
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring co-doping of cobalt and nitrogen in a fullerene-based carbon composite and its effect on the supercapacitive performance†
Bohong
Jiang‡
a,
Guangpu
Zhang‡
b,
Qin
Tang
c,
Fancang
Meng
a,
Dechun
Zhou
a,
Wenli
Zhao
a,
Wei
Jiang
 *b and
Qingmin
Ji
*a
*b and
Qingmin
Ji
*a
aHerbert Gleiter Institute for Nanoscience, School of Materials Science and Engineering, Nanjing University of Science & Technology, 200 Xiaolingwei, Nanjing, 210094, China. E-mail: jiqingmin@njust.edu.cn
bNational Special Superfine Powder Engineering Technology Research Center, Nanjing University of Science and Technology, 200 Xiaolingwei, Nanjing, 210094, China. E-mail: superfine_jw@126.com
cSchool of Chemistry and Chemical Engineering, Yancheng Institute of Technology, No. 1 Mid. Xiwang Avenue, Yancheng, China
First published on 7th December 2021
Abstract
Fullerene-based carbons have attracted great attention in applications of energy storage and conversion due to their unique properties and tunable architectures. However, fullerene's poor long-range conductivity limits their application performance for supercapacitors. Therefore, developing active fullerene-based carbons with high capacitances is expected. In this work, mesoporous carbon composites with various doping amounts of cobalt (Co) and nitrogen (N) were fabricated via manipulation of fullerene self-assembly with a cobalt tetramethoxy phenylporphyrin (CoTMPP) and pyrolysis. The confined state of CoTMPPs within the fullerene superstructures facilitates the fixation of Co and N during the carbonation and leads to a highly homogeneous distribution state within fullerene-based carbon composites. When used as electrodes for supercapacitors, the doped carbon composite showed significantly improved electrochemical performance. The specific capacitance could be nearly ten times higher than that of the fullerene carbons without doping and they have excellent cyclic stability. The work presents new insight into controllable functional doping of carbon materials. It may also further promote the development of fullerene superstructures for advanced electrochemical applications.
Introduction
Unlike other kinds of carbon materials, fullerene could be used as building blocks to assemble into multi-dimensional superstructures with various morphologies and transform into stable mesoporous carbon materials after thermal treatments.1–6 Due to their unique properties and tunable architectures at the molecular level, these fullerene-based carbons have attracted great attention for application in solar cells, energy storage and conversion.7–9 However, when used as electrode materials for supercapacitors, the specific capacitance of the fullerene-based carbons was still relatively low. Therefore, the search for an active fullerene-based supercapacitor with improved performance is still needed.Doping heteroatoms in the structure is one of the best solutions to overcome the shortcomings of fullerene's poor long-range conductivity.10–12 It may also improve the structural features of the fullerene superstructures for electrochemical applications.13,14 Almost all the species, metals or non-metals could be doped in fullerenes. Particularly, metal doping may effectively improve the electronic properties of fullerene-derived carbons.15–17 The doped metals could form a binary electron acceptor–donor system and provide more active sites in the structures.18
Various strategies have been applied for the metal doping of fullerene superstructures. For example, Peng et al. fabricated three-dimensional cross-type Fe-doped fullerene structures by self-assembly of ferrocene-modified fullerene and achieved enhanced electrochemical performance.19 Saianand et al. introduced copper into fullerene superstructures by a hard template and used them as effective catalysts for the oxygen reduction reaction (ORR).20 We also successfully prepared fullerene carbon materials with enhanced supercapacitive properties by regulating the self-assembled structures of ferrocene-modified fullerenes in our previous work.21 However, these studies still cannot control the doping ratio of metals. The relationship between the metal doping content and application performance of fullerene-derived carbons is thus unclear.
Herein, we fabricated cobalt (Co) and nitrogen (N) doped mesoporous carbon composites based on the liquid–liquid interfacial precipitation (LLIP) process of C60 fullerenes with a cobalt tetramethoxy phenylporphyrin (CoTMPP) and pyrolysis. By manipulating the mixing ratios of C60 and CoTMPP, we obtained various porous carbon materials with controllable doping amounts of Co and N (totally 0.30–1.52%) (Fig. 1). Co, a transition metal, has excellent energy storage capability for supercapacitors. But unfavorable aggregation may easily occur during the doping process or their usage.22 The confined state of CoTMPP within C60 crystalline structures could prevent Co agglomeration in the carbon structure and successfully dope functional Co and N simultaneously. With the homogenous distribution of Co and N, the resultant mesoporous carbon composites exhibit excellent energy storage performance, the specific capacitance value of which could be about ten times higher than that of the fullerene carbons without doping and also well maintained 99.5% specific capacitance at least after 5000 cycles.
 | ||
| Fig. 1 Scheme for the preparation of Co and N doped carbon composites by manipulating C60 assembly with CoTMPP and pyrolysis. | ||
Experimental part
The assembly process of fullerene superstructures
C60s are dissolved in toluene (about 2.5 mg mL−1) by sonication for 1 hour. The undissolved C60s in the toluene solution are removed by filtration. The self-assembly of fullerenes is based on the liquid–liquid interfacial precipitation (LLIP) process. For details, 4 ml C60 solution is placed in a glass bottle (15 mL), and 8 mL IPA is slowly added on the top of the solution to form an interface. The mixture solution is left undisturbed for 5 minutes and then stored at room temperature for 12 hours after shaking. The sediment from the process is collected by centrifugation. In the C60CoTMPP assembly process, C60 and CoTMPP with different mixing ratios (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10:1, 20:1) are dissolved in toluene by sonication, and then similar procedures mentioned above are followed. The C60CoTMPP crystals obtained from mixing ratios of 5:1, 10:1, and 20:1 are named C60CoTMPP-0.2, C60CoTMPP-0.1 and C60CoTMPP-0.05, respectively.
:1, 10:1, 20:1) are dissolved in toluene by sonication, and then similar procedures mentioned above are followed. The C60CoTMPP crystals obtained from mixing ratios of 5:1, 10:1, and 20:1 are named C60CoTMPP-0.2, C60CoTMPP-0.1 and C60CoTMPP-0.05, respectively.
Formation of mesoporous carbons from fullerene superstructures
The collected precipitate from the LLIP process is dried under vacuum. The dry samples are then heated to 900 °C with a heating speed of 10 °C min−1 and kept for 2 hours at 900 °C in a tubular pyrolysis furnace under a constant nitrogen flow (120 cm3 min−1). The carbonized C60CoTMPP crystals from C60CoTMPP-0.2, C60CoTMPP-0.1 and C60CoTMPP-0.05 are named C60CoTMPP-0.2_900, C60CoTMPP-0.1_900 and C60CoTMPP-0.05_900, respectively.Electrochemical measurements
The electrochemical performance is measured in a three-electrode system in 1 M H2SO4 aqueous electrolyte. A glassy carbon electrode is used as the working electrode, with a Ag/AgCl electrode as the reference electrode and Pt wire as the counter electrode. For the preparation of the working electrode, one drop of the IPA–water suspension of the porous carbon samples (5 μL, 5 mg mL−1) is spread on the surface of the glassy carbon electrode and dry at room temperature. 2.5 μL Nafion solution (0.05%) is dropped on the glassy carbon electrode before the measurement. The specific capacitance (Cs) is calculated from the cyclic voltammograms (CV) the galvanostatic charge/discharge (CD) curves according to the following equations:| Cs(weight) = Q/(2ΔE·v·m) |
| or Cs(area) = Q/(2ΔE·v·A) |
| Cs = I/(m·ΔE/Δt) |
Characterization
Field emission scanning electron microscopy (FE-SEM) is performed at 15 kV on a FEI Quanta 250FEG. Transmission electron microscopy (TEM) is performed by using a Tecnai G220 at 200 kV. Nitrogen sorption measurements are conducted on powder samples at 77 K using an Autosorb-1 surface area and pore size analyzer (Gemini VII 2390). X-Ray diffraction (XRD) patterns are measured using a Bruker-AXS D8 Advance diffractometer. Raman spectra are recorded on a Raman scattering spectrometer (Renishaw-invia) using excitation radiation at 514 nm from an Ar+ ion laser and at a power of 10 mW. Fourier transform infrared (FTIR) spectra are obtained using a FTIR spectrometer Nicolet S10 (Thermo Fisher). The Co contents in the carbon samples are determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) using Agilent 5110.Results and discussion
Fullerene superstructures from the mixture of C60 and CoTMPP
According to the solubility of C60 in solvents, C60s may self-assemble into crystalline superstructures at the interface of good and bad solvents.23 Therefore, various solvent types could affect the packing of C60s and lead to different morphologies of C60 superstructures. As the driving force of the fullerene assembly is mainly based on π–π interactions, other π-molecules may easily intercalate into the C60s superstructures from the co-assembly.8 In the system with CoTMPP, a metalloporphyrin molecule, as its metal ion center could coordinate with carbon atoms in fullerene,24,25 it may thus further promote the insertion into C60 superstructures.In this work, we use toluene (PhMe)–isopropyl alcohol (IPA) as the solvent system to carry out the self-assembly of C60s based on the LLIP process. Without mixing CoTMPP, the assembly of C60s at the toluene–IPA interface leads to the formation of nanorods. As shown in Fig. S1 (ESI†), the nanorods are ∼400 nm in diameter, ∼10 μm in length and possess smooth surfaces. The co-assembly with CoTMPP alters the packing of C60s and leads to the formation of C60CoTMPPs with completely different morphologies (Fig. 2 and Table S1, ESI†). C60CoTMPP-0.2 shows rod morphology with polyprism structures (Fig. 2a). Its length is ∼2.5 μm and its diameter is ∼1 μm. C60CoTMPP-0.1 seems to possess microparticles with lamellar structures (about ∼1.9 μm in diameter) and a rough surface (Fig. 2b). While for C60CoTMPP-0.05 with the lowest mixing amount, similar rod morphology like the nanorods from pure C60s is obtained (Fig. 2c), but it shows shorter lengths (∼2.3 μm) and larger diameters (∼1.2 μm). We suggest that the influence of the CoTMPP amount on the morphologies of C60CoTMPP superstructures should be caused by the strong packing inclination between CoTMPPs. CoTMPPs can form flaky crystals in the solvent mixture of PhMe–IPA (Fig. S2, ESI†). This aggregation inclination of CoTMPPs may change their insertion state within C60s and might also interfere with the packing of C60s during the co-assembly process. It thus leads to distinct structural features of the C60CoTMPP superstructures. It should be mentioned that even higher content of CoTMPP is possible to assemble with C60s. For example, crystalline nanosheets of C60/CoTMPP can be formed at a mixing ratio of 1:1. However, the C60/CoTMPP hybrid structure with mixing ratios higher than 5:1 may cause serious fusion of Co components and destroy the morphology during the following heat treatment. As the resultant blocks can hardly be used for further applications, we thus focus on the C60/CoTMPP superstructures with the mixing ratio lower than 5:1.
 | ||
| Fig. 2 SEM and TEM images of (a, d and g) C60CoTMPP-0.2; (b, e and h) C60CoTMPP-0.1; and (c, f and i) C60CoTMPP-0.05. | ||
The elemental mapping analyses indicate the homogenous distributions of Co, N, and C elements across the microstructures for all C60CoTMPP superstructures (Fig. S3, ESI†). HR-TEM observations prove the changes of C60s packing after the intercalation of CoTMPPs (Fig. 2g–i). The nanorods from bare C60s (C60-rod) show regular lattices of 1.21 nm and 0.927 nm, which correspond to the (110) and (101) planes (Fig. S4, ESI†). The lattice spacing of C60CoTMPP superstructures is inclined to shrink with the increase of the CoTMPP amount. As shown in Fig. 2g, C60CoTMPP-0.2 shows a lattice spacing of 1.16 nm for the (110) plane, while the spacing of C60CoTMPP-0.1 and C60CoTMPP-0.05 turns to be 0.863 nm and 0.882 nm for the (101) plane, respectively.
The detailed crystalline structures of C60CoTMPPs are analyzed by the X-ray diffraction (XRD) technique (Fig. 3). Similar to C60 nanorods without CoTMPP, the XRD patterns of C60CoTMPPs also show the characteristic peaks of the HCP phase. However, with the presence of CoTMPP, a new diffraction peak at 6.2°, which should arise from the packing of CoTMPPs, appeared. The diffraction peak for the (110) plane (around 7.4°) also shifts to a little higher degree for C60CoTMPPs, which is consistent with the TEM observations. The intercalation of CoTMPPs within C60s might strengthen the interactions between molecules and lead to the spacing shrinkage of C60s.
 | ||
| Fig. 3 XRD patterns of C60CoTMPP structures. (i) C60CoTMPP_0.2, (ii) C60CoTMPP_0.1, (iii) C60CoTMPP_0.05, (iv) C60-rods, and (v) CoTMPPs. | ||
To understand the molecular interactions between C60 and CoTMPP, FTIR spectra of pure C60 rods, CoTMPP, and C60CoTMPPs were measured (Fig. S5, ESI†). The peaks around 550 cm−1, 1185 cm−1 and 1430 cm−1 for C60 nanorods and C60CoTMPPs can be attributed to the ring stretching and deformation vibrations of C60s.26 The characteristic peaks of CoTMPP at 750–900 cm−1 (ρ-substituted phenyl groups and N–H bending), 1510 cm−1 (C–C bonds in the pyrrole ring), 1254 cm−1 (C–N stretching vibration), and 1000 cm−1 (breathing modes of the pyrrole ring) are found to shift to lower wavenumbers for C60CoTMPPs.27 This implies the interactions between the porphyrin moieties and C60s in the C60CoTMPPs.
Mesoporous carbon from C60/CoTMPP crystals
As solvent molecules exist within the fullerene superstructures from the LLIP assembly process, carbonization of fullerene superstructures may induce the formation of meso- or micropores in the carbon structures, which is beneficial for their applications for energy storage. In this work, C60CoTMPP crystals were carbonized through heat treatment at 900 °C under N2 flow.The Raman spectra of the resultant C60CoTMPP_900s exhibit two strong bands around 1360 and 1580 cm−1, corresponding to the D band from defect-containing or disordered graphitic carbon and the G band from the in-plane vibration of sp2 carbons (Fig. 4a). The typical peaks from C60 (two Ag bands and six Hg bands), which are observed in Raman spectra of C60CoTMPPs, disappeared (Fig. S6, ESI†). This indicates the successful structural transformation to carbon materials after the heat treatment (Fig. 4a). The intensity ratios of the D band and G band (ID/IG) for C60_900 (without CoTMPP), C60CoTMPP-0.05_900, C60CoTMPP-0.1_900 and C60CoTMPP-0.2_900 are 0.76, 1.09, 0.92 and 1.18. This result suggests that the intercalation of CoTMPPs in the fullerene superstructures may induce the formation of more defects in the carbon structures. But the orders of ID/IG for C60CoTMPP_900s are not exactly the same as the CoTMPP amount in C60CoTMPPs. This implies that the intercalation state of CoTMPP might also influence the transforming structures of carbons. In C60CoTMPP_900s, the A1g vibration mode (690 cm−1) of Co components appears and its intensity is increased with the additional CoTMPP amount in C60CoTMPPs.28,29 This suggests the consistence of the Co doping amount of C60CoTMPP_900s with the intercalated CoTMPPs in C60CoTMPPs.
 | ||
| Fig. 4 (a) Raman spectra and (b) XRD patterns of C60_900s and C60CoTMPP_900s. (i) C60_900s (ii) C60CoTMPP-0.05_900, (iii) C60CoTMPP-0.1_900, and (iv) C60CoTMPP-0.2_900. | ||
The XRD patterns of C60CoTMPP_900s also indicate that the characteristic diffraction peaks of C60CoTMPP disappeared after carbonation (Fig. 4b). Unlike the carbonized C60_900 (without doping) with only a wide peak in the range of 20–30°, two additional diffraction peaks appeared at 25.8° and 44.2° for C60CoTMPP_900s. The diffraction at 25.8° is ascribed to the (002) plane of graphite,30 which suggests that the existence of CoTMPP might promote the stacking of graphene sheets during the structural transformation of fullerenes into carbons. While the diffraction at 44.2° should arise from the planes of cobalt nitride and cobalt oxide,31,32 which can be attributed to the (111) plane of CoN4 (JCPDS No. 41-0943) and the (400) plane of Co3O4 (JCPDS No. 42-1467) (Fig. S7, ESI†). Its peak intensity is shown to increase with the mixing amount of CoTMPP in C60CoTMPPs, indicating again varied doping amounts of Co and N in C60CoTMPP_900s.
The TEM observations directly indicate the formation of the porous matrix of C60CoTMPP_900s from the dense crystalline nature of C60CoTMPPs (Fig. S8, ESI† and Fig. 5). Compared with various C60CoTMPP_900s, we can find that C60CoTMPP-0.1_900 has a much looser networked structure, which means more pore spaces and larger exposure surfaces for active sites. The elemental mapping analyses based on TEM prove the existence of Co, N, and C elements (Fig. 5). This also reveals the maintenance of the dispersion state of Co and N after the high-temperature heat treatment. With different doping amounts, C60CoTMPP_900s possess varied Co distribution states in the structure (Fig. S8, ESI†). C60CoTMPP-0.01_900 shows tiny nanoparticles of Co components with relatively scattered distributions. We can also observe tiny nanoparticles in the structure of C60CoTMPP-0.1_900, but it has much more dense distributions. While for C60CoTMPP-0.2_900, nanoparticles can be hardly found, and metal components seem to aggregate as blocks in the structure.
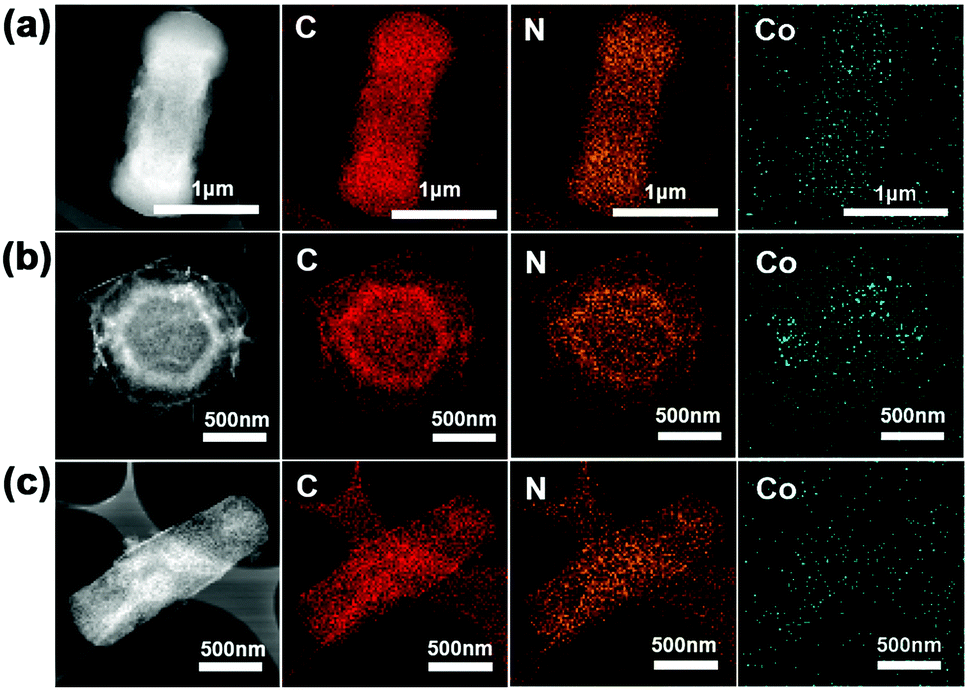 | ||
| Fig. 5 STEM images and the corresponding elemental mapping images of C60CoTMPP_900s. (a) C60CoTMPP-0.2_900, (b) C60CoTMPP-0.1_900, and (c) C60CoTMPP-0.05_900. | ||
To understand the molecular transformation during the pyrolysis, we use thermogravimetric analysis (TGA) to gain information (Fig. S9, ESI†). The thermograms of C60CoTMPPs show a small weight loss of 5–8% below 150 °C, which should be attributed to the removal of solvent molecules within the superstructures. The weak mass loss that occurred between 450 and 600 °C is related to the gradual decomposition of CoTMPP. A significant mass loss at a temperature higher than 700 °C is observed, which should be associated with molecular changes from C60s. C60s are transferred into carbon networks or decomposed by the oxygen within the structures. Comparing the weight loss ratios of C60CoTMPPs and C60-rod, we can find that the presence of CoTMPPs in the C60 superstructures may effectively restrain the decomposition of C60s during the pyrolysis. The oxygen within the structure might prefer to combine with Co. This leads to the formation of more oxidized Co and lower decomposition of C60s for C60CoTMPPs with a large mixing amount of CoTMPP.
The chemical states of Co and N in C60CoTMPP_900s are further analyzed based on XPS measurement (Fig. 6 and Fig. S10, ESI†). Fig. 6a shows the Co 2p spectrum in the binding energy range from 770 to 810 eV by the Gaussian method. It contains split spin–orbit 2p3/2 (778–785 eV) and 2p1/2 (793–798 eV) regions. The spectrum is fitted into two states, whose peaks at 779 and 795 eV should belong to Co3+ (Co–O); peaks at 782 and 797 eV are attributed to Co2+ (Co–Nx) with satellite peaks at 786 and 805 eV.33,34 The N 1s spectra are deconvoluted into five peaks, which can be assigned to primary pyridine N (398.5 eV), metal N (399.2 eV), pyrrole N (400.2 eV), graphite N (401.5 eV), and oxidized N (404.5 eV) (Fig. 6b).35,36 The C 1s spectrum shows the existence of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond (285.8 eV) with CC (284.4 eV) and a C–O bond (289.2 eV) (Fig. 6c).37 These results indicate that the doped Co and N atoms may form Co–N and Co–O in the carbon network of the fullerene-derived carbons.
N bond (285.8 eV) with CC (284.4 eV) and a C–O bond (289.2 eV) (Fig. 6c).37 These results indicate that the doped Co and N atoms may form Co–N and Co–O in the carbon network of the fullerene-derived carbons.
 | ||
| Fig. 6 XPS spectra of C60CoTMPP-0.1_900. (a) Co 2p spectrum, (b) N 1s spectrum, and (c) C 1s spectrum. | ||
According to the relative area of each component peak in XPS spectra, the proportions of different Co states in C60CoTMPP_900s are compared (Table S2, ESI†). The Co–N:Co–O ratio is 1.52 (C60CoTMPP-0.05_900), 3.66 (C60CoTMPP-0.1_900), and 0.5 (C60CoTMPP-0.2_900), respectively. These results indicate that the formation of Co–N becomes greatly restrained in the carbonization of C60CoTMPPs when a large amount of CoTMPP is involved. Since CoTMPP in large amounts tends to aggregate themselves during the co-assembly with C60s, the densely packed state of CoTMPPs in the superstructure and the oxygen in CoTMPPs may promote the oxidation of Co during carbonization and lead to the formation of less Co–N. ICP measurement was further employed to confirm the Co content in C60CoTMPP_900s (Table S3, ESI†). The result indicates that the mass fraction of the Co element is 1.52% for C60CoTMPP-0.2_900, 0.71% for C60CoTMPP-0.1_900, and 0.30% for C60CoTMPP-0.05_900, respectively. In comparison of Co ratios in C60CoTMPP (before carbonation), which are 1.26% (C60CoTMPP-0.2), 0.57% (C60CoTMPP-0.1) and 0.26% (C60CoTMPP-0.05), there is almost no loss of doped Co during the heating process. Compared with the theoretical content of Co in C60CoTMPP (Table S3, ESI†), there are also no big differences. These results demonstrate that we could precisely control the doping amount of Co in the fullerene carbon materials through this assembly process.
The doping influence on the porous structures of C60CoTMPP_900s is analyzed based on nitrogen adsorption/desorption isotherms (Fig. S11, ESI†). Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods are adapted to calculate the specific surface area and pore size distributions (Table S4, ESI†). The isotherms of C60CoTMPP_900s are shown to be of type IV with a H3 hysteresis loop. This indicates the existence of mesopores and a small part of micropores in C60CoTMPP_900s. C60CoTMPP-0.1_900 has the highest BET surface area of about 496 m2 g−1, while those of C60CoTMPP-0.2_900 and C60CoTMPP-0.05_900 are 365 m2 g−1 and 446 m2 g−1, respectively. As the intercalation of CoTMPP in C60s may bring more packing disorders in the structure, it might facilitate the production of more pores from the decomposition during the heat treatment. This thus leads to a higher surface area of C60CoTMPP_900s than the porous carbon from pure C60s (C60_900), the surface area of which is 285 m2 g−1 (Fig. S12, ESI†). However, when more CoTMPPs are involved, the presence of more CoTMPP aggregates and their effect on promoting the stacking of graphene sheets may diminish part of pores in the carbon structures, resulting in lower surface areas and also narrower distribution of pore size for C60CoTMPP-0.2_900.
Electrochemical performances of C60/CoTMPP_900s
The porous carbons derived from C60CoTMPP_900s are applied as electrode materials for supercapacitors. Their electrochemical supercapacitive properties are measured based on cyclic voltammetry (CV) and galvanostatic charge–discharge cycles (GCD) by using a three-electrode configuration in 1 M H2SO4 aqueous electrolyte. C60CoTMPP_900s are deposited at the surface of the glassy carbon electrode. SEM observation of the C60CoTMPP_900 layers indicates their uniform coverage states on the electrode surface with porous nature accessible for electrolytes (Fig. S13, ESI†). The CV curves of C60CoTMPP_900s at 10 mV s−1 over the range of 0–0.8 V are shown in Fig. 7a. All samples display a rectangular-like shape curve, which proves that C60CoTMPP_900s could act like a typical double-layer capacitor. In contrast to the low specific capacitance of C60_900 without doping (32.5 F g−1 at 10 mV s−1 and 46.9 F g−1 at 1 A g−1) (Fig. S14, ESI†),21 C60CoTMPP_900s with Co and N doping all show enhanced capacitive property of fullerene-based carbons. C60CoTMPP-0.1_900 possesses the highest specific capacitance of 296 F g−1 or 0.1 F cm−2. C60CoTMPP-0.05_900 and C60CoTMPP-0.2_900 have a specific capacitance of 168 F g−1 (0.06 F cm−2) and 55 F g−1 (0.02 F cm−2), respectively. That is to say, the capacitance order of C60CoTMPP_900s is C60CoTMPP-0.1_900 > C60CoTMPP-0.2_900 > C60CoTMPP-0.05_900. This order is consistent with that of the surface area of C60CoTMPP_900s and Co–N content in the carbons. This means that both structural and chemical factors may affect the performance. The surface area of the electrode is demonstrated as one major factor for supercapacitor performance.38 It is related to the ion diffusion ability in the carbon electrode and the formation of an ion-electron double layer. The double-layer capacitance is known to be directly proportional to the surface area.39 But for this system, the capacitance difference of C60CoTMPP_900s is much larger than that of their BET surface areas. This implies that the doping state of Co–N should bring great influence on their effective surface area for ion diffusion and conversion, which leads to a large difference in the electrochemical performance. However, the distinct effects from the porous structure and doping state cannot be evaluated separately. The assembly structures of fullerenes are sensitive to the assembly conditions. Small changes in the type and volume of solvents or additives may alter the structural features of the fullerene superstructures.23,40 Therefore, the intercalation of functional molecules within the C60s superstructures will inevitably change the assembled morphologies. The advantage of tunable architectures for fullerene assemblies always accompanies the functions.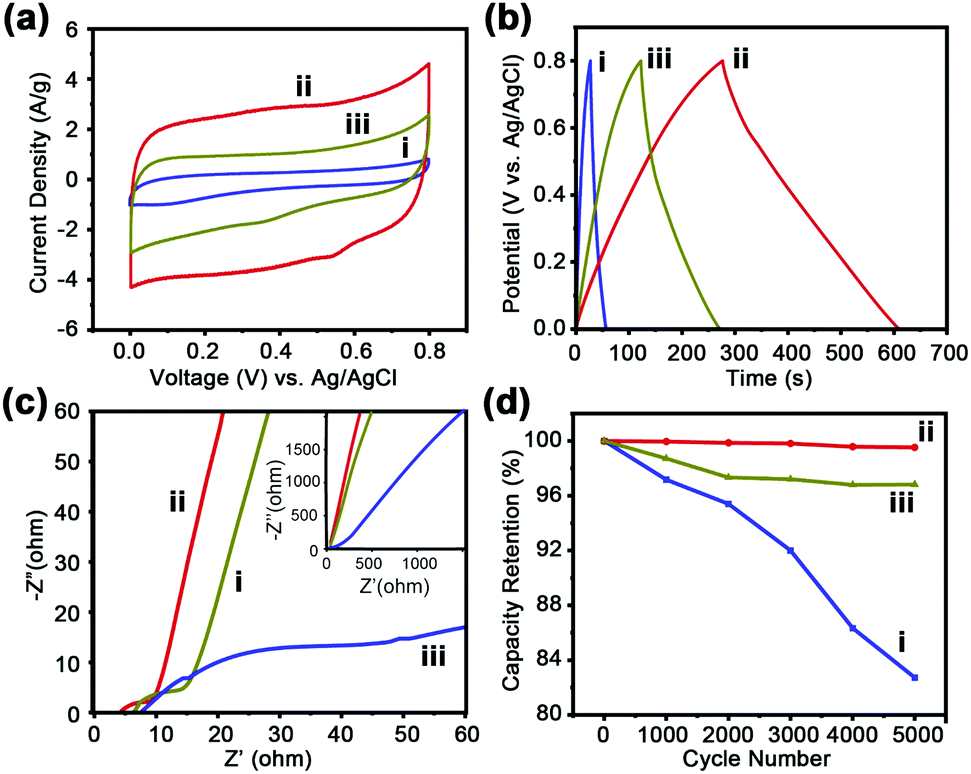 | ||
| Fig. 7 (a) CV curves of C60CoTMPP_900s at a scan rate of 10 mV s−1; (b) CD curves of C60CoTMPP_900s at a current density of 1 A g−1; (c) Nyquist plots of C60CoTMPP_900s; and (d) cycling stability test of C60CoTMPP_900s. (i) C60CoTMPP_0.2-900, (ii) C60CoTMPP_0.1-900, and (iii) C60CoTMPP_0.05-900. | ||
The GCD curves of C60CoTMPP_900s are all triangular shapes, which also indicates a typical double-layer capacitor of the samples. The longer discharge time of C60CoTMPP-0.1_900 means a higher specific capacitance of 416.31 F g−1 at 1 A g−1 (Fig. 7b). Compared with other reported fullerene-derived carbon materials (Table S5, ESI†), the performance of C60CoTMPP-0.1_900 could exceed those of most of the reported fullerene carbon materials.
To further investigate the mechanism of charge storage for supercapacitors, electrochemical impedance spectroscopy (EIS) is performed. The Nyquist plots of C60CoTMPP_900s reveal good capacitive behaviors (Fig. 7c), which possess a semicircle in the high-frequency area and inclined linear plot at low frequencies. The width of the semicircle at high frequencies can determine charge transfer resistance (Rct), which is associated with the charge transfer across the electrode–electrolyte interface. C60CoTMPP-0.1_900 displays the shortest diameter of the semicircle, which means the lowest Rct. This indicates more effective charge transfer and better capacitive contribution in C60CoTMPP-0.1_900. The slope inclination in the low-frequency area is related to internal resistance (Rs). As C60CoTMPP-0.1_900 exhibits the highest inclination degree, a higher electrical conductivity of C60CoTMPP-0.1_900 is suggested. The calculated Rct values for C60CoTMPP-0.05_900, C60CoTMPP-0.1_900, and C60CoTMPP-0.2_900 are 11.2, 5.6, and 45.4 Ω, and the Rs values are 6.2, 4.6, and 8.2 Ω, respectively. C60CoTMPP-0.2_900 shows the highest Rct and Rs values, which suggests that besides the Co doping amount, the doping state and structural features may also have a large influence on the electron transfer in the carbon composites.
From the above results, it is observed that the content of Co–N in the carbon composites plays the main role in improving the electrochemical performance. C60CoTMPP-0.1_900 possesses unique morphology and highest content of Co–N, and thus exhibits better capacitance than C60CoTMPP-0.05_900 and C60CoTMPP-0.2_900. Although C60CoTMPP-0.2_900 has the highest Co doping amount, more doped Co in the carbon structure exists as the Co–O state with the least content of Co–N, which brings an adverse effect on the conductivity of the carbon composite. The doping influence on the structural features also affects the electrochemical performance. The doping ratio of CoTMPP for C60CoTMPP-0.1 completely changes the inclination of forming the rod morphology. The homogeneous distribution of CoTMPP in C60CoTMPP-0.1 also facilitates the formation of Co–N and the transformation of more porous structures during the carbonation process. With less doping of CoTMPP, the assembled structure largely maintains the features of the C60 superstructures without doping and shows limited influence on the formation of pores during the carbonation. While for C60CoTMPP-0.2 with large doping of CoTMPP, CoTMPPs are dispersed in the superstructure as molecular aggregates and may more easily induce the formation of Co–O and dense packing of graphite carbons during the heat treatment. These factors make C60CoTMPP-0.2_900 exhibit the worst performance. It should be noted that besides the doping content in C60CoTMPP_900s, the existence of graphitic carbons is also important for electrochemical performance. Based on the TGA result, C60s will not begin carbon transformation at a temperature lower than 700 °C. This means that no graphitic carbons formed for the carbonized C60CoTMPPs by 700 °C. The resultant carbons also show poor electrochemical performance (Fig. S15, ESI†), in which C60CoTMPP-0.1_700 only has a specific capacitance of 35 F g−1, similar to fullerene-carbons without doping.
Considering the best electrochemical performance of C60CoTMPP-0.1, we further evaluate its electrochemical stability. Fig. S16 (ESI†) shows the CV curves of C60CoTMPP-0.1_900 at different scan rates from 10 to 100 mV s−1. All curves at different scan rates display quasi-rectangular shape, indicating the good maintenance of double-layer capacitor behavior as well as good rate capability. The capacitance retention is also excellent, which is shown to keep >99% of the initial capacitance after 5000 GCD cycles (Fig. 7d). In contrast, C60CoTMPP-0.05_900 could maintain about 97% of the capacitance, but C60CoTMPP-0.2_900 shows a big decrease with the cycles. The worse electrochemical stability for C60CoTMPP-0.2_900 should be due to the existence of more Co–O in the carbon composite, since cobalt oxides have disadvantages of poor electrical conductivity, easy polarization, and volumetric expansion during the charge and discharge processes.41,42 Therefore, this result further proves that the control of the CoTMPP intercalation state in C60 structures is essential to both the porous feature and the doping states of Co in the carbon composites.
Conclusions
In summary, we successfully manipulated the intercalation of CoTMPP in the C60 self-assembled structure through the LLIP process and fabricated C60 composite crystals with various microscopic morphologies. After carbonization, the CoTMPP confined in the C60 crystals transformed into active Co–N in the carbon structures. The content of Co–N is shown to be not only directly related to the mixing ratio of CoTMPP in C60 crystals but also the distribution state in the superstructures. The presence of Co–N in the porous carbon composites improves the electrochemical properties greatly. The resultant carbon composite showed an enhanced specific capacitance of 416.31 F g−1 at 1 A g−1, which is almost ten times higher than that of the C60 carbons without doping, and could almost show no activity loss after at least 5000 cycles. This work brings new insight into controllable functional doping of carbon materials by fullerene interfacial self-assembly. This may also further promote the development of fullerene superstructures for advanced electrochemical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by Instrument & Equipment Open Funding of Nanjing University of Science and Technology, by the National Natural Science Foundation of China No. 21875108 and by the Fundamental Research Funds for the Central Universities (No. 30921013106).Notes and references
- M. R. Benzigar, S. Joseph, H. Ilbeygi, D. H. Park, S. Sarkar, G. Chandra, S. Umapathy, S. Srinivasan, S. N. Talapaneni and A. Vinu, Angew. Chem., Int. Ed., 2020, 57, 569–573 CrossRef PubMed.
- T. Xu, W. Shen, W. Huang and X. Lu, Mater. Today Nano, 2020, 11, 100081 CrossRef.
- L. K. Shrestha, Q. Ji, T. Mori, K. Miyazawa, Y. Yamauchi, J. P. Hill and K. Ariga, Chem. – Asian. J., 2013, 8, 1662–1679 CrossRef CAS PubMed.
- F. Lu, E. A. Neal and T. Nakanishi, Acc. Chem. Res., 2019, 52, 1834–1843 CrossRef CAS PubMed.
- R. G. Shrestha, L. K. Shrestha, M. Abe and K. Ariga, J. Nanosci. Nanotechnol., 2015, 15, 2394–2399 CrossRef CAS PubMed.
- Q. Tang, P. Bairi, R. G. Shrestha, J. P. Hill, K. Ariga, H. Zeng, Q. Ji and L. K. Shrestha, ACS Appl. Mater. Interfaces, 2017, 9, 44458–44465 CrossRef CAS PubMed.
- Q. Tang, S. Maji, B. Jiang, J. Sun, W. Zhao, J. P. Hill, K. Ariga, H. Fuchs, Q. Ji and L. K. Shrestha, ACS Nano, 2019, 13, 14005–14012 CrossRef CAS PubMed.
- Q. Tang, S. Zhang, X. Liu, M. Sumita, S. Ishihara, H. Fuchs, Q. Ji, L. K. Shrestha and K. Ariga, Phys. Chem. Chem. Phys., 2017, 19, 29099–29105 RSC.
- T. Anusha, K. S. Bhavani, J. S. Kumar and P. K. Brahman, Diamond Relat. Mater., 2020, 104, 107761 CrossRef CAS.
- P. Bairi, T. Tsuruoka, S. Acharya, Q. Ji, J. P. Hill, K. Ariga, Y. Yamauchi and L. K. Shrestha, Mater. Horiz., 2018, 5, 285–290 RSC.
- L. K. Shrestha, R. G. Shrestha, Y. Yamauchi, J. P. Hill, T. Nishimura, K. Miyazawa, T. Kawai, S. Okada, K. Wakabayashi and K. Ariga, Angew. Chem., Int. Ed., 2015, 127, 965–969 CrossRef.
- P. Bairi, R. G. Shrestha, J. P. Hill, T. Nishimura, K. Ariga and L. K. Shrestha, J. Mater. Chem. A, 2016, 4, 13899–13906 RSC.
- T. Wakahara, P. D’Angelo, K. Miyazawa, Y. Nemoto, O. Ito, N. Tanigaki, D. D. Bradley and T. D. Anthopoulos, J. Am. Chem. Soc., 2012, 134, 7204–7206 CrossRef CAS PubMed.
- T. Wakahara, M. Sathish, K. Miyazawa, C. Hu, Y. Tateyama, Y. Nemoto, T. Sasaki and O. Ito, J. Am. Chem. Soc., 2009, 131, 9940–9944 CrossRef CAS PubMed.
- S. Saha, P. Samanta, N. C. Murmu and T. Kuila, J. Energy Storage, 2018, 17, 181–202 CrossRef.
- Z. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 8, 702–730 RSC.
- M. A. Lebedeva, T. W. Chamberlain and A. N. Khlobystov, Chem. Rev., 2015, 115, 11301–11351 CrossRef CAS PubMed.
- A. L. Balch and K. Winkler, Coord. Chem. Rev., 2021, 438, 213623 CrossRef CAS.
- Z. Peng, Y. Hu, J. Wang, S. Liu, C. Li, Q. Jiang, J. Lu, X. Zeng, P. Peng and F. F. Li, Adv. Energy Mater., 2019, 9, 1802928 CrossRef.
- G. Saianand, A. I. Gopalan, J. C. Lee, C. Sathish, K. Gopalakrishnan, G. E. Unni, D. Shanbhag, V. D. Dasireddy, J. Yi and S. Xi, Small, 2020, 16, 1903937 CrossRef CAS PubMed.
- B. Jiang, Q. Tang, W. Zhao, J. Sun, R. An, T. Niu, H. Fuchs and Q. Ji, CrystEngComm, 2020, 22, 6287–6294 RSC.
- S. J. Uke, V. P. Akhare, D. R. Bambole, A. B. Bodade and G. N. Chaudhari, Front. Mater., 2017, 4, 21 CrossRef.
- J. Minato and K. Miyazawa, Carbon, 2005, 43, 2837–2841 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov and R. N. Lyubovskaya, Coord. Chem. Rev., 2014, 262, 16–36 CrossRef CAS.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- G. Andrievsky, V. Klochkov, A. Bordyuh and G. Dovbeshko, Chem. Phys. Lett., 2002, 364, 8–17 CrossRef CAS.
- G. Zuo, X. Lu, Z. Xue, B. Lv, Y. Wang and J. Kang, Synth. React. Inorg. Met.-Org. Chem., 2006, 36, 589–594 CrossRef CAS.
- S. Bhattacharyya, B. Konkena, K. Jayaramulu, W. Schuhmann and T. K. Maji, J. Mater. Chem. A, 2017, 5, 13573–13580 RSC.
- C. Liu, G. Bai, X. Tong, Y. Wang, B. Lv, N. Yang and X.-Y. Guo, Electrochem. Commun., 2019, 98, 87–91 CrossRef CAS.
- L. Fu, Y. Chen, S. Zhao, Z. Liu and R. Zhu, RSC Adv., 2016, 6, 19482–19491 RSC.
- J. Sheng, L. Wang, L. Deng, M. Zhang, H. He, K. Zeng, F. Tang and Y.-N. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 7191–7200 CrossRef CAS PubMed.
- Y. Wu, Q. Shi, Y. Li, Z. Lai, H. Yu, H. Wang and F. Peng, J. Mater. Chem. A, 2015, 3, 1142–1151 RSC.
- S.-H. Cho, K. R. Yoon, K. Shin, J.-W. Jung, C. Kim, J. Y. Cheong, D.-Y. Youn, S. W. Song, G. Henkelman and I.-D. Kim, Chem. Mater., 2018, 30, 5941–5950 CrossRef CAS.
- K. Niu, B. Yang, J. Cui, J. Jin, X. Fu, Q. Zhao and J. Zhang, J. Power Sources, 2013, 243, 65–71 CrossRef CAS.
- J. M. Ziegelbauer, T. S. Olson, S. Pylypenko, F. Alamgir, C. Jaye, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2008, 112, 8839–8849 CrossRef CAS.
- Y. Jiang, Y. Lu, X. Wang, Y. Bao, W. Chen and L. Niu, Nanoscale, 2014, 6, 15066–15072 RSC.
- S. S. Park, S.-W. Chu, C. Xue, D. Zhao and C.-S. Ha, J. Mater. Chem., 2011, 21, 10801–10807 RSC.
- E. J. Lee, L. Lee, M. A. Abbas and J. H. Bang, Phys. Chem. Chem. Phys., 2017, 19, 21140–21151 RSC.
- E. Taer, A. Agustino, R. Farma, R. Taslim, Awitdrus, M. Paiszal, A. Ira, S. D. Yardi, Y. P. Sari, H. Yusra, S. Nurjanah, S. D. Hartati, Z. Aini and R. N. Setiadi, J. Phys.: Conf. Ser., 2018, 1116, 032040 CrossRef.
- K. Ariga and L. K. Shrestha, Mater. Adv., 2021, 2, 582–597 RSC.
- S. Wang, L. Li, Y. Shao, L. Zhang, Y. Li, Y. Wu and X. Hao, Adv. Mater., 2019, 31, 1806088 CrossRef PubMed.
- D. Qiu, X. Ma, J. Zhang, B. Zhao and Z. Lin, Chem. Phys. Lett., 2018, 710, 188–192 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and TEM images, FTIR spectra, Raman spectra, XPS spectra, nitrogen isotherms and CV curves of fullerene superstructures and related fullerene-based carbons. See DOI: 10.1039/d1ma00937k |
| ‡ B. J and G. Z contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |