
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of blood and synthetic matrix-matched calibrations using manual and inline sample preparation methods†
C. Derrick
Quarles
Jr
 *,
Nick
Bohlim
,
Kevin
Wiederin
,
Nathan
Saetveit
and
Patrick
Sullivan
*,
Nick
Bohlim
,
Kevin
Wiederin
,
Nathan
Saetveit
and
Patrick
Sullivan
Elemental Scientific, Inc., 7277 World Communications Dr, Omaha, NE, USA. E-mail: derrick.quarles@icpms.com
First published on 1st June 2022
Abstract
Biomonitoring and clinical testing are important for improving human health. These tests help public health officials or medical doctors monitor the levels of essential elements and assess exposure to toxic or potentially toxic elements within the human body. While a great deal of work has been published on biomonitoring and clinical analyses, the majority of the work has been performed with manual sample preparation. This work will explore the use of two different automation platforms for clinical analyses, one for high-throughput sampling of manually prepared blood samples and the second an inline whole blood preparation method with micro-volume sampling. The comparison and validation of these systems was carried out by analyzing 2019 New York Department of Health Proficiency Testing samples that had known reference ranges for the analytes of interest (Cd, Hg, Mn, Se, and Pb). In addition, the ICP-MS methods were calibrated using two different matrix options, purchased base blood and synthetic clinical matrix, which were compared for background levels, detection limits, and accuracy relative to the reported reference values for the New York Department of Health Proficiency Testing samples.
Introduction
Biomonitoring, or the measurement of biomarkers in biological samples, is an important aspect of public health policy. Within the United States, the National Health and Nutrition Examination Survey (NHANES) conducted by the Centers for Disease Control and Prevention (CDC) uses biomonitoring information to better assess public health.1,2 Other countries, such as Canada, Germany, and France, also have implemented biomonitoring programs at the national level.3 Smaller-scale studies are also conducted at a national or local level to evaluate exposure over a specific time period or exposure for specific age groups.4–8 In addition to biomonitoring programs, clinical testing laboratories may also analyze biological samples to assess industrial exposures, monitor therapeutic levels, or to aid physicians in diagnostic evaluations of patients. Advancements in this field over the last decade have led to better understanding of how the association of metals may affect thyroid levels, prenatal exposures may lead to learning or social disabilities, the degradation of metal-on-metal implants changes over time, and increased blood lead levels may be correlated to multiple sclerosis patients.9–13Lead, cadmium, and mercury are well-known heavy metals, toxic to humans, with the severity dependent on the route and duration of exposure. Blood Pb measurements can be conducted to determine exposure levels, which will be indicative of acute and/or chronic exposure.14 There are numerous health effects associated with lead exposure for both children and adults;15 however, due to the neurological effects associated with exposure, the main testing focus has been on children. While no safe level of blood concentration has been identified for children, the CDC has set the current reference value for blood lead to 5 μg dL−1.16–18 Blood Cd measurements provide data related to recent exposure; however, if chronic exposure is suspected, analysis of a patient's urine is required.19 The health effects from cadmium exposure typically are related to kidney or lung damage in both children and adults.20,21 Mercury exposure may come from metallic, inorganic, or organic forms of mercury, with the form playing a role in the severity of the exposure. Blood Hg levels are a good indicator of body burden, as it relates to organic Hg exposure, but also can be indicative of recent exposure to metallic or inorganic mercury.1,22 Blood Hg levels do not provide long-term exposure information.23 In addition, most methods used for blood Hg do not provide information about the form of Hg, which requires more advanced techniques involving elemental speciation.
Elements such as chromium, copper, iron, manganese, nickel, selenium, and zinc are essential elements that are important to biological functions in the human body (e.g., metal cofactors for metalloproteins). When these elements are in excess or deficient compared to normal levels or present in specific forms/species which are toxic instead of beneficial, there can be negative health impacts. Expected normal levels can be altered due to nutritional intake, disease, or environmental/industrial exposure. For example, Wilsons disease causes excess copper accumulation within the body's organs, resulting in cell damage and necrosis.24–27 Iron deficiency is generally a result of inadequate nutritional intake, leading to iron-deficient anemia.28 Any environmental exposure to both heavy metals and essential elements alike comes with a chance to elevate levels within the body. Exposures come from numerous sources, such as energy production and/or metals processing, which releases chromium and other elements into the environment, or additives in gasoline such as methylcyclopentadienyl manganese tricarbonyl (MMT).29,30 Concern regarding increased chromium, nickel, or cobalt levels within the body due to metal-on-metal implant degradation has led to the need for increased monitoring.13,31 Manganese and selenium are typically measured in whole blood, whereas chromium, copper, iron, nickel, and zinc are typically measured in serum.
Inductively coupled plasma-mass spectrometry (ICP-MS) has become the preferred analytical technique for measuring biological samples due to superior detection limits, wide dynamic range, speed of analysis, and multi-element measurement capability as compared to other atomic spectroscopy techniques.13,32 The analysis of whole blood by ICP-MS has been well characterized in the literature; however, the methods by which samples are prepared for analysis vary greatly.1,32–42 A review of the literature by Ivanenko et al. reported three main sample treatment techniques: decomposition by acid digestion, dilution with an acidic matrix, or dilution with an alkaline matrix.36 Lu et al. compared digestion pretreatment to alkaline dilution for the analysis of Se, Cd, Pb, and Mn in blood and found that the alkali method provided better precision and required less sample (200 μL of whole blood) than the digestion technique (500 μL of whole blood).39 The alkali methods reported in the literature vary greatly in both the amount of whole blood required (50 μL up to 2 mL) and the amount of dilution (5× to 50×).1,35,37–40,43–45 As such, analytical methods conducted using 50 or 100 μL of whole blood are better able to accommodate limited sample volumes, such as those obtained from infants.
The analysis of whole blood samples is almost always performed with the use of matrix-matched calibration standards. Biomonitoring methods need to be optimized for low-level determinations, so blood pools used for matrix-matching of standards require screening before use. Once the blood has been screened, it is typically packaged into smaller containers and stored at −80 or 4 °C.32,40 Gajek et al. reported a blood method using synthetic matrix-matched standards as an alternative method to using screened blood pools and validated it using standard reference materials (SRMs) and proficiency testing (PT) materials.44 It was determined that a 50× dilution was not enough to overcome the matrix-effects for the blood samples without some type of matrix-matching.
In all of the aforementioned literature references, the blood samples were manually prepared for analysis. The work presented here will compare manual preparation of blood samples to automated inline sample preparation. In addition, a new intelligent in-line flow injection valve will be presented for the analysis of the manually prepared samples. In the previous synthetic matrix validation, the synthetic matrix was compared to no matrix. Here we will present the comparison of results from a set of PT samples from the New York Department of Health (NYDOH) that were analyzed using matrix-matched calibrations of both blood and synthetic matrix.
Methods
Materials and reagents
All reagents, diluents, and rinse solutions were prepared using 18.2 MΩ cm water from an EMD Millipore high-purity filtration system (Millipore Sigma, Burlington, MA, USA). Nitric acid (70%, Seastar, Sidney, BC, CAN), methanol (OmniSolv® LC-MS grade, Supelco, Bellefonte, PA, USA), Triton X-100 (laboratory grade, Millipore Sigma), ethylenediaminetetraacetic acid disodium salt (EDTA-Na2, Millipore Sigma), butyl alcohol (99.9%, Millipore Sigma) and sodium hydroxide (ACS reagent grade, Millipore Sigma) were used to prepare the following solutions. The diluent, transfer, and rinse (for the prepFAST M5 in-line automation module) were prepared with 0.5% (v/v) nitric acid, 2% (v/v) methanol, 0.05% (v/v) Triton X-100, 0.01% (w/v) EDTA-Na2, and 0.002% (w/v) sodium hydroxide. The internal standard (inline sample preparation method) was prepared with 0.5% (v/v) nitric acid, 0.1% (v/v) Triton X-100, 0.1% (v/v) butyl alcohol, and 100 μg L−1 Ga, Ir, and Rh (Elemental Scientific, Inc., Omaha, NE, USA). The sample carrier (a syringe-driven carrier flow is supplied by the prepFAST IC chromatography module) and rinse station 1 of the autosampler were prepared with 0.1% Triton X-100. Rinse station 2 and the working solution (used for syringe-driven sample loading) consisted of DI water. Synthetic clinical matrix (CLIN-0500, Elemental Scientific, Inc.) and blood base matrix (bovine whole blood in sodium heparin, 7200811, LAMPIRE Biological Laboratories, Inc, Pipersville, PA, USA) were used to matrix match all calibration curves by manual or inline sample preparation methods.Sample preparation
Calibration curves were prepared using a stock solution containing Cd, Pb, Mn, Se (100 mg L−1, M1-CLIN-B-A-100, Elemental Scientific, Inc.) and a Hg stock standard (10 mg L−1, S1–Hg-10 × 100, Elemental Scientific, Inc.). The calibration standards were certified based on gravimetric and volumetric preparation, and verified against NIST SRM 3100 series. Each of the methods investigated used the same calibration range: Cd 0.5–10 μg L−1, Mn 0.5–10 μg L−1, Hg 0.5–10 μg L−1, Se 2–40 μg L−1, and Pb 5–100 μg dL−1. For the manual sample preparation, blood or synthetic clinical matrix was added at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio, 20 μg L−1 of Rh, Ga, and Ir were added to each sample, and the remainder of the standard was prepared using diluent solution. For the inline sample preparation method, the stock standard was taken up and diluted inline using dilution factors of 200, 100, 50, 20, and 10×. For each standard, the clinical matrix was automatically spiked in at a 1:50 dilution. NYDOH (Albany, NY, USA) PT blood reference samples were used to evaluate the performance of the analytical methods. For the manual sample preparation, 50 μL of blood was aliquoted into a 15 mL vial and diluted with the diluent + internal standard to a final volume of 2.5 mL (50× dilution). For the inline sample preparation method, 70 μL of the original blood sample (blood samples placed in autosampler vials with no prior sample preparation) were syringe-loaded into a 50 μL sample loop. This blood sample is then transferred into a second dilution loop (500 μL) while it is diluted at a ratio of 1 part blood: 49 parts diluent (50×). For the inline sample preparation method, the blood samples must be visually inspected for blood clots. If clots are present, the samples cannot be analyzed with the inline technique. For all calibration methods, any contamination in the calibration matrix was accounted for when determining the sample concentrations.
:50 ratio, 20 μg L−1 of Rh, Ga, and Ir were added to each sample, and the remainder of the standard was prepared using diluent solution. For the inline sample preparation method, the stock standard was taken up and diluted inline using dilution factors of 200, 100, 50, 20, and 10×. For each standard, the clinical matrix was automatically spiked in at a 1:50 dilution. NYDOH (Albany, NY, USA) PT blood reference samples were used to evaluate the performance of the analytical methods. For the manual sample preparation, 50 μL of blood was aliquoted into a 15 mL vial and diluted with the diluent + internal standard to a final volume of 2.5 mL (50× dilution). For the inline sample preparation method, 70 μL of the original blood sample (blood samples placed in autosampler vials with no prior sample preparation) were syringe-loaded into a 50 μL sample loop. This blood sample is then transferred into a second dilution loop (500 μL) while it is diluted at a ratio of 1 part blood: 49 parts diluent (50×). For the inline sample preparation method, the blood samples must be visually inspected for blood clots. If clots are present, the samples cannot be analyzed with the inline technique. For all calibration methods, any contamination in the calibration matrix was accounted for when determining the sample concentrations.
Sample preparation and introduction
 | ||
| Fig. 1 Displays a simplified schematic of the sample introduction systems: (a) SampleSense Clinical and (b) prepFAST IC Clinical. The column valve shown in (b) is not utilized in the methods presented in this work. | ||
ICP-MS
Two different platforms were used to measure the blood samples, a NexION 2000 ICP-MS (PerkinElmer, Shelton, CT, USA) and an iCAP TQ ICP-MS (Thermo Fisher Scientific, Bremen, Germany).Results and discussion
The most common way to calibrate for biological samples is to use matrix-matched standard calibrations. Three different calibration strategies were compared: manual preparation matrix-matched with blood, manual preparation matrix-matched with synthetic matrix, and inline preparation matrix-matched with synthetic matrix. The response function and linearity for each calibration strategy can be found in Tables S1–S3.† The sensitivity (slope) of the three calibrations were roughly equivalent, which was to be expected since the amount of matrix was held constant (1:50) across the three calibrations strategies. There was no significant difference in the linearity of the three methods, with all calibration curves resulting in an R2 ≥ 0.9996. There were two exceptions; the MnO and Hg calibration curves prepared in the blood matrix were found to have an R2 of 0.9987 and 0.9986, respectively. The calibration curve slopes had relatively the same standard error (SE), except for Se (both KED and QQQ modes) and Pb (Tables S1–S3†). The SE for the blood-matrix was 1.6 and 2.4, for the synthetic matrix (manual) was 0.58 and 1.0, and for the synthetic matrix (inline) was 0.58 and 1.1, for Se and SeO respectively. The SE for Pb also showed a distinct difference: 4.8 for blood matrix, 0.31 for synthetic matrix (manual), and 0.27 for synthetic matrix (inline). The limits of detection and quantification for the three calibration strategies varied between the blood matrix and the synthetic matrix (Table 1). The biggest differences were noticed for Mn, Se, and Pb, with the blood matrix-matched calibration resulting in worse detection limits. This is most likely a result of the synthetic matrix having lower backgrounds compared to the blood matrix. In a routine laboratory setting, the blood matrix (purchased by laboratory) must be screened prior to use, and a decision is made whether the elements of interest have low enough concentrations in that purchased blood supply to be used as a calibration matrix. On the other hand, the synthetic matrix used in these experiments has been purified for elements of interest, resulting in lower backgrounds.
| Element | Measurement mode | m/z | Manual preparation | Manual preparation | Inline preparation | |||
|---|---|---|---|---|---|---|---|---|
| Blood matrix-matched calibration | Synthetic matrix-matched calibration | Synthetic matrix-matched calibration | ||||||
| LOD (μg L−1) | LOQ (μg L−1) | LOD (μg L−1) | LOQ (μg L−1) | LOD (μg L−1) | LOQ (μg L−1) | |||
| a LOD = (3 × σblank)/m. b LOQ = (10 × σblank)/m. c m = slope. | ||||||||
| Mn | KED (He) | 55 | 0.016 | 0.530 | 0.004 | 0.013 | 0.006 | 0.020 |
| Mn (MnO) | QQQ (O2) | 71 | 0.397 | 1.32 | 0.016 | 0.053 | 0.019 | 0.063 |
| Se | KED (He) | 78 | 0.152 | 0.506 | 0.022 | 0.073 | 0.025 | 0.083 |
| Se (SeO) | QQQ (O2) | 96 | 0.335 | 1.12 | 0.015 | 0.050 | 0.005 | 0.017 |
| Cd | KED (He) | 113 | 0.015 | 0.050 | 0.010 | 0.033 | 0.010 | 0.033 |
| Hg | KED (He) | 202 | 0.017 | 0.057 | 0.010 | 0.033 | 0.008 | 0.027 |
| Pb | KED (He) | 208 | 0.070 (μg dL−1) | 0.233 (μg dL−1) | 0.005 (μg dL−1) | 0.017 (μg dL−1) | 0.002 (μg dL−1) | 0.007 (μg dL−1) |
The three calibration strategies were used to evaluate fifteen 2019 NYDOH PT blood samples (BE19-01 to BE19-15). These PT samples were analyzed over three different analytical runs and on two different ICP-MS platforms, for six separate measurements in total. Each PT sample has a reference value and reference range (except for Se) based on the 2019 clinical laboratory proficiency testing program results. Table 2 displays the Pb results from each calibration strategy. Overall, the measured values fall within the specified reference ranges. Fig. 2 displays linear regressions comparing the measured values of Pb to the target values for each of the calibration strategies. The linear regressions all show excellent agreement between the expected values and the measured values, with all slopes determined to be between 0.95 and 1.05. No significant difference, for the Pb results, was determined for the blood matrix as compared to the synthetic matrix or between the manual and inline sample preparation. In total, 18 different analytical measurements were performed for the 3 calibration strategies. Excluding any outliers, the precision across the 3 different calibration methods including two different ICP-MS's, ranged from 4.5 to 12.5%. The worst precision (12.5%) was from BE19-04 which had a result of 1.8 ± 0.2 μg dL−1, whereas the best precision was reported for BE19-07 which had a result of 72.0 ± 3.2 μg dL−1.
| NYDOH PT sample | NYDOH reference value (μg dL−1 Pb) | Reference range (μg dL−1 Pb) | Manual preparation (μg dL−1 Pb) | Manual preparation (μg dL−1 Pb) | Inline preparation (μg dL−1 Pb) |
|---|---|---|---|---|---|
| Blood matrix-matched calibration | Synthetic matrix-matched calibration | Synthetic matrix-matched calibration | |||
| a *Significant difference, t-test (p < 0.05). Comparison between manual preparation methods. | |||||
| BE19-01 | 0.84 | 0–2.84 | 1.4 ± 0.4 | 1.06 ± 0.4 | 0.76 ± 0.05 |
| BE19-02 | 5.5 | 3.5–7.5 | 5.7 ± 0.4 | 5.6 ± 0.3 | 5.2 ± 0.3 |
| BE19-03 | 2.29 | 0.29–4.29 | 1.96 ± 0.03 | 2.29 ± 0.18 | 2.22 ± 0.37 |
| BE19-04 | 1.8 | 0–3.8 | 1.6 ± 0.1 | 1.8 ± 0.2 | 1.7 ± 0.3 |
| BE19-05 | 9.3 | 7.3–11.3 | 8.9 ± 0.2 | 9.0 ± 0.2 | 9.1 ± 1.1 |
| BE19-06 | 11.4 | 9.4–13.4 | 11.3 ± 0.1 | 11.4 ± 0.4 | 11.4 ± 1.7 |
| BE19-07 | 71 | 64–78 | 72 ± 3 | 71 ± 6 | 72 ± 3 |
| BE19-08 | 31.5 | 28.4–34.7 | 31.8 ± 0.2 | 31.1 ± 1.1 | 31.8 ± 1.6 |
| BE19-09 | 21.3 | 19.2–23.4 | 21.9 ± 0.6 | 21.3 ± 1.2 | 22.2 ± 0.8 |
| BE19-10 | 2.35 | 0.35–4.35 | 1.82 ± 0.40 | 2.41 ± 0.28 | 2.22 ± 0.37 |
| BE19-11 | 26.3 | 23.7–28.9 | 26.6 ± 1.0 | 26.8 ± 0.8 | 26.6 ± 0.8 |
| BE19-12 | 7.7 | 5.7–9.7 | 7.1 ± 0.4 | 7.6 ± 0.4 | 7.1 ± 0.6 |
| BE19-13 | 17.7 | 15.7–19.7 | 18.9 ± 0.6 | 18.1 ± 1.2 | 17.9 ± 0.3 |
| BE19-14 | 3.38 | 1.38–5.38 | 2.76 ± 0.59 | 3.48 ± 0.27 | 3.30 ± 0.45 |
| BE19-15 | 0.61 | 0–2.61 | <LOQ* | 0.59 ± 0.03 | 0.57 ± 0.07 |
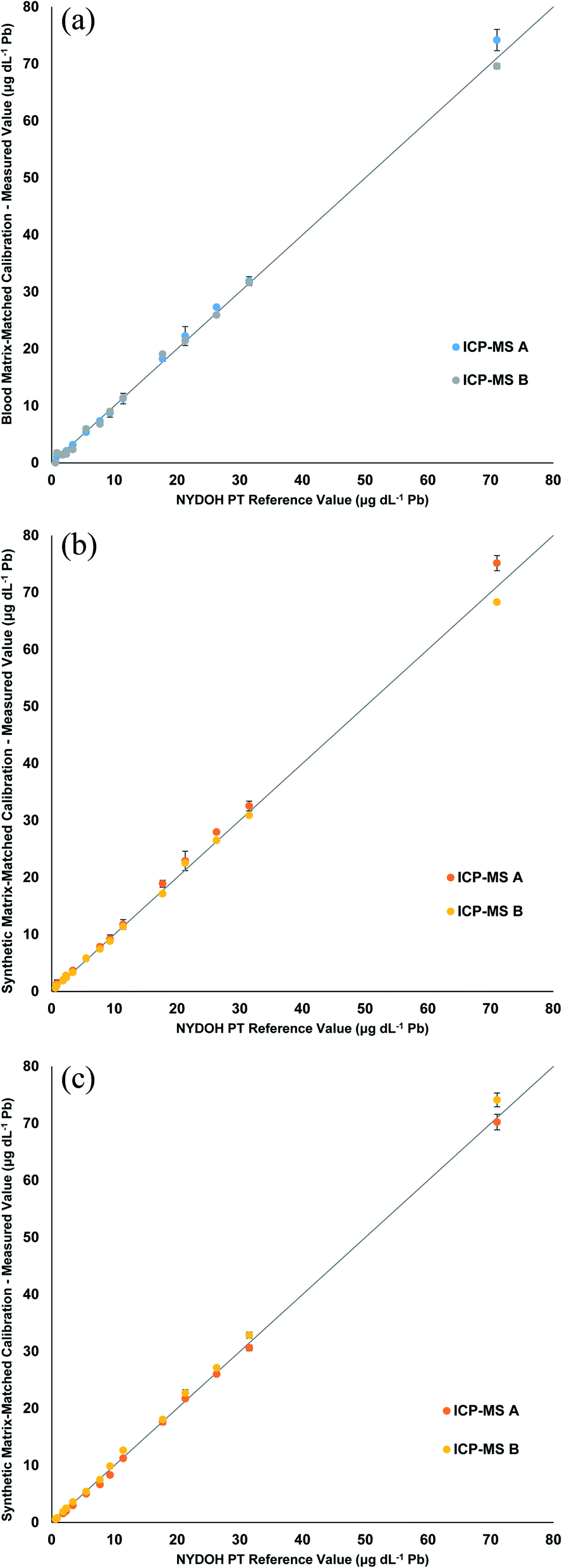 | ||
| Fig. 2 Linear regressions comparing the measured values of Pb from the NYDOH PT samples to the target values for the (a) blood matrix calibration with manual preparation, (b) synthetic matrix calibration with manual preparation, and (c) synthetic matrix calibration with inline preparation. Values reported are the average of 3 measurements from 2 different ICP-MS instruments (n = 6). | ||
Table 3 displays the Se results for the three calibration strategies. The blood matrix results show a clear low bias that ranges from 32–73 μg L−1 lower than the reference values (Fig. 3a). Two different ICP-MS platforms were used; one measured Se (m/z = 78) in collision mode (He), while the other was equipped with a triple quadrupole and measured Se as SeO (m/z = 96) using reaction mode (O2) with a mass shift. The two different measurements showed the same trend. The manual preparation with synthetic matrix (Fig. 3b) and the inline preparation with synthetic matrix (Fig. S1†) both show excellent correlations (0.98 to 1.05) to the reference values. Table S4† displays the blank intensity counts for the blood and synthetic matrix. The blood matrix has a higher background of Se compared to that of the synthetic matrix, which suggests this is the cause for the bias in the PT results.
| NYDOH PT sample | NYDOH reference value (μg L−1 Se) | Manual preparation (μg L−1 Se) | Manual preparation (μg L−1 Se) | Inline preparation (μg L−1 Se) |
|---|---|---|---|---|
| Blood matrix-matched calibration | Synthetic matrix-matched calibration | Synthetic matrix-matched calibration | ||
| a *Significant difference, t-test (p < 0.05). Comparison between manual preparation methods. | ||||
| BE19-01 | 158 ± 15 | 86 ± 14* | 151 ± 1 | 159 ± 5 |
| BE19-02 | 312 ± 32 | 253 ± 9* | 303 ± 3 | 327 ± 15 |
| BE19-03 | 252 ± 10 | 183 ± 7* | 237 ± 8 | 240 ± 10 |
| BE19-04 | 468 ± 37 | 395 ± 10* | 468 ± 28 | 484 ± 23 |
| BE19-05 | 154 ± 7 | 100 ± 6* | 147 ± 2 | 151 ± 11 |
| BE19-06 | 274 ± 8 | 225 ± 8* | 271 ± 2 | 273 ± 5 |
| BE19-07 | 449 ± 18 | 378 ± 4* | 451 ± 33 | 457 ± 11 |
| BE19-08 | 196 ± 5 | 157 ± 11* | 193 ± 8 | 196 ± 5 |
| BE19-09 | 309 ± 10 | 276 ± 5 | 298 ± 22 | 324 ± 14 |
| BE19-10 | 149 ± 3 | 117 ± 7* | 144 ± 9 | 157 ± 13 |
| BE19-11 | 303 ± 22 | 245 ± 1* | 316 ± 10 | 303 ± 1 |
| BE19-12 | 278 ± 14 | 224 ± 1* | 275 ± 8 | 274 ± 13 |
| BE19-13 | 143 ± 14 | 100 ± 9* | 147 ± 5 | 139 ± 6 |
| BE19-14 | 380 ± 30 | 323 ± 6* | 382 ± 20 | 383 ± 16 |
| BE19-15 | 189 ± 15 | 149 ± 10* | 192 ± 6 | 191 ± 1 |
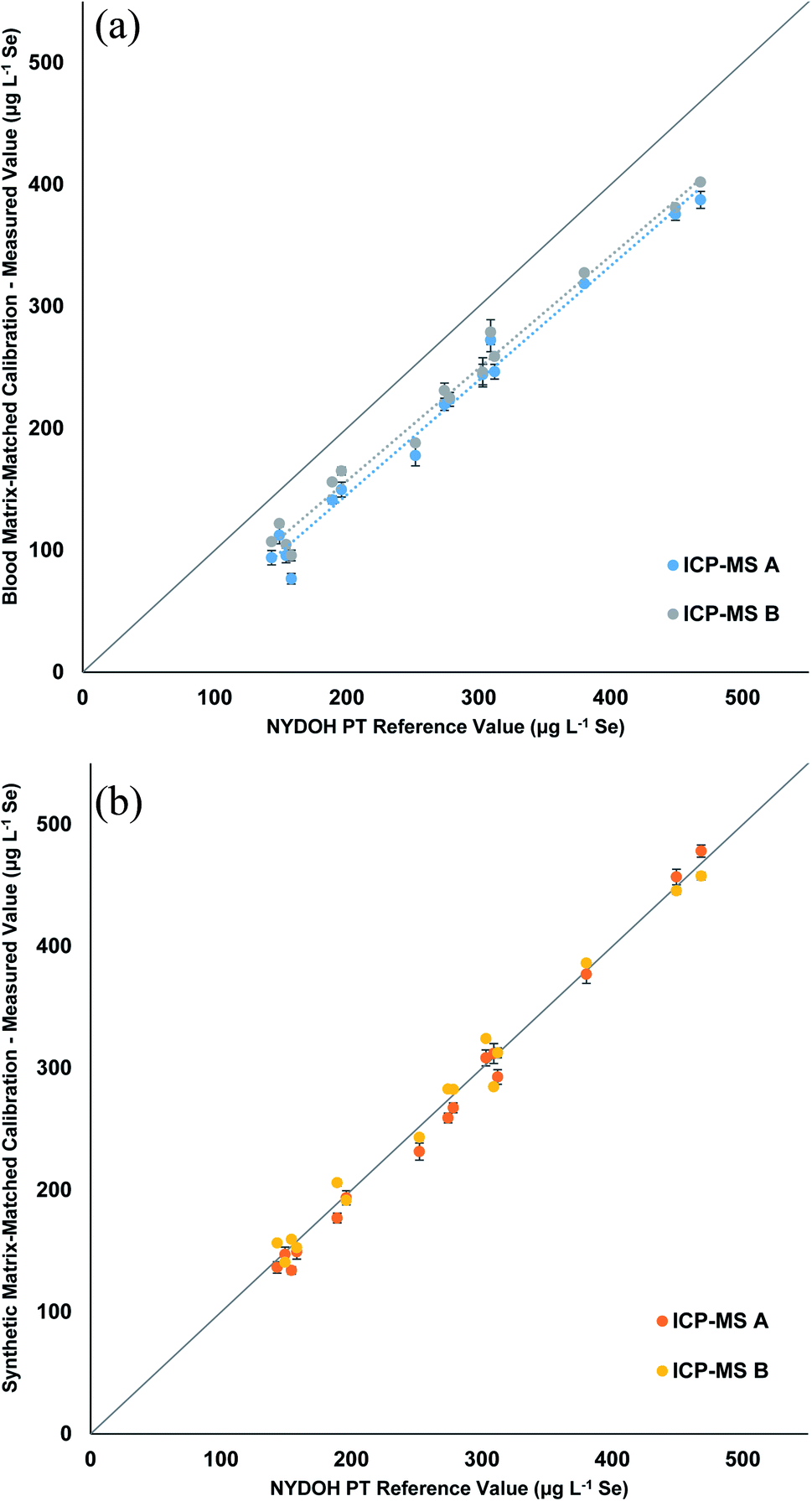 | ||
| Fig. 3 Linear regressions comparing the measured values of Se from the NYDOH PT samples to the target values for the (a) blood matrix calibration with manual preparation and (b) synthetic matrix calibration with manual preparation. Values reported are the average of 3 measurements from 2 different ICP-MS instruments (n = 6). | ||
The Mn results shown in Table 4 reveal a similar pattern to the Se results for the blood matrix calibration. The overall bias is low and ranges from 1.2–5.4 μg L−1 lower than the reference values. However, the response for the two different ICP-MS methods show that there is a significant difference in the results for the two ICP-MS methods (Fig. 4a). The blank intensities for the blood and synthetic matrix do not differ as drastically for the collision cell method (Mn) as compared to the significant drop in intensities seen for the mass shifting method (MnO). When operating in collision cell mode, the abundance sensitivity (peak broadening/tailing from an adjacent mass) is worse (higher), which can lead to neighboring masses interfering (e.g., 56Fe, 40Ar16O, and/or 40Ca16O on 55Mn) when concentrations are high enough. This could explain why the background is still high for Mn in the synthetic matrix when operating in collision cell mode. When the synthetic matrix (manual and inline preparation) is used, the linear correlation is very good (Fig. 4b and S2†); however, there is a slightly high recovery with the collision cell method, likely due to the aforementioned abundance sensitivity. These data suggest that the mass shift method using the synthetic matrix provides more accurate results.
| NYDOH PT sample | NYDOH reference value (μg L−1 Mn) | Reference range (μg L−1 Mn) | Manual preparation (μg L−1 Mn) | Manual preparation (μg L−1 Mn) | Inline preparation (μg L−1 Mn) |
|---|---|---|---|---|---|
| Blood matrix-matched calibration | Synthetic matrix-matched calibration | Synthetic matrix-matched calibration | |||
| a *Significant difference, t-test (p < 0.05). Comparison between manual preparation methods. b **Significant difference, t-test (p < 0.05). Comparison between manual and inline synthetic preparation methods. | |||||
| BE19-01 | 15.6 | 12.6–18.6 | 10.2 ± 1.1* | 15.4 ± 0.7 | 15.6 ± 1.4 |
| BE19-02 | 20.8 | 17.3–24.3 | 19.3 ± 2.8 | 21.3 ± 2.1 | 21.8 ± 2.2 |
| BE19-03 | 30.1 | 25–35.2 | 27.0 ± 2.9 | 31.3 ± 3.1 | 31.3 ± 1.8 |
| BE19-04 | 12.9 | 9.9–15.9 | 9.42 ± 0.82 | 10.8 ± 0.1** | 13.3 ± 0.6 |
| BE19-05 | 34 | 28.2–39.8 | 30 ± 1 | 34 ± 1 | 35 ± 1 |
| BE19-06 | 31.8 | 26.4–37.2 | 28.4 ± 2.8 | 31.5 ± 1.8 | 32.2 ± 1.5 |
| BE19-07 | 23.3 | 19.3–27.3 | 20.8 ± 2.8 | 23.7 ± 2.6 | 23.5 ± 2.6 |
| BE19-08 | 19 | 15.8–22.2 | 16 ± 3 | 20 ± 3 | 18 ± 2 |
| BE19-09 | 34.7 | 28.8–40.6 | 33.5 ± 0.7 | 34.4 ± 0.1 | 36.0 ± 0.1 |
| BE19-10 | 16.4 | 13.4–19.4 | 14.9 ± 2.8 | 16.3 ± 0.9 | 15.7 ± 1.4 |
| BE19-11 | 33.8 | 28.1–39.5 | 30.4 ± 1.4* | 37.3 ± 1.6 | 34.4 ± 0.9 |
| BE19-12 | 20.2 | 16.8–23.6 | 16.9 ± 2.6 | 19.2 ± 0.7 | 19.6 ± 0.5 |
| BE19-13 | 23.1 | 19.2–27.0 | 19.4 ± 2.8 | 22.0 ± 1.1 | 22.9 ± 0.9 |
| BE19-14 | 17.1 | 14.1–20.1 | 14.1 ± 2.7 | 16.0 ± 1.1 | 16.5 ± 1.0 |
| BE19-15 | 11.9 | 8.9–14.9 | 10.2 ± 1.4 | 11.9 ± 0.9 | 12.8 ± 0.1 |
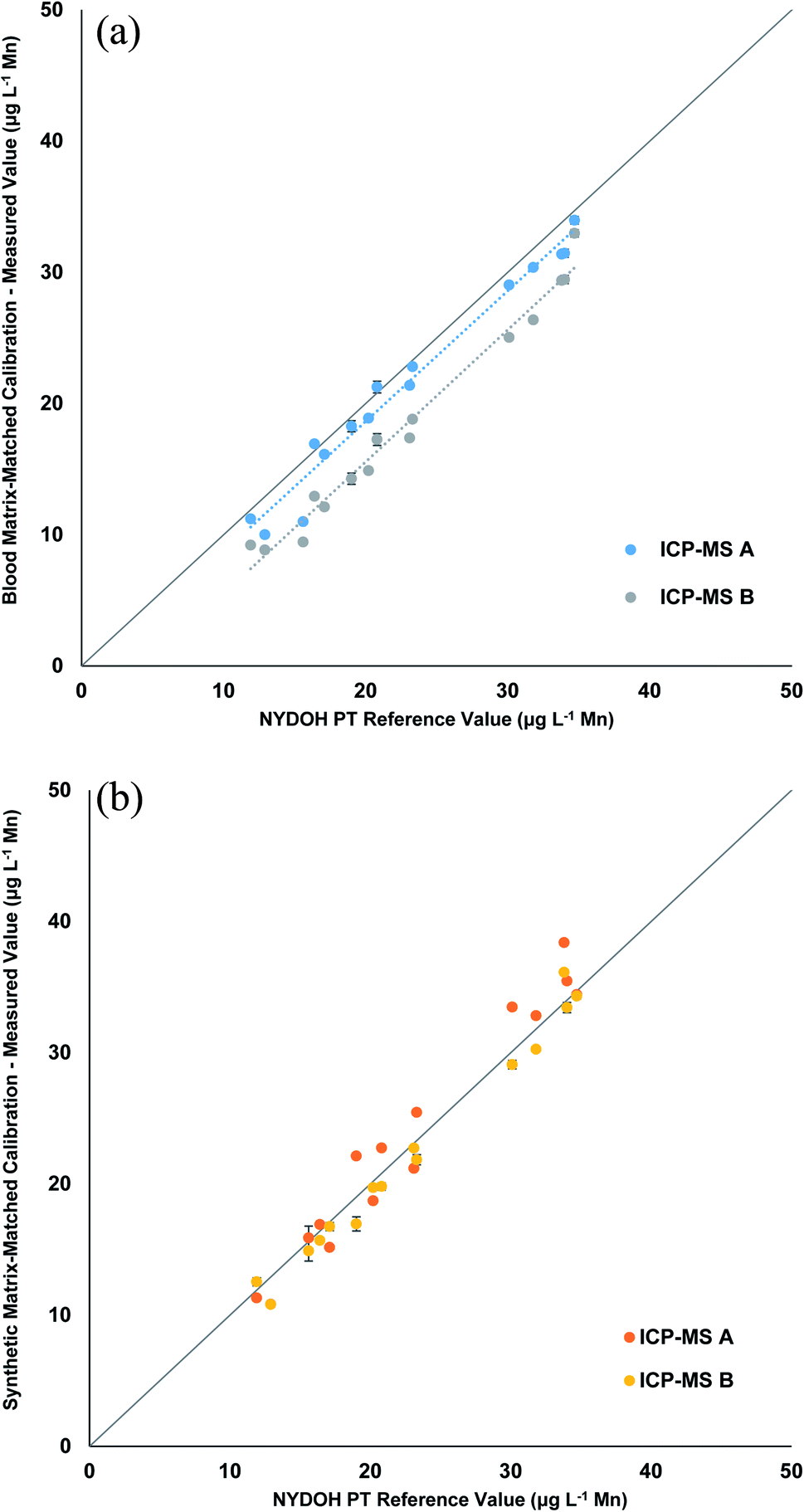 | ||
| Fig. 4 Linear regressions comparing the measured values of Mn from the NYDOH PT samples to the target values for the (a) blood matrix calibration with manual preparation and (b) synthetic matrix calibration with manual preparation. Values reported are the average of 3 measurements from 2 different ICP-MS instruments (n = 6). | ||
Table 5 displays the Hg results for the three different calibration strategies. The results for the blood matrix reveal a slightly low bias compared to the reference values, whereas the synthetic results show a better correlation. Fig. S3† shows that the ICP-MS A method trends to a lower bias as the concentration increases (slope = 0.89) which reduces the reported average for the manually prepared blood matrix samples in Table 5. In comparison the slope (0.97) for the ICP-MS B method (collision cell) has a better correlation. The reported values are all well within the reported reference ranges, so the observed low bias could be considered negligible. Fig. S4 and S5† show excellent correlation (0.95 to 1.02) for the synthetic matrix prepared manually or inline.
| NYDOH PT sample | NYDOH reference value (μg L−1 Hg) | Reference range (μg L−1 Hg) | Manual preparation (μg L−1 Hg) | Manual preparation (μg L−1 Hg) | Inline preparation (μg L−1 Hg) |
|---|---|---|---|---|---|
| Blood matrix-matched calibration | Synthetic matrix-matched calibration | Synthetic matrix-matched calibration | |||
| a *Significant difference, t-test (p < 0.05). Comparison between manual preparation methods. b **Significant difference, t-test (p < 0.05). Comparison between manual and inline synthetic preparation methods. | |||||
| BE19-01 | 1.28 | 0–4.28 | 1.39 ± 0.14 | 1.42 ± 0.08 | 1.39 ± 0.53 |
| BE19-02 | 25.2 | 17.6–32.8 | 24.4 ± 3.4 | 25.6 ± 0.1** | 24.0 ± 0.3 |
| BE19-03 | 37 | 26–48 | 34 ± 3 | 37 ± 2 | 37 ± 4 |
| BE19-04 | 16 | 11.2–20.8 | 15 ± 1 | 16 ± 1 | 17 ± 2 |
| BE19-05 | 8.8 | 5.8–11.8 | 8.0 ± 1.2 | 9.4 ± 0.7 | 9.1 ± 1.3 |
| BE19-06 | 21.5 | 15.1–28 | 20.0 ± 1.4 | 22.3 ± 1.9 | 21.1 ± 1.3 |
| BE19-07 | 11.4 | 8.0–14.8 | 10.5 ± 0.4 | 12.5 ± 1.5 | 12.0 ± 1.5 |
| BE19-08 | 3.35 | 0.35–6.35 | 2.87 ± 0.06* | 3.61 ± 0.17 | 3.35 ± 0.47 |
| BE19-09 | 7.3 | 4.3–10.3 | 6.6 ± 0.5* | 7.9 ± 0.1 | 7.8 ± 1.3 |
| BE19-10 | 32 | 22.4–41.6 | 29 ± 1 | 30 ± 1 | 30 ± 1 |
| BE19-11 | 5.7 | 2.7–8.7 | 4.7 ± 0.1* | 6.0 ± 0.2 | 5.7 ± 1.0 |
| BE19-12 | 14.2 | 9.9–18.5 | 11.8 ± 0.4 | 14.1 ± 1.3 | 14.1 ± 1.8 |
| BE19-13 | 30 | 21–39 | 27 ± 1 | 30 ± 2 | 30 ± 2 |
| BE19-14 | 0.95 | 0–3.95 | 0.24 ± 0.19* | 0.88 ± 0.13 | 0.88 ± 0.20 |
| BE19-15 | 2.15 | 0–5.15 | 0.89 ± 0.51* | 2.26 ± 0.42 | 1.87 ± 0.41 |
The Cd results showed excellent correlation between the expected values and the measured values for all three calibration strategies (Table 6). One note of caution, however, is that across all 15 PT samples measured, the spread of values only spans from 0.18 to 11.6 μg L−1 Cd and only four data points are over 1.5 μg L−1 Cd. Fig. S6–S8† display the linear regressions for each calibration method. In addition, there were no significant differences noticed between ICP-MS methods.
| NYDOH PT sample | NYDOH reference value (μg L−1 Cd) | Reference range (μg L−1 Cd) | Manual preparation (μg L−1 Cd) | Manual preparation (μg L−1 Cd) | Inline preparation (μg L−1 Cd) |
|---|---|---|---|---|---|
| Blood matrix-matched calibration | Synthetic matrix-matched calibration | Synthetic matrix-matched calibration | |||
| a *Significant difference, t-test (p < 0.05). Comparison between manual preparation methods. | |||||
| BE19-01 | 0.29 | 0–1.29 | 0.68 ± 0.32 | 0.37 ± 0.01 | 0.26 ± 0.05 |
| BE19-02 | 0.85 | 0–1.85 | 0.87 ± 0.09 | 0.92 ± 0.06 | 0.88 ± 0.04 |
| BE19-03 | 0.36 | 0–1.36 | 0.60 ± 0.16 | 0.41 ± 0.06 | 0.36 ± 0.05 |
| BE19-04 | 1.34 | 0.34–2.34 | 1.34 ± 0.27 | 1.35 ± 0.04 | 1.43 ± 0.05 |
| BE19-05 | 0.92 | 0–1.92 | 1.1 ± 0.2 | 0.93 ± 0.09 | 0.93 ± 0.09 |
| BE19-06 | 1.09 | 0.09–2.09 | 1.14 ± 0.09 | 1.20 ± 0.04 | 1.14 ± 0.25 |
| BE19-07 | 8.40 | 7.1–9.7 | 8.58 ± 0.66 | 8.60 ± 0.17 | 8.73 ± 0.28 |
| BE19-08 | 2.91 | 1.91–3.91 | 2.90 ± 0.20 | 2.94 ± 0.16 | 3.07 ± 0.13 |
| BE19-09 | 5.10 | 4.1–6.1 | 5.44 ± 0.24 | 5.09 ± 0.63 | 5.27 ± 0.23 |
| BE19-10 | 11.60 | 9.9–13.3 | 11.19 ± 0.86 | 11.57 ± 0.60 | 11.30 ± 0.01 |
| BE19-11 | 0.83 | 0–1.83 | 0.91 ± 0.02 | 0.92 ± 0.03 | 0.81 ± 0.07 |
| BE19-12 | 0.96 | 0–1.96 | 0.93 ± 0.03 | 0.97 ± 0.01 | 1.0 ± 0.1 |
| BE19-13 | 0.63 | 0–1.63 | 0.69 ± 0.03 | 0.77 ± 0.09 | 0.79 ± 0.14 |
| BE19-14 | 0.60 | 0–1.06 | 0.65 ± 0.02* | 0.70 ± 0.01 | 0.73 ± 0.07 |
| BE19-15 | 0.18 | 0–1.182 | 0.19 ± 0.01 | 0.24 ± 0.04 | 0.18 ± 0.01 |
The overall %BIAS (or % difference) for the obtained results can be viewed in Table S5.† The %BIAS for all 5 elements is approximately the same for the synthetic matrix-matched calibrations regardless of whether it was manually or inline prepared. The biggest %BIAS differences are noticed when comparing the blood matrix-matched calibration to the synthetic matrix-matched calibrations, Pb is 2.5× higher, Se is 8.8× higher, Cd is 1.4× higher, Hg is 1.9× higher, and Mn is 3.6× higher.
Matrix-matched standards were used to make sure the plasma conditions remained as constant as possible when samples are analyzed. Fig. 5 displays the average internal standard (71Ga, 103Rh, and 193Ir) recovery for the fifteen PT samples from each calibration strategy, as compared to the calibration standard internal standard response. The individual internal standard recoveries for each method per element can be found in Fig. S9–S11.† The recovery for the synthetic matrix (manual) matches that of the blood matrix (manual) method, with an overall value of 97.6% and 97.5%, respectively. The inline sample preparation method resulted in an overall average recovery of 100.8%, slightly higher than the other two methods. The overall spread of recoveries is excellent for all three methods, between 94% and 103%, which suggests that the clinical matrix provides a suitable option for matrix matching blood samples for clinical analysis.
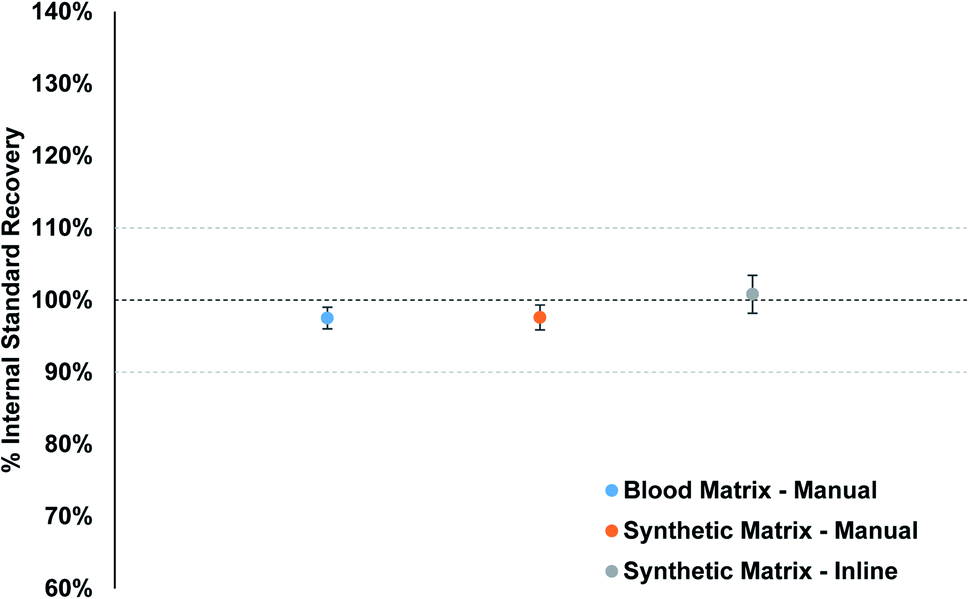 | ||
| Fig. 5 Average internal standard recovery for the 3 calibration strategies. The average value represents the average recovery from Ga, Rh, and Ir from each calibration method over the 15 PT samples analyzed. | ||
Conclusion
Three different blood sample preparation and sample introduction techniques were compared in this study. The blood matrix used in this study had higher Se and Mn levels than the synthetic matrix, raising the background and reducing the quality of the analytical results. The use of synthetic matrix provided more accurate results when analyzing the NYDOH PT samples since the synthetic matrix is purified for the elements of interest. The lower backgrounds in the synthetic matrix led to better detection limits for all elements, but the effect was most evident for Mn and Se. The internal standard recoveries for the synthetic matrix were between 94–103% when analyzing the NYDOH PT samples, confirming that the synthetic matrix is mimicking the blood matrix conditions in the plasma. The linear regressions for Cd, Hg, Mn, Se, and Pb had excellent correlation using the synthetic matrix. As a whole, the study suggests that using synthetic matrix is superior to using blood due to the convenience (not having to screen pooled blood for contamination), safety (no need for an analyst to handle pooled blood), and most importantly, analytical benefits.Both the SampleSense Clinical and prepFAST IC Clinical sample introduction systems accurately determined the elements of interest in the NYDOH PT samples. To our knowledge, this is the first report of micro-volume (50 μL) blood sampling using an inline sample preparation technique (prepFAST IC Clinical). This method offers less human interaction, with automated sample preparation leading to time savings and reduced potential health exposure for the analyst.
Conflicts of interest
The authors of this manuscript work for Elemental Scientific, Inc. which manufactures and sells the SampleSense Clinical and prepFAST IC used in this work.References
- D. R. Jones, J. M. Jarrett, D. S. Tevis, M. Franklin, N. J. Mullinix, K. L. Wallon, C. D. Quarles Jr, K. L. Caldwell and R. L. Jones, Talanta, 2017, 162, 114–122 CrossRef CAS PubMed.
- J. L. Pirkle, J. Osterloh, L. L. Needham and E. J. Sampson, Int. J. Hyg Environ. Health, 2005, 208, 1–5 CrossRef CAS PubMed.
- G. Saravanbhavan, K. Werry, M. Walker, D. Haines, M. Malowany and C. Khoury, Int. J. Hyg Environ. Health, 2017, 220, 189–200 CrossRef PubMed.
- A. Bazzi, J. O. Nriagu and A. M. Linder, J. Environ. Monit., 2008, 10, 1226–1232 RSC.
- S. E. Cusick, E. G. Jaramillo, E. C. Moody, A. S. Ssemata, D. Bitwayi, T. C. Lund and E. Mupere, BMC Public Health, 2018, 18, 717 CrossRef PubMed.
- J.-P. Goulle, P. L. Roux, M. Castanet, L. Mahieu, S. Guyet-Job and M. Guerbet, J. Anal. Toxicol., 2015, 39, 707–713 CrossRef CAS PubMed.
- D. C. Rice, R. Lincoln, J. Martha, L. Parker, K. Pote, S. Xing and A. E. Smith, J. Expo. Sci. Environ. Epidemiol., 2010, 20, 634–643 CrossRef CAS PubMed.
- B. Schultze, P. M. Lind, A. Larsson and L. Lind, Scand. J. Clin. Lab. Invest., 2014, 74, 143–148 CrossRef CAS PubMed.
- K. A. Campbell, R. Hickman, M. D. Fallin and K. M. Bakulski, Curr. Opin. Toxicol., 2021, 26, 39–48 CrossRef.
- K. L. Y. Christensen, Int. J. Hyg Environ. Health, 2013, 216, 624–632 CrossRef PubMed.
- M. d. Oliveira, T. M. R. Gianeti, F. C. G. d. Rocha, P. N. Lisboa-Filho and M. Piacenti-Silva, Sci. Rep., 2020, 10, 13112 CrossRef PubMed.
- R. P. Sidaginamale, T. J. Joyce, J. K. Lord, R. Jefferson, P. G. Blain, A. V. F. Norgol and D. J. Langton, Bone Jt. Res., 2013, 2, 84–95 CrossRef CAS PubMed.
- H. S. Yang, D. R. LaFrance and Y. Hao, Am. J. Clin. Pathol., 2021, 156, 167–175 CrossRef CAS PubMed.
- F. Barbosa Jr, J. E. Tanus-Santos, R. F. Gerlach and P. J. Parsons, Environ. Health Perspect., 2005, 113, 1669–1674 CrossRef PubMed.
- Toxicological profile for lead, U.S. Department of Heath and Human Services, Public Health Service, https://www.atsdr.cdc.gov/toxprofiles/tp13.pdf, accessed November 11, 2021 Search PubMed.
- K. L. Caldwell, P.-Y. Cheng, J. M. Jarrett, A. Makhmudov, K. Vance, C. Ward, R. L. Jones and M. E. Mortensen, Pediactrics, 2017, 140, e20170272 CrossRef PubMed.
- Council on Environmental Health, Pediactrics, 2016, 138, e20161493 CrossRef PubMed.
- H. Nakata, S. M. M. Nakayama, J. Yabe, K. Muzandu, H. Toyomaki, Y. B. Yohannes, A. Kataba, G. Zyambo, Y. Ikenaka, K. Choongo and M. Ishizuka, Chemosphere, 2021, 271, 129832 CrossRef CAS PubMed.
- Toxicological profile for cadmium, U.S. Department of Heath and Human Services, Public Health Service, https://www.atsdr.cdc.gov/toxprofiles/tp5.pdf, accessed November 11, 2021 Search PubMed.
- W. H. Hallenbeck, Experientia, 1984, 40, 136–142 CrossRef CAS PubMed.
- D. Il'yasova and G. G. Schwartz, Toxicol. Appl. Pharmacol., 2005, 207, 179–186 CrossRef PubMed.
- J. C. Clifton II, Pediatr. Clin. North Am., 2007, 54, 237–269 CrossRef PubMed.
- Toxicological profile for mercury, U.S. Department of Heath and Human Services, Public Health Service, https://www.atsdr.cdc.gov/toxprofiles/tp46.pdf, accessed November 11, 2021 Search PubMed.
- M. Bost, S. Houdart, M. Oberli, E. Kalonji, J.-F. Huneau and I. Margaritis, J. Trace Elem. Med. Biol., 2016, 35, 107–115 CrossRef CAS PubMed.
- G. Crisponi, V. M. Nurchi, D. Fanni, C. Gerosa, S. Nemolato and G. Faa, Coord. Chem. Rev., 2010, 254, 876–889 CrossRef CAS.
- C. M. Saporito-Magrina, R. N. Musacco-Sebio, G. Andrieux, L. Kook, M. T. Orrego, M. V. Tuttolomondo, M. F. Desimone, M. Boerries, C. Borner and M. G. Repetto, Metallomics, 2018, 10, 1743–1754 CrossRef CAS PubMed.
- H. Zischka and C. Einer, Int. J. Biochem. Cell Biol., 2018, 102, 71–75 CrossRef CAS PubMed.
- M. B. Zimmermann and R. F. Hurrell, Lancet, 2007, 370, 511–520 CrossRef CAS.
- J. M. Lyznicki, M. S. Karlan and M. K. Khan, J. Occup. Environ. Med., 1999, 41, 140–143 CrossRef CAS PubMed.
- M. Tumolo, V. Ancona, D. De Paola, D. Losacco, C. Campanale, C. Massarelli and V. F. Uricchio, Int. J. Environ. Res. Publ. Health, 2020, 17, 5438 CrossRef CAS PubMed.
- M. Kovochich, B. L. Finley, R. Novick, A. D. Monnot, E. Donovan, K. M. Unice, E. S. Fung, D. Fung and D. J. Paustenbach, Crit. Rev. Toxicol., 2018, 48, 853–901 CrossRef CAS PubMed.
- C. D. Palmer, M. E. Lewis Jr, C. M. Geraghty, F. Barbosa Jr and P. J. Parsons, Spectrochim. Acta, Part B, 2006, 61, 980–990 CrossRef.
- C. V. Watson, M. Lewin, A. Ragin-Wilson, R. L. Jones, J. M. Jarrett, K. L. Wallon, C. Ward, N. Hilliard and E. Irvin-Barnwell, Environ. Res., 2020, 183, 109208 CrossRef CAS PubMed.
- S. D'Ilio, N. Violante, M. Di Gregorio, O. Senofonte and F. Petrucci, Anal. Chim. Acta, 2006, 579, 202–208 CrossRef PubMed.
- P. Heitland and H. D. Koster, J. Trace Elem. Med. Biol., 2006, 20, 253–262 CrossRef CAS PubMed.
- N. B. Ivanenko, A. A. Ganeev, N. D. Solovyev and L. N. Moskvin, Anal. Chem., 2011, 66, 784–799 CrossRef CAS.
- C. S. Kira, A. M. Sakuma and N. D. C. Gouveia, J. Appl. Pharm. Sci., 2014, 4, 39–45 CAS.
- N. Laur, R. Kinscherf, K. Pomytkin, L. Kaiser, O. Knes and H.-P. Deigner, PLoS One, 2020, 15, e0233357 CrossRef CAS PubMed.
- Y. Lu, M. Kippler, F. Harari, M. Grander, B. Palm, H. Nordqvist and M. Vahter, Clin. Biochem., 2015, 48, 140–147 CrossRef CAS PubMed.
- W. J. McShane, R. S. Pappas, V. Wilson-McElprang and D. Paschal, Spectrochim. Acta, Part B, 2008, 63, 638–644 CrossRef.
- D. E. Nixon, K. R. Neubauer, S. J. Eckdahl, J. A. Butz and M. F. Burritt, Spectrochim. Acta, Part B, 2004, 59, 1377–1387 CrossRef.
- M. L. Praamsma, J. G. Arnason and P. J. Parsons, J. Anal. At. Spectrom., 2011, 26, 1224–1232 RSC.
- I. D. B. Bravo, R. S. Castro, N. L. Riquelme, C. T. Diaz and D. A. Goyenaga, J. Trace Elem. Med. Biol., 2007, 21, 14–17 CrossRef PubMed.
- R. Gajek, F. Barley and J. She, Anal. Methods, 2013, 5, 2193–2202 RSC.
- J. M. Harrington, D. J. Young, A. S. Essader, S. J. Sumner and K. E. Levine, Biol. Trace Elem. Res., 2014, 160, 132–142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ja00056c |
| This journal is © The Royal Society of Chemistry 2022 |