
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlyme-based electrolytes: suitable solutions for next-generation lithium batteries
Daniele
Di Lecce
 a,
Vittorio
Marangon
ab,
Hun-Gi
Jung
c,
Yoichi
Tominaga
de,
Steve
Greenbaum
f and
Jusef
Hassoun
*abeg
a,
Vittorio
Marangon
ab,
Hun-Gi
Jung
c,
Yoichi
Tominaga
de,
Steve
Greenbaum
f and
Jusef
Hassoun
*abeg
aGraphene Labs, Istituto Italiano di Tecnologia, via Morego 30, Genova, 16163, Italy. E-mail: jusef.hassoun@iit.it; jusef.hassoun@unife.it
bUniversity of Ferrara, Department of Chemical, Pharmaceutical and Agricultural Sciences, Via Fossato di Mortara 17, 44121, Ferrara, Italy
cCenter for Energy Storage Research, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul, 02792, Republic of Korea
dTokyo University of Agriculture and Technology, Graduate School of Bio-Applications and Systems Engineering (BASE), 2-24-16, Naka-cho, Koganei-shi, Tokyo 184-8588, Japan
eInstitute of Global Innovation Research (GIR), Tokyo University of Agriculture and Technology, Tokyo, Japan
fHunter College of CUNY, 695 Park Avenue, NY 10065, New York, USA
gNational Interuniversity Consortium of Materials Science and Technology (INSTM) University of Ferrara Research Unit, University of Ferrara, Via Fossato di Mortara, 17, 44121, Ferrara, Italy
First published on 7th January 2022
Abstract
The concept of green in a battery involves the chemical nature of electrodes and electrolytes as well as the economic sustainability of the cell. Although these aspects are typically discussed separately, they are deeply interconnected: indeed, a new electrolyte can allow the use of different cathodes with higher energy, lower cost or more pronounced environmental compatibility. In this respect, we focus on an alternative class of electrolyte solutions for lithium batteries formed by dissolving LiX salts in glyme solvents, i.e., organic ethers with the molecular formula CH3O[CH2CH2O]nCH3 differing by chain length. The advantages of these electrolytes with respect to the state-of-the-art ones are initially illustrated in terms of flammability, stability, toxicity, environmental compatibility, cell performances and economic impact. A particular light is shed on the stability of these systems, particularly in the polymer state, and in various environments including oxygen, sulfur and high-energy lithium metal. Subsequently, the most relevant studies on the chemical–physical features, characteristic structures, favorable properties, and electrochemical behavior of glyme-based solutions are discussed, and the most recent technological achievements in terms of cell design and battery performance are described. In the final sections, the use of glyme-based electrolytes in high-energy cells arranged by coupling a lithium-metal anode with conventional insertion cathodes as well as in alternative and new batteries exploiting the Li–S and Li–O2 conversion processes is described in detail. The various paragraphs actually reveal the advantages, including safety, low cost and sustainability, which can be achieved by employing the glyme-based electrolytes with respect to the commercially available ones, in particular taking into account future and alternative applications. Particular relevance is given to the glymes with long chains that show remarkable stability, high safety and very low toxicity. Therefore, this review is expected to shed light on the potentialities, the actual advantages compared to the state-of-the-art batteries, and the possible applications of electrolytes based on glyme solvents in next-generation energy storage systems.
1. Introduction
Several literature papers proposed improvement of the sustainability of electrochemical energy storage systems focused on the green nature of various cell components. On the other hand, the most significant approach for achieving sustainability and the green concept is the demonstration of actual advances in comparison with the existing technologies.1 In this respect, the widespread use of lithium-ion (Li-ion) batteries over the past decades has been driving a notable technological change in our society and is currently enabling a gradual transition to a more environmentally sustainable automotive mobility.2 Smartphones, laptops, and tablets powered by lithium-ion cells are among the most used devices in our daily life, and hybrid and full electric vehicles are becoming widely adopted, thanks to several programs launched worldwide to decrease environmental pollution.3 Furthermore, Li-ion batteries could play a key role in developing smart grids coupled with renewable energy sources,4 as suggested by the already installed and operating battery packs for stationary storage.5 Driven by the undeniable success of the Li-ion technology, ambitious targets have been set by several countries to further boost cell performance by increasing the energy density, and the lithium–metal battery design has been identified as one of the most promising new systems.6 Despite a few practical examples of commercial lithium–metal batteries, various challenges still have to be overcome to develop high-energy cells with a suitable safety level and a reliable long-term behavior.6 In this regard, the characteristic properties of the most common electrolyte solutions for Li-ion batteries pose some issues.7 These solutions typically consist of mixtures of a lithium salt (e.g., LiPF6) and organic ester solvents, such as alkyl carbonates, and indeed exhibit high conductivity which enable a satisfactory battery performance in terms of capacity, energy density, and rate capability.7 On the other hand, these electrolytes are volatile, flammable, and poorly stable when in contact with lithium metal, in particular during prolonged operation.8–10 Therefore, several research studies have been focusing since nineties on optimizing alternative electrolyte solutions,11–15 such as those prepared by dissolving a lithium salt in a lowly volatile and modestly flammable ether oligomer with a –(CH2CH2O)– unit, that is, ethylene oxide (EO)-glymes.16–24 EO-based glymes can be synthesized from ethylene epoxide by various large scale methods, including reaction in an alcoholic environment with sodium following the Williamson mechanism, methylation of glycol ether with methyl sulfate, Lewis acid-catalyzed cleavage of ethylene oxide by ether, and reaction of ethylene glycol with alcohol catalyzed polyperfluorosulfonic acid resin at high temperature and pressure.25 Instead, aliphatic carbonates presently used in battery electrolyte formulations may be industrially achieved by more complex organic pathways including phosgenation, oxidative carbonylation, reaction of urea with alcohols, reaction of oxiranes with carbon dioxide, through metal carbonates, and by carbonate interchange reaction.26 In particular, dialkyl carbonates such as DMC and DEC, may be prepared from halohydrins, and from alcohols and carbon monoxide with elemental sulfur, while alicyclic carbonates such as EC may be obtained from the corresponding halogenated carbonates.26 For readers’ convenience, Table 1 reports the acronyms of the several chemical species discussed herein.| Chemical species | Acronym |
|---|---|
| nGlyme, n(ethylene glycol) dimethyl ether, CH3-O-(CH2-CH2-O)n-CH3 | Gn |
| Poly(ethylene glycol) dimethyl ether | PEGDME |
| Poly(ethylene oxide) | PEO |
| Poly(methyl methacrylate) | PMMA |
| Poly(butyl acrylate) | PBA |
| Polypyrrole | PPy |
| 1,3-Dioxolane | DOL |
| 1,2-Dimethoxy ethane | DME (or G1) |
| Ethylene glycol diethyl ether | EG |
| Dimethylacetamide | DMA |
| Tetrahydrofuran | THF |
| 1,1,2,2-Tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether, hydrofluoroether | HFE |
| Propylene carbonates | PC |
| Ethylene carbonates | EC |
| Dimethyl carbonates | DMC |
| Methylethylcarbonate, ethylmethylcarbonate | EMC |
| Diethyl carbonate | DEC |
| Fluoroethylene carbonate | FEC |
| Methylethylcarbonate | MEC |
| Acetonitrile | ACN |
| Dimethyl sulfoxide | DMSO |
| Dimethylformamide | DMF |
| Dimethylacetamide | DMA |
| Γ-Butyrolactone | GBL |
| Tetraethylsulfamide | TESA |
| Lithium-bis-(trifluormethanesulfonyl)-imide, LiN(SO2CF3)2, often LiTFSA | LiTFSI |
| Lithium bis(fluoro sulfonyl)imide, LiN(SO2F)2, often LiFSA | LiFSI |
| Cyclic imide, lithium 1,2,3-dithiazolidine-4,4,5,5-tetrafluoro-1,1,3,3-tetraoxide, LiN(C2F4S2O4) | LiCTFSI |
| Lithium hexafluorophosphate | LiPF6 |
| Lithium tetrafluoroborate | LiBF4 |
| Lithium bis(perfluoroethanesulfonyl)imide, often LiBETA | LiBETI |
| Lithium tetrafluoroborate | LiTFB |
| Lithium trifluoromethylsulfonate, lithium triflate, LiSO3CF3 | LiTf |
| Lithium trifluoroacetate | LiTFA |
| Lithium 2-trifluoromethyl-4,5-dicyanoimidazole | LiTDA |
| Lithium tetra(trifluoromethanesulfonyl)propene | LiTFSP |
| Lithium bis(nonafluorobutanesulfonyl)imide, LiN(C4F9SO2)2 often LiNFSA | LiNFSI |
| Lithium nitrate | LiNO3 |
| Lithium bis(pentafluoroethylsulfonyl)imide, LiN(SO2C2F5)2 | LiBETI |
| Lithium bis(oxalato)borate, LiC4BO8 | LiBOB |
Dilithium dodeca![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) luorododecaborate, Li2B12F12 luorododecaborate, Li2B12F12 |
Li2DFB |
| N,N-Diethyl-N-methyI-N-(2-methoxyethyl) ammonium bis(trifluoromethanesulfonyl)imide | DEMETFSI |
| Methyl butyl pyrrolidinium-bis-(trifluoromethanesulfonyl)-imide | Pyr14TFSI |
| 1,1,2,2-Tetrafluoroethyl 2,2,2-trifluoroethyl ether | TFTFE |
| LiNixMnyCozO2 | NMC |
| Solid electrolyte interphase | SEI |
| Ionic liquid | IL |
| Solvate ionic liquid | SIL |
| Solvent separated ion pair | SSIP |
| Contact ion pair | CIP |
| Vinylene carbonate | VC |
| Vinylethylene carbonate | VEC |
| 1,3-Propane sultone | 13PS |
| Hard carbon | HC |
| Trimethyl phosphate | TMP |
| Triethyl phosphate | TEP |
| Tripropyl phosphate | TPrP |
| 2-(2,2,2-Trifluoroethoxy)-1,3,2-dioxaphospholane 2-oxide | TFEP |
| 2,2,2-Trifluoroethyl methyl carbonate | FEMC |
| Di-(2,2,2-trifluoroethyl)carbonate | DFDEC |
| 1,1,2,2-Tetrafluoroethyl-2′,2′,2′-trifluoroethyl ether | TTFE |
| Ethoxy(pentafluoro)cyclotriphosphazene | PFPN |
| Polystyrene | PS |
| Poly(ethylene glycol) methacrylate | PEGMA |
| poly(ethylene glycol) dimethacrylate | PEGDMA |
| poly(ethylene glycol) methyl ether acrylate | PEGA |
In addition to the simplest and more environmentally friendly preparation pathway compared to those of the common electrolyte solvents used in batteries (i.e., EC, DMC, EMC, and DEC), glymes, and particularly those with longer chain lengths (G3, G4, and PEGDME), are characterized by a more relevant safety content, lower flammability and flash point as well as less relevant toxicity except for possible issues on fertility expected for glymes with lower chain lengths. Table 2 displays the physical–chemical properties of the conventional carbonate solvents and glymes used for battery application, including the related safety hazards as indicated by the SDS data (Sigma-Aldrich) and confirmed by the literature.25,27–31 PC and EC are the only carbonate solvents without flammability; however EC has an elevated toxicity. Instead, EC and DMC which are typically combined as the electrolyte solvents for LIBs are highly flammable, thus suggesting the need for a safer alternative.32 On the other hand, glymes show a favorable trend associated with the lengthening of the –CH2CH2O– chains. Indeed, DME (G1) and DEGDME (G2) are the only glyme species to show relevant flammability, while TREGDME (G3) and TEGDME (G4) exhibit low toxicity and liquid PEGDMEs (250 and 500 g mol−1) are classified as non-dangerous for humans. Despite DME being usually employed in Li–S batteries due to its enhanced stability towards lithium polysulfide intermediates formed by the conversion electrochemical process, the safety issues related to its relevant volatility and flammability are by now acknowledged, and the search for safer electrolyte solutions to achieve Li–S devices of practical interest is a deeply investigated topic.33 Moreover, a decrease of the electrolyte flashpoint may be achieved by using non-flammable co-solvents and flame retardant additives, such as phosphorus-containing (e.g., TMP, TEP, TPrP, and TFEP) and fluorinated (e.g., FEMC, FEC, DFDEC, TTFE, and PFPN) species,34 or by functionalization of the glyme solvent.35 Among the various approaches, solid electrolyte configurations may represent a viable strategy to increase the safety content of the battery due to enhanced chemical, thermal and mechanical stability. Ceramic electrolytes such as garnet-type Li7La3Zr2O12 (LLZO) or NASICON-derived structures, e.g., Li1.5Al0.5Ge1.5(PO4)3 (LAGP), provide fast Li+ transport and suitable ionic conductivity at room temperature, despite the cycling behavior likely to be affected by the interphase stability due to poor contact between the electrodes and electrolyte in the cell.36 On the other hand, solid polymer electrolytes benefit from suitable electrode/electrolyte contact and remarkable conductivity which, however, are reached at medium-high operative temperatures. Indeed, PEO-based electrolytes usually require temperatures above 65 °C to allow proper amorphization of the crystalline structure and satisfactory battery performance,37 which may be improved through the introduction of copolymer blocks such as PS, PEGMA, PEGDMA or PEGA,36,38 or ceramic fillers such as SiO2, ZrO2 or TiO2 in the PEO matrix.37,39,40 The substitution of PEO with polycarbonate species was recently considered due to the lower crystallinity and good oxidative stability, even though their low stability towards lithium metal may limit their application.36 Furthermore, alternative polymer chemistries can be employed to stabilize the lithium anode and synthesize effective separators in order to enhance the Li+ exchange between the electrodes.41,42
| Solvents | Safety hazards (SDS) | Melting point [°C] | Boiling point [°C] | Flash point [°C] | Density [g cm−3] | Viscosity [mPa s] |
|---|---|---|---|---|---|---|
| DMC | - Flammable | 2 | 90 (ref. 27) | 18 (ref. 27) | 1.069 | 0.59 (ref. 28) |
| EC | - Elevated toxicity | 35 | 248 (ref. 27) | 160 (ref. 27) | 1.321 | 2.56 (ref. 28) |
| - Irritating | ||||||
| PC | - Irritating | −55 | 242 (ref. 27) | 132 (ref. 27) | 1.204 | 2.5 (ref. 28) |
| EMC | - Flammable | −55 | 109 (ref. 27) | 23 (ref. 27) | 1.006 | 0.65 (ref. 29) |
| DEC | - Flammable | −43 | 126 (ref. 27) | 33 (ref. 27) | 0.975 | 0.753 (ref. 28) |
| DME | - Flammable | −58 | 84 (ref. 27) | −2 (ref. 27) | 0.867 | 0.42–0.46 (ref. 25) |
| - Irritating | ||||||
| - May affect fertility | ||||||
| DEGDME | - Flammable | −64 | 162 | 57 | 0.94 | 0.98–1.0 (ref. 25) |
| - May affect fertility | ||||||
| TREGDME | - Irritating | −45 | 225 | 113 | 0.985 | 1.95–2.16 (ref. 25) |
| - May affect fertility | ||||||
| TEGDME | - May affect fertility | −30 | 276 (ref. 27) | 140 (ref. 27) | 1.009 | 3.3–3.7 (ref. 25) |
| PEGDME (250 g mol−1) | — | −23 | 240 | 135 | 1.03 | 7.2 (20 °C) (ref. 30) |
| PEGDME (500 g mol−1) | — | 13 | >250 | 254 | 1.07 | 28 (20 °C) (ref. 30) |
Along with the initial conceptualization and pioneering studies of glyme-based electrolytes, Li–metal batteries using the most common intercalation electrodes, such as LiCoO2 and graphite, as well as other insertion cathodes such as LiFePO4, were proposed with promising results, in spite of several issues which were firstly identified.43–60 Afterwards, “high-concentration” glyme-based electrolytes have attracted a great deal of attention due to their favorable properties, and there is an intriguing dilemma on the actual nature of these mixtures, which have been described as either solvent-in-salt solutions or solvated ionic liquids (SILs).61–82 Moreover, glyme-based electrolytes have been gaining renewed interest due to a possible suitability for the emerging high-energy lithium–sulfur (Li–S) battery, which is formed by combining a lithium–metal anode and a sulfur-based cathode.83–102 This new technology is nowadays considered close to practical applications and holds promise of a breakthrough in storable energy per unit mass.103,104 Furthermore, glyme-based solutions have been selected as the electrolytes of choice for the lithium–oxygen (Li–O2) cell, in which a lithium–metal anode is coupled with a gas diffusion layer electrode enabling O2 electrochemical conversion thanks to an open design. Notably, this system could store even more energy than the Li–S cell and has been suggested as a possible battery for future applications.105–130 In this review, we discuss with chronological details various developments in the research on glyme-based electrolytes for lithium batteries, which are summarized in the scheme in Fig. 1 (panel a). For the reader's convenience the various techniques and expressions cited throughout the following sections are listed and explained in Table 3. Furthermore, panel b of Fig. 1 reports a comparison between the present Li-ion battery and the two emerging energy storage systems (i.e., Li–S and Li–O2 cells), which can allow the use of glyme-based electrolytes for possible application in electric vehicles. The figure suitably reveals the advances potentially achieved by these new systems both in terms of the driving range (km by a single charge)131 and in terms of the economic impact (USD per kW h of battery pack).132
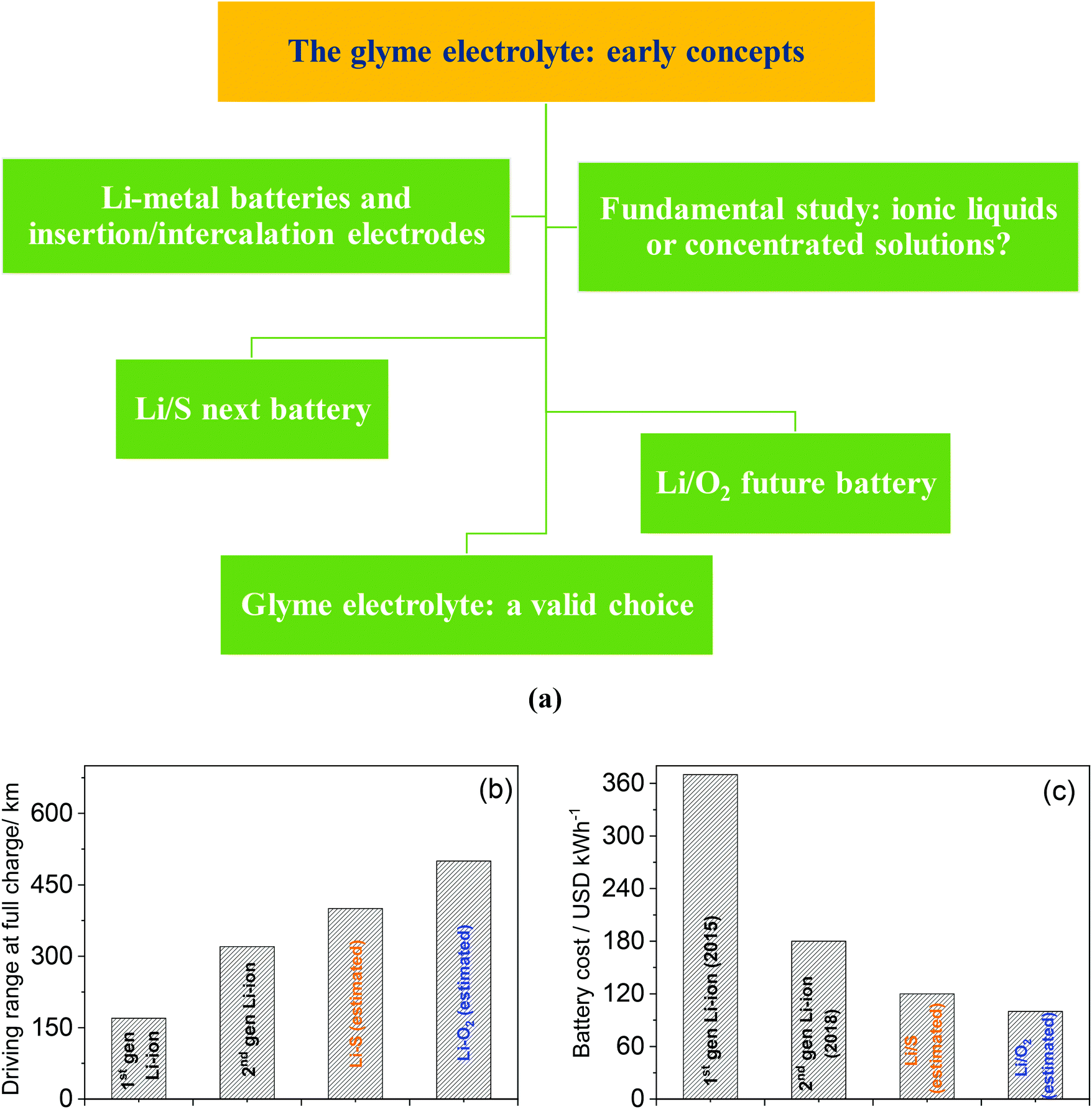 | ||
| Fig. 1 (a) Schematic of the main research topics involving the fundamental and technological investigations of glyme-based electrolytes for lithium batteries carried out over the past 25 years. (b and c) Comparison between the present Li-ion battery and the two emerging energy storage systems (i.e., Li–S and Li–O2 cells) which can be allowed by the use of a glyme-based electrolyte for possible application in electric vehicles in terms of (b) the driving range (km by a single charge) and (c) economic impact (USD per kW h of battery pack). | ||
| Techniques and other acronyms | Acronym |
|---|---|
| Electrochemical quartz crystal microbalance | EQCM-A |
| Cyclic voltammetry | CV |
| Electrochemical impedance spectroscopy | EIS |
| Very-low-frequency electrochemical impedance spectroscopy | VLF-EIS |
| Galvanostatic cycling | GC |
| Nuclear magnetic resonance | NMR |
| Pulse field gradient nuclear magnetic resonance | PFG-NMR |
| Pulsed-gradient spin-echo nuclear magnetic resonance | PGSE-NMR |
| Electrophoretic NMR | eNMR |
| First-principles molecular dynamics | FPMD |
| Electrochemical mass spectrometry | ECMS |
| Linear sweep voltammetry | LSV |
| Solvate ionic liquid | SIL |
| Contact ion pair | CIP |
| Scanning electron microscopy | SEM |
| Transmission electron microscopy | TEM |
| X-Ray diffraction | XRD |
| Small-angle X-ray scattering | SAXS |
| In situ subtractively normalized Fourier transform infrared spectroscopy | SNIFTIRS |
| X-ray photoelectron spectroscopy | XPS |
| Thermogravimetric analysis | TGA |
| Open circuit voltage | OCV |
| Gas diffusion layer | GDL |
| Oxygen reduction reaction | ORR |
| Oxygen evolution reaction | OER |
| Rotating ring disk electrode | RRDE |
| Mass spectrometry | MS |
| High-energy X-ray total scattering | HEXTS |
| Molecular dynamics | MD |
| Molecular weight | MW |
| Molecular orbital | MO |
| Density functional theory | DFT |
| Potentials of mean force | PMF |
| Infrared | IR |
| X-ray absorption spectroscopy | XAS |
| X-ray absorption near edge structure | XANES |
| Oxygen reduction reaction | ORR |
| Oxygen evolution reaction | OER |
| Donor number | DN |
| Highest occupied molecular orbital | HOMO |
| Lowest unoccupied molecular orbital | LUMO |
2. The “glyme electrolyte”: initial studies and fundamental concepts
Pioneering studies on the possible energy-storage applications of glymes were carried out in the late 1990s, mostly driven by encouraging results on the lithium–metal polymer battery employing PEO.6 Indeed, electrolytes using low-molecular-weight glymes exhibited promising characteristics in terms of chemical stability and viscosity, thereby holding the promise of an enhanced safety level as compared to that of conventional alkyl-carbonate-based solutions, along with suitable Li+ transport properties at room temperature.19,53,86 On the other hand, understanding the complex Li-electrode/electrolyte interface in glyme-based cells required several studies, which are still ongoing to fully clarify the characteristic features of such an intriguing and intrinsically safe system. Initial works opened a debate on the actual suitability of this class of electrolyte solutions in high-energy batteries owing to doubtful results on the compatibility of glyme solvents with lithium metal. Accordingly, a first comparative investigation of the Li electrode in solutions of various salts, that is, LiAsF6, LiClO4, LiTf, LiTFSI, LiBF4, LiBr and LiI, in DME, EG, and G1 solvents, with DOL, PC, EC and DMC as co-solvents, suggested poor surface chemistry and a rough morphology of the metal electrode after electrodeposition–dissolution, which lead to a low cycling efficiency in rechargeable batteries. In this regard, cyclic voltammetry provided insights into the surface films deposited in the cell, as shown in Fig. 2a, which reports a steady-state profile of a polycrystalline gold electrode in solutions using the above-mentioned solvents and LiTf as salt. Li underpotential deposition (UPD) and stripping are marked in the figure along with the non-Faradaic region.16 However, other studies demonstrated promising characteristics of glyme-based solutions of LiTFSI (Gn with n from 1 to 4), revealing for this salt high compatibility with various solvents and high electrochemical stability. Notably, LiTFSI appeared strongly associated in glymes and moderately associated in TESA at low concentrations, formed stable solvates in G1 at intermediate concentrations, and displayed thermodynamic properties approaching those of molten salts at high concentrations. Fig. 2b shows the trend of specific conductivity as a function of the concentration of LiTFSI in Gn solvents. Reasonably high specific conductivity was observed for both LiTFSI and LiClO4 in various solvents, which indicated that the ionic conductance at high concentration in solvents of low dielectric constant was limited by a charge transfer process rather than by the migration of free ions.17 To verify the hypothesis of stable solvates that persist in the solution influencing its properties, phase diagrams and Raman spectra have been measured for mixtures of LiTFSI and ACN, PC or glymes (Gn, with n = 1, 4, and 10). In fact, the systems without solvates show relative intensities of the solvent and salt Raman bands which are proportional to the concentration. On the other hand, important changes in the relative intensities of these bands reflect the presence of stable solvates in the electrolyte, which are additionally detected in the phase diagrams. Moreover, X-ray crystallography reveals free ions, SSIPs and CIPs in the solutions, suggesting that no stable solvates are formed in (Gn)x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LiTFSI electrolytes for n > 2.18 The interplay between the lithium-ion solvation state, ionic conductivity, and charge–discharge cycling efficiency of the Li–metal anode was further investigated. In particular, a ternary mixed solvent consisting of Gn (n from 1 to 4), EC, and EMC, dissolving LiPF6 (1 M), was comparatively studied. These glyme solutions exhibited higher conductivity and higher lithium cycling efficiency than EC/EMC, while both the conductivity and viscosity typically increased as the ethylene-oxide chain length decreased (decrease in n). Notably, this decrease in viscosity was associated with a change in the lithium-ion solvation structure, which occurred when a glyme was added to EC/EMC and was caused by a selective solvation of the glyme with respect to lithium ions as demonstrated by 13C-NMR measurements. The lithium cycling efficiency value depended on the charge–discharge current (Ips), and the ethylene-oxide number, n, affected this trend. In particular, when n increased there was a decrease in the Ips exhibiting the maximum value of efficiency (Effmax, see Fig. 2c). A similar change in conductivity with n was observed (see Fig. 2c). Among the various glymes taken into account, G2 or G3 exhibited very promising characteristics, namely high conductivity and suitable charge–discharge cycling behavior at a high current.19 In a subsequent report,20 the crystal structures of glyme solvates with LiTFSI and LiBETI were determined, and order–disorder solid phase transitions in many of the solvates were identified. Further work was carried out to shed light on the molecular interactions in glyme-based solutions. Accordingly, phase diagrams of mixtures of Gn (n = 1, 2, 3, and 4) and LiBETI, LiAsF6, LiI, LilO4, LiBF4, LiTf, LiBr, LiNO3, and LiCF3CO2 were proposed, and the relationships between the ionic association strength of the salt (that is, anion characteristics), chain length of the glyme, solvate formation, and thermophysical properties of the mixture were investigated, thereby providing a comprehensive model of solvate formation and ionic interactions in these electrolyte systems. Fig. 2d illustrates an approximate ordering for increasing the ionic association strength of LiX salts in glymes.21 Glymes were suggested to form solid complexes with lithium salts, which would be suitable as “soft” solid electrolytes for Li batteries exhibiting a wide range of ion transport properties. Thus, complexes between LiAsF6 and Gn (n = 3 and 4) showed significantly different cation transference numbers, resulting from the presence of channels for Li+ migration in G3 and from weak binding of AsF6− in the structure of G4.22 Considerable efforts were devoted to understanding the ion–solvent arrangements in the glyme-based electrolyte solutions. For instance, 19F NMR spectroscopy and conductivity data were collected to determine the ion pair formation constants of LiTFB and LiTf solutions in mixtures of DOL and G1, G2, or water, and the obtained results were interestingly similar to those of liquid PEGDME and solid PEO.23 Furthermore, the formation constants of ionic pair solutions of lithium salts in G1, G2, and G3 were estimated using 7Li, 11B and 19F NMR analyses, which demonstrated that even in solvents with very similar coordination (in terms of the donor and acceptor number values) and dielectric properties, the ionic pair formation constant depended on effects related to ion agglomerate formation, non-covalent interactions between ions and the liquid matrix, as well as the number of interacting centers in the solvent molecules.24
:LiTFSI electrolytes for n > 2.18 The interplay between the lithium-ion solvation state, ionic conductivity, and charge–discharge cycling efficiency of the Li–metal anode was further investigated. In particular, a ternary mixed solvent consisting of Gn (n from 1 to 4), EC, and EMC, dissolving LiPF6 (1 M), was comparatively studied. These glyme solutions exhibited higher conductivity and higher lithium cycling efficiency than EC/EMC, while both the conductivity and viscosity typically increased as the ethylene-oxide chain length decreased (decrease in n). Notably, this decrease in viscosity was associated with a change in the lithium-ion solvation structure, which occurred when a glyme was added to EC/EMC and was caused by a selective solvation of the glyme with respect to lithium ions as demonstrated by 13C-NMR measurements. The lithium cycling efficiency value depended on the charge–discharge current (Ips), and the ethylene-oxide number, n, affected this trend. In particular, when n increased there was a decrease in the Ips exhibiting the maximum value of efficiency (Effmax, see Fig. 2c). A similar change in conductivity with n was observed (see Fig. 2c). Among the various glymes taken into account, G2 or G3 exhibited very promising characteristics, namely high conductivity and suitable charge–discharge cycling behavior at a high current.19 In a subsequent report,20 the crystal structures of glyme solvates with LiTFSI and LiBETI were determined, and order–disorder solid phase transitions in many of the solvates were identified. Further work was carried out to shed light on the molecular interactions in glyme-based solutions. Accordingly, phase diagrams of mixtures of Gn (n = 1, 2, 3, and 4) and LiBETI, LiAsF6, LiI, LilO4, LiBF4, LiTf, LiBr, LiNO3, and LiCF3CO2 were proposed, and the relationships between the ionic association strength of the salt (that is, anion characteristics), chain length of the glyme, solvate formation, and thermophysical properties of the mixture were investigated, thereby providing a comprehensive model of solvate formation and ionic interactions in these electrolyte systems. Fig. 2d illustrates an approximate ordering for increasing the ionic association strength of LiX salts in glymes.21 Glymes were suggested to form solid complexes with lithium salts, which would be suitable as “soft” solid electrolytes for Li batteries exhibiting a wide range of ion transport properties. Thus, complexes between LiAsF6 and Gn (n = 3 and 4) showed significantly different cation transference numbers, resulting from the presence of channels for Li+ migration in G3 and from weak binding of AsF6− in the structure of G4.22 Considerable efforts were devoted to understanding the ion–solvent arrangements in the glyme-based electrolyte solutions. For instance, 19F NMR spectroscopy and conductivity data were collected to determine the ion pair formation constants of LiTFB and LiTf solutions in mixtures of DOL and G1, G2, or water, and the obtained results were interestingly similar to those of liquid PEGDME and solid PEO.23 Furthermore, the formation constants of ionic pair solutions of lithium salts in G1, G2, and G3 were estimated using 7Li, 11B and 19F NMR analyses, which demonstrated that even in solvents with very similar coordination (in terms of the donor and acceptor number values) and dielectric properties, the ionic pair formation constant depended on effects related to ion agglomerate formation, non-covalent interactions between ions and the liquid matrix, as well as the number of interacting centers in the solvent molecules.24
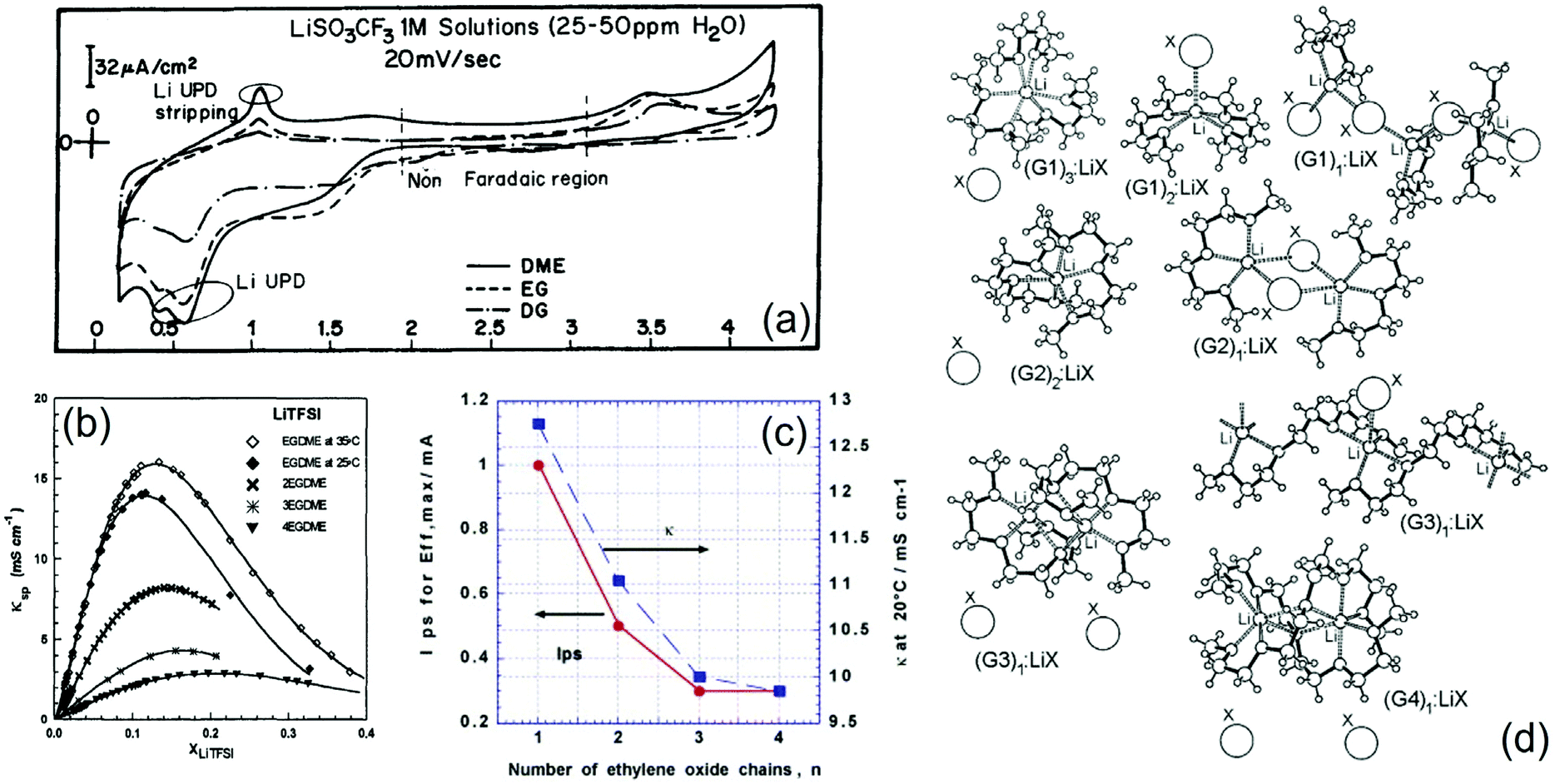 | ||
| Fig. 2 (a) Typical steady-state voltammograms obtained from polycrystalline gold electrodes in 1 M LiTf solutions of G1 (DME), ethyl glyme (EG) and G2 (DG). Scan rate: 20 mV s−1. Li underpotential deposition and stripping peaks and the non-Faradaic region are marked. Reproduced with permission.16 Copyright © 1996 Elsevier Science Ltd. (b) Specific conductivity (κ) at 25 °C of LiTFSI in glymes, that is, DME (EGDME; κ at 35 °C is also shown), G2 (2EGDME), G3 (3EGDME), and G4 (4EGDME). Reproduced with permission.17 Copyright © 1998 Plenum Publishing Corporation. (c) Relationship between electrolyte conductivity (κ) and charge–discharge cycling current (Ips) for the maximum value of cycling efficiency of lithium (Effmax) and n for 1 M LiPF6–EM80/nG20, where EM100−x/nGx represents the mixed solvent of EC/MEC (3:7) and n-glyme (mixing volume ratio = 100 − x:x), with plating charge (Qp) of 0.025 mA h. Reproduced with permission.19 Copyright © 2003 Elsevier Ltd. All rights reserved. (d) Schematic illustrations of various glyme–LiX solvate structures with G1, G2, G3, and G4. Reproduced with permission.21 Copyright © 2006 American Chemical Society. | ||
Therefore, glymes are proposed for LIBs because of the promising features of PEO-based electrolytes, with a stability depending on the specific glyme–salt (G–S) combination. In particular, the salt concentration appears to remarkably influence the electrolyte characteristics in terms of the solvation ability, dissociation degree, and ionic conductivity. The formation of stable G–S solvates is hypothesized and preliminarily verified for specific compositions, while it is excluded for others. Furthermore, the Li/electrolyte interphase characteristics and the charge/discharge efficiency apparently depend on the adopted G–S combination, due to the effects of the electrolyte viscosity and the structure of the ion–solvent species. Possibly, the better efficiency is ascribed to electrolytes formed by using glymes with increased chain lengths (i.e., with increasing n in Gn). In addition, phases with order–disorder structures are observed in some S–G solvates and suggested to affect the ion association degree of the salts, thus modifying the performance of the electrolyte in lithium cells.
3. Li-Metal batteries using “glyme electrolytes” and insertion/intercalation electrodes
Glyme-based electrolytes have shown promising performances in lithium–metal batteries using insertion cathodes. A Li|LiCoO2 cell employing the [Li(G4)][CTFSI] complex was assembled and tested, revealing a stable galvanostatic response during 50 cycles. LiCTFSI may form solid and liquid complexes with G3 and G4, respectively. Notably, the electrolyte was stable in a wide potential range from 0 to 4.5 V vs. Li/Li+ and had a much higher thermal stability as compared to that of pure G4, whilst the vapor pressure of [Li(G4)][CTFSI] was negligible at temperatures lower than 100 °C. Furthermore, the [Li(G4)][CTFSI] complex exhibited an ionic conductivity of 0.8 mS cm−1 at 30 °C, which was slightly lower than that of conventional alkyl-carbonate-based electrolyte solutions and higher than that of the [Li(G3)][CTFSI] complex (see Fig. 3a), in spite of a relatively high viscosity due to the high molar concentration (ca. 3 mol dm−3). The pulsed gradient spin-echo NMR (PGSE-NMR) method was employed to measure the self-diffusion coefficients of Li+ cations, CTFSI− anions, and glyme molecules, and the ionicity (dissociativity) of [Li(G4)][CTFSI] at 30 °C was estimated to be ca. 0.5.43 Another example showed that increasing the amount of glyme in G4–LiTFSI complexes (where the molar ratio of G4 ranged from 40 mol% to 60 mol%) decreases the viscosity and increases the ionic conductivity, thereby improving the rate capability of Li|LiCoO2 cells. In addition, Li|graphite cells with SEI forming additives, such as VC, VEC, and 13PS, showed a cycling performance comparable to that of conventional carbonate-based electrolytes. Lithium cells using Li4Ti5O12 and LiFePO4 along with the [Li(G4)][TFSI] complex exhibited excellent reversibility due to an optimal working voltage range.44 Besides, an electrolyte formed by G3 and LiFSI in a 1:1 molar ratio showed relatively high thermal stability and stable cycling using LiFePO4 and graphite electrodes, with 82% of capacity retention at 100 cycles.45
 | ||
| Fig. 3 (a) Temperature dependence of the ionic conductivity of [Li(G3)][CTFSI] and [Li(G4)][CTFSI] glyme–Li salt complexes. Reproduced with permission.43 Copyright © 2009 Elsevier B.V. All rights reserved. (b and c) Linear sweep voltammograms of [Li(glyme)x][TFSI] complexes (x = 1, 4, 8, and 20) at a scan rate of 1 mV s−1 at 30 °C, where the glyme is (b) G3 and (c) G4. Each inset depicts a magnification of current density. Adapted with permission (https://pubs.acs.org/doi/10.1021/ja203983r).46 Copyright © 2011 American Chemical Society. Further permission related to the material excerpted should be directed to the American Chemical Society. (d and g) Electrochemical behavior of a Li|G3–LiTFSI|LiFePO4 cell in terms of (d) voltage profiles at the 1st, 50th, 100th, 200th, 300th, 400th, 500th, and 600th cycle and (g) trend of charge and discharge capacities per positive electrode as a function of the cycle number; temperature: 30 °C; voltage range: 2.5–4.0 V; and current rate: C/8. (e and h) Electrochemical behavior of a Li|G3–LiTFSI|LiNi1/3Mn1/3Co1/3O2 cell in terms of (e) voltage profiles at the 1st, 10th, 50th, 100th, 200th, 300th, and 400th cycle (voltage range: 2.7–4.2 V) and (h) trend of discharge capacity per positive electrode as a function of the cycle number for various upper charge cutoff voltages (4.2 V, 4.4 V, and 4.6 V): temperature: 30 °C; current: C/8. Adapted with permission.48 Copyright © 2013 The Authors. Published by Elsevier B.V. CC BY-NC-ND license. (f and i) Electrochemical behavior of the Li|LiTf–LiNO3–G4|LiMn0.5Fe0.5PO4 cell in terms of (f) voltage profiles and (i) trend of charge and discharge capacities per positive electrode and coulombic efficiency as a function of the cycle number; temperature: 25 °C; voltage range: 2.0–4.3 V; and current rate: C/5 rate (1C = 170 mA g−1 with reference to the cathode mass); test performed after an electrochemical activation of the cell. Reproduced with permission.53 Copyright © 2016 Elsevier B.V. All rights reserved. | ||
A literature work suggested that the oxidative stability of glyme molecules can be enhanced by complex formation with alkali metal cations, such as [Li(Gn)1][TFSI] with n = 3 and 4, which were classified as a room-temperature SILs consisting of a [Li(Gn)1]+ complex cation and a TFSI− anion due to their liquid state maintained over a wide temperature range and a high self-dissociativity (ionicity) at room temperature. The increase in salt concentration in the [Li(Gn)1][TFSI] equimolar SIL remarkably enhanced the oxidative stability of the solution to a potential as high as 5 V vs. Li+/Li, likely owing to the donation of lone pairs of ether oxygen atoms to the Li+ cation, which resulted in the lowering of the HOMO of the glyme as suggested by ab initio molecular orbital calculations. On the other hand, [Li(Gn)x][TFSI] solutions with x > 1 were stable up to 4 V vs. Li+/Li+ (see Fig. 3b and c). NMR data indicated Li+ transport in the equimolar complex via migration of the [Li(G3)1]+ solvate, although the ligand exchange mechanism occurred as a result of the electrochemical reaction at the electrode/electrolyte interface in the Li|[Li(G3)1][TFSI]|LiCoO2 cell. This battery exhibited a steady behavior for more than 200 charge–discharge cycles with a voltage range of 3.0–4.2 V.46 Further studies of the Li|LiCoO2 cell using molten [Li(Gn)1][TFSI] equimolar complexes were carried out to elucidate the relationship between the Li+ limiting current density under one-dimensional finite-diffusion conditions and the rate capability. Voltage drops and incomplete discharge of the cell were observed when the applied current was higher than the limiting current density, which may reflect a depletion of the lithium salt in proximity to the cathode or saturation at the anode interphase. Comparative tests of single-particle LiCoO2 and an electrode sheet in a typical electrolyte solution based on PC, in a binary LiTFSI-(DEME)TFSI ionic liquid, and in a [Li(Gn)1][TFSI] molten complex suggested that the Li+ transport in the solution controls the rate capability of the cells using the latter electrode.47 Notably, the [Li(G3)][TFSI] complex demonstrated a very promising performance in lithium–metal cells using LiFePO4 and LiNi1/3Mn1/3Co1/3O2 cathodes. The battery using the former positive electrode operated at about 3 V (Fig. 3d), delivering a rather stable capacity over 600 charge–discharge cycles (Fig. 3g), whilst that using the latter had a working voltage of about 4 V (Fig. 3e) and exhibited a cycling trend strongly depending on the upper voltage cutoff (Fig. 3h). Accordingly, the cell charged up to 4.2 V showed a capacity retention approaching 60% after 400 cycles. Further tests revealed the formation of a favorable Li/electrolyte interphase, thereby suggesting the possible applicability of this electrolyte formulation in lithium batteries.48 In this regard, EQCM-A was effectively employed to measure the product of the viscosity (ηL) and density (ρL) of the electrolyte near the electrode surface, i.e., ηLρL, along with changes in mass. The collected data showed a decrease in the ηLρL value during lithium deposition and a sharp increase during lithium dissolution, which were ascribed to changes in the concentration and dissolved state of Li+ in proximity to the electrode. The latter increase with the Li dissolution may adversely affect the cation transference number, leading to a decrease in the anodic current in the battery.49 Highly concentrated electrolytes have been extensively investigated due to their suitable properties for use in lithium metal cells with insertion cathodes. A comparative study of molten mixtures of LiTFSI and various ether solvents (THF, G1, G2 and G3), where the ratio of ether-oxygen atoms to Li+ (i.e., O/Li) was fixed at four, revealed that the capacity of the Li|LiCoO2 cell with [Li(THF)4][TFSI] dramatically decreased during cycling, whereas a similar cell employing [Li(G3)1][TFSI] displayed a stable behavior with a coulombic efficiency higher than 99% over 100 cycles. These results were related to the oxidative decomposition of the solvents and a persistent Al corrosion occurring in [Li(THF)4][TFSI] and [Li(G1)2][TFSI], which contain shorter ethers, whilst the use of [Li(G3)1][TFSI] ensured effective suppression of the side reactions.50 The literature also suggested that the ionic conductivity of glyme–LiTFSI solvate ionic liquids (SILs) may be inversely proportional to the viscosity, in agreement with Walden's rule. Thus, a decrease in ionic conductivity with increasing concentration of LiTFSI was observed and associated with the formation of a bulky lithium species, i.e., [Li(TFSI)2]−.51 The [Li(G4)][TFSI] complex was employed in a quasi-solid-state electrolyte with fumed silica nanoparticles. This electrolyte was used in double-layered and triple-layered high-voltage bipolar stacked batteries, which showed a working voltage of 6.7 and 10.0 V, respectively, that is, two and three times that of the single-layered device (3.4 V). The double-layered device showed a capacity retention of 99% after 200 cycles at C/2.52
The characteristic features of the positive electrodes have crucial effects on the cell performance as they may influence the working voltage, the properties of the electrode/electrolyte interphase, and the rate capability. Accordingly, an electrolyte formed by dissolving 1 mol LiTf and 1 mol LiNO3 in 1 kg of G4 exhibited different behaviors in lithium–metal cells employing LiFePO4 and LiMn0.5Fe0.5PO4 cathodes. The cell using LiFePO4 operated at 3.5 V with the capacity ranging from 150 mA h g−1 at C/10 to 110 mA h g−1 at 2C, while the one employing LiMn0.5Fe0.5PO4 showed two plateaus at 4.1 V and 3.5 V (Fig. 3f) with the capacity ranging from 160 mA h g−1 at C/10 to 75 mA h g−1 at 2C, and a stable value of about 125 mA h g−1 for 100 cycles at C/5 (Fig. 3i), where both the specific capacity and current rate were with reference to the mass of the cathode. Notably, the higher working voltage of LiMn0.5Fe0.5PO4 as compared to that of LiFePO4 may have a detrimental effect on the coulombic efficiency of the battery.53
We remark that the presence of LiNO3 in the formulation significantly affects the stability of the electrode/electrolyte interface, as demonstrated in two consecutive studies.54,55 Indeed, the cycle life of Li|LiFePO4 cells with PEGDME (MW 500 g mol−1) dissolving LiTf was significantly enhanced by the addition of LiNO3 to the electrolyte solution, leading to a stable performance over 60 cycles with a capacity with reference to the cathode of 150 mA h g−1 and a flat working voltage of 3.5 V, which were reflected as a theoretical energy density of 520 W h kg−1 (normalized to the mass of the positive electrode). Furthermore, PFG-NMR measurements suggested suitable ionic conductivity, lithium transference number, ionic-association degree, and self-diffusion coefficient for energy-storage application. It is worth mentioning that the high thermal stability of this electrolyte may remarkably improve the safety of the lithium–metal battery.54 In agreement with the above described results, a G3–LiTf solution was upgraded by adding LiNO3, which enhanced the electrode/electrolyte interface and widened the electrochemical stability window of the solution, thus enabling its application in a battery with the LiFePO4 cathode. An electrochemical activation procedure of this cell leading to the formation of stable interfaces at the electrode surface was optimized and thoroughly investigated. Fig. 4 shows the related voltage profiles (a) and the corresponding SEM images of the LiFePO4/electrolyte interphase upon cycling (b).55 Unsymmetrical glymes having different end groups were also studied (Fig. 4c). These glymes had ethyl and butyl end groups and were used to prepare liquid solvates with LiTFSI and LiFSI (glyme:salt ratios of 1:1, 2:1, and 3:1), which exhibited high ionic conductivity at room temperature (on the order of 10−3 S cm−1), were stable up to 4.5 V, and ensured a steady galvanostatic performance with LiFePO4, with a capacity of 145 mA h g−1 (with reference to the cathode) and good rate capability up to 2C at room temperature. Furthermore, preliminary cycling tests with NMC suggested possible applicability in high-voltage batteries and DSC studies indicated a low crystallization temperature between −60 °C and −75 °C for the LiFSI-based electrolyte.56 Therefore, both the salt and the glyme chain length may substantially affect the ion transport, the lithium/electrolyte interphase characteristics, and the electrochemical stability window, thereby determining the lithium cell response. Indeed, solutions of LiFSI, LiTFSI, or LiBETI in Gn (n = 2 and 3) had different properties depending on the selected formulation. Thus, decreasing the chain length increased the ionic conductivity from ca. 10−3 to ca. 10−2 S cm−1, despite having detrimental effects on the lithium transference number. Galvanostatic lithium stripping-deposition and EIS measurements (Fig. 4d–i) suggested that all the electrolytes may form an interphase at the metal anode suitable for a few charge/discharge cycles, with low values of polarization and rather constant resistance values. However, the use of LiBETI led to poor lithium-passivation properties over long-term cycling, whilst widening the anodic stability window to 4.6 V vs. Li+/Li. Among these various formulations, LiTFSI-based electrolytes were the most adequate for application in Li|LiFePO4 batteries that ensured a capacity between 134 and 144 mA h g−1 in galvanostatic tests with over 100 cycles at a C/3 rate (1C was 170 mA g−1; both the capacity and C-rate were with reference to the mass of LiFePO4 in the cathode).57 The electrochemical performance of the Li|LiFePO4 battery was further enhanced as described above, by adding LiNO3 to these solutions. A thorough investigation of such improved solutions showed evidence of fast ion transport, a wide stability window, suitable lithium–metal passivation, and cathode/electrolyte interphase characteristics that depended on the Gn chain length (n = 2 and 3). An optimized Li|LiFePO4 battery using a G3–LiTFSI–LiNO3 electrolyte delivered 154 mA h g−1 at C/3 without any decay after 200 cycles, as well as a retention above 70% after 500 cycles at 1C and 5C with a coulombic efficiency approaching 100% (Fig. 4j), which benefitted from a stable, ionically conductive electrode/electrolyte interphase (Fig. 4k–n).59 Relevantly, a solid composite-electrolyte based on PEGDME (MW 2000 g mol−1), dissolving LiTFSI and LiNO3, and incorporating nanometric silica (SiO2) particles was prepared by solvent casting and investigated in Li|LiFePO4 polymer batteries. This electrolyte had an ionic conductivity higher than 10−4 S cm−1 at temperatures above 40 °C after subsequent heating and cooling cycles, exhibited a lithium transference number ranging from 0.22 at 45 °C to 0.27 at 70 °C and a low resistance at the interphase with lithium metal, and was stable up to ca. 4.4 V. When used in the Li|LiFePO4 battery at 50 °C, this composite electrolyte enabled a coulombic efficiency of about 100%, a capacity retention approaching 99% after 300 cycles at a C/3 rate, and a maximum capacity of 150 mA h g−1.58 Furthermore, G2 and G3 dissolving LiTFSI and LiNO3 in concentrations approaching the solvent saturation limit were used in lithium cells employing the LiFePO4 cathode, which exhibited a promising cycling performance. Notably, an additional reduction step at a low voltage cutoff (i.e., 1.2 V) during the first discharge allowed the formation of a suitable SEI, as mentioned above (see Fig. 4a and b), thereby leading to a coulombic efficiency of ca. 100%, a capacity approaching 160 mA h g−1, and low capacity fading over cycling.60
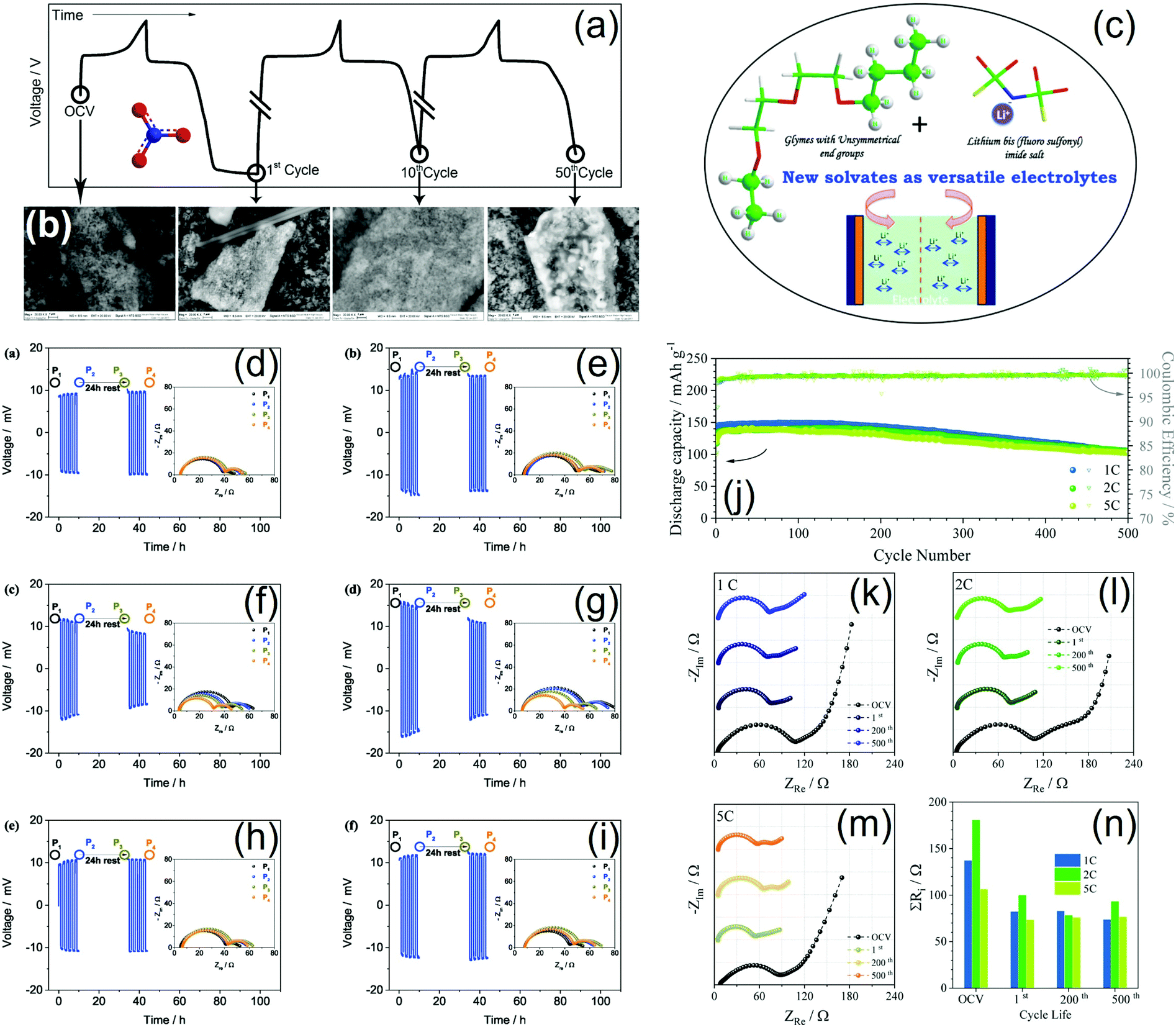 | ||
| Fig. 4 (a and b) Ex situ SEM investigation of LiFePO4 electrodes after cycling in a Li|G3–LiTf–LiNO3|LiFePO4 cell under the OCV conditions (just after cell assembly and stabilization) and after the 1st, 10th, and 50th cycles; (a) voltage profiles and (b) SEM images. First discharge performed by decreasing the voltage below 2 V at a C/5 rate and limiting the time to 5.15 h; subsequent cycles within the 2–4 V voltage range at a C/5 rate (1C = 170 mA g−1 with reference to the cathode mass). Reproduced with permission.55 Copyright © 2017 American Chemical Society. (c) Schematic exemplifying the promising properties of solvates based on glymes with unsymmetrical (ethyl and butyl) end groups as electrolytes for lithium batteries characterized by high conductivity. Reproduced with permission.56 Copyright © 2017 American Chemical Society. (d–i) Voltage profiles of lithium stripping/deposition galvanostatic cycling tests of symmetrical Li|Li cells and corresponding Nyquist plots of the EIS before the test (P1), after 5 cycles (P2), upon 24 h of rest after cycling (P3), and after additional 5 cycles (P4) for (d) G2–LiTFSI, (e) G3–LiTFSI, (f) G2–LiFSI, (g) G3–LiFSI, (h) G2–LiBETI, and (i) G3–LiBETI. Cycling test at a constant current density of 0.1 mA cm−2. Step time: 1 h; EIS carried out by applying an AC signal with an amplitude of 10 mV in the frequency range from 500 kHz to 100 mHz; temperature: 25 °C. Reproduced with permission.57 Copyright © 2019 Elsevier Ltd. All rights reserved. (j) Galvanostatic cycling trend in terms of discharge capacity and coulombic efficiency over 500 cycles of a Li|LiFePO4 cell using the LiTFSI–LiNO3–G3 electrolyte at 1C, 2C and 5C rates (1C = 170 mA g−1 with reference to the cathode mass). (k–m) Nyquist plots of EIS measurements performed on the same cell during the cycling tests at (k) 1C, (l) 2C, and (m) 5C rates; impedance spectra were recorded under the OCV conditions, after the 1st, 200th, and 500th cycles. (n) Electrode/electrolyte interphase resistance for the same cell as extracted from the EIS data of panels k–m. Reproduced with permission.59 Copyright © 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
We can reasonably evaluate the glyme-based electrolytes (in particular using G3 and G4 and PEGDME) as possible electrolyte solvents for lithium battery application, since they can have a stable electrode/electrolyte interphase, particularly when ad hoc additives are used, low volatility, and sufficient dissociation degree. In particular, efficient operation is observed with typical electrodes used in batteries such as those based on Li-intercalation (LiCoO2 and graphite layered materials) and Li-insertion (LiFePO4 olivine and Li2Ti5O12 spinel). Possible formation of SILs due to salt concentration changes is suggested to remarkably improve the oxidative stability of glyme electrolytes up to 5 V vs. Li/Li+, and the migration of G–S complexes in specific formulations is indicated to improve cell performances, depending on the current value which is the limiting factor due to the depletion of the lithium salt in the electrode proximity. New electrodes such as LiNi1/3Mn1/3Co1/3O2 and LiMn0.5Fe0.5PO4 can also be employed in lithium cells using a glyme-based electrolyte with a stability and efficiency depending on the upper voltage cutoff. All the measurements indicate that the control of the salt concentration at the electrode/electrolyte interphase during cell operation and the formation of a favorable SEI are the key factors for achieving the optimal operation of the lithium cells using insertion or intercalation electrodes. In particular, the addition of LiNO3 to the electrolyte formulation as a sacrificial film-forming agent appears to significantly improve the SEI at the electrodes, in particular using glymes having longer chains such as PEGDME and adopting suitable electrochemical activation steps. We remark again that the glyme electrolyte ionic transport, electrochemical stability and interphase characteristics are substantially governed both by the salt nature and solvent chain length, and by the specific combination and experimental setup used for allowing efficient cell operation. Therefore, targeted studies are suggested for investigating the various systems and further evaluating the actual battery applicability.
4. Lithium salts in glymes: ionic liquids or concentrated solutions?
Electrolytes formed by dissolving various lithium salts in glymes differing by the ether chain length were initially considered as simple solutions, mainly characterized by the solute concentration.16–24 Subsequently, the concept of liquid solvated salt-complexes with similar characteristics to those of ionic liquids, i.e., molten salts, was proposed.133 In this regard, Raman spectroscopy may shed light on the various solvate structures occurring in the liquid phase at room temperature, whilst PFG-NMR may reveal information on the ion transport properties that can be crucial to understanding the ion–solvent interactions. Accordingly, a literature work investigated solutions of LiFSI in G3 or G4, where the Gn:salt molar ratio was 1:1, which were identified as SILs comprising a cationic [Li(Gn)]+ complex and the FSI− anion based on Raman data. For Gn:LiFSI ratios higher than 1, anionic LixFSIy complexes were formed in addition to the cationic ones. Moreover, PFG-NMR revealed that the self-diffusion coefficients of Li+ (DLi) and glyme (Dglyme) were the same when Gn:LiFSI was 1:1, which indicated that Li+ and glyme diffuse together as a cationic [Li(Gn)]+ complex. The ratio of the self-diffusion coefficients of anions and cations, DFSI/DLi, was constant at ca. 1.1–1.3 when Gn:LiFSI was 1:1 and increased as the amount of LiFSI in the solution was raised, suggesting a change in the ion transport mechanism. Furthermore, the increase in LiFSI concentration enhanced the oxidative stability of the electrolyte and mitigated the Al corrosion.61 The local structure of Li+ ions in equimolar mixtures of glymes (G3, G4) was investigated in solutions of LiTFSI, LiBETI, LiTf, LiBF4, LiClO4, LiNO3, and LiTFA. Raman spectra and ab initio molecular orbital calculations revealed a crown-ether like conformation of the glyme molecules to form a monomeric [Li(Gn)]+ complex in the molten state. Raman spectroscopic data identified the fraction of the free glyme in the [Li(Gn)]X solutions (where X is the anion; see Fig. 5a), which was estimated to be a small percent in [Li(Gn)]X with perfluorosulfonylimide type anions. Equimolar mixtures characterized by a low concentration of free glyme were regarded as SILs, while those containing a substantial amount of free glyme were classified as concentrated solutions. It is worth mentioning that the concentration of free glyme decreases as the concentration of salt increases, leading to a notable increase in the lithium–metal electrode potential. The significantly high electrode potential in SILs suggested the presence of stable [Li(Gn)]+ complexes in the molten state.62 In another work, the [Li(G4)1][TFSI] SIL was mixed with various polymers, i.e., PEO, PMMA, and PBA, to prepare quasi-solid electrolytes for lithium batteries. The stability of the [Li(G4)]+ complex in the various solutions was assessed by comparing the ratio of the self-diffusion coefficient of glyme and Li+ ions (DG/DLi). Thus, the [Li(Gn)1][TFSI] solution and the composites based on PMMA and PBA behaved as ILs since DG/DLi = 1 (Li+ and Gn diffuse together), whilst the PEO-based electrolyte was characterized by DG/DLi > 1, indicating the existence of free G4 molecules (ligand exchange between G4 and PEO). The highly stable [Li(G4)1]+ complex (PMMA- and PBA-based solutions) led to high thermal stability, high Li+ transference number, and a wide electrochemical stability window. Among the investigated composites, the PBA-based solutions showed the lowest glass transition temperature, little affinity towards Li+ ions, and favorable lithium transport properties.63 SAXS, Raman spectroscopy, and computational modeling provided further insight into the structure of G4–LiTFSI SILs. Indeed, a peak at Q 0.95 Å−1 in the SAXS spectra indicated structural correlations of typical ILs. This peak grew in intensity as the concentration of salt increased, reaching a maximum at the equimolar ratio (G4:LiTFSI 1:1; see Fig. 5b) due to the effective solvation of each Li+ ion by one G4 molecule forming a [Li(G4)]+ complex. These data were confirmed by Raman spectroscopy analyses, which suggested the occurrence of SSIPs in the solution. However, it is worth considering that even at the equimolar concentration not all Li+ ions were solvated and minor interactions between cations and anions were observed.64 A further report suggested that changing the G3:LiTFSI ratio may significantly affect the electrode/electrolyte interphase and, therefore, the cycle life of the cell. Indeed, SILs containing excess LiTFSI ensured notable mitigation of detrimental reactions between the LiCoO2 cathode and electrolyte.65
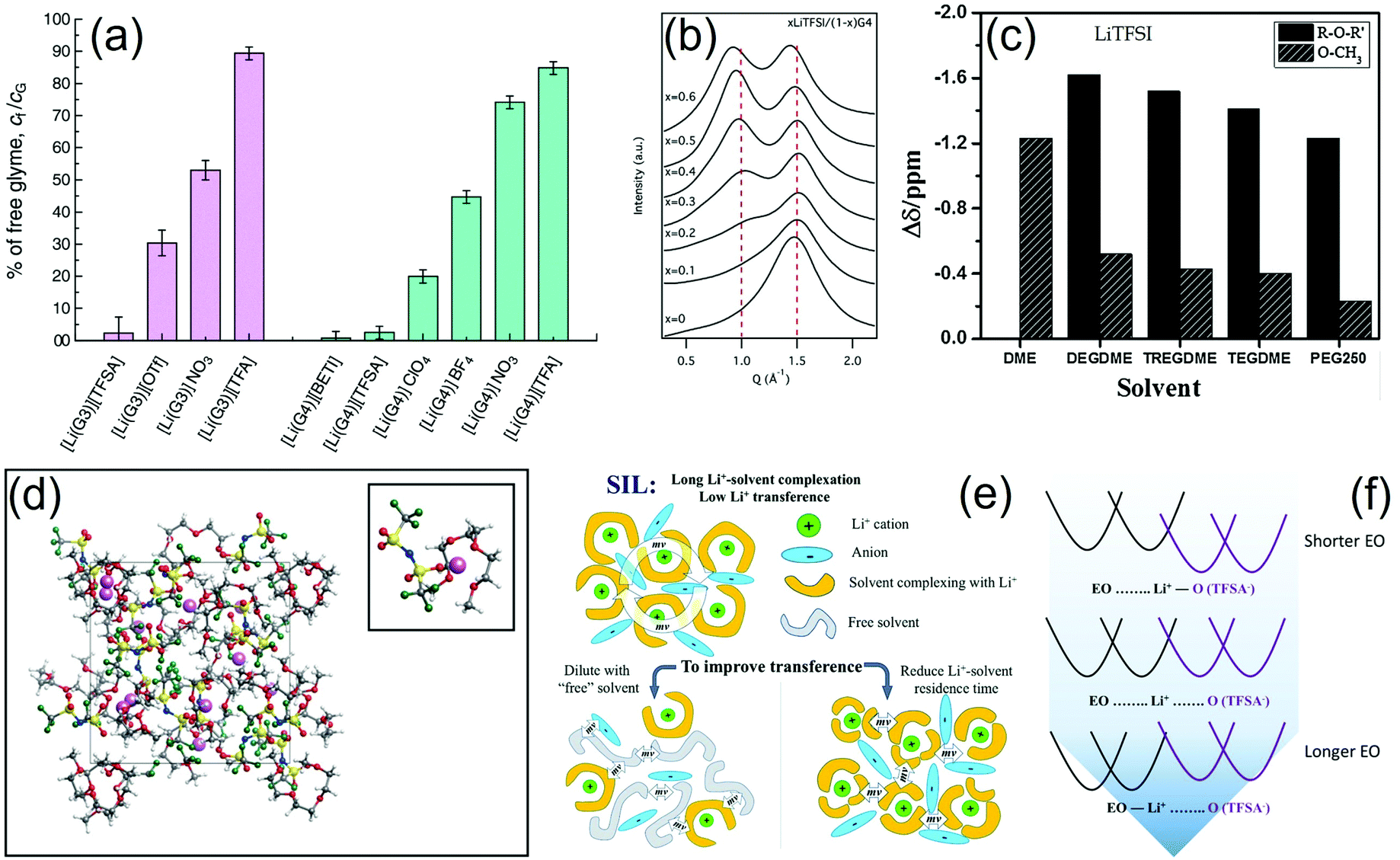 | ||
| Fig. 5 (a) Estimated percentages of free glyme (cf/cG, where cf and cG are the concentration of free glyme and the total concentration of glyme in the mixture, respectively) in equimolar molten mixtures [Li(glyme)1]X at 30 °C (X is TFSI, Tf, NO3, TFA, BETI, TFSA, ClO4, and BF4). Reproduced with permission.62 This journal is © the Owner Societies 2015. (b) SAXS diffraction patterns of different mole fractions of LiTFSI dissolved in G4. The dotted vertical lines represent guidelines to the eye. Reproduced with permission.64 Copyright © 2015 Elsevier B.V. All rights reserved. (c) Chemical shift difference of 17O resonance of glyme between Li salt solution and pure solvent (Δδ = δelectrolyte − δsolvent) as determined by NMR experiments. Adapted with permission.68 Copyright © 2016 Elsevier Ltd. All rights reserved. (d) Snapshot of the structure for the LiTFSI–G3 electrolyte solution; CIP in which a Li ion is solvated by one G3 molecule and one TFSI anion; the inset shows the representative solvation structures of the Li ion. Adapted with permission.69 Copyright © 2016 American Chemical Society. (e) Schematic illustration of an SIL with a low Li+ transference number due to momentum conservation constraint and two possible strategies to facilitate the momentum exchange in the electrolyte and to increase Li+ transference. Reproduced with permission.73 This journal is © the Owner Societies 2018. (f) Schematic illustration of the potentials of mean force (PMF = −RTlng(r), where g(r) is the pair correlation function for the Li–O atom pair) in glyme–Gn solutions, suggesting the predominance of the CIP formation for short glyme chains (n < 4) and preference in the Li+–solvate complexes for long glyme chains (n > 4). Reproduced with permission.78 Copyright © 2019 American Chemical Society. | ||
Glyme-based SILs typically exhibit a high viscosity, which may adversely impact the ion mobility, so various solvent additives able to enhance the conductivity of the solution have been studied. Among the diluents proposed so far, HFE demonstrated suitable properties for use in batteries. Hence, the liquid structure of HFE diluted [Li(G4)]1TFSI was investigated combining Raman spectroscopy and DFT data, which suggested that the additive does not directly coordinate the Li+ ion. SSIPs along with monodentate and bidentate CIPs were found in the neat and HFE diluted [Li(G4)]1[TFSI], and the monodentate CIP decreased whilst the SSIP increased when diluting with HFE. HEXTS experiments were performed and supported by data of MD simulations, where the intermolecular force-field parameters, mainly partial atomic charges, were newly proposed for the HFE and glymes. A new peak ascribed to the correlation between the [Li(G4)][TFSI] ion pairs was found at ca. 0.6–0.7 Å−1 in the X-ray structure factors, and it was suggested that the terminal oxygen atoms of G4 in the [Li(G4)]+ complex frequently repeat coordinating/uncoordinating, although almost all of the G4 molecules coordinate the Li+ ions.66 The effects of dilution with various solvents on the viscosity and the ionic conductivity of [Li(Gn)]1[TFSI] molten complexes were further studied. Nonpolar solvents, such as toluene, DEC, and HFE, formed stable [Li(G3)]+ and [Li(G4)]+ solvates, whilst ligand exchange indicating competitive solvation took place between glyme and polar solvents, such as water and PC. On the other hand, ACN exhibited intermediate properties, as it participated in the lithium solvation to form mixed [Li(G3)(ACN)]+ and [Li(G4)(ACN)]+ complexes. Furthermore, [Li(G4)][TFSI] was found to be more conductive than [Li(G3)][TFSI] when diluted with nonpolar solvents due to higher ionic dissociativity.67
Natural abundance 17O NMR spectroscopy provided information on the solvation behavior of LiTf and LiTFSI in glymes with different chain lengths, that is, Gn with n = 1, 2, 3, and 4 and PEGDME with MW of 250 and 500 g mol−1. The chemical shifts of the glyme oxygen in the solutions and in the neat solvents were compared, revealing a more pronounced effect of the salt addition on the chemical shift of ether oxygens compared to that of terminal oxygens, which suggested a preferential coordination of Li+ with the ether oxygen. The NMR data showed a mitigation of the chemical-shift changes as the glyme chain length increased (see Fig. 5c), which was attributed to the increased number of ether oxygens coordinating each Li+. Moreover, the chemical shift of anion oxygen suggested that the long chains may decrease the ion association.68 The solvation structure of lithium ions in the G3–LiTFSI was determined by performing MO and MD simulations based on DFT. According to these analyses, the Li+ ions in the equimolar mixture were solvated mainly by crown-ether-like curled G3 molecules and in direct contact with a TFSI− anion (see the snapshot in Fig. 5d). The aggregate formed with Li+ and TFSI− anions and/or G3 chains was equally stable, thereby suggesting that a small fraction of cations may form aggregates.69 Several characteristics of the anion, such as its flexibility and its ligand properties, influence the solvate structure, as demonstrated in a computational investigation of glyme-based solutions of LiTFSI and LiTDI. The related results revealed that TFSI− anions favor the formation of SILs, whilst TDI− anions preferably give rise to ionic aggregates. In particular, the latter anions ensured the presence of “free” cationic species even at extremely high salt concentration.70 Important parameters may be extracted from PGSE-NMR measurements to provide a large dataset describing various electrolyte solutions. For example, a comparative analysis of several systems formed by dissolving LiPF6, LiBF4, LiTFSI, LiBETI, LiBOB, LiTf or Li2DFB in PC, EC, GBL, DEC, Gn (where n = 2, 3, 4, and 5), or PEGDME of average MW of 400 and 1000 g mol−1 was carried out by plotting the Li+ and anion diffusion constants (DLi and Danion) versus the ionic conductivity (σ). The Nernst–Einstein (NE) relationship was employed to calculate the degree of apparent ion dissociation (α) from the DLi, Danion and σ parameters. Furthermore, the apparent lithium transference number (tLi) was determined from DLi and Danion, and the number of charge-carrying ions (Ncarrier) was estimated from α and the salt concentration. Additional relationships were considered, such as σ vs. α, σ vs. Ncarrier, and σ vs. tLi.71 The Li+ transport in glyme-based electrolytes was also investigated by comparing the characteristic features of various solutions, that is, 1.0 M LiTf, LiTFSI, or LiFSI in G4 as well as 0.5, 2.0, or 2.7 M LiTFSI in G4. Ionic conductivity (σ), viscosity (η), and density were measured, and the self-diffusion coefficients (D) of Li+, anions, and solvent were determined via PGSE-NMR. This study suggested that the mobility (μ) in the solution is controlled by the salt, as the Lewis basicity and hardness of anions affect the Li+–anion and Li+–G4 interactions, as well as η and D. The interaction energies (ΔE) determined by DFT calculations based on the supermolecule method were found to be in the order LiTf > LiTFSI > LiFSI, which is consistent with the dissociation degree of these salts in solutions. The study demonstrated that increasing μ and the number of carrier ions (n) is an effective way to enhance σ for glyme-based electrolytes with low dielectric constants. In this regard, suitable properties may be achieved using DME.72 VLF-EIS analyses of Li|Li symmetrical cells and MD simulations using atomistic, polarizable force field provided additional insight into the Li+ transport of equimolar SILs, enabling the calculation of the three Onsager coefficients. Accordingly, a recent study of a G4–LiTFSI mixture (1:1 ratio) showed that even though the ionic conductivity and the Li+ transport number (i.e., the transport parameter extracted from PFG-NMR data) were acceptably high, the Li+ transference number under anion-blocking conditions was extremely low. This observation was related to the strong complexation of Li+ by glyme molecules, which leads to a long residence time of the solvent near the cation and causes significant anti-correlation of cation and anion motion due to the constraint of momentum conservation (see Fig. 5e). Moreover, it is worth mentioning that any transport number determined by PFG-NMR would substantially differ from the relevant electrochemical transference number in the presence of significant ion pairing. We also remark that strongly anti-correlated motion of the cation and anion is typical of ionic liquids and, according to the structural model described above, glyme molecules complexed with Li+ cannot ensure momentum exchange between ions. Therefore, reducing the residence time between Li+ and solvent molecules or diluting the solution (adding excess solvent) may increase the Li+ transference number, as depicted in Fig. 5e.73 On the other hand, an unusual LiTFSP salt in a salt-in-glyme-based (salt-in-solvent) electrolyte solution exhibited predominant Li+ conductivity with very high lithium transference numbers (70% from the polarization experiments), as well as an ionic conductivity that was three times higher than that of a solution of LiTf in G2. This salt-in-solvent electrolyte was studied by PFG-NMR and EIS in symmetrical Li|Li cells, which suggested a suitable lithium-conduction behavior for applications in a battery, as ascribed to the reduced mobility of large, solvated anions along with improved ionic dissociation.74
Recently, FPMD simulations identified the solvate structure in G4–LiTFSI electrolyte solutions. If the salt and solvent were in an equimolar ratio, the simulations showed a positive correlation between the total coordination number of Li+ ions and the phase stability. At the ground state of the equimolar G4–LiTFSI mixture, curled G4 molecules and TFSI− anions coordinated most of the Li+ ions through 4 and 1 O atoms, respectively, and [Li(G4)1][TFSI] was the second most stable CIP in gas-phase cluster calculations. If the concentration of LiTFSI was low, Li+ ions were not in direct contact with TFSI− anions and coordinated by two G4 molecules. As expected, pairing between Li(G4)+ and TFSI− was likely to occur at an equimolar ratio between the salt and solvent, leading to properties typical of ionic liquids.75 Cell temperature and salt concentration were shown to have effects on the ionic conductivity and the Li+ transference number of liquid–solid, glyme-based electrolytes containing nanoporous alumina. Adsorption of the anion on the surface of the alumina particles had a beneficial impact on the cation transference number, mostly at lower salt concentration and elevated temperature. Even though the lithium transference number was high, unfavorable mossy lithium deposits were observed.76 In this regard, the performance of the lithium anode was investigated also by comparing the effect of the concentration, ether chain length, molecular structure, and electrolyte formulation of various solutions, namely, Gn–EC–LiFSI (where n = 1, 2, 3, and 4), DOL–EC–LiFSI and DEC–EC–LiPF6. Conventional carbonate-based electrolytes using LiPF6 gave rise to needle-like lithium dendrites, whereas high-concentration ether-based solutions favored knot-like and rounded lithium structures. Enhanced Li+–solvent interactions and fewer free molecules were found in high-concentration EC-based electrolytes. These latter solutions showed fewer side reactions with the lithium-metal anode along with significant mitigation of the dendrite formation.77 A recent study suggested a dynamic chelating effect, which was found to be related to solvent exchange and/or contact ion-pair formation/dissociation, having significant effects on Li+ transport. Data on the pair correlation functions for the Li–O(Gn) and Li–O(TFSI−) pairs enabled to propose the PMF trend depicted in the schematic in Fig. 5f, which suggested the predominance of CIP and Li+–glyme complex formation for short-chain and long-chain solvents, respectively. According to this work, G4 had intermediate characteristics, showing solvent exchange and CIP association/dissociation at a similar rate, which ensured high conductivity and low viscosity.78 In agreement with these observations, a comparison of NMR diffusion and EIS data on glyme-based solutions of LiPF6 revealed stronger ion pairing with decreasing glyme chain length. Furthermore, a decrease in the dielectric constant of the solvent with increasing temperature was suggested to further increase the ion association, thereby adversely affecting the ionic conductivity.134
As mentioned above, the lithium transference number in glyme–salt mixtures is influenced by ion–ion anticorrelations. This aspect was further demonstrated by eNMR measurements, which revealed the migration of the species forming G4–LiTFSI or G4–LiBF4 electrolytes in an electric field. Relevant data on the electrophoretic mobility and the self-diffusion coefficients for the 1H, 7Li, and 19F nuclei provided insight into transference numbers, effective charges, and ionicities in various solutions characterized by a different salt concentration. In agreement with other reports, eNMR suggested that the G4 molecule migrates along with the cation due to the formation of a stable solvate complex. Furthermore, NMR showed an increase in effective charges as the solution approached the equimolar ratio between glyme and salt and a notable difference in effective charge for lithium and anions, as well as between the G4–LiTFSI and G4–LiBF4 systems. A schematic representation of the ion–ion and ion–solvent interactions in these solutions as a function of concentration and salt composition is reported in Fig. 6a. Anticorrelations between solvate cations and the anionic complexes due to momentum conservation were identified as a crucial phenomenon possibly affecting the behavior of these mixtures to a different extent, depending on the employed electrolyte formulation. In this regard, high salt concentration along with the use of smaller anions that may form large asymmetric clusters may lead to strong anticorrelation.79 Therefore, a clear understanding of the solvate structure may elucidate the Li+ transport mechanisms in highly concentrated solutions, thereby providing crucial insight for electrolyte design. Concerning this point, a study of speciation in concentrated Gn–LiX and aqueous electrolyte systems identified the solvent activity and the activity coefficient in the gas phase at equilibrium with the solution as suitable parameters to classify the mixtures as SILs or superconcentrated solutions. Thus, analyses performed with a Raman/IR spectral analysis technique, which may reveal the free-solvent concentration, showed that the solution can be regarded as an SIL when the activity coefficient (f) is lower than 0.01. Accordingly, electrolyte solutions with the compositions of (H2O)1−x–LiTFSIx, (G3)1−x–LiTFSIx, and G3–LiX (where X is NO3−, TFA−, or TFSI−) can be classified as shown in Fig. 6b, by reporting f as a function of x, that is, the salt molar fraction.80 As mentioned, the Li+ transference number estimated using the potentiostatic polarisation method of typical SILs, such as the G4–LiTFSI equimolar mixture, is considerably lower than the corresponding transport number estimated via PFG-NMR measurements of the self-diffusion coefficient. This experimental evidence was interpreted by considering the dynamic ion correlations (i.e., cation–cation, anion–anion, and cation–anion cross-correlations) in the Onsager transport formalism, which describes the ionic conductivity σion as given by eqn (1):
| σion = σ++ + σ−− − 2σ+− | (1) |
| σdistinct++ = σ++ − σself+ and σdistinct−− = σ−− − σself− | (2) |
| σion = σself+ + σdistinct++ + σself− + σdistinct−− − 2σ+− | (3) |
 | ||
| Fig. 6 (a) Schematic representation of speciation: suggested dominant ion clusters in the low and high salt concentration regimes for [Li(G4)x][TFSI] (top) and [Li(G4)x][BF4] (bottom). Anionic species are depicted in blue, cationic species in red, and neutral species in gray. Reproduced with permission.79 Copyright © 2020 American Chemical Society. (b) Plots of activity coefficients vs. Li salt mole fraction. Blue and red denote [LiTFSI]x(H2O)1−x and [LiTFSI]x(G3)1−x, respectively, and green denotes [LiX][G3] (X = NO3−, TFA−, or TFSA−); f < 0.01 is herein used as a thermodynamic criterion for categorizing the mixture as an SIL instead of a concentrated electrolyte solution. Reproduced with permission.80 © 2020 American Chemical Society. (c) Five transport coefficients according to the Onsager formalism [σ+(self), σ−(self), σ++(distinct), σ−−(distinct), and σ+−: left to right] normalized by the ionic conductivity (σion) of the molten Li salt solvates for the [Li(G4)][TFSI] complex. Adapted with permission.81 This journal is © the Owner Societies 2020. (d) Plots of the Li+ transference number based on potentiostatic polarization measurements (tPPLi) vs. ionic conductivity for [Li(Gn)]X complexes (n = 3 and 4; X = TFA, NO3, Tf, BF4, NFSA, ClO4, BETI, TFSI, and FSI). Reproduced with permission.82 This journal is © the Owner Societies 2021. | ||
Notably, the sign of σdistinct++, σdistinct−−, and 2σ+− reflects the above-mentioned dynamic cross correlations. Therefore, the parameters calculated by normalizing the terms of eqn (3) to σion describe the contribution of these dynamic correlations to the ionic conductivity, as shown in Fig. 6c for the [Li(G4)][TFSI] system. In this equimolar mixture, cation–cation, anion–anion, and cation–anion migrations are anti-correlated, so that σdistinct++ and σdistinct−− negatively contribute to the ionic conductivity, whilst 2σ+− positively contributes to the ionic conductivity. Hence, Fig. 6c suggests that the momentum conservation of the [Li(G4)]+ complex and the TFSI− anion is achieved by momentum exchange of the ions, as is typical of ILs. Notably, conventional concentrated solutions would instead exhibit significant momentum exchange between the ions and solvent.81 These interactions between the various species in the solution have major effects on the relationship between the Li+ transference number estimated via the potentiostatic polarization method (tPPLi) and the ionic conductivity (Fig. 6d). Accordingly, high tPPLi and low ionic conductivity were observed in electrolytes containing anions with high Lewis basicity, which showed a strongly coupled, collective migration of Li+ cations and anions forming clusters. An example of these solutions is the [Li(G3)][TFA] system, which is in fact characterized by all positive Onsager coefficients in eqn (3), thereby leading to a tPPLi as high as 0.90, in spite of an ionic conductivity below 0.1 mS cm−1. On the other hand, the σdistinct++/σion, σdistinct−−/σion, and σ+−/σion coefficients were all negative for the [Li(G3)][TFSI] system, which is reflected as a relatively high conductivity and low tPPLi (i.e., about 1.1 mS cm−1 and 0.028, respectively). Furthermore, the five Onsager transport coefficients for [Li(G3)][TFA] were larger than those of [Li(G3)][TFSA] by about two order of magnitudes due to the low ionic conductivity of the former electrolyte.81,82
The data reported above clarify in part the features of electrolytes formed by dissolving salts in glymes. Under specific concentrations, the G–S systems were classified as liquid solvated salts (SILs), similar to ionic liquids (ILs), in which both cations and anions contribute to the ionic conductivity. The free-glyme fraction is the parameter taken into account for rationalizing the G–S characteristics. Indeed, solutions with a low free-glyme fraction belong to the SILs, particularly using G3 and G4, whilst those with a high free-glyme fraction belong to the concentrated solution class. The stability of [Li(Gn)]+ complexes, strongly depending on the glyme chain length and the anion nature, affects the thermal stability and Li-transference number of the electrolyte. Furthermore, the formation of SILs can increase the electrolyte viscosity and hinder ion mobility and cell performances. The use of nonpolar additives in the glyme solutions can actually increase the ion mobility without altering the Li-solvent coordination and structure, which is instead changed by polar additives. In addition, the coordination of Li+ ions to the ether-oxygen which plays a key role in allowing the formation of complexes is correlated with the salt concentration. For example, the formation of crown complexes and aggregates compatible with SILs is observed when salts having the TSFI− anion are used at low concentration. Interestingly, both the mobility and viscosity of the glyme electrolyte are controlled by the Lewis basicity and hardness of the anions, while the number of ion carriers and the ionic conductivity increase using glymes with relatively low dielectric constants. Moreover, the strong interactions between glyme chains and Li+ occurring in concentrated solutions can increase the residence time of the solvent near the ions, thus decreasing the Li+-transference number. Therefore, diluted solutions have typically a higher Li+-transference number than concentrated ones, except for some particular cases (e.g., the solvent-in-salt electrolytes in which very large anions are used to remarkably increase the tLi+). The temperature affects the formation of SILs that, in turn, govern the growth of the lithium dendrites at the metal surface. Indeed, diluted solutions may favor the formation of noodle-like dendrites, and instead knot-like and rounded lithium structures can be formed in concentrated solutions. Interestingly, predominant CIP formation and stronger ion pairing are observed in electrolytes using short-chain glymes compared to those using long-chain glymes, in which Li+–glyme complexes are preferentially formed, whilst the ion association degree is modified by changing the temperature value which directly affects the dielectric constant. Furthermore, ion–ion anticorrelation with the migration of G–S clusters within the electric field is affected by the salt concentration and anion size, where high concentrations and small ions lead to large asymmetric clusters with strong anticorrelation. Most likely, the glyme solutions can be regarded as SILs when the activity coefficient (f) is lower than 0.01, and the increase of the anion's Lewis basicity favors the lithium transference number, however depressing the ionic conductivity. Taking into account the incomplete scenario depicted by the several findings reported above, we can reasonably suggest further more systematic studies aimed at fully rationalizing the complex features of the glyme-based electrolyte since they are actually assuming an increasing importance in view of their possible application in a new generation of energy storage systems, as will be illustrated in the subsequent paragraphs.
5. “Glyme electrolyte” in a Li–S cell: a battery close to practical applications
Glymes dissolving lithium salts appeared very promising candidates as lowly flammable electrolytes for application in efficient and high-energy Li–S batteries. These cells react via a multielectron conversion process delivering a theoretical specific capacity as high as 1675 mA h g−1 (with reference to the mass of sulfur), which is reflected as a gravimetric energy density of ca. 3600 or 2600 W h kg−1 with reference to the mass of sulfur or Li2S, respectively.135 The electrochemical conversion of sulfur and lithium occurs at about 2.4 and 2.1 V vs. Li+/Li according to eqn (4):| 16Li + S8 ⇄ 8Li2S | (4) |
This reaction involves lithium polysulfide intermediates (Li2Sx with 2 ≤ x ≤ 8), which dissolve in common electrolyte media for x > 2, giving rise to complex, potential-dependent equilibria between various species with different oxidation states.136 Accordingly, Li–S cells undergo a gradual cathode loss in the electrolyte solution upon the electrochemical process, which may worsen the cycling performance and typically requires the optimization of a suitable cell design differing from that of conventional lithium-ion batteries. So far, a great deal of effort has been devoted to mitigating the detrimental effects of polysulfide dissolution, which has been regarded as one of the most challenging issues presently hindering practical applications of Li–S batteries. In this regard, the electrolyte formulation can radically affect the pathways of electrochemical reaction and, thus, the cell performance.137
A pioneering work reported the characteristic features of two lithium cells employing a pitch carbon-coated Li2S cathode and a solid-state PEO20LiTf–Li2S–ZrO2 electrolyte (LiTf/EO molar ratio of 1:20) or a G4–LiTf liquid electrolyte (G4/LiTf molar ratio of 1:4), respectively. The former battery delivered a stable capacity of 500 mA h gLi2S−1 when cycled at a C/3 rate and at 80 °C, while the latter battery exhibited a lower capacity, which is about 300 mA h gLi2S−1, when tested at a C/6 rate and at 30 °C.83 A more recent work reported a lithium–sulfur polymer battery using solid PEGDME (MW 2000 g mol−1) operating at 50 °C with the capacity approaching 700 mA h gS−1 over 90 charge/discharge cycles. The polymer electrolyte showed high thermal stability and a stable interphase during the Li–S conversion process at 2.2 V vs. Li+/Li.104 Besides, [Li(G3)4][TFSI] and [Li(G3)1][TFSI] complexes were investigated as electrolyte solutions possibly able to mitigate the polysulfide dissolution. Indeed, a Li–S cell using the [Li(G3)1][TFSI] complex delivered a discharge capacity higher than 700 mA h gS−1 with a coulombic efficiency above 98% over more than 400 cycles. Further evidence suggested that the addition of a nonflammable fluorinated solvent that preserves the solvate complex structure may improve the rate capability of the battery.84 Following this trend, Gn–LiX equimolar mixtures (where n = 3 or 4 and X is BETI−, TFSI−, Tf−, BF4−, or NO3−) were investigated in Li–S cells and the dissolution of lithium polysulfides (Li2Sx) was measured. According to this study, electrolytes exhibiting SIL characteristics such as [Li(Gn)][BETI] and [Li(Gn)][TFSI] may effectively ensure the mitigation of the Li2Sx dissolution, as shown in Fig. 7a. We remark that equimolar SILs would not contain “free” solvent molecules that can solvate the Li2Sx species, as widely discussed in the previous section. On the other hand, lithium polysulfides were highly soluble in concentrated solutions, such as Gn–LiTf and Gn–LiNO3 (Fig. 7a). Therefore, the batteries employing SILs ensured a stable response in galvanostatic cycling test, delivering a capacity between 600 and 700 mA h gS−1 with a coulombic efficiency above 98% over 100 cycles, whereas those using concentrated solutions displayed the worst performances. Moreover, the typical irreversible reduction of NO3− anions at the positive electrode during discharge was observed, whilst BF4 anions gave rise to detrimental side reactions with the polysulfide anions leading to undesired byproducts.85 Alongside these remarkable effects of the anions, the Gn chain length was shown to have a significant influence on the mobility of the various species in the electrolyte solution, thereby remarkably impacting the Li–S cell behavior. Furthermore, the beneficial effects on the “polysulfide-shuttle” process of the addition of LiNO3 along with LiTf to the electrolyte formulation were widely demonstrated. This adverse process consists in an apparent charging without actual energy storage due to the simultaneous oxidation of polysulfides at the cathode and their direct reduction at the anode upon migration in the solution across the cell separator. In this regard, Fig. 7(b and c) shows that the ineffective charge of a Li–S cell employing a G2–LiTf electrolyte (see panel b) is actually suppressed after the addition of LiNO3 to the solution (see panel c).86 Another work proposed that the dissociation of polysulfide dianions to radicals, and particularly the trisulfur radical (S3˙−), may play a crucial role in the electrochemical behavior of the Li–S cell. Accordingly, operando XANES measurements revealed the presence of these radicals in batteries using solutions of LiClO4 and LiNO3 in either G4 or DMA, thereby suggesting that S3˙− existed in minor concentration in glymes and in relatively high concentration in electron pair donor solvents. Notably, a high degree of dissociation of the anion precursor, S62−, to the trisulfur radical in the latter solvents allowed the full utilization of both S and Li2S. As shown in this study, catholyte-based batteries would benefit from low-volatility and electron pair donor solvents, and the combination of a highly adsorptive cathode with a highly dissociative solvent could be extremely effective in ensuring a superior performance. On the other hand, electron pair donor solvents would be detrimental to achieving polysulfide-entrapping in the cathode.87 It is worth mentioning that the Li–S chemistry would affect also the lithium-passivation properties of the glyme based solution, as shown by a literature report which provided relevant XPS and electrochemical data on G4–LiTf solutions charged by various polysulfide species, that is, Li2S2, Li2S4, Li2S6, and Li2S8. The results of this work demonstrated the presence of S-containing species in the SEI layer deposited over the lithium–metal anode and showed that the presence of polysulfides in the solution decreased the resistance of this layer. The addition of Li2Sx in the electrolyte stabilized the electrode/electrolyte interphase resistance and had a buffer effect which mitigated the cathode dissolution, leading to an improvement of the Li–S cell performance.88
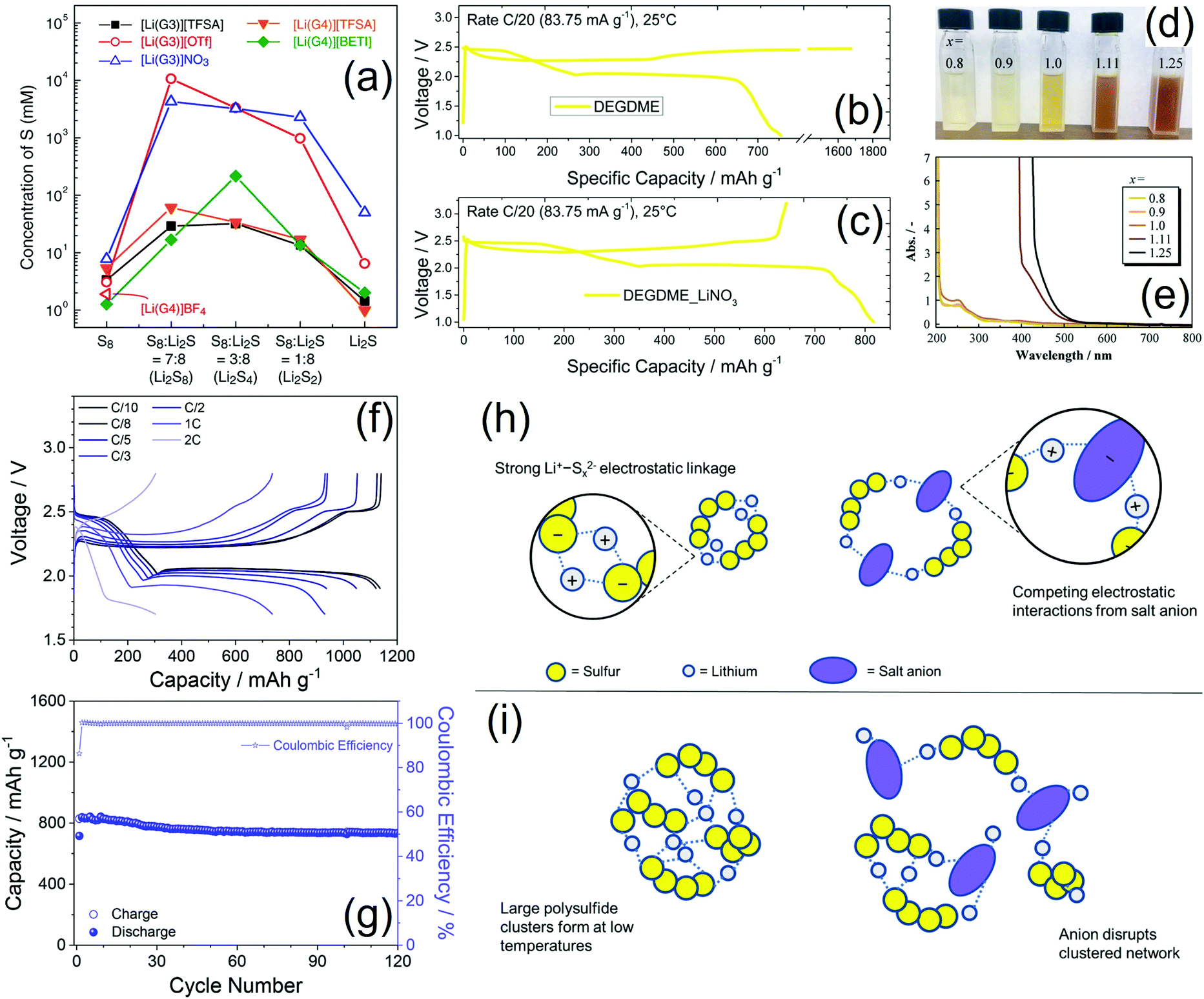 | ||
| Fig. 7 (a) Solubility limits of S8, Li2S8, Li2S4, Li2S2, and LiS2, where Li2Sm (m = 8, 4, 2) is the nominal formula of the mixture prepared using stoichiometric amounts of S8 and Li2S; the nominal formula assumes a complete reaction between S8 and Li2S without occurrence of disproportion reactions; [Li(glyme)]X represented by empty and full symbols are categorized as SILs and concentrated solutions, respectively. Reproduced with permission.85 Copyright © 2013 American Chemical Society. (b and c) Voltage profiles of Li–S cells using (b) G2–LiTf (LiTf in a concentration of 1 mol kgsolvent−1) and (c) G2–LiTf–LiNO3 (LiTf and LiNO3 in concentrations of 1 mol kgsolvent−1 and 0.4 mol kgsolvent−1, respectively); temperature: 25 °C; voltage range: 1.2–3.2 V; current rate: C/20 (83.75 mA g−1 with reference to the cathode mass). Adapted with permission.86 Copyright © 2015 American Chemical Society. (d) Photograph and (e) UV–vis spectra of [Li(G3)x][TFSI] solutions of Li2S8 (x = 0.8, 0.9, 1.0, 1.11, and 1.25) diluted four times with HFE at room temperature. Reproduced with permission.98 Copyright © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (f and g) Electrochemical behavior of a catholyte-based, Li|G2–Li2S8–LiNO3–LiTf|C cell in terms of (f) voltage profiles at C/10, C/8, C/5, C/3, C/2, 1C and 2C rates (1C = 1675 mA gS−1) and (g) trend of charge and discharge capacities per positive electrode and coulombic efficiency at a C/3 rate (1C = 1675 mA gS−1) as a function of the cycle number (both LiTf and LiNO3 in concentrations of 1 mol kgsolvent−1); voltage range: 1.8–2.8 V from C/10 to C/3 and 1.7–2.8 V from C/2 to 2C. Adapted with permission.99 Copyright © 2018 Elsevier B.V. All rights reserved. (h and i) Illustration of competing interactions between lithium species in electrolyte solutions for Li–S batteries; (h) strong Li+–Sx2− bond networks disrupted from competing electrostatic interactions between lithium ions and lithium salt anions; and (i) lithium polysulfides that naturally form at low temperatures, disrupted from the influence of competing lithium salt anions. Reproduced with permission.100 Copyright © 2020 American Chemical Society. | ||
Further approaches to reduce the polysulfide shuttling involved the design of electrolyte membranes with intrinsic microporosity, which would ensure size- and ion-selective transport. Polymeric membranes were optimized via MD simulations of the solvate structures of LiTFSI with lithium polysulfides (Li2Sx, where x = 8, 6, and 4) in glymes of different chain lengths. These simulations suggested that a pore size lower than 1.2–1.7 nm might bock the polysulfide crossover, thus possibly enabling a suitable electrochemical behavior of redox-flow, lithium–sulfur batteries, even in the absence of LiNO3 in the electrolyte formulation.89 Alternative strategies recently explored to improve the cycle life and rate capability of Li–S cells included the addition of support solvents to SILs based on Gn–salt complexes. An example is represented by a novel fluorinated ether derivative, i.e., TFTFE, which was added to the [Li(G4)1][TFSI] equimolar mixture, thereby enhancing the ionic transport across the electrolyte and, thus, the rate capability of the Li–S battery. Moreover, TFTFE was shown to decrease the solubility of lithium polysulfides in the electrolyte due to its low donor ability which limits the Li+ solvation.90 Another study comparing several equimolar mixtures of either G3 or G4 with various salts (LiBETI, LiBF4, LiTf and LiNO3) revealed that the dissolution of lithium polysulfides may be suppressed when SILs are employed.91 On the other hand, the G4–LiTf solution with the lithium salt in a 1 mol kg−1 concentration was considered the preferred electrolyte for performing a comparative evaluation of the electrochemical performance of several cathode composites prepared by mixing sulfur with carbon materials of various characters (namely, graphite, mesocarbon microbeads, and multi-walled carbon nanotubes). Besides, the electrolyte characteristics in terms of 1H, 7Li, and 19F nucleus self-diffusion coefficients, ionic conductivity, and ionic association degree were investigated by combining NMR and impedance spectroscopy measurements. Notably, the best composite in the Li–S cell using this electrolyte achieved a capacity higher than 500 mA h g−1 over 140 cycles, with no sign of dendrite formation or any shuttle reaction.92 Glymes were also employed in combination with micropore-rich activated carbon incorporating sulfur,93,96 as well as with S-Ketjenblack composites showing promising results.94 Other cathode chemistries have been explored, such as a TiS2–S composite reacting via intercalation of Li+ ions into TiS2 along with Li–S conversion.95 Furthermore, a SIL consisting of a [Li(G4)][TFSI] equimolar mixture was investigated as a possible electrolyte for lithium-ion and silicon–sulfur batteries. In detail, pre-cycling in a FEC-containing solution before the test in the glyme-based electrolyte was shown to be an effective strategy to form a stable SEI incorporating fine LiF grains and organo-fluorine compounds over a Si-flake anode, which ensured a remarkable enhancement of the capacity retention.97
A recent work provided additional evidence of the effect of the lithium-salt concentration on the solubility of polysulfides in the electrolyte solution, which has been generally attributed to the presence of “free” glyme molecules favoring the solvation of Li2Sx species, as indeed mentioned above. In this regard, [Li(Gn)]X complexes in equimolar mixtures (where LiX is a typical salt) can be locally destroyed to form “free” glyme chains upon the electrochemical process in Li–S cells. Therefore, non-equimolar mixtures (where the Gn:LiX molar ratio is higher than 1) may actually decrease the concentration of dissolved Li2S8, thereby improving the reversible capacity and the cycling stability, whilst increasing the coulombic efficiency. Photographic images (Fig. 7d) and UV/vis spectra (Fig. 7e) of various Li2S8-containing [Li(G3)x][TFSI] SILs with x values of 0.8, 0.9, 1.0, 1.11, and 1.25, which were diluted with HFE, are consistent with a decrease in the solubility of Li2S8 at increasing concentration of LiTFSI.98 Although the mitigation of the dissolution of lithium polysulfides in the electrolyte has been regarded as a valuable strategy to enable high-performance Li–S batteries, several works have suggested that long cycle life and high coulombic efficiency may be achieved even in catholyte-based, semi-liquid configurations, provided that the lithium–metal anode is effectively protected against parasitic reactions with dissolved Li2Sx. Catholyte solutions of polysulfides formed by dissolving either LiTFSI or LiTf as salts along with LiNO3 as an anode-protection additive in G2 were lately employed in Li–S cells exhibiting notable performances in terms of specific capacity, capacity retention, and coulombic efficiency. These solutions had suitable characteristics for applications, that is, satisfactory Li+ transport ability, a wide electrochemical stability window, and good Li-passivation properties. Semi-liquid cells using dissolved Li2S8 with overall sulfur loading ranging from 3 to 6 mg cm−2 displayed a high rate capability, delivered a maximum capacity of ca. 1100 mA h gS−1 at a C/10 rate (Fig. 7f), and ensured a stable capacity of about 800 mA h gS−1 at a C/3 rate with the coulombic efficiency exceeding 99% (Fig. 7g).99 Notably, the electrostatic attraction forces between the Li+ cation, salt anion, and Sx2− anion would play a significant role in determining the solvate structures in the electrolyte solution, thus significantly affecting the behavior of the cell. Testing under low-temperature conditions shed light on this point, revealing that lithium polysulfides may form clusters that adversely impact the Li–S conversion kinetics. Therefore, a low testing temperature may lead to poor performance, despite the low freezing point and high ionic conductivity of the glyme-based electrolyte. More abundant Li+–Sx2− bonds and polysulfide clusters are formed whenever the cation has a higher affinity towards the Sx2− anion rather than the salt anion. On the other hand, it was suggested that a strongly bound lithium salt would disrupt polysulfide aggregates ensuring a substantially improved low-temperature performance. A graphical schematic of the polysulfide clustering mechanism based on competing forces in the electrolyte solution is shown in Fig. 7(h and i).100 Another study of the cathode dissolution in the electrolyte during operation of the Li–S cell showed that lithium polysulfides are formed in both glyme-based and fluorinated-ether-based electrolytes, in which the polysulfides are highly soluble and lowly soluble, respectively. However, lithium polysulfides were trapped in the cathode pores when the latter solutions were employed, and the polysulfide concentration in the separator was below the limit of detection of HPLC analyses, thereby suggesting a decrease of interaction between dissolved polysulfides in fluorinated-ether-based electrolytes. The different behavior of glyme-based and fluorinated-ether-based electrolytes affected the length and potential of the voltage plateaus of the battery upon cycling.101 The presence of Li2Sx in the electrolyte solution may have additional major effects on the cell response by influencing the composition of the SEI, as mentioned above and recently investigated.102
The Li–S battery is presently considered the most energetic and promising energy storage system for future practical applications. However, this alternative battery requires suitable electrolytes for allowing an adequate electrode/electrolyte interface and, at the same time, compatibility with lithium metal and low flammability. A further characteristic required is the compatibility of the electrolytes with the polysulfides, which are unavoidably formed in the cell. Alongside PEO-based electrolytes, those using glymes, in particular with the longer chain-length, appear the most promising candidates for achieving the abovementioned targets. Hence, solutions based on G2, G3, and G4 with the addition of LiNO3 as the sacrificial film-forming agent to suppress the polysulfide shuttle, as well as liquid and solid PEGDME can be successfully employed as the electrolyte media for Li–S cells. Interestingly, electrolytes with SIL characteristics described above appear to limit the polysulfide dissolution due to the absence of free solvent molecules, while polysulfides are soluble in concentrated solutions. Furthermore, formation of radicals during the electrochemical process of the battery affecting the stability may be limited by using glymes with long chains. The presence of polysulfides in combination with LiNO3 as the additive actually stabilizes the lithium metal interphase, and therefore solutions of polysulfides are proposed as the catholyte to operate in Li–S batteries. It is worth mentioning that polymeric and porous membranes are also proposed with certain success of mitigating the polysulfide shuttle effect even in the absence of LiNO3; however the use of the former is suggested alongside the improvement of the cathode material. Lithium salt concentration and its nature modify the structure of G–S and change the polysulfide clustering, thus remarkably affecting the performance of the cell. An interesting example is that of the non-equimolar G–S mixtures that decrease the solubility of the Li2S8 intermediate and enhance the battery stability; despite this, dissolved Li2S8 can be actually used as the liquid cathode in Li–S cells with LiNO3.
6. “Glyme electrolyte” in a Li–O2 battery: the upcoming future
The Li–O2 cell is an appealing next-generation battery system which may possibly ensure a notable breakthrough in energy density. This technology is based on reaction (5) of oxygen with lithium in the electrochemical cell to produce lithium peroxide, occurring at 2.96 V vs. Li+/Li and delivering a formal specific capacity of 1168 mA h g−1 with reference to the mass of Li2O2.| 2Li + O2⇄Li2O2 | (5) |
However, the development of Li–O2 batteries presents several challenges to overcome, which are mostly associated with parasitic reactions upon cell operation limiting the reversibility of the electrochemical process.138,139 Furthermore, the carbonate-based electrolytes employed in conventional lithium-ion batteries decompose in contact with the electrochemical process intermediates, that is, superoxide radical and peroxide,140,141 so they cannot be used in Li–O2 cells. On the other hand, excessively volatile ether solvents would pose serious limitations for a system designed for potentially operating in open air.142 In this regard, glymes are considered suitable solvents for Li–O2 batteries,143 since they show high chemical stability as well as a boiling point and volatility that can be modulated by modifying the chain length.19
The applicability of G4–LiTf in Li–O2 cells was initially demonstrated by elucidating the reaction mechanism in a PEO–G4–LiTf plasticized system. The results suggested that the highly solvating, base-resistant PEO plasticized by the low-molecular-weight, end-capped G4 is a very good medium to study the electrochemical processes attributed to peroxide and oxide species in water-free Li–O2 cells.105 Subsequently, another work demonstrated that the use of a liquid G4–LiTf electrolyte solution and an appropriate cell design can allow the Li–O2 system to reversibly cycle at a current rate as high as 3 A g−1 delivering an outstanding capacity of 5000 mA h g−1 (Fig. 8a). As is widely established in the Li–O2 community, these values are with reference to the mass of carbon in the electrode film, that is, A gcarbon−1 and mA h gcarbon−1, respectively.106 The electrolyte formulation significantly affects the electrochemical performance and the discharge products of Li–O2 cells. Indeed, in a further study the use of glymes as solvents led to a large amount of Li2O2 in the positive electrodes after discharge, while only a small amount of Li2O2 was detected after discharge in electrolytes based on nitrile, ionic liquids, phosphate, and sulfoxide. The employed solvent also influences the relative amount of Li2CO3 and LiF, which are formed as byproducts via oxidation and decomposition of the solvent and via the attack of superoxide radical anions on the binder and/or the F-containing imide salt, respectively. This work suggested the dibutyl diglyme as the most suitable solvent among those taken into account.107 Therefore, glyme-based solutions have been so far extensively investigated as electrolytes for Li–O2 cells with promising results, and alternative chemistries have also been suggested. Accordingly, there is a pioneering report on the behavior of a lithium-ion-air battery where the metal anode is replaced by a lithiated silicon–carbon composite effectively employing a LiTf–G4 electrolyte solution.108 Besides the salt and solvent nature, the salt/solvent molar ratio plays a crucial role in the oxygen-conversion mechanism in the cell, as demonstrated by comparing a series of solutions of LiTFSI in G3 and G4 with various concentrations. According to this study, the O2˙− radical and solvated Li+ would form an intermediate complex at the electrode/electrolyte interphase, that is, Li+(Gx)n⋯O2˙− (Fig. 8b), leading to the formation of CIP solvates containing one Li+ ion which is coordinated with four or five oxygen atoms of one glyme molecule in concentrated solutions (route a). A decomposition pathway via reaction of O2˙− with the glyme was therefore proposed (route a in Fig. 8b) to elucidate the performance degradations of Li–O2 cells employing concentrated solutions. Dilution would gradually produce a mixture of (i) CIP and SSIP solvates, (ii) SSIP solvates (route b in Fig. 8b), and eventually a mixture of SSIP solvates and free glyme molecules (route c in Fig. 8b). Thus, intermediate concentrations would favor the accessibility of O2˙− to Li+ and decrease the interactions between O2˙− and the glyme molecule that lead to detrimental decompositions (route b), whilst low concentrations would reasonably increase the frequency of collision between O2˙− and glyme, thereby enhancing the parasitic processes (route c). Therefore, the glyme/salt molar ratio of 5 to 1 was found to be critical for the cycling performance of the cell, and high stability over 20 cycles at 500 mA gcarbon−1 was demonstrated using (Gn)5–LiTFSI electrolytes with n = 3 and 4.109 The electrolyte purity is another crucial aspect that should be taken into account, as shown in a qualitative and quantitative investigation of the reaction of KO2 with glymes of various chain lengths via1H NMR, FTIR, and UV-Vis spectroscopy, which demonstrated major effects on the cell performance.110 Notably, glyme-based electrolytes would also ensure a wide temperature range of applicability, as demonstrated in a further report. As expected, the polarization and rate capability of a Li–O2 cell using G4–LiTf were shown to be influenced by the operating temperature (Fig. 8c). Indeed, low temperatures would slow down the diffusion of Li+ ions, whilst elevated temperatures would decrease the electrolyte viscosity and consequently increase the oxygen mobility. Thermal effects on the crystallinity of Li2O2 formed upon cell discharging were also observed.111 It is worth considering that the reversibility of the Li–O2 process upon full-capacity cycling is rather poor, and the outstanding long-term performances reported so far have been often obtained by limiting the capacity below 1000 mA h gcarbon−1. On the other hand, it has been demonstrated that extended full-capacity cycling of Li–O2 batteries using glymes would be ensured by selecting appropriate electrode materials (carbon source and catalyst) and cycling protocols. In particular, the formation of a stable interfacial layer on the cathode surface during the initial cycling may stabilize the subsequent cycling stages. Whilst the initial cell operation was characterized by the predominant formation of Li2O2, the subsequent cycles led to the predominant formation of side products and the eventual stabilization of the yield of Li2O2 at about 33–40%.112 Besides, interesting results have been obtained by limiting the capacity of a Li–O2 battery to 1000 mA h gcarbon−1 using an electrolyte with very low volatility. The electrolyte was formed by dissolving LiTFSI in PEGDME (MW 500 g mol−1) and then mixing this solution with the Pyr14TFSI ionic liquid.113 Moreover, a new lithium-ether-derived chelate IL showed promising characteristics for applications in Li–O2 batteries, namely, high stability against the lithium metal anode and against superoxide-initiated hydrogen abstraction when compared to DME. This electrolyte chemistry ensured a decrease in the amount of parasitic species formed during cycling, such as formate, as well as a ten-fold decrease in CO2 evolution upon charge as compared to that observed in DME-based solutions.114 Among the various possible lithium salts for aprotic solutions for Li–O2 batteries, LiNO3 dissolved in polyether solvents exhibited beneficial properties on both the ORR and OER. The anion enhanced the former reaction by enabling the formation of submicrometric Li2O2 particles via a reaction pathway involving superoxide radicals and acted as a redox mediator by producing at the negative electrode NO2− ions which, in turn, formed NO2 at 3.6–3.8 V vs. Li+/Li. This latter species catalyze the Li2O2 oxidation forming NO2−, which is then oxidized again, thereby decreasing the OER overpotentials within the electrochemical stability window of glyme-based electrolytes.115 1 M solutions of LiTf, LiTFSI, and LiFSI in G4 were further studied in Li–O2 batteries to elucidate the interplay between ion transport and lithium stripping/plating, thereby revealing for the G4–LiTFSI electrolyte mixture a high rate capability, a fast electrochemical reaction at the anode side, and a promising performance. Notably, the performance of the Li–O2 cell depended on lithium oxide layers formed on the negative electrode. These results suggested that a suitable anode-surface oxidation may enhance the electrode/electrolyte interface characteristics.116,120 The electrolyte formulation and the electrochemical conditions may affect both kinetics and thermodynamics of the ORR, for instance by altering the solvation in high DN solvents. Along with solvent DN, the level of dissociation of the salt may have additional contributions in determining the characteristics of the ORR and controlling the morphology of Li2O2 deposits, as schematically illustrated in Fig. 8d.117 Besides fundamental studies aiming to shed light on the electrochemical reactions occurring in the cell, further works focused on developing alternative battery designs, such as the metal-free Li-ion oxygen system which was mentioned above. For example, an effective approach to achieve excellent cycling stability and low cell polarization over 100 cycles involved the use of silicon particles at the negative electrode. In this regard, a stable SEI over the anode surface would mitigate oxygen crossover effects, which would improve the long-term cyclability of the battery.118 Another lithium-ion oxygen cell using a lithiated hard carbon (HC) anode, a monolithic carbon cathode, and a G2-based electrolyte was reported with promising results.121 Additional evidence of the role played by the anion nature in the amount of Li2O2 precipitated on the separator in the Li–O2 cell using glyme-based electrolytes suggested a relationship between the stability of the discharge intermediate (LiO2) in the electrolyte and the anion. Accordingly, electrolyte solutions characterized by a high intermediate solubility would favor the precipitation of Li2O2 across the cell, thereby adversely affecting the reversibility of the oxygen conversion process. Fig. 8e shows how the fraction of Li2O2 in the separator, with reference to the theoretical total amount of Li2O2, changes with the anion composition.119
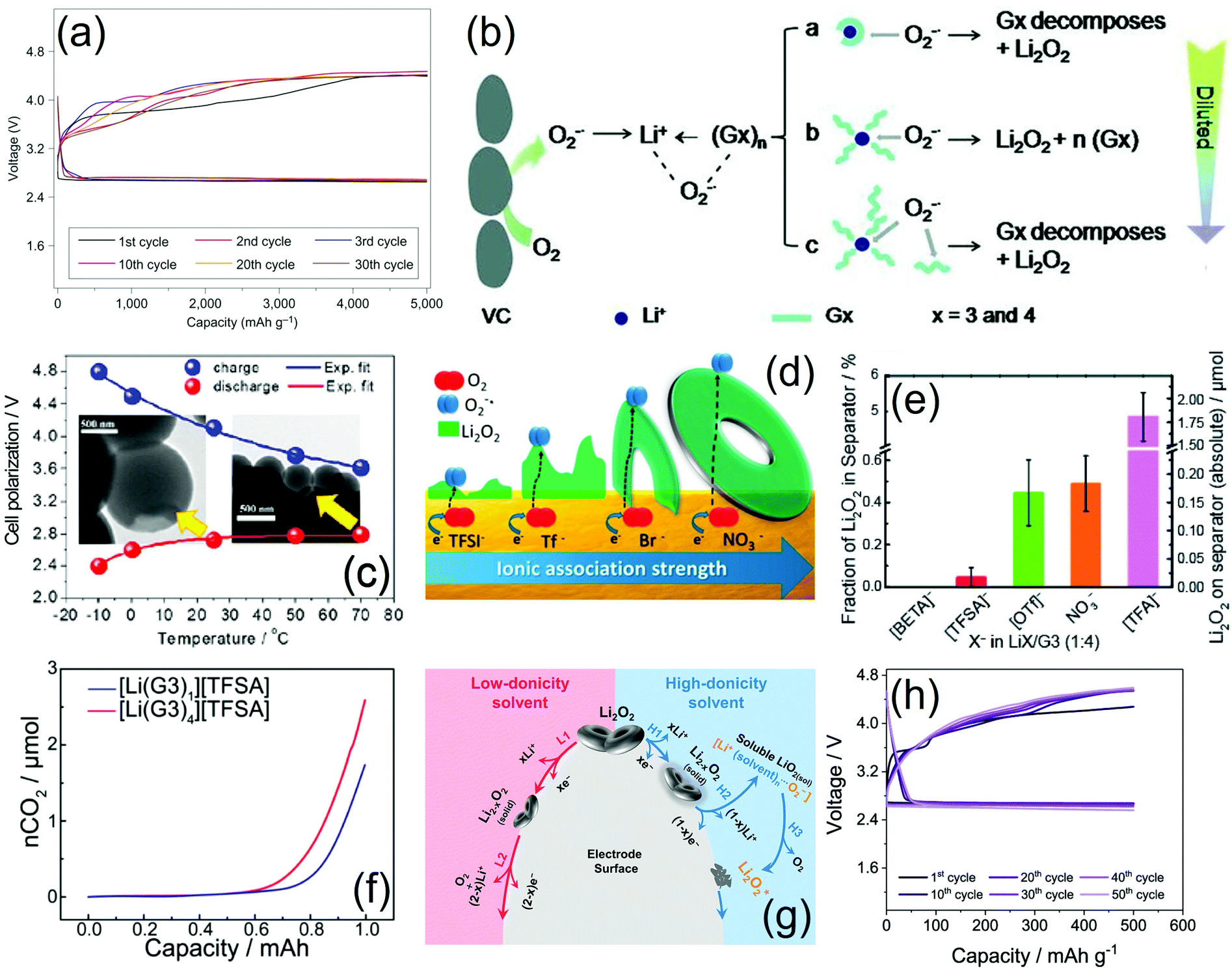 | ||
| Fig. 8 (a) Voltage profiles of a Li|G4–LiTf|O2 battery under a 5000 mA h g−1 specific-capacity limit with reference to the mass of carbon cast on the cathode GDL; current rate: 500 mA g−1 (current rate with reference to the mass of carbon cast on the cathode GDL). Adapted with permission.144 Copyright © 2012 Macmillan Publishers Limited. All rights reserved. (b) Mechanism of the O2 reduction reaction in a Li–O2 cell at the interface between carbon black and glyme–LiTFSI electrolyte solution during discharge as proposed elsewhere. Reproduced with permission.109 Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) Trends of charge (blue) and discharge (red) voltage vs. temperature for a Li–O2 cell using a G4–LiTf electrolyte solution. Insets: ex situ TEM images of the cathode of the same cell after galvanostatic discharge at 25 °C (left-hand side) and at 70 °C (right-hand side); capacity limited to 10000 mA h g−1 with reference to the mass of carbon cast on the cathode GDL; current rate: 500 mA g−1 (current rate with reference to the mass of carbon cast on the cathode GDL). Reproduced with permission.111 Copyright © 2013 American Chemical Society. (d) Schematic representation of the effect of salt anions (i.e., TFSI−, Tf−, Br−, and NO3−) in electrolyte solutions based on G2 on the kinetics and thermodynamics of the ORR, as well as on the morphology of electrodeposited Li2O2 in Li–O2 batteries. Reproduced with permission.117 Copyright © 2016 American Chemical Society. (e) Results of quantitative determination of Li2O2 on the separator in a Li–O2 cell using LiX/G3 (1:4) electrolytes, where X− = BETI−, TFSI−, Tf−, NO3−, and TFA−. The values on the left y-axis are the percentage of the amount detected in the separator with respect to the amount of total expected (theoretical) Li2O2 present in the cell after discharge to 1 mA h cm−2, whilst the corresponding absolute values (μmol) are shown on the right y-axis; error bars denote the standard error. Adapted with permission.119 Copyright © 2017 The Chemical Society of Japan. (f) Integrated CO2 evolution during charge of Li–O2 cells using either [Li(G3)1][TFSI] (blue curve) or [Li(G3)4][TFSI] (red curve) electrolytes, which was calculated from electrochemical MS data, after galvanostatic discharge to 1 mA h. Adapted with permission (https://pubs.acs.org/doi/pdf/10.1021/acsami.6b14449).122 Copyright © 2017 American Chemical Society. Further permission related to the material excerpted should be directed to the American Chemical Society. (g) Solvent-controlled Li2O2 decomposition mechanism in Li–O2 batteries as proposed elsewhere; “H” denotes high-donicity solvent and “L” denotes low-donicity solvent. Li2O2* denotes the Li2O2 generated by LiO2(sol) disproportionation, where LiO2(sol) is a soluble discharge intermediate product of the ORR. Reproduced with permission.125 Copyright © 2018 Elsevier Inc. (h) Voltage profiles of a Li–O2 cell using G3–LiTFSI–LiNO3 (both LiTFSI and LiNO3 at concentrations of 2 mol kgsolvent−1); specific capacity limited to 500 mA h g−1 with reference to the mass of carbon cast on the cathode GDL; current rate: 100 mA g−1 (current rate with reference to the mass of carbon cast on the cathode GDL); carbon loading over the cathode GDL: 0.65 mg cm−2 (carbon mass: 1.3 mg, geometric electrode area: 2 cm2) voltage range: 1.5–4.6 V; cell aged for 7 days before cycling. Adapted with permission.130 Copyright © 2020 American Chemical Society. | ||
As discussed in the previous sections, literature reports have described various beneficial properties of SILs when compared to conventional electrolytes for applications in lithium batteries employing insertion and sulfur-conversion cathodes. SILs may improve the Li–O2 cell too, as demonstrated by a study of G3–LiTFSI mixtures where the solvent and salts were in an equimolar ratio or contained excess glyme. The related results suggested that SILs would have a higher oxidative stability than conventional electrolytes, as well as a lower volatility, which would suit the open Li–O2 cell configuration. High salt concentrations would mitigate the parasitic reactions that lead to CO2 evolution, as shown in Fig. 8f, although either type of solution did not ensure the full theoretical value of O2 evolution, and side processes were detected upon charging. The discharge product morphology was also related to the solubility of the superoxide intermediate, which is in turn affected by the salt concentration.122 Moreover, Raman spectroscopy analyses of glyme-based electrolytes suggested that the increase in LiTFSI concentration favors the formation of cationic and anionic complexes that stabilize the G4 molecules against degradation. High-concentration electrolytes enabled an improvement as high as 300% in charge/discharge cycling tests with a limited capacity of 500 mA h g−1, for solutions containing higher LiTFSI concentrations. A better cyclability at a low G4:LiTFSI molar ratio was associated with a decrease in the growth rate of lithium carbonate species deriving from glyme degradation.123 On the other hand, a G4–LiTFSI solution with a 1 mol kg−1 concentration enabled the achievement of hundreds of cycles without signs of decay to a Li–O2 cell using a multiwalled carbon nanotube electrode. The reversibility of the electrochemical process in this cell was demonstrated by detecting the reversible formation and dissolution of Li2O2 during the electrochemical process.124 Regarding this point, the Li2O2 oxidation pathway was associated with the solvent donicity according to a strong solvent-controlled mechanism. Thus, a solution route forming a soluble LiO2 intermediate after Li2O2 oxidation was suggested to occur in high-donicity solvents (Fig. 8g, right-hand side pathway), whilst a solid-solution route forming a solid Li2−xO2 intermediate was identified in low-donicity solvents (Fig. 8g, left-hand side pathway). Notably, the former oxidation mechanism was causally related to an observed poor cycling stability of the relevant Li–O2 cells.125 A key aspect to consider when increasing the salt concentration in Gn–LiX solutions is the change in Li+ transport properties and ionic conductivity, as indeed extensively discussed in section 4. We remark herein that viscosity and Li+ transference number measurements on solutions using either G1 or G2 and either LiTf or LiTFSI suggested a failure of Walden's rule, in spite of a qualitative correlation with the association constant of the salts.126
The electrolyte composition may affect the crucial parameters for application in a Li–O2 battery using a carbon-cloth GDL, such as the conductivity, viscosity, contact angle, and decomposition temperature. Among various formulations, G4–LiTFSI was often selected as the electrolyte of choice due to its suitable properties for use in the open metal–oxygen cell design. In this regard, a recent work demonstrated an enhanced performance as compared to those obtained with other formulations, that is, a longer cycle life at a discharge capacity limit of 2000 mA h g−1 with reference to the mass of the Pt catalyst in the GDL.127 As mentioned above, LiNO3-based solutions have been investigated as electrolytes for Li–O2 batteries with promising results, which were attributed to the role of NO3− as a redox mediator for the electrochemical process. Furthermore, LiNO3 was shown to improve the lithium–electrolyte interphase by favoring the formation of a Li2O layer which mitigates the lithium dendrite growth and the electrolyte decomposition. Dual solvent systems based on DMSO and G4 were suggested as effective solutions characterized by an increased number per volume and mobility of Li+ and NO3−. The former solvent has a relatively high dielectric constant and a low viscosity, which allowed a decrease in the overpotential of the charge process along with an enhancement of the power density of the battery. Such a mixed formulation mitigated the low dissociation degree of LiNO3 typically leading to low ionic conductivity.128 As for the effects of the glyme chain length, SNIFTIRS and electrochemical data on Li–O2 cells using gold electrodes lately suggested that G2 may be more stable than G4 between 3.6 and 3.9 V vs. Li+/Li, although water impurities may reduce its stability.129 On the other hand, a recent comparative study of concentrated solutions of LiTFSI and LiNO3 salts in either G2 or G3 demonstrated that the former solvent would have volatility issues limiting its applicability in Li–O2 cells with open design. Gradual solvent evaporation during cycling was observed along with a rapid cell failure. In contrast, the use of the G3-based electrolyte ensured stability over time as well as a specific capacity ranging from 500 to 1000 mA h gcarbon−1. Such a concentrated G3–LiTFSI–LiNO3 was characterized by a high conductivity, a high Li+ transference number, a wide electrochemical stability window, and favorable lithium–metal passivation properties.130
Despite still being in a very early stage, the Li–O2 battery is the most appealing energy storage system due to the highest theoretical potentialities among the various candidates. Parasitic reaction of the electrolyte solvents with the radicals formed during the Li–O2 electrochemical process as well as the necessity of operating in an open environment (i.e., O2 or dry air) represent the major drawbacks to overcome for enabling an efficient battery. Glyme electrolytes, which are modulable in terms of stability and volatility by changing the chain length, as well as the salt nature and concentration represent the most suitable electrolyte for allowing cell operation. Furthermore, the use of the lithium metal in a practical battery exposed to oxygen or air may be actually granted by long chain glymes such as highly viscous or solid PEGDME. In this regard, alternative lithium metal-free batteries are proposed in analogy with the Li-ion ones by using alloying electrodes at the negative side, however with partial success. A solution based on G4 represents the first reported example of an electrolyte in a lithium oxygen cell with high capacity and a stable trend. G3 may be also adequate for Li–O2 cells, but G2 and G1 appear unsuitable at the moment due to excessive volatility, and consequent evaporation during cell cycling in an open environment. The salt-to-solvent molar ratio is crucial for the lithium–oxygen conversion mechanism: indeed, intermediate concentrations limit the glyme decomposition due to O2˙− radicals, but low concentrations lead to detrimental decomposition. Decomposition reaction is in fact governed by the intermediate Li+(Gn)O2− complex which can form coordinated Li+ ions, CIP, SSIP solvates, and free glyme molecules. The latter species are principally formed at low concentrations and can react with O2˙−, thus leading to the electrolyte decomposition. In addition, solvent chemistry controls the reaction mechanism: solvents with high DN lead to the formation of a soluble LiO2 intermediate, but solvents with low DN favor the formation of a Li2−xO2 intermediate through a solid-solution mechanism. It is worth mentioning that the performances of the Li–O2 cell are also affected by the temperature, the use of adequate GDLs, the cell setup and the experimental conditions, as well by the presence of LiNO3 even though with less remarkable effects compared to Li–S batteries. Furthermore, SIL formation may improve the Li–O2 battery performance, while high salt concentration can mitigate the parasitic reactions due to the formation of cationic and anionic complexes, particularly using G4.
7. Remarks and conclusions
Extensive works have been conducted over the past 25 years to assess the possible applicability of glymes in lithium batteries and shed light on the chemical–physical properties of their mixtures with lithium salts. Indeed, the various advantages of glyme-based electrolytes against common alkyl-carbonate-based solutions have been often highlighted, although some questions on their high-power performance when compared to their conventional, ester-based counterpart are still open. So far, glyme-based solutions have been thoroughly studied by using a wide portfolio of experimental methods, modelling approaches, and techniques, which have provided comprehensive description of their highly versatile characteristics, tunable by changing the glyme chain length along with the salt composition and concentration. Notably, glyme-based solutions typically exhibit a higher thermal stability as well as lower flammability and volatility than common lithium-battery electrolytes, particularly at middle and moderately high chain lengths, thereby possibly enabling the use of the high-energy lithium–metal anode. Thanks to such tailored properties, glymes can be effectively employed in lithium batteries with insertion cathodes working below 4 V, such as LiFePO4, as well as with 4 V-class cathodes such as LiCoO2, NMC and LiMnxFe1−xPO4 when used in concentrated formulations as salt-in-solvent mixtures or SILs. Furthermore, glymes are especially suited for high-energy Li–S batteries and considered to be the solvents of choice for Li–O2 batteries, due to a high chemical stability in the cell along with a suitable volatility for the open design. Among the various lithium salts for glyme-based electrolytes that have been investigated, LiTFSI, LiTf, LiFSI, and LiBETI have shown favorable characteristics in the above-mentioned cell designs based on insertion or conversion chemistries. Furthermore, the crucial role of LiNO3 as a film-forming additive to achieve reversible lithium–metal plating and anode protection in conventional Li–metal and Li–S batteries has been clearly demonstrated, and its additional redox-mediator properties in Li–O2 cells have been suggested.Interestingly, the electrolyte properties may vary between an SIL and a conventional electrolyte, depending on the concentration and nature of the salt, along with the glyme chain length. As extensively discussed in the previous sections, an SIL has been described as a [Li(Gn)]X system, where Li+ is mainly solvated by crown-ether-like curled Gn molecules forming a complex in contact with the TFSI− anion, and negligible “free” solvent is detected in the mixture. In this regard, the stability of the [Li(Gn)]+ complex may be revealed by comparing the ratio between the self-diffusion coefficients of glyme and Li+ ions (DG/DLi). Therefore, when designing a high-concentration glyme-based electrolyte, the peculiar features of the anion should be considered along with the solvent molecular weight. As an example, we remark that TFSI− would favor the formation of SILs, whilst TDI− would promote ionic aggregation. Herein, we point out that the Li+ transference number of SILs under anion-blocking conditions is typically low because of the Li+–Gn complexation, which causes anti-correlation of cation and anion motion due to momentum conservation. In contrast, conventional concentrated solutions exhibit momentum exchange between ions and solvent molecules. Strong Lewis base anions ensure collective migration of ion pairs, which leads to a high Li+ transference number and a low ionic conductivity. Instead, anions with low Lewis basicity typically lead to a high conductivity and a low Li+ transference number. The increase in viscosity might be another drawback of highly concentrated Gn–LiX mixtures, which may require the use of diluents to increase the ionic conductivity. Lithium dendrite formation and anode passivation properties are other key aspects to consider when optimizing an electrolyte formulation enhanced by additives.
Several works have suggested that SILs may improve the long-term cycling ability of Li–S cells by preventing the shuttle effect. According to this approach, the polysulfide dissolution would be significantly decreased by limiting the amount of “free” solvent molecules able to participate in the solvation process, thereby leading to a quasi-solid sulfur reaction mechanism in the cell. On the other hand, glyme-based catholytes have shown a promising performance in a wide range of current rates, benefiting from a highly reversible polysulfide conversion on the positive electrode and from suitable Li+ transport properties. Concerning this point, the addition of LiNO3 to the glyme-based electrolyte can actually mitigate the detrimental reaction of dissolved polysulfide with lithium by enabling efficient protection of the metal anode against this parasitic process. As for the Li–O2 system, the electrolyte formulation controls the Li–O2 conversion mechanism and the morphology of the Li2O2 discharge product, thus influencing the cell performance. Indeed, solvent DN and dissociation level of the salt may drive the pathway of the ORR. Moreover, literature works have suggested favorable properties of SILs compared to conventional electrolyte solutions.
Fig. 9 summarizes the main concepts discussed herein and the main conclusions drawn by reviewing the most relevant studies of glyme-based electrolytes for lithium batteries. The figure shows that the glyme-based electrolytes can allow the change from the intercalation chemistry to the more energetic and alternative conversion chemistry (top-side scheme) and the use of the lithium metal in batteries (bottom-side scheme). In particular, PEGDME in the liquid and solid states is nontoxic, nonflammable, and safe and can actually allow scalable Li–S and Li–O2 batteries for future applications. Fig. 9 also reveals that the increase of the salt concentration in the glyme-based electrolytes unlikely leads to the decrease of the lithium transference number; however it increases at the same time the safety and stability of the battery (right-hand arrows). In this scenario the electrolyte viscosity may potentially represent advantages in terms of scalability, and disadvantages in terms of conductivity and cell performances. The fundamental investigation and applied research have indeed enabled a deep understanding of the Gn–LiX structure along with substantial technological improvements of the lithium cell. A critical analysis of the extensive experimental evidence collected over the last two decades demonstrates the suitability of glymes as electrolyte solvents for lithium–metal batteries, and suggests viable strategies to achieve enhanced, next-generation energy-storage systems.
 | ||
| Fig. 9 Schematic representation of the promising characteristics of glyme-based electrolyte solutions for application in lithium–metal batteries using insertion (e.g., olivine-based compounds) as well as conversion cathodes (e.g., S-based and O2-based electrodes); the typical relationship between LiX salt concentration, Li+ transport properties, safety, and stability of the glyme-based solutions is also shown. | ||
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This project/work has received funding from the European Union's Horizon 2020 research and innovation programme Graphene Flagship under grant agreement no. 881603. The authors are also thankful for the grant “Fondo di Ateneo per la Ricerca Locale (FAR) 2021”, University of Ferrara, and the Institute of Global Innovation Research (GIR) in Tokyo University of Agriculture and Technology.Notes and references
- P. Jessop, Green Chem., 2020, 22, 13–15 RSC.
- D. Di Lecce, R. Verrelli and J. Hassoun, Green Chem., 2017, 19, 3442–3467 RSC.
- M. Marinaro, D. Bresser, E. Beyer, P. Faguy, K. Hosoi, H. Li, J. Sakovica, K. Amine, M. Wohlfahrt-Mehrens and S. Passerini, J. Power Sources, 2020, 459, 228073 CrossRef CAS.
- A. A. Kebede, T. Coosemans, M. Messagie, T. Jemal, H. A. Behabtu, J. Van Mierlo and M. Berecibar, J. Energy Storage, 2021, 40, 102748 CrossRef.
- T. Chen, Y. Jin, H. Lv, A. Yang, M. Liu, B. Chen, Y. Xie and Q. Chen, Trans. Tianjin Univ., 2020, 26, 208–217 CrossRef.
- A. Varzi, K. Thanner, R. Scipioni, D. Di Lecce, J. Hassoun, S. Dörfler, H. Altheus, S. Kaskel, C. Prehal and S. A. Freunberger, J. Power Sources, 2020, 480, 228803 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- Y. Zhang, T.-T. Zuo, J. Popovic, K. Lim, Y.-X. Yin, J. Maier and Y.-G. Guo, Mater. Today, 2020, 33, 56–74 CrossRef CAS.
- K. Qin, K. Holguin, M. Mohammadiroudbari, J. Huang, E. Y. S. Kim, R. Hall and C. Luo, Adv. Funct. Mater., 2021, 31, 2009694 CrossRef CAS.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 CrossRef CAS.
- M. Ue and K. Uosaki, Curr. Opin. Electrochem., 2019, 17, 106–113 CrossRef CAS.
- D. Campanella, D. Belanger and A. Paolella, J. Power Sources, 2021, 482, 228949 CrossRef CAS.
- K. Liu, Z. Wang, L. Shi, S. Jungsuttiwong and S. Yuan, J. Energy Chem., 2021, 59, 320–333 CrossRef.
- I. Osada, H. De Vries, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS PubMed.
- P. Ding, Z. Lin, X. Guo, L. Wu, Y. Wang, H. Guo, L. Li and H. Yu, Mater. Today, 2021, 51, 449–474 CrossRef CAS.
- D. Aurbach and E. Granot, Electrochim. Acta, 1997, 42, 697–718 CrossRef CAS.
- D. Brouillette, G. Perron and J. E. Desnoyers, J. Solution Chem., 1998, 27, 151–182 CrossRef CAS.
- D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski and J. E. Desnoyers, Phys. Chem. Chem. Phys., 2002, 4, 6063–6071 RSC.
- S. Tobishima, H. Morimoto, M. Aoki, Y. Saito, T. Inose, T. Fukumoto and T. Kuryu, Electrochim. Acta, 2004, 49, 979–987 CrossRef CAS.
- W. A. Henderson, F. McKenna, M. A. Khan, N. R. Brooks, V. G. Young and R. Frech, Chem. Mater., 2005, 17, 2284–2289 CrossRef CAS.
- W. A. Henderson, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS PubMed.
- C. Zhang, D. Ainsworth, Y. G. Andreev and P. G. Bruce, J. Am. Chem. Soc., 2007, 129, 8700–8701 CrossRef CAS PubMed.
- A. Plewa, M. Kalita and M. Siekierski, Electrochim. Acta, 2007, 53, 1527–1534 CrossRef CAS.
- A. Plewa-Marczewska, M. Kalita, M. Marczewski and M. Siekierski, Electrochim. Acta, 2010, 55, 1389–1395 CrossRef CAS.
- S. Tang and H. Zhao, RSC Adv., 2014, 4, 11251 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- F. Guo, W. Hase, Y. Ozaki, Y. Konno, M. Inatsuki, K. Nishimura, N. Hashimoto and O. Fujita, Exp. Therm. Fluid Sci., 2019, 109, 109858 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- E. R. Logan, E. M. Tonita, K. L. Gering, L. Ma, M. K. G. Bauer, J. Li, L. Y. Beaulieu and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A705–A716 CrossRef CAS.
- A. Conesa, S. Shen and A. Coronas, Int. J. Thermophys., 1998, 19, 1343–1358 CrossRef CAS.
- K. Schoenhammer, H. Petersen, F. Guethlein and A. Goepferich, Pharm. Res., 2009, 26, 2568–2577 CrossRef CAS PubMed.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 CrossRef.
- R. Gond, W. van Ekeren, R. Mogensen, A. J. Naylor and R. Younesi, Mater. Horiz., 2021, 8, 2913–2928 RSC.
- Y. Liu, S. Fang, P. Shi, D. Luo, L. Yang and S. Hirano, J. Power Sources, 2016, 331, 445–451 CrossRef CAS.
- S. Li, F. Lorandi, H. Wang, T. Liu, J. F. Whitacre and K. Matyjaszewski, Prog. Polym. Sci., 2021, 122, 101453 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- Y. H. Jo, S. Li, C. Zuo, Y. Zhang, H. Gan, S. Li, L. Yu, D. He, X. Xie and Z. Xue, Macromolecules, 2020, 53, 1024–1032 CrossRef CAS.
- X.-L. Wang, A. Mei, X.-L. Li, Y.-H. Lin and C.-W. Nan, J. Power Sources, 2007, 171, 913–916 CrossRef CAS.
- G. Derrien, J. Hassoun, S. Sacchetti and S. Panero, Solid State Ionics, 2009, 180, 1267–1271 CrossRef CAS.
- S. Li, F. Lorandi, J. F. Whitacre and K. Matyjaszewski, Macromol. Chem. Phys., 2020, 221, 1900379 CrossRef CAS.
- C. Li, S. Liu, C. Shi, G. Liang, Z. Lu, R. Fu and D. Wu, Nat. Commun., 2019, 10, 1363 CrossRef PubMed.
- T. Tamura, T. Hachida, K. Yoshida, N. Tachikawa, K. Dokko and M. Watanabe, J. Power Sources, 2010, 195, 6095–6100 CrossRef CAS.
- A. Orita, K. Kamijima, M. Yoshida, K. Dokko and M. Watanabe, J. Power Sources, 2011, 196, 3874–3880 CrossRef CAS.
- S. Seki, K. Takei, H. Miyashiro and M. Watanabe, J. Electrochem. Soc., 2011, 158, A769 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Electrochem. Soc., 2012, 159, A1005–A1012 CrossRef CAS.
- S. Seki, N. Serizawa, K. Takei, K. Dokko and M. Watanabe, J. Power Sources, 2013, 243, 323–327 CrossRef CAS.
- N. Serizawa, S. Seki, K. Takei, H. Miyashiro, K. Yoshida, K. Ueno, N. Tachikawa, K. Dokko, Y. Katayama, M. Watanabe and T. Miura, J. Electrochem. Soc., 2013, 160, A1529–A1533 CrossRef CAS.
- C. Zhang, A. Yamazaki, J. Murai, J.-W. Park, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2014, 118, 17362–17373 CrossRef CAS.
- H. Hirayama, N. Tachikawa, K. Yoshii, M. Watanabe and Y. Katayama, Electrochemistry, 2015, 83, 824–827 CrossRef CAS.
- Y. Gambe, Y. Sun and I. Honma, Sci. Rep., 2015, 5, 8869 CrossRef CAS PubMed.
- D. Di Lecce, L. Carbone, V. Gancitano and J. Hassoun, J. Power Sources, 2016, 334, 146–153 CrossRef CAS.
- L. Carbone, M. Gobet, J. Peng, M. Devany, B. Scrosati, S. Greenbaum and J. Hassoun, J. Power Sources, 2015, 299, 460–464 CrossRef CAS.
- L. Carbone, D. Di Lecce, M. Gobet, S. Munoz, M. Devany, S. Greenbaum and J. Hassoun, ACS Appl. Mater. Interfaces, 2017, 9, 17085–17095 CrossRef CAS PubMed.
- D. Shanmukaraj, S. Lois, S. Fantini, F. Malbosc and M. Armand, Chem. Mater., 2018, 30, 246–251 CrossRef CAS.
- S. Wei, Z. Li, K. Kimura, S. Inoue, L. Pandini, D. Di Lecce, Y. Tominaga and J. Hassoun, Electrochim. Acta, 2019, 306, 85–95 CrossRef CAS.
- V. Marangon, Y. Tominaga and J. Hassoun, J. Power Sources, 2020, 449, 227508 CrossRef CAS.
- S. Wei, S. Inoue, D. di Lecce, Z. Li, Y. Tominaga and J. Hassoun, ChemElectroChem, 2020, 7, 2344–2344 CrossRef CAS.
- V. Marangon, L. Minnetti, M. Adami, A. Barlini and J. Hassoun, Energy Fuels, 2021, 35, 10284–10292 CrossRef CAS PubMed.
- S. Terada, K. Ikeda, K. Ueno, K. Dokko and M. Watanabe, Aust. J. Chem., 2019, 72, 70 CrossRef CAS.
- K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2015, 17, 8248–8257 RSC.
- R. Kido, K. Ueno, K. Iwata, Y. Kitazawa, S. Imaizumi, T. Mandai, K. Dokko and M. Watanabe, Electrochim. Acta, 2015, 175, 5–12 CrossRef CAS.
- L. Aguilera, S. Xiong, J. Scheers and A. Matic, J. Mol. Liq., 2015, 210, 238–242 CrossRef CAS.
- S. Seki, N. Serizawa, K. Takei, S. Tsuzuki, Y. Umebayashi, Y. Katayama, T. Miura, K. Dokko and M. Watanabe, RSC Adv., 2016, 6, 33043–33047 RSC.
- S. Saito, H. Watanabe, K. Ueno, T. Mandai, S. Seki, S. Tsuzuki, Y. Kameda, K. Dokko, M. Watanabe and Y. Umebayashi, J. Phys. Chem. B, 2016, 120, 3378–3387 CrossRef CAS PubMed.
- K. Ueno, J. Murai, K. Ikeda, S. Tsuzuki, M. Tsuchiya, R. Tatara, T. Mandai, Y. Umebayashi, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2016, 120, 15792–15802 CrossRef CAS.
- J. Peng, L. Carbone, M. Gobet, J. Hassoun, M. Devany and S. Greenbaum, Electrochim. Acta, 2016, 213, 606–612 CrossRef CAS.
- M. Callsen, K. Sodeyama, Z. Futera, Y. Tateyama and I. Hamada, J. Phys. Chem. B, 2017, 121, 180–188 CrossRef CAS PubMed.
- P. Jankowski, M. Dranka, W. Wieczorek and P. Johansson, J. Phys. Chem. Lett., 2017, 8, 3678–3682 CrossRef CAS PubMed.
- K. Hayamizu, Electrochim. Acta, 2017, 254, 101–111 CrossRef CAS.
- M. Saito, S. Yamada, T. Ishikawa, H. Otsuka, K. Ito and Y. Kubo, RSC Adv., 2017, 7, 49031–49040 RSC.
- D. Dong, F. Sälzer, B. Roling and D. Bedrov, Phys. Chem. Chem. Phys., 2018, 20, 29174–29183 RSC.
- J. Popovic, D. Höfler, J. P. Melchior, A. Münchinger, B. List and J. Maier, J. Phys. Chem. Lett., 2018, 9, 5116–5120 CrossRef CAS PubMed.
- Y. Sun and I. Hamada, J. Phys. Chem. B, 2018, 122, 10014–10022 CrossRef CAS PubMed.
- M. Nojabaee, J. Popovic and J. Maier, J. Mater. Chem. A, 2019, 7, 13331–13338 RSC.
- J.-D. Xie, W.-J. Liu, C. Li, J. Patra, Y. A. Gandomi, Q.-F. Dong and J.-K. Chang, Electrochim. Acta, 2019, 319, 625–633 CrossRef CAS.
- N. Arai, H. Watanabe, T. Yamaguchi, S. Seki, K. Ueno, K. Dokko, M. Watanabe, Y. Kameda, R. Buchner and Y. Umebayashi, J. Phys. Chem. C, 2019, 123, 30228–30233 CrossRef CAS.
- F. Schmidt and M. Schönhoff, J. Phys. Chem. B, 2020, 124, 1245–1252 CrossRef CAS PubMed.
- N. Arai, H. Watanabe, E. Nozaki, S. Seki, S. Tsuzuki, K. Ueno, K. Dokko, M. Watanabe, Y. Kameda and Y. Umebayashi, J. Phys. Chem. Lett., 2020, 11, 4517–4523 CrossRef CAS PubMed.
- K. Shigenobu, K. Dokko, M. Watanabe and K. Ueno, Phys. Chem. Chem. Phys., 2020, 22, 15214–15221 RSC.
- K. Shigenobu, M. Shibata, K. Dokko, M. Watanabe, K. Fujii and K. Ueno, Phys. Chem. Chem. Phys., 2021, 23, 2622–2629 RSC.
- J. Kim, J. Hassoun, S. Panero, Y. K. Sun and B. Scrosati, Green, 2011, 1, 323–328 CAS.
- K. Dokko, N. Tachikawa, K. Yamauchi, M. Tsuchiya, A. Yamazaki, E. Takashima, J.-W. Park, K. Ueno, S. Seki, N. Serizawa and M. Watanabe, J. Electrochem. Soc., 2013, 160, A1304–A1310 CrossRef CAS.
- K. Ueno, J.-W. Park, A. Yamazaki, T. Mandai, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2013, 117, 20509–20516 CrossRef CAS.
- L. Carbone, M. Gobet, J. Peng, M. Devany, B. Scrosati, S. Greenbaum and J. Hassoun, ACS Appl. Mater. Interfaces, 2015, 7, 13859–13865 CrossRef CAS PubMed.
- M. Cuisinier, C. Hart, M. Balasubramanian, A. Garsuch and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1401801 CrossRef.
- M. Agostini, S. Xiong, A. Matic and J. Hassoun, Chem. Mater., 2015, 27, 4604–4611 CrossRef CAS.
- C. Li, A. L. Ward, S. E. Doris, T. A. Pascal, D. Prendergast and B. A. Helms, Nano Lett., 2015, 15, 5724–5729 CrossRef CAS PubMed.
- H. Lu, Y. Yuan, Z. Hou, Y. Lai, K. Zhang and Y. Liu, RSC Adv., 2016, 6, 18186–18190 RSC.
- K. Ueno, Electrochemistry, 2016, 84, 674–680 CrossRef CAS.
- L. Carbone, J. Peng, M. Agostini, M. Gobet, M. Devany, B. Scrosati, S. Greenbaum and J. Hassoun, ChemElectroChem, 2017, 4, 209–215 CrossRef CAS.
- S. Usuki, S. Uchida, Y. Matsui, M. Yamagata, H. Hinago and M. Ishikawa, Electrochemistry, 2017, 85, 650–655 CrossRef CAS.
- S. Seki, N. Serizawa, K. Takei, Y. Umebayashi, S. Tsuzuki and M. Watanabe, Electrochemistry, 2017, 85, 680–682 CrossRef CAS.
- Q. Zhang, D. C. Bock, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, J. Electrochem. Soc., 2017, 164, A897–A901 CrossRef CAS.
- S. Okabe, S. Uchida, Y. Matsui, M. Yamagata and M. Ishikawa, Electrochemistry, 2017, 85, 671–674 CrossRef CAS.
- M. Haruta, T. Moriyasu, A. Tomita, T. Takenaka, T. Doi and M. Inaba, J. Electrochem. Soc., 2018, 165, A1874–A1879 CrossRef CAS.
- Y. Ishino, K. Takahashi, W. Murata, Y. Umebayashi, S. Tsuzuki, M. Watanabe, M. Kamaya and S. Seki, Energy Technol., 2019, 7, 1900197 CrossRef CAS.
- D. Di Lecce, V. Marangon, A. Benítez, Á. Caballero, J. Morales, E. Rodríguez-Castellón and J. Hassoun, J. Power Sources, 2019, 412, 575–585 CrossRef CAS.
- A. Gupta, A. Bhargav, J.-P. Jones, R. V. Bugga and A. Manthiram, Chem. Mater., 2020, 32, 2070–2077 CrossRef CAS PubMed.
- U. Košir, I. K. Cigić, J. Markelj, S. D. Talian and R. Dominko, Electrochim. Acta, 2020, 363, 137227 CrossRef.
- M. Nojabaee, K. Küster, U. Starke, J. Popovic and J. Maier, Small, 2020, 16, 2000756 CrossRef CAS PubMed.
- S. Dörfler, H. Althues, P. Härtel, T. Abendroth, B. Schumm and S. Kaskel, Joule, 2020, 4, 539–554 CrossRef.
- V. Marangon, D. di Lecce, L. Minnetti and J. Hassoun, ChemElectroChem, 2021, 8, 3971–3981 CrossRef CAS.
- J. Hassoun, F. Croce, M. Armand and B. Scrosati, Angew. Chem.,– Int. Ed., 2011, 50, 2999–3002 CrossRef CAS PubMed.
- H. G. Jung, J. Hassoun, J. B. Park, Y. K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS PubMed.
- W. Xu, J. Hu, M. H. Engelhard, S. A. Towne, J. S. Hardy, J. Xiao, J. Feng, M. Y. Hu, J. Zhang, F. Ding, M. E. Gross and J. G. Zhang, J. Power Sources, 2012, 215, 240–247 CrossRef CAS.
- J. Hassoun, H.-G. Jung, D.-J. Lee, J.-B. Park, K. Amine, Y.-K. Sun and B. Scrosati, Nano Lett., 2012, 12, 5775–5779 CrossRef CAS PubMed.
- F. Li, T. Zhang, Y. Yamada, A. Yamada and H. Zhou, Adv. Energy Mater., 2013, 3, 532–538 CrossRef CAS.
- K. U. Schwenke, S. Meini, X. Wu, H. A. Gasteiger and M. Piana, Phys. Chem. Chem. Phys., 2013, 15, 11830 RSC.
- J. B. Park, J. Hassoun, H. G. Jung, H. S. Kim, C. S. Yoon, I. H. Oh, B. Scrosati and Y. K. Sun, Nano Lett., 2013, 13, 2971–2975 CrossRef CAS PubMed.
- E. N. Nasybulin, W. Xu, B. L. Mehdi, E. Thomsen, M. H. Engelhard, R. C. Massé, P. Bhattacharya, M. Gu, W. Bennett, Z. Nie, C. Wang, N. D. Browning and J.-G. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 14141–14151 CrossRef CAS PubMed.
- G. A. Elia, R. Bernhard and J. Hassoun, RSC Adv., 2015, 5, 21360–21365 RSC.
- B. D. Adams, R. Black, Z. Williams, R. Fernandes, M. Cuisinier, E. J. Berg, P. Novak, G. K. Murphy and L. F. Nazar, Adv. Energy Mater., 2015, 5, 1400867 CrossRef.
- D. Sharon, D. Hirsberg, M. Afri, F. Chesneau, R. Lavi, A. a. Frimer, Y.-K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2015, 7, 16590–16600 CrossRef CAS PubMed.
- M. Saito, S. Yamada, T. Fujinami, S. Kosaka, Y. Tachikawa, K. Ito and Y. Kubo, ECS Trans., 2017, 75, 53–58 CrossRef CAS.
- D. Sharon, D. Hirsberg, M. Salama, M. Afri, A. A. Frimer, M. Noked, W. Kwak, Y.-K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 5300–5307 CrossRef CAS PubMed.
- S. Wu, K. Zhu, J. Tang, K. Liao, S. Bai, J. Yi, Y. Yamauchi, M. Ishida and H. Zhou, Energy Environ. Sci., 2016, 9, 3262–3271 RSC.
- H.-M. Kwon, M. L. Thomas, R. Tatara, A. Nakanishi, K. Dokko and M. Watanabe, Chem. Lett., 2017, 46, 573–576 CrossRef CAS.
- M. Saito, T. Fujinami, S. Yamada, T. Ishikawa, H. Otsuka, K. Ito and Y. Kubo, J. Electrochem. Soc., 2017, 164, A2872–A2880 CrossRef CAS.
- D. Hirshberg, D. Sharon, E. De La Llave, M. Afri, A. A. Frimer, W.-J. Kwak, Y.-K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2017, 9, 4352–4361 CrossRef CAS PubMed.
- H.-M. Kwon, M. L. Thomas, R. Tatara, Y. Oda, Y. Kobayashi, A. Nakanishi, K. Ueno, K. Dokko and M. Watanabe, ACS Appl. Mater. Interfaces, 2017, 9, 6014–6021 CrossRef CAS PubMed.
- A. Chamaani, M. Safa, N. Chawla, M. Herndon and B. El-Zahab, J. Electroanal. Chem., 2018, 815, 143–150 CrossRef CAS.
- L. Carbone, P. T. Moro, M. Gobet, S. Munoz, M. Devany, S. G. Greenbaum and J. Hassoun, ACS Appl. Mater. Interfaces, 2018, 10, 16367–16375 CrossRef CAS PubMed.
- Y. Wang, N.-C. Lai, Y.-R. Lu, Y. Zhou, C.-L. Dong and Y.-C. Lu, Joule, 2018, 2, 2364–2380 CrossRef CAS.
- G. Horwitz, C. Rodríguez, M. Factorovich and H. R. Corti, J. Phys. Chem. C, 2019, 123, 12081–12087 CrossRef CAS.
- M. Tang, J.-C. Chang, S. R. Kumar and S. J. Lue, Energy, 2019, 187, 115926 CrossRef CAS.
- Y. Hayashi, S. Yamada, T. Ishikawa, Y. Takamuki, M. Sohmiya, H. Otsuska, K. Ito, Y. Kubo and M. Saito, J. Electrochem. Soc., 2020, 167, 020542 CrossRef CAS.
- G. Horwitz, E. J. Calvo, L. P. Méndez De Leo and E. de la Llave, Phys. Chem. Chem. Phys., 2020, 22, 16615–16623 RSC.
- V. Marangon, C. Hernandez-Rentero, S. Levchenko, G. Bianchini, D. Spagnolo, A. Caballero, J. Morales and J. Hassoun, ACS Appl. Energy Mater., 2020, 3, 12263–12275 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 RSC.
- L. Goldie-Scot, https://about.bnef.com/blog/behind-scenes-take-lithium-ion-battery-prices/, BloombergNEF, March, 5, 2019.
- D. J. Eyckens and L. C. Henderson, Front. Chem., 2019, 7, 263 CrossRef CAS.
- D. Morales, R. E. Ruther, J. Nanda and S. Greenbaum, Electrochim. Acta, 2019, 304, 239–245 CrossRef CAS.
- L. Carbone, S. G. Greenbaum and J. Hassoun, Sustainable Energy Fuels, 2017, 1, 228–247 RSC.
- J. B. Robinson, K. Xi, R. V. Kumar, A. C. Ferrari, H. Au, M.-M. Titirici, A. Parra-Puerto, A. Kucernak, S. D. S. Fitch, N. Garcia-Araez, Z. L. Brown, M. Pasta, L. Furness, A. J. Kibler, D. A. Walsh, L. R. Johnson, C. Holc, G. N. Newton, N. R. Champness, F. Markoulidis, C. Crean, R. C. T. Slade, E. I. Andritsos, Q. Cai, S. Babar, T. Zhang, C. Lekakou, N. Kulkarni, A. J. E. Rettie, R. Jervis, M. Cornish, M. Marinescu, G. Offer, Z. Li, L. Bird, C. P. Grey, M. Chhowalla, D. Di Lecce, R. E. Owen, T. S. Miller, D. J. L. Brett, S. Liatard, D. Ainsworth and P. R. Shearing, J. Phys.: Energy, 2021, 3, 031501 CAS.
- W. Chen, T. Lei, C. Wu, M. Deng, C. Gong, K. Hu, Y. Ma, L. Dai, W. Lv, W. He, X. Liu, J. Xiong and C. Yan, Adv. Energy Mater., 2018, 8, 1702348 CrossRef.
- N. Mahne, O. Fontaine, M. O. Thotiyl, M. Wilkening and S. A. Freunberger, Chem. Sci., 2017, 8, 6716–6729 RSC.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- W. Xu, V. V. Viswanathan, D. Wang, S. A. Towne, J. Xiao, Z. Nie, D. Hu and J.-G. Zhang, J. Power Sources, 2011, 196, 3894–3899 CrossRef CAS.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- S. A. Freunberger, Y. Chen, F. Bardé, K. Takechi, F. Mizuno and P. G. Bruce, in The Lithium Air Battery, eds. N. Imanishi, A. C. Luntz and P. Bruce, Springer New York, New York, NY, 2014, pp. 23–58 Search PubMed.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS.
- H.-G. Jung, J. Hassoun, J.-B. Park, Y.-K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |