matter. The reuse of membrane tools rather than their direct incineration should be encouraged to drive membrane technology to a greener level.261
4. Continuous chromatography
4.1. Simulated moving bed chromatography
The conventional four-zone simulated moving bed (SMB) chromatography unit consists of a closed system with several packed columns connected in a loop. The system is divided into four zones by two inlets named feed and eluent/desorbent and two outlets named extract and raffinate.A racemic solution feed is continuously fed into the process through the feed stream, and an eluent solution (usually the same solvent composition as feed solution) is fed into the process through the eluent stream. The enantioseparation occurs in the packed columns containing a chiral stationary phase and the solutions are collected at two outlets (raffinate: less retained enantiomer, extract: more retained enantiomer). The remaining mixture of enantiomers that has not been separated can be recycled through the columns while the feed racemic solution is feed.263 A SMB process does not reach the steady-state but the cyclic steady state because of the periodic repetition of port switching.263 The key advantages of an SMB process over batch chromatography is the cost-effective potential to enhance productivities and to reduce the solvent consumption compared to conventional discontinuous processes.264,265
The first application of a countercurrent SMB chromatography was described in Broughton's patent from 1961 and was widely adopted in the petrochemical industry and for complex separations.266 In 1992, the application of SMB for enantioseparation was reported for the resolution of 1-phenylethanol.267 Negawa and Shoji used an eight-column SMB unit packed with Chiralcel OD and a mixture of hexane and isopropanol as eluent and developed a purification process with great advantages when compared to the batch process, namely a minor solvent consumption by using more concentrated solutions and increased productivity since it is performed in continuous mode. Since then, several patents about enantioseparation of active pharmaceutical ingredients and their analogs were published, namely for (S)-levetiracetam,268 sertraline,269 oxetine,270 pharmaceutically desired chiral tetralones,271 and Schiff bases.272 Diehl et al.273 developed the resolution of cis-8-benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane, a quinolone derivative by SMB chromatography in a kilogram scale with enantiomeric excesses above 98%.
In 1993, Ching et al.274 published the continuous purification of racemic Praziquantel, an anthelminthic API, by SMB chromatography with a four-column configuration. The co-authors optimized the process275,276 having obtained maximum purity of 97.5% for the (L)-enantiomer and from the obtained raffinate solution, it was possible to concentrate it and crystallize the desired enantiomer with an overall yield of 80%. In 2016, Andrade Neto et al.277 reported a theoretical study about adaptive nonlinear model predictive control (NMPC) strategies to separate racemic praziquantel using SMB chromatography. Finally, in 2019, Cunha et al.278 described the resolution of praziquantel with an SMB built-in-house unit with [1-2-2-1] column configuration with 100% chiral purity for the (L)-enantiomer (Table 11).
| Product | Chiral stationary phase | Eluent | Purity | Ref. |
|---|---|---|---|---|
| (R)-1-Phenylethanol | Chiralcel OD | Hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IPA :IPA |
99% | 267 |
| (S)-1-Phenylethanol | 92% | |||
| (L)-Praziquantel | Microcrystalline cellulose triacetate | MeOH | 93.7% | 274 |
| (D)-Praziquantel | 90.1% | |||
| (L)-Praziquantel | Microcrystalline cellulose triacetate | MeOH | 97.5% | 275 and 276 |
| (D)-Praziquantel | 85% | |||
| 1,2,3,4-Tetrahydro-1-naphthol | Chiralpak AD | Heptane, isopropanol and TFA 95:5:0.2 |
98% | 279 |
| (S)-Levetiracetam | Chiralpak AD | EtOH:ciclohexane |
98% | 268 |
| (S)-Ketoprofen | Kromasil TBB | Hexane, MTBE, AcOH (85:15:0.1) |
96% | 293 |
| Phenylpropanolamine | Chiralpak AD | MeOH | 99% | 294 and 295 |
| (S)-Pindolol | Chiral-AGP | Water:ACN |
99% | 296 |
| (S)-Ibuprofen | Kromasil TBB | Hexane:TBME:AcOH |
100% | 297 |
| Guaifenesin | Chiralpak AD | Heptane, EtOH (85:15) |
98% | 298 |
| Quinolone derivatives | Chiralpak AD | Acetonitrile | 98.9% | 273 |
| (S)-Ketoprofen | Chiralpak AD | EtOH:TFA 0.01% |
98.6% | 299 |
| (R)-Ketoprofen | 99.8% | |||
| (R)-Aminoglutethimide | Chiralcel OD | Hexane:EtOH:ethanolamine |
99% | 264 |
| (2R,3S,2R)-Nadolol | Chiralpak IA | MeOH:ACN:diethylamine (25:75:0.1) |
100% | 300 |
| (L)-Praziquantel | Chiralcel OZ | MeOH | 100% | 278 |
| (D)-Praziquantel | 97% | |||
| (R)-Guaifenesin | Chiralcel OD | Hexane:EtOH (70:30) |
99.0% | 301 and 302 |
| (S)-Guaifenesin | 99.0% | |||
| (2S,3S)-Butanediol | Chromalite-PCG600M | Water | 99% | 303 and 304 |
In 2000, Ludemann-Hombourger et al.279 reported a new model of a continuous chromatographic process called “Varicol” based on a non-synchronous shift of the inlet/outlet valves in a multicolumn system as opposed to the classical SMB process. Consequently, the zone lengths are continuously oscillating by one column, with the increase of one zone being compensated by the decrease of an adjacent one delivering continuous streams without reaching a steady state. The differences concerning the SMB are that: the zone lengths are not constant in time which means the number of columns per zone is not constant, the inlet/outlet lines are not shifted simultaneously, and lastly, there is no constant solid flow rate concerning the inlet/outlet lines. The authors performed the enantioseparation of 1,2,3,4-tetrahydro-1-naphthol and compared the efficiency of both Varicol and SMB approaches. It was used Chiralpak AD packed columns and a mixture of heptane, isopropanol, and trifluoroacetic acid (TFA) 95/5/0.2 (v/v/v) as eluent. By using theoretical and experimental results, the co-workers showed with specific column configurations and solid flow rate, that the performance of a 6-columns SMB can be obtained with a 5-columns Varicol, with this last operation mode presenting higher productivity and solvent consumption.
Several studies on the comparison of operation strategies for enantiomers separation by SMB chromatography have been carried out and demonstrated numerically.263,280–284 Including temperature gradient,285 solvent gradient265,286–288 time variability289 known as PowerFeed mode and modulation of the feed concentration during the switching cycles (ModiCon).290 Also, different types of SMB apparatus were patented by Vroon et al.291 and Michel Anton.292 Zhang et al.281 investigated numerically four different modes of SMB: Varicol, PowerFeed, ModiCon, and classical SMB in three to five column units and compared the obtained results. The authors also considered the possibility of combining two operating modes in a new hybrid process and evaluated the feasibility. The conclusions taken on the studied scenario involving chiral separations, the PowerFeed, and ModiCon modes allow one to achieve even better performance than Varicol and classical SMB.
Lee et al.294,295,304 developed a standing-wave design method for nonlinear simulated moving bed (SMB) systems with significant mass-transfer effects and an operating pressure limit. It was also possible to determine ideal column lengths for process optimization. The enantioseparation of phenylpropanolamine was used to test the design method. Purities of 99% were obtained. The separation of racemic pindolol is of great commercial value because S-pindolol is the effective component for the treatment of hypertension.
Zhang et al.296 reported theoretical and experimental investigations of the enantioseparation of racemic pindolol by classical SMB and Varicol approach with a five-column setup. In the first study, the co-workers selected the operating conditions based on a mathematical model, and finally, they validated the model predictions. The chosen eluent was water and acetonitrile (ACN) 90:10, respectively. The effect of column configuration was explored and the configuration [1-2-1-1] was superior for the classical SMB approach. For the Varicol approach, a configuration of [1.5-1.5-1-1] was used. The authors also studied the effect of eluent recycling on the product's final purity. In both approaches, 100% purity was obtained.
Park et al.297 separated racemic ibuprofen in an SMB four-column unit and performed experiments based on simulation results from ASPEN chromatography software. The authors studied the effect of feed concentration/flow rate on the final purity of the raffinate stream. Final purities of 100% for (S)-ibuprofen (raffinate stream) were obtained. Ketoprofen enantiomers were first separated by SMB chromatography in 2004 by Yoon et al.293 in a six-column (1-2-2-1) configuration packed with Kromasil TBB stationary phase and a mixture of n-hexane, methyl-tert-butyl ether (MTBE), and acetic acid (AcOH) as eluent. For the same purpose, Ribeiro et al.305 carried on an exploratory study on how eluent composition influences the adsorption behavior of ketoprofen enantiomers in packed Chiralpak AD columns. Later, the same co-workers299 set up a six-column (1-2-2-1) configuration packed with Chiralpak AD as stationary phase and utilized ethanol with 0.01% trifluoroacetic acid (TFA) as eluent. The maximum obtained productivity was 3.84 g L−1 h−1 and purities of 98.6% and 99.8% for (R)-ketoprofen and (S)-ketoprofen, respectively.
Jiang et al.306 theoretically investigated the feasibility of an internal temperature gradient established by a difference between feed and eluent temperatures for the separation of ketoprofen enantiomers based on a built model. The separation of aminoglutethimide enantiomers by a continuous multicolumn chromatographic process was investigated theoretically and experimentally by Lin et al.264 A mathematical model was built to design and optimize the operation conditions, and experiments were run using a five-column Varicol process with the column configuration of (1/1.5/1.5/1) and a classical six-column SMB process with (1-2-2-1) configuration. By using the Varicol approach, the (R)-aminoglutethimide enantiomer was obtained with a purity of 99.3% and productivity of 2.45 g L−1 h−1 (59.1 g L−1 d−1). When comparing the enantioseparation performance it was found that the Varicol process has a better performance than the SMB process, with a 20% increase of (R)-aminoglutethimide productivity.
Arafah et al.300 carried on the enantioseparation of Nadolol in a pilot SMB unit with a classic (1-2-2-1) configuration. The results obtained were remarkable with 100% purity, 100% yield, and system productivity of 0.77 g L−1 h−1.
Guaifenesin is a worldwide prescribed drug for cough and cold labeled as an expectorant. Gomes et al.298 explored the separation of a racemic mixture of guaifenesin onto a chiral stationary phase Chiralpak AD by altering mobile phase solvents and proportions, by modeling and designing an SMB unit. The authors obtained purities up to 98%. Some years later, Gong et al.301 reported the resolution of Guaifenesin racemic mixture with the obtention of both enantiomers with 99% purity. The six-column SMB process with 1-2-2-1 column configuration, divided into four zones by two inlet ports (feed and desorbent) and two outlet ports (raffinate and extract) was performed in countercurrent and by switching the position of the inlet and outlet ports under constant feed composition and flow rate. The (R) enantiomer was obtained from the raffinate stream as it had weak adsorption to the stationary phase and the (S) enantiomer was obtained from the extract stream.
In a second study,302 the authors determined the separation performance of the SMB unit in four operation modes (conventional SMB, VariCol, PowerFeed, and ModiCon). When compared with the traditional six-column SMB process and with the same column configuration, the PowerFeed mode improved the productivity by 10.6%, and solvent consumption was reduced by 8.7% but also increased the pressure drop and pressure fluctuations which can limit the application. The VariCol mode with a five-column configuration increased the productivity by 26%. Lastly, the ModiCon mode presented the best results, increasing by 27.5% the productivity and decreasing solvent consumption by 21.7% – Fig. 17.
 | ||
| Fig. 17 Set-up for the separation of guaifenesin enantiomers by SMB process with [1-2-2-1] configuration. (feed: racemic solution of guaifenesin; raffinate: solution of (R)-guaifenesin; eluent: hexane/ethanol 70/30; extract: S-guaifenesin.) Adapted from ref. 301. | ||
The continuous separation of racemic 2,3-butanediol was reported by Lee et al.303,304 In a first study, the adsorption behaviors of the enantiomers in Chromalite-PCG600 M as stationary phase were investigated and numerical computations were carried out in Aspen Chromatography simulator. Both 2,3-butanediol enantiomers could be separated with high purities up to 97% and yield in a continuous countercurrent mode. In a second study, to increase productivity and meet the quality requirements, a simulated process optimization was carried out based on a Langmuir isotherm standing-wave-design method to explore the effects of feed concentration and/or column length on the production rate. The great advantages of using an SMB process are that the purification can be performed in continuous, better product purities and yield can be achieved, and it was not necessary any complementary reactions for product derivatization and isolation.
In 2016, Fuereder et al.307 reported the integration of simulated moving bed chromatography and enzymatic racemization enabling the synthesis of single enantiomers from a racemic mixture is theoretically 100% yield overcoming the limitation of conventional SMB processes by only separating the enantiomers and not racemizing one into the other. The system contained Chirobiotic TAG columns-SMB unit, an amino acid racemase-containing enzyme membrane reactor, and a nanofiltration unit for the concentration of the SMB raffinate stream before racemization on lab-scale to produce enantiopure (D)-methionine. The process was running for 30 h without significant variations in product concentration and purity and with a global yield of 93.5% (Fig. 18).
 | ||
| Fig. 18 Integration of simulated moving bed chromatography, enzymatic racemization and nanofiltration to produce (D)-methionine. Extract: (D)-methionine solution; raffinate: (L)-methionine solution; nanofiltration system: concentration of the raffinate solution; enzyme membrane reactor: racemization of (L)-methionine.307 | ||
Recently, Harriehausen et al.308 reported the synthesis of enantiopure mandelic acid and methionine enantiomers exploiting enzymatic racemization coupled with SMB chromatography. The key idea is to recycle the undesired enantiomer from the SMB process, racemize this recycling solution, and concentrate it in a solvent removal unit. The authors tested two different types of SMB unit configurations (1) a closed-loop four-zone and (2) an open-loop three-zone SMB separation process. The main outcome was that configuration (2) outperformed the conventional (1) setup. The configuration (2) could save 25% of the stationary phase, leading to a productivity enhancement of 33%. Moreover, the co-workers concluded that for the obtention of high productivities it is essential to use highly selective stationary phases, and high feed concentrations, which are limited by solubilities of the compounds in the eluent. Also, the SMB unit should be designed for less retention of the target enantiomer to elute it at the raffinate outlet (Fig. 19).
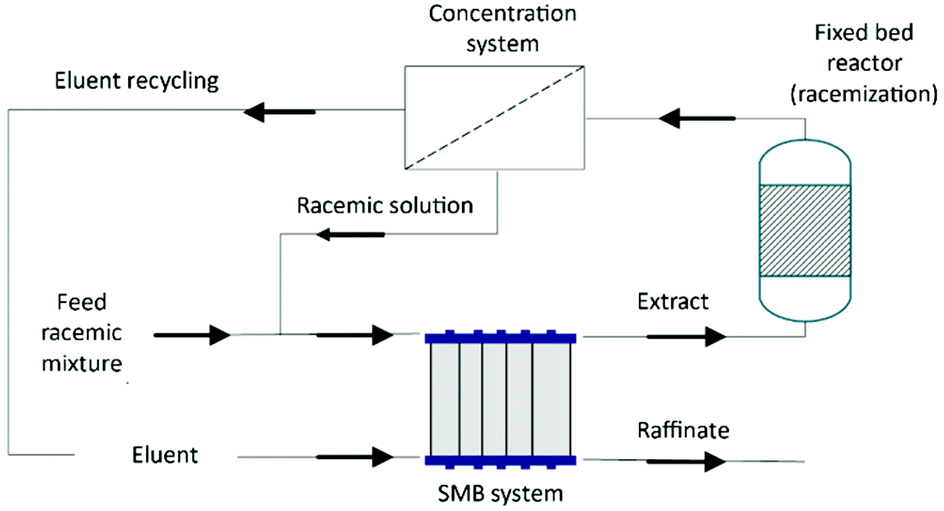 | ||
| Fig. 19 Integration of simulated moving bed chromatography, enzymatic racemization, and a concentration system to produce pure enantiomers. Extract: undesired enantiomer solution; raffinate: desired enantiomer solution; fixed bed reactor containing an immobilized enzyme to racemize the undesired enantiomer. Concentration system: concentration of the extract solution. Both streams (racemic solution and eluent) are recycled and feed into the SMB system. | ||
4.2. Supercritical fluid simulated moving bed
Supercritical fluid chromatography (SFC) is a powerful tool for the separation of compounds with very similar structure and volatility, as the case of enantiomers.309 The use of a supercritical fluid as a mobile phase presents some advantages over liquid chromatography such as higher separation efficiency and reduced purification time (due to higher flow rates).310 Consequently, it reduces solvent consumption and cost and increases productivity defined as racemate processed per unit mass of stationary phase and per unit time.311 Moreover it is not necessary to concentrate the solute solution from the eluent.310 The advantages and applications of supercritical fluid chromatography for enantioseparation are described in the following reviews.311–320When merging simulated moving bed with supercritical fluid chromatography technologies it leads to a purification procedure with unique characteristics with advantages of both techniques, named Supercritical fluid simulated moving bed (SFC-SMB). In 1996, Clavier et al.321 described for the first time the fusion between the two chromatography methods. The system consisted of a simulated moving bed unit containing SFC separation columns and supercritical CO2 as eluent. Essentially, the elution strength of supercritical eluents varies with pressure and temperature gradients.313 One year later, Mazzotti et al.322 described an isocratic mode (pressure constant along the zones) and a pressure gradient mode (a gradient is imposed along the zones) for the development of optimal design and procedures to SFC-SMB units.
In 2001, Denet et al.323 described the first enantioseparation in an SFC-SMB pilot unit with eight columns and four sections. Complete resolution of two tetralol enantiomers was achieved in pressure-gradient mode at 200 bar at zones 1 and 2 and 150 bar at zones 3 and 4. Peper et al.310 developed an SFC-SMB process with eight custom-made columns packed with Kromasil CHI-TBB for the resolution of a racemic mixture of ibuprofen (Table 12).
| Product | Chiral stationary phase | Eluent | Temp. (°C) | Pressure (bar) | Purity | Ref. |
|---|---|---|---|---|---|---|
| Tetralol enantiomers | Chiralcel OD | CO2 modified with ethanol (5.4 wt%) | 40 | 200/150 | >97% | 323 |
| (S)-Ibuprofen | Kromasil CHI-TBB | CO2 modified with isopropanol | 40 | 171 | >98% | 310 |
| Bi-naphthol | Kromasil CHI-DMB | CO2 modified with isopropanol | 40 | 160 | — | 325 |
| 1-Phenyl-1-propanol | Chiralcel OD | CO2 modified with methanol (2.6 wt%) | 30 | 180 | >98% | 326 |
The authors tested 2/2/2/2 and the 2/2/3/1 columns configuration while column length, operating pressure, operating temperature, and content of modifier were kept constant. The feed concentration was also studied. Purities higher than 98% were obtained. In another publication, the co-workers324 compared batch-SFC and SFC-SMB methods for racemic mixtures of tocopherol and ibuprofen from an economical point of view (e.g. product price, cost of process). Johannsen et al.325 published the resolution development of bi-naphthol enantiomers. The influence of the modifier, modifier content, and column configuration on the productivity of the SFC-SMB process was investigated by simulation. The authors found that a six-column configuration (1/2/2/1) is adequate for the separation of bi-naphthol on Kromasil CHI-DMB.
In 2005, resolution of 1-phenyl-1-propanol by SFC-SMB was described by Rajendran et al.326 in an eight Chiralcel-OD columns pilot unit. Complete separation was achieved at low feed concentrations. The solvent consumption was substantially lower than in SMB chromatographic systems.
In 2007, Kaemmerer et al.327 determined that with an SFC-SMB, most of the carbon dioxide is recycled, and the solvent consumption is 76% lower. Since the desired compound is collected in a concentrated solution because the main part of the mobile phase is CO2, the liquified gas is evaporated by decreasing the system pressure. Consequently, the time and the energy spent to evaporate the gas are lower than distilling the liquid eluent from liquid chromatographic systems.
In 2018, Johannsen and Brunner309 used an SCF-SMB system to separate cis-/trans-phytol and α/δ-tocopherol isomers.
Most chromatographic methods are not considered environmentally friendly and do not follow the principles of green chemistry. Improvements by eliminating hazardous reagents and reducing the use of flammable and toxic solvents must be put into practice.
Improvements should be done by replacing hazardous solvents with greener alternative ones (e.g., acetonitrile and methanol by ethanol; ionic liquids).
The use of carbon dioxide in the form of a supercritical fluid offers a substitute for organic solvent. Also, it has low viscosity and high diffusivity that allows high flow rates and faster separations. Consequently, carbon dioxide promotes the reduction of organic solvent usage, elimination of waste, and reduction of treatment costs.328
5. Other resolution techniques
5.1. Continuous enzyme-mediated kinetic resolution
Kinetic resolution consists of the consumption of one of the two enantiomers in a chemical reaction. Through this difference, an excess of the less-reactive enantiomer is created, the concentration of which goes through a maximum before it disappears on full completion of the reaction.11Enzymes began to be frequently used as catalysts on an industrial scale due to their enantioselectivity, substrate specificity, and ability to promote reactions under mild conditions.329 To minimize the environmental impact of a chemical transformation, it has been explored the use of scCO2 as the reaction solvent.
ScCO2 has been identified as a green solvent with significant potential for industrial use as it provides a clean, non-toxic, non-flammable, and tunable solvent system that is easily removed.330,331
Continuous-flow kinetic resolution of racemic 1-phenylethanol (99.7 eeS% and 47% yield) in scCO2, was described by Matsuda et al.332 with productivities 400 times higher than the batch procedure. The authors used an immobilized lipase (Novozym) to perform the kinetic resolution. Hobbs et al.329 also carried out the resolution of 1-phenylethanol in scCO2 system but using cross-linked enzyme aggregates (93 eeS%). The interesting results showed that it is possible to expand the use of a continuous flow system by the incorporation of two reactors linked in series. The first reactor was used for the metal-catalyzed hydrogenation of acetophenone and the second reactor for immobilized lipase to catalyze the kinetic resolution of the resulting (S)-1-phenylethanol.
Water-immiscible ionic liquids (ILs) have recently emerged as exceptionally interesting green non-aqueous reaction media for bio-transformations (P5). Biphasic systems based on ILs and scCO2 provide an integral green bioprocess in non-aqueous media, where both the resolution and extraction steps are coupled in efficient reaction/separation processes.333
Likewise, Reetz et al.331 described the application of ILs as solvents and scCO2 as extractants of the desired products for lipase-catalyzed kinetic resolution of chiral secondary alcohols. The co-workers demonstrated how the approach allows the combination of enzymatic catalytic esterification with highly efficient enantiomer separation in a continuous flow operation avoiding the use of organic solvents.
In 2009, Lozano et al.333 also applied the IL-scCO2 system to the continuous-flow kinetic resolution of racemic 1-phenylethanol using immobilized lipase (Novozym 435) and acidic zeolite catalysts having obtained enantioselectivities up to 97%.
Continuous-flow kinetic resolution of amines can be performed with immobilized transaminases as described by Molnár et al.334 The authors immobilized the whole-cell (E. coli containing overexpressed transaminases) and filled it in packed columns. The racemic solution was fed into the packed bed and a minimum of 98 ee% was obtained.
Enzyme mediated kinetic resolution presents many advantages namely the high stereoselectivity, the possibility of operation in continuous mode, the inherent ease of scalability, and the energy-saving due to the used mild conditions. The main drawback of the enzyme-mediated kinetic resolution is that the activity decreases after a cycle period that must be determined.
The use of scCO2 as solvent at least follows the first principle of green chemistry. It prevents waste (P1), it is an energy-efficient operation (P6), and do not use of organic solvents (P12) minimizing the risks of explosions and fires.335
5.2. Multistage enantioselective liquid–liquid extraction
Liquid–liquid extraction is a very known unit operation that might be operated in a continuous countercurrent mode to fractionate the racemate into its enantiomers, which is advantageous when scaling up.336 Enantioselective liquid–liquid extraction (ELLE) is an attractive alternative for commercial operation but based on the available literature, it still needs development towards commercialization and improvement of efficiency.337 Such as liquid membrane separation, ELLE is based on the selective recognition of one of the enantiomers by a chiral selector.338Holbach et al. reported the continuous countercurrent flow experiments using a process intensified extraction column to separate a racemic mixture of phenylsuccinic acid in water and hydroxypropyl-β-cyclodextrin as the extractant in n-decanol.338 Both enantiomers were separated with an enantiomeric excess of up to 60% and yields up to 80%. However, the high number of required extraction stages for a complete separation reveals the necessity of efficient equipment with many equilibrium stages.
Combining efficient mixing of two immiscible liquids with fast phase separation, the centrifugal contact separator (CCS) is a compact continuous flow device that seems perfectly fit for continuous operation. In addition, high centrifugal forces and preeminent mass-transfer characteristics of CCS make it suitable for use in process intensification of ELLE.339
Schuur et al.337,340 reported the use of centrifugal contactor separator (CCS) equipment for continuous ELLE. In a CCS, two immiscible liquids are contacted and subsequently separated. These two-unit operations combined in one allows a more energy-efficient separation. The authors applied a countercurrent operated pilot-scale cascade of six CCS devices to a system composed of racemic 3,5-dinitrobenzoyl-(R,S)-leucine in water and a cinchona alkaloid as the extractant dissolved in 1,2-dichloroethane.
Continuous CCS seems highly suitable for ELLE, moreover, only a small amount of the host/extractant needs to be available to separate larger amounts of enantiomers in a continuous flow mode (60 g to separate 17.7 kg racemate). In a multistage countercurrent process approach is not necessary to achieve a complete separation of enantiomers in a single stage and therefore the selectivity requirements of the extractant/host are not so narrow.336
Tang et al. described the continuous ELLE of 4-nitro-D,L-phenylalanine in water using PdCl2{(S)-BINAP} as an extractant in 1,2-dichloroethane,341 the continuous ELLE of amino-(4-nitro-phenyl)-acetic acid enantiomers in water using CuPF6{(S)-BINAP} in 1,2-dichloroethane339 in a countercurrent cascade of 10 CCSs. The minimum number of stages for full separation of both racemic mixtures was calculated and a minimum value of 14 was obtained for >97% enantiomeric excess at both streams.
The co-workers also performed the continuous separation of α-cyclohexyl-mandelic acid342 and phenylsuccinic acid343 enantiomers using hydroxyphenyl-β-cyclodextrin as extractant dissolved in 1,2-dichloroethane.
Environmentally friendly and versatile extractants should be considered during development and host recycling should be performed to not generate any waste. Moreover, in 2007, alternatives to halogenated solvents were identified by the ACS Green Chemistry Institute Pharmaceutical Roundtable (ACS GCIPR) as a key subject in which more research was required.344 The motivation for identifying alternatives to halogenated solvents is diverse and includes environmental health and safety concerns, economic and disposal costs, and regulatory and legal frameworks.345
5.3. Countercurrent chromatography
Countercurrent chromatography (CCC) is an effective separation technique based on the liquid–liquid partitioning of solutes in a solution of two non-miscible solvents.346,347 Therefore, the mobile and stationary phases in the chromatographic process are liquids. The conventional column we are used to thinking in traditional liquid chromatography is substituted by an instrument that produces a centrifugal field used to maintain stationary one of the liquid phases called a centrifugal partition chromatograph.346 Another technique would be high-speed CCC where it is used a specific apparatus. An advantage of CCC is that the superficial area of the liquid stationary phase is much higher than the one found in chromatography with solid support.348Regarding the separation of enantiomers, the application of CCC can be of great interest since this technique offers the possibility to produce enantiomerically pure compounds at a lower cost compared to conventional liquid chromatography and had the advantages of high load capacity, low solvent consumption, and easy scaling-up to preparative scale.349,350
As for other enantioselective separation techniques, in CCC a chiral host is needed to establish an enantioselective environment and it is chosen based on the solubility properties in the stationary phase. The racemic mixture is partitioned between the two phases of the biphasic solvent system.346
Commonly, the two-phase solvent systems used in CCC for enantioseparation are constituted by an organic solvent, and a strongly polar solution, often aqueous. The enantio-recognition occurs in the stationary phase, either organic or aqueous, where the chiral selector is retained. Exhaustive reviews on the enantiomeric separations in CCC were published by Foucault348 and Huang et al.351
The attention of some related researchers was concentrated on looking for ways of improving the separation efficiency of chiral separation by CCC. Novel methods such as multiple dual mode elution352 that consist of switching alternatively between reversed and normal phase operation during the experiment was applied for the resolution of (S)-naproxen derivative.353 The recycling elution mode (or closed-loop recycling elution mode) consists in recycling the effluent that leaves the chromatograph was applied for the separation of amlodipine besilate,354 oxybutynin,355 and naproxen356 enantiomers. This last methodology has the greener advantage that would not consume extra solvent because the mobile phase is recycled in a closed-loop. These methods partly mitigate the resolution limitation of CCC in enantioseparation, but more studies must be performed in the future.
As far as we know, no continuous countercurrent chromatography protocol was described for enantiomeric separation, all described examples were performed in discontinuous mode. Therefore, the envisagement of a semi-continuous or continuous CCC technology would improve the capacity and the productivity of the technique.346 A new system was patented by Couillard et al.,357 which describes a continuous centrifugal partition chromatography process that might apply to the separation of enantiomers.346 This system is characterized by the injection of the feed solution at an intermediate point of the centrifugal chromatograph followed by sequential feeding of a dense solvent phase and a light solvent phase from opposite ends of the column.
Likewise, intermittent counter-current extraction was described by Sutherland et al.358 consisting of a quasi-continuous counter-current chromatography process used to separate caffeine, vanillin, naringenin, and carvone and might be used for enantiomeric separation.
6. General conclusion and outlook
Over the last decades, the continuous mode has gained the interest of industry and academia due to its many advantages over discontinuous procedures including better reproducibility, yields,49,51–53 shorter development period, smaller equipment footprint, and precise control of process parameters. Even with these advantages, the continuous chiral resolution is not frequently used as often as it should be. This review was aimed to introduce the different continuous approaches – and highlight some case studies – Fig. 20.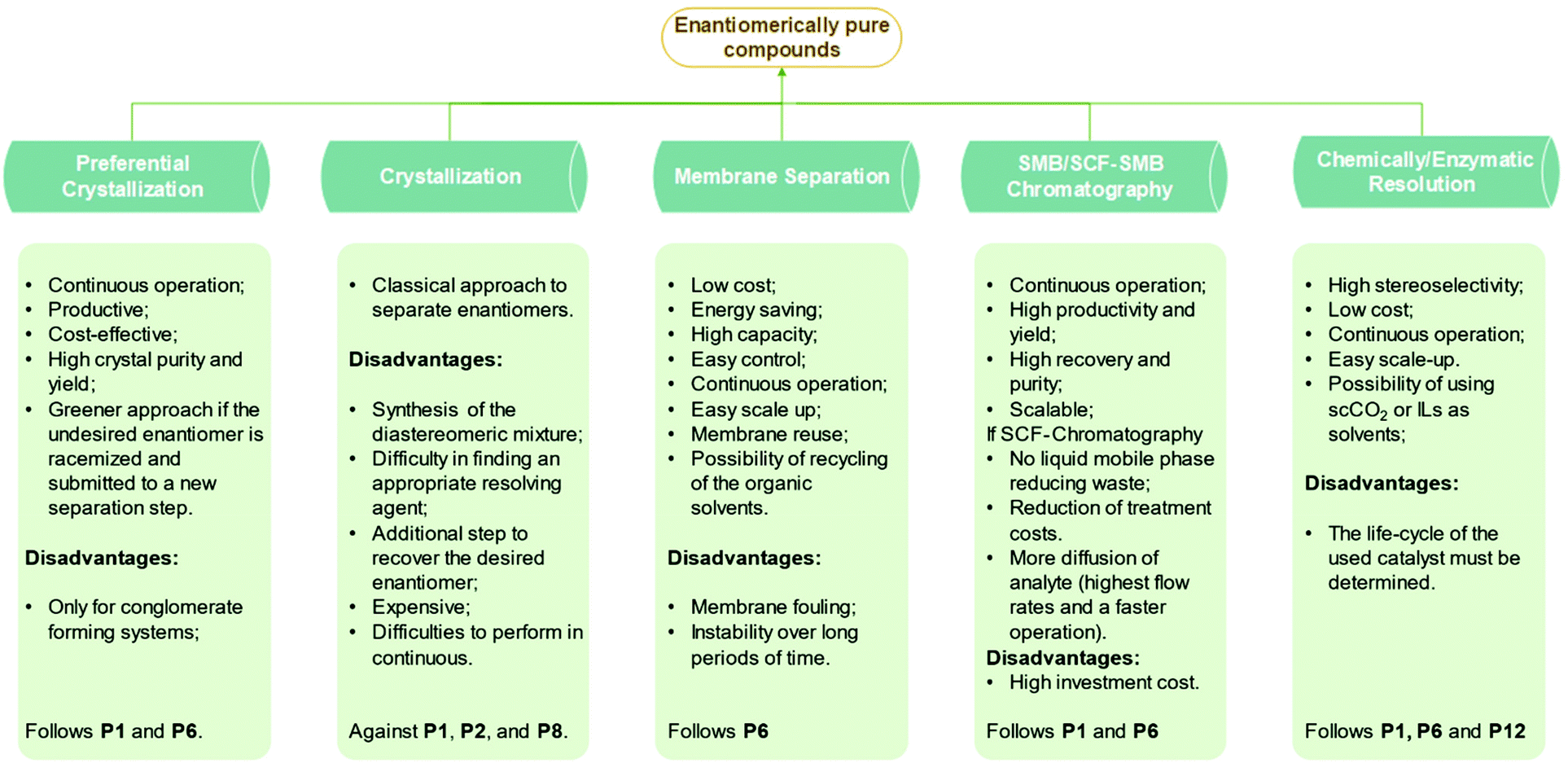 | ||
| Fig. 20 Semi-continuous and continuous approaches to obtain enantiomerically pure compounds. | ||
A preferential crystallization process is only feasible for conglomerate-forming systems, by seeding of a racemic or enantiomerically enriched solution with crystals of the preferred pure enantiomer, control over the crystallizing enantiomer is obtained. It is a cost-effective method to obtain crystals of a single enantiomer. Unfortunately, only 10% of the racemic mixtures belong to the conglomerate forming group, consequently, the scope of this technique is limited.
It was discussed different possible setups that can be explored during process development. There is no better setup, all should be considered depending on the project, availability of equipment, and level of expertise. In general, two-coupled crystallizers and a feed tank offer more possibilities since they can continuously feed racemic solution and, in separate vessels, isolate individual crystalline enantiomers. It also offers the possibility to recover from a failed resolution simply by heating the crystallizers to a temperature that would dissolve all the formed crystals (P1). Also, the isolated undersired enantiomer might be racemized and submitted to a new preferential crystallization procedure to obtain higher yields processes (P1). This possibility may be the best advantage of continuous preferential crystallization. In summary, continuous preferential crystallization offers higher productivities and improved process control.
If the racemic mixture does not belong to the conglomerate forming group, continuous crystallization (following the classical approach where a diastereomer is previously prepared) can be done although it does not follow P1, P2 and P8.
By developing a continuous crystallization procedure, more control on the morphology and particle size distribution is obtained, the development can be faster, and the reproducibility can be enhanced.
The membrane-based enantiomeric resolution has attracted attention due to its inherent advantages namely ease of scale-up, low footprint, energy consumption, and continuous mode of operation (P6). Membranes can be separated into two main groups: liquid membranes and solid membranes. The liquid membranes have been extensively investigated for the separation of enantiomers due to their high separation factor and increased mass transfer. Hollow fiber membrane modules present high surface area per volume resulting in a relatively compact system, consequently very easy for scaling-up. The literature demonstrates how the solute exchange between two crystallizers separated by a hollow-fiber membrane module results in both high purity and high yield enantiomeric separation process. This option combines the high selectivity and mass transfer of liquid membranes with the stability and mechanical strength of solid membranes. The results gathered in this review show how efficient supported liquid membranes can be for enantiomeric resolution. Moreover, there are HF modules available in the market which turns this option easier for industrial application and continuous or semi-continuous operation. Solid membranes with inherent chiral polymers and polymeric membranes functionalized with a chiral selector are the most classical type. In recent years, composite, graphene oxide, and MOFs membranes have been studied to find an optimal combination of membrane stereo-selectivity, permeability, and mechanical stability. The results show that they are a promising type of membrane material but more studies on the preparation, stereo-selectivity, waste management, membrane recycling, and use on a large scale must be carried on. Molecularly imprinted membranes are a promising field in which a polymer network is prepared with specific recognition sites for a target template molecule. However, it would be valuable to see this technology being applied at a higher scale and in a cGMP setting for enantiomeric resolution.
Looking out at all the studies described herein, it seems that complete enantioseparation of a racemic mixture through one cycle process with membranes is not feasible. Multi-cycle operation processes may be the path forward to achieve better separation, even though the number of cycles required to achieve it will likely affect the productivity of the operation. The combination of two technologies as continuous crystallization and membrane separation, as described by Svang-Ariyaskul,126 might be a good setup to achieve an efficient and greener semi-continuous enantiomeric separation.
The combination of the simulated moving bed technique with supercritical fluid chromatography leads to a separation unit with great advantages. Namely, the increase of mass transfer due to the higher diffusivities of solutes in supercritical fluids increases the separation efficiency and decreases pressure drop (P1). Faster cycles (by using higher flow rates), and a reduction of organic solvents (2–10 times lower)313 do not only contribute to a greener, environmentally friendly separation process but also reduce the costs in theory.311,324 We may consider the fact that an SFC system requires additional heating and cooling not required for HPLC systems, and thus, additional energy requirements. At the inlet of the unit, the feed containing carbon dioxide must be cooled to a liquid, at the outlet during depressurization it is necessary to heat. Finally, to recycle carbon dioxide is required additional cooling to condense the resulting gas. In total, an operating SFC unit is heated and cooled simultaneously at several locations.312 Despite the advantages here mentioned to operating an SMB unit under supercritical conditions, not many studies about enantiomer resolution in pilot scales have been reported in the literature. This may be due to the high cost of assembling an SMB pilot unit under supercritical conditions.
There is no better approach for enantiomeric separation, it depends on each case. But we firmly believe that with this review paper, the readers might have an overall idea of each possible technique, when to use each technique, and which green chemistry principles it follows.
More studies must be performed to improve the greenness of enantiomeric separation since it is still not a green operation. The mindset of chemists and engineers is changing, and the environmental impact of the developed processes is a real preoccupation. Therefore, there is a trend on developing processes as much greener as it can be. With this review paper it is possible to identify what has already been achieved and what should be kept in mind when developing new processes to improve the greener impact on enantiomeric separation.
Abbreviations
| API | Active pharmaceutical ingredient |
| (D)-DTTA | Di-p-toluyl-(D)-tartaric acid |
| ACN | Acetonitrile |
| AcOH | Acetic acid |
| ATR | Attenuated total reflectance |
| BINAP | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl |
| BLM | Bulk liquid membrane |
| CCC | Countercurrent chromatography |
| CD | Cyclodextrin |
| CNT | Carbon nanotubes |
| COFs | Covalent organic frameworks |
| CPC | Coupled preferential crystallizer |
| CPP | Critical process parameters |
| CQAs | Critical quality attributes |
| D2EHPA | Di(2-ethylhexyl)phosphoric acid |
| DACH | (S,S)-1,2-Diaminocyclohexane |
| DBTA | O,O-Dibenzoy(L)-(2S,3S)-tartaric acid |
| DFT | Density functional theory |
| DMAC | Dimethylacetamide |
| DMSO | Dimethylsulfoxide |
| E a | Activation energy |
| ee | Enantiomeric excess |
| EGDMA | Ethylene glycol dimethacrylate |
| ELM | Emulsion liquid membrane |
| EtOH | Etanol |
| FBC | Fluidized bed crystallizer |
| FDA | Food and Drugs Administration |
| Glu | Glutamic acid |
| GO | Graphene oxide |
| HF | Hollow fiber |
| HOFs | Hydrogen-bonded organic frameworks |
| ILs | Ionic liquids |
| IPA | Isopropanol |
| MAA | Methacrylic acid |
| MeOH | Metanol |
| MIM | Molecularly imprinted membranes |
| MMM | Mixed matrix membranes |
| MOFs | Metal–organic frameworks |
| MPD | m-Phenylene diamine |
| MSMPR | Mixed-suspension mixed-product-removal |
| MWCO | Molecular weight cut-off |
| NMP | N-Methylpyrrolidone |
| PAT | Process Analytical Technologies |
| PC | Preferential crystallization |
| PFC | Plug flow crystallizers |
| Phe | Phenylalanine |
| PLGA | Poly-(L)-glutamic acid |
| poly-PSPA | Poly-[dimethyl(10-pinanyl)silyl]phenylacetylene |
| PVDF | Polyvinylidene fluoride |
| ScCO2 | Supercritical carbon dioxide |
| SFC | Supercritical fluid chromatography |
| SLM | Supported liquid membrane |
| SMB | Simulated moving bed |
| TFA | Trifluoroacetic acid |
| TMC | 1,3,5-Trimesoyl chloride |
| Trp | Tryptophan |
| Tyr | Tyrosine |
| α | Separation factor |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Hovione Farmaciência S.A. for funding this project. C. A. M. A acknowledge Fundação para a Ciência e Tecnologia (FCT) for financial support (PTDC/QUIQOR/32008/2017, PTDC/QUI-QOR/1131/2020, UIDB/04138/2020 and UIDP/04138/2020). The project leading to this application has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 951996.Notes and references
- M. L. Pasteur, C. R. Seances Acad. Sci., 1848, 26, 535–539 Search PubMed.
- Food and Drugs Administration (FDA), Development of New Stereoisomeric Drugs, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs, (accessed 24 January 2021).
- Eur. Med. Agency, Investigation of Chiral Active Substances Guideline, 1994, pp. 381–391 Search PubMed.
- B. L. He and D. K. Lloyd, Chiral methods, Elsevier Ltd, 2020 Search PubMed.
- Nextmsc, Chiral Chemicals Market by Technology (Traditional Separation Method), Asymmetric Preparation Method (Asymmetric Synthesis Method and Asymmetric Catalysis Method), Biological Separation Method, and Other Separation Methods, by Application (Pharmaceuticals, https://www.nextmsc.com/report/chiral-chemicals-market, (accessed 29 January 2021).
- FactMR, Chiral Chemicals Market's Reliance on Demand from Pharmaceutical Industry Continues Unabated: Fact.MR's Study, https://www.accesswire.com/623438/Chiral-Chemicals-Markets-Reliance-on-Demand-from-Pharmaceutical-Industry-Continues-Unabated-FactMRs-Study, (accessed 29 January 2021).
- S. W. Smith, Toxicol. Sci., 2009, 110, 4–30 CrossRef CAS PubMed.
- J. M. Daniels, E. R. Nestmann and A. Kerr, Ther. Innov. Regul. Sci., 1997, 31, 639–646 Search PubMed.
- S. K. Branch and A. J. Hutt, in Drug Stereochemistry - Analytical Methods and Pharmacology, CRC Press, 3rd edn, 2012, pp. 240–273 Search PubMed.
- N. R. Srinivas, R. H. Barbhaiya and K. K. Midha, J. Pharm. Sci., 2001, 90, 1205–1215 CrossRef CAS PubMed.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed.
- L. A. Nguyen, H. He and C. Pham-huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CAS.
- J. Tomaszewski and M. M. Rumore, Drug Dev. Ind. Pharm., 1994, 20, 119–139 CrossRef CAS.
- X. Yuan, L. Wang, G. Liu, G. Dai and K. Tang, Chirality, 2019, 31, 445–456 CrossRef CAS PubMed.
- C. Rougeot and J. E. Hein, Org. Process Res. Dev., 2015, 19, 1809–1819 CrossRef CAS.
- T. Vetter, C. L. Burcham and M. F. Doherty, AIChE J., 2015, 61, 2810–2823 CrossRef CAS.
- W. Bi, M. Tian and K. H. Row, Analyst, 2011, 136, 379–387 RSC.
- M. Kaspereit, S. Swernath and A. Kienle, Org. Process Res. Dev., 2012, 16, 353–363 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- J. Orehek, D. Teslić and B. Likozar, Org. Process Res. Dev., 2021, 25, 16–42 CrossRef CAS.
- J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 887–895 CrossRef CAS.
- G. Coquerel, Chem. Soc. Rev., 2014, 43, 2286–2300 RSC.
- T. Vetter, C. L. Burcham and M. F. Doherty, Chem. Eng. Sci., 2014, 106, 167–180 CrossRef CAS.
- M. L. Pasteur, C. R. Seances Acad. Sci., 1853, 37, 162–166 Search PubMed.
- G. Levilain and G. Coquerel, CrystEngComm, 2010, 12, 1983–1992 RSC.
- K. S. Kim, WO1998/13344, 1998.
- P. G. Holton, US4515811, 1980.
- A. S. Dunn, V. Svoboda, J. Sefcik and J. H. Ter Horst, Org. Process Res. Dev., 2019, 23, 2031–2041 CrossRef CAS.
- N. Sato, T. Uzuki, K. Toi and T. Akashi, Agric. Biol. Chem., 1969, 33, 1107–1108 CAS.
- C. Viedma, Phys. Rev. Lett., 2005, 94, 1–4 CrossRef PubMed.
- M. Iggland and M. Mazzotti, Cryst. Growth Des., 2011, 11, 4611–4622 CrossRef CAS.
- C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS PubMed.
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS PubMed.
- I. Baglai, M. Leeman, M. Kellogg and W. L. Noorduin, Org. Biomol. Chem., 2019, 17, 35–38 RSC.
- L. C. Sogutoglu, R. R. E. Steendam, H. Meekes, E. Vlieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC.
- H. Lorenz, A. Perlberg, D. Sapoundjiev, M. P. Elsner and A. Seidel-Morgenstern, Chem. Eng. Process., 2006, 45, 863–873 CrossRef CAS.
- S. Srisanga and J. H. Ter Horst, Cryst. Growth Des., 2010, 10, 1808–1812 CrossRef CAS.
- K. Petruševska-Seebach, A. Seidel-Morgenstern and M. P. Elsner, Cryst. Growth Des., 2011, 11, 2149–2163 CrossRef.
- G. Coquerel, M. N. Petit and R. Bouaziz, US6022409, 2000.
- E. Ndzié, P. Cadinael, A. Schoofs and G. Coquerel, Tetrahedron: Asymmetry, 1997, 8, 2913–2920 CrossRef.
- G. Coquerel, G. Tauvel and M. N. Petit, US8034948B2, 2011.
- L. Courvoisier, E. Ndzié, M. Petit, U. Hedtmann, U. Sprengard and G. Coquerel, Chem. Lett., 2001, 30, 364–365 CrossRef.
- F. X. Gendron, J. Mahieux, M. Sanselme and G. Coquerel, Cryst. Growth Des., 2019, 19, 4793–4801 CrossRef CAS.
- F. Czapla, H. Haida, M. P. Elsner, H. Lorenz and A. Seidel-Morgenstern, Chem. Eng. Sci., 2009, 64, 753–763 CrossRef CAS.
- C. Gervais, P. Cardinae and S. Petit, J. Phys. Chem. B, 2002, 106, 646–652 CrossRef CAS.
- S. Bellies, P. Cardinael, E. Ndzié, S. Petit and G. Coquerel, Chem. Eng. Sci., 2001, 56, 2281–2294 CrossRef.
- J. Li, T. C. Lai, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2017, 17, 1000–1007 CrossRef CAS.
- Y. Ma, S. Wu, E. Genito, J. Macaringue, T. Zhang, J. Gong and J. Wang, Org. Process Res. Dev., 2020, 24, 1785–1801 CrossRef CAS.
- K. Galan, M. J. Eicke, M. P. Elsner, H. Lorenz and A. Seidel-morgenstern, Cryst. Growth Des., 2015, 15, 1808–1818 CrossRef CAS.
- E. Temmel, J. Gänsch, A. Seidel-morgenstern and H. Lorenz, Crystals, 2020, 10, 394 CrossRef CAS.
- J. H. Chaaban, K. Dam-Johansen, T. Skovby and S. Kiil, Org. Process Res. Dev., 2013, 17, 1010–1020 CrossRef CAS.
- F. Cameli, C. Xiouras and G. D. Stefanidis, CrystEngComm, 2020, 22, 3519–3525 RSC.
- R. Lakerveld, B. Benyahia, R. D. Braatz and P. I. Barton, AIChE J., 2013, 59, 3671–3685 CrossRef CAS.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed.
- S. Ferguson, G. Morris, H. Hao, M. Barrett and B. Glennon, Chem. Eng. Sci., 2012, 77, 105–111 CrossRef CAS.
- J. L. Quon, H. Zhang, A. Alvarez, J. Evans, A. S. Myerson and B. L. Trout, Cryst. Growth Des., 2012, 12, 3036–3044 CrossRef CAS.
- A. J. Alvarez and A. S. Myerson, Cryst. Growth Des., 2010, 10, 2219–2228 CrossRef CAS.
- D. Zhang, S. Xu, S. Du, J. Wang and J. Gong, Engineering, 2017, 3, 354–364 CrossRef.
- C. Darmali, S. Mansouri, N. Yazdanpanah and M. W. Woo, Ind. Eng. Chem. Res., 2019, 58, 1463–1479 CrossRef CAS.
- B. Wood, K. P. Girard, C. S. Polster and D. M. Croker, Org. Process Res. Dev., 2019, 23, 122–144 CrossRef CAS.
- M. P. Elsner, D. F. Menéndez, E. A. Muslera and A. Seidel-Morgenstern, Chirality, 2005, 17, 183–195 CrossRef PubMed.
- A. A. Rodrigo, H. Lorenz and A. Seidel-morgenstern, Chirality, 2004, 16, 499–508 CrossRef CAS PubMed.
- S. Qamar, K. Galan, M. Peter, I. Hussain and A. Seidel-morgenstern, Chem. Eng. Sci., 2013, 98, 25–39 CrossRef CAS.
- A. Majumder and Z. K. Nagy, Pharmaceutics, 2017, 9, 1–19 CrossRef PubMed.
- S. Y. Wong, A. P. Tatusko, B. L. Trout and A. S. Myerson, Cryst. Growth Des., 2012, 12, 5701–5707 CrossRef CAS.
- I. Radoljub, J. J. De Yoreo and D. Robert, Cryst. Growth Des., 2004, 4, 1045–1052 CrossRef.
- D. Zhang, S. Xu, S. Du, J. Wang and J. Gong, Engineering, 2017, 3, 354–364 CrossRef.
- M. Jiang and R. D. Braatz, CrystEngComm, 2019, 21, 3534–3551 RSC.
- M. Midler, US3510266, 1970.
- M. Midler, US3892539, 1975.
- E. J. J. Grabowski, Chirality, 2005, 17, 249–259 CrossRef PubMed.
- D. Binev and H. Lorenz, Chem. Eng. Sci., 2015, 133, 116–124 CrossRef CAS.
- T. Kollges and T. Vetter, Cryst. Growth Des., 2018, 18, 1686–1696 CrossRef CAS.
- K. Ito, T. Akashi, S. Tatsumi, Midarebashi, K. Shi and K. Ken, US3260744, 1966.
- M. Eicke, A. Seidel-Morgenstern and M. P. Elsner, Chem. Eng. Trans., 2009, 17, 651–656 Search PubMed.
- S. Qamar, M. Peter Elsner, I. Hussain and A. Seidel-Morgenstern, Chem. Eng. Sci., 2012, 71, 5–17 CrossRef CAS.
- J. H. Chaaban, K. Dam-johansen, T. Skovby and S. Kiil, Org. Process Res. Dev., 2014, 18, 601–612 CrossRef CAS.
- T. Köllges and T. Vetter, Cryst. Growth Des., 2017, 17, 233–247 CrossRef.
- M. Mangold, D. Khlopov, E. Temmel, H. Lorenz and A. Seidel-Morgenstern, Chem. Eng. Sci., 2017, 160, 281–290 CrossRef CAS.
- M. Mangold, N. Huskova, J. Gänsch and A. Seidel-morgenstern, Processes, 2020, 8, 1–16 CrossRef.
- M. P. Elsner, G. Ziomek and A. Seidel-Morgenstern, Chem. Eng. Sci., 2007, 62, 4760–4769 CrossRef CAS.
- M. P. Elsner, G. Ziomek and A. Seidel-Morgenstern, Chem. Eng. Sci., 2011, 66, 1269–1284 CrossRef CAS.
- M. P. Elsner, G. Ziomek and A. Seidel-Morgenstern, AIChE J., 2009, 55, 640–649 CrossRef CAS.
- S. Qamar, K. Galan and A. Seidel-morgenstern, Chem. Eng. Trans., 2013, 32, 2053–2058 Search PubMed.
- G. Levilain, M. J. Eicke and A. Seidel-Morgenstern, Cryst. Growth Des., 2012, 12, 5396–5401 CrossRef CAS.
- M. J. Eicke, G. Levilain and A. Seidel-Morgenstern, Cryst. Growth Des., 2013, 13, 1638–1648 CrossRef CAS.
- E. Temmel, M. J. Eicke, F. Cascella, A. Seidel-morgenstern and H. Lorenz, Cryst. Growth Des., 2019, 19, 3148–3157 CrossRef CAS PubMed.
- D. Binev and H. Lorenz, Cryst. Growth Des., 2016, 16, 1409–1419 CrossRef CAS.
- A. Majumder, Processes, 2018, 6, 1–17 CrossRef CAS.
- J. E. Hein, B. H. Cao, M. W. Meijden, M. Leeman and R. M. Kellogg, Org. Process Res. Dev., 2013, 17, 946–950 CrossRef CAS.
- J. E. Hein, B. Huynh Cao, C. Viedma, R. M. Kellogg and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 12629–12636 CrossRef CAS PubMed.
- H. D. Baumgard, B. Haedke, J. Himmelreich and H. Judat, US6315966B1, 2001.
- R. Xie, L. Y. Chu and J. G. Deng, Chem. Soc. Rev., 2008, 37, 1243–1263 RSC.
- J. T. F. Keurentjes, L. J. W. M. Nabuurs and E. A. Vegter, J. Membr. Sci., 1996, 113, 351–360 CrossRef CAS.
- C. Fernandes, M. E. Tiritan and M. M. M. Pinto, Symmetry, 2017, 9, 1–19 CrossRef.
- M. Ulbricht, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 804, 113–125 CrossRef CAS PubMed.
- E. van der Ent, J. Membr. Sci., 2001, 185, 207–221 CrossRef CAS.
- M. Yoshikawa and A. Higuchi, in Encyclopedia of Membrane Science and Technology, John Wiley & Sons, Inc., 2013 Search PubMed.
- Y. Lu, J. Y. Chan, H. Zhang, X. Li, Y. Nolvachai, P. J. Marriott, X. Zhang, G. P. Simon, M. M. Banaszak Holl and H. Wang, J. Membr. Sci., 2021, 620, 1–9 Search PubMed.
- J. J. Keating, S. Bhattacharya and G. Belfort, J. Membr. Sci., 2018, 555, 30–37 CrossRef CAS.
- S. Peacock, D. Walba, F. C. A. Gaeta, R. C. Helgeson and D. J. Cram, J. Am. Chem. Soc., 1980, 102, 2043–2052 CrossRef CAS.
- D. J. Cram and J. M. Cram, Science, 1974, 183, 803–809 CrossRef CAS PubMed.
- M. Newcomb, J. L. Toner, R. C. Helgeson and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 4941–4947 CrossRef CAS.
- T. J. Ward and K. D. Ward, Anal. Chem., 2012, 84, 626–635 CrossRef CAS PubMed.
- W. H. Pirkle and E. M. Doherty, US5080795, 1992.
- A. Gössi, W. Riedl and B. Schuur, J. Chem. Technol. Biotechnol., 2018, 93, 629–644 CrossRef.
- J. Ramkumar and S. Chandramouleeswaran, Indian J. Adv. Chem. Sci., 2015, 3, 293–298 CAS.
- B. J. V. Verkuijl, A. J. Minnaard, J. G. De Vries and B. L. Feringa, J. Org. Chem., 2009, 74, 6526–6533 CrossRef CAS PubMed.
- H. M. Krieg, J. Lotter, K. Keizer and J. C. Breytenbach, J. Membr. Sci., 2000, 167, 33–45 CrossRef CAS.
- P. J. Pickering and J. B. Chaudhuri, Chirality, 1997, 9, 261–267 CrossRef CAS.
- P. Dzygiel and P. Wieczorek, J. Membr. Sci., 2000, 172, 223–232 CrossRef CAS.
- E. V. Yurtov and M. Y. Koroleva, Pet. Chem., 2014, 54, 581–594 CrossRef CAS.
- P. J. Pickering and J. B. Chaudhuri, J. Membr. Sci., 1997, 127, 115–130 CrossRef CAS.
- D. Huang, Y. Long, W. Liu, Y. Hong, M. Zhong and S. Xu, in Proceedings of the 5th International Conference on Information Engineering for Mechanics and Materials, 2015, pp. 533–538 Search PubMed.
- D. Kong, Z. Zhou, H. Zhu, Y. Mao, Z. Guo, W. Zhang and Z. Ren, J. Membr. Sci., 2016, 499, 343–351 CrossRef CAS.
- Y. Zhao, X. Zhu, W. Jiang, H. Liu and B. Sun, Molecules, 2021, 26, 1–30 Search PubMed.
- P. Dzygiel, P. Wieczorek, J. Å. Jonsson, M. Milewska and P. Kafarski, Tetrahedron, 1999, 55, 9923–9932 CrossRef CAS.
- J. D. Clark, B. Han, A. S. Bhown and S. R. Wickramasinghe, Sep. Purif. Technol., 2005, 42, 201–211 CrossRef CAS.
- P. Dzygiel, P. Wieczorek and P. Kafarski, J. Sep. Sci., 2003, 26, 1050–1056 CrossRef CAS.
- E. Miyako, T. Maruyama, N. Kamiya and M. Goto, Chem. Commun., 2003, 3, 2926–2927 RSC.
- R. M. C. Viegas, C. A. M. Afonso, J. G. Crespo and I. M. Coelhoso, J. Membr. Sci., 2007, 305, 203–214 CrossRef CAS.
- F. Jiao, W. Yang, D. Huang, J. Yu, X. Jiang, F. Jiao, W. Yang, D. Huang and J. Yu, DOI:10.1080/01496395.2012.659784.
- C. A. M. Afonso and J. G. Crespo, Angew. Chem., Int. Ed., 2004, 43, 5293–5295 CrossRef CAS PubMed.
- A. Svang-Ariyaskul, W. J. Koros and R. W. Rousseau, Chem. Eng. Sci., 2012, 77, 35–41 CrossRef CAS.
- L. Zeng, Q. Liu, Q. Yi, K. Tang and B. Van Der Bruggen, Ind. Eng. Chem. Res., 2020, 59, 13735–13743 CrossRef CAS.
- A. Maximini, H. Chmiel, H. Holdik and N. W. Maier, J. Membr. Sci., 2006, 276, 221–231 CrossRef CAS.
- H. B. Ding, P. W. Carr and E. L. Cussler, AIChE J., 1992, 38, 1493–1498 CrossRef CAS.
- T. Takeuchi, R. Horikawa and T. Tanimura, Anal. Chem., 1984, 56, 1152–1155 CrossRef CAS.
- P. Hadik, L. P. Szabó and E. Nagy, Desalination, 2002, 148, 193–198 CrossRef CAS.
- P. Hadik, L. Kotsis, M. Eniszné-Bódogh, L. P. Szabó and E. Nagy, Sep. Purif. Technol., 2005, 41, 299–304 CrossRef CAS.
- Z. Wang, C. Cai, Y. Lin, Y. Bian, H. Guo and X. Chen, Sep. Purif. Technol., 2011, 79, 63–71 CrossRef CAS.
- T. Prapasawat, A. W. Lothongkum and U. Pancharoen, Chem. Pap., 2014, 68, 180–189 CAS.
- A. Svang-Ariyaskul, Dissertation Thesis, Georgia Institute of Technology, 2010.
- A. Svang-Ariyaskul, W. J. Koros and R. W. Rousseau, Chem. Eng. Sci., 2009, 64, 1980–1984 CrossRef CAS.
- J. L. Lopez and S. L. Matson, J. Membr. Sci., 1997, 125, 189–211 CrossRef CAS.
- T. Börner, G. Rehn, C. Grey and P. Adlercreutz, Org. Process Res. Dev., 2015, 19, 793–799 CrossRef.
- N. Sunsandee, N. Leepipatpiboon, P. Ramakul and U. Pancharoen, Chem. Eng. J., 2012, 180, 299–308 CrossRef CAS.
- N. Sunsandee, N. Leepipatpiboon and P. Ramakul, Korean J. Chem. Eng., 2013, 30, 1312–1320 CrossRef CAS.
- S. Lai, S. Tang, J. Xie, C. Cai, X. Chen and C. Chen, J. Chromatogr. A, 2017, 1490, 63–73 CrossRef CAS PubMed.
- T. Aoki, S. Tomizawa and E. Oikawa, J. Membr. Sci., 1995, 99, 117–125 CrossRef CAS.
- C. Thoelen, M. De, E. Theunissen, Y. Kondo, I. F. J. Vankelecom, P. Grobet, M. Yoshikawa and P. A. Jacobs, J. Membr. Sci., 2001, 186, 153–163 CrossRef CAS.
- K. Taki, I. Arita, M. Satoh and J. Komiyama, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1035–1041 CrossRef CAS.
- J. H. Kim, J. Jegal, J. H. Kim, K. Lee and Y. Lee, J. Appl. Polym. Sci., 2003, 89, 3046–3051 CrossRef CAS.
- J. H. Kim, J. H. Kim, J. Jegal and K. Lee, J. Membr. Sci., 2003, 213, 273–283 CrossRef CAS.
- S. Xie, W. Wang, P. Ai, M. Yang and L. Yuan, J. Membr. Sci., 2008, 321, 293–298 CrossRef CAS.
- A. Higuchi, M. Tamai, Y. A. Ko, Y. I. Tagawa, Y. H. Wu, B. D. Freeman, J. T. Bing, Y. Chang and Q. D. Ling, Polym. Rev., 2010, 50, 113–143 CrossRef CAS.
- T. Aoki, T. Fukuda, T. Kaneko, M. Teraguchi and M. Yagi, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4502–4517 CrossRef CAS.
- S. Hazarika, J. Membr. Sci., 2008, 310, 174–183 CrossRef CAS.
- C. Ma, X. Xu, P. Ai, S. Xie, Y. Lv, H. Shan and L. Yuan, Chirality, 2011, 23, 379–382 CrossRef CAS PubMed.
- L. M. Yuan, W. Ma, M. Xu, H. L. Zhao, Y. Y. Li, R. L. Wang, A. H. Duan, P. Ai and X. X. Chen, Chirality, 2017, 29, 315–324 CrossRef CAS PubMed.
- X. Weng, J. E. Baez, M. Khiterer, M. Y. Hoe, Z. Bao and K. J. Shea, Angew. Chem., Int. Ed., 2015, 54, 11214–11218 CrossRef CAS PubMed.
- R. J. Petersen, J. Membr. Sci., 1993, 83, 81–150 CrossRef CAS.
- P. G. Ingole, K. Singh and H. C. Bajaj, Desalination, 2011, 281, 413–421 CrossRef CAS.
- P. G. Ingole, H. C. Bajaj, D. N. Srivastava, B. Rebary and K. Singh, Sep. Sci. Technol., 2013, 48, 1777–1787 CrossRef CAS.
- P. G. Ingole, H. C. Bajaj and K. Singh, Desalination, 2014, 343, 75–81 CrossRef CAS.
- K. Singh, S. Devi, H. C. Bajaj, P. Ingole, J. Choudhari and H. Bhrambhatt, Sep. Sci. Technol., 2014, 49, 2630–2641 CrossRef CAS.
- L. Miao, Y. Yang, Y. Tu, S. Lin, J. Hu, Z. Du, M. Zhang and Y. Li, React. Funct. Polym., 2017, 115, 87–94 CrossRef CAS.
- J. Gaálová, F. Yalcinkaya, P. Cuřínová, M. Kohout, B. Yalcinkaya, M. Koštejn, J. Jirsák, I. Stibor, J. E. Bara, B. Van der Bruggen and P. Izák, J. Membr. Sci., 2020, 596, 1–9 CrossRef.
- Z. Zhou, D. Li, Q. Wu, T. Zheng, H. Yuan and N. Peng, Chem. Eng. Res. Des., 2020, 160, 437–446 CrossRef CAS.
- M. Gogoi, R. Goswami, P. G. Ingole and S. Hazarika, Sep. Purif. Technol., 2020, 233, 116061 CrossRef CAS.
- J. Ke, Y. Zhang, X. Zhang, Y. Liu, Y. Ji and J. Chen, J. Membr. Sci., 2020, 597, 117635 CrossRef CAS.
- G. Liu, J. Wanqin and X. Nanping, Chem. Soc. Rev., 2015, 44, 5016–5030 RSC.
- Z. P. Smith and B. D. Freeman, Angew. Chem., Int. Ed., 2014, 53, 10286–10288 CrossRef CAS PubMed.
- C. Meng, Y. Sheng, Q. Chen, H. Tan and H. Liu, J. Membr. Sci., 2017, 526, 25–31 CrossRef CAS.
- C. Meng, Q. Chen, H. Tan, Y. Sheng and H. Liu, J. Membr. Sci., 2018, 555, 398–406 CrossRef CAS.
- F. Qie, J. Guo, B. Tu, X. Zhao, Y. Zhang and Y. Yan, Chem. – Asian J., 2018, 13, 2812–2817 CrossRef CAS PubMed.
- C. Meng, Q. Chen, X. Li and H. Liu, J. Membr. Sci., 2019, 582, 83–90 CrossRef CAS.
- C. Meng, S. Zhang, Q. Chen, X. Li and H. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 10893–10901 CrossRef CAS PubMed.
- W. Shi and D. L. Plata, Green Chem., 2018, 20, 5237–5406 RSC.
- M. Xue, B. Li, S. Qiu and B. Chen, Mater. Today, 2016, 19, 503–515 CrossRef CAS.
- C. Li and L. Heinke, Symmetry, 2020, 12, 1–13 Search PubMed.
- S. Mukherjee, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2018, 367, 82–126 CrossRef CAS.
- S. Xie, Z. Zhang, Z. Wang and L. Yuan, J. Am. Chem. Soc., 2011, 133, 11892–11895 CrossRef CAS PubMed.
- Z. G. Gu, W. Q. Fu, X. Wu and J. Zhang, Chem. Commun., 2016, 52, 772–775 RSC.
- P. Peluso, V. Mamane and S. Cossu, J. Chromatogr. A, 2014, 1363, 11–26 CrossRef CAS PubMed.
- J. Hou, H. Zhang, G. P. Simon and H. Wang, Adv. Mater., 2020, 32, 1–13 Search PubMed.
- J. M. Holcroft, K. J. Hartlieb, P. Z. Moghadam, J. G. Bell, G. Barin, D. P. Ferris, E. D. Bloch, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, K. M. Thomas, J. R. Long, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 5706–5719 CrossRef CAS PubMed.
- J. Zhang, S. Chen, A. Zingiryan and X. Bu, J. Am. Chem. Soc., 2008, 130, 17246–17247 CrossRef CAS PubMed.
- M. J. Ingleson, J. Bacsa and M. J. Rosseinsky, Chem. Commun., 2007, 3036–3038 RSC.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed.
- Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4135 CrossRef CAS PubMed.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- K. J. Hartlieb, J. M. Holcroft, P. Z. Moghadam, N. A. Vermeulen, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 2292–2301 CrossRef CAS PubMed.
- P. Li, Y. He, J. Guang, L. Weng, J. C. G. Zhao, S. Xiang and B. Chen, J. Am. Chem. Soc., 2014, 136, 547–549 CrossRef CAS PubMed.
- W. Wang, X. Dong, J. Nan, W. Jin, Z. Hu, Y. Chen and J. Jiang, Chem. Commun., 2012, 48, 7022–7024 RSC.
- Y. Lu, H. Zhang, J. Y. Chan, R. Ou, H. Zhu, M. Forsyth, E. M. Marijanovic, C. M. Doherty, P. J. Marriott, M. M. B. Holl and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 16928–16935 CrossRef CAS PubMed.
- D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321–14323 CrossRef CAS PubMed.
- F. Song, C. Wang, J. M. Falkowski, L. Ma, W. Lin, N. Carolina and U. States, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS PubMed.
- M. Banerjee, S. Das, M. Yoon, H. J. Choi and M. H. Hyun, J. Am. Chem. Soc., 2009, 131, 7524–7525 CrossRef CAS PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
- C. J. Kepert, T. J. Prior and M. J. Rosseinsky, J. Am. Chem. Soc., 2000, 122, 5158–5168 CrossRef CAS.
- B. Liu, O. Shekhah, H. K. Arslan, J. Liu, C. Wöll and R. A. Fischer, Angew. Chem., Int. Ed., 2012, 51, 807–810 CrossRef CAS PubMed.
- Z. Kang, M. Xue, L. Fan, J. Ding, L. Guo, L. Gao and S. Qiu, Chem. Commun., 2013, 49, 10569–10571 RSC.
- Z. Qiao, Z. Wang, C. Zhang, S. Yuan, Y. Zhu and J. Wang, AIChE J., 2012, 59, 4364–4372 Search PubMed.
- P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737–2738 CrossRef CAS.
- J. Zhao, H. Li, Y. Han, R. Li, X. Ding, X. Feng and B. Wang, J. Mater. Chem. A, 2015, 3, 12145–12148 RSC.
- S. Y. Zhang, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2015, 137, 12045–12049 CrossRef CAS PubMed.
- X. Hou, T. Xu, Y. Wang, S. Liu, J. Tong and B. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 32264–32269 CrossRef CAS PubMed.
- X. Hou, T. Xu, Y. Wang, S. Liu, J. Tong and B. Liu, ACS Appl. Mater. Interfaces, 2017, 9, S1–S15 CrossRef PubMed.
- X. Hou, T. Xu, Y. Wang, S. Liu, R. Chu, J. Zhang and B. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 26365–26371 CrossRef CAS PubMed.
- J. Y. Chan, H. Zhang, Y. Nolvachai, Y. Hu, H. Zhu, M. Forsyth, Q. Gu, D. E. Hoke, X. Zhang, P. J. Marriot and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 17130–17134 CrossRef CAS PubMed.
- X. Wang, Y. Zhu, J. Liu, C. Liu, C. Cao and W. Song, Chem. – Asian J., 2018, 13, 1535–1538 CrossRef CAS PubMed.
- A. Abbas, Z. X. Wang, Z. Li, H. Jiang, Y. Liu and Y. Cui, Inorg. Chem., 2018, 57, 8697–8700 CrossRef CAS PubMed.
- Y. Peng, T. Gong and Y. Cui, Chem. Commun., 2013, 49, 8253–8255 RSC.
- H. Wang, S. Zhao, Y. Liu, R. Yao, X. Wang, Y. Cao, D. Ma, M. Zou, A. Cao, X. Feng and B. Wang, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- T. Rajkumar, D. Kukkar, K. Kim, J. Ryeul and A. Deep, J. Ind. Eng. Chem., 2019, 72, 50–66 CrossRef CAS.
- A. W. Peters, T. C. Wang and P. Deria, Chem. Commun., 2017, 53, 7561–7564 RSC.
- C. Yang, Y.-Z. Zheng and X.-P. Yan, RSC Adv., 2017, 7, 36297–36301 RSC.
- R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630–8634 CrossRef CAS PubMed.
- T. Chen, H. Tan, Q. Chen, L. Gu, Z. Wei and H. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 48402–48411 CrossRef CAS PubMed.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- D. R. Kryscio and N. A. Peppas, Acta Biomater., 2012, 8, 461–473 CrossRef CAS PubMed.
- S. Lai, S. Tang, J. Xie, C. Cai, X. Chen and C. Chen, J. Chromatogr. A, 2017, 1490, 63–73 CrossRef CAS PubMed.
- W. J. Cheong, F. Ali, J. H. Choi, J. O. Lee and K. Yune Sung, Talanta, 2013, 106, 45–59 CrossRef CAS PubMed.
- S. Yang, Y. Wang, Y. Jiang, S. Li and W. Liu, Polymers, 2016, 8, 1–16 Search PubMed.
- M. Yoshikawa, K. Tharpa and S. Dima, Chem. Rev., 2016, 116, 11500–11528 CrossRef CAS PubMed.
- M. Yoshikawa, Bioseparation, 2001, 10, 277–286 CrossRef CAS PubMed.
- A. S. Michaels, R. F. Baddour, H. J. Bixler and C. Y. Choo, Ind. Eng. Chem. Process Des. Dev., 1962, 1, 14–25 CrossRef CAS.
- G. Wulff, A. Sarhan and K. Zabrocki, Tetrahedron Lett., 1973, 14, 4329–4332 CrossRef.
- G. Wulff and G. Kirstein, Angew. Chem., Int. Ed. Engl., 1990, 29, 684–686 CrossRef.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- M. Yoshikawa, J. Izumi, T. Ooi and T. Kitao, Polym. Bull., 1998, 40, 517–524 CrossRef CAS.
- Y. Kondo, M. Yoshikawa and H. Okushita, Polym. Bull., 2000, 44, 517–524 CrossRef CAS.
- M. Yoshikawa, N. Hotta, J. Kyoumura, Y. Osagawa and T. Aoki, Sens. Actuators, B, 2005, 104, 282–288 CrossRef CAS.
- M. Yoshikawa, K. Murakoshi and T. Kogita, Eur. Polym. J., 2006, 42, 2532–2539 CrossRef CAS.
- M. Yoshikawa, J. Izumi and T. Kitao, Macromolecules, 1996, 29, 8197–8203 CrossRef CAS.
- M. Yoshikawa, T. Fujisawa, J. Izumi, T. Kitao and S. Sakamoto, Anal. Chim. Acta, 1998, 365, 59–67 CrossRef CAS.
- M. Yoshikawa, T. Fujisawa and J. Izumi, Macromol. Chem. Phys., 1999, 365, 1458–1465 CrossRef.
- M. Yoshikawa, J. Izumi and T. Kitao, React. Funct. Polym., 1999, 42, 93–102 CrossRef CAS.
- M. Yoshikawa, A. Shimada and J. Izumi, Analyst, 2001, 126, 775–780 RSC.
- M. Yoshikawa, J. Izumi, T. Kitao and S. Koya, J. Membr. Sci., 1995, 108, 171–175 CrossRef CAS.
- M. Yoshikawa, K. Koso, K. Yonetani, S. Kitamura and S. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 385–396 CrossRef CAS.
- M. Yoshikawa, J. Izumi and T. Kitao, Chem. Lett., 1996, 25, 611–612 CrossRef.
- Y. Kondo, Y. Morita, A. Fujimoto, M. Tounai, S. Kimura and M. Yoshikawa, Chirality, 2003, 14, 498–503 CrossRef PubMed.
- J. Izumi, M. Yoshikawa and T. Kitao, Maku, 1997, 22, 149–154 CAS.
- M. Hatanaka, Y. Nishioka and M. Yoshikawa, J. Appl. Polym. Sci., 2013, 128, 123–131 CrossRef CAS.
- M. Hatanaka, Y. Nishioka and M. Yoshikawa, J. Membr. Sep. Technol., 2013, 2, 109–119 CAS.
- M. Hatanaka, Y. Nishioka and M. Yoshikawa, J. Mater. Sci. Res., 2012, 1, 114–122 CAS.
- M. Hatanaka, Y. Nishioka and M. Yoshikawa, Macromol. Chem. Phys., 2011, 212, 1351–1359 CrossRef CAS.
- Y. Sueyoshi, C. Fukushima and M. Yoshikawa, J. Membr. Sci., 2010, 357, 90–97 CrossRef CAS.
- M. Yoshikawa, T. Ooi and J. Izumi, J. Appl. Polym. Sci., 1999, 72, 493–499 CrossRef CAS.
- Y. Sueyoshi, A. Utsunomiya, M. Yoshikawa, G. P. Robertson and M. D. Guiver, J. Membr. Sci., 2012, 401–402, 89–96 CrossRef CAS.
- M. Yoshikawa, J. Izumi and T. Kitao, Polym. J., 1997, 29, 205–210 CrossRef CAS.
- A. Dzgoev and K. Haupt, Chirality, 1999, 11, 465–469 CrossRef CAS.
- P. Screenivasulu Reddy, T. Kobayashi, M. Abe and N. Fujii, Eur. Polym. J., 2002, 38, 521–529 CrossRef CAS.
- Z. Jiang, Y. Yu and H. Wu, J. Membr. Sci., 2006, 280, 876–882 CrossRef CAS.
- S. Son and J. Jegal, J. Appl. Polym. Sci., 2006, 104, 1866–1872 CrossRef.
- N. Bing, Z. Xu, X. Wang, Z. Yang and H. Yang, J. Appl. Polym. Sci., 2007, 106, 71–76 CrossRef CAS.
- Y. Lei, F. Chen and Y. Luo, Iran. Polym. J., 2014, 23, 679–687 CrossRef CAS.
- B. Gao, Y. Li and K. Cui, Int. J. Polym. Mater. Polym. Biomater., 2018, 67, 517–527 CrossRef CAS.
- H. Li, Q. Huang, D. Li, S. Li, X. Wu, L. Wen and C. Ban, Org. Process Res. Dev., 2018, 22, 278–285 CrossRef CAS.
- T. Masawaki, M. Sasai and S. Tone, J. Chem. Eng. Jpn., 1992, 25, 33–39 CrossRef CAS.
- R. Viveiros, S. Rebocho and T. Casimiro, Polymer, 2018, 10, 1–27 Search PubMed.
- L. Madikizela, S. Ncube and L. Chimuka, in Mip Synthesis, Characteristics and Analytical Application, Elsevier B.V., 1st edn, 2019, vol. 86, pp. 337–364 Search PubMed.
- R. A. Lorenzo, A. M. Carro, C. Alvarez-lorenzo and A. Concheiro, Int. J. Mol. Sci., 2011, 12, 4327–4347 CrossRef CAS PubMed.
- E. Drioli, A. Brunetti, G. Di Profio and G. Barbieri, Green Chem., 2012, 14, 1561–1572 RSC.
- S. H. Park, A. Alammar, Z. Fulop, B. A. Pulido, S. P. Nunes and G. Szekely, Green Chem., 2021, 23, 1175–1184 RSC.
- W. Xie, T. Li, A. Tiraferri, E. Drioli, A. Figoli, J. C. Crittenden and B. Liu, ACS Sustainable Chem. Eng., 2021, 9, 50–75 CrossRef CAS.
- W. J. Lee, P. S. Goh, W. J. Lau, A. F. Ismail and N. Hilal, Membranes, 2021, 11, 1–35 Search PubMed.
- J. W. Lee, Ind. Eng. Chem. Res., 2020, 59, 9619–9628 CrossRef CAS.
- X. Lin, R. Gong, J. Li, P. Li, J. Yu and A. E. Rodrigues, J. Chromatogr. A, 2016, 1467, 347–355 CrossRef CAS PubMed.
- D. Antos and A. Seidel-Morgenstern, Chem. Eng. Sci., 2001, 56, 6667–6682 CrossRef CAS.
- D. B. Broughton and C. G. Gerhold, US2985589, 1961.
- M. Negawa and F. Shoji, J. Chromatogr. A, 1992, 590, 113–117 CrossRef CAS.
- T. Futagawa, J. P. Canvant, E. Cavoy, M. Deleers, M. Hamende and V. Zimmermann, US6107492, 2000.
- H. A. Zinnen and M. J. Gattuso, US6455736, 2002.
- M. J. Gattuso, US5889186, 1999.
- H. A. Zinnen and M. J. Gattuso, US6410794, 2002.
- A. L. L. Duchateau, R. Gebhard and Q. B. Broxterman, WO2006/0122430, 2006.
- H. Diehl, A. Krebs, E. Krebs, W. Lange, H. Paul, D. Seidel, R. Grosser and T. Reichelt, US7709644B2, 2010.
- C. B. Ching, B. G. Lim, E. J. D. Lee and S. C. Ng, J. Chromatogr., 1993, 634, 215–219 CrossRef CAS.
- B. Lim, C. Ching, R. B. H. Tan and S. C. Ng, Chem. Eng. Sci., 1995, 50, 2289–2298 CrossRef CAS.
- B. G. Lim and C. B. Ching, J. Chromatogr. A, 1996, 734, 247–258 CrossRef CAS.
- A. S. Andrade Neto, A. R. Secchi, M. B. Souza Jr. and A. G. Barreto Jr., J. Chromatogr. A, 2016, 1470, 42–49 CrossRef CAS PubMed.
- F. C. Cunha, A. R. Secchi, M. B. de Souza and A. G. Barreto, Chirality, 2019, 31, 583–591 CrossRef CAS PubMed.
- O. Ludemann-Hombourger, R. M. Nicoud and M. Bailly, Sep. Sci. Technol., 2000, 35, 1829–1862 CrossRef CAS.
- L. S. Pais, J. M. Loureiro and A. E. Rodrigues, AIChE J., 1998, 44, 561–569 CrossRef CAS.
- Z. Zhang, M. Mazzotti and M. Morbidelli, Korean J. Chem. Eng., 2004, 21, 454–464 CrossRef CAS.
- I. B. R. Nogueira, A. M. Ribeiro, R. Requião, K. V. Pontes, H. Koivisto, A. E. Rodrigues and J. M. Loureiro, Appl. Soft Comput. J., 2018, 67, 29–47 CrossRef.
- P. Suvarov, J. W. Lee, A. Vande Wouwer, A. Seidel-Morgenstern and A. Kienle, J. Chromatogr. A, 2019, 1602, 266–272 CrossRef CAS PubMed.
- J. Matos, R. P. V. Faria, I. B. R. Nogueira, J. M. Loureiro and A. M. Ribeiro, Comput. Chem. Eng., 2019, 123, 344–356 CrossRef CAS.
- C. Migliorini, M. Wendlinger, M. Mazzotti and M. Morbidelli, Ind. Eng. Chem. Res., 2001, 40, 2606–2617 CrossRef CAS.
- S. Abel, M. Mazzotti and M. Morbidelli, J. Chromatogr. A, 2002, 944, 23–39 CrossRef CAS PubMed.
- J. Houwing, T. B. Jensen, S. H. Van Hateren, H. A. H. Billiet and L. A. M. Van der Wielen, AIChE J., 2003, 49, 665–674 CrossRef CAS.
- T. B. Jensen, T. G. P. Reijns, H. A. H. Billiet and L. A. M. van der Wielen, J. Chromatogr. A, 2000, 873, 149–162 CrossRef CAS PubMed.
- M. M. Kearney and K. L. Hieb, US5102553, 1992.
- H. Schramm, M. Kaspereit, A. Kienle and A. Seidel-Morgenstern, Chem. Eng. Technol., 2002, 25, 1151–1155 CrossRef CAS.
- R. C. Vroon, M. A. Boon and P. J. T. Bussman, WO2015/0314218, 2015.
- M. Anton, WO2017/199031, 2017.
- T. H. Yoon, B. H. Chung and I. H. Kim, Biotechnol. Bioprocess Eng., 2004, 9, 285–291 CrossRef CAS.
- K. B. Lee, C. Y. Chin, Y. Xie, G. B. Cox and N. H. L. Wang, Ind. Eng. Chem. Res., 2005, 44, 3249–3267 CrossRef CAS.
- K. B. Lee, S. Mun, F. Cauley, G. B. Cox and N. H. L. Wang, Ind. Eng. Chem. Res., 2006, 45, 739–752 CrossRef CAS.
- Y. Zhang, K. Hidajat and A. K. Ray, Chem. Eng. Sci., 2007, 62, 1364–1375 CrossRef CAS.
- J. S. Park, W. S. Kim, J. M. Kim and I. H. Kim, J. Chem. Eng. Jpn., 2008, 41, 624–626 CrossRef CAS.
- P. S. Gomes, M. Zabkova, M. Zabka, M. Minceva and A. E. Rodrigues, AIChE J., 2009, 56, 125–142 Search PubMed.
- A. E. Ribeiro, P. S. Gomes, L. S. Pais and A. E. Rodrigues, Sep. Sci. Technol., 2011, 46, 1726–1739 CrossRef CAS.
- R. S. Arafah, A. E. Ribeiro, A. E. Rodrigues and L. S. Pais, Chirality, 2019, 31, 62–71 CrossRef CAS PubMed.
- R. Gong, X. Lin, P. Li, J. Yu and A. E. Rodrigues, J. Chromatogr. A, 2014, 1363, 242–249 CrossRef CAS PubMed.
- Y. Yang, K. Lu, R. Gong, D. Qu, P. Li, J. Yu and A. E. Rodrigues, Adsorption, 2019, 25, 1227–1240 CrossRef CAS.
- C. G. Lee, Y. J. Song, K. B. Lee and S. Mun, J. Ind. Eng. Chem., 2019, 80, 677–685 CrossRef CAS.
- C. G. Lee, C. Y. Jo, K. B. Lee and S. Mun, Sep. Purif. Technol., 2021, 254, 117597 CrossRef CAS.
- A. E. Ribeiro, N. S. Graça, L. S. Pais and A. E. Rodrigues, Sep. Purif. Technol., 2008, 61, 375–383 CrossRef CAS.
- X. Jiang, L. Zhu, B. Yu, Q. Su, J. Xu and W. Yu, J. Chromatogr. A, 2018, 1531, 131–142 CrossRef CAS PubMed.
- M. Fuereder, C. Femmer, G. Storti, S. Panke and M. Bechtold, Chem. Eng. Sci., 2016, 152, 649–662 CrossRef CAS.
- I. Harriehausen, K. Wrzosek, H. Lorenz and A. Seidel-Morgenstern, Adsorption, 2020, 26, 1199–1213 CrossRef CAS.
- M. Johannsen and G. Brunner, J. Supercrit. Fluids, 2018, 134, 61–70 CrossRef CAS.
- S. Peper, M. Lübbert, M. Johannsen and G. Brunner, Sep. Sci. Technol., 2002, 37, 2545–2566 CrossRef CAS.
- D. Speybrouck and E. Lipka, J. Chromatogr. A, 2016, 1467, 33–55 CrossRef CAS PubMed.
- C. J. Welch, W. R. Leonard, J. O. DaSilva, M. Biba, J. Albaneze-Walker, D. W. Henderson, B. Laing and D. J. Mathre, LC-GC Asia Pac., 2005, 18, 24–31 Search PubMed.
- L. Miller, J. Chromatogr. A, 2012, 1250, 250–255 CrossRef CAS PubMed.
- K. Kalíková, T. Šlechtová, J. Vozka and E. Tesařová, Anal. Chim. Acta, 2014, 821, 1–33 CrossRef PubMed.
- A. Ghinet, Y. Zehani and E. Lipka, J. Pharm. Biomed. Anal., 2017, 145, 845–853 CrossRef CAS PubMed.
- E. R. Francotte, Practical Aspects and Applications of Preparative Supercritical Fluid Chromatography, Elsevier Inc., 2017 Search PubMed.
- S. H. Yip, D. Wu, J. Kempson, A. Hernandez, H. Zhang, P. Li, D. Sun and A. Mathur, Sep. Sci. Plus, 2019, 2, 343–352 CrossRef CAS.
- C. West, TrAC, Trends Anal. Chem., 2019, 120, 1–9 CrossRef.
- D. Speybrouck, M. Howsam and E. Lipka, Trends Anal. Chem., 2020, 133, 1–16 CrossRef.
- R. M. Nicoud, Ind. Eng. Chem. Res., 2014, 53, 3755–3765 CrossRef CAS.
- J. Y. Clavier, R. M. Nicoud and M. Perrut, Process Technol. Proc., 1996, 12, 429–434 CAS.
- M. Mazzotti, G. Storti and M. Morbidelli, J. Chromatogr. A, 1997, 786, 309–320 CrossRef CAS.
- F. Denet, W. Hauck, R. M. Nicoud, O. Di Giovanni, M. Mazzotti, J. N. Jaubert and M. Morbidelli, Ind. Eng. Chem. Res., 2001, 40, 4603–4609 CrossRef CAS.
- S. Peper, M. Johannsen and G. Brunner, J. Chromatogr. A, 2007, 1176, 246–253 CrossRef CAS PubMed.
- M. Johannsen, S. Peper and A. Depta, J. Biochem. Biophys. Methods, 2002, 54, 85–102 CrossRef CAS PubMed.
- A. Rajendran, S. Peper, M. Johannsen, M. Mazzotti, M. Morbidelli and G. Brunner, J. Chromatogr. A, 2005, 1092, 55–64 CrossRef CAS PubMed.
- H. Kaemmerer, G. Brunner and M. Johannsen, J. Supercrit. Fluids, 2007, 43, 204–213 CrossRef CAS.
- M. A. Korany, H. Mahgoub, R. S. Haggag, M. A. A. Ragab and O. A. Elmallah, J. Liq. Chromatogr. Relat. Technol., 2017, 40, 839–852 CrossRef CAS.
- H. R. Hobbs, B. Kondor, P. Stephenson, R. A. Sheldon, N. R. Thomas and M. Poliakoff, Green Chem., 2006, 8, 816–821 RSC.
- T. Matsuda, T. Harada and K. Nakamura, Green Chem., 2004, 6, 440–444 RSC.
- M. T. Reetz, W. Wiesenhöfer, G. Franciò and W. Leitner, Adv. Synth. Catal., 2003, 345, 1221–1228 CrossRef CAS.
- T. Matsuda, K. Watanabe, T. Harada, K. Nakamura, Y. Arita, Y. Misumi, S. Ichikawa and T. Ikariya, Chem. Commun., 2004, 2286–2287 RSC.
- P. Lozano, T. De Diego, C. Mira, K. Montague, M. Vaultier and J. L. Iborra, Green Chem., 2009, 11, 538–554 RSC.
- Z. Molnár, E. Farkas, Á. Lakó, B. Erdélyi, W. Kroutil, B. G. Vértessy, C. Paizs and L. Poppe, Catalysts, 2019, 9, 1–11 CrossRef.
- S. Mane, Anal. Methods, 2016, 8, 7567–7586 RSC.
- B. Schuur, B. J. V. Verkuijl, A. J. Minnaard, J. G. De Vries, H. J. Heeres and B. L. Feringa, Org. Biomol. Chem., 2011, 9, 36–51 RSC.
- B. Schuur, J. G. M. Winkelman, J. G. de Vries and H. J. Heeres, Chem. Eng. Sci., 2010, 65, 4682–4690 CrossRef CAS.
- A. Holbach, J. Godde, R. Mahendrarajah and N. Kockmann, AIChE J., 2015, 61, 266–276 CrossRef CAS.
- K. Tang, P. Wen, P. Zhang and Y. Huang, Sep. Purif. Technol., 2014, 134, 100–109 CrossRef CAS.
- B. Schuur, A. J. Hallett, J. G. M. Winkelman, J. G. De Vries and H. J. Heeres, Org. Process Res. Dev., 2009, 13, 911–914 CrossRef CAS.
- K. Tang, P. Wen, P. Zhang and Y. Huang, Chirality, 2015, 27, 75–81 CrossRef CAS PubMed.
- K. Tang, H. Zhang and P. Zhang, Ind. Eng. Chem. Res., 2013, 52, 3893–3902 CrossRef CAS.
- K. Tang, H. Zhang and Y. Liu, AIChE J., 2013, 59, 2594–2602 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- A. Jordan, P. Stoy and H. F. Sneddon, Chem. Rev., 2021, 121, 1582–1622 CrossRef CAS PubMed.
- N. Rubio and C. Minguillón, in Chiral Recognition in Separation Methods: Mechanisms and Applications, 2010, pp. 241–274 Search PubMed.
- G. K. E. Scriba, Chiral Separations - Methods and Protocols, Humana Press, 3rd edn, 2019 Search PubMed.
- A. P. Foucault, J. Chromatogr. A, 2001, 906, 365–378 CrossRef CAS PubMed.
- A. Berthod, M. J. Ruiz-Ángel and S. Carda-Broch, J. Chromatogr. A, 2009, 1216, 4206–4217 CrossRef CAS PubMed.
- X. Y. Huang, D. Pei, J. F. Liu and D. L. Di, J. Chromatogr. A, 2018, 1531, 1–12 CrossRef CAS PubMed.
- X. Y. Huang and D. L. Di, TrAC, Trends Anal. Chem., 2015, 67, 128–133 CrossRef CAS.
- A. E. Kostanyan and A. A. Erastov, J. Chromatogr. A, 2015, 1406, 118–128 CrossRef CAS PubMed.
- N. Rubio, S. Ignatova, C. Minguillón and I. A. Sutherland, J. Chromatogr. A, 2009, 1216, 8505–8511 CrossRef CAS PubMed.
- P. Zhang, G. Sun, K. Tang, C. Zhou, C. Yang and W. Yang, Sep. Purif. Technol., 2015, 146, 276–283 CrossRef CAS.
- P. Zhang, G. Sun, K. Tang, W. Yang, G. Sui and C. Zhou, J. Sep. Sci., 2014, 1–8 Search PubMed.
- S. Tong, Y. Guan, J. Yan, B. Zheng and L. Zhao, J. Chromatogr. A, 2011, 1218, 5434–5440 CrossRef CAS PubMed.
- F. Couillard, A. Foucault and D. Durand, US7422685B2, 2008.
- I. Sutherland, P. Hewitson and S. Ignatova, J. Chromatogr. A, 2009, 1216, 8787–8792 CrossRef CAS PubMed.
Footnote |
| † The authors picked the best-case scenario reported in each article to present here. |
| This journal is © The Royal Society of Chemistry 2022 |