
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of π-spacer in regulating the photovoltaic performance of copper electrolyte dye-sensitized solar cells using triphenylimidazole dyes†
Palivela Siva
Gangadhar‡
ab,
Anooja
Jagadeesh‡
bc,
Manne Naga
Rajesh
a,
Andrew Simon
George
bc,
Seelam
Prasanthkumar
ab,
Suraj
Soman
 *bc and
Lingamallu
Giribabu
*ab
*bc and
Lingamallu
Giribabu
*ab
aPolymer and Functional Materials Division, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, Telangana, India. E-mail: giribabu@iict.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, New Delhi 201002, India
cPhotosciences and Photonics Section, Chemical Sciences and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Thiruvananthapuram 695019, India. E-mail: suraj@niist.res.in
First published on 1st December 2021
Abstract
In dye-sensitized solar cells (DSSCs), π-spacers play a critical role in regulating photovoltaic accomplishment. This is more pronounced when we use alternate redox mediators like cobalt and copper, which are mass transport limited. Previously we showed that triphenylimidazole dyes with non-planar donor units and phenyl spacers having D–A–π–A architecture are more suitable for [Cu(dmp)2]1+/2+ electrolytes for realizing improved power conversion efficiencies compared to the D–D–π–A counterpart. Interestingly when we changed the π-spacer from phenyl (LG-P1 and LG-P3 dyes) to thiophene (LG-P2 and LG-P4 dyes) we observed a reversal of the trend in photovoltaic performance. By attaining a more planar architecture and with properly placed energetics, the D–D–π–A dye LG-P2 with anthracene as an auxiliary donor having thiophene π-spacer outperformed the LG-P4 sensitizer having benzothiadiazole as an auxiliary acceptor in D–A–π–A architecture with thiophene π-spacer. Detailed studies carried out using electrochemical impedance spectroscopy (EIS), charge extraction (CE), open-circuit voltage decay (OCVD), and transient photovoltage and photocurrent decay measurements revealed interesting behaviour in photovoltaic performance when employing an alternate [Cu(dmp)2]1+/2+ electrolyte.
1. Introduction
The enhancing exploitation of non-renewable energy resources like fossil fuels to meet the growing global energy demand has resulted in an ecological imbalance.1 Because of the progressive energy deficiency problems, there is a real need for development of alternative energy sources. In this respect, solar power is the most feasible option of the worldwide research community. The researchers’ predominant attention is on the utilization of solar energy owing to its sustainable advantages like global accessibility and ecological friendliness. In the past three decades, advances in numerous techniques or architectures have been made to harness the solar energy.2 Among them, dye-sensitized solar cells (DSSCs) have been developed rapidly due to their flexibility, transparency, high power-conversion efficiencies (PCEs) under artificial light conditions and lower manufacturing cost.3–7 Therefore, they are considered to be one among the most promising solutions for future green energy demands and low-carbon economy. In DSSCs, the dye is one of the main components which absorbs light and generates photo-electrons. A large variety of dyes, such as ruthenium(II), porphyrin and metal-free organic dyes, have been explored so far.8–11 Among these dyes, metal-free organic dyes have the advantage of their easily modulated structures, compatible with various redox couples, and recorded highest efficiencies (14.3%).12A great diversity of organic scaffolds have been used in DSSCs based on the donor–π–acceptor (D–π–A) approach and device efficiencies of more than 10% have been reported in many cases.13–18 The advantage of the D–π–A architecture is displayed in the effective intra-molecular charge transfer (ICT) properties from the donor (D) to the acceptor (A) via the π-conjugated bridge. In addition to this, after injection of the electron to the TiO2 conduction band, the hole formed on the π-spacer will migrate to the donor part of the sensitizer and it minimizes the recombination of electrons from the TiO2 conduction band with the excited dye. There have been continued efforts to further improve the efficiency of DSSCs using diverse structural frames such as D–D–π–A and D–A–π–A systems.19–22 With the same strategy, our group reported four D–π–A system (D1–D4) based metal-free organic sensitizers, where phenanthroimidazole acts as a donor and either cyanoacrylic acid or rhodanine-3-acetic acid acts as an acceptor, which showed impressive PCEs of up to 10.2% in the presence of the co-adsorbent chenodeoxycholic acid (CDCA).23 In extension of our efforts towards the development of metal-free organic dyes, very recently we explored other types of structural frames, viz. D–D–π–A (LG-P1) and D–A–π–A (LG-P3), where we introduced triphenylimidazole as the donor unit, anthracene and benzothiadiazole as auxiliary donor and auxiliary acceptor units, respectively, and cyanoacrylic acid as the binding group/accepter. The performance was evaluated with the new generation [Cu(dmp)2]1+/2+ electrolyte. Of the two dyes, the D–A–π–A dye (LG-P3) exhibited better PCE with 1.96% and 9.63% under AM 1.5G 1 sun and 1000 lux indoor illumination, respectively.24
As per the previous literature reports, the presence of the electron rich thiophene π-linker not only facilitates improved light absorption and dipole moment, but also decreases the dihedral angle resulting in better orbital overlap between adjacent donor/accepter and the π-spacer plane effectively promoting the electron injection to TiO2.25–29 Consequently, most of the thiophene π-linker based dyes show better efficiency than their phenyl π-linker based counterparts. Based on these aspects we modified our earlier reported dyes (LG-P1and LG-P3) by introducing thiophene as the π-linker instead of the phenyl moiety as shown in Fig. 1, which are named LG-P2 and LG-P4, to explore further possibilities to improve their photovoltaic performance. Herein, we investigated the structure–property–performance relationships of the synthesized dyes and studied the effect of the thiophene π-linker extensively using a range of perturbation tools. The corresponding photophysical, theoretical and photovoltaic properties of LG-P2 and LG-P4 were investigated and compared (Fig. 1). The results revealed that the dihedral angle between phenyl or thiophene units and the auxiliary donor/acceptor functionalities (anthracene or benzothiadiazole) plays a pivotal role and affects the light harvesting ability, energy levels and interfacial charge transfer as well as recombination in devices.
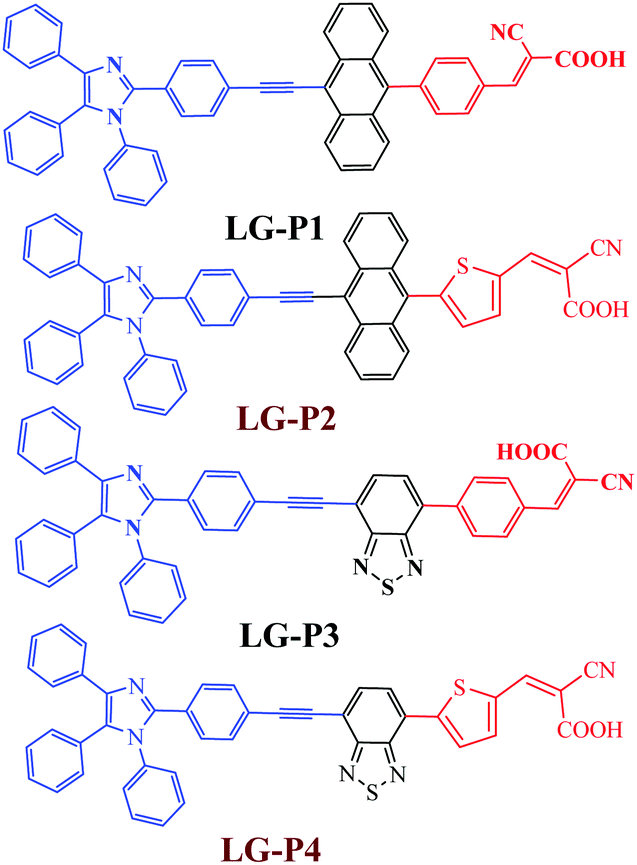 | ||
| Fig. 1 Molecular structure of LG-P sensitizers. | ||
It is well established that sensitizers based on the thiophene spacer have shown better efficiency when compared to their phenyl spacer based counterparts.30,31 On the other hand, in some other cases, molecules with the phenyl spacer have shown better efficiency than the thiophene derivatives.32 In the present study, when we replaced the phenyl group with the thiophene group in triphenylimidazole dyes, there was an improvement in the planarity of the molecules. The DSSC using the molecule having anthracene as the auxiliary acceptor performed better when the π-spacer was switched from phenyl (in LG-P1) to thiophene (in LG-P2). On the other hand, the change of the phenyl spacer (in LG-P3) to thiophene (in LG-P4) had a detrimental effect on the performance of the device employing the molecule with benzothiadiazole as the auxiliary acceptor. The present manuscript deals with the study of the reason behind this opposite behavior of the two molecules when the spacer is switched from phenyl to the thiophene group.
2. Experimental section
2.1 Synthesis
Benzil, 4-ethynylbenzaldehyde, aniline, 5-formyl thiophene-2-boronic acid, 9,10-dibromoanthracene, and 4,7-dibromobenzo[c][1,2,5]thiadiazole were obtained commercially and used without further purification. 1, 2, and 3 were synthesized as per literature methods.30,33,34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v). To this 2-(4-ethynylphenyl)-1,4,5-triphenyl-1H-imidazole (1) (1.07 g, 2.724 mmol), Pd(PPh3)2Cl2 (0.1 g, 0.1362 mmol), and CuI (25 mg, 0.1362 mmol) were added and then the mixture was subjected to purging for one hour. After completion of purging the reaction mixture was stirred at room temperature overnight, and the solvent was removed under reduced pressure. Silica gel column chromatography was adopted to purify the obtained solid material (hexane/CH2Cl2 = 10:1, v/v), recrystallized from chloroform/pentane to give the required product 4 (yield 35%) as a dark yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.06 (s, 1H), 8.68 (dd, J = 16.0, 8.6 Hz, 2H), 7.99 (d, J = 3.7 Hz, 1H), 7.78 (d, J = 8.7 Hz, 2H), 7.70–7.66 (m, 2H), 7.62 (ddd, J = 5.1, 3.8, 1.8 Hz, 4H), 7.59 (d, J = 1.1 Hz, 1H), 7.57–7.53 (m, 2H), 7.48 (ddd, J = 8.7, 6.6, 1.2 Hz, 2H), 7.39 (d, J = 5.2 Hz, 1H), 7.34 (dd, J = 4.9, 2.4 Hz, 2H), 7.30 (dd, J = 4.6, 2.7 Hz, 2H), 7.28 (d, J = 1.4 Hz, 1H), 7.25–7.20 (m, 3H), 7.16 (dd, J = 7.8, 1.7 Hz, 2H), 7.14–7.10 (m, 2H). 13C NMR (CDCl3, 400 MHz) δ (ppm) = 182.89, 145.16, 136.53, 134.27, 131.94, 131.48, 131.16, 130.81, 129.33, 128.88, 128.61, 128.43, 128.26, 128.15, 127.47, 127.07, 126.78, 126.66, 126.37. MALDI-TOF: m/z, calcd. For C48H30N2OS 682.21, found (683.21, [M]+): FT-IR (neat, cm−1): 670, 720, 772, 966, 1020, 1074, 1151, 1219, 1272, 1378, 1462, 1599, 1672, 1734, 2852, 2920, 2957.
:1, v/v), recrystallized from chloroform/pentane to give the required product 5 (yield 30%) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ (ppm) 1H NMR (500 MHz, CDCl3) δ 9.99 (s, 1H), 8.24 (d, J = 4.0 Hz, 1H), 7.97 (d, J = 4.0 Hz, 1H), 7.84 (dd, J = 12.8, 5.8 Hz, 2H), 7.60 (d, J = 7.1 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.48 (d, J = 8.6 Hz, 2H), 7.34–7.25 (m, 4H), 7.25–7.19 (m, 5H), 7.15 (dd, J = 1.4 Hz, 2H), 7.08 (dd, J = 1.3 Hz, 2H). 13CNMR (CDCl3, 400 MHz) δ (ppm) = 182.98, 136.70, 132.98, 132.93, 132.25, 131.13, 129.94, 129.32, 129.19, 128.96, 128.42, 128.39, 128.24, 128.07, 128.01, 127.51, 126.74. MALDITOF: m/z, calcd. For C40H24N4OS2 640.14, found (641.10, M + 1): FT-IR (neat, cm−1): 722, 754, 802, 852, 1030, 1067, 1128, 1283, 1387, 1464, 1548, 1737, 2854, 2924, 2960.
:1, v/v), recrystallized from MeOH/ether to give the required sensitizer LG-P2 (yield 50%) as a yellow solid. Anal. calcd for C51H31N3O2S % (749.21): C, 81.69; H, 4.18; N, 5.60; found: C, 81.75; H, 4.20; N, 5.58.1H NMR (DMSO-d6, 400 MHz) δ 8.67 (d, J = 8.7 Hz, 2H), 8.34 (s, 1H), 8.01 (d, J = 3.2 Hz, 1H), 7.91 (q, J = 8.7 Hz, 4H), 7.80 (d, J = 8.8 Hz, 2H), 7.76–7.67 (m, 5H), 7.67–7.54 (m, 4H), 7.44 (dd, J = 12.3, 5.5 Hz, 6H), 7.33 (dt, J = 22.4, 7.9 Hz, 4H), 7.16 (t, J = 7.4 Hz, 1H). 13C NMR (DMSO-d6, 400 MHz) δ (ppm) = 135.07, 133.85, 133.03, 132.52, 131.88, 131.71, 130.82, 130.31, 130.10, 129.83, 129.52, 129.46, 129.31, 129.07, 128.87, 128.81, 128.12, 127.80, 126.94. MALDI-TOF: m/z calcd. For C51H31N3O2S 749.21, found (750.18, M + 1): FT-IR (neat, cm−1): 690, 772, 970, 1022, 1220, 1425, 1476, 1512, 1556, 1698, 2225, 2236, 2897, 2961, 3044, 3385, 3473.
:1, v/v), recrystallized from MeOH/ether to give the required sensitizer LG-P4 (yield 60%) as a pale red solid. Anal. calcd for C43H25N5O2S2% (749.21): C, 72.97; H, 3.56; N, 9.89; found: C, 73.00; H, 3.60; N, 9.87. 1H NMR (DMSO-d6, 400 MHz) δ 8.43 (s, 1H), 8.30 (td, J = 7.6, 3.7 Hz, 2H), 8.06–8.01 (m, 2H), 7.75–7.63 (m, 2H), 7.61–7.55 (m, 2H), 7.54–7.46 (m, 3H), 7.41–7.35 (m, 3H), 7.34–7.30 (m, 4H), 7.30–7.24 (m, 4H), 7.20 (dd, J = 8.4, 6.1 Hz, 1H). 13CNMR(DMSO-d6, 400 MHz) δ (ppm) = 132.62, 132.38, 131.88, 131.60, 131.43, 130.65, 130.02, 129.81, 129.46, 129.25, 129.16, 128.98, 128.80, 128.69, 127.07, 126.86, 126.37. MALDI-TOF: m/z, calcd. For C43H25N5O2S2 707.14, found (708.25, M + 1): FT-IR (neat, cm−1): 655, 720, 955, 1025, 1202, 1277, 1372, 1430, 1518, 1698, 2218, 2374, 2926, 2977, 3450.
:3 v/v). To this 2-(4-ethynylphenyl)-1,4,5-triphenyl-1H-imidazole (1) (1.07 g, 2.724 mmol), Pd(PPh3)2Cl2 (0.1 g, 0.1362 mmol), and CuI (25 mg, 0.1362 mmol) were added and then the mixture was subjected to purging for one hour. After completion of purging the reaction mixture was stirred at room temperature overnight, and the solvent was removed under reduced pressure. Silica gel column chromatography was adopted to purify the obtained solid material (hexane/CH2Cl2 = 10:1, v/v), recrystallized from chloroform/pentane to give the required product 4 (yield 35%) as a dark yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.06 (s, 1H), 8.68 (dd, J = 16.0, 8.6 Hz, 2H), 7.99 (d, J = 3.7 Hz, 1H), 7.78 (d, J = 8.7 Hz, 2H), 7.70–7.66 (m, 2H), 7.62 (ddd, J = 5.1, 3.8, 1.8 Hz, 4H), 7.59 (d, J = 1.1 Hz, 1H), 7.57–7.53 (m, 2H), 7.48 (ddd, J = 8.7, 6.6, 1.2 Hz, 2H), 7.39 (d, J = 5.2 Hz, 1H), 7.34 (dd, J = 4.9, 2.4 Hz, 2H), 7.30 (dd, J = 4.6, 2.7 Hz, 2H), 7.28 (d, J = 1.4 Hz, 1H), 7.25–7.20 (m, 3H), 7.16 (dd, J = 7.8, 1.7 Hz, 2H), 7.14–7.10 (m, 2H). 13C NMR (CDCl3, 400 MHz) δ (ppm) = 182.89, 145.16, 136.53, 134.27, 131.94, 131.48, 131.16, 130.81, 129.33, 128.88, 128.61, 128.43, 128.26, 128.15, 127.47, 127.07, 126.78, 126.66, 126.37. MALDI-TOF: m/z, calcd. For C48H30N2OS 682.21, found (683.21, [M]+): FT-IR (neat, cm−1): 670, 720, 772, 966, 1020, 1074, 1151, 1219, 1272, 1378, 1462, 1599, 1672, 1734, 2852, 2920, 2957.
:1, v/v), recrystallized from chloroform/pentane to give the required product 5 (yield 30%) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ (ppm) 1H NMR (500 MHz, CDCl3) δ 9.99 (s, 1H), 8.24 (d, J = 4.0 Hz, 1H), 7.97 (d, J = 4.0 Hz, 1H), 7.84 (dd, J = 12.8, 5.8 Hz, 2H), 7.60 (d, J = 7.1 Hz, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.48 (d, J = 8.6 Hz, 2H), 7.34–7.25 (m, 4H), 7.25–7.19 (m, 5H), 7.15 (dd, J = 1.4 Hz, 2H), 7.08 (dd, J = 1.3 Hz, 2H). 13CNMR (CDCl3, 400 MHz) δ (ppm) = 182.98, 136.70, 132.98, 132.93, 132.25, 131.13, 129.94, 129.32, 129.19, 128.96, 128.42, 128.39, 128.24, 128.07, 128.01, 127.51, 126.74. MALDITOF: m/z, calcd. For C40H24N4OS2 640.14, found (641.10, M + 1): FT-IR (neat, cm−1): 722, 754, 802, 852, 1030, 1067, 1128, 1283, 1387, 1464, 1548, 1737, 2854, 2924, 2960.
:1, v/v), recrystallized from MeOH/ether to give the required sensitizer LG-P2 (yield 50%) as a yellow solid. Anal. calcd for C51H31N3O2S % (749.21): C, 81.69; H, 4.18; N, 5.60; found: C, 81.75; H, 4.20; N, 5.58.1H NMR (DMSO-d6, 400 MHz) δ 8.67 (d, J = 8.7 Hz, 2H), 8.34 (s, 1H), 8.01 (d, J = 3.2 Hz, 1H), 7.91 (q, J = 8.7 Hz, 4H), 7.80 (d, J = 8.8 Hz, 2H), 7.76–7.67 (m, 5H), 7.67–7.54 (m, 4H), 7.44 (dd, J = 12.3, 5.5 Hz, 6H), 7.33 (dt, J = 22.4, 7.9 Hz, 4H), 7.16 (t, J = 7.4 Hz, 1H). 13C NMR (DMSO-d6, 400 MHz) δ (ppm) = 135.07, 133.85, 133.03, 132.52, 131.88, 131.71, 130.82, 130.31, 130.10, 129.83, 129.52, 129.46, 129.31, 129.07, 128.87, 128.81, 128.12, 127.80, 126.94. MALDI-TOF: m/z calcd. For C51H31N3O2S 749.21, found (750.18, M + 1): FT-IR (neat, cm−1): 690, 772, 970, 1022, 1220, 1425, 1476, 1512, 1556, 1698, 2225, 2236, 2897, 2961, 3044, 3385, 3473.
:1, v/v), recrystallized from MeOH/ether to give the required sensitizer LG-P4 (yield 60%) as a pale red solid. Anal. calcd for C43H25N5O2S2% (749.21): C, 72.97; H, 3.56; N, 9.89; found: C, 73.00; H, 3.60; N, 9.87. 1H NMR (DMSO-d6, 400 MHz) δ 8.43 (s, 1H), 8.30 (td, J = 7.6, 3.7 Hz, 2H), 8.06–8.01 (m, 2H), 7.75–7.63 (m, 2H), 7.61–7.55 (m, 2H), 7.54–7.46 (m, 3H), 7.41–7.35 (m, 3H), 7.34–7.30 (m, 4H), 7.30–7.24 (m, 4H), 7.20 (dd, J = 8.4, 6.1 Hz, 1H). 13CNMR(DMSO-d6, 400 MHz) δ (ppm) = 132.62, 132.38, 131.88, 131.60, 131.43, 130.65, 130.02, 129.81, 129.46, 129.25, 129.16, 128.98, 128.80, 128.69, 127.07, 126.86, 126.37. MALDI-TOF: m/z, calcd. For C43H25N5O2S2 707.14, found (708.25, M + 1): FT-IR (neat, cm−1): 655, 720, 955, 1025, 1202, 1277, 1372, 1430, 1518, 1698, 2218, 2374, 2926, 2977, 3450.
Methods
1H-NMR spectra were recorded on a 500 MHz INOVA spectrometer. The elemental analysis was performed on an Elementar, Vario MICRO CUBE analyzer. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry was performed on Shimadzu Biotech Axima Performance 2.9.3.20110624: Mode Reflectron-HiRes, Power: 85. Major fragmentations are given as percentages relative to the base peak intensity. Cyclic and differential pulse voltammetric measurements were performed on a PC-controlled electrochemical analyzer (CH Instruments model CHI620C). All these experiments were performed with 1 mM concentration of compounds in dichloromethane at a scan rate of 100 mV s−1 in which tetrabutyl ammonium perchlorate (TBAP) was used as a supporting electrolyte as documented in our previous reports.UV-visible spectroscopy studies were carried out on a Shimadzu (Model UV-3600) spectrophotometer by using 1 mM concentration solutions. Steady-state fluorescence spectra were recorded on a Fluorolog-3 spectrofluorometer (Spex model, JobinYvon) for solutions with optical density at the wavelength of excitation (λex) ≈ 0.05. Time-resolved fluorescence measurements were carried out using a HORIBA JobinYvon spectrofluorometer. The details of DFT calculations are given in the ESI.† The detailed optimization studies are presented in the ESI.†
Device fabrication and characterization
Fabrication of the LG-P2 and LG-P4 dye based DSSC was done using FTO substrates TEC 15 Ω cm−2 and TEC 7 Ω cm−2 (GreatCell Solar) as the working electrode (WE) and the counter electrode (CE) respectively. Both WEs and hole drilled CEs were subjected to thorough cleaning using detergent solution, distilled water, acetone, and isopropanol in an ultrasonic bath. The cleaned electrodes were annealed at 500 °C for 30 minutes. The annealed CEs were deposited with poly(3,4-ethylenedioxithiophene) (PEDOT) via electrochemical deposition to enhance its catalytic property while the annealed WEs were deposited with TiO2 pre-blocking layers by immersing them in aqueous TiCl4 solution (53 mM) at 70 °C for 30 min. 18-NRT paste and 18 NR-AO paste (GreatCell Solar) were subsequently screen printed on the TiCl4 treated substrates to obtain the TiO2 active layer and scattering layer, respectively, to obtain a final thickness of ∼6 μm. The WEs were again annealed at 500 °C, before immersing them in the respective dye solutions (0.3 mM LG-P2 and LG-P4, in a 1:1 mixture of acetonitrile and t-butanol) for 16 h at room temperature. The standard liquid copper electrolyte [Cu(dmp)2+/2+] with the standard composition of 0.2 M bis-(2,9-dimethyl-1,10-phenanthroline)copper(I)bis(trifluoromethane sulfonyl)imide, 0.04 M bis-(2,9-dimethyl-1,10-phenanthroline)copper(II)bis(trifluoromethane sulfonyl)imide chloride, 0.1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and 0.5 M 4-tert-butylpyridine (TBP) in acetonitrile was filled via the drilled holes in the CE after assembling the electrodes using a UV curable epoxy. A microscopic cover slide was used for the final sealing of the holes.
Photocurrent density–voltage (J–V) characterization of the fabricated DSSCs was done under 0.1–1 sun conditions (10–100 mW cm−2 at AM 1.5G) using Oriel 3A – class AAA solar simulator (Model PVIV-94043A, Newport). All measurements were carried out using a circular mask with an effective area of 0.1256 cm2. The J–V measurement under indoor light conditions was carried out using a custom-made setup with a daylight light emitting diode (DL-LED) as the light source. The incident photon-to-current conversion efficiency (IPCE) measurements of the cells were done using a 350 W Xenon lamp coupled with a monochromator setup (Newport). The electrochemical impedance spectroscopy (EIS), charge extraction (CE) and open-circuit voltage decay (OCVD) measurements were performed using the Autolab-PGSTAT 302N (Metrohm) electrochemical workstation. The EIS plots were recorded under dark conditions, at an applied forward bias of 0.45–0.75 V for various cells, with a perturbation amplitude of 10 mV and frequency ranging from 100 mHz to 100 kHz (on a logarithmic scale). The OCVD measurement was done by illuminating the device under open circuit conditions with a white LED, followed by switching off the light and recording the potential decay. Toolbox (Dyenamo) was used to perform transient photovoltage and photocurrent decay measurements with an LED source for bias and a modulation of less than 10% of the steady state value.
3. Results and discussion
The synthetic steps involved for the synthesis of LG-P2 and LG-P4 sensitizers are demonstrated in Scheme 1. The vital starting material 2-(4-ethynylphenyl)-1,4,5-triphenyl-1H-imidazole (1) was accomplished as per the methods reported in the literature.14 We adopted the Suzuki coupling reaction for the intermediates 2 and 3 while the Sonogashira coupling reaction was adopted for formyl intermediates 4 and 5. Finally, the targeted dyes LG-P2 and LG-P4 were accomplished by using the Knoevenagel condensation reaction.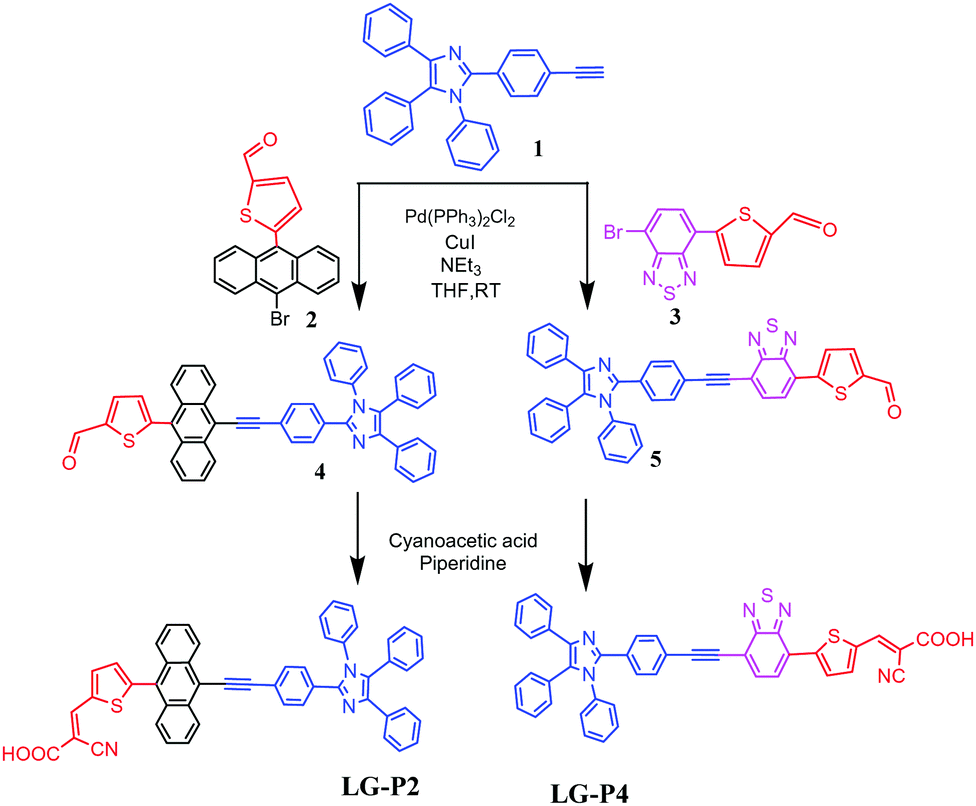 | ||
| Scheme 1 Synthetic scheme of LG-P2 and LG-P4 sensitizers. | ||
Both the sensitizers were characterized by various spectroscopic techniques that include elemental analysis, MALDITOF-MS, 1H NMR, 13C NMR and IR. The elemental analysis of both the dyes is presented in the experimental section and is found to be satisfactory. The MALDI-MS spectrum of LG-P2 showed a peak at m/z = 750.18 (C51H31N3O2S), while that of LG-P4 showed a peak at m/z = 708.25 (C43H25N5O2S2) ascribable to the molecular ion peak (see Fig. S1–S12, ESI†). Further the molecular integrity of both the dyes is confirmed from 1H NMR, 13C NMR and IR spectral analysis (Fig. S2–S13, ESI†).
Quantum mechanical studies
We adopted density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations using the Gaussian 09 package with the functional basis set of the B3LYP/6-31 G(d,p) level in order to optimize the structural, electronic and optical properties of both D–D–π–A and D–A–π–A dyes. Fig. 2a presents the optimized structure of both the dyes which reveals that three flexible phenyl ring appended triphenylimidazole reduces the planarity in solution which results in the minimization of aggregation for both the dyes. The highest occupied molecular orbital (HOMO) of LG-P2 is located on the donor triphenylimidazole and anthracene, whereas the HOMO of LG-P4 is located exclusively on the donor triphenylimidazole. On the other hand, the LUMO is present over the π-spacer (thiophene) and anchoring cyanoacrylic acid in the LG-P2 dye. In the case of LG-P4, the LUMO is present on the auxiliary acceptor (benzothiadiazole moiety), π-spacer (thiophene) and anchoring cyanoacrylic acid (Fig. 2b and Table S1, ESI†). Thus, the theoretical calculation of LG-P2 and LG-P4 revealed appropriate delocalization of electron density on donor and acceptor moieties for both the sensitizers. Subsequently, the band gap was calculated from the difference between HOMO/LUMO of the two dyes and the resultant values of LG-P2 and LG-P4 were 2.51 eV and 2.14 eV, respectively.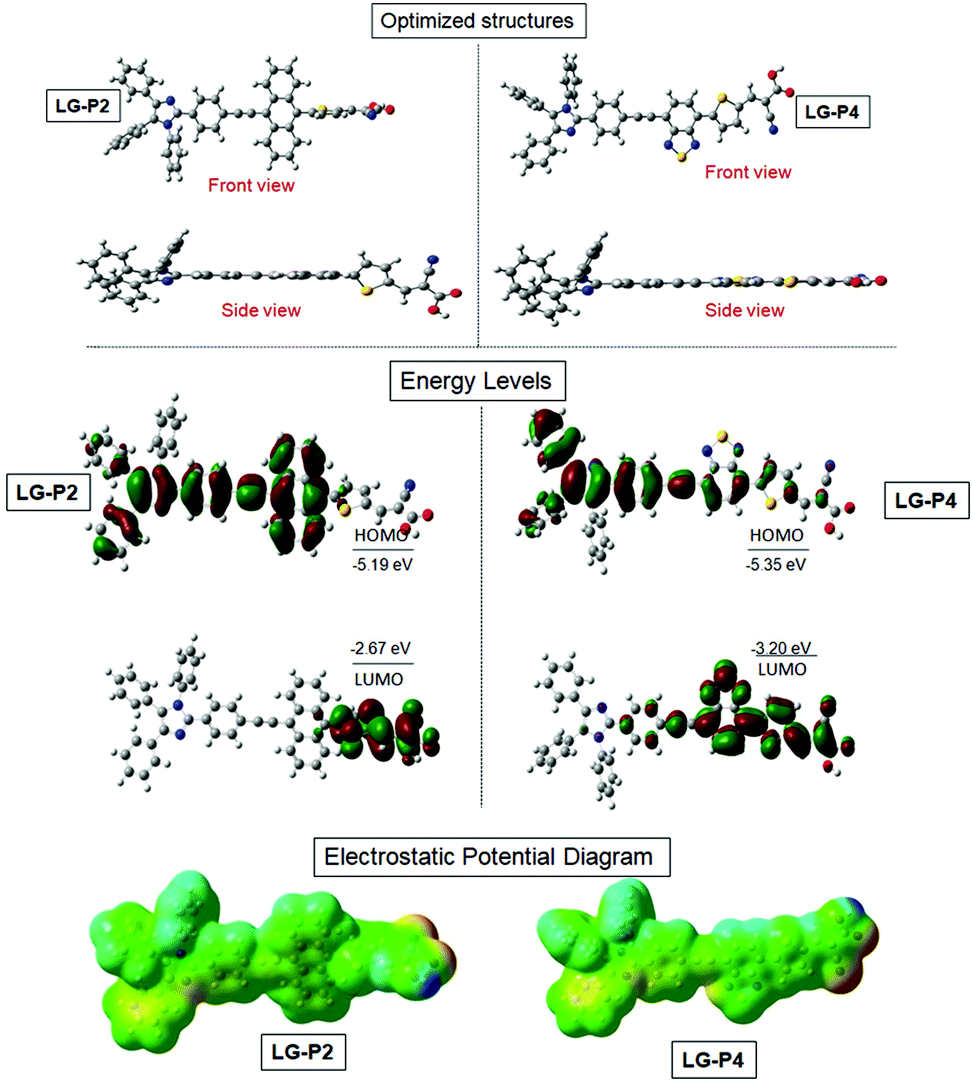 | ||
| Fig. 2 B3LYP/6-31G(d,p)-calculated (a) optimized structures, (b) frontier HOMO, frontier LUMO and (c) electrostatic maps of LG-P2 and LG-P4 dyes. | ||
Furthermore, single-point TD-DFT studies of LG-P2 and LG-P4 were performed by CPCM methodology with the CAM-B3LYP functional and utilizing tetrahydrofuran as a solvent. Fig. 3a depicts the UV-visible absorption spectra of dyes carried out by both theoretical and experimental analysis. It was observed that dyes have shown first 10 vertical singlet–singlet transitions and the corresponding absorption spectrum, very well matching with the experimental studies. The calculated vertical excitation energies for singlet together with calculated oscillator strengths are listed in Table S1 (ESI†). In addition, electrostatic potentials (ESP) of the dyes were recorded and their map displayed that the positive electrostatic potential was at the donor triphenylimidazole while the negative potential was concentrated at the anchoring cyanoacrylic acid group, for both the dyes (Fig. 2c). Later, the theoretical values of dipole moments of LG-P2 and LG-P4 dyes were estimated to be 8.49 D and 11.95 D, respectively. The dihedral angle between the anthracene (auxiliary donor) and thiophene groups in the LG-P2 dye was estimated to be 85.85° while that between BTD (auxiliary acceptor) and the thiophene moiety in LG-P4 was 2.21°. Hence, LG-P2 exhibits a more twisted structure when compared to the LG-P4 dye. In comparison to the corresponding LG-P1 dye with a dihedral angle of 108.04° between anthracene (auxiliary donor) and the phenyl spacer and with a dipole moment of 4.16 D, there is considerable improvement in both dihedral angle and dipole moment on exchanging the π-spacer with the thiophene unit, contributing towards improved performance of LG-P2 compared to LG-P1.24
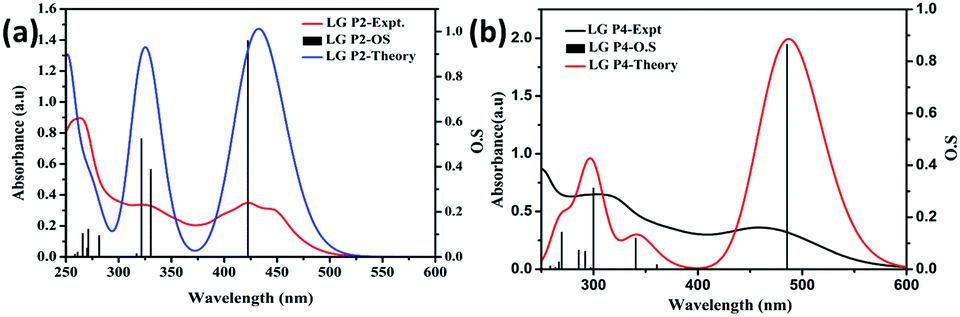 | ||
| Fig. 3 Theoretical and experimental optical absorption spectra of metal free organic dyes in dichloromethane. (a) LG-P2 and (b) LG-P4. | ||
Optical and redox properties
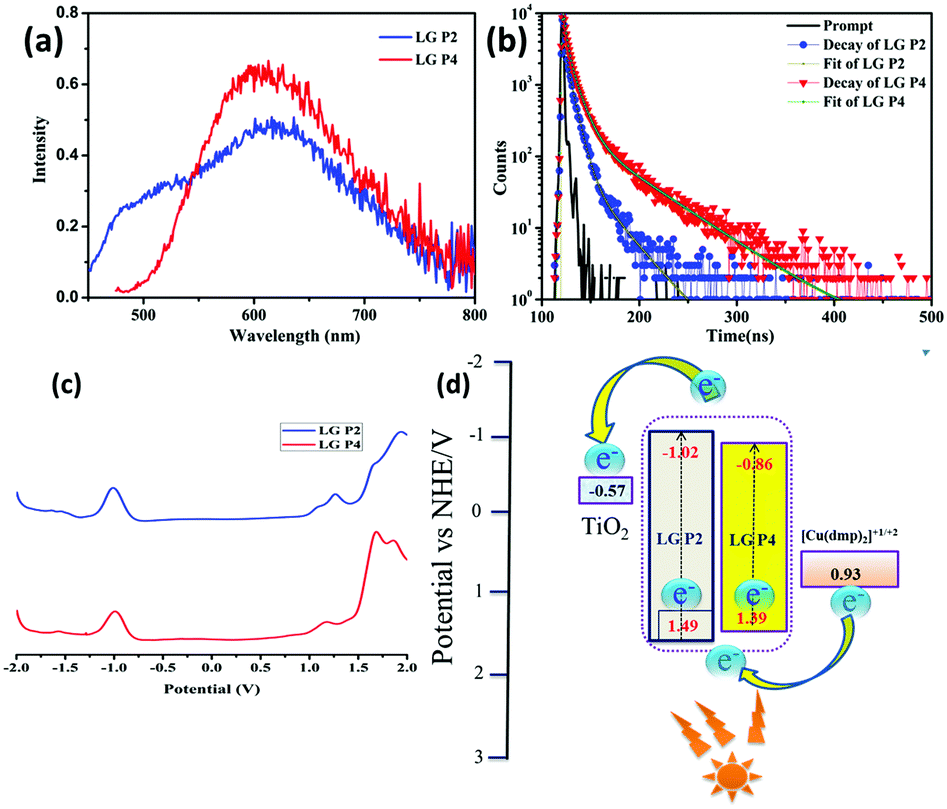
We measured the optical absorption properties of both the sensitizers in acetonitrile solvent as they are essential to gain more understanding on the intra-molecular charge transfer processes with the newly synthesized dyes. Fig. 3a and b illustrates the absorption spectra of both the sensitizers and the respective optical data are presented in Table 1. The peaks in the 250–350 nm region belong to the donor triphenylimidazole and are due to π–π* transitions. The absorption peaks of π-spacer anthracene in the LG-P2 dye appeared in the 400 nm region. On the other hand, for the LG-P4 sensitizer the peak in this region is broad and the onset of absorption extends up to the 600 nm region that arises due to intramolecular charge transfer (ICT). In addition, we measured the absorption properties of both the sensitizers in toluene, acetronitrile and DMF to understand the effect of solvatochromism on absorption properties. Further, in both LG-P2 and LG-P4 sensitizers, the absorption maxima bathochromically shifted by 3 to 20 nm compared to their corresponding phenyl spacer sensitizers (LG-P1 and LG-P3) probably due to the presence of the electron withdrawing thiophene group.24 The DCM solution emission measurements were carried out by excitation at 410 nm in the case of the LG-P2 dye and at 500 nm in the case of the LG-P4 dye at room temperature. Both the dyes showed emission maxima at ∼610 nm (Fig. 4a). Singlet state lifetimes of both LG-P2 and LG-P4 dyes in DCM were estimated from the fluorescence decay curves (λex = 405 nm) and were found to be 0.12 ns and 1.13 ns, respectively (Fig. 4b). Both steady-state emission and lifetime quenched when the dyes were adsorbed onto TiO2 films. The quenched properties might be due to the electron transfer from the excited state of the sensitizer to the conduction band of TiO2. Finally, we estimated the singlet state energies (E0–0) of both the sensitizers from the intersection point of emission and absorption spectra, as 2.51 and 2.25 eV for LG-P2 and LG-P4, respectively.| Dye | Absorption λmax, nm (logε, M−1 cm−1)a |
Emission (nm) λem,maxb | τ (ns), A (%)c | E 1/2 vs. NHEd | E 0–0 (eV) |
|
|---|---|---|---|---|---|---|
a Absorption spectra were recorded in DCM solution. Error limits λmax = ±1 nm, ε ±10%.
b Solvent: DCM, λmax = ±1 nm.
c All lifetimes are in nanoseconds (ns) at λex = 410 nm.
d Solvent: THF, error limits: E1/2 ±0.03 V, 0.1 M TBAP. e ±0.05 eV.
e
E
0–0 was determined from the intersection of absorption and emission spectra, as shown in Fig. 3 and 4a.
f
 was determined as EOX − E0–0. was determined as EOX − E0–0.
|
||||||
| LG-P2 | 325 (4.05) | 619 | 0.78 (42%) | 1.49 | 2.51 | −1.02 |
| 424 (4.06) | 3.26 (5%) | |||||
| 444 (4.01) | 0.12 (53%) | |||||
| LG-P4 | 308 (4.33) | 606 | 1.13 (44%) | 1.39 | 2.25 | −0.86 |
| 459 (4.08) | 0.29 (42%) | |||||
| 5.77 (14%) | ||||||
 | ||
| Fig. 4 (a) Emission spectra of LG-P2 and LG-P4 in dichloromethane at an excitation wavelength of 410 nm. (b) Fluorescence decay curves of LG-P2 and LG-P4. (c) Differential pulse voltammogram (DPV) of LG-P2 and LG-P4 in dichloromethane solvent and 0.1 M TBAP. (d) The improved donor–acceptor interactions and enhanced absorption for LG-P2 sensitizer might have resulted in better injection which in turn led to better IPCE for LG-P2 compared to LG-P1 device (Fig. 3). | ||
The redox properties of both the sensitizers were measured by differential pulse voltammetry in order to understand the possibility of electron transfer from the excited state of the sensitizer (LUMO) to the conduction band of TiO2 and the regeneration of the oxidized dye (HOMO) by taking electron from the Cu(II/I) redox mediator. Fig. 4c illustrates the DPV of both the sensitizers in DCM solvent and the corresponding redox data are presented in Table 1. The LG-P2 dye shows reversible oxidation at 1.49 V vs. NHE whereas in the case of LG-P4 it appears at 1.39 V vs. NHE and these are more positive when compared to the Cu(II/I) redox mediator. The expression 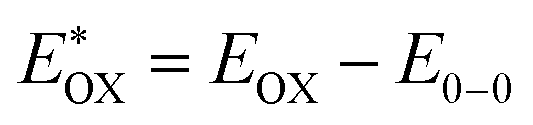 is used to evaluate the excited-state oxidation potential
is used to evaluate the excited-state oxidation potential  and is found to be −1.02 and −0.86 V for LG-P2 and LG-P4. On the other hand, in the case of LG-P1 and LG-P3 the excited state oxidation potentials were found to be −1.13 and −1.06 V and the HOMO–LUMO gap for these sensitizers was 2.65 and 2.53 eV, respectively. Based on
and is found to be −1.02 and −0.86 V for LG-P2 and LG-P4. On the other hand, in the case of LG-P1 and LG-P3 the excited state oxidation potentials were found to be −1.13 and −1.06 V and the HOMO–LUMO gap for these sensitizers was 2.65 and 2.53 eV, respectively. Based on 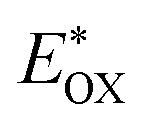 and the HOMO–LUMO gap, LG-P4 is injection limited which can hamper the realization of higher PCE compared to its corresponding LG-P2 sensitizer. Optical and electrochemical properties suggest that the present class of sensitizers are broad absorbing when compared to LG-P1 and LG-P3 sensitizers.24
and the HOMO–LUMO gap, LG-P4 is injection limited which can hamper the realization of higher PCE compared to its corresponding LG-P2 sensitizer. Optical and electrochemical properties suggest that the present class of sensitizers are broad absorbing when compared to LG-P1 and LG-P3 sensitizers.24
Photovoltaic characterization
The photovoltaic performance of DSSCs employing LG-P2 and LG-P4 dyes along with the [Cu(dmp)2]1+/2+ redox couple was measured under the one sun condition (Fig. 5a) and the corresponding parameters are listed in Table 2. In contrast to our previous report, the device employing the LG-P2 dye having anthracene as the auxiliary donor in D–D–π–A architecture displayed a better power conversion efficiency (PCE) of 1.41 ± 0.19%, with an open circuit voltage (Voc) of 0.74 ± 0.01 V, a short circuit current density (Jsc) of 2.71 ± 0.17 mA cm−2, and a fill factor (FF) of 70.5 ± 4.32% in comparison to the corresponding D–A–π–A sensitizer LG-P4 having benzothiadiazole as the auxiliary acceptor unit. The LG-P4 based device exhibited a PCE of 0.43 ± 0.03%, a Voc of 0.5 ± 0.06 V, a Jsc of 1.82 ± 0.18 mA cm−2 and a FF of 47.3 ± 3.59%. From the measured PV parameters, it is obvious that the D–D–π–A dye (LG-P2) based devices exhibited better Voc as well as Jsc when compared to D–A–π–A dye (LG-P4) based devices. The higher Jsc of the LG-P2 based device in comparison to the LG-P4 device is in accordance with the IPCE spectra (Fig. 5b). The red shifted spectrum for LG-P4 above 500 nm may be attributed to its highly coplanar structure, owing to its low dihedral angle and lower bandgap.35 However, the IPCE maximum for LG-P4 is low compared to the LG-P2 device, which aligns with the corresponding absorption spectra (Fig. 3). Also a more positively shifted LUMO for LG-P4 might have resulted in a reduced injection efficiency and hence lowered the IPCE for this dye (Fig. 4d). This reduced IPCE is the major reason for lower Jsc in DSSCs employing LG-P4 as the sensitizer.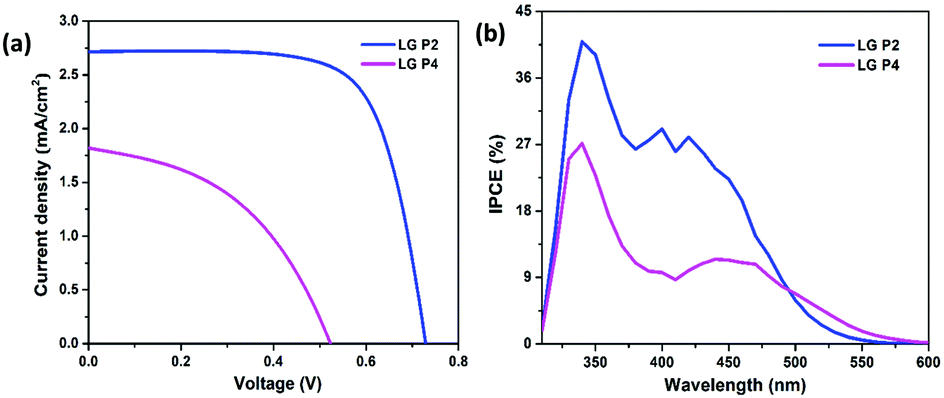 | ||
| Fig. 5 (a) J–V characteristics and (b) photocurrent action spectra of LG-P2 and LG-P4 dye based DSSCs with the [Cu(dmp)2]1+/2+ electrolyte. | ||
| Dye | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| LG-P2 | 0.74 ± 0.01 | 2.71 ± 0.17 | 70.5 ± 4.32 | 1.41 ± 0.19 |
| LG-P4 | 0.50 ± 0.06 | 1.82 ± 0.18 | 47.3 ± 3.59 | 0.43 ± 0.03 |
Further investigation of the improved performance of the LG-P2 based device was performed using various optical and electrical perturbation methods. It is very interesting to observe a reversal in the performance of DSSCs employing triphenylimidazole based organic dyes with the change in π-spacer from phenyl in our previous work to thiophene in the present study.24 A comparison of the results obtained for LG-P1 and LG-P3 based devices with LG-P2 and LG-P4 is given in the ESI† (Table S2). The switching of π-spacer from phenyl to thiophene led to an increment in PCE for DSSCs using the D–D–π–A dye, which is attributed to the enhanced conjugation between the auxiliary donor and acceptor moieties, owing to its better planarity contributing towards better optoelectronic properties. This contribution to improved donor–acceptor interactions and enhanced absorption for the LG-P2 sensitizer in turn resulted in better injection leading to better IPCE for LG-P2 compared to the LG-P1 device (Fig. 3). This improved injection resulted in the improvement of both Jsc and Voc of the device employing the LG-P2 dye leading to its better performance. On the other hand, this π-spacer switching in the case of the D–A–π–A dye having benzothiadiazole as the auxiliary acceptor unit resulted in a decreased device performance, even with an improved planarity for LG-P4 when compared to the LG-P3 dye. This decreased performance of LG-P4 based devices may be due to the injection limitation for the LG-P4 dye due to the closer LUMO to the conduction band with reduced bandgap. Additionally the chance of aggregation of the highly co-planar LG-P4 dye molecules on the TiO2 surface, leading to a decreased injection, also cannot be ignored. We have studied this aspect in detail in our previous contributions using indolo[3,2-b]indole D–π–A sensitizers.36
The J–V characteristics of DSSCs fabricated with LG-P2 and LG-P4 dyes and [Cu(dmp)2]1+/2+ electrolyte were also measured under 0.5 and 0.1 sun conditions and the corresponding device parameters are shown in Table S3 (ESI†). The photovoltaic parameters were also obtained under low intensity illumination (1000 lux DLLED) as presented in Table S4 (ESI†). Under indoor light conditions, the LG-P2 device exhibited better performance than LG-P4. The better PCE of the LG-P2 device (7.46%) could be attributed to its enhanced Voc (0.5 V), Jsc (93 μA cm−2) and FF (51.6%) compared to the LG-P4 device with a PCE of 0.97%, a Voc of 0.12 V, a Jsc of 87 μA cm−2 and a FF of 28.7%.
The interfacial charge transfer kinetics in the DSSCs was studied in detail for exploring the reason for the reduced performance in the LG-P4 dye, in spite of its better planarity, smaller bandgap and broader absorption. In DSSCs, Voc is the difference between the Fermi energy level of the semiconductor (EFermi) and the redox potential of the electrolyte (Eredox). EFermi is determined by the conduction band edge position and the charge density in the semiconductor layer. Voc is also affected by the recombination processes. Higher the recombination, lower will be the Voc of the device. The higher dark current in the LG-P4 based device is indicative of the larger recombination rate in these devices compared to the one using the LG-P2 dye (Fig. 6a). Fig. 6b shows the dependence of Voc on the light intensity (Iin). The slope of this semi-logarithmic plot, (dVoc)/(dlogIin), gives the inverse of the diode ideality factor, also known as the recombination factor, as β = (2.303kbT/q)[(dVoc)/(dlogIin)]−1, where kb is Boltzmann's constant, T is the temperature, and q is the electronic charge. If the slope of the Vocvs. logIin plot becomes 59 mV decade−1, an ideal β value of 1 is obtained, which indicates that recombination is occurring from the TiO2 CB alone.37,38β values calculated for LG-P2 and LG-P4 sensitized DSSCs are 0.59 and 0.23 respectively, as shown in Fig. 6b. Deviation of both the devices from the ideal β value symbolizes the contribution of trap states in the recombination mechanism. However, the LG-P4 based device with larger deviation from the ideal β value might have a higher rate of trap-state mediated recombination, which contributed to its reduced Voc. The voltage vs extracted charge plot, depicted in Fig. 7a, also reveals the reason for higher recombination for the LG-P4 device, as the CB edge of the device employing this dye is seen to be positively shifted with respect to the LG-P2 based device. This causes more surface trap states to overlap with the energy levels of oxidized dye molecules, contributing towards more trap state mediated recombination.39,40 This shift in the CB of TiO2 may be attributed to the difference in the dipole moment of the two dye molecules being attached to the TiO2 surface.
 | ||
| Fig. 6 (a) I–V characteristics under dark conditions and (b) open circuit voltage versus intensity (Vocvs. logIin) plot for LG-P2 and LG-P4 based DSSCs using the [Cu(dmp)2]1+/2+ electrolyte. | ||
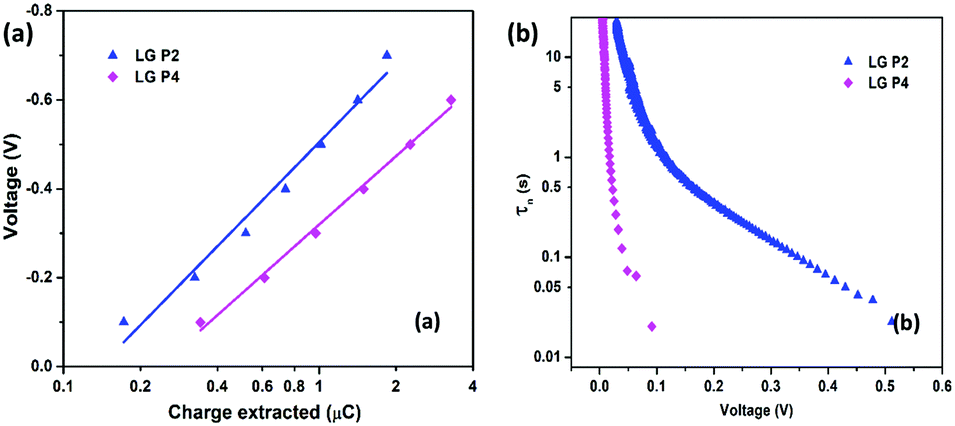 | ||
| Fig. 7 (a) Charge extraction and (b) lifetime (τn) versus voltage plot (obtained from OCVD measurements) for LG-P2 and LG-P4 dye based DSSCs using the [Cu(dmp)2]1+/2+ electrolyte. | ||
The lifetime of electrons in the TiO2 CB is directly influenced by the rate of recombination. The lifetime (τn) plot obtained from OCVD measurements (Fig. 7b) shows that the LG-P4 based device possesses a lower τn, i.e. a higher recombination rate, when compared to the LG-P2 based device. Also, higher recombination in LG-P4 devices might have adversely affected the FF of these devices. The τn plot obtained from EIS measurements as well as transient photovoltage decay measurements also exhibits the same trend (Fig. 8). The representative Nyquist plot obtained from EIS measurements under dark conditions (Fig. 8a) exhibits one large semicircle for each device, in the intermediate frequency range, which corresponds to charge transfer at the TiO2/dye/electrolyte interface. The corresponding Bode plots are shown in Fig. S15 (ESI†). The τn values are obtained from Bode plots by using the relation τn = 1/(2πfmax), where fmax is the frequency corresponding to the peak in the bode plot. These τn values plotted against applied voltage are displayed in Fig. 8b. The smaller semicircle for LG-P4 based devices indicates higher recombination in these devices. The higher recombination for the LG-P4 dye may be attributed to the lower driving force for electron injection from the excited state of the dye to the TiO2 CB associated with the LG-P4 dye, hence reducing the electron concentration in the TiO2 layer. Additionally, with a highly planar geometry (dihedral angle of 2.21°) LG-P4 dye molecules are prone to aggregation which might have inhibited the injection of electrons leading to more recombinations.36 The DSSC based on the LG-P4 dye hence possesses lower Voc, owing to its more positively shifted TiO2 conduction band, higher recombination and lower electron concentration. The diffusion coefficient of electrons in the semiconducting layer of DSSCs is related to their transport time as Dn = d2/τd, where d is the thickness of the TiO2 layer (∼6 μm). The lower electron density in the semiconductor layer for the LG-P4 based device results in slower diffusion of electrons leading to a lower diffusion coefficient (Dn) as shown in Fig. 8d, which is calculated from the τd obtained from transient photocurrent decay measurements (Fig. S16, ESI†). This in turn leads to lower charge collection efficiency (ηcc) in devices employing the LG-P4 dye compared to LG-P2 (Fig. S16, ESI†). The lower absorption of the LG-P4 dye along with lower ηinj and ηcc leads to lower IPCE and hence lower Jsc in the case of DSSCs employing the D–A–π–A LG-P4 sensitizer in comparison to the D–D–π–A dye (LG-P2) employing the [Cu(dmp)2]1+/2+ electrolyte. This observation is opposite to that we observed in our previous work, wherein the D–A–π–A dye with phenyl as π-spacer (LG-P3) showed better photovoltaic performance compared to the D–D–π–A sensitizer (LG-P1). Thus, the change of π-spacer from phenyl to thiophene led to considerable variations in optoelectronic properties leading to differences in photovoltaic performance using the [Cu(dmp)2]1+/2+ electrolyte.
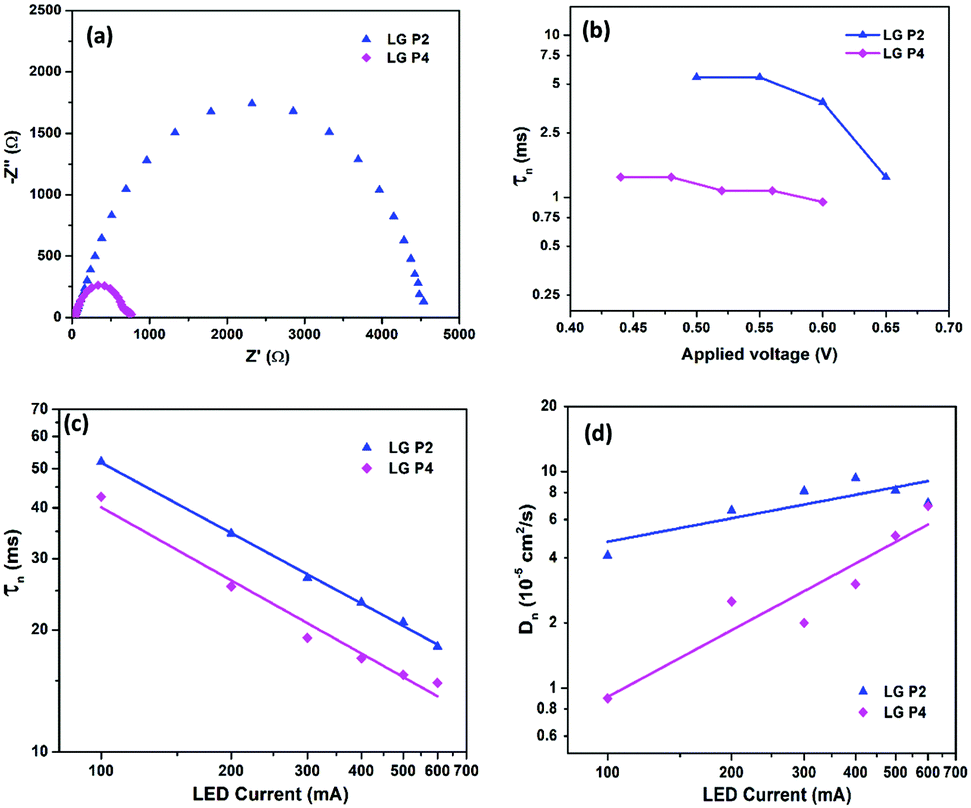 | ||
| Fig. 8 (a) Representative Nyquist plot (at a bias potential of 0.6 V), (b) lifetime (τn) versus applied voltage plot (from EIS measurements), (c) lifetime (τn) versus LED current plot obtained from transient photovoltage decay measurements, and (d) diffusion coefficient (Dn) versus LED current plot obtained from transient photocurrent decay measurements of LG-P2 and LG-P4 dye based DSSCs using the [Cu(dmp)2]1+/2+ electrolyte. | ||
4. Conclusion
Two new organic dyes with D–D–π–A (LG-P2) and D–A–π–A (LG-P4) architectures were synthesized, with non-planar triphenylimidazole as the donor, thiophene as the π-bridge and cyano acrylic acid as the anchoring group. The onset of the absorption spectra extends up to 600 nm probably due to the presence of the electron withdrawing benzothiadiazole group in LG-P4. These were employed as sensitizers in [Cu(dmp)2]1+/2+ electrolyte based DSSCs and the corresponding photovoltaic performance was evaluated. In the present work, by carefully tuning the π-spacer we succeeded in improving the photovoltaic performance of the anthracene auxiliary donor based D–D–π–A dye from 0.29% to 1.41%. In contrast to our previous report,24 using thiophene as π-spacer the dye with anthracene as the auxiliary donor (LG-P2) exhibited better PCE compared to the D–A–π–A dye with benzothiadiazole as the auxiliary acceptor (LG-P4), under both outdoor and indoor conditions. Under the one sun condition, the LG-P2 based device showed a PCE of 1.41%, with a Voc of 0.74 V, a Jsc of 2.71 mA cm−2, and a FF 70.5%, whereas the LG-P4 dye based device resulted in a lower PCE of 0.43% with a Voc of 0.5 V, a Jsc of 1.82 mA cm−2 and a FF of 47.3%. More than the efficiency values, the genesis of the present contribution is to unveil the care that needs to be taken in designing molecular systems suitable for use with new generation alternate electrolytes like copper. In addition to the donor units, alkyl side chains, and auxiliary donors/acceptors, π-spacer also plays a very critical role and needs to be handpicked through appropriate optimizations accounting for the changes in donor and other functionalities to fine-tune the molecular backbone and energetics realizing improved performance. The introduction of thiophene as π-spacer endowed a highly co-planar structure to the D–A–π–A dye, leading to more aggregation of these planar molecules which inhibited electron injection to the TiO2 conduction band besides the position of the LUMO closer to the CB resulting in lower driving force for injection. This not only affected the Voc but also diminished the Jsc of the DSSC incorporating the LG-P4 dye. The D–D–π–A LG-P2 dye showcased a better lifetime, diffusion coefficient and charge collection efficiency contributing to improved PCE. This points to the care that needs to be taken while designing and synthesizing molecular systems to be used along with alternate redox systems like the [Cu(dmp)2]1+/2+ electrolyte to realize good charge transfer and photovoltaic performance.Author contributions
Palivela Siva Gangadhar: validation, investigation, resources. Anooja Jagadeesh: validation, investigation, resources. Manne Naga Rajesh: validation, investigation, resources. Andrew Simon George: validation, investigation, resources. Seelam Prasanthkumar: resources, software. Suraj Soman: conceptualisation, methodology, writing – review and editing. Lingamallu Giribabu: conceptualisation, methodology, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SS would like to thank the SERB CRG project (CRG/2020/001406) and the CSIR-FIRST (MLP65) project. SS also acknowledges CSIR Mission (HCP30) and FTT (MLP38) Projects for financial support. PSG, MNR and AJ acknowledge CSIR for the research fellowship. ASG thanks the SERB CRG project (CRG/2020/001406) for a fellowship. We thank the CSIR-IICT director for the support (IICT/Pubs./2021/271).References
- N. Abas, A. Kalair and N. Khan, Futures, 2015, 69, 31–49 CrossRef.
- L. Giribabu, R. K. Kanaparthi and V. Velkannan, Chem. Rec., 2012, 12, 306–328 CrossRef CAS PubMed.
- C. Dragonetti and A. Colombo, Molecules, 2021, 26, 2461 CrossRef CAS PubMed.
- W. Naim, V. Novelli, I. Nikolinakos, N. Barbero, I. Dzeba, F. Grifoni, Y. Ren, T. Alnasser, A. Velardo, R. Boreelli, S. Haacke, S. M. Zakeeruddin, M. Gratzel, C. Barolo and F. Sauvage, JACS Au, 2021, 1, 409–426 CrossRef CAS.
- K. S. Srivishnu, S. Prasanthkumar and L. Giribabu, Mater. Adv., 2021, 2, 1229–1247 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS PubMed.
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- V. K. Singh, R. K. Kanaparthi and L. Giribabu, RSC Adv., 2014, 4, 6970–6984 RSC.
- P. S. Gangadhar, G. Reddy, S. Prasanthkumar and L. Giribabu, Phys. Chem. Chem. Phys., 2021, 23, 14969–14996 RSC.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J.-I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- L. Han, H. Yu, Q. Chen, Y. Lu, J. He and S. Jiang, Dyes Pigm., 2021, 187, 109092 CrossRef CAS.
- J. V. S. Krishna, N. V. Krishna, T. H. Chowdhury, S. Singh, I. Bedja, A. Islam and L. Giribabu, J. Mater. Chem. C, 2018, 6, 11444–11456 RSC.
- M. V. Vinayak, T. M. Lakshmykanth, M. Yoosuf, S. Soman and K. R. Gopidas, Sol. Energy, 2016, 124, 227–241 CrossRef CAS.
- N. Duvva, R. K. Kanaparthi, J. Kandhadi, G. Marotta, P. Salvatori, F. De Angelis and L. Giribabu, J. Chem. Sci., 2015, 127, 383–394 CrossRef CAS.
- S. Soman, M. Rahim, S. Lingamoorthy, C. H. Suresh and S. Das, Phys. Chem. Chem. Phys., 2015, 17, 23095–23103 RSC.
- R. K. Kanaparthi, J. Kandhadi and L. Giribabu, Tetrahedron, 2012, 68, 8383–8393 CrossRef CAS.
- K. Devulapally, G. Reddy, S. Prasanthkumar, A. Jagadeesh, S. Soman and L. Giribabu, J. Porphyrins Phthalocyanines, 2021, 25, 407–417 CrossRef CAS.
- P. Nitha, V. Jayadev, S. C. Pradhan, V. V. Divya, C. H. Suresh, J. John, S. Soman and A. Ajayaghosh, Chem. – Asian J., 2020, 15, 3503–3512 CrossRef CAS PubMed.
- B. Hemavathi, V. Jayadev, P. C. Ramamurthy, R. K. Pai, K. N. Narayanan Unni, T. Ahipa, S. Soman and R. G. Balakrishna, New J. Chem., 2019, 43, 15673–15680 RSC.
- C.-P. Lee, R. Y.-Y. Lin, L.-Y. Lin, C.-T. Li, T.-C. Chu, S.-S. Sun, J. T. Lin and K.-C. Ho, RSC Adv., 2015, 5, 23810–23825 RSC.
- N. Duvva, Y. K. Eom, G. Reddy, K. S. Schanze and L. Giribabu, ACS Appl. Energy Mater., 2020, 3, 6758–6767 CrossRef CAS.
- P. S. Gangadhar, A. Jagadeesh, A. S. George, G. Reddy, S. Prasanthkumar, S. Soman and L. Giribabu, Mol. Syst. Des. Eng., 2021, 6, 779–789 RSC.
- D. E. Almenningen, H. E. Hansen, M. F. Vold, A. F. Buene, V. Venkatraman, S. Sunde, B. H. Hoff and O. R. Gautun, Dyes Pigm., 2021, 185, 108951 CrossRef CAS.
- J. M. dos Santos, E. Tanaka, A. A. Wiles, G. Cooke and N. Robertson, Mol. Sys. Des. Eng., 2021, 6, 381–389 RSC.
- X. Ren, S. Jian, M. Cha, G. Zhou and Z. S. Wang, Chem. Mater., 2012, 24, 3493–3499 CrossRef CAS.
- J. V. S. Krishna, D. Koteshwar, T. H. Chowdhury, S. P. Singh, I. Bedja, A. Islam and L. Giribabu, J. Mater. Chem. C, 2019, 7, 13594–13605 RSC.
- W. L. Fan, Y.-Z. Chang, I.-L. Zhao, Z.-N. Xu, D.-Z. Tan and Y.-G. Chen, New J. Chem., 2018, 42, 20163–20170 RSC.
- N. V. Krishna, J. V. S. Krishna, S. P. Singh, L. Giribabu, A. Islam and I. Bedja, J. Phys. Chem. C, 2017, 121, 25691–25704 CrossRef.
- N. V. Krishna, J. V. S. Krishna, S. P. Singh, L. Giribabu, L. Han, I. Bedja, R. K. Gupta and A. Islam, J. Phys. Chem. C, 2017, 121, 6464–6477 CrossRef CAS.
- A. Mishra, M. K. R. Fischer and P. Bauerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- L. T. Thuy, P. Ho, L. QuocBao, C. T. T. Thuy, R. Cheruku, H.-J. Jo, S. Thogiti and J. H. Kim, J. Nanosci. Nanotechnol., 2017, 17, 8053–8060 CrossRef CAS.
- Y. Geng, F. Pop, C. Yi, N. Avarvari, M. Gratzel, S. Decurtinsa and S.-X. Liu, New J. Chem., 2014, 38, 3269–3274 RSC.
- K.-M. Kim and J.-I. Hong, Tetrahedron, 2016, 72, 8387–8392 CrossRef CAS.
- P. V. Santhini, V. Jayadev, S. C. Pradhan, S. Lingamoorthy, P. R. Nitha, M. V. Chaithanya, R. K. Mishra, K. N. Narayanan Unni, J. John and S. Soman, New J. Chem., 2019, 43, 862–873 RSC.
- S. Soman, S. C. Pradhan, M. Yoosuf, M. V. Vinayak, S. Lingamoorthy and K. R. Gopidas, J. Phys. Chem. C, 2018, 122, 14113–14127 CrossRef CAS.
- M. V. Vinayak, M. Yoosuf, S. C. Pradhan, T. M. Lakshmykanth, S. Soman and K. R. Gopidas, Sustainable Energy Fuels, 2018, 2, 303–314 RSC.
- S. Rühle, M. Greenshtein, S.-G. Chen, A. Merson, H. Pizem, C. S. Sukenik, D. Cahen and A. Zaban, J. Phys. Chem. B, 2005, 109, 18907–18913 CrossRef PubMed.
- E. Ronca, M. Pastore, L. Belpassi, F. Tarantelli and F. D. Angelis, Energy Environ. Sci., 2013, 6, 183–193 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00852h |
| ‡ These authors contributed equally towards the completion of this work. |
| This journal is © The Royal Society of Chemistry 2022 |