
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVaccine adjuvant platform and fluorescence imaging of amphiphilic γ-PGA-IMQ-LA-FL conjugates†
Wenzhu
Yin
 ab,
Dechun
Xuan
c,
Bihua
Deng
a,
Mingxu
Zhou
a,
Fang
Ma
a,
Jinqiu
Zhang
*ac and
Yu
Lu
*abc
ab,
Dechun
Xuan
c,
Bihua
Deng
a,
Mingxu
Zhou
a,
Fang
Ma
a,
Jinqiu
Zhang
*ac and
Yu
Lu
*abc
aInstitute of Veterinary Immunology & Engineering, National Research Center of Engineering and Technology for Veterinary Biologicals, Jiangsu Key Laboratory for Food Quality and Safety-State Key Laboratory Cultivation Base, Ministry of Science and Technology, Jiangsu Academy of Agricultural Sciences, Nanjing 210014, China
bJiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious and Zoonoses, Yangzhou, 225009, China
cSchool of Pharmacy, Jiangsu University, Zhenjiang 212013, China
First published on 20th January 2022
Abstract
Adjuvants have proven to be integral components in most vaccines for promoting appropriate immune responses at both innate and adaptive levels. Imiquimod (IMQ, R837), an agonist for Toll-like receptors 7 (TLR7), is a potent molecular activator of the innate immune system and has been developed as an adjuvant for immunotherapy. However, due to its pharmacokinetic profile, IMQ may induce strong local and systemic inflammatory reactions and is poorly tolerated. To overcome these hurdles, herein, we conjugate IMQ and lauryl alcohol (LA) to the backbone of biodegradable poly (γ-glutamic acid) (γ-PGA) to prepare an amphiphilic polymer (shortened to γ-PGA-IMQ-LA) with improved stability and biodistribution of IMQ. In order to investigate the distribution of the amphiphilic polymer in vitro and in vivo, fluorescein was grafted to the lateral chain to synthesize fluorescent-labeled γ-PGA-IMQ-LA-FL (shortened to FIP). The novel amphiphilic FIP polymer was characterized via1H NMR, FI-IR, UV-vis, fluorescence spectra and zeta potential. Using ovalbumin (OVA) as a model antigen and FIP as an adjuvant for immunization, the formulations of FIP + OVA in different dosages could significantly promote the production of OVA-specific IgG in mice and prolong immune responses with acceptable safety.
Introduction
Among the various health interventions, vaccination is undoubtedly one of the most successful methods and continues to have a huge impact on the prevention of deaths caused by global infectious diseases.1 Since the first introduction of the smallpox vaccine by Edward Jenner in 1798, hundreds of vaccines have been developed to protect against a range of infectious diseases.2 Although traditional vaccines, including inactivated pathogens or attenuated variants, can induce potent immune responses and provide protection against infections, safety concerns have limited their clinical application, especially in individuals with compromised immune systems.3,4 To solve these problems, subunit and recombinant vaccines generated from modern biotechnology have been explored because of their higher purities and better tolerance compared with inactivated or live-attenuated pathogens. Unfortunately, these new vaccines have insufficient immunogenicity and do not induce effective immune responses when used alone and, therefore, must be delivered with immunostimulatory adjuvants to enhance the vaccine potency.5,6Adjuvants are usually considered to be integral components that are capable of enhancing and/or shaping antigen-specific immune responses effectively7 and reducing antigen dosage. Adjuvants in development, or in use currently, include aluminum salts, oil emulsions, saponins, immune stimulating complexes, liposomes, microparticles, nonionic block copolymers, polysaccharides, cytokines and bacterial derivatives.8 However, many adjuvants may cause headaches, inflammation and other adverse effect, which restricts their uses in vaccines. An ideal vaccine adjuvant should be compatible with antigens, biologically safe, capable of eliciting the expected immune responses, easy to use, easily prepared and inexpensive. With the growing understanding of the important role of adjuvants in vaccines, there is an urgent need to develop novel adjuvants that can enhance and shape vaccine-elicited responses.9
Toll-like receptor (TLR) agonists are key to the successful design of potent vaccines and cancer immunotherapeutics. TLR agonists are a class of transmembrane pattern recognition receptors (PRRs) and are widely distributed amongst innate immune cell subsets over a broad range of species.10,11 TLR 7 agonists, such as imiquimod (IMQ, R837), have been examined as vaccine adjuvants in a number of clinical trials.12–15 However, their pharmacokinetic profiles are characterized by rapid systemic dissemination upon local (e.g. subcutaneous or intramuscular) administration, thereby causing unwanted innate immune activation at multiple distal tissues.16 In addition, they are water-insoluble drugs and exhibit unacceptable systemic toxicity, which is currently a strong limitation in applying IMQ in mass immunization campaigns. In the context of vaccination, the rapid diffusion of molecular adjuvants dramatically lowers the ability of antigen and TLR agonists to reach the same antigen-presenting cells in the draining lymph node (DLN), which results in suboptimal immunity to the delivered antigen and in consequence wasted inflammation.17 To mitigate these limitations, it has become an attractive target for us to utilize covalent ligation of TLR agonists to polymeric nanoparticles, which might provide chemically better-defined alternatives to physical (co)encapsulation systems to improve its biodistribution and pharmacokinetics.18–20
Poly(γ-glutamic acid) (γ-PGA) is a water soluble anionic bioderived polypeptide polymer that has been used in a diverse range of applications including in the food industry, cosmetics, agriculture, carrier and drug delivery.21,22 Biodegradable nanoparticles (NPs) composed of γ-PGA with L-phenylalanine ethyl ester have been shown to generate antigen-specific cellular and humoral immune responses after immunization with various antigens.23,24 The structure of liposomes allows them to interact with various cells and components of the immune system, rendering them ideal candidates for antigen delivery.25–27 The conjugation of a small molecule TLR7 agonist to macromolecular constructs provides the possibility to impede systemic clearance, positively affecting the pharmacokinetic profiles.28,29 In addition, lipid amphiphiles exhibit an affinity to albumin and lipoproteins and hence promote lymphatic translocation to lymphoid tissues by “hitching a ride” along the interstitial albumin flow.15 Although adjuvant effects of TLR7/8 agonists and water-soluble γ-PGA derivatives are well-documented, respectively, only limited studies have attempted to incorporate these TLR7/8 agonists into water-soluble γ-PGA derivative nanoparticles as adjuvants.30–33
In the present work, we firstly grafted IMQ to the γ-PGA skeleton through covalent bonding to improve its lymphatic delivery and pharmacokinetics. Furthermore, we designed an amphiphilic covalently bound fluoresceinamine, IMQ and lauryl alcohol (LA) to γ-PGA to generate an amphiphilic polymer γ-PGA-IMQ-LA with improved stability and biodistribution of IMQ. It is well known that water-soluble drugs find it difficult to penetrate the cell membrane to get into the cell. So it requires the modification of hydrophobic groups, such as alkyl chains, to increase its lipophilicity for membrane permeability. At the same time, the amphiphile molecules can self-assemble into nanostructures such as fibrils, micelles, and other structures for vaccine adjuvants.34 To assess the cell uptake of the amphiphilic polymer γ-PGA-IMQ-LA, we grafted fluorescein to the lateral chain to synthesise fluorescent labeling γ-PGA-IMQ-LA-FL (shortened to FIP) (Scheme 1). The novel amphiphilic FIP polymer was characterized using 1H NMR, UV-vis, fluorescence, FI-IR and zeta potential. Using ovalbumin (OVA) as a model antigen and FIP as an adjuvant in mice for immunization, the formulation of FIP + OVA could enhance the production of OVA-specific IgG and promote immune responses with acceptable safety. In contrast to pure lipidation, the conjugated amphiphilic polymer is very water-soluble which is beneficial for injection. The amphiphilic FIP conjugates showed antigen-specific humoral and cellular immune response synergistic effects. Moreover, the FIP adjuvant could be prepared into different formulations for commercialization and used for fluorescence tracing.
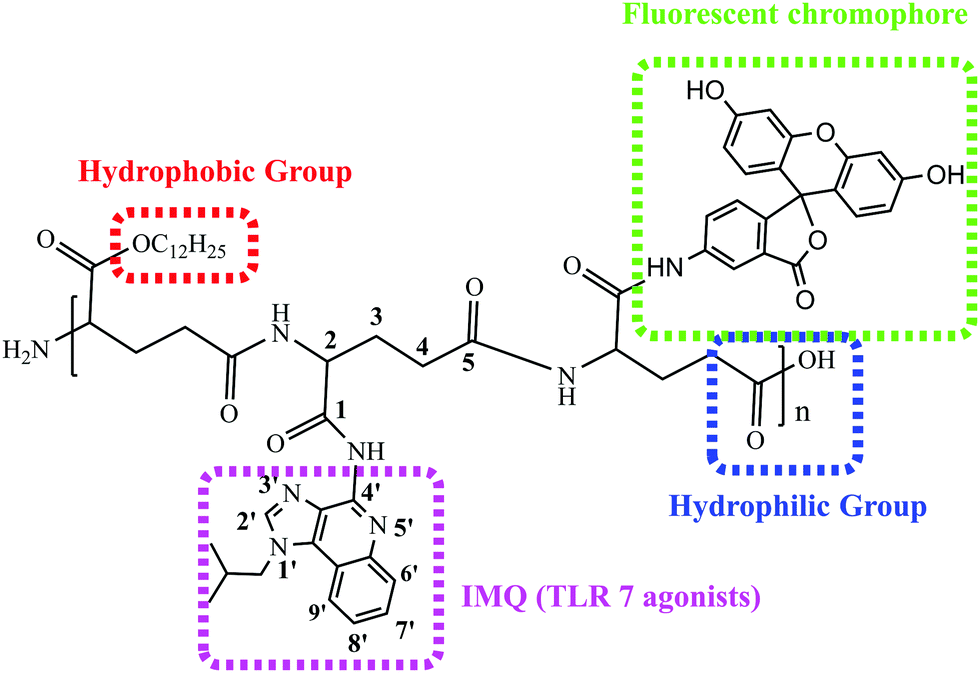 | ||
| Scheme 1 The structure of the amphiphilic polymer FIP. | ||
Experimental
Materials
Poly(γ-glutamic acid) (γ-PGA, MW = 700 kDa) was purchased from Shineking Biotechnology (Nanjing, China), imiquimod (IMQ) was obtained from meilunpharm (China), and ovalbumin (OVA, MW = 40 kDa) was purchased from Sigma. The reagents (1-dodecanol, DCM, DMSO, DMF, SOCl2, EtOH, HCl, Et3N, HOAc, and Na2SO4) were obtained from Aladdin, fluoresceinamine (5-NH2-FL) was purchased from Asianchem (Nanjing, China). Doubly distilled water from a Millipore system (Milli-Q Academic A10) was used for all analysis and purification steps.Synthesis of γ-PGA-IMQ-LA-FL (FIP)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v-v) for 3 days to remove small molecular impurities, and then lyophilized as a pale yellow solid. Spectral data are in accordance with the reported data.
:1, v-v) for 3 days to remove small molecular impurities, and then lyophilized as a pale yellow solid. Spectral data are in accordance with the reported data.
Characterization of FIP
The 1H NMR spectra were recorded using a Bruker DMX 400 spectrometer with D2O or DMSO-d6 solvent. UV-vis (UV-2700, SHIMADZU, Japan), fluorescence (F-4600, HITACHI, Japan) and FI-IR (ALPHA, Bruker, Germany) were used to analyzie IMQ and fluoresceinamine whether grafted to γ-PGA. The zeta potential values of FIP were analyzed using a Zetasizer-Nano (Malvern, UK).Animals and cells
In vivo experiments were conducted using 6–8 week old female BALB/c mice with weights of 18–20 g (purchased from the Comparative Medicine Center of Yangzhou University). Mice were housed in the laboratory animal facilities under specific pathogen-free conditions with a normal schedule and with free access to food and water. All experiments were strictly performed in accordance with the Institutional guidelines. RAW264.7 cells (ATCC) were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM, Transgen Biotech), supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin with 5% CO2 at 37 °C.Safety study
where Asample and Acontrol are the absorbance values of the wells (FIP) and control wells (PBS), respectively. For each sample, the final absorbance was an average of those measured from six wells in parallel.
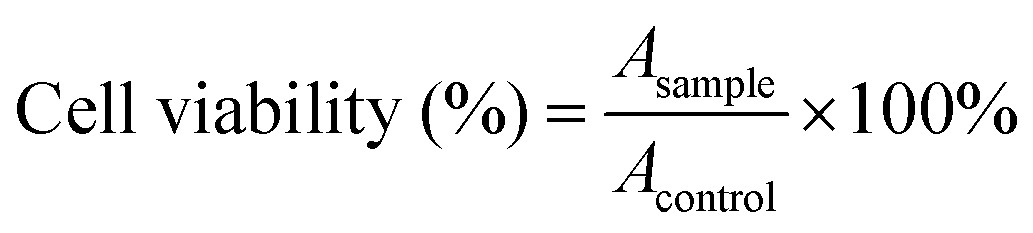
Flow cytometry experiments
To determine FIP uptake by the cells quantitatively, RAW264.7 cells were seeded in a 24-well plate at a density of 2 × 105 cells per mL before the cells were pulsed with FIP at different concentrations. After 24 h of incubation, the macrophages were dissociated using PBS followed by centrifugation for 15 min at 1000 rpm and resuspended with ice PBS for detection. Flow cytometry analysis was performed on a BD Accuri. Data were processed using the FlowJo software package.Confocal microscopy
RAW264.7 cells were pulsed overnight at 37 °C with 40 μL of a 1 mg mL−1 FIP solution in PBS. Next, the culture medium was aspirated and washed with PBS three times. Thirdly, cells were fixed with 4% paraformaldehyde (PFA) solution for 30 min followed by washing with PBS. Fourthly, the cells were stained with phalloidin (red, labeling cell membrane) in a dark environment at room temperature for 1 h and then washed with PBS three times. Finally, the DAPI (blue, labeling cell nucleus) dye was added using the same method for 15 min, washed with PBS three times and fluorescently confocal imaged using an Ultra View VOX (PerkinElmer) microscope (60×).Histological examination
In vivo toxicity of FIP was evaluated by Histological examination in BALB/c mice. Briefly, the PBS-dissolved FIP (500 μg mL−1) was injected through hypodermic injection at a dose of 200 μL per mouse (n = 3) and they were sacrificed 28 days later. The major organs (spleen, liver and kidneys) and the subcutaneous tissues at the injection sites were excised and fixed in 4% PFA solution. Then, the tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. The histological sections were photographed under an Olympus microscope (Olympus IX71, Tokyo, Japan).Preparation of the related vaccines
| Group | Antigen | Dose/mouse | Form |
|---|---|---|---|
| PBS | — | 200 μL | Water |
| OVA | OVA (100 μg) | 200 μL | Water |
| FIP (500 μg mL−1) | — | 200 μL | Water |
| γ-PGA (500 μg mL−1) | OVA (100 μg) | 200 μL | Water |
| IMQ (500 μg mL−1) | OVA (100 μg) | 200 μL | Water |
| FIP (500 μg mL−1) | OVA (100 μg) | 200 μL | Water |
| Group | Antigen | Form | Volume (μL) | |
|---|---|---|---|---|
| Control | Negative control (PBS) | — | W | 200 |
| Postive control (OVA) | OVA (100 μg) | W | 200 | |
| O/W | ||||
| W/O | ||||
| W/O/W | ||||
| Experiment | FIP | OVA | W | 200 |
| O/W | ||||
| W/O | ||||
| W/O/W | ||||
Protocol 3#
To reduce the cost of the vaccine, we investigated the dosage effect of FIP on OVA-specific IgG. Three groups of OVA + FIP-O/W vaccines with different concentrations (5, 100 or 500 μg mL−1) OVA-O/W, the fluidic OVA and PBS as the control groups were carried out to assay OVA-specific IgG. Female BALB/c mice (n = 6) were immunized twice in the six groups as shown in Table 3.| Group | Antigen | Dose/mouse | Form | |
|---|---|---|---|---|
| PBS | — | 200 μL | Water | |
| OVA | OVA (100 μg) | 200 μL | Water | |
| FIP | 5 μg mL−1 | OVA (100 μg) | O/W | |
| 100 μg mL−1 | OVA (100 μg) | 200 μL | O/W | |
| 500 μg mL−1 | OVA (100 μg) | O/W | ||
OVA-specific IgG antibody analysis
Serum was collected 14 days and 28 days after the first immunization. OVA-specific IgG antibody levels were determined using an enzyme-linked immunosorbent assay (ELISA). Briefly, a 96-well ELISA plate was coated with 100 μL of 10 μg mL−1 OVA antigen in carbonate buffer (pH 9.6) at 4 °C overnight and then blocked with blocking buffer (PBS containing 5% BSA, pH 7.4) at 37 °C for 2 h. After washing with PBST (PBS containing 0.1% tween-20, pH 7.4), serially diluted serum was added into each well and then incubated for 1.5 h at 37 °C. After washing with PBST five times, the plates were incubated with anti-mouse IgG-HRP antibody for another hour at 37 °C and then developed with TMB substrate for 30 min. The reaction was terminated by adding 2 M H2SO4, and then the absorbance at 450 nm was recorded using a microplate reader.Statistical analysis
Data for cell viability, antibody and cytokine secretion assay were presented as mean ± SD and were statistically analyzed using the two-tailed Student's t test.*p < 0.05,**p < 0.01 was considered to be significant.Results and discussion
Polyglutamate conjugation of a long carbon-chain and IMQ (TLR7 agonist)
γ-PGA is a kind of environmentally-friendly macromolecular substance with hydrophilicity, easy biodegradation and strong absorbability. Furthermore, the hydrophilic bioinspired synthetic polymer with an abundancy of carboxylic acid groups makes it is well suited for the design of water-soluble sustained release agents and drug carriers.19,20 γ-PGA has already been employed in a phase III trial polymer-anticancer drug conjugate, Opaxio, consisting of paclitaxel conjugated to glutamic acid residues applied in ovarian or fallopian tube cancer treatment.37 An ideal adjuvant needs to dissolve in water and/or oil. To circumvent these issues De Geest's group designed lipid-polymer amphiphiles consisting of a lipid motif as the hydrophobic part and a hydrophilic polymer connected to the lipid moiety at one of its chain ends.15 With these in our minds, we conjugated IMQ, a small-molecule TLR7 agonist, and an LA motif as the hydrophobic part onto the γ-PGA polymer backbone to gain an amphiphilic polymer. This concept is illustrated in Scheme 2.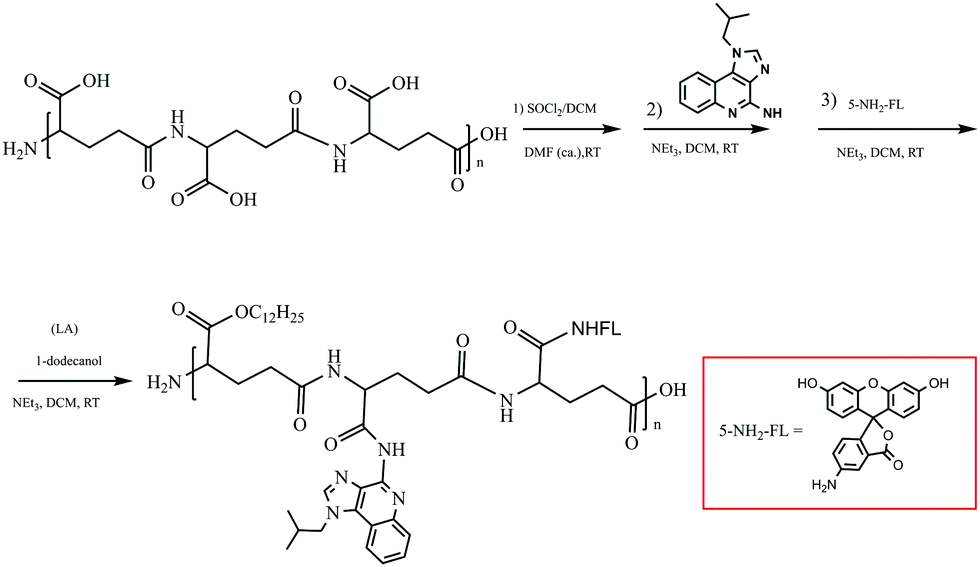 | ||
| Scheme 2 The synthesis route to FIP. | ||
Amphiphilic FIP was synthesized by the reaction of γ-PGA with SOCl2 to generate a reactive intermediate of acyl chloride polymer backbone that can later on be used for conjugation of IMQ, LA, as well as for fluorescent labelling. The purified FIP was characterized in DMDO-d6 using 1H NMR spectroscopy. From Fig. 1, we can see that the IMQ (in D2O + CF3CO2D, blue box) and LA (in D2O, red box) were attached to γ-PGA units via covalent bonds. The degree of substitution of the IMQ was determined from the ratio between the integrated area resonances of the hydrogen at the C-2 position (Scheme 1) in the γ-PGA (in D2O) groups (δ 4.15 ppm) and the aromatic ring hydrogen in IMQ units (δ 6.68 ppm). Likewise, the degree of esterification with LA was calculated using the ratio between the integrated area resonances of the hydrogen at the C-12 in the LA methyl group (δ 0.85 ppm) and the hydrogen at C-2 (δ 4.15 ppm) in the γ-PGA units. The results illuminated that the grafting degrees of IMQ and LA were about 8% and 5.6% in mole % (12% and 6% in wt%), respectively. Due to the content of fluorescein being very low, its peaks barely show in 1H NMR (Single 1H NMR in Fig. S1, ESI†).
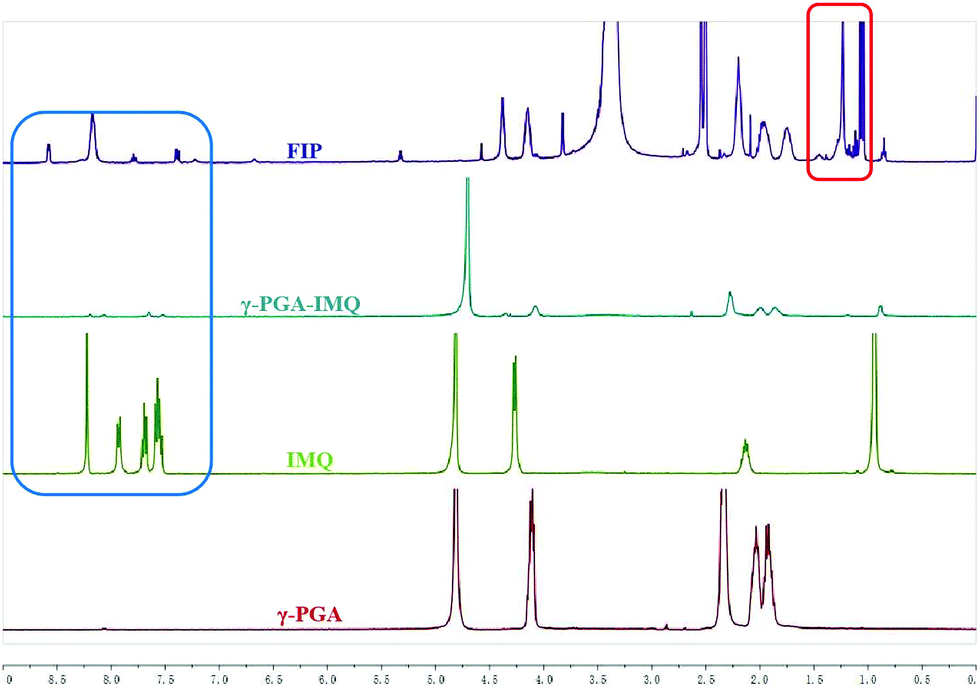 | ||
| Fig. 1 1H NMR image of FIP, γ-PGA-IMQ, IMQ and γ-PGA. | ||
To investigate whether IMQ and fluorescein conjugated to the γ-PGA framework, we carried out spectroscopic studies to examine the effectiveness of FIP. The UV-vis spectrum of FIP showed distinct IMQ peaks at about 306 and 318 nm (Fig. 2a). The absorbance from 400 to 530 nm was ascribed to fluorescein absorbance. To further verify the grafting of IMQ and fluorescein to the γ-PGA framework, a fluorescence assay was conducted with Ex 280 (Fig. 2b) and 455 nm (Fig. 2c) for IMQ and fluorescein, respectively. The results suggested that IMQ and fluorescein were successfully graft to the γ-PGA framework.
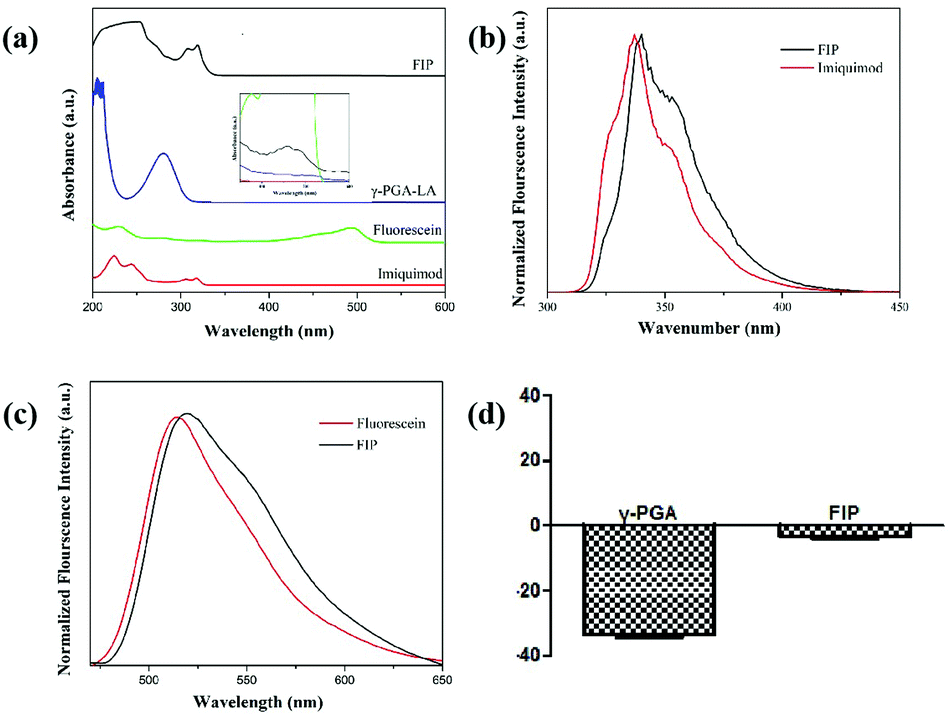 | ||
| Fig. 2 (a) UV-Vis absorbance spectra. The fluorescence spectra of FIP with Ex 280 nm (b) and Ex 455 nm (c) for IMQ, fluorescein and FIP, and (d) the zeta potential value. | ||
As shown in Fig. 2d, the hydrodynamic diameter of FIP was about 284.7 nm with a polydispersity index (PDI) of 0.265 (Fig. S2, ESI†). The low PDI indicates excellent and uniform dispersion of FIP. The zeta potential value of FIP was −4.00 ± 0.91 mV, while the value for γ-PGA was −35.10 ± 1.49 mV, which suggests that γ-PGA was successfully conjugated with IMQ and fluorescein.
Fourier-transform infrared (FT-IR) spectroscopy was used to confirm the IMQ and LA grafting to the γ-PGA skeleton (Fig. 3). Compared with γ-PGA, the absorption peaks at 3078 and 3113 cm−1 (purple box) were assigned as sp2 hybridization C–H stretching from the IMQ aromatic ring. The peak at 3277 cm−1 was ascribed to the N–H vibration of IMQ. The peaks at 1742 and 1765 cm−1 (red box) were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of an ester. The peaks between 2854–2954 cm−1 (blue box) were enhanced due to sp3 hybridization C–H stretching of the LA chain. The new peak at 1666 cm−1 (gray box) compared with that of crude γ-PGA was due to amide carbonyl stretching vibration, in which IMQ reacted with γ-PGA. These results demonstrate that IMQ and LA were successfully modified onto γ-PGA (single FI-IR of FIP and γ-PGA-LA is shown in Fig. S3, ESI†).
O stretching of an ester. The peaks between 2854–2954 cm−1 (blue box) were enhanced due to sp3 hybridization C–H stretching of the LA chain. The new peak at 1666 cm−1 (gray box) compared with that of crude γ-PGA was due to amide carbonyl stretching vibration, in which IMQ reacted with γ-PGA. These results demonstrate that IMQ and LA were successfully modified onto γ-PGA (single FI-IR of FIP and γ-PGA-LA is shown in Fig. S3, ESI†).
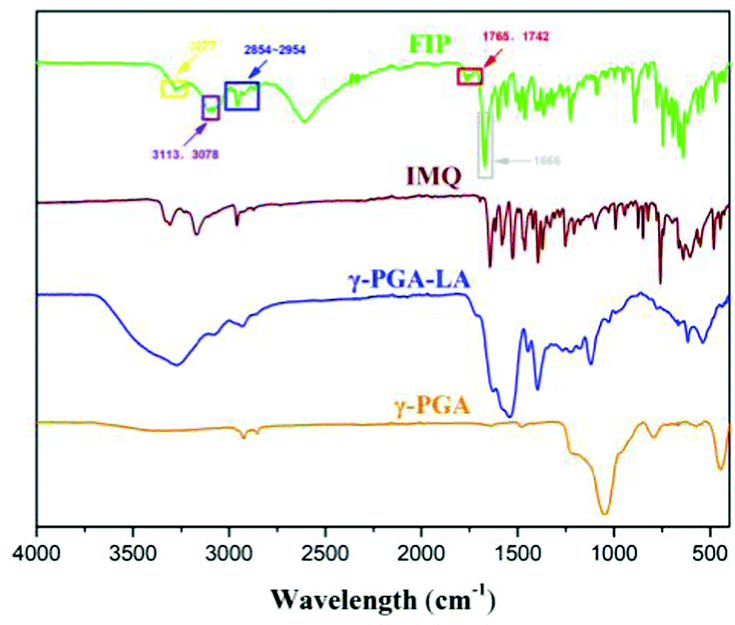 | ||
| Fig. 3 The FI-IR images of FIP and related substrates. | ||
Cellular cytotoxicity
An ideal vaccine adjuvant should have excellent biocompatibility and safety. The good cell viability of FIP was evidenced by the CCK-8 assay. RAW264.7 cells were treated with different concentrations (5, 100 or 1000 μg mL−1) of FIP (containing about 12% IMQ, w/w) or IMQ (100 or 1000 μg mL−1), acetic acid (HOAc, pH 6), and PBS for 24 h. As shown in Fig. 4, the FIP hardly caused a loss in the proliferation ability of the cells and the survival rate of the cells treated with FIP (5 or 100 μg mL−1) had no significant difference to that of the PBS-treated cells (p > 0.05). However, the cell viabilities showed a significant decline when treated with plain IMQ acid solution (pH 6) at the concentration of 100 μg mL−1, or HOAc solution (p < 0.05). IMQ demonstrated high toxicity to RAW264.7 cells at 100 μg mL−1 (cell viability < 17%). Therefore, the FIP could be considered as a nontoxic, safety-acceptable and biocompatible adjuvant.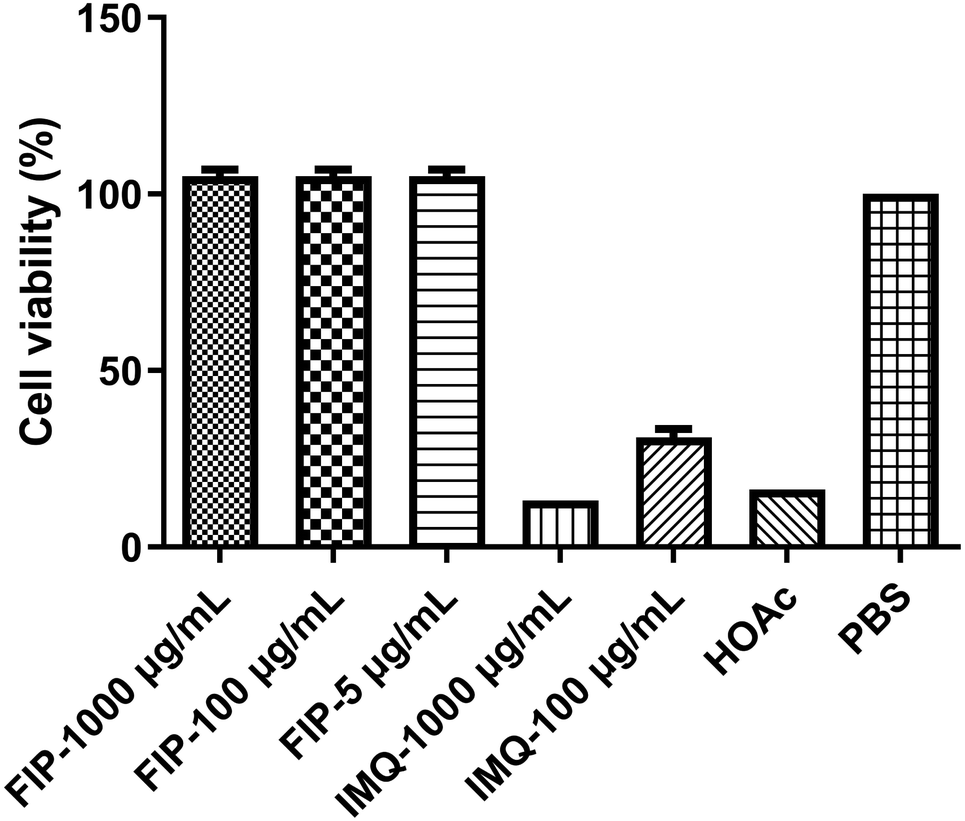 | ||
| Fig. 4 The cell viability of each sample with different concentrations. | ||
Pure IMQ has been reported to cause many adverse reactions, such as local skin inflammation, erosion and ulcers when injected into skin.38,39 Also, IMQ can dissolve in acid solution, then further stimulate the body and pose some safety risks. An effective solution to the problem is covalent ligation of IMQ to a larger carrier, which prevents uncontrolled systemic distribution.40 As shown in Fig. 5, after subcutaneous injection of mice with FIP (1000 μg mL−1) or IMQ (100 μg mL−1), the IMQ group showed significant skin reactions after 2 days, however, no adverse skin reaction was observed in the FIP group up to 14 days after immunization. The FIP did not trigger any skin redness, swelling or ulceration. The mice in the FIP group were fine with a normal diet and daily routine.
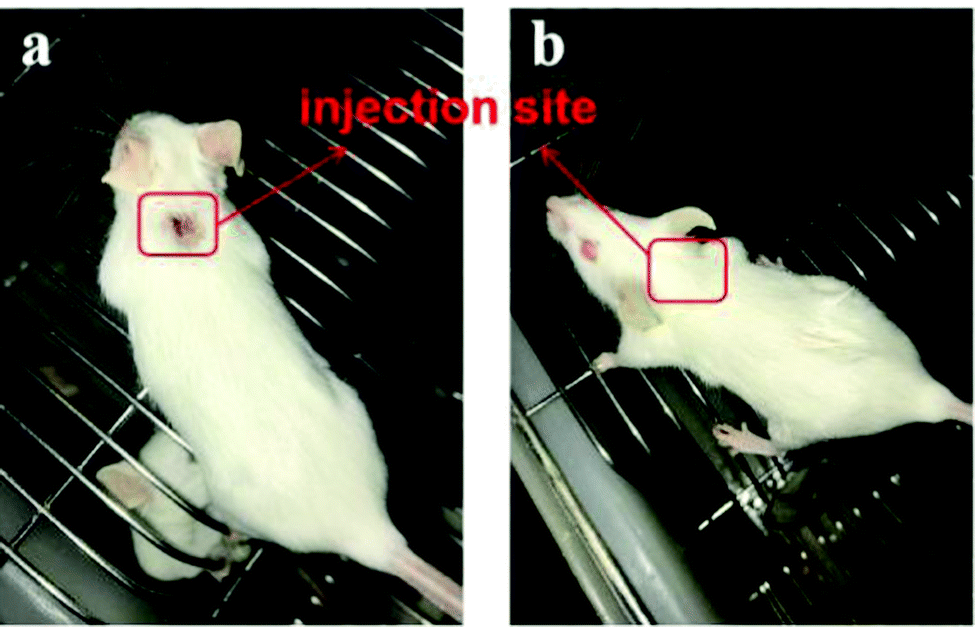 | ||
| Fig. 5 The skin reaction pf mice subjected to subcutaneous injection with (a) IMQ and (b) FIP in PBS solution after 2 days. | ||
Histological examination
To further evaluate the toxicity of FIP in vivo, histological examination was performed using the standard hematoxylin and eosin (H&E) staining method. The FIP were injected through hypodermic injection at a concentration of 500 μg mL−1 (200 μL) per mouse (n = 3) for 28 d, and then the mice were sacrificed and the major organs (spleen, liver and kidneys) and the subcutaneous tissues at the injection sites were removed and taken out for pathological analysis. The histopathological alterations were shown in Fig. 6. For the FIP group mice, the spleen, liver, kidneys and the subcutaneous tissues at the injection sites showed no marked pathological abnormalities and adverse effects as compared to the control group (Fig. 6b). The results suggest that the FIP adjuvant-based vaccine has good safety. | ||
| Fig. 6 H&E staining images from the major organs and injection sites of BALB/c mice after 28 days of immunization. Data are representative of at least 3 mice. The scale bar is 50 μm. | ||
Cellular uptake measurement by flow cytometry
The activation or polarization of macrophages is an integral part in innate and adaptive immune responses.41,42 Macrophages can be polarized into M1 (classically activated) and M2 (alternatively activated) phenotypes. Activated macrophages may be induced by various cytokines.43 To further qualitatively evaluate the effect of FIP on RAW 264.7 activation, the concentration of cytokines in the supernatants of RAW264.7 cells stimulated with OVA or OVA + FIP was measured by ELISA kits. The results showed that FIP significantly promoted the secretion of inflammatory cytokines IL-2, 4, 6 and IFN-γ, which may be an important indicator for M1 macrophage activation (Fig. S4, ESI†).To investigate the cellular uptake of FIP by RAW264.7 cells, a flow cytometer assay was conducted. The median fluorescence intensity (MFI) of cells treated with different concentrations of FIP was determined. As shown in Fig. 7, RAW 264.7 cells incubated with FIP showed a high fluorescence intensity, which indicated that FIP can be engulfed effectively by RAW 264.7 cells. The data in Fig. 7b also demonstrate that the cellular uptake of FIP by RAW 264.7 cells (the percentages of fluorescent cells) increased in a dose dependent manner.
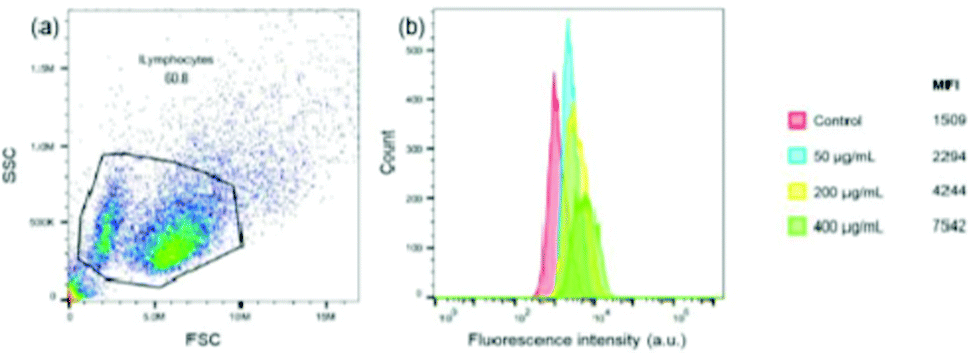 | ||
| Fig. 7 Flow cytometry analysis of RAW264.7 cell uptake of FIP. (a) Aspect ratio; (b) MFI image of RAW264.7 cells incubated with different FIP concentrations. | ||
To corroborate the uptake of FIP, the cellular localization of FIP in RAW264.7 cells was also observed by confocal microscopy. Cells were stained with DAPI (for cell nucleus) and Phalloidin (for membrane), respectively. Images of the RAW264.7 cells without (Fig. 8a) and with FIP (Fig. 8b) show that FIP did not cause significant cytotoxicity and did not cause cells apoptosis in RAW264.7 cells. Meanwhile, the green fluorescein signals could only be obtained in RAW264.7 cells incubated with FIP. This indicated that FIP could be engulfed by RAW264.7 cells and packaged in the cell membrane, which would be beneficial for antigen delivery.
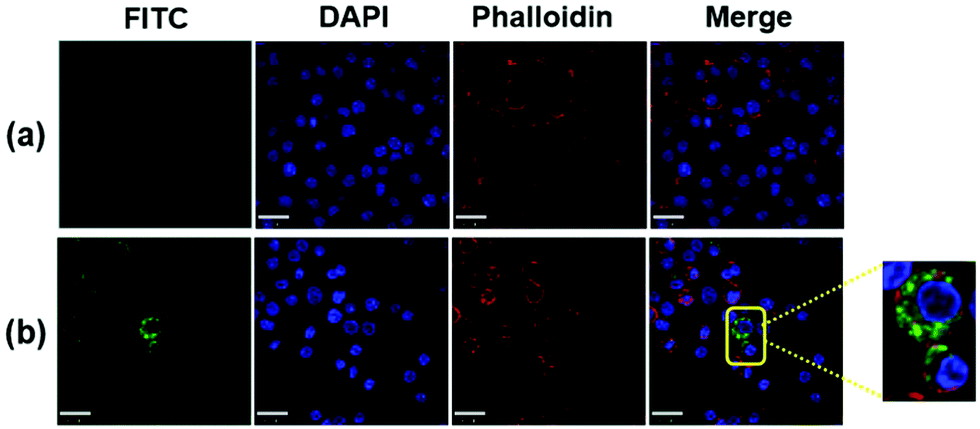 | ||
| Fig. 8 Confocal microscopy images of RAW264.7 cells incubated with (a) DMEM and (b) FIP (40 μL, 1 mg mL−1). | ||
In vivo effect of FIP on the immune system
After verifying the improved safety profile as well as the efficiency of cell uptake with FIP in vitro and in vivo, we then evaluated the adaptive immune responses in mice immunized with different components, formulations and concentrations.IMQ has been used as a small molecule immunomodulator in anti-virus and anti-tumor treatments. To elucidate the possible adjuvant properties of FIP, we evaluated the immunostimulating efficacy of FIP, IMQ or γ-PGA with OVA as a model antigen in PBS solution.
With respect to the humoral responses, IMQ, which was the main framework for immune stimulation, induced the strongest serum OVA-specific IgG among those groups (Fig. 9). The dose of vaccine needs to be determined in the practical use process. Therefore, we chose the protocol of keeping the fixed adjuvant dosage (Table 1). Mice immunized with OVA + IMQ appeared to have obvious redness, swelling, festering at the injection sites and other adverse reactions (Fig. 5a). However, there were no side-effects when using OVA + IMQ (Fig. 5b). This phenomenon may be attributed to the fact that IMQ in an unformulated state rapidly distributes throughout the whole body and causes systemic inflammation. Covalent conjugation of IMQ to macromolecular γ-PGA carriers strongly abrogates systemic dissemination due to controlled release of IMQ.44 Interestingly, as shown in Fig. 9, OVA + FIP demonstrated a significantly stronger immune response than its backbone OVA + γ-PGA but less than OVA + IMQ (p < 0.05). The results indicated that IMQ can effectively enhance the immune response. According to 1H NMR (Fig. S1a, ESI†) the content (mass ratio) of IMQ in FIP was about 12%. 500 μg of FIP (containing approximately 60 μg of IMQ) had a slightly lower immune-stimulating potentiality than that of 500 μg pristine IMQ. As an amphiphilic polymer, FIP can self-assemble into nanoparticles (ANPs). Particles with a size of 20–200 nm undergo endocytosis and are preferentially ingested by dendritic cells (DCs).45,46 In this study, we obtained an average Z-average diameter of about 284.7 nm (Fig. S2, ESI†). Owing to an increase in the surface hydrophobicity of ANPs, an enhanced adhesion force between ANPs and cell membranes facilitated internalization of FIP and antigens into cells. Furthermore, ANPs were drained into lymphoid tissue,47 degraded to release IMQ slowly, and enhanced immune reactions. Emulsions are liquid dispersions of two immiscible phases and have a long history of use as adjuvants in both human and veterinary vaccines.48 There are mainly three types of emulsion vaccines according to the inner water phase or outer water phase: water-in-oil (W/O), oil-in-water (O/W) and water-oil–water (W/O/W). Due to the reactogenicity of W/O emulsions (traditional vaccines), O/W or W/O/W emulsions are considered as better alternatives. However, the latter two types of emulsions are evaluated as less effective in affording high antibody levels and long antibody duration. Incorporating molecular immunopotentiators within emulsions could overcome the drawbacks of molecular adjuvants (such as biodistribution and pharmacokinetics) and thus improve the adjuvanticity of emulsions.
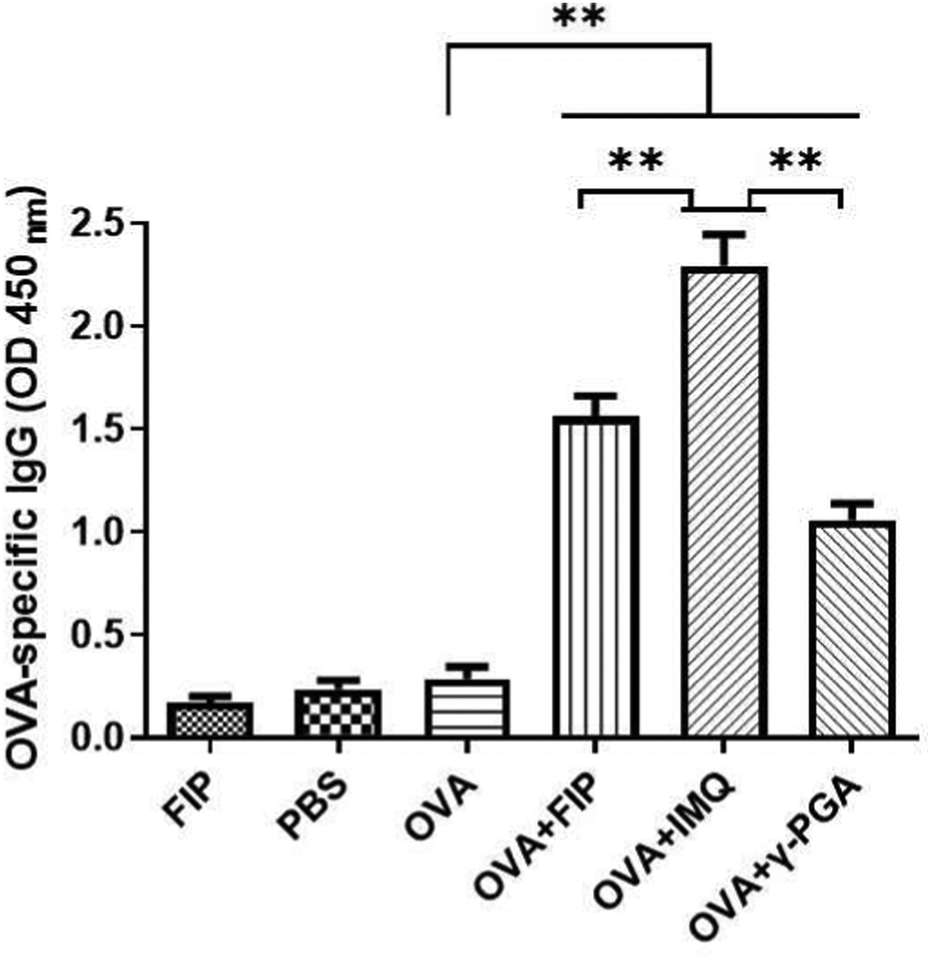 | ||
| Fig. 9 The OVA special-IgG of mice immunized different components of FIP in PBS solution. | ||
We next determined whether FIP would exert any adaptive immune enhancing effects when incorporated into different emulsions. Mice in each group were immunized with OVA + FIP-O/W, OVA + FIP-W/O/W, OVA + FIP-W/O, OVA-O/W, OVA-W/O/W, OVA-W/O, OVA alone, OVA + FIP or PBS, respectively. Sera samples were collected at 14 and 28 dpv after the first immunization and OVA-specific IgG was detected by ELISA (Fig. S5, ESI† and Fig. 10). As shown in Fig. 10, all emulsion formulations elicited significantly higher humoral immune responses than that of OVA alone at 28 dpv (p < 0.05). The OVA + FIP-O/W and OVA + FIP-W/O vaccinated groups had significantly higher OVA-specific antibodies than those of the different formations of OVA + FIP vaccinated groups at 2 weeks after the first and second immunizations (p < 0.05). Furthermore, various OVA + FIP emulsion formulations induced significantly enhanced OVA-specific antibodies compared to OVA-W/O/W, OVA-O/W or OVA-W/O. The results suggested that incorporation of FIP into O/W or W/O emulsion formulations could significantly improve the adjuvanticity of these emulsions.
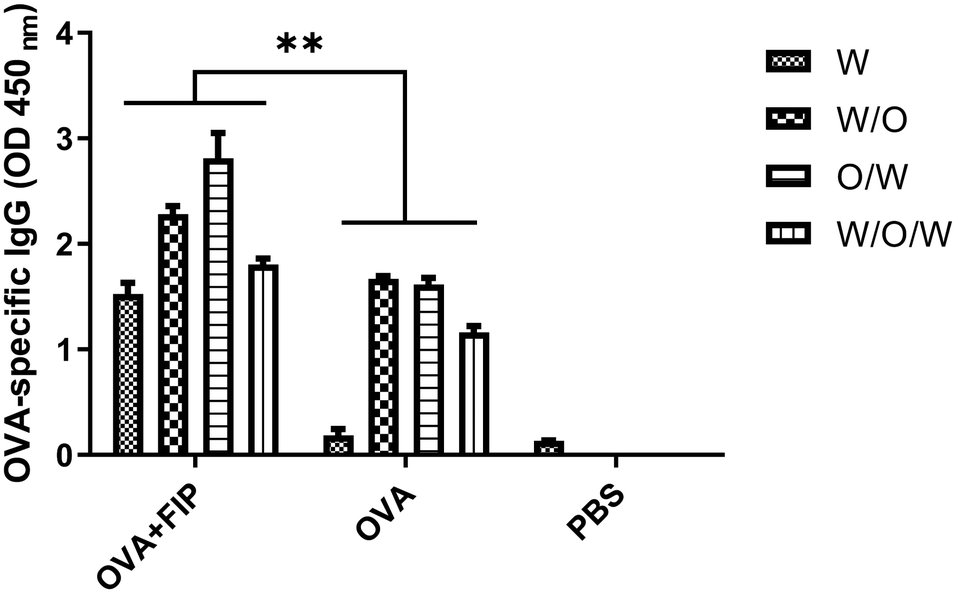 | ||
| Fig. 10 The OVA specific-IgG of mice immunized with different formulations at 28 dpv. | ||
Interestingly, compared with the different formulations of pure OVA, our material has a stronger immune stimulating potentiality. After 28 days of immunization to determine the OD450nm of OVA + FIP comparison by ELSIA it can be found that O/W > W/O > W/O/W ≈ W. The data indicate that the FIP material could be applied to multi-formulations as an ideal adjuvant, especially as a water formulation adjuvant.
Considering the practical application of FIP adjuvant, we then tested the effect of adjuvant dose dependence on immune potentiality. Fig. 11 shows the immune responses induced by different adjuvant dosages for OVA, and the results demonstrate that the OVA antibody titer was the highest when the concentration of FIP was 5 μg mL−1 compared with other higher concentrations of FIP. The experiments indicate that low doses of FIP were adequate for eliciting effective immune responses. Future comprehensive work involving the incorporation of FIP into other antigens or formulations for different animals are needed to obtain a synergetic effect.
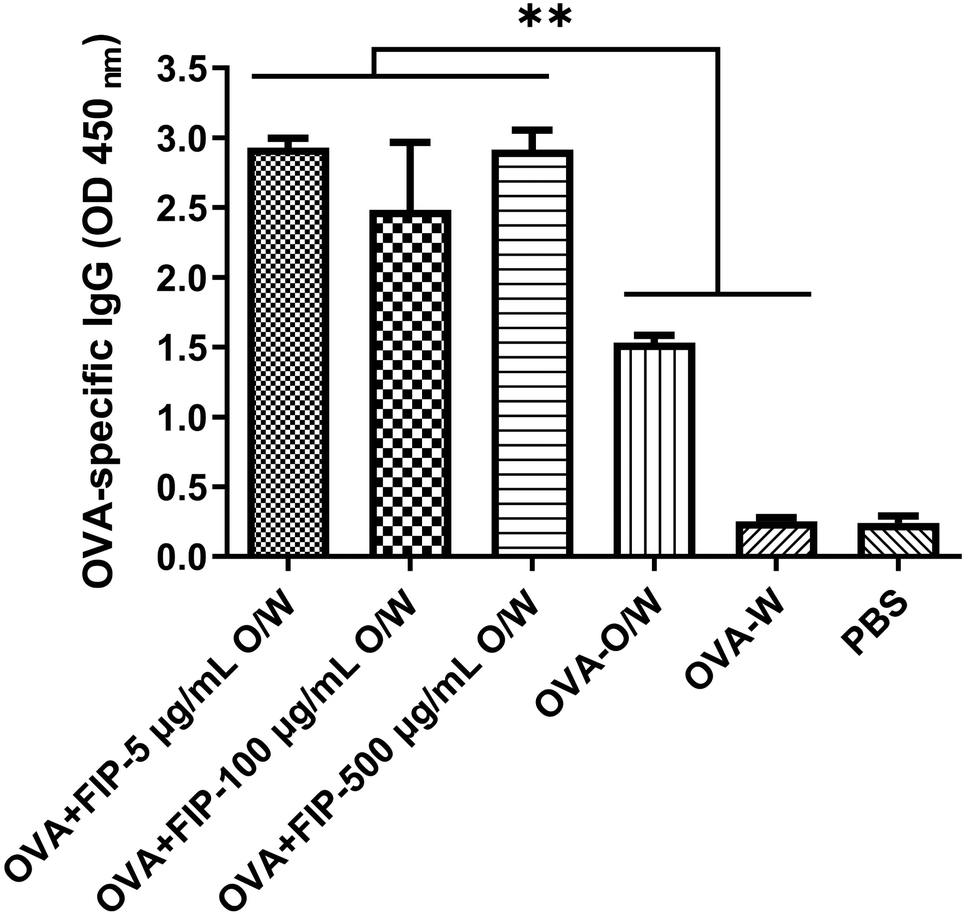 | ||
| Fig. 11 The OVA specific-IgG of mice immunized with OVA + FIP-O/W at different concentrations in 28 dpv. | ||
Conclusions
In summary, we report a simple yet efficient amphiphilic anionic FIP as a vaccine adjuvant for potent immunostimulatory effects after subcutaneous administration. Flow cytometry analysis of RAW264.7 cells demonstrated that FIP can be effectively taken up by the cells. Confocal fluorescent images further confirmed FIP distributed into the cell membrane. The CCK-8 assay and H&E staining revealed that FIP had low toxicity and good biocompatibility. These results indicate that the FIP adjuvant may have good safety in vivo. Low dose (5 μg mL−1) in oil-in-water (O/W) formulation can induce high immune responses and enhanced higher OVA-specific antibodies. Furthermore, the FIP maintained long-time immune memory and lasting immunity effectivity. The FIP provided a novel protein-based vaccine delivery platform, which was capable of inducing strong humoral and cellular immunity for effective vaccine prevention. Controlling the pharmacokinetics of IMQ through conjugation to the γ-PGA skeleton, therefore, enables the development of potent adjuvants in vaccines to drive durable, high-quality immunity responses and long immune memory with strongly reducing systemic toxicity of IMQ. We believe that the strategy explored here will be facile and effective toward generating subunit vaccines for improving the immune stimulating effect and will have immense potentiality in the development of immunotherapy for various diseases.Author contributions
Synthesis (W. Z. Yin). Characterization (W. Z. Yin and D. C. Xuan). Animals and cells (W. Z. Yin, D. C. Xuan, F. Ma, B. H. Deng and M. X. Zhou). Supervision (Y. Lu and J. Q. Zhang). Writing – original draft (W. Z. Yin and J. Q. Zhang). Writing – review & editing (W. Z. Yin, J. Q. Zhang and Y. Lu).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32172895, No. 31772701, No. 31902243) and the Special Fund for Independent Innovation of Agricultural Science and Technology in Jiangsu Province of China (No. CX (17) 3027).Notes and references
- M. C. Hanson, M. P. Crespo, W. Abraham, K. D. Moynihan, G. L. Szeto, S. H. Chen, M. B. Melo, S. Muerller and D. J. Irvine, J. Clin. Invest., 2015, 125(6), 2532–2546 CrossRef PubMed
.
- S. Plotkin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(34), 12283–12287 CrossRef CAS PubMed
- H. J. Larson, C. Jarrett, E. Eckersberger, D. M. Smith and P. Paterson, Vaccine, 2014, 32(19), 2150–2159 CrossRef PubMed
- K. Hoelzer, L. Bielke, D. P. Blake, E. Cox, S. M. Cutting, B. Devriendt, E. Erlacher-Vindel, E. Goossens, K. Karaca, S. Lemiere, M. Metzner, M. Raicek, M. C. Suriñach, N. M. Wong, C. Gay and F. V. Immerseel, Vet. Res., 2018, 49, 70 CrossRef PubMed
- S. Sevimli, F. C. Knight, P. Gilchuk, S. Joyce and J. T. Wilson, ACS Biomater. Sci. Eng., 2017, 3(2), 179–194 CrossRef CAS PubMed
- L. Yang, W. Li, M. Kirberger, W. Z. Liao and J. Y. Ren, Biomater. Sci., 2016, 4, 785–802 RSC
- S. G. Reed, M. T. Orr and C. B. Fox, Nat. Med., 2013, 19, 1597–1608 CrossRef CAS PubMed
- M. L. Mbow, E. D. Gregorio, N. M. Valiante1 and R. Rappuoli, Curr. Opin. Immunol., 2010, 22(3), 411–416 CrossRef CAS PubMed
- A. Comberlato, K. Paloja and M. M. C. Bastings, J. Mater. Chem. B, 2019, 7, 6321–6346 RSC
- B. J. Ignacio, T. J. Albin, A. P. Esser-Kahn and M. Verdoes, Bioconjugate Chem., 2018, 29(3), 587–603 CrossRef CAS PubMed
- A. Iwasaki and R. Medzhitov, Science, 2010, 327(5963), 291–295 CrossRef CAS PubMed
- J. Kim, T. S. Griffith and J. Panyam, J. Pharmacol. Exp. Ther., 2019, 370(3), 715–724 CrossRef PubMed
- P. Redondo, J. del Olmo, A. Lopez-Diaz de Cerio, S. Inoges, M. Marquina, I. Melero and M. Bendandi, J. Invest. Dermatol., 2007, 127(7), 1673–1680 CrossRef CAS PubMed
- J. P. Vasilakos and M. A. Tomai, Expert Rev. Vaccines, 2013, 12, 809–819 CrossRef CAS PubMed
- J. D. Vrieze, B. Louage, K. Deswarte, Z. F. Zhong, R. D. Coen, S. V. Herck, L. Nuhn, C. K. Frich, A. N. Zelikin, S. Lienenklaus, N. N. Sanders, B. N. Lambrecht, S. A. David and B. G. D. Geest, Angew. Chem., Int. Ed., 2019, 58(43), 15390–15395 CrossRef PubMed
- L. Nuhn, N. Vanparijs, A. De Beuckelaer, L. Lybaert, G. Verstraete, K. Deswarte, S. Lienenklaus, N. M. Shukla, A. C. D. Salyer, B. N. Lambrecht, J. Grooten, S. A. David, S. D. Koker and B. G. D. Geest, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(29), 8098–8103 CrossRef CAS PubMed
- T. Y.-H. Wu, M. Singh, A. T. Miller, E. De Gregorio, F. Doro, U. D’Oro, D. A. G. Skibinski, M. L. Mbow, S. Bufali, A. E. Herman, A. Cortez, Y. K. Li, B. P. Nayak, E. Tritto, C. M. Filippi, G. R. Otten, L. A. Brito, E. Monaci, C. Li, S. Aprea, S. Valentini, S. Calabró, D. Laera, B. Brunelli, E. Caproni, P. Malyala, R. G. Panchal, T. K. Warren, S. Bavari, D. T. O’Hagan, M. P. Cooke and N. M. Valiante, Sci. Transl. Med., 2014, 6(263), 263ra160 Search PubMed
- N. Pippa, M. Gazouli and S. Pispas, Vaccines, 2021, 9(6), 558 CrossRef CAS PubMed
- S. H. Kelly, L. S. Shores, N. L. Votaw and J. H. Collier, Adv. Drug Delivery Rev., 2017, 114, 3–18 CrossRef CAS PubMed
- W. G. Kim, B. Choi, H.-J. Yang, J.-A. Han, H. Jung, H. Cho, S. Kang and S. Y. Hong, Bioconjugate Chem., 2016, 27, 2007–2013 CrossRef CAS PubMed
- A. Seth, M. B. Heo and Y. T. Lim, Biomaterials, 2014, 35, 7992–8001 CrossRef CAS PubMed
- Y. H. Lin, C. K. Chung, C. T. Chen, H. F. Liang, S. C. Chen and H. W. Sung, Biomacromolecules, 2005, 6(2), 1104–1112 CrossRef CAS PubMed
- T. Uto, M. Toyama, Y. Nishi, T. Akagi, F. Shima, M. Akashi and M. Baba, Results Immunol., 2013, 3, 1–9 CrossRef PubMed
- F. Shima, T. Uto, T. Akagi, M. Baba and M. Akashi, Acta Biomater., 2013, 9, 8894–8901 CrossRef CAS PubMed
- M. Henriksen-lacey, K. S. Korsholm, P. Andersen, Y. Perrie and D. Christensen, Expert Opin. Drug Delivery, 2011, 8(4), 505–519 CrossRef CAS PubMed
- M. Mockey, E. Bourseau, V. Chandrashekhar, A. Chaudhuri, S. Lafosse, E. Le Cam, V. F. J. Quesniaux, B. Ryffel, C. Pichon and P. Midoux, Cancer Gene Ther., 2007, 14(9), 802–814 CrossRef CAS PubMed
- J. Wang, H. Zope, M. A. Islam, J. Rice, S. Dodman, K. Lipert, Y. H. Chen, B. R. Zetter and J. J. Shi, Front Bioeng. Biotech., 2020, 8, 787 CrossRef PubMed
- B. Wang, S. V. Herck, Y. Chen, X. Y. Bai, Z. F. Zhong, K. Deswarte, B. N. Lambrecht, N. N. Sanders, S. Lienenklaus, H. W. Scheeren, S. A. David, F. Kiessling, T. Lammers, B. G. De Geest and Y. Shi, J. Am. Chem. Soc., 2020, 142, 12133–12139 CrossRef CAS PubMed
- C. S. Liong, A. A. A. Smith, J. L. Mann, G. A. Roth, E. C. Gale, C. L. Maikawa, B. S. Ou and E. A. Appel, Adv. Ther., 2021, 4(4), 2000081 CrossRef CAS
- J. S. Wu, J. X. Li, N. Shu, Q. J. Duan, Q. S. Tong, J. Y. Zhang, Y. C. Huang, S. Y. Yang, Z. B. Zhao and J. Z. Du, Nano Res., 2022, 15, 510–518 CrossRef CAS
- S. M. Jin, S. N. Lee, J. E. Kim, Y. J. Yoo, C. Song, H. S. Shin, H. Phuengkham, C. H. Lee, S. H. Um and Y. T. Lim, Adv. Sci., 2021, 8, 2102043 CrossRef CAS PubMed
- S. B. Park, M. H. Sung, H. Uyama and D. K. Han, Prog. Polym. Sci., 2021, 113, 101341 CrossRef CAS
- D. V. Lysebetten, A. Malfanti, K. Deswarte, K. Koynov, B. Golba, T. T. Ye, Z. F. Zhong, S. Kasmi, A. Lamoot and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 6011–6022 CrossRef PubMed
- I. W. Hamley, Bioconjugate Chem., 2021, 32, 1472–1490 CrossRef CAS PubMed
- C. C. Meng, X. Zhi, C. Li, C. F. Li, Z. Y. Chen, X. S. Qiu, C. Ding, L. J. Ma, H. M. Lu, D. Chen, G. Q. Liu and D. X. Cui, ACS Nano, 2016, 10(2), 2203–2213 CrossRef CAS PubMed
- J. Q. Zhang, J. F. Miao, X. G. Han, Y. Lu, F. Lv, B. H. Deng, Y. H. Zhao, C. Ding and J. B. Hou, BMC Vet. Res., 2018, 14, 415 CrossRef CAS PubMed
- L. Paz-Ares, H. Ross, M. O'Brien, A. Riviere, U. Gatzemeier, J. Von Pawel, E. Kaukel, L. Freitag, W. Digel, H. Bischoff, R. García-Campelo, N. Iannotti, P. Reiterer, I. Bover, J. Prendiville, A. J. Eisenfeld, F. B. Oldham, B. Bandstra, J. W. Singer and P. Bonomi, Br. J. Cancer, 2008, 98, 1608–1613 CrossRef CAS PubMed
- K. LBarr, T. H. Konia and M. A. Fung, J. Cutan. Pathol., 2011, 38(4), 346–350 Search PubMed
- J. Conde, K. Davis, E. Nituen, N. Balmer, D. Jones and A. Mcmichael, J. Dermatol. Treat, 2010, 21(2), 122–124 CrossRef CAS
- L. Nuhn, L. V. Hoecke, K. Deswarte, B. Schepens, Y. P. Li, B. Lambrecht, S. D. Koker, S. A. David, X. Saelens and B. D. Geest, Biomaterials, 2018, 178, 643–651 CrossRef CAS PubMed
- F. R. Bertani, P. Mozetic, M. Fioramonti, M. Iuliani, G. Ribelli, F. Pantano, D. Santini, G. Tonini, M. Trombetta, L. Businaro, S. Selci and A. Rainer, Sci. Rep., 2017, 7(1), 8965 CrossRef PubMed
- J. Wei and G. E. Besner, J. Surg. Res., 2015, 197(1), 126–138 CrossRef CAS PubMed
- D. P. Vasconcelos, A. C. Fonseca, M. Costa, I. F. Amaral, M. A. Barbosa, A. P. Águas and J. N. Barbosa, Biomaterials, 2013, 34, 9952–9959 CrossRef CAS PubMed
- B. J. Ignacio, T. J. Albin, A. P. Esser-Kahn and M. Verdoes, Bioconjugate Chem., 2018, 29(3), 587–603 CrossRef CAS PubMed
- R. Bastola, S. Lee and J. Pharm, Investigation, 2019, 49, 279–285 CAS
- S. Yi, S. D. Allen, Y. G. Liu, B. Z. Ouyang, X. Li, P. Augsornworawat, E. B. Thorp and E. A. Scott, ACS Nano, 2016, 10(12), 11290–11303 CrossRef CAS PubMed
- T. K. Kishimoto and R. A. Maldonado, Front. Immunol., 2018, 9, 230 CrossRef PubMed
- Y. J. Lin, C. N. Wen, Y. Y. Lin, W. C. Hsieh, C. C. Chang, Y. H. Chen, C. H. Hsu, Y. J. Shih, C. H. Chen and C. T. Fang, Nat. Commun., 2020, 11, 315 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01169c |
| This journal is © The Royal Society of Chemistry 2022 |