
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid automated total arsenic and arsenic speciation by inductively coupled plasma mass spectrometry†
C. Derrick
Quarles
Jr
 *,
Patrick
Sullivan
,
Nick
Bohlim
and
Nathan
Saetveit
*,
Patrick
Sullivan
,
Nick
Bohlim
and
Nathan
Saetveit
Elemental Scientific, Inc., 7277 World Communications Dr., Omaha, NE, USA. E-mail: derrick.quarles@icpms.com
First published on 20th May 2022
Abstract
This work focuses on providing fast and reliable separations of arsenobetaine (AsB), trimethylarsine oxide (TMAO), dimethylarsinic acid (DMA), monomethylarsonic acid (MMA), arsenocholine (AsC), arsenite (As(III)), and arsenate (As(V)). Two different methods are presented: (1) a one-column method for the determination of AsB, DMA, MMA, AsC, As(III), and As(V) with a separation time of ∼2 minutes and (2) a two-column method for the determination of AsB, TMAO, DMA, MMA, AsC, As(III), and As(V) with a separation time of ∼4.5 minutes. Recovery of the two methods falls between 94 and 107%. Methods were evaluated for accuracy by analyzing proficiency samples from Centre de Toxicologie du Québec (CTQ) and New York Department of Health (NYDOH). Correlation between the measured values and reference values was very good, with a <4.5% difference in results. Limits of detection in a urine matrix ranged from 2.8–6.0 ng L−1 As and 4.1–9.1 ng L−1 As for the one- and two-column methods, respectively.
Introduction
Determining the total amount of a particular element or suite of elements in specific samples is an important and routine method in all analytical laboratories today. However, to truly assess the level of toxicity, bioavailability, and/or stability of some elements, the chemical species present also needs to be identified.1,2 The chemical species can be defined as an element with a specific isotopic composition, oxidation state, and/or molecular structure.3 Some of the most common elements that are monitored for their exact chemical species are arsenic, mercury, and chromium, however, many other elements are also studied. Advancements in analytical instrumentation over the years has led to elemental speciation methodologies that are important to toxicological, clinical, environmental, food, pharmaceutical, and geochemistry fields.Arsenic is perhaps one of the most studied elements for understanding its chemical species and potential toxicity. Arsenic is a naturally occurring element in the Earth's crust and is found in soil around the world at varying amounts, 0.1–40 mg kg−1.4 Arsenic is or has been used in pesticides, herbicides, food additives, drugs, poisons, and chemical weapons.4–6 Arsenic can be found in many different chemical forms. The inorganic forms, arsenite (As(III)) or arsenate (As(V)), are more toxic due to higher bioavailability. Some organic forms (dimethylarsinic acid (DMA) and monomethylarsonic acid (MMA)) are less toxic due to a lower bioavailability, while other organic forms of arsenic such as arsenobetaine (AsB), arsenocholine (AsC), and trimethylarsine oxide (TMAO) are considered non-toxic.7–11
The most common routes of arsenic exposure are from drinking water, food consumption (such as rice or seafood), or industrial exposures.2,8,12 Arsenic is excreted in the urine, therefore measuring urinary arsenic levels can help identify any exposure that had occurred within the previous 24–48 h.13 However, simply measuring the total arsenic levels will not reveal the full impact of the potential exposure. To fully assess the overall health implications for individuals with elevated levels of arsenic, the chemical form of the arsenic species must be identified. The most common methodology for measuring total arsenic is performed using an inductively coupled plasma-mass spectrometer (ICP-MS), whereas determining the arsenic species is typically done by chromatographic separation prior to introduction to the ICP-MS. A large amount of work has been dedicated to this topic in recent decades, however the resulting methods are generally lengthy, offer only a sub-set of the arsenic species, and/or have high operational costs.2,11,14–26 Wegwerth et al. presented the fastest method (2 minutes) for arsenic speciation to date but it did not include TMAO.25 Ciardullo et al. presented a method for seven arsenic species (AsB, AsC, DMA, MMA, TMAO, As(III), and As(V)), however 25 minutes were needed to complete the separation.15
In this work, the sample introduction system for total arsenic and the chromatographic separation of the arsenic species are performed within a single platform automation system (prepFAST IC) connected to a single ICP-MS. This provides automation in the sample preparation and delivery, but also reduces the potential bias of having two completely different setups for these measurements. Two different arsenic speciation methods were developed and evaluated for AsB, AsC, DMA, MMA, As(III), As(V), and TMAO. The methods were evaluated for column recovery, accuracy, precision, and limits of detection. Accuracy of the methods were evaluated by analyzing proficiency testing samples from the Centre de Toxicologie du Québec (CTQ) and New York Department of Health (NYDOH).
Methods
Materials and reagents
All solutions were prepared using 18.2 MΩ cm water from an EMD Millipore high-purity filtration system (Millipore Sigma, Burlington, MA, USA). Nitric acid (70%, Seastar, Sidney, BC, CAN), Triton X-100 (Laboratory Grade, Millipore Sigma), sodium hydroxide (ACS Reagent Grade, Millipore Sigma), and ammonium carbonate (ACS Reagent Grade, JT Baker, Avantor Performance Materials, LLC, Radnor, PA, USA) were used to prepare the following solutions. For total arsenic measurements, the prepFAST carrier, diluent, and rinse were prepared with 2% (v/v) nitric acid. The internal standard was prepared with 2% (v/v) nitric acid and 100 μg L−1 Ga (Elemental Scientific, Inc., Omaha, NE, USA). The sample carrier, syringe-driven carrier supplied by the prepFAST IC chromatography module, and the autosampler rinse 1 consisted of 2% nitric and 0.05% Triton X-100. Autosampler rinse 2 and the working solution consisted of DI water. Synthetic clinical matrix (CLIN-0500, Elemental Scientific, Inc.) was used to matrix-match the calibration curves for direct mode analysis of urine samples; no matrix-matching was used for the urine As speciation analysis.Sample preparation
Calibration curves for the direct measurements were prepared using a stock solution containing 100 μg L−1 As (1000 mg L−1 As, Elemental Scientific). The stock standard was diluted inline using the dilution factors of 200, 100, 50, 20, 10, and 5×, resulting in a calibration of 0.5–20 μg L−1 As. Calibration curves for the six arsenic speciation measurements were prepared using a stock standard of 100 μg L−1 AsB, DMA, As(III), AsC, MMA, and As(V) (10 mg L−1 per individual species, Elemental Scientific). Calibration curves for the seven arsenic speciation measurements were prepared using a stock standard of 100 μg L−1 AsB, TMAO, DMA, As(III), AsC, MMA, and As(V) (10 mg L−1 per individual species, Elemental Scientific). Proficiency testing (PT) samples were obtained from the Centre de Toxicologie du Québec (CTQ) and New York Department of Health (NYDOH). The PT samples were stored at 4 °C until used. Prior to analysis the samples were allowed to warm up to room temperature. Urine spike samples were prepared by collecting anonymous urine and these were manually spiked with 10 μg L−1 of each arsenic species. The spiked urine samples were used to evaluate recovery on a per species basis and provide samples with known TMAO levels since the available reference materials used in this work did not contain TMAO.Sample introduction
Standards and samples were introduced to the ICP-MS using a prepFAST IC Clinical system (Elemental Scientific), which contains 8 quartz syringes. The prepFAST IC is an automated total metals and chromatography system, which includes an autosampler and the ability to dilute standards and samples inline to the ICP-MS. The prepFAST IC system can operate in total metals or elemental speciation modes with no physical changes required. The sample or standard is taken up from the autosampler deck, prepared inline, and then passes through a column valve that is either inline or offline depending on the method selected. For example, the column is in the offline position when analyzing for total metals and inline when performing elemental speciation such as arsenic speciation. The prepFAST module syringes perform inline dilutions: syringe 1 used for cleaning loops, syringe 2 is used as a carrier to move the sample into the diluted sample loop, syringe 3 is used to dilute samples/standards, syringe 4 adds clinical matrix, and syringe 5 is used for sample loading of precise volumes. The speciation module syringes contain 3 high-pressure syringes (≤1500 psi or ≤100 bar) that are used as carrier (direct mode) or eluent 1 (speciation mode) to the ICP-MS (syringe 6), eluent 2 (syringe 7), and post-column internal standard (syringe 8). In direct mode, the carrier (flow rate = 300 μL min−1) was 2% (v/v) nitric acid and in speciation mode eluent 1 was 0.5 mM ammonium carbonate, pH = 9.5. These two modes are automatically switched between in the software. When in speciation mode, eluent 2 consisted of 80 mM ammonium carbonate, pH = 9.2. The chromatographic separation was performed using a flow rate of 1.0 mL min−1 for syringe 6 (eluent 1) and syringe 7 (eluent 2). The standard or sample is syringe loaded into a 200 μL sample loop and then transferred into a 1000 μL dilution loop prior to being introduced to the ICP-MS. When operating in direct mode, column valves A and B are bypassed such that the standard or sample is introduced directly to the ICP-MS with no chromatographic separation (Fig. 1a). Urine samples were diluted inline by 10× for all measurements. When operating in speciation mode, the standard or sample is introduced to column A (one-column method) or to both column A and then column B (two-column method) before being introduced to the ICP-MS (Fig. 1b). Column A is an anion exchange column (Elemental Scientific, CF-As-01, 4 × 50 mm) made up of quaternary amine groups, while column B is a C18 column (Elemental Scientific, CF-As-03, 4.6 × 125 mm). Dilution just before the column has been shown to eliminate any arsenic species interconversion that could take place.27 In speciation mode, 50 μL of sample is injected onto the column using a valve toggle method rather than the entire loop for the direct method.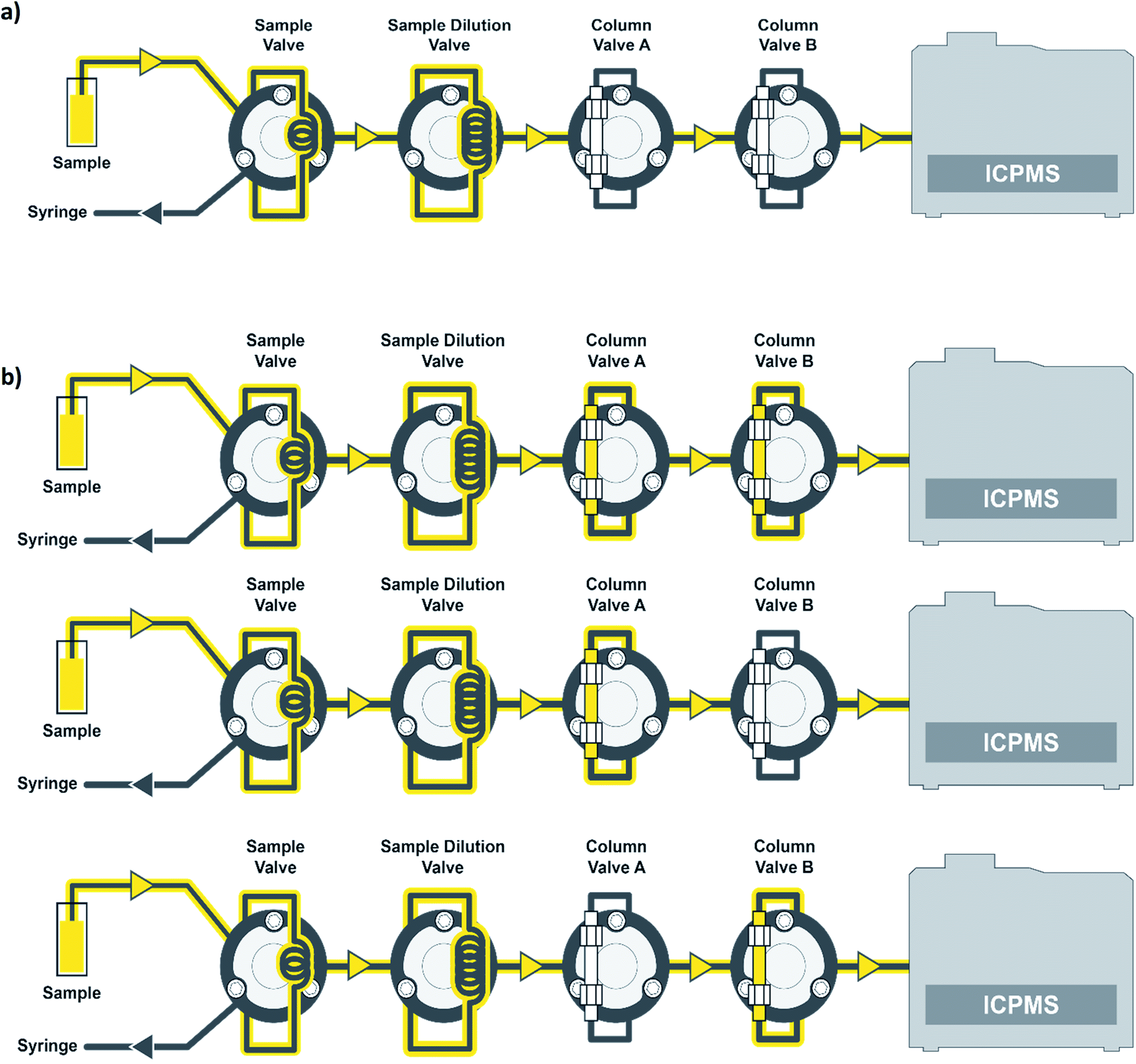 | ||
| Fig. 1 Simplified schematic of the prepFAST IC: (a) liquid flow path for total arsenic measurements and (b) liquid flow path for the one- and two-column arsenic species methods. The one-column method utilizes just column valve A, while the two-column method utilizes column valve A and B. | ||
ICP-MS
An Agilent 7900 ICP-MS (Agilent Technologies, Santa Clara, CA, USA) was utilized for all measurements. The plasma gas was set to 15 L min−1 Ar, auxiliary gas set to 0.9 L min−1 Ar, nebulizer gas set to 0.7 L min−1 Ar, and make-up gas set to 0.34 L min−1 Ar. The RF power was set to 1.5 kW. A PFA prepFAST ST nebulizer (PF-2040, Elemental Scientific), a Scott spray chamber, and a 2.5 mm torch injector were employed on the ICP-MS. The collision cell gas was set to 4.0 mL min−1 of He. The analytes (m/z) monitored were 71Ga and 75As for direct mode. Dwell times were set to 100 ms with 3 replicates for direct mode measurements. For the speciation measurements, the ICP-MS method was set to TRA mode, with 250 ms dwell time, and only 75As was monitored. All of the ICP-MS data was processed using Xceleri (Elemental Scientific).Results and discussion
We previously reported the separation of AsB, DMA, MMA, As(III), and As(V) utilizing ammonium phosphate buffer as the eluents.27 This method did not provide an adequate separation when AsC was present in the sample. Additionally, ammonium phosphate buffers could compromise the analysis of phosphorous for future measurements. Therefore, ammonium carbonate was selected as the eluent for the separation of AsB, As(III), DMA, AsC, MMA, and As(V). This separation was optimized to ∼2 minutes using a gradient step of 0.5 mM ammonium carbonate followed by 80 mM ammonium carbonate with an anion exchange column (Fig. 2a). In this separation AsB and TMAO both eluted in the void volume (Fig. 2a). Thus, if distinguishing between AsB and TMAO is critical a second method is required. Fig. 2b displays the separation of AsB, TMAO, As(III), DMA, AsC, MMA, and As(V) performed using the combination of an anion exchange column and a C18 column. The separation is done by sending the sample through the anion exchange column first; the AsB/TMAO peak elutes off of the column and passes into the C18 column. Both columns are on switching valves, which allows for the C18 column to be then switched offline at this point in the method, trapping the AsB and TMAO species. The As(III), DMA, AsC, MMA, and As(V) are eluted off of the anion exchange column, followed by a switch back to eluent 1 and the C18 column brought back online. Eluent 1 is then used to elute TMAO and AsB off of the C18 column. TMAO elutes first, due to the As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond that makes it slightly more polar than AsB. Two options are possible with this method: to bypass column 1 or keep column 1 inline. The advantage of having column 1 inline would be that eluent 1 passes through the anion exchange column providing extra conditioning prior to the next sample. No obvious differences in data quality were detected between these two options when eluting TMAO and AsB. The total separation time for the two-column method was optimized to ∼4.5 minutes. Table 1 displays how these two methods compare to recent published literature. While there are few publications on TMAO, whereas arsenic speciation methods have become very common in recent years.
O bond that makes it slightly more polar than AsB. Two options are possible with this method: to bypass column 1 or keep column 1 inline. The advantage of having column 1 inline would be that eluent 1 passes through the anion exchange column providing extra conditioning prior to the next sample. No obvious differences in data quality were detected between these two options when eluting TMAO and AsB. The total separation time for the two-column method was optimized to ∼4.5 minutes. Table 1 displays how these two methods compare to recent published literature. While there are few publications on TMAO, whereas arsenic speciation methods have become very common in recent years.
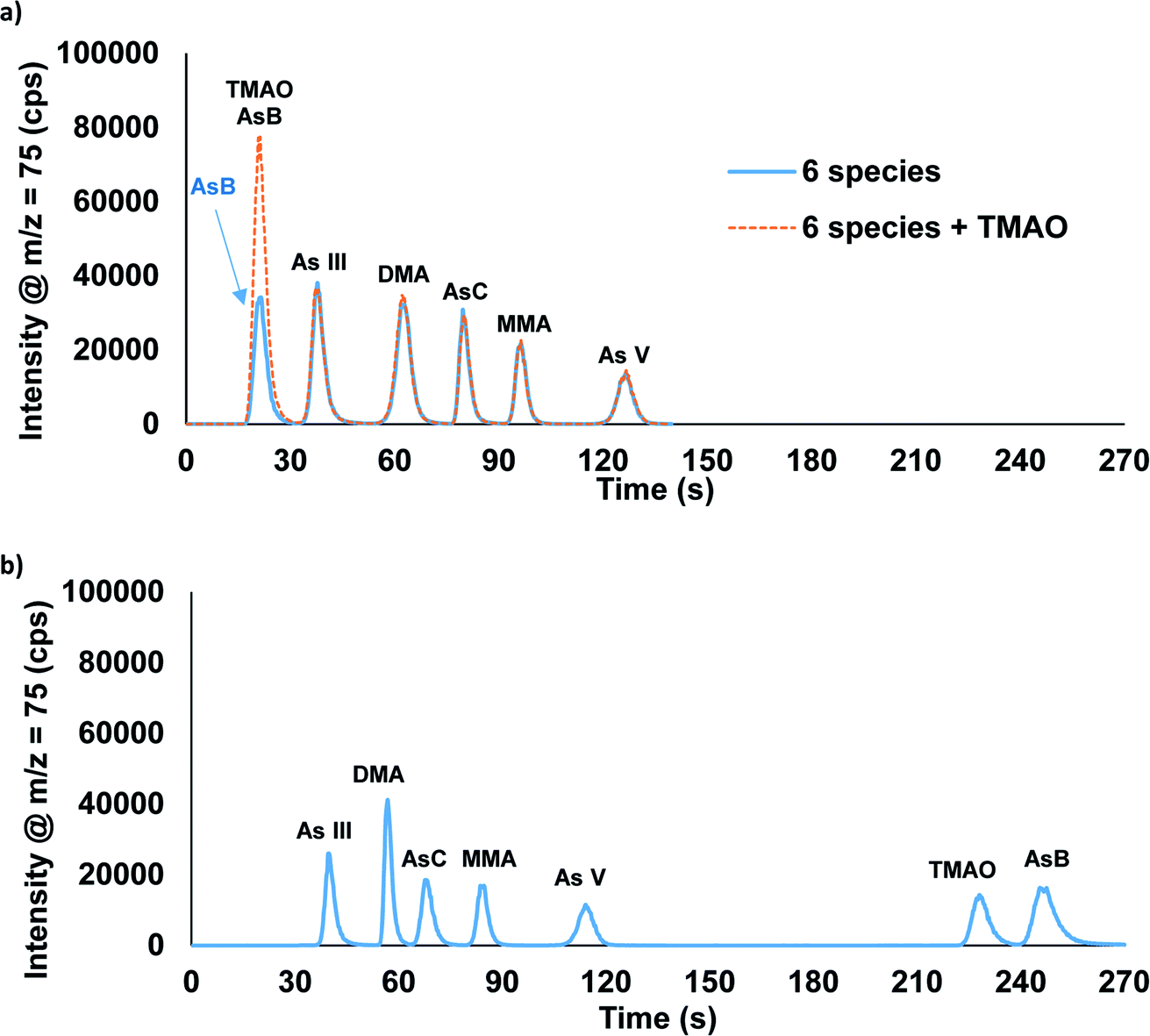 | ||
| Fig. 2 Chromatographic separation of AsB, TMAO, As(III), DMA, AsC, MMA, and As(V) using the (a) one-column and (b) two-column methods. Each species was spiked into the urine sample at 50 μg L−1 As. | ||
| Author | Year | Matrix | As species | Number of species | Time (min) |
|---|---|---|---|---|---|
| a Methods presented in this publication. | |||||
| Quarlesa | 2022 | Urine | AsB, As(III), DMA, AsC, MMA, As(V) | 6 | 2 |
| Quarlesa | 2022 | Urine | As(III), DMA, AsC, MMA, As(V), AsB, TMAO | 7 | 4.5 |
| Langasco19 | 2022 | Rice | As(III), DMA, MMA, As(V) | 4 | 10 |
| Barnet14 | 2021 | Rice | As(III), DMA, MMA, As(V) | 4 | 7 |
| Hwang17 | 2021 | Fish | AsC, AsB, As(III), DMA, MMA, As(V) | 6 | 7.5 |
| Kara18 | 2021 | Rice | AsC, AsB, As(III), DMA, MMA, As(V) | 6 | 35 |
| Montoro-Leal20 | 2021 | Urine | AsB, cacodylate, As(III), As(V) | 4 | 8 |
| Wegwerth25 | 2021 | Urine | AsB, As(III), DMA, AsC, MMA, As(V), Rox | 7 | 2 |
| Rodriguez11 | 2021 | Urine | AsC, AsB, As(III), DMA, MMA, As(V) | 6 | 28 |
| Song23 | 2021 | Urine | AsC/AsB, DMA, As(III), MMA, As(V) | 5 | 11 |
| Herath16 | 2020 | Rice | As(III), DMA, MMA, As(V) | 4 | 4.5 |
| Quarles27 | 2018 | Urine | AsB, DMA, As(III), MMA, As(V) | 5 | 5 |
| Savage22 | 2017 | Water | AsB/TMAO, iAs | 2 | 10 |
| Ciardullo15 | 2010 | Fish | As(III), As(V), MMA, DMA, AsB, TMAO, AsC | 7 | 25 |
| Tian24 | 2009 | Plants | As(V), As(III), MMA, DMA, TMAO | 5 | 8.5 |
| Ruiz-Chancho21 | 2008 | Plants | As, TMAO | 2 | 6 |
| Zhao26 | 2006 | Plants, soils | As(III)/As(V), MMA, DMA, TMAO | 4 | 1.2 |
The one- and two-column methods were evaluated for recovery. Table 2 displays the results from the analysis of a urine sample that had been spiked with 10 μg L−1 of all 7 species being studied. TMAO was not included in the one-column method. Both methods had very good recovery that ranged from 94–107% (one-column) and from 97–105% (two-column). The precision ranged from 0.9 to 9.9% RSD and 2.1 to 8.0% RSD, for the one- and two-column methods, respectively (n = 3). While not shown here, urine was spiked individually with each species which resulted in recoveries that ranged from 94–105% for both methods.
| As species | 6 species – one-column method | 7 species – two-column method |
|---|---|---|
| Measured value (μg L−1) | Measured value (μg L−1) | |
| As(III) | 10.5 ± 0.4 | 9.7 ± 0.2 |
| DMA | 9.4 ± 0.4 | 10.0 ± 0.8 |
| AsC | 10.7 ± 0.1 | 10.2 ± 0.6 |
| MMA | 10.0 ± 0.7 | 9.9 ± 0.3 |
| As(V) | 10.1 ± 1.0 | 10.4 ± 0.3 |
| TMAO | n/a | 9.8 ± 0.6 |
| AsB | 9.9 ± 0.7 | 10.5 ± 0.6 |
The limits of detection (LOD) were calculated for both the one- and two-column methods (Table 3). The LODs were determined by analyzing urine blanks (n = 10) and applying a 3σ criteria.28 The LODs for these two methods are slightly higher than the previously reported method (0.3–1.7 ng L−1),27 however the values are comparable or lower than the published values using similar methods which range from 3–100 ng L−1.25,29,30 The LODs for the one- and two-column methods are comparable, with the average LOD for the one-column method (4 ng L−1) slightly lower than the two-column method (7 ng L−1). The limit of quantification (LOQ), using a 10σ criteria for these two methods, ranges from 15–30 ng L−1. The linearity of both methods was excellent (R2 ≥ 0.9995) and the slopes were comparable between methods.
| One column method | Two-column method | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Response function | R 2 | SESlope | SEInt | LOD (ng L−1) | Response function | R 2 | SESlope | SEInt | LOD (ng L−1) | |
| a LOD = (3 × σblank)/m. m = slope. SE = standard error. | ||||||||||
| AsB | y = 7453x − 139 | 0.9995 | 0.038 | 0.020 | 3 | y = 7267x − 247 | 0.9999 | 0.014 | 0.008 | 9 |
| As(III) | y = 7499x − 326 | 0.9998 | 0.047 | 0.024 | 5 | y = 7418x − 256 | 0.9999 | 0.045 | 0.024 | 8 |
| DMA | y = 7853x − 309 | 0.9998 | 0.068 | 0.036 | 5 | y = 7940x − 637 | 0.9996 | 0.038 | 0.021 | 5 |
| AsC | y = 7271x − 236 | 0.9997 | 0.054 | 0.029 | 3 | y = 7503x − 277 | 0.9999 | 0.040 | 0.021 | 9 |
| MMA | y = 8055x − 441 | 0.9997 | 0.066 | 0.035 | 5 | y = 7991x − 59 | 0.9998 | 0.072 | 0.044 | 6 |
| As(V) | y = 8061x − 288 | 0.9996 | 0.050 | 0.027 | 6 | y = 8155x − 36 | 0.9999 | 0.057 | 0.031 | 4 |
| TMAO | — | — | — | — | — | y = 8358x − 455 | 0.9999 | 0.042 | 0.023 | 7 |
These methods were validated by analyzing proficiency testing samples from the NYDOH (5) and CTQ (11) programs. The PT samples were analyzed for total arsenic first to ensure the correct value was obtained for each sample. Table S1† displays the total arsenic reference and measured values for the 16 proficiency samples. There was excellent correlation between the targeted and measured values (Fig. S1†), which is supported by the linear regression slope of 0.9938 (SEslope = 7.12), where a perfect correlation would be equal to 1.0000.
Validation of the one-column and two-column methods were then performed following confirmation that the total arsenic values were correct. The proficiency samples can be separated into three groups: CTQ PC samples which provide a range of inorganic arsenic and total arsenic, CTQ QM samples which provide target values for each arsenic species, and the NYDOH UE samples which only provide target values for total arsenic. There was a fourth group included which were in-house spiked urine samples to ensure that there were target values for each species in the method since none of the proficiency testing samples provided values for AsC or TMAO.
Table S2† displays the reference values for total arsenic and inorganic arsenic for a direct comparison to the measured values for the six arsenic species via the one-column method. The measured values are reported by species, sum of the species, and total inorganic arsenic per sample. Fig. S2† displays a linear regression for the measured sum of arsenic species to the reference values. The correlation is very good (m = 1.0109, SEslope = 7.54) over a fairly wide range (4–631 μg L−1 total arsenic) of arsenic samples. Fig. S3† displays the inorganic reference values reported to the measured values. The slope for this linear regression is 0.9548 (SEslope = 1.70) which is being lowered by the highest concentration point. This sample had a reference value of 153 μg L−1 inorganic arsenic and a measured value of 146 μg L−1 inorganic arsenic, which equates to a −4.5% BIAS which is acceptable. If this point is removed the slope would be 0.9754 further supporting the method has good correlation. The final comparison was done between the reference and measured values for each arsenic species (Fig. S4†), which revealed a correlation that was close to perfect (m = 1.0008, SEslope = 1.35).
Table 4 displays the reference values for total arsenic and inorganic arsenic providing a direct comparison to the measured values for the seven arsenic species, two-column method. The measured values are reported by species, sum of the species, and total inorganic arsenic per sample, however in this experiment TMAO was also included. One noticeable difference from the previous study is that six of the proficiency testing samples had detectable levels of TMAO. Fig. S5† displays the comparison between the sum of the arsenic species measured and the reference total arsenic values. The correlation is very good (m = 0.9954, SEslope = 12.38) with only one data point that appears clearly off of the trend line. That data point was from PC-U-S2008 which had a reference value of 378 μg L−1 total arsenic and a measured value of 338 μg L−1 sum of arsenic species, which equates to a % BIAS of −10.6. When removing this data point the standard error of the slope goes from 12.38 to 4.96. Fig. S6† displays the comparison between the sum of the inorganic species measured to the reference inorganic arsenic values. The correlation between the inorganic species is excellent (m = 1.0018, SEslope = 2.98). Fig. S7† displays the comparison between the individual arsenic species measured using the two-column method and the reported values, with a slope of 0.9988 (SEslope = 1.20). The correlation between the measured values and the reference values were very good for both the one- and two-column methods, suggesting that either method can be used for reliable and accurate arsenic species measurements. Table S3† displays the CTQ QM reference value for each arsenic species and how it compares to the one- and two-column measured values. Two samples had reportable amounts of As(V) and MMA that were not on the provided reference values. The QM-U-Q2013 proficiency testing sample had 2.56 ± 0.19 μg L−1 As(V) and 2.91 ± 0.31 μg L−1 As(V) measured by the one- and two-column methods. The QM-U-Q2005 proficiency testing sample had 2.64 ± 0.23 μg L−1 MMA and 1.17 ± 0.09 μg L−1 MMA by the one- and two-column methods. These two samples were produced in 2005 and 2013, so it is not unreasonable to have some species interconversion over time which may be the cause for these species being measured. The fact that both methods detected As(V) and MMA further confirms the existence of each species in these samples.
| Total As target (ref. range) | iAs trget (ref. range) | Two-column method (μg L−1 As) | Sum | iAs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| As III | DMA | AsC | MMA | As V | TMAO | AsB | |||||
| PC-U-S1907 | 128 (108–148) | 2.44 (0.475–4.41) | 0.628 ± 0.035 | 1.92 ± 0.12 | 0 | 0 | 0.503 ± 0.025 | 1.67 ± 0.02 | 134 ± 11 | 139 | 1.13 |
| PC-U-S1908 | 172 (145–199) | 153 (122–184) | 0 | 2.05 ± 0.27 | 0 | 0.68 ± 0.07 | 148 ± 7 | 0 | 12.2 ± 0.6 | 163 | 148 |
| PC-U-S1912 | 631 (535–726) | 2.08 (0.123–4.03) | 2.32 ± 0.12 | 2.01 ± 0.08 | 0 | 0 | 0 | 2.35 ± 0.15 | 652 ± 32 | 659 | 2.32 |
| PC-U-S1913 | 26.2 (20.6–31.8) | 23.9 (17.5–30.3) | 19.4 ± 1.1 | 5.85 ± 0.43 | 0 | 0 | 1.42 ± 0.15 | 0 | 0 | 26.7 | 20.8 |
| PC-U-S2008 | 378 (326–430) | 2.73 (1.08–4.38) | 0.397 ± 0.047 | 6.04 ± 0.11 | 0 | 0.25 ± 0.03 | 1.40 ± 0.06 | 8.39 ± 0.55 | 322 ± 26 | 338 | 1.80 |
| QM-U-Q2004 | 93.3 (92.2–94.4) | 80.4 | 82.7 ± 4.9 | 2.39 ± 0.19 | 0 | 0 | 5.15 ± 0.61 | 0 | 6.87 ± 0.52 | 97.1 | 87.9 |
| QM-U-Q2005 | 49.9 (49.4–50.4) | 40.2 | 0 | 1.78 ± 0.07 | 0 | 1.17 ± 0.09 | 46.1 ± 6.0 | 0 | 3.20 ± 0.19 | 52.5 | 46.1 |
| QM-U-Q2006 | 32.4 (32.0–32.8) | 28.7 | 27.8 ± 2.4 | 1.89 ± 0.03 | 0 | 0 | 1.89 ± 0.21 | 0 | 1.49 ± 0.08 | 33.1 | 29.7 |
| QM-U-Q2013 | 42.3 (41.8–42.8) | 6.12 | 5.37 ± 0.25 | 22.0 ± 2.9 | 0 | 3.8 ± 0.4 | 2.91 ± 0.31 | 0 | 5.71 ± 0.34 | 39.8 | 8.28 |
| QM-U-Q2014 | 378 (374–382) | 0 | 0 | 2.95 ± 0.21 | 0 | 0 | 0 | 2.37 ± 0.49 | 374 ± 21 | 380 | 0 |
| QM-U-Q2015 | 86.0 (85.0–87.0) | 0 | 0 | 73.5 ± 5.1 | 0 | 0 | 0 | 0 | 9.88 ± 0.74 | 83.4 | 0 |
| UE19-10 | 188 (150–226) | — | 121 ± 8 | 1.88 ± 0.21 | 0.845 ± 0.100 | 0 | 55.7 ± 2.7 | 0 | 7.03 ± 0.38 | 186 | 177 |
| UE19-11 | 3.70 (0.0–9.7) | — | 0.308 ± 0.025 | 0.487 ± 0.033 | 1.28 ± 0.14 | 0 | 0.764 ± 0.076 | 0 | 3.01 ± 0.28 | 5.85 | 1.07 |
| UE20-06 | 61.0 (49–73) | — | 18.9 ± 1.8 | 7.75 ± 0.66 | 8.96 ± 0.79 | 3.24 ± 0.33 | 6.59 ± 0.52 | 2.28 ± 0.40 | 23.5 ± 2.3 | 71.2 | 25.5 |
| UE20-08 | 21.0 (15–27) | — | 19.7 ± 0.6 | 0.415 ± 0.040 | 1.92 ± 0.08 | 0 | 1.48 ± 0.01 | 0 | 2.11 ± 0.14 | 25.6 | 21.2 |
| UE20-10 | 101 (81–121) | — | 46.9 ± 4.1 | 9.94 ± 0.46 | 5.75 ± 0.55 | 0 | 17.6 ± 0.6 | 2.40 ± 0.20 | 22.7 ± 1.7 | 105 | 64.5 |
| Urine spike 1 | 70 | 20 | 9.35 ± 0.45 | 9.21 ± 0.51 | 10.5 ± 0.4 | 10.2 ± 0.1 | 10.3 ± 0.5 | 9.41 ± 0.33 | 9.99 ± 0.41 | 69.0 | 19.7 |
| Urine spike 2 | 70 | 20 | 9.59 ± 0.27 | 10.1 ± 0.4 | 10.2 ± 0.2 | 10.5 ± 0.6 | 10.8 ± 0.4 | 11.2 ± 0.8 | 10.7 ± 0.7 | 73.1 | 20.4 |
Conclusion
Two arsenic speciation methods were developed and validated in this study. The one-column, six arsenic species method provides a rapid and reliable method for samples where TMAO is of no importance or not present. When TMAO is of importance the two-column, seven arsenic species method provides a reliable method to distinguish the levels of TMAO and AsB. The column recovery of each arsenic species was found to range between 94 and 107% for the two methods presented in this work. The correlation for the total arsenic measurements from the reference values from the CTQ and NYDOH proficiency testing samples were excellent. Additionally, the correlation between the arsenic species sum, inorganic, and individual species measured values and the reference values was determined to be very good (<4.5% and <0.5% difference for the one- and two-column methods, respectively). Limits of detection in a urine matrix ranged from 2.8–6.0 ng L−1 As and 4.1–9.1 ng L−1 As for the one- and two-column methods, respectively.Conflicts of interest
The authors of this manuscript work for Elemental Scientific, Inc. which manufactures and sells the prepFAST IC used in this work.Acknowledgements
The authors would like to thank Andrew Toms for his support and guidance.References
- P. Apostoli, J. Anal. Chem., 1999, 363, 499–504 CAS.
- M. M. Nearing, I. Koch and K. J. Reimer, Spectrochim. Acta, Part B, 2014, 99, 150–162 CrossRef CAS.
- D. M. Templeton, F. Ariese, R. Cornelis, L.-G. Danielsson, H. Muntau, H. P. Van Leeuwen and R. Lobinski, Pure Appl. Chem., 2000, 72, 1453–1470 CrossRef CAS.
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201–235 CrossRef CAS PubMed.
- S. Chauhan, S. Chauhan, R. D'Cruz, S. Faruqi, K. K. Singh, S. Varma, M. Singh and V. Karthik, Environ. Toxicol. Pharmacol., 2008, 26, 113–122 CrossRef CAS PubMed.
- W. Lorenc, A. Hanc, A. Sajnog and D. Baralkiewicz, Mass Spectrom. Rev., 2022, 41, 32–50 CrossRef CAS PubMed.
- Fourth National Report on Human Exposure to Enviromental Chemicals, https://www.cdc.gov/exposurereport/pdf/FourthReport_UpdatedTables_Volume4_Mar2021-508.pdf, accessed February 9, 2022 Search PubMed.
- ATSDR Public Health Statement – Arsenic, https://www.atsdr.cdc.gov/ToxProfiles/tp2-c1-b.pdf, accessed February 9, 2022 Search PubMed.
- L. Benramdane, M. Accominotti, L. Fanton, D. Malicier and J.-J. Vallon, Clin. Chem., 1999, 45, 301–306 CrossRef CAS.
- V. W. Lai, Y. Sun, E. Ting, W. R. Cullen and K. J. Reimer, Toxicol. Appl. Pharmacol., 2004, 198, 297–306 CrossRef CAS PubMed.
- P. F. Rodriguez, R. M. Martin-Aranda, J. L. Lopez Colon and J. H. de Mendoza, Talanta, 2021, 221, 121494 CrossRef CAS PubMed.
- EPA Drinking Water Standard for Arsenic, https://nepis.epa.gov/Exe/ZyPdf.cgi?Dockey=20001XXC.txt, accessed February 9, 2022 Search PubMed.
- L. L. Yu, C. P. Verdon, W. C. Davis, G. C. Turk, K. L. Caldwell, R. L. Jones, B. Buckley and R. Xie, Anal. Methods, 2011, 3, 1107 RSC.
- L. S. Barnet, D. Pozebon, V. L. Dressler and D. Cioata, J. Food Compos. Anal., 2021, 99, 103849 CrossRef CAS.
- S. Ciardullo, F. Aureli, A. Raggi and F. Cubadda, Talanta, 2010, 81, 213–221 CrossRef CAS PubMed.
- I. Herath, P. Kumarathilaka, J. Bundchuh, A. Marchuk and J. Rinklebe, Talanta, 2020, 208, 120457 CrossRef CAS PubMed.
- I. M. Hwang, H. M. Lee, H.-W. Lee, J.-H. Jung, E. W. Moon, N. Khan and S. H. Kim, ACS Omega, 2021, 6, 19427–19434 CrossRef CAS PubMed.
- S. Kara, D. S. Chormey, A. Saygilar and S. Bakirdere, Food Chem., 2021, 356, 129706 CrossRef CAS PubMed.
- I. Langasco, F. Barracu, M. A. Deroma, J. F. Lopez-Sanchez, A. Mara, P. Meloni, M. I. Pilo, A. S. Estrugo, G. Sanna, N. Spano and A. Spanu, J. Environ. Manage., 2022, 302, 114105 CrossRef CAS PubMed.
- P. Montoro-Leal, J. C. Garci-Mesa, I. Morales-Benitez, A. Garcia de Torres and E. Vereda Alonso, Talanta, 2021, 235, 122769 CrossRef CAS PubMed.
- M. J. Ruiz-Chancho, J. F. Lopez-Sanchez, E. Schmeisser, W. Goessler, K. A. Francesconi and R. Rubio, Chemosphere, 2008, 71, 1522–1530 CrossRef CAS PubMed.
- L. Savage, M. Carey, M. Hossain, M. R. Islam, P. M. C. S. de Silva, P. N. Williams and A. A. Meharg, Environ. Sci. Technol., 2017, 51, 12210–12218 CrossRef CAS PubMed.
- X. Song, L. Huiling, C. Ma, W. Duan, Y. Sun, Y. Li, T. Huang and B. Zhou, At. Spectrosc., 2021, 42, 278–281 CAS.
- Y. Tian, M.-L. Chen, X.-W. Chen, J.-H. Wang, Y. Hirano, H. Sakamoto and I. Setsu, J. Anal. At. Spectrom., 2010, 25, 48–54 RSC.
- P. J. Wegwerth, S. A. Erdahl, M. L. Wermers, M. M. Hanley, S. J. Eckdahl and P. J. Jannetto, J. Appl. Lab. Med., 2021, 846–857 CrossRef PubMed.
- R. Zhao, M. Shao, H. Wang, Y. Taneike and X. Zhang, Sci. Total Environ., 2006, 371, 293–303 CrossRef CAS PubMed.
- C. D. Quarles, P. Sullivan, M. P. Field, S. Smith and D. R. Wiederin, J. Anal. At. Spectrom., 2018, 33, 745–751 RSC.
- A. Hubaux and G. Vos, Anal. Chem., 1970, 42, 849–855 CrossRef CAS.
- V. M. O. Carioni, J. A. McElroy, J. M. Guthrie, R. A. Ngwenyama and J. D. Brockman, Talanta, 2017, 165, 76–83 CrossRef CAS PubMed.
- E. Leese, J. Morton, E. Tan, P. H. Gardiner and V. A. Carolan, J. Anal. Toxicol., 2014, 38, 24–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ja00055e |
| This journal is © The Royal Society of Chemistry 2022 |