
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh valence state metal-ion doped Fe–Ni layered double hydroxides for oxygen evolution electrocatalysts and asymmetric supercapacitors†
Wenke
Shao
,
Qiufan
Wang
 *,
Can
Huang
and
Daohong
Zhang
*
*,
Can
Huang
and
Daohong
Zhang
*
Key Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, Hubei R&D Center of Hyperbranched Polymers Synthesis and Applications, South-Central University for Nationalities, Wuhan 430074, China. E-mail: Zhangdh27@163.com
First published on 5th January 2022
Abstract
The delicate design of nanostructures consisting of multiple components is important for dual-function electrode materials for energy storage. In this work, cobalt-doped nickel–iron layered double hydroxide (Fe–Ni3Co2 LDH) assembled from one-dimensional (1D) nanoneedle subunits and FeSe2/C is synthesized via a facile, one-pot self-template method. FeNi3Co2 LDH exhibits remarkable activity for catalyzing the oxygen evolution reaction with a low onset overpotential of 224 mV and a low Tafel slope of 73 mV dec−1, much better than those of FeNi LDH. Significantly, the Fe–Ni3Co2 LDH//FeSe2/C asymmetric supercapacitor (ASC) delivers an ultra-high energy density (22.3 μW h cm−2), high power density (2.07 mW cm−2), and outstanding cycling stability (84.8% capacitance retention after 5000 galvanostatic charge–discharge cycles at 0.3 mA). The density functional theory (DFT) calculation reveals that the high electrochemical activity of Fe–NiCo LDHs is mainly attributed to cobalt doping which can modify the electronic structure and narrow the bandgap, thereby bringing enhanced conductivity, facile electron transfer, and abundant active sites. Our work could provide new insight into the synthesis of novel multifunctional nanomaterials.
1. Introduction
Among the variety of energy storage and conversion devices, supercapacitors (SCs) and water-splitting cells are necessary to overcome the increasingly serious fossil energy shortage.1,2 SCs possess several merits such as a fast charge and discharge rate, high power delivery, and excellent cycling lifespan, but suffer from relatively low energy density, which limit their application fields. Increasing the energy density calls for an electrode with high capacitance and stable operability when working at high voltage, as well as a high mass or volume ratio of the electrode inside the device. Similar to SCs, the process of water splitting, especially for the oxygen evolution reaction (OER), commonly shows a large overpotential due to its four-electron transfer process with slow dynamics.3 To accelerate the process, a highly efficient electrocatalyst is needed to reduce the reaction barrier. Pseudocapacitive materials including transition metallic/bi-metallic hydroxides, and oxides, have been employed to boost the energy density of devices owing to their multiple oxidation states, excellent electron relay systems, and tunable morphologies. Such properties make them multifunctional for redox SCs as well as the OER.4 To date, layered double hydroxides (LDHs) with high theoretical capacitance, tunable composition, varying oxidation states of transition metals, and unique layered structures stand out among the aforementioned materials.5 The structure of LDHs consists of positively charged host layers and exchangeable hydrated anions located in the interlayer gallery for charge balance.6 Interestingly, the host layers and the interlayer negative ions can exchange flexibly without structural change. Thus, the LDHs present a variable nanostructure.7 However, the LDH electrodes have a relatively large diameter (0.2–5 μm) and thickness (6–40 nm), with fewer surface atoms exposed, causing capacity fading and resulting in poor electrochemical performance.5To solve the problems mentioned above, one approach is to design a tri-metallic LDH via a third type of metal cation doping; the flexibility of cations in the LDH layers could directly change the structure of the metal hydroxide layer in the LDH. High valence state dopants tune the electronic structure of bimetallic LDHs, producing strong synergistic effects between the doping cations and the host cations and creating more active sites at the same time. Yang et al.8 demonstrated that the Cr metal has several oxidation states (from +1 to +6) and Cr3+ and Cr4+ are very common and stable oxidation states and could have a positive effect on the active species (either Fe or Ni sites) for the oxidation process. Wu et al.9 developed W doped Ni–Fe LDH with the decreased thickness of nanosheets being only 5.2 nm, which effectively increases the active sites. Chen et al.10 explained that the substitution of Ru dopants into Fe centers of the NiFe-LDH efficiently reduced the kinetic energy barrier of the Volmer step and thereby accelerated the electrochemical process. Another way is to design micro/nanoarchitectures with large specific surface area and suitable pore size distribution, where all electroactive species can participate in the faradaic redox reaction process, ensuring rapid mass and electron transport. Metal–organic frameworks (MOFs) with a uniform structure, steady pore size distribution and large specific area have been found to be suitable precursor materials toward the construction of well-defined electrode materials.11,12 MOF-derived LDHs for SC electrodes usually exhibit high specific capacitance.13,14 Besides, metal ions in MOF-derived LDHs (such as Fe3+ in MIL-88A-derived LDHs) as the active sites have fast OER kinetics because they have optimum bond energetics for the adsorption of OER intermediates.15,16
Herein, we present a facile, one-pot self-templated strategy to synthesize Fe–Ni3Co2 LDH nanospheres assembled using a 1D nanoneedle and FeSe2 nanoparticles with carbon from an aminophenolic resin (AF) shell (FeSe2/C). Starting from spindle-like particles of MIL-88A (a MOF; MIL stands for Materials from Institute Lavoisier), Fe–Ni3Mx LDHs (M = V, Al, Cr, Mo, and Mn) with other valence state metal ions are obtained through the simultaneous etching and coprecipitation reactions. The electrochemical and OER performances of Fe–Ni3Mx LDHs have been analyzed and optimized with changes in the molar ratio M during the synthesis process. This indicates that the high valence state dopants like Co, V, Al, Cr, Mo and Mn lead to enhanced conductivity, favorable kinetics, and modification of the electronic structure of both the Ni and Fe sites, producing favorable synergistic effects for sustainable supercapacitors and water splitting applications. The all-solid-state asymmetric supercapacitor device assembled using the synthesized Fe–Ni3Co2 LDH as the positive electrode and FeSe2/C as the negative electrode reached a specific capacitance of 83.9 mF cm−2 at 0.3 mA and energy density (22.3 μW h cm−2) at high power density (2.07 mW cm−2). It also showed outstanding cycling stability and outstanding mechanical flexibility. DFT calculations were also carried out to simulate the change of the electronic structure and bandgap caused by transition metal-ion doping.
2. Experimental
2.1 Synthesis of MIL-88A
For the synthesis of diamond-shaped MIL-88A crystals, 5 mmol FeCl3·6H2O and 5 mmol fumaric acid were dissolved in 25 mL DMF. Then the mixture was transferred into a Teflon-lined stainless autoclave and heated to 100 °C for 4 h. Finally, the product was washed with ethanol several times and dried at 60 °C.2.2 Synthesis of Fe–Ni3Mx LDHs
66 mg of MIL-88A, 900 mg (3 mmol) Ni(NO3)2·6H2O, x mmol (x = 0, 1, 2, and 3) Co(NO3)2·6H2O and 900 mg (15 mmol) urea were dissolved in 60 mL DI water and stirred for 10 min to form a homogeneous solution. Then the solution was heated in an oil bath at 90 °C under seal without stirring for 6 h. The precipitate was collected by centrifugation and washed with ethanol several times. For comparison, pristine Fe–Ni3 LDH was synthesized without the addition of Co(NO3)2·6H2O (x = 0). Fe–Ni3Mn2 LDH and Fe–Ni3Mo2 LDH were synthesized using the same procedure except that a manganese source (2 mmol MnCl2·4H2O) and a molybdenum source (2 mmol Na2MoO4·2H2O) were used, respectively. The Fe–Ni6V1 LDH, Fe–Ni6Al1 LDH, and Fe–Ni6Cr1 LDH were synthesized by replacing the cobalt source with a vanadium source (1 mmol VCl3), an aluminum source (1 mmol Al(NO3)3·9H2O), and a chromium source (1 mmol Cr(NO3)3·9H2O) and heated in an oil bath at 120 °C under seal without stirring for 12 h.2.3 Synthesis of FeSe2/C
MIL-88A particles were dispersed in a mixture solution of ethanol (16 mL) and deionized water (14 mL) by stirring for 30 min at room temperature. 230 mg of cetyltrimethylammonium bromide (CTAB), concentrated ammonia solution (0.1 mL, 28 wt%) and 35 mg 4-aminophenol were added into the resultant dispersion consecutively. After stirring for 30 min, 0.06 mL formaldehyde (37 wt%) were added and the mixed dispersion was mechanically stirred for 8 h at 30 °C. The obtained core–shell MIL-88A@AF was collected and washed with deionized water and ethanol three times, respectively. A ceramic boat with MIL-88A@AF was put downstream of the tube furnace and Se powder was put upstream (the mass ratio of MIL-88A@AF and Se powder is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The tube was heated from room temperature to 400 °C at a rate of 2 °C min−1 and kept for 30 min in an Ar atmosphere.
:2). The tube was heated from room temperature to 400 °C at a rate of 2 °C min−1 and kept for 30 min in an Ar atmosphere.
3. Results and discussion
The synthetic processes of the Fe–Ni and Fe–Ni3Mx LDH (M = Co, V, Mo, and Mn) electrodes and the FeSe2/C negative electrode are schematically illustrated in Fig. 1. First, uniform diamond-like MIL-88A particles with a smooth surface are synthesized as the sacrificial template through a hydrothermal method. After the etching and co-precipitation process, the diamond gradually transforms to the sphere and grows up to different LDHs with all kinds of surface topography because of the different hydrolysis rates of various metal-ions. The simulated crystal structures are shown on the right; the third metal-ions replace the position of Fe3+, which may change the electro-structure of Ni2+ and Fe3+ sites and bestow high conductivity to improve the electrochemical activity as energy storage electrodes and electrocatalysts. As a template, MIL-88A could also be coated with AF to synthesize an FeSe2/C electrode via a gas-phase selenization process (the mass ratio of MIL-88A@AF and Se powder is 1:2).
 | ||
| Fig. 1 Schematic illustration for the formation of MIL-88A, Fe–Ni3Mx LDH (M = Co, V, Mo, and Mn), and FeSe2/C derived from MIL-88A. | ||
The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images show the smooth surface and solid nature of the hexagonal prisms MIL-88A with an average diameter and length of 400 nm and 800 nm, respectively (Fig. 2a and b). The X-ray diffraction (XRD) pattern of the as-prepared MIL-88A matches well with the previous result (Fig. S1, ESI†). Then, the urchin-like Fe–Ni3Mx LDHs were synthesized based on the hydrolysis of MIL-88A. MIL-88A particles chemically convert into an Fe–Ni LDH hollow polyhedral structure assembled with nanosheet shells. During the process, OH− ions generated from the hydrolysis of urea can gradually etch MIL-88A templates, and the released Fe3+ ions will co-precipitate with Ni2+ and OH− ions to form thin layered Fe–Ni LDH shells.17 The simultaneous etching and growth processes could gradually change the solid precursor into hollow Fe–Ni LDH polyhedra (Fig. 2c and d). However, the solid Fe–Ni3Co2 LDH nanospheres appeared when Co2+ was added with Ni2+ at the same time, and the nanosheets were transformed into nanoneedles (Fig. 2e). The process of this transformation is shown in Fig. S2 (ESI†), water as the solvent and the introduction of the second metal-ions lead to fast hydrolysis which could produce nanospheres because of the fast etching of the template.18 The nanosheets on the surface are larger and then transformed into a more dense nanoneedles over time. The thin tips weaved in multiple directions and the open porous architecture of the Fe–Ni3Co2 LDH (Fig. 2f) can provide a large specific surface area and abundant active sites, which are beneficial for providing high electrochemical activity. Moreover, the TEM image in Fig. 2g indicates the solid structure of the Fe–Ni3Co2 LDH, and the thickness of the tips is approximately 5 nm. The high-resolution TEM (HRTEM) image (Fig. S3a, ESI†) shows that the interplanar distances of the nanoneedles are 0.154 nm, which coincide with the interlayer spacing of the (110) planes of the LDH. The well-defined selected area electron diffraction (SAED) pattern (the inset image in Fig. S3b, ESI†) has two relatively bright diffraction rings, attributed to the characteristic (012) and (110) facets of the Fe–Ni3Co2 LDH. The corresponding elemental mapping images (Fig. 2g) validates the distribution of Fe, Ni and Co in the whole Fe–Ni3Co2 LDH. The XRD result in Fig. S4 (ESI†) reflected the successful synthesis of the Fe–Ni3Co2 LDH. With increasing content of Co2+, no other diffraction peaks can be detected.
 | ||
| Fig. 2 (a) SEM and (b) TEM image of MIL-88A, (c) SEM and (d) TEM image of Fe–Ni LDH, SEM (e and f), TEM (g) and the corresponding elemental mapping images of Fe, Ni, and Co of Fe–Ni3Co2 LDH. | ||
Other Fe–Ni3Mx LDHs were synthesized via the same procedure using different metal ion sources. Fe–Ni6V1 LDH (Fig. 3a) and Fe–Ni3Mo2 LDH (Fig. 3e) nanospheres have similar structures assembled with nanosheets. The crystal structure and phases were consistent with iron–nickel carbonate hydroxide (JCPDS#40-0215) (Fig. S5, ESI†). The result reflects that the change of metal salt may affect the insertion of the anion (NO3−, Cl−), which may cause the crystal plane to shift or even disappear. For the Fe–Ni6V1 LDH in Fig. 3b, the dense nanosheets were vertically grown on the nanosphere, and these nanosheets interconnect with each other to form an open porous structure which is beneficial for the connection between the active material and the electrolyte. The Fe–Ni6V1 LDH nanosphere was larger than that of Fe–Ni3Co2 LDH. The HRTEM image shows that the interplanar distances of the nanoneedles are 0.154 nm, which coincide with the interlayer spacing of the (110) planes of the LDH. The selected area electron diffraction (SAED) patterns (inset of Fig. 3c) of Fe–Ni6V1 LDH nanosheets revealed that the diffraction rings indexed the (100) and (110) lattices, which was consistent with the X-ray diffraction (XRD) pattern. The uniform distribution of these elements shown in Fig. 3d not only confirms the existence of Fe–Ni6V1 LDH but might also be highly beneficial for the synergistic effects among these cations. In addition, metal salt with different hydrolysis rates could found unequal nucleation and bring to varying nanosheets on the surface of the nanospheres.18 As shown in Fig. 3e and f, the nanosheets are thinner on the surface of the Fe–Ni3Mo2 LDH, and it is speculated to be because MoO42− has a slower hydrolysis rate and no other complementary anions inserting into metal layers; it also causes the disappearance of (003) and (006) reflections. The HRTEM image (Fig. 3g) shows that the interplanar distances of the nanosheets are 0.197 nm, which coincide with the interlayer spacing of the (018) planes of the LDH. The selected area electron diffraction (SAED) patterns (inset of Fig. 3g) of Fe–Ni3Mo2 LDH nanosheets, revealed that the diffraction rings indexed the (110) lattices, which was consistent with the X-ray diffraction (XRD) pattern. The corresponding elemental mapping images (Fig. 3h) validates the distribution of Fe, Ni and Mo in the Fe–Ni3Mo2 LDH. Besides, Mn ions with a faster rate of hydrolysis lead to a hollow structure of the LDH (Fig. 3i and j), which has much smaller and thinner nanosheets. The HRTEM image (Fig. 3k) shows that the interplanar distances of the nanosheets are 0.154 nm, which coincide with the interlayer spacing of the (110) planes of the LDH. The selected area electron diffraction (SAED) patterns (inset of Fig. 3k) of Fe–Ni3Mn2 LDH nanosheets, revealed the diffraction rings indexed the (101) lattices, which was consistent with the X-ray diffraction (XRD) pattern. The corresponding elemental mapping images (Fig. 3l) validates the distribution of Fe, Ni and Mn in the Fe–Ni3Mn2 LDH. For preparing FeSe2 as the negative electrode, an aminophenol-formaldehyde (AF) was used as a coating layer for MIL-88A to keep the completeness of the diamond-like structure. After the high-temperature gas-phase selenization process, the diamond-like materials were kept (Fig. 3m) and transformed into the bulk-nanoparticle structure (Fig. 3n). Besides, the high-resolution TEM image in Fig. 3o and the intensive and sharp diffraction XRD peaks (Fig. 3p) revealed the growth of FeSe2/C crystallites. To investigate the influence of different valence elements on the surface chemical states and composition of Fe–Ni LDH nanosheets, X-ray photoelectron spectroscopy (XPS) of Fe–Ni3Co2 LDH, Fe–Ni6V1 LDH and FeSe2/C was carried out (Fig. S6 and S7, ESI†). And the percentage of Fe, Co and Ni atoms in the materials obtained from XPS is listed in Table S1 (ESI†), the ratios of Ni and Co/V are approximately consistent with the feed ratio in the synthesis process.
 | ||
| Fig. 3 SEM (a and b) and TEM images (c and d) of Fe, Ni, and V of the Fe–Ni6V1 LDH. The inset image of (c) shows the SAED pattern of the Fe–Ni6V1 LDH. SEM (e and f) and TEM images (g and h) of Fe, Ni, and Mo of the Fe–Ni3Mo2 LDH. The inset image of (g) shows the SAED pattern of the Fe–Ni3Mo2 LDH. SEM (i and j) and TEM images (k and l) of Fe, Ni, and Mn of the Fe–Ni3Mn2 LDH. The inset image of (k) shows the SAED pattern of the Fe–Ni3Mn2 LDH. SEM (m) and TEM images (n and o) of FeSe2/C. The inset image (o) shows the SAED pattern of FeSe2/C. (p) XRD of FeSe2/C. | ||
The electrocatalytic performance of the Fe–Ni3Cox (x = 0, 1, 2, and 3) LDH electrode for the OER in 1 M KOH electrolyte was firstly evaluated by linear sweep voltammetry (LSV) curves at 5 mV s−1 (Fig. 4a). The Fe–Ni3Co2 LDH displays the best activity with the smallest overpotential of 224 mV to drive a current density of 10 mA cm−2 compared to those of pristine Fe–Ni LDH (227 mV), Fe–Ni3Co1 LDH (230 mV) and Fe–Ni3Co3 LDH (237 mV), and the smallest overpotential of 298 mV to drive a current density of 50 mA cm−2 than those of pristine Fe–Ni LDH (305 mV), Fe–Ni3Co1 LDH (314 mV) and Fe–Ni3Co3 LDH (314 mV) (Fig. 4b). To obtain some insights into the OER kinetics, the corresponding Tafel plots circulated from LSV curves of these catalysts are investigated. As shown in Fig. 4c, Fe–Ni3Co2 LDH exhibits enhanced reaction kinetics with the smallest Tafel slope (73 mV dec−1) among all LDH electrodes, indicating its higher transfer coefficient and enhanced electrocatalytic kinetics. The electrochemically active surface area (ECSA) was illustrated to evaluate the improved OER activity, which was estimated from the double-layer capacitances (Cdl) (Fig. 4d) of the cyclic voltammetry (CV) curves (Fig. S8, ESI†). The Fe–Ni3Co2 LDH has the largest Cdl of 7.11 mF cm−2, and the ECSA value (177.75 cm2) is much higher than those of Fe–Ni LDH (77.75 cm2), Fe–Ni3Co1 LDH (46 cm2) and Fe–Ni3Co3 LDH (141 cm2) shown in Fig. S9 (ESI†), suggesting that more electrochemically active sites are created by the introduction of Co. The electrochemical properties compared with state-of-the-art and other electrocatalysts are summarized in Table S2 (ESI†). The electrochemical impedance spectroscopy (EIS) results (Fig. 4e) indicate that Fe–Ni3Co2 LDH exhibits the lowest charge transfer resistance among these samples, and too much Co doping (Fe–Ni3Co3 LDH) results in increased charge-transfer resistance, which might be another reason for the degraded performance as shown by the LSV curves.
 | ||
| Fig. 4 (a) OER LSV curves of Fe–Ni3Cox (x = 0, 1, 2, and 3) LDH samples in 1.0 M KOH electrolyte. (b) Overpotentials required by these four samples to drive current densities of 10 mA cm−2 and 50 mA cm−2. (c) Tafel plots derived from the polarization curves in (a). (d) Double-layer capacitance (Cdl) plots and (e) Nyquist plots of Fe–Ni3Cox (x = 0, 1, 2, and 3) LDH. (f) A chronoamperometry curve of Fe–Ni3Co2 LDH at the overpotential (50 mA cm−2). | ||
The stability of the Fe–Ni3Co2 LDH electrode (Fig. 4f) and the Fe–Ni LDH, Fe–Ni3Co1 LDH, Fe–Ni3Co3 LDH (Fig. S10, ESI†) was confirmed by the results of a long-term electrochemical durability test performed at an overpotential (50 mA cm−2) for 12 h. This revealed that the Co-ion doping provides higher stability of electrocatalysts during the OER process. The characterization of TEM, XRD and XPS after a 12 h durability test has been provided in the ESI.† The 3D structure of the Fe–Ni3Co2 LDH was still maintained in the TEM images (Fig. S11, ESI†). Meanwhile, the carbon black nanoparticles added in the electrode preparation process can be clearly seen in the enlarged view of Fig. S11a (ESI†). Fig. S12a (ESI†) shows the XRD pattern of the Fe–Ni3Co2 LDH after a long-term OER test. No obvious change could be found in the XRD pattern except for the new peak of carbon black (2θ = 25.0°), which indicates excellent structural stability.13 Meanwhile, Fig. S12b and c (ESI†) show the XPS spectrum of Ni 2p and Fe 2p after the OER. The valence states of Ni and Fe remained almost unchanged. The increased ratio of Co2+ proved the occurrence of electron transfer in Fig. S12d (ESI†). And the assigned ratio of the M–O group and the M–OH group in O 1s has increased after OER processes (Fig. S12e and f, ESI†), which is related to the oxidation of M(OH)2 to MOOH.19 The related OER mechanism in an alkaline environment is generally described by the following steps:20,21
| FeNiCo + OH− → FeNiCoOH + e− |
| FeNiCoOH + OH− → FeNiCoO + H2O + e− |
| FeNiCoO + OH− → FeNiCoOOH + e− |
| FeNiCoOOH + OH− → FeNiCoO + O2 + H2O + e- |
“FeCoNi” represents the metal active site. Metals as the active sites have the electronic gains and losses, and they are partially oxidized after the OER process, which are consistent with the above characterization. LSV curves of the Fe–Ni3Co2 LDH before (black) and after (red) 1000 CV cycles are shown in Fig. S13 (ESI†); the polarization curve shows negligible change after 1000 CV scans. These results manifest the advantages of Fe–Ni3Cox LDHs in composition and structure in terms of activity and stability for the OER. The facile reaction method used here has also been demonstrated to be versatile and was applied to the synthesis of Fe–Ni6V1 LDH, Fe–Ni3Mn2 LDH, and Fe–Ni3Mo2 LDH with good OER activity by changing the dopants. The OER performance of these catalysts in 1.0 M KOH is indicated by the LSV curves shown in Fig. S14 (ESI†).
To understand the doping effect in-depth, the electronic structures of Fe–Ni LDH and Fe–NiCo LDH were calculated using density functional theory plus U (DFT+U) method. According to the reaction process of the LDH in alkaline electrolyte, the H desorption from surficial LDH will be a critical step in the redox reaction.22,23Fig. 5a and b show the optimized structures of pure Fe–Ni LDH and the Fe–NiCo LDH, respectively. The hydrogen desorption energies (EH) decrease with the cobalt ion doping, and EH (−1.52 eV) of the Fe–NiCo LDH and −2.62 eV of the Fe–Ni LDH demonstrated the positive effect of Co on the hydrogen desorption. As shown in the partial densities of states (PDOSs) and the total densities of states (TDOSs) curves (Fig. 5c and d), the bandgap of the Fe–Ni LDH between the valence and conduction bands of about 2.6 eV and a narrow bandgap were observed after doping cobalt ions, indicating a more conductive electronic structure.24 Therefore, the theory and experiment were in great agreement that the redox reactions and OER kinetics on the electrode could be facilitated by cobalt doping to improve the electric conductivity along with the significantly enhanced intrinsic electrochemical activity.
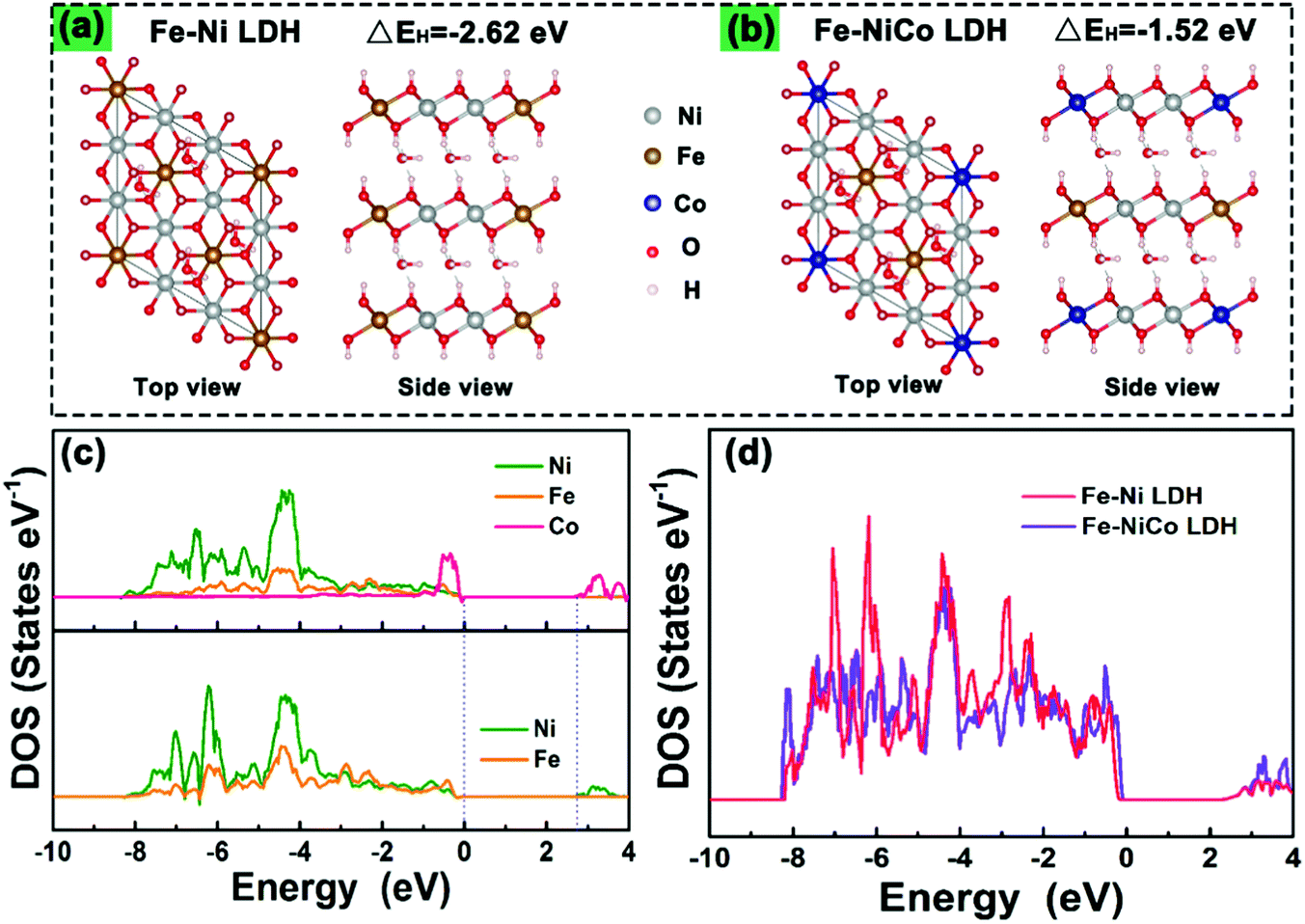 | ||
| Fig. 5 Optimized structures of (a) the pure Fe–Ni LDH and (b) the Fe–NiCo LDH for the calculation of hydrogen desorption energies (EH). Partial DOS (c) and total DOS (d) of the Fe–NiCo LDH composite and pure Fe–Ni LDH. | ||
The cyclic voltammetry (CV), the galvanostatic charge–discharge curves (GCD) and the EIS of Fe–Ni3Cox LDH (x = 0, 1, 2, and 3) electrodes were tested using a three-electrode system in 3 M LiCl electrolyte (Fig. S15 and S16, ESI†). The comparison diagrams of Fe–Ni3Cox LDH electrodes with different reactant ratios are shown in Fig. S17 (ESI†). The electrochemical performance of Fe–Ni3Mx LDHs (M = V, Al, Cr, Mo and Mn) were also systematically evaluated (Fig. S18 and S19, ESI†), which indicated the higher electrochemical activity of the high valence state metal ion doping LDH than that of pristine Fe–Ni LDH. To gain an in-depth insight into the charge storage mechanism of the Fe–Ni3Mx LDH electrodes, the pseudocapacitive analysis was carried out based on the CV measurements at different scan rates (Fig. 6a). Generally, there are two main storage components: diffusion-controlled faradaic contribution and surface pseudocapacitive contribution (referred to as pseudocapacitance). The ratio of surface capacitive contribution can be quantified by the following equation:25i (V) = k1v + k2v1/2, where i(V) is the total current response at a given potential V and v is the scan rate. By determining the k1 values, the fraction of the current arising from the surface capacitive process can be distinguished from the diffusion process. The dependence of i/v1/2 on v1/2 demonstrated obvious linear behavior in Fig. 6b. The calculated result of pseudocapacitive contribution is 62% at a scan rate of 8 mV s−1 based on the above quantitative analysis (Fig. 6c), suggesting that the electrochemical capacity is mainly controlled by pseudocapacitive capacity. Moreover, the pseudocapacitive contributions with different scan rates are also illustrated in Fig. 6d. As identified, the surface capacitive contribution at 1 mV s−1 is around 36% and gradually enlarges as the scan rate increases, demonstrating that the pseudocapacitive contribution becomes dominant, especially during high-rate cycling. Fig. 6e shows that the capacitance of Fe–Ni3Co2 LDH electrode is maintained at 116.6% of the initial value after 5000 cycles, indicating excellent stability. Nyquist plots (Fig. 6f) at initial and after 5000 cycles manifest the small and decreased Rct values of the Fe–Ni3Co2 LDH electrode. Therefore, the high surface capacitive contribution of Fe–Ni3Co2 LDH accounts for the excellent rate capability and long-term cycling performance.
 | ||
| Fig. 6 Electrochemical performance of Fe–Ni3Co2 LDH electrode. (a) CV curves at different scan rates of 1–8 mV s−1. (b) i/v1/2vs. v1/2 plots at different potentials. (c) Capacitive charge storage contributions at 8 mV s−1. (d) Contribution of diffusion and capacitive control at different scan rates. (e) The capacitance retention after 5000 cycles. (f) Nyquist plots at initial and after 5000 cycles. | ||
An all-solid-state asymmetric supercapacitor device was constructed from the Fe–Ni3Co2 LDH as the positive electrode and FeSe2/C as the negative electrode in PVA/LiCl gel electrolyte (Fig. 7a). The potential windows of the Fe–Ni3Co2 LDH and FeSe2/C electrodes tested at 80 mV s−1 in a three-electrode cell lay in the range of 0–1 V and −1.0–0 V (Fig. 7b). To determine the optimal working potential window, the CV curves of ASC at various voltages from 0.8 to 1.4 V were employed. It can be seen that the CV curve exhibits no obvious polarization when the voltage window reaches 1.4 V (Fig. 7c). Thus, the optimal potential window was selected to be 1.4 V. Fig. 7d displays the CV curves of ASC in a working potential window of 0–1.4 V at different scan rates. No apparent redox peaks can be observed in the CV curves of ASC and the shape of curves can be well retained even at a scan rate of 200 mV s−1. Fig. 7e shows the GCD profiles of ASC at different current densities from 0.3 to 1.0 mA. All GCD curves exhibit a nonlinear and triangular shape, indicating good electrochemical reversibility and the coexistence of EDLC and pseudocapacitance during the charge/discharge process. Notably, the Fe–Ni3Co2 LDH//FeSe2/C device delivers a high specific capacitance of 83.9 mF cm−2 at 0.3 mA (Fig. 7f). The decreased capacitance at higher current density can be related to the inadequate redox reactions. Also, the ASC device exhibits a high energy density of 22.3 μW h cm−2 (22.3 W h Kg−1) at a power density of 2.08 mW cm−2 (2076 W Kg−1) and retains 11.6 μW h cm−2 (11.6 W h Kg−1) at a power density of 6.76 mW cm−2 (6760 W Kg−1) (Fig. 7g), which is comparable to and much higher than that of previously reported asymmetric devices, such as NiO@MnCo-LDH//AC ASC (9.8 μW h cm−2 at 0.38 mW cm−2),26 Ni-Co DHs//pen ink/nickel/CF ASC (9.57 μW h cm−2 at 492.17 μW cm−2),27 NiCo2O4//AC (9.46 μW h cm−2 and 2041 μW cm−2),28 Ni-Mn LDH and MnO2 nano-composite//AC (16 W h Kg−1 at 15 KW Kg−1),29 Ni3S2-nanorod//CoNi2S4 (6.6 W h Kg−1 at 820 W Kg−1),30 CoMn LDH//AC (4.4 W h Kg−1 at 2500 W Kg−1).31 Furthermore, the ASC device maintains approximately 84.8% capacitance retention of its initial cell capacitance after 5000 cycles at 0.3 mA (Fig. 7h), indicating good long-term cycling stability. To further explore the response of electrochemical performances to mechanical strains (Fig. S20, ESI†), CV measurements at 30 mV s−1 and GCD measurements at 0.8 mA cm−2 are performed on the ASC under different bending conditions (Fig. S20a and b, ESI†), which demonstrate excellent flexibility and the desirable aptitude of the active materials for the deformation of the conductive matrix.
 | ||
| Fig. 7 (a) Schematic illustration of an asymmetric supercapacitor based on the Fe–Ni3Co2 LDH positive electrode and FeSe2/C negative electrode. CV curves of (b) Fe–Ni3Co2 LDH and FeSe2/C, and (c) Fe–Ni3Co2 LDH//FeSe2/C device in different voltage windows at a scan rate of 30 mV s−1. (d) CV curves of ASC device at different scan rates. (e) GCD curves of the ASC device. (f) The specific capacity of the device at various currents. (g) Comparison of the areal and mass-energy density between Fe–Ni3Co2 LDH//FeSe2/C ASC and other solid-state ASCs. (h) Cycling performance of the device at 0.3 mA. | ||
4. Conclusions
In summary, a strategy of doping high valence metal elements into Fe–Ni LDH was used to successfully fabricate novel Fe–Ni3Mx trimetallic LDH nanospheres. With an optimized metal ratio, Fe–Ni3Mx LDH exhibits much better electrochemical activity than pristine Fe–Ni LDH. The fabricated Fe–Ni3Co2 LDH//FeSe2/C all-solid-state ASC device exhibits a high areal capacity of 83.9 mF cm−2 at 0.3 mA, a high energy density of 22.3 μW h cm−2 at a power density of 2076 μW cm−2 and outstanding cycling stability (84.8% retention after 5000 cycles), and also has outstanding mechanical flexibility after bending from 0° to 180°. When used as an OER electrode, Fe–Ni3Co2 LDH can achieve 10 mA cm−2 for alkaline at an overpotential of 224 mV. This strategy for the design of high capacitance electrodes and excellent OER electrocatalysts broadens the feasibility for application in various fields.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 51702369 and 51873233), the innovation group of the National Ethnic Affairs Commission of China (MZR20006), the Key R&D Plan of Hubei Province (2020BAB077), and the Fundamental Research Funds for the Central Universities (CZZ21009 and CZP20006).References
- H. Qian, B. Wu, Z. Nie, T. Liu, P. Liu, H. He, J. Wu, Z. Chen and S. Chen, A flexible Ni3S2/Ni@CC electrode for high-performance battery-like supercapacitor and efficient oxygen evolution reaction, Chem. Eng. J., 2020, 127646 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355(6321), aad4998 CrossRef PubMed.
- Q. Qin, L. Chen, T. Wei and X. Liu, MoS2/NiS yolk-shell microsphere-based electrodes for overall water splitting and asymmetric supercapacitor, Small, 2019, 15(29), 1803639 CrossRef PubMed.
- V. Sharma, U. N. Pan, T. Ibomcha Singh, A. Kumar Das, N. Hoon Kim and J. Hee Lee, Pragmatically designed tetragonal copper ferrite super-architectures as advanced multifunctional electrodes for solid-state supercapacitors and overall water splitting, Chem. Eng. J., 2020, 415, 127779 CrossRef.
- M. Zhao, Q. Zhao, B. Li, H. Xue, H. Pang and C. Chen, Recent progress in layered double hydroxide based materials for electrochemical capacitors: design, synthesis and performance, Nanoscale, 2017, 9(40), 15206–15225 RSC.
- L. Mohapatra and K. Parida, A review on the recent progress, challenges and perspective of layered double hydroxides as promising photocatalysts, J. Mater. Chem. A, 2016, 4(28), 10744–10766 RSC.
- G. M. Tomboc, J. Kim, Y. Wang, Y. Son, J. Li, J. Y. Kim and K. Lee, Hybrid layered double hydroxides as multifunctional nanomaterials for overall water splitting and supercapacitor applications, J. Mater. Chem. A, 2021, 9(8), 4528–4557 RSC.
- Y. Yang, L. Dang, M. J. Shearer, H. Sheng, W. Li, J. Chen, P. Xiao, Y. Zhang, R. J. Hamers and S. Jin, Highly active trimetallic NiFeCr layered double hydroxide electrocatalysts for oxygen evolution reaction, Adv. Energy Mater., 2018, 8(15), 1703189 CrossRef.
- L. Wu, L. Yu, F. Zhang, D. Wang, D. Luo, S. Song, C. Yuan, A. Karim, S. Chen and Z. Ren, Facile synthesis of nanoparticle-stacked tungsten-doped nickel iron layered double hydroxide nanosheets for boosting oxygen evolution reaction, J. Mater. Chem. A, 2020, 8(16), 8096–8103 RSC.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Accelerated hydrogen evolution kinetics on NiFe-layered double hydroxide electrocatalysts by tailoring water dissociation active sites, Adv. Mater., 2018, 30(10), 1706279 CrossRef PubMed.
- W. Liu, M. Shao, W. Zhou, B. Yuan, C. Gao, H. Li, X. Xu, H. Chu, Y. Fan, W. Zhang, S. Li, J. Hui, D. Fan and F. Huo, Hollow Ni-CoSe2 embedded in nitrogen-doped carbon nanocomposites derived from metal-organic frameworks for high-rate anodes, ACS Appl. Mater. Interfaces, 2018, 10(45), 38845–38852 CrossRef CAS PubMed.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Bifunctional electrocatalysts for overall water splitting from an iron/nickel-based bimetallic metal-organic framework/dicyandiamide composite, Angew. Chem., Int. Ed., 2018, 57(29), 8921–8926 CrossRef CAS PubMed.
- F. Li, Z. Sun, H. Jiang, Z. Ma, Q. Wang and F. Qu, Ion-exchange synthesis of ternary feconi-layered double hydroxide nanocage toward enhanced oxygen evolution reaction and supercapacitor, Energy Fuels, 2020, 34(9), 11628–11636 CrossRef CAS.
- J. Li, S. Lu, H. Huang, D. Liu, Z. Zhuang and C. Zhong, ZIF-67 as continuous self-sacrifice template derived NiCo2O4/Co,N-CNTs nanocages as efficient bifunctional electrocatalysts for rechargeable Zn–Air batteries. ACS Sus, Chem. Eng., 2018, 6(8), 10021–10029 CAS.
- J. Zhang, L. Yu, Y. Chen, X. F. Lu, S. Gao and X. W. Lou, Designed formation of double-shelled Ni–Fe layered-double-hydroxide nanocages for efficient oxygen evolution reaction, Adv. Mater., 2020, 32(16), 1906432 CrossRef CAS PubMed.
- S. Anantharaj, S. Kundu and S. Noda, “The Fe Effect”: A review unveiling the critical roles of Fe in enhancing OER activity of Ni and Co based catalysts, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- J. Zhang, Z. Li, Y. Chen, S. Gao and X. W. Lou, Nickel-iron layered double hydroxide hollow polyhedrons as a superior sulfur host for lithium-sulfur batteries, Angew. Chem., Int. Ed., 2018, 57(34), 10944–10948 CrossRef CAS PubMed.
- Z. Jiang, Z. Li, Z. Qin, H. Sun, X. Jiao and D. Chen, LDH nanocages synthesized with MOF templates and their high performance as supercapacitors, Nanoscale, 2013, 5(23), 11770–11775 RSC.
- M. Han, N. Wang, B. Zhang, Y. Xia, J. Li, J. Han, K. Yao, C. Gao, C. He, Y. Liu, Z. Wang, A. Seifitokaldani, X. Sun and H. Liang, High-Valent Nickel Promoted by Atomically Embedded Copper for Efficient Water Oxidation, ACS Catal., 2020, 10(17), 9725–9734 CrossRef CAS.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, Recent advances in layered double hydroxide electrocatalysts for the oxygen evolution reaction, J. Mater. Chem. A, 2019, 7(10), 5069–5089 RSC.
- U. Aftab, A. Tahira, R. Mazzaro, V. Morandi, M. I. Abro, M. M. Baloch, J. A. Syed, A. Nafady and Z. H. Ibupoto, Facile NiCo2S4/C nanocomposite: an efficient material for water oxidation, Tungsten, 2020, 2(4), 403–410 CrossRef.
- Z. Lu, L. Qian, Y. Tian, Y. Li, X. Sun and X. Duan, Ternary NiFeMn layered double hydroxides as highly-efficient oxygen evolution catalysts, Chem. Commun., 2016, 52(5), 908–911 RSC.
- J. Zhao, J. Chen, S. Xu, M. Shao, Q. Zhang, F. Wei, J. Ma, M. Wei, D. G. Evans and X. Duan, Hierarchical NiMn layered double hydroxide/carbon nanotubes architecture with superb energy density for flexible supercapacitors, Adv. Funct. Mater., 2014, 24(20), 2938–2946 CrossRef CAS.
- Y. Yang, Y. Ou, Y. Yang, X. Wei, D. Gao, L. Yang, Y. Xiong, H. Dong, P. Xiao and Y. Zhang, Modulated transition metal-oxygen covalency in the octahedral sites of CoFe layered double hydroxides with vanadium doping leading to highly efficient electrocatalysts, Nanoscale, 2019, 11(48), 23296–23303 RSC.
- Q. Wang, X. Ran, W. Shao, M. Miao and D. Zhang, High performance flexible supercapacitor based on metal-organic-framework derived CoSe2 nanosheets on carbon nanotube film, J. Power Sources, 2021, 490, 229517 CrossRef CAS.
- L. Gao, R. Fan, R. Xiao, K. Cao, P. Li, W. Wang and Y. Lu, NiO-bridged MnCo-hydroxides for flexible high-performance fiber-shaped energy storage device, Appl. Surf. Sci., 2019, 475, 1058–1064 CrossRef CAS.
- L. Gao, J. U. Surjadi, K. Cao, H. Zhang, P. Li, S. Xu, C. Jiang, J. Song, D. Sun and Y. Lu, Flexible fiber-shaped supercapacitor based on nickel-cobalt double hydroxide and pen ink electrodes on metallized carbon fiber, ACS Appl. Mater. Interfaces, 2017, 9(6), 5409–5418 CrossRef CAS PubMed.
- S. T. Senthilkumar, N. Fu, Y. Liu, Y. Wang, L. Zhou and H. Huang, Flexible fiber hybrid supercapacitor with NiCo2O4 nanograss@carbon fiber and bio-waste derived high surface area porous carbon, Electrochim. Acta, 2016, 211, 411–419 CrossRef CAS.
- W. Quan, C. Jiang, S. Wang, Y. Li, Z. Zhang, Z. Tang and F. Favier, New nanocomposite material as supercapacitor electrode prepared via restacking of Ni-Mn LDH and MnO2 nanosheets, Electrochim. Acta, 2017, 247, 1072–1079 CrossRef CAS.
- S. J. Patil, J. H. Kim and D. W. Lee, Self-assembled Ni3S2//CoNiS4 nanoarrays for ultra high-performance supercapacitor, Chem. Eng. J., 2017, 322, 498–509 CrossRef CAS.
- A. D. Jagadale, G. Guan, X. Li, X. Du, X. Ma, X. Hao and A. Abudula, Ultrathin nanoflakes of cobalt-manganese layered double hydroxide with high reversibility for asymmetric supercapacitor, J. Power Sources, 2016, 306, 526–534 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01125a |
| This journal is © The Royal Society of Chemistry 2022 |