
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of a push–pull arylene–vinylene terpyridyl conjugate: multifunctional behaviors exhibited by a single molecule†
Amit
Sil
 ,
Utsav
Ghosh
,
Suman
Dolai
,
Soumitra
Manna
,
Apurba
Maity
and
Sanjib K.
Patra
*
,
Utsav
Ghosh
,
Suman
Dolai
,
Soumitra
Manna
,
Apurba
Maity
and
Sanjib K.
Patra
*
Department of Chemistry, IIT Kharagpur, Kharagpur 721302, WB, India. E-mail: skpatra@chem.iitkgp.ac.in; Tel: +91 3222 283338
First published on 10th May 2022
Abstract
A multifunctional D–π–A push–pull arylene–vinylene conjugated terpyridine, 4′-(4-{2-[4-[bis(4-thiophen-2-yl-phenyl)amino]phenyl-ethenyl]}phenyl)-2,2′:6′,2′′-terpyridine, has been designed and successfully developed. Its photophysical properties were systematically explored, revealing its tunable multifunctional behaviors including solvatochromism, vapochromism, piezofluorochromism and remarkable chemosensing properties. The push–pull congener exhibits strong emission which is highly sensitive to solvent polarity, showing blue to yellow fluorescence in the presence of volatile organic compounds (VOCs). The compound also exhibits reversible mechanochromic behavior with high color contrast between yellow and green in its pristine and ground form. Moreover, the synthesized push–pull probe shows remarkable sensitivity towards NACs at the ppb level (especially for picric acid) in solution, vapor and contact mode as a ‘turn-off’ sensor which can be visualized by the naked eye. The fluorescence quenching through supramolecular complexation has been further supported by DFT calculations, time-resolved fluorescence and 1H NMR titration. Furthermore, the conjugated terpyridine can efficiently detect toxic Hg2+ at the submicromolar level with high selectivity and sensitivity without interference by other competing metal ions. The selectivity, sensitivity, reversibility, and recyclability allowed us to demonstrate its practical utility as a solid state kit for onsite detection of toxic Hg2+, NACs and volatile organic solvents.
Introduction
Multifunctional π-conjugated D–π–A type organic congeners emitting strong fluorescence have received much attention due to their various fascinating stimuli-responsive properties including piezofluorochromism,1–4 vapofluorochromism,5–8 solvatochromism,5–7,9 and thermofluorochromism.10–12 As compared to the inorganic materials, one of the prominent characteristics of π-conjugated organic materials is their structural flexibility and very high molecular polarizability resulting strong electron delocalization and intramolecular charge transfer (ICT) in the “push–pull” molecular backbone.13,14 A subtle change in π-conjugated congeners could produce a dramatic change in the configuration, crystal stacking, electronic structure, and furthermore in the optoelectronic property. Multifunctional D–π–A type organic materials have been the subject of active research and have attracted a great deal of attention because of their potential application in organic light emitting diodes,15–20 field-effect transistors,21–25 organic solid-state lasers,26–29 as well as sensing probes for detection of environmentally concerned toxic metal ions,30–40 solvent vapors41 and explosive nitroaromatic compounds (NACs).42,43 However, it is rare to witness all of the above-mentioned combined and fascinating properties in a single π-conjugated congener because of the lack of clear guidelines for designing molecular structures simultaneously possessing multiple features.Arylene–vinylene conjugated terpyridine molecules represent a new category of highly luminescent D–π–A type chromophores with chelation functionality and relatively long lived excited states due to their extended π-conjugation.44–51 The extended π-conjugation in the arylene–vinylene conjugated terpyridine is beneficial to reduce the energy of LUMO and hence the band gap of the probes, enhancing detection sensitivity. 2,2′:6′,2′′-Terpyridine (tpy), having three pyridyl nitrogen atoms, has been preferred for its extremely strong binding affinity towards most of the transition metal ions. In addition, D–π–A conjugated compounds linked with a vinylene bridge usually exhibit strong ICT emission and structural flexibility, which is favorable to induce piezofluorochromism along with solvatochromism and vapochromism properties.44–51 Studies of excited state behavior of these categories of D–π–A type congeners have been utilized to develop not only the fluorescent sensors for toxic metal ions and NACs but also highly luminescent materials for organic light emitting devices (OLEDs). Furthermore, these systems can also exhibit solvatochromic behavior which is beneficial for chemical sensing of solvents and could be potentially used to detect volatile organic compounds (VOCs).44–54 Inspired by all the important fascinating properties shown by arylene–vinylene conjugated terpyridines we have aimed to develop a single multifunctional molecule keeping all these structural features in a single molecule.
Designing a single molecule for multiple applications is always a demanding and attractive strategy. Nevertheless, multifunctional D–π–A type arylene–vinylene conjugated terpyridine is not reported to the best of our knowledge. Our aim in this work is to design and develop such a multifunctional terpyridyl based single D–π–A congener which will offer multiple fascinating properties such as solvatochromism, vapochromism, piezofluorochromism and sensing. By taking all these aspects into consideration, in the present work, we report facile and easy synthesis of multifunctional stimuli-responsive D–π–A structured push–pull arylene–vinylene conjugated terpyridine with tunable photophysical properties such as solvatochromism, vapochromism, piezofluorochromism and most importantly the selective sensing ability of environmentally toxic Hg2+ ions and explosive NACs.
Results and discussion
Design, synthesis and characterization
D–π–A based arylene–vinylene conjugated terpyridine having functional triarylamine (TAA) as the donor moiety, phenylene vinylene as the π-conjugated bridge, and 2,2′:6′,2′′-terpyridine as the acceptor was designed and synthesized following our previously reported procedure.51 The electron rich functional triarylamine unit with thienylphenyl (D) has been judiciously chosen to access its extended π-conjugation and tunable ICT (intramolecular charge transfer) emission in the visible region.55–57 Furthermore, the propeller geometry of the triphenylamine moiety will induce steric hindrance preventing its molecular packing in solid state. This will have an advantageous effect to gain enhanced emission of the conjugated chromophore in solid state. Moreover, the structural variation was extended by incorporating two thienyl moieties at the para-position of the core triphenylamine unit. On the other hand, 2,2′:6′,2′′-terpyridyl unit has been installed due to its promising π-acceptor (A) character and most remarkably for its ability to serve as a tridentate chelating ligand for toxic transition metal ions.The D–π–A arylene–vinylene conjugated terpyridine (6) was prepared following the synthetic protocol as shown in Scheme 1. 4-(Bis(4-bromophenyl)amino)benzaldehyde (3) was synthesized according to the literature reported procedure.58 The synthesis starts from triphenylamine which undergoes a Vilsmeier–Haack formylation reaction, using DMF and POCl3 at 60 °C, affording 4-(diphenylamino)benzaldehyde (2) with 79% yield. Later, 4-(bis(4-bromophenyl)amino)benzaldehyde (3) was synthesized in 82% yield by reacting 4-(diphenylamino)benzaldehyde (2) with NBS (2 eq.) in chloroform under refluxing conditions. Subsequently, compound 3 was further reacted with thiophene-2-boronic acid pinacol ester through the Suzuki coupling protocol to afford the targeted aryl-aldehyde, 4-[bis(4-thiophen-2-yl-phenyl)amino]benzaldehyde (4), for the Wittig condensation reaction in 68% yield. In the 1H NMR (CDCl3) spectrum of 4, the characteristic signal of the aldehyde proton appears as a singlet at 9.85 ppm and the other aromatic protons resonate in the region of 7.72–7.09 ppm. The HRMS study further confirms the formation of 4 by showing its molecular ion peak, [M + H]+, at 438.0942 which is in good agreement with the calculated value. On the other hand the phosphonium ylide, 4-(2,2′:6′,2′′-terpyridyl-4′)-benzyl triphenylphosphonium bromide (5), was synthesized following our previously reported procedure starting from 4′-(tolyl)-2,2′:6′,2′′-terpyridine which was synthesized via one pot modified Kröhnke annulation reaction.59 Finally, arylene vinylene conjugated terpyridine, tpy-C6H4–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C6H4–N(C6H4–C4H3S)2 (6), was achieved employing Wittig condensation reaction between aryl-aldehyde (4) and phosphonium ylide (5) in the presence of tBuOK under solvent free mild conditions by grinding in a mortar. Purification by silica gel column chromatography (60–120 mesh) using ethyl acetate and hexanes as the eluent was performed to achieve analytically pure bright yellow and highly luminescent arylene–vinylene conjugated terpyridine (6). It was unambiguously characterized by multinuclear NMR, IR and HRMS (ESI+) analyses (Fig. S4, S5 and S7, ESI†). The 1H NMR spectrum exhibits one multiplet at 8.84–8.72 ppm corresponding to the pyridyl protons and the other aromatic protons resonate in the region of 8.69–7.06 ppm whereas the aromatic carbons resonated at 156.5–118.9 ppm. The HRMS study further confirms the formation of compound 6 by showing its molecular ion peak, [M + H]+, at 743.2349 and the experimental isotopic distribution is in good agreement with the simulated one.
CH–C6H4–N(C6H4–C4H3S)2 (6), was achieved employing Wittig condensation reaction between aryl-aldehyde (4) and phosphonium ylide (5) in the presence of tBuOK under solvent free mild conditions by grinding in a mortar. Purification by silica gel column chromatography (60–120 mesh) using ethyl acetate and hexanes as the eluent was performed to achieve analytically pure bright yellow and highly luminescent arylene–vinylene conjugated terpyridine (6). It was unambiguously characterized by multinuclear NMR, IR and HRMS (ESI+) analyses (Fig. S4, S5 and S7, ESI†). The 1H NMR spectrum exhibits one multiplet at 8.84–8.72 ppm corresponding to the pyridyl protons and the other aromatic protons resonate in the region of 8.69–7.06 ppm whereas the aromatic carbons resonated at 156.5–118.9 ppm. The HRMS study further confirms the formation of compound 6 by showing its molecular ion peak, [M + H]+, at 743.2349 and the experimental isotopic distribution is in good agreement with the simulated one.
 | ||
| Scheme 1 Synthetic route for 6 (yield in parentheses). | ||
Solvatochromism
Typically, D–π–A type push–pull congeners with strong dipoles have the greatest potential to exhibit significant dependence on solvents through intramolecular charge transfer state (ICT) or twisted intramolecular charge-transfer (TICT) transitions from the electron-donating unit (D) to the electron-withdrawing unit (A) through the π-conjugated bridges.60–62 To get a clear insight of the ICT of the push–pull D–π–A nature of compound 6, the change in absorption and emission spectra was investigated in different solvents by varying polarity. The good solubility of 6 in a broad range of solvents allowed us to investigate its photophysical properties in different solvents. A slightly positive solvatochromism was observed showing a red shift of the absorption maxima (λmax) from 360 nm to 371 nm (Table 1) with a change in solvent polarity from hexanes (ET(30) = 31.0) to DMSO (ET(30) = 45.1).63–65 In contrast, a remarkable bathochromic shift in the emission spectra of 6 was observed with increasing solvent polarity manifesting efficient positive solvatochromism (Fig. 1). The strong red-shift of fluorescence in different solvents suggests that arylene–vinylene conjugated terpyridine (6) forms different ICT/TICT states in different solvents which could be the reason for the fluorescence tuning.3,66–68 In low to medium polar solvents (such as hexanes, benzene, toluene, diethyl ether and 1,4-dioxane) the emission maximum was red shifted from 441 nm to 485 nm whereas with increasing solvent polarity from THF to DMSO, the emission maximum was markedly shifted to longer wavelength to 567 nm from 507 nm. Interestingly, for low to medium polar solvents from hexanes to chloroform (except THF) a relatively high energy emission band (shoulder) was also observed in the region of 397 nm to 425 nm. In contrast, this high energy emission band was gradually red shifted from 436 to 460 nm with increasing solvent polarity (from DCM to DMSO) with the emergence of two clearly shifted dual fluorescence emission bands in the region of 436–460 nm and 524–567 nm respectively (Table 1) suggesting the presence of two different emissive components in high polar solvents presumably induced by ICT or TICT as also observed in other specially designed dual-emissive fluorophores.69–72 It is well known in the literature that fluorophores containing D–π–A structure show solvent dependent changes in their absorption and emission spectra due to the interaction between the solvent and fluorophore molecules.4,6,7,10,65,73–76 The solvatochromic behaviour of 6 could be illustrated owing to its D–π–A arrangement having a functional triphenylamine group (donor), phenylene vinylene linkage (π-conjugate bridge) and a terpyridyl group (acceptor). The bathochromic shift in the emission spectra with increasing solvent polarity suggests that arylene–vinylene conjugated terpyridine (6) has enlarged dipole moment and charge transfer characteristics in excited states. The charge transfer plausibly occurs in the excited state from the electron donating triarylamine core to the electron accepting terpyridyl moiety through the π-conjugated core.4,65,73–75 Solvent polarity dependent dual emission in terpyridyl and similar conjugates due to the presence of closely lying excited states is reported in the literature.69–72 The low energy band is highly solvatochromic in nature, and is stabilized in polar medium due to charge separation, and has been attributed to a low-lying 1ICT state.69,72 The full width at half maximum (FWHM) becomes wider (55 nm to 210 nm) from low to high polar solvents (hexanes to DMSO) indicating the stabilization of the emissive center by the polar solvent molecules (Table 1). The fluorescence quantum yield (Φ) in most of the solvents was reasonably high demonstrating strong fluorescence with Φ in the range of 0.13 to 0.44 as calculated using quinine sulphate (0.1 M H2SO4; Φ = 0.54) as a reference.68 Inspired by the remarkable solvatochromic behavior of the arylene–vinylene conjugated terpyridine (6), the visual change in fluorescence colour was also investigated in the presence of different volatile organic compounds (VOCs). To demonstrate this application, filter paper (Whatman 42) test strips were prepared by dipping in the concentrated DCM solution of 6 and dried in air. The paper strips showed remarkable fluorescence color change (under 365 nm light) from cyan (in hexane vapour) to yellow (in methanol vapour) when exposed (for 5 min) to the different solvent vapours, establishing efficient vapochromic behaviour as shown in Fig. 2. It is noteworthy to mention here that the solution state PL spectra in methanol could not be recorded due to insolubility of 6 in methanol. The visual change in fluorescence color was monitored under the illumination of 365 nm light (Fig. 2). It is interesting to note that the change in the fluorescence colour of the test paper strips is nearly the same as evidenced in their corresponding pure solvent medium.| Solvent | E T (30)a | λ ab (nm) | ε max × 104 (M−1 cm−1) | λ em (nm) | Δλstc (cm−1) | FWHM (nm) | Φ F |
|---|---|---|---|---|---|---|---|
| a Polarity parameter. b Molar extinction coefficient. c Stokes shift. d Quantum yield was determined using quinine sulphate (0.1 M H2SO4; Φ = 0.54) as a reference. e Shoulder emission band. | |||||||
| Hexanes | 31.0 | 360 | 1.7 | 397,e 441, 466e | 5102 | 55 | 0.41 |
| Toluene | 33.9 | 361 | 4.2 | 410,e 463 | 6102 | 79 | 0.36 |
| Benzene | 34.3 | 362 | 4.1 | 411,e 466 | 6165 | 72 | 0.44 |
| Ether | 34.5 | 363 | 8.0 | 412,e 482 | 6801 | 87 | 0.29 |
| Dioxane | 36.0 | 364 | 9.8 | 413,e 485 | 6853 | 84 | 0.23 |
| THF | 37.4 | 365 | 3.9 | 430, 507 | 7673 | 109 | 0.34 |
| EtOAc | 38.1 | 365 | 5.6 | 425,e 506 | 7634 | 95 | 0.28 |
| CHCl3 | 39.1 | 366 | 3.2 | 415,e 507 | 7598 | 87 | 0.36 |
| CH2Cl2 | 40.7 | 367 | 3.7 | 436, 524 | 8163 | 103 | 0.32 |
| 1,2 DCE | 41.3 | 368 | 4.4 | 441, 530 | 8305 | 110 | 0.36 |
| Acetone | 42.2 | 369 | 3.7 | 445, 543 | 8684 | 130 | 0.19 |
| DMF | 43.2 | 369 | 7.8 | 456, 556 | 9114 | 197 | 0.17 |
| DMSO | 45.1 | 371 | 6.4 | 460, 567 | 9317 | 210 | 0.13 |
 | ||
| Fig. 1 Visual appearance of 6 in different solvents under 365 nm light. | ||
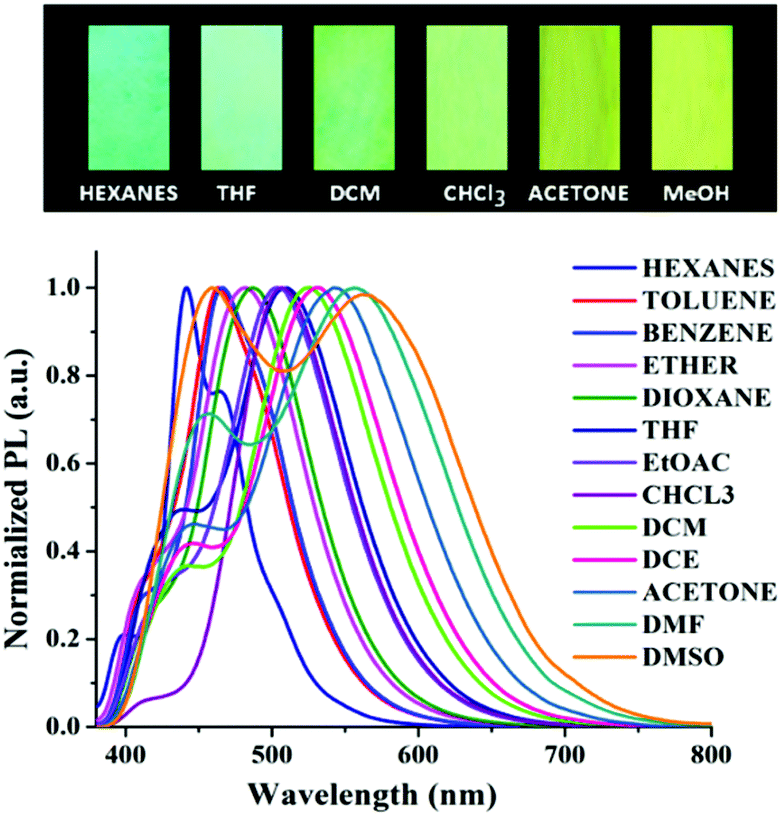 | ||
| Fig. 2 Emission spectra of 6 in various solvents of different polarity. (Top) Visual appearance of the test paper strips made of 6 upon exposure to different solvent vapours under the illumination of 365 nm light. | ||
Piezofluorochromism
The strong ICT emission of the arylene–vinylene conjugated terpyridine (6) with a propeller-like functionalized triarylamine terminus encouraged us to investigate the piezofluorochromic behavior. Compound 6 was found to be highly emissive even in its solid state, exhibiting yellow emission under illumination at 365 nm. Interestingly, the yellow emissive solid sample changed to green emissive after grinding with a pestle.1,2 To get more insight and to further confirm the piezofluorochromic behavior, the solid state fluorescence spectrum of 6 was recorded before and after grinding, revealing blue shift of the emission maxima to 512 nm from the emission maxima (λem = 556 nm) of the unground solid (Fig. 3).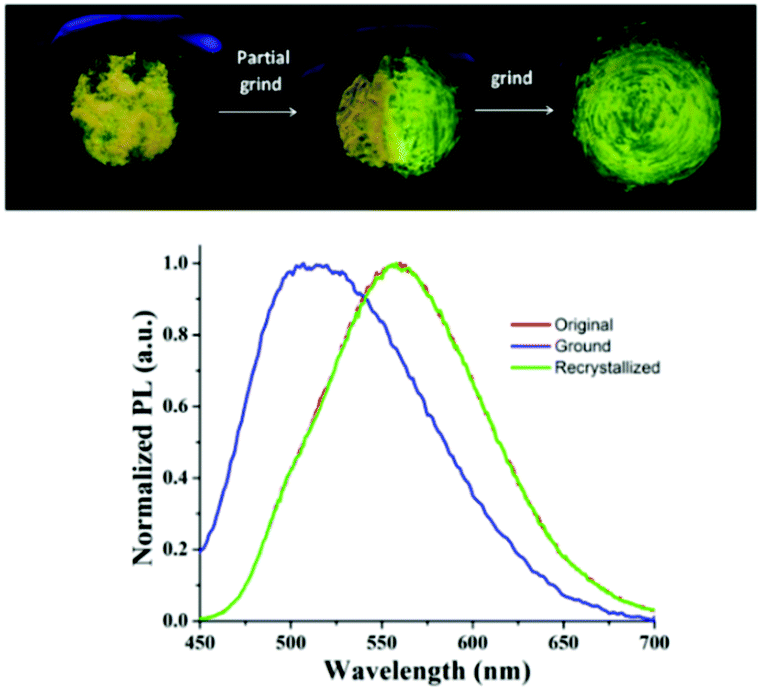 | ||
| Fig. 3 Solid state emission profile of 6 in the unground, ground and recrystallized form (top). Piezofluorochromic behavior of 6 under the illumination of 365 nm light (bottom). | ||
Moreover, recrystallization of the ground sample reproduced the initial yellow emissive state illustrating the reversible nature of the piezofluorochromism. The blue shift in the emission spectra of the ground sample suggests that different intermolecular π–π interaction modes presumably exist in these two states. The powder X-ray diffraction (PXRD) analysis was performed for 6 in its ground, unground and recrystallized forms to understand the origin of mechanochromism (Fig. S8, ESI†). The sharp diffraction peaks were noticed in its unground and recrystallized state manifesting the crystalline character of the arylene–vinylene conjugated terpyridine (6). In contrast, the sharp diffraction peaks disappeared and a diffused band was generated indicating the transition from the crystalline to amorphous state upon grinding compound 6. Unfortunately, harvesting of single crystals of 6 suitable for SCXRD studies was not successful. The PXRD analysis revealed the change in morphology from the crystalline state to the amorphous state illustrating the change in intermolecular aggregation mode inducing mechanochromism.1–4
Detection of nitroaromatics (NACs)
Nitroaromatic compounds are essential energetic materials mostly used as components for the preparation of landmines and explosive materials as well as in the dye industry, rocket fuel manufacturing, and the pharmaceutical industry.77,78 Furthermore, because of their high solubility in water, they can easily contaminate soil and groundwater through short-term or long-term exposure. As a result, they can cause hazardous effects on human health such as headache, respiratory disorders, weakness, anemia, skin irritation, liver injury and carcinogenicity.79–81 Nitroaromatic compounds are known to be highly electron deficient molecules and undergo strong π–π and supramolecular interactions with electron rich π-conjugated fluorescent probes resulting in change in emission properties. A considerable number of fluorescent probes based on polymers,82,83 gels,84–86 small molecule sensors,42,43,87 nanoparticles,88–90 nano-fibers91,92 and MOFs93–95 have been developed for the detection of NACs. Among them, novel, simple, economic and efficient sensing probes based on small molecules such as pyrene, coumarins, purpurin, fluorescein, triphenylamine, heterooligophenylene, pentacenequinone, fluoranthene and iptycene are of current interest for their well-defined structure and convenient synthetic method allowing a clear understanding of the structure–property relationship with superior performance.42,96 Prompted by the highly emissive and π-electron rich nature of the arylene–vinylene conjugated terpyridine (6), fluorescence chemosensing properties for the detection of nitroaromatic explosives (NACs) have been explored at ambient temperature. Compound 6 exhibited a low energy absorption band at 366 nm as a result of the π–π* transition of the conjugated molecular backbone and a relatively high energy absorption band at 280 nm due to the intra-ligand charge transfer (ICT) transition (Fig. S9, ESI†). The solid state absorption maxima of 6 (recorded on a thin film sample on a quartz plate) followed a similar trend as in solution state exhibiting absorption maxima at 286 nm and 380 nm (Fig. S10, ESI†) and a red shift (by 14 nm) with relatively broad absorption in comparison to solution ascribed to the presence of intermolecular π–π stacking interactions. The coexistence of different degrees of aggregation in the thin film is supported by the broad nature of the absorption spectra. The optical energy band gap of 6 was calculated to be 2.74 eV by inspecting the edge of the solid state absorption spectra, using the equation Eoptg (eV) = 1240/λcut off. Upon excitation at 366 nm (λex) compound 6 shows a strong green emission band centered at 507 nm (λem). The solution fluorescence quantum yield of 6 was found to be 0.36 when measured in CHCl3. The time-resolved fluorescence spectra of 6 in chloroform were also recorded to understand the nature of the excited states (Fig. S15, ESI†). The fluorescence lifetime of arylene–vinylene conjugated terpyridine (6) was found to be 2.27 ns as measured by the time-correlated single photon counting (TCSPC) method (Table S2, ESI†).As it is well known that planar polyaromatic compounds induce intermolecular aggregation in solution as a result of π–π interaction,97–100 we have recorded concentration dependent emission spectra to evaluate the optimum concentration in which self-aggregation of 6 is minimum. With decreasing concentration from 10−3 to 10−5 M, the intensity of the emission band increased with a blue shift in the emission maxima from 523 to 507 nm (λem) demonstrating the formation of self-assembled aggregates at higher concentration (∼10−3 M), whereas the aggregation was minimum forming unimers at relatively lower concentration (∼10−5 M) (Fig. S11, ESI†). When the solution was further diluted to 10−6 M, the fluorescence intensity decreased. Hence, the NAC sensing was explored in dilute solution (in the concentration range of ca. 10−5 M in CHCl3) not only to nullify the self-association behavior and loss of sensitivity but also to minimize the inner-filter effect. Moreover, the absorption spectra of PA do not overlap significantly with the emission spectra of the probe (Fig. S14, ESI†) implying the minimal secondary hetero-inner filter effect.101
The CHCl3 solution of compound 6 was treated with different nitroaromatics such as nitrobenzene (NB), nitrotoluene (NT), 4-hydroxy nitrobenzene (HNB), 4-nitrobenzoic acid (NBA), 2,6-dinitrotoluene (DNT), and picric acid (PA). Interestingly, the emission intensity of 6 was readily quenched upon addition of NACs as a result of facile electron transfer from the π-electron rich fluorophore to the electron deficient NACs. Moreover, the emission intensity of 6 was quenched substantially with different percentage after the addition of various nitroaromatics based on the electron deficient nature of the NACs. Amongst the various NACs tested, PA being the most electron deficient induced maximum fluorescence quenching demonstrating the efficient detection sensitivity of 6 towards explosive picric acid. To examine the selectivity towards NACs, compound 6 was treated with other non-nitro aromatic compounds such as chlorinated (such as 1,2-dichlorobenzene and 1,4-dichlorobenzene) and alkylated aromatic compounds (such as toluene and mesitylene) and no significant fluorescence quenching was observed (Fig. 4).
 | ||
| Fig. 4 (a) Percentage of emission quenching of 6 (1 × 10−5 M) towards different nitroaromatics and non-nitro aromatic compounds (1 × 10−3 M) in CHCl3. (b) Quenching efficiency of 6 in the presence of different NACs in CHCl3. Probe (6), toluene (TOL), dichlorobenzene (DCB), nitrobenzene (NB: N1), nitrotoluene (NT: N2), p-hydroxy nitrobenzene (HNB: N3), nitrobenzoic acid (NBA: N4), 2,6-dinitrobenzene (DNT: N5), and picric acid (PA: N6). | ||
To understand the sensing mechanism and to gain more profoundness of the physical process, fluorescence spectroscopic titrations were executed with a continuous variation of concentrations of NACs. The titration study showed that the emission steadily quenched without showing any residual emission band upon incremental addition of NACs (Fig. S13, ESI†). Amongst all the NACs, picric acid showed maximum PL quenching and after the addition of 32 equivalents of PA almost complete quenching was observed (Fig. 5). Prompted by the excellent ‘turn-off’ sensing behavior, solution mode visual detection of PA was performed which is important for real time application. Interestingly, upon addition of PA, the color of the solution of 6 immediately turned to intense yellow from light green, probably due to the formation of a ground state charge transfer complex with PA, whereas the intense green fluorescent solution became non-emissive under the illumination of 365 nm light demonstrating the easy colorimetric detection of PA by the naked eye (Fig. S12, ESI†).
 | ||
| Fig. 5 Fluorescence titration of 6 upon incremental addition of PA; (inset) visual appearance of 6 before and after the addition of PA under the illumination of 365 nm light. | ||
To gain insight into the rate of fluorescence quenching, the quenching behavior of 6 was examined by the Stern–Volmer (SV) equation expressed as I0/I = 1 + KSV·[Q], where I0 and I are the fluorescence intensities in the absence and presence of the analyte (NACs), [Q] is the analyte concentration and KSV is the Stern–Volmer rate constant.102–104 The plot of relative intensities (I0/I) against concentrations of NACs revealed the KSV value in the order of 104 M−1 for PA (Fig. 6). From the Stern–Volmer plot, the quenching response was found to be almost linear against incremental concentration of NACs. The Stern–Volmer constants of 6 for different NACs are summarized in Table S1 (ESI†). To evaluate the sensitivity of the probes, the limit of detection (LOD) of 6 towards PA was calculated from the fluorescence titration studies and was found to be 7.05 × 10−7 M (166 ppm) signifying its applicability even in the sub-micromolar range (Fig. 6). To gain more insight into the quenching mechanism, the fluorescence lifetime of 6 was measured before and after step-wise addition of PA. The fluorescence lifetime was found to be nearly invariant with different equivalents of PA suggesting that the fluorescence quenching follows mainly a static mechanism through ground-state charge transfer complex formation (Fig. S15, ESI†).
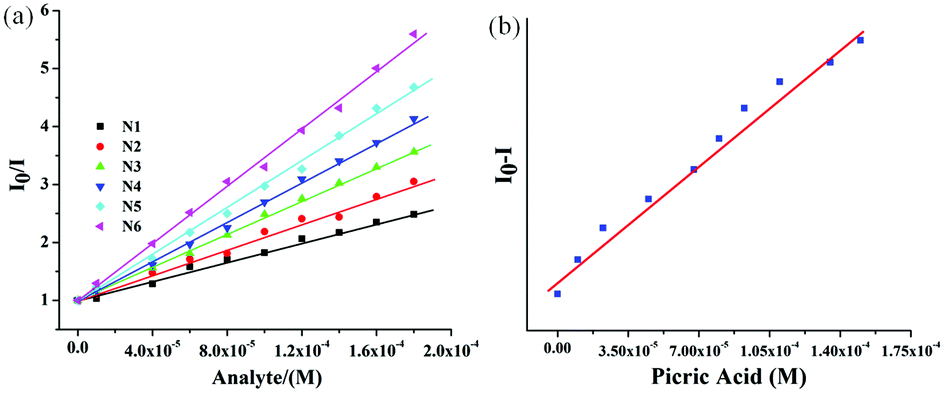 | ||
| Fig. 6 (a) The Stern–Volmer plot for 6 toward different nitroaromatics: N1: NB, N2: NT, N3: HNB, N4: NBA, N5: DNT, N6: PA. (b) Plot for the evaluation of LOD for 6 toward PA. | ||
In order to elucidate the sensing mechanism of the probe, it is important to understand the possible interaction between the probe molecule and PA. As this information could not be retrieved from the experimental data completely, detailed DFT and TD-DFT studies have been carried out. In probe 6, the electron density of HOMO is distributed over the whole molecule except the three pyridine rings of the terpyridine part, whereas the LUMO electron density is distributed over the middle phenyl rings and one pyridine ring of the terpyridine part. The HOMO electron densities of the complexes are exactly the same as that of HOMO of the probe. The LUMO density, unlike in the probe, is localized over the PA moiety (Fig. S29, ESI†). It is well known that the π–π stacking interaction105 between the aromatic rings of the probe and PA plays a crucial role in the formation of supramolecular interactions.106 From DFT calculations, three interaction modes were predicted, namely, 6-PA-1, 6-PA-2 and 6-PA-3, corresponding to the three interaction sites in the probe such as the middle phenyl group, the terpyridyl group and the arylene group, respectively, as presented in Fig. 7. In all of the three interaction modes, the PAs are parallel to the planes of the active site of the probe, confirming the π–π stacking interaction, and the corresponding distances are 3.31 Å, 3.26 Å and 3.29 Å, respectively. To identify the most stable adduct, the Gibbs free energy profile of all the three interaction modes was computed. The relative energy of each interaction mode was evaluated with respect to the energy sum of the probe (6) and PA as presented in Table S5 (ESI†). The relative energy values suggest that 6-PA-2 is the most favorable interaction mode involving π–π stacking with the terpyridyl moiety.
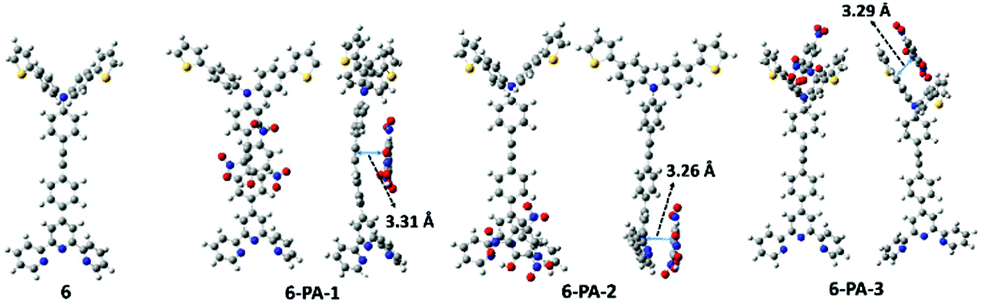 | ||
| Fig. 7 Optimized structures of the probe (6) and three possible interaction modes with PA. | ||
The electronic spectra of the probe and 6-PA-2 have been computed (Fig. S30, ESI†) and the important transition parameters are tabulated in Table S4 (ESI†). The mechanism of fluorescence quenching has been analyzed from the theoretical calculations and is presented in Fig. 8. The S0 → S1 transition in the probe is corresponding to electron excitation from the HOMO to LUMO. S1 being a bright state shows strong absorption and emission profiles as reported in Table S3 (ESI†). The principal absorption in the supramolecular complex, on the other hand, is contributed by the S0 → S4 transition, resulting from transition from the HOMO to the LUMO+3 frontier orbital. The excited electron in the S4 state relaxes back to the first-excited S1 state via an ultrafast internal conversion obeying Kasha's rule.107 The S1 state is a dark state and is accessed by the occupation of the LUMO orbital where the electron density is completely localized on the PA moiety. The transfer of electron density from the donor 6 to the acceptor PA can be attributed to the darkness of the state which results in quenching of fluorescence. Thus the fluorescence quenching in 6-PA-2 can be ascribed to the photo-induced electron transfer (PET) process from the donor 6 to acceptor PA.96 The possibility of excited state proton transfer from PA to the terpyridyl moieties can be ruled out as no residual emission band appeared after the addition of excess PA.
 | ||
| Fig. 8 Proposed electron transfer mechanism leading to PL quenching; HOMO and LUMO energy levels for 6 and PA were calculated by theoretical calculations. | ||
The static quenching mechanism as suggested by the Stern–Volmer plot and fluorescence lifetime measurements (vide supra) allowed us to further investigate the mode of supramolecular interaction and to identify experimentally the binding mode of PA between the π-conjugated arylene–vinylene conjugated terpyridine and PA by 1H NMR spectroscopic titration studies with incremental addition of PA as presented in Fig. 9. The 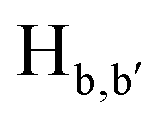 and
and  pyridyl protons and
pyridyl protons and 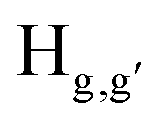 phenylene protons resonating at 7.93, 7.89 and 7.36 ppm for free 6 experienced downfield shift by 0.03, 0.01 and 0.08 ppm respectively in the 6-PA supramolecular complex. This downfield behavior is presumably due to the withdrawal of electron density from 6 by PA indicating the possibility of weak H-bonding interaction. In contrast, the signals centered at 8.79
phenylene protons resonating at 7.93, 7.89 and 7.36 ppm for free 6 experienced downfield shift by 0.03, 0.01 and 0.08 ppm respectively in the 6-PA supramolecular complex. This downfield behavior is presumably due to the withdrawal of electron density from 6 by PA indicating the possibility of weak H-bonding interaction. In contrast, the signals centered at 8.79 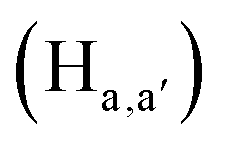 ppm, 8.75 ppm
ppm, 8.75 ppm 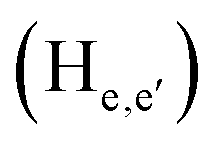 and 8.69
and 8.69 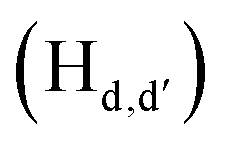 ppm for the terpyridyl protons experienced upfield shift to 8.77, 8.72 and 8.68 ppm respectively. Similarly, the arylene (in the NAr3 moiety) protons were shifted upfield to 7.78 ppm from 7.83 ppm upon addition of PA. This observation suggests π–π intermolecular interactions between the aromatic ring of PA and the terpyridyl core and as well as with the arylene unit.106,108,109
ppm for the terpyridyl protons experienced upfield shift to 8.77, 8.72 and 8.68 ppm respectively. Similarly, the arylene (in the NAr3 moiety) protons were shifted upfield to 7.78 ppm from 7.83 ppm upon addition of PA. This observation suggests π–π intermolecular interactions between the aromatic ring of PA and the terpyridyl core and as well as with the arylene unit.106,108,109
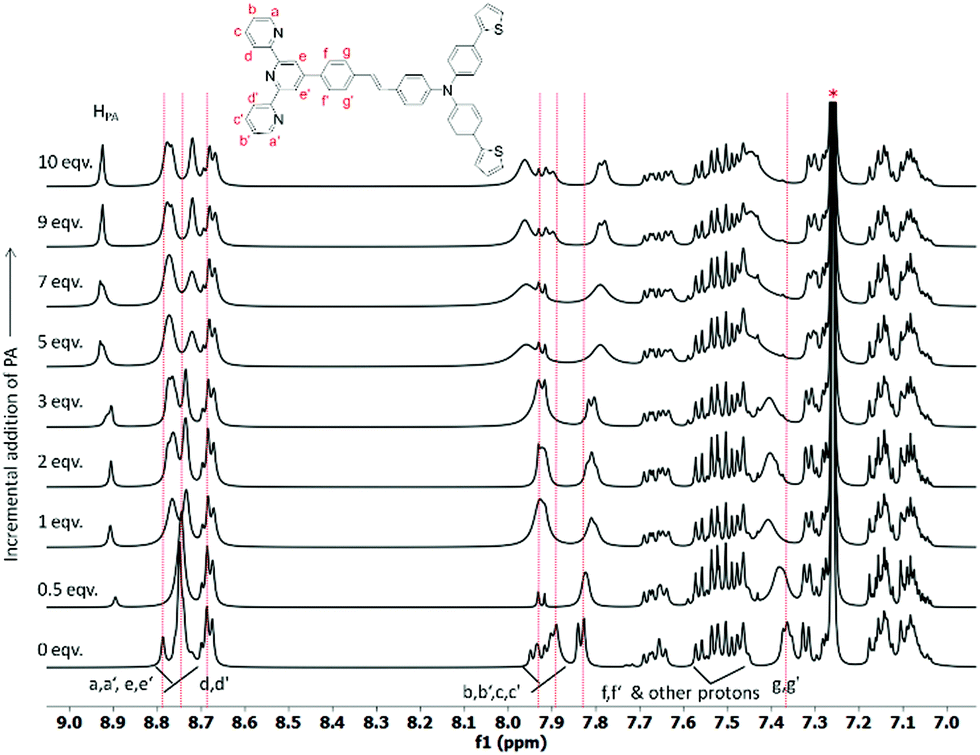 | ||
| Fig. 9 Change in the chemical shift of the protons in 6 during 1H NMR titration in CDCl3 on incremental addition of PA (* = residual CHCl3). | ||
The excellent ‘turn-off’ sensing behavior motivated us to explore the contact mode sensing as the on-site detection kit of picric acid. In order to manifest the potential utility, it was investigated by recording the fluorescence spectra of the thin-film of 6 after treating with PA vapors with respect to the unexposed film (SI). It was observed that the initial emission of 6 was quenched gradually with increasing exposure time and 77% quenching was observed after 540 s of exposure time and ultimately reached equilibrium (Fig. S17a, ESI†). To check the reusability of the thin films, the emission spectrum was recorded after exposing the thin film to the vapor of picric acid for 420 s, and consequently washing the film with Milli-Q water and drying in hot air. The whole process was repeated multiple times. Interestingly, the initial fluorescence intensity was significantly retained after repeated usage of the probe film even after five cycles of exposure, demonstrating the high reusability and photo-stability of the thin film (Fig. S17b, ESI†). It is well known that excessive usage of nitroaromatic compounds also pollutes soil and drinking water due to short-term or long-term exposure in the environment. Considering the fact and prompted by the reusability nature of the thin film, the trace detection ability was measured towards aqueous solution of picric acid. The change in emission was monitored by separately immersing the thin film in different concentrations of picric acid in aqueous medium. It was observed that the initial fluorescence intensity of 6 was quenched ca. 71% when the concentration of picric acid reached 100 μM (Fig. S18, ESI†), which was clearly visible to the naked eye.
To understand the change in the surface morphology of 6 in the presence and absence of PA, the scanning electron microscopic image analysis was conducted. Sample of 6, prepared by drop casting 5 × 10−4 M chloroform solution on an aluminum substrate, showed porous nanofiber-like surface morphology which may be advantageous to detect nitroaromatics as a thin film sensor. Interestingly, upon treatment with PA the nanofibers of 6 changed to irregular shape morphology with porous nature as investigated by FESEM analysis (Fig. S19, ESI†). Inspired by the porous surface nature of 6 and the efficient PA vapour detection by the thin film, the contact mode sensing approach was adopted to check the efficiency of 6 for the detection of trace amounts of PA. Considering the factors of portability and cost-efficiency, the filter paper test strips were prepared using Whatman-42 filter paper. To test the possibility, the chloroform solution of PA of varying concentration (10−3–10−9 M) was prepared and 10 μL of each solution was drop-cast on each fresh test paper strip. The visual fluorescence response at different concentration levels was monitored under the illumination of 365 nm light. Dark black spots were observed and it was noticed that the spots were prominent for the concentrated analyte (PA) solution and faded upon dilution showing that the minimum detection limit of picric acid by the naked eye is even up to the 10−9 M level (Fig. 10). Thus, these results establish that compound 6 is an excellent chemosensor for instant visual detection of trace amounts of picric acid in solution and as well as in solid state.
 | ||
| Fig. 10 Visual detection of PA of different concentrations by the paper test strips coated with probe 6 (under 365 nm light) demonstrating remarkable sensitivity toward trace PA. | ||
Toxic metal ion recognition
A good chelating or binding site attached to fluorophores is necessary to be an efficient and sensitive fluorescent sensor for the detection of toxic metal ions. The high emissive nature of the π-electron rich arylene–vinylene conjugate having terpyridyl binding sites further encouraged us to explore its multifunctional application as a fluorescence chemosensor for the selective detection of toxic metal ions. Development of highly selective and sensitive chemosensory materials for the detection of toxic metal ions such as Cd2+, Pb2+, Hg2+, Zn2+, Ni2+ and Cu2+ has attracted extensive scientific interest.30–40 Among the various toxic metal ions, mercury is one of the most prevalent toxic and hazardous pollutants causing serious fatal threat to human health by crossing through biological membranes, causing severe impairment to the brain, nervous system, kidneys, capillary vessels and alimentary system.45,110,111 Moreover, mercury is extensively used in batteries, paints and electric equipment, resulting in a high level of contamination of mercury across the atmosphere and surface water. Thus, Hg2+ can be accumulated in human bodies through food and drink and ultimately lead to several human diseases such as acrodynia (pink disease), Minamata diseases and Hunter-Russell syndrome.112 Inevitably, the development of highly efficient methods to monitor and detect mercury levels in the environment or in vivo biological media is highly desirable, and is our prime interest. It is noteworthy to mention that the United States Environmental Protection Agency (EPA) has fixed the upper limit of Hg2+ in drinking water at 10 nM (2 ppb).113The systematic metal ion recognition study was carried out qualitatively in semi-aqueous THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O medium. The absorption and emission spectra of 6 were recorded in the 1:1 THF:H2O solvent system at 5 × 10−6 M concentration at room temperature (300 K) (Fig. S21, ESI†). As observed in other organic solvents, 6 exhibited a low energy absorption maximum centered at 361 nm (ε = 5.6 × 104 M−1cm−1) with a second band centered at 282 nm in the high energy region. Compound 6 exhibited strong yellowish green emission with dual emission bands centered at 440 nm and 530 nm (λex = 361 nm) due to the presence of the extended π-conjugated backbone of the arylene–vinylene conjugated terpyridine. To investigate the metal ion recognition qualitatively, probe 6 was treated with various biologically and environmentally relevant metal ions including alkali metal ions (K+, Na+), alkaline-earth metal ions (Ca2+, Mg2+, Sr2+, Ba2+), and heavy/transition metal ions (such as Hg2+, Pb2+, Cd2+, Ni2+, Zn2+, Ag+, Co2+, Fe3+, and Cr3+) and the corresponding absorption and emission spectra were recorded in 1:1 THF:H2O. However, upon addition of various metal ions, no significant change in the absorption or emission spectra of 6 was observed. In contrast, upon addition of the Hg2+ metal ion, the intensity of the absorption band at 286 nm was considerably decreased and a new absorption band centered at 290 nm (ε = 2.32 × 104 M−1 cm−1) appeared concomitantly accompanied by a slight red shift of 4 nm indicating the metal induced intraligand charge transfer (Fig. S22a, ESI†). Moreover, the intensity of the absorption band centered at 361 nm (ε = 2.15 × 104 M−1 cm−1) disappeared with a prominent blue shift of 23 nm exhibiting a new absorption band at 338 nm (ε = 2.65 × 104 M−1 cm−1) with a shoulder at 423 nm (ε = 6.8 × 103 M−1 cm−1). More interestingly, upon treatment with the Hg2+ metal ion, the emission band at 530 nm completely disappeared whereas the emission band at 440 nm displayed no significant change (Table S3, ESI†). The pronounced disappearance of fluorescence response at λmax of 530 nm resulted in a decrease in PL quantum yield from 0.24 to 0.10 with a change in the emissive color from green to blue (Fig. S22b, ESI†). Other metal ions only caused very slight variations in emission intensity (at λmax = 440 nm and 530 nm) but disappearance of the low energy emission band at 530 nm was not observed in contrast to the Hg2+ metal ion, validating the efficient selectivity of probe 6 towards the Hg2+ ion as depicted in Fig. 11. The disappearance of the emission band centred at 530 occurs through chelation quenched fluorescence (CHQF) in the presence of Hg2+, presumably by the PET process as depicted in Fig. S21 (ESI†).114
:H2O medium. The absorption and emission spectra of 6 were recorded in the 1:1 THF:H2O solvent system at 5 × 10−6 M concentration at room temperature (300 K) (Fig. S21, ESI†). As observed in other organic solvents, 6 exhibited a low energy absorption maximum centered at 361 nm (ε = 5.6 × 104 M−1cm−1) with a second band centered at 282 nm in the high energy region. Compound 6 exhibited strong yellowish green emission with dual emission bands centered at 440 nm and 530 nm (λex = 361 nm) due to the presence of the extended π-conjugated backbone of the arylene–vinylene conjugated terpyridine. To investigate the metal ion recognition qualitatively, probe 6 was treated with various biologically and environmentally relevant metal ions including alkali metal ions (K+, Na+), alkaline-earth metal ions (Ca2+, Mg2+, Sr2+, Ba2+), and heavy/transition metal ions (such as Hg2+, Pb2+, Cd2+, Ni2+, Zn2+, Ag+, Co2+, Fe3+, and Cr3+) and the corresponding absorption and emission spectra were recorded in 1:1 THF:H2O. However, upon addition of various metal ions, no significant change in the absorption or emission spectra of 6 was observed. In contrast, upon addition of the Hg2+ metal ion, the intensity of the absorption band at 286 nm was considerably decreased and a new absorption band centered at 290 nm (ε = 2.32 × 104 M−1 cm−1) appeared concomitantly accompanied by a slight red shift of 4 nm indicating the metal induced intraligand charge transfer (Fig. S22a, ESI†). Moreover, the intensity of the absorption band centered at 361 nm (ε = 2.15 × 104 M−1 cm−1) disappeared with a prominent blue shift of 23 nm exhibiting a new absorption band at 338 nm (ε = 2.65 × 104 M−1 cm−1) with a shoulder at 423 nm (ε = 6.8 × 103 M−1 cm−1). More interestingly, upon treatment with the Hg2+ metal ion, the emission band at 530 nm completely disappeared whereas the emission band at 440 nm displayed no significant change (Table S3, ESI†). The pronounced disappearance of fluorescence response at λmax of 530 nm resulted in a decrease in PL quantum yield from 0.24 to 0.10 with a change in the emissive color from green to blue (Fig. S22b, ESI†). Other metal ions only caused very slight variations in emission intensity (at λmax = 440 nm and 530 nm) but disappearance of the low energy emission band at 530 nm was not observed in contrast to the Hg2+ metal ion, validating the efficient selectivity of probe 6 towards the Hg2+ ion as depicted in Fig. 11. The disappearance of the emission band centred at 530 occurs through chelation quenched fluorescence (CHQF) in the presence of Hg2+, presumably by the PET process as depicted in Fig. S21 (ESI†).114
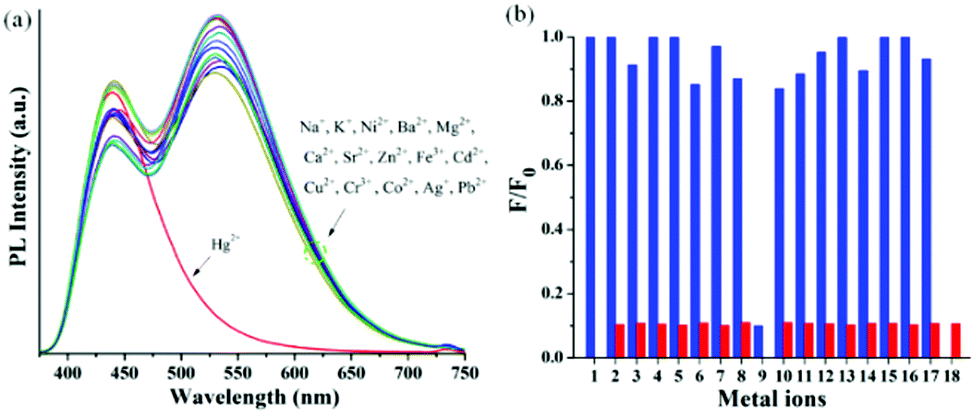 | ||
| Fig. 11 (a) Change in the PL response of 6 after the addition of various metal ions (1 equiv.); (b) selectivity profile; blue bars: free sensor or treated with metal ions. Red bars: Treated with competing metal ions followed by 1 equiv. of Hg2+; 1–18 represent (1) 6, (2) K+, (3) Ni2+, (4) Ag+, (5) Ba2+, (6) Zn2+, (7) Sr2+, (8) Fe3+, (9) Hg2+, (10) Cu2+, (11) Cd2+, (12) Cr3+, (13) Ca2+, (14) Co2+, (15) Na+, (16) Mg2+, (17) Pb2+, and (18) all. | ||
To be an efficient probe for the detection of Hg2+, selectivity is one of the important aspects and hence, the selectivity profile of 6 in response to other biologically and environmentally relevant metal ions was also investigated by the naked eye. Unlike other metal ions, the Hg2+ ion immediate turned the yellowish green emissive probe into a blue emissive solution, which can be easily visualized by the naked eye under the illumination of 365 nm light, indicating compound 6 as an efficient colorimetric probe for the detection of Hg2+ (Fig. S23, ESI†). However, the most important criterion of a selective probe is the ability to sense a specific metal ion in the vicinity of other ions. In the competitive binding experiment, no obvious interference was observed in the presence of other metal ions and even in more complex mixtures (Fig. 11).
Furthermore, spectroscopic titration study was executed with the continuous variation of concentration of the metal ion to gain an insight into the sensing mechanism and to find the binding stoichiometry of 6 with the Hg2+ ion. The emission spectra showed a gradual decrease in the intensity of the emission band centered at 530 nm whereas negligible change in intensity was observed for the emission band centered at 440 nm upon incremental addition of the Hg2+ metal ion (Fig. 12). On the other hand, upon addition of incremental equivalents of the Hg2+ ion, the absorption band at 361 nm shifted to 338 nm with a significant blue shift by 23 nm whereas the absorption band at 286 nm underwent a slight red shift by 4 nm, along with a new shoulder band at 423 nm (Fig. S24, ESI†). The existence of a single equilibrium between the probe and the in situ generated Hg2+-coordinated probe molecule was established by the formation of the isosbestic point at 300 nm and 417 nm respectively. Interestingly, the change of the absorption and emission spectra became saturated when one equivalent of Hg2+ ions was added indicating the presence of 1:1 metal:ligand binding stoichiometry. Furthermore, Job's plot constructed from emission titration studies at λmax of 530 nm also indicated 1:1 Hg2+–probe stoichiometry as depicted in Fig. 13a. The formation of the 1:1 6-Hg2+ complex was further confirmed from MALDI-TOF analysis showing the molecular ion peak ([M-Cl]+) at 979.324. The experimental and simulated isotopic distribution pattern for the molecular ion peak is in perfect agreement as illustrated in Fig. 13b.
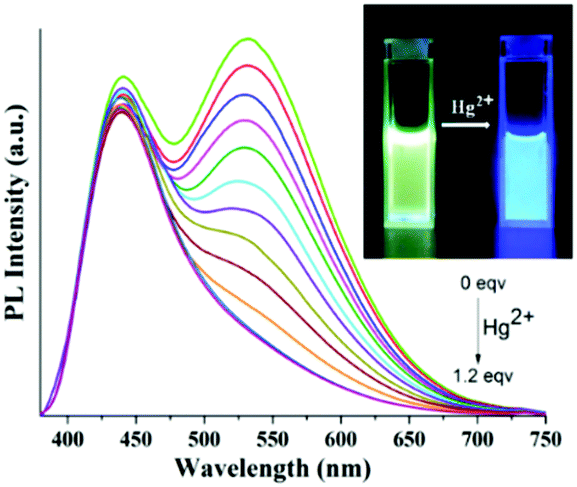 | ||
| Fig. 12 PL response of 6 in 1:1 THF:H2O upon incremental addition of Hg2+ ions (0–1.2 equiv); inset: visual color change of 6 upon addition of Hg2+ metal ions under 365 nm light. | ||
 | ||
| Fig. 13 (a) Job's plot of 6-Hg2+ complexes in 1:1 THF:H2O medium (where the mol fraction of Hg2+ = [Hg2+]/[6] + [Hg2+]). (b) Simulated and experimental isotopic distribution pattern for the molecular ion peak of the 6-Hg2+ complex obtained from MALDI-TOF mass spectrometric analysis. | ||
To understand the nature of the fluorescence quenching process, the emission response of 6 towards the Hg2+ metal ion was investigated by constructing the Stern–Volmer plot (vide supra) from the fluorescence titration experiment (Fig. S26, ESI†). The association constant of Hg2+ against 6 was found to be 4.1(±0.2) × 105 M−1 as estimated from the Stern–Volmer plot. The lowest detectable concentration level of Hg2+ was measured using linear response to evaluate the sensitivity of 6 towards the Hg2+ metal ion. The limit of detection (LOD) was found to be 1.54 × 10−7 M (31 ppb) suggesting its remarkable sensitivity in sub-micromolar level of detection towards the Hg2+ metal ion (Fig. S27, ESI†).
To gain an insight on coordination of the terpyridyl conjugate to the Hg2+ ion, 1H NMR spectroscopic titration studies were conducted with incremental addition of the Hg2+ metal ion to a solution of probe 6 in DMSO-d6. Upon addition of Hg2+ ions, the signals for pyridyl protons (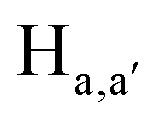 ,
,  ,
,  ,
, 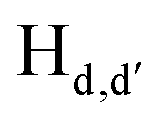 and
and 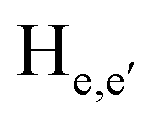 ) and 4′-phynelene ring protons (Hf,f′) were broadened and shifted to the downfield region due to the coordination of the Hg2+ metal ion with the terpyridyl chelating receptor site of probe 6 as shown in Fig. 14. No significant shift was observed in the 1H NMR spectrum after the addition of more than one equivalent of Hg2+ ions, further confirming 1:1 binding stoichiometry between 6 and Hg2+.
) and 4′-phynelene ring protons (Hf,f′) were broadened and shifted to the downfield region due to the coordination of the Hg2+ metal ion with the terpyridyl chelating receptor site of probe 6 as shown in Fig. 14. No significant shift was observed in the 1H NMR spectrum after the addition of more than one equivalent of Hg2+ ions, further confirming 1:1 binding stoichiometry between 6 and Hg2+.
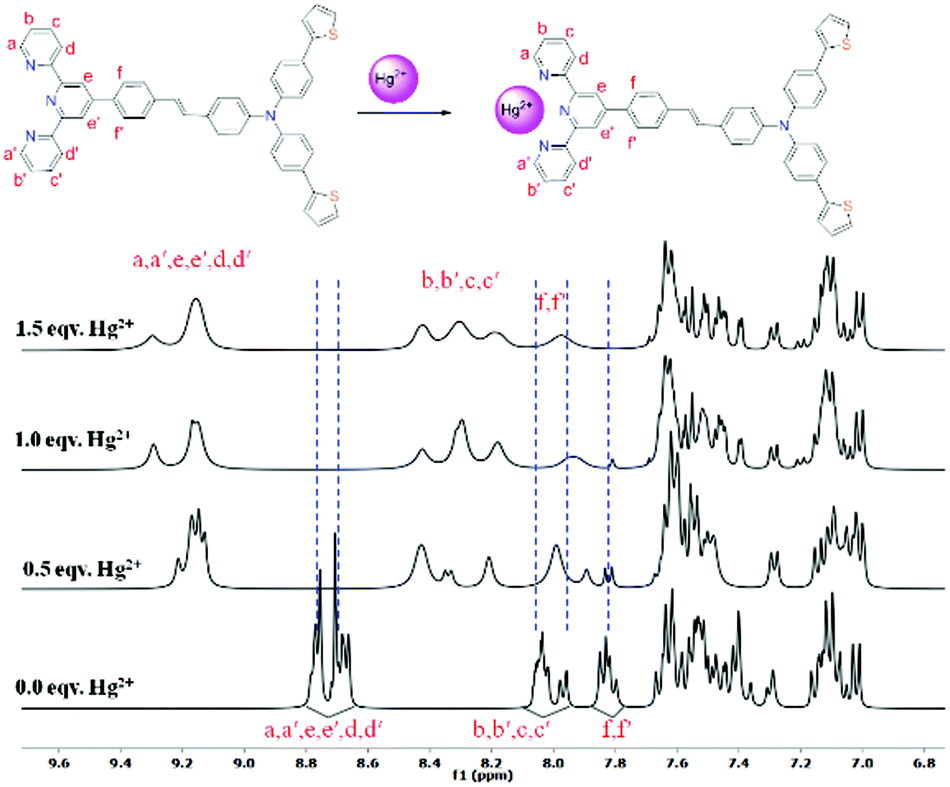 | ||
| Fig. 14 Change in the chemical shift of the protons in 6 after successive addition of Hg2+ in DMSO-d6 (* = H2O). | ||
To check whether the sensing process is reversible or not, S2− was added into the solution of 6 pre-incubated with the Hg2+ metal ion, and the changes in the emission spectra were monitored. After the addition of S2− into the solution of 6 pre-incubated with the Hg2+ metal ion, the emission band at 530 nm reappeared as observed for the free probe. This fact was also supported by visual naked eye observation under the illumination of 365 nm light. The blue fluorescence immediately turned to yellowish green due to decomplexation of the Hg2+ ion and formation of stable HgS, leading to regain of the free probe as shown in Fig. 15. To illustrate the sensitivity of S2− towards 6-Hg2+, the corresponding fluorescence titration study was carried out with incremental addition of the S2− aliquot and the concomitant appearance of the emission band at 530 nm suggests 6 to be a reversible sensor for the Hg2+ ion (Fig. S28, ESI†). The excellent sensing ability of the sensing probe towards the Hg2+ ion encouraged us to check its practical applicability as an on-site detection kit. Test kits were prepared by immersing filter papers in the DCM solution of 6 (1 mM) and then drying thoroughly in air. The test-kits coated with 6 were dipped in aqueous Hg2+ solution of various concentrations (1 μM to 1 mM). When illuminated under a 365 nm UV lamp, an obvious color change from bright green to blue fluorescence was visualized with the increasing concentration of the Hg2+ ion (1 μM to 1 mM) as shown in Fig. 16. This distinct color change can easily be detected by the naked eye and therefore, the sensing probe can be successfully applied as a test paper-kit to detect the toxic Hg2+ ion.
 | ||
| Fig. 15 (a) PL reversibility study of 6 with Hg2+ ions and S2− ions; (b) visual fluorescence colour changes of 6 (2 × 10−4 M) after successive addition of Hg2+ ions and S2− ions (under 365 nm light). | ||
 | ||
| Fig. 16 Color change (under 365 nm light) of test paper strips made of 6 after treating with different samples of Hg2+ of varying concentration (1–1000 μM). | ||
Conclusions
In conclusion, we have successfully synthesized a multifunctional D–π–A type push–pull arylene–vinylene terpyridyl conjugate with a functionalized triarylamine moiety with stimuli responsive photophysical properties such as solvatochromism, vapochromism, piezofluorochromism and chemosensing behavior towards environmentally toxic Hg2+ ions and explosive NACs. The push–pull chromophore showed outstanding solvatochromic behavior with tunable emission in the range of λem 397 nm (for hexanes) to 567 nm (for DMSO) respectively induced by ICT and/or TICT. Moreover, the arylene–vinylene conjugated terpyridine (test paper strips) exhibited remarkable vapochromism under 365 nm light showing cyan (in hexane vapour) to yellow (in methanol vapour) fluorescent colour when exposed to different VOCs. More interestingly, the push–pull congener showed different solid state emission with high color contrast between yellow and green in its pristine and ground form manifesting reversible mechanochromic behavior due to the phase transformation between the crystalline and amorphous states. Furthermore, the highly emissive arylene–vinylene conjugated terpyridine exhibited efficient chemosensing properties for the detection of nitroaromatics in both solution and solid state. The favorable supramolecular complexation as evidenced by TCSPC measurement, 1H NMR titration and DFT studies facilitates the ‘turn off’ fluorescence sensing through static quenching induced by PET from the photo-excited π-electron rich probe to the electron-deficient nitroaromatics having favorable HOMO–LUMO energy levels. The arylene–vinylene conjugated terpyridine offers excellent sensitivity toward picric acid (PA), exhibiting contact mode trace detection of PA at a nanomolar level and thus can be utilized as a reversible, reusable solid state kit for the onsite detection of NACs. Furthermore, the arylene–vinylene conjugated terpyridyl probe showed an outstanding fluorescence response for the recognition of environmentally toxic Hg2+ with high selectivity and sensitivity. The reusability and cost-effectiveness of the probe were further demonstrated successfully by multiple cycles of alternative addition of Hg2+ ions followed by S2− ions. The sensing ability of the probe towards Hg2+ ions as filter paper strips revealed its practical utility as an on-site detection kit for Hg2+ in a sample. This work clearly brings out a novel strategy to access tunable multifunctional properties from a single but simple, easily processable arylene–vinylene conjugated terpyridine. For further improvement of the multifunctional behavior, sensitivity and efficacy, efforts to structurally alter the arylene–vinylene conjugates by fine-tuning of electronic properties are underway in our laboratory.Experimental section
Synthesis and characterization
Compounds 1–3 and 5 were synthesized following the literature procedure (SI).51,58:hexanes, 5:95) on silica gel (60–120 mesh) to achieve analytically pure lemon yellow solid product 4; yield: 0.204 g (68%). 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H, -CHO), 7.72 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 7.9 Hz, 4H), 7.27 (d, J = 10.7 Hz, 4H), 7.22–7.13 (m, 4H), 7.09 (t, J = 9.3 Hz, 4H); 13C{1H} NMR (101 MHz, CDCl3) δ 190.7 (-CHO), 153.0, 145.5, 143.8, 133.1, 131.6, 130.0, 128.4, 127.4, 126.5, 125.1, 123.3, 120.6, 118.2; HRMS (ESI+): C27H19NOS2, calculated value 438.0986 ([M + H]+); experimental 438.0942 ([M + H]+); FTIR (KBr, cm−1): 1718 (![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) CO str), 1578 (CC str).
: hexanes, 5:95) on silica gel (60–120 mesh) to achieve analytically pure bright yellow solid product 6; yield: 0.44 g (52%). 1H NMR (600 MHz, CDCl3) δ 8.84–8.72 (m, 4H, py), 8.69 (t, J = 7.7 Hz, 2H, py), 7.99–7.87 (m, 4H, py), 7.83 (d, J = 7.6 Hz, 2H), 7.66 (dd, J = 19.4, 10.2 Hz, 2H), 7.59–7.52 (m, 2H), 7.52–7.45 (m, 4H), 7.43–7.34 (m, 2H), 7.32 (d, J = 7.7 Hz, 2H), 7.30–7.26 (m, 2H), 7.22–7.12 (m, 4H), 7.11–7.06 (m, 4H); 13C{1H} NMR (150 MHz, CDCl3) δ 156.5, 156.0, 150.4, 149.3, 146.9, 144.3, 143.9, 139.3, 137.1, 135.7, 132.4, 132.3, 132.1, 129.9, 129.6, 128.9, 128.2, 127.4, 127.1, 126.1, 124.9, 124.5, 124.0, 123.1, 122.7, 121.7, 120.9, 118.9; HRMS (ESI+): C49H34N4S2, calculated value 743.2303 ([M + H]+); experimental 743.2349 ([M + H]+); Anal. Calcd for C49H34N4S2: C, 79.21; H 4.61; N, 7.54. Found: C, 77.66; H 4.40; N, 8.06. mp 108 °C. FTIR (KBr, cm−1): 2928 (C–H str), 1590 (CC str), 1498 (CN str); λmax(ε) in CHCl3: 366 nm (ε = 3.2 × 104 M−1 cm−1), 286 nm (ε = 6.5 × 104 M−1 cm−1); λem (CHCl3): 507 nm (λex: 366 nm).
CO str), 1578 (CC str).
: hexanes, 5:95) on silica gel (60–120 mesh) to achieve analytically pure bright yellow solid product 6; yield: 0.44 g (52%). 1H NMR (600 MHz, CDCl3) δ 8.84–8.72 (m, 4H, py), 8.69 (t, J = 7.7 Hz, 2H, py), 7.99–7.87 (m, 4H, py), 7.83 (d, J = 7.6 Hz, 2H), 7.66 (dd, J = 19.4, 10.2 Hz, 2H), 7.59–7.52 (m, 2H), 7.52–7.45 (m, 4H), 7.43–7.34 (m, 2H), 7.32 (d, J = 7.7 Hz, 2H), 7.30–7.26 (m, 2H), 7.22–7.12 (m, 4H), 7.11–7.06 (m, 4H); 13C{1H} NMR (150 MHz, CDCl3) δ 156.5, 156.0, 150.4, 149.3, 146.9, 144.3, 143.9, 139.3, 137.1, 135.7, 132.4, 132.3, 132.1, 129.9, 129.6, 128.9, 128.2, 127.4, 127.1, 126.1, 124.9, 124.5, 124.0, 123.1, 122.7, 121.7, 120.9, 118.9; HRMS (ESI+): C49H34N4S2, calculated value 743.2303 ([M + H]+); experimental 743.2349 ([M + H]+); Anal. Calcd for C49H34N4S2: C, 79.21; H 4.61; N, 7.54. Found: C, 77.66; H 4.40; N, 8.06. mp 108 °C. FTIR (KBr, cm−1): 2928 (C–H str), 1590 (CC str), 1498 (CN str); λmax(ε) in CHCl3: 366 nm (ε = 3.2 × 104 M−1 cm−1), 286 nm (ε = 6.5 × 104 M−1 cm−1); λem (CHCl3): 507 nm (λex: 366 nm).
Author contributions
A. S.: Conceptualization, investigation, methodology, formal analysis, visualization, writing – original draft; U. G., A. M., S. D.: investigation and methodology; S. M.: theoretical investigation; SKP: conceptualization, supervision, resources, validation, funding acquisition, writing – original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. and S. D. acknowledge IIT KGP, UG acknowledge CSIR and A. M. thanks UGC for doctoral fellowship. SKP acknowledges the support from CSIR, India (01(2986)/19/EMRII), and DST, Govt. of India (SR/FST/CSII-026/2013). Central Research Facility, IIT Kharagpur is gratefully acknowledged for the analytical facility. Paramshakti Supercomputer of IIT Kharagpur (supported by CDAC Pune under the National Supercomputing Mission of the Govt. of India) is gratefully acknowledged for the computational facility. The authors thank Prof. Nilmoni Sarkar's group (IIT KGP) for TCSPC measurement facilities. Prof. Alakesh Bisai's group (IISER Kolkata, India) is acknowledged for elemental and HPLC analyses. The authors are thankful to the reviewers for their constructive suggestions and comments to improve the manuscript.References
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC.
- P. S. Hariharan, N. S. Venkataramanan, D. Moon and S. P. Anthony, J. Phys. Chem. C, 2015, 119, 9460–9469 CrossRef.
- C. Y.-K. Chan, J. W.-Y. Lam, Z. Zhao, S. Chen, P. Lu, H. H.-Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4320–4327 RSC.
- R. I. Stock, L. G. Nandi, C. R. Nicoleti, A. D.-S. Schramm, S. L. Meller, R. S. Heying, D. F. Coimbra, K. F. Andriani, G. F. Caramori, A. J. Bortoluzzi and V. G. Machado, J. Org. Chem., 2015, 80, 7971–7983 CrossRef PubMed.
- K. Hofmann and S. Spange, J. Org. Chem., 2012, 77, 5049–5055 CrossRef PubMed.
- E. Benedetti, L. S. Kocsis and K. M. Brummond, J. Am. Chem. Soc., 2012, 134, 12418–12421 CrossRef PubMed.
- E. J. Rivera, C. Barbosa, R. Torres, L. Grove, S. Taylor, W. B. Connick, A. Clearfield and J. L. Colón, J. Mater. Chem., 2011, 21, 15899–15902 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef.
- S. G. Esteban, P. De La Cruz, A. Aljarilla, L. M. Arellano and F. Langa, Org. Lett., 2011, 13, 5362–5365 CrossRef.
- B. Xu, Z. Chi, X. Zhang, H. Li, C. Chen, S. Liu, Y. Zhang and J. Xu, Chem. Commun., 2011, 47, 11080–11082 RSC.
- A. J. Zucchero, P. L. McGrier and U. H.-F. Bunz, Acc. Chem. Res., 2010, 43, 397–408 CrossRef PubMed.
- E. V. Verbitskiy, G. L. Rusinov, O. N. Chupakhin and V. N. Charushin, Dyes Pigm., 2020, 180, 108414 CrossRef.
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef.
- S. M. Kelly and S. M. Kelly, Flat panel displays: advanced organic materials, Royal Society of Chemistry, 2000, vol. 2 Search PubMed.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D.-C. Bradley, D. A. Dos Santos, J.-L. Bredas and M. Lögdlund, Nature, 1999, 397, 121–128 CrossRef.
- Y. Shirota, J. Mater. Chem., 2005, 15, 75–93 RSC.
- K. R. Justin Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Chem. Mater., 2004, 16, 5437–5444 CrossRef.
- F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421–3434 CrossRef.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef PubMed.
- P.-T. Wu, F. S. Kim and S. A. Jenekhe, Chem. Mater., 2011, 23, 4618–4624 CrossRef.
- F. S. Kim, X. Guo, M. D. Watson and S. A. Jenekhe, Adv. Mater., 2010, 22, 478–482 CrossRef.
- Y. Song, C. Di, X. Yang, S. Li, W. Xu, Y. Liu, L. Yang, Z. Shuai, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2006, 128, 15940–15941 CrossRef.
- I. D.-W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef PubMed.
- M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef.
- G. Kranzelbinder and G. Leising, Rep. Prog. Phys., 2000, 63, 729 CrossRef CAS.
- N. Tessler, Adv. Mater., 1999, 11, 363–370 CrossRef CAS.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- L. He, B. Dong, Y. Liu and W. Lin, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC.
- J. A. Cotruvo Jr, A. T. Aron, K. M. Ramos-Torres and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC.
- H. Zhu, J. Fan, B. Wang and X. Peng, Chem. Soc. Rev., 2015, 44, 4337–4366 RSC.
- X. Qian and Z. Xu, Chem. Soc. Rev., 2015, 44, 4487–4493 RSC.
- R. D. Hancock, Chem. Soc. Rev., 2013, 42, 1500–1524 RSC.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed.
- E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed.
- S. Vaidya, C. Johnson, X.-Y. Wang and R. H. Schmehl, J. Photochem. Photobiol., A, 2007, 187, 258–262 CrossRef CAS.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- L. Mosca, S. Karimi Behzad and P. Anzenbacher, J. Am. Chem. Soc., 2015, 137, 7967–7969 CrossRef CAS PubMed.
- X. Y. Wang, A. Del Guerzo and R. H. Schmehl, Chem. Commun., 2002, 2344–2345 RSC.
- S. K. Chung, Y. R. Tseng, C. Y. Chen and S. S. Sun, Inorg. Chem., 2011, 50, 2711–2713 CrossRef CAS PubMed.
- P. F. Shi, Q. Jiang, H. C. Duan and D. Q. Wang, Chin. Chem. Lett., 2014, 25, 586–588 CrossRef CAS.
- L. S. Natrajan, A. Toulmin, A. Chew and S. W. Magennis, Dalton Trans., 2010, 39, 10837–10846 RSC.
- S. Righetto, S. Rondena, D. Locatelli, D. Roberto, F. Tessore, R. Ugo, S. Quici, S. Roma, D. Korystov and V. I. Srdanov, J. Mater. Chem., 2006, 16, 1439–1444 RSC.
- A. Sil, A. Maity, D. Giri and S. K. Patra, Sens. Actuators, B, 2016, 226, 403–411 CrossRef CAS.
- W. Jun, X. U. Quan-qing, C. H.-I. Shao-ming, Z. Hui-miao and F. U. Wen-fu, IMAGING Sci. Photochem., 2009, 27, 8 Search PubMed.
- A. Sil, D. Giri and S. K. Patra, J. Mater. Chem. C, 2017, 5, 11100–11110 RSC.
- P. Song, S. G. Sun, S. Wang, F. C. Ma, Y. Q. Xu and X. J. Peng, Spectrochim. Acta, Part A, 2011, 81, 283–289 CAS.
- S. Vaidya, C. Johnson, X. Y. Wang and R. H. Schmehl, J. Photochem. Photobiol., A, 2007, 187, 258–262 CrossRef CAS.
- Z. J. Hu, J. X. Yang, Y. P. Tian, H. P. Zhou, X. T. Tao, G. B. Xu, W. T. Yu, X. Q. Yu and M. H. Jiang, J. Mol. Struct., 2007, 839, 50–57 CrossRef CAS.
- A. Chowdhury and P. S. Mukherjee, J. Org. Chem., 2015, 80, 4064–4075 CrossRef CAS PubMed.
- E. Matteucci, A. Baschieri, L. Sambri, F. Monti, E. Pavoni, E. Bandini and N. Armaroli, ChemPlusChem, 2019, 84, 1353–1365 CrossRef CAS PubMed.
- Y. Zhang, J. Sun, G. Zhuang, M. Ouyang, Z. Yu, F. Cao, G. Pan, P. Tang, C. Zhang and Y. Ma, J. Mater. Chem. C, 2014, 2, 195–200 RSC.
- J.-F. Lefebvre, X.-Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50, 5258–5260 RSC.
- F. Kroehnke, Synthesis, 1976, 1–24 CrossRef CAS.
- Z. Q. Liang, X. M. Wang, G. L. Dai, C. Q. Ye, Y. Y. Zhou and X. T. Tao, New J. Chem., 2015, 39, 8874–8880 RSC.
- P. Das, A. Kumar, A. Chowdhury and P. S. Mukherjee, ACS Omega, 2018, 3, 13757–13771 CrossRef CAS PubMed.
- P. S. Hariharan, D. Moon and S. P. Anthony, J. Mater. Chem. C, 2015, 3, 8381–8388 RSC.
- O. A. Kucherak, L. Richert, Y. Mély and A. S. Klymchenko, Phys. Chem. Chem. Phys., 2012, 14, 2292–2300 RSC.
- R. S. Moog, D. D. Kim, J. J. Oberle and S. G. Ostrowski, J. Phys. Chem. A, 2004, 108, 9294–9301 CrossRef CAS.
- Y. Yang, B. Li and L. Zhang, Sens. Actuators, B, 2013, 183, 46–51 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- C. Cao, X. Liu, Q. Qiao, M. Zhao, W. Yin, D. Mao, H. Zhang and Z. Xu, Chem. Commun., 2014, 50, 15811–15814 RSC.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, 3rd Edition, 2006 Search PubMed.
- T. Soujanya, A. Philippen, S. Leroy, M. Vallier and F. Fages, J. Phys. Chem. A, 2000, 104, 9408–9414 CrossRef CAS.
- W. Schuddeboom, S. A. Jonker, J. M. Warman, U. Leinhos, W. Kuehnle and K. A. Zachariasse, J. Phys. Chem., 1992, 96, 10809–10819 CrossRef CAS.
- G. Brancato, G. Signore, P. Neyroz, D. Polli, G. Cerullo, G. Abbandonato, L. Nucara, V. Barone, F. Beltram and R. Bizzarri, J. Phys. Chem. B, 2015, 119, 6144–6154 CrossRef CAS PubMed.
- A. D’Aléo, E. Cecchetto, L. De Cola and R. M. Williams, Sensors, 2009, 9, 3604–3626 CrossRef PubMed.
- Z. Q. Liang, X. M. Wang, G. L. Dai, C. Q. Ye, Y. Y. Zhou and X. T. Tao, New J. Chem., 2015, 39, 8874–8880 RSC.
- A. Jiménez-Sánchez, M. Rodríguez, R. Métivier, G. Ramos-Ortíz, J. L. Maldonado, N. Réboles, N. Farfán, K. Nakatani and R. Santillan, New J. Chem., 2014, 38, 730–738 RSC.
- B. Wang and Y. Qian, New J. Chem., 2013, 37, 1402–1407 RSC.
- A. Bolduc, Y. Dong, A. Guérin and W. G. Skene, Phys. Chem. Chem. Phys., 2012, 14, 6946–6956 RSC.
- J. Shen, J. Zhang, Y. Zuo, L. Wang, X. Sun, J. Li, W. Han and R. He, J. Hazard. Mater., 2009, 163, 1199–1206 CrossRef CAS PubMed.
- S. Letzel, T. Göen, M. Bader, J. Angerer and T. Kraus, Occup. Environ. Med., 2003, 60, 483–488 CrossRef CAS PubMed.
- Y. Salinas, R. M. Mañez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC.
- K. L. Diehl and E. V. Anslyn, Chem. Soc. Rev., 2013, 42, 8596–8611 RSC.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- T. Wang, N. Zhang, W. Bai and Y. Bao, Polym. Chem., 2020, 11, 3095–3114 RSC.
- V. Bhalla, A. Gupta, M. Kumar, D. S.-S. Rao and S. K. Prasad, ACS Appl. Mater. Interfaces, 2013, 5, 672–679 CrossRef CAS PubMed.
- K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834–4841 CrossRef CAS PubMed.
- S. Barman, J. A. Garg, O. Blacque, K. Venkatesan and H. Berke, Chem. Commun., 2012, 48, 11127–11129 RSC.
- Y. Chandrasekaran, N. Venkatramaiah and S. Patil, Chem. – Eur. J., 2016, 22, 5288–5294 CrossRef CAS PubMed.
- S. S.-R. Dasary, A. K. Singh, D. Senapati, H. Yu and P. C. Ray, J. Am. Chem. Soc., 2009, 131, 13806–13812 CrossRef CAS PubMed.
- Y. Jiang, H. Zhao, N. Zhu, Y. Lin, P. Yu and L. Mao, Angew. Chem., Int. Ed., 2008, 47, 8601–8604 CrossRef CAS PubMed.
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- Y. Yang, H. Wang, K. Su, Y. Long, Z. Peng, N. Li and F. Liu, J. Mater. Chem., 2011, 21, 11895–11900 RSC.
- Y. Z. Liao, V. Strong, Y. Wang, X. G. Li, X. Wang and R. B. Kaner, Adv. Funct. Mater., 2012, 22, 726–735 CrossRef CAS.
- C. L. Liu, L. P. Zhou, D. Tripathy and Q. F. Sun, Chem. Commun., 2017, 53, 2459–2462 RSC.
- Y. Zhang, B. Li, H. Ma, L. Zhang and W. Zhang, J. Mater. Chem. C, 2017, 5, 4661–4669 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- J. L. Banal, J. M. White, K. P. Ghiggino and W. W.-H. Wong, Sci. Rep., 2014, 4, 1–5 Search PubMed.
- A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019–1027 CrossRef CAS.
- Y. Okazawa, K. Kondo, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2015, 137, 98–101 CrossRef CAS PubMed.
- Y. Hong, J. W.-Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- S. Kumar Panigrahi and A. Kumar Mishra, J. Photochem. Photobiol., C, 2019, 41, 100318 CrossRef CAS.
- V. Vajpayee, H. Kim, A. Mishra, P. S. Mukherjee, P. J. Stang, M. H. Lee, H. K. Kim and K. W. Chi, Dalton Trans., 2011, 40, 3112–3115 RSC.
- S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, Inorg. Chem., 2011, 50, 11736–11745 CrossRef CAS PubMed.
- T. Liu, L. Ding, G. He, Y. Yang, W. Wang and Y. Fang, ACS Appl. Mater. Interfaces, 2011, 3, 1245–1253 CrossRef CAS PubMed.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9195 RSC.
- M. Lu, X. Zhang, P. Zhou, Z. Tang, Y. Qiao, Y. Yang and J. Liu, Chem. Phys. Lett., 2019, 725, 45–51 CrossRef CAS.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- T. S. Lobana, A. K. Sandhu, A. Kaur and J. P. Jasinski, J. Organomet. Chem., 2014, 751, 519–524 CrossRef CAS.
- V. Venkatramaiah, S. Kumar and S. Patil, Chem. Commun., 2012, 48, 5007–5009 RSC.
- P. B. Tchounwou, W. K. Ayensu, N. Ninashvili and D. Sutton, Environ. Toxicol. An Int. J., 2003, 18, 149–175 CrossRef CAS PubMed.
- A. Mousavi, R. D. Chávez, A.-M. S. Ali and S. E. Cabaniss, Environ. Forensics, 2011, 12, 14–18 CrossRef CAS.
- J. F. Fisher and W. H. Organization, Elemental mercury and inorganic mercury compounds: human health aspects, World Health Organization, 2003 Search PubMed.
- M. Update, EPA Fact Sheet EPA-823-F-01-011.
- A. T. Afaneh and G. Schreckenbach, J. Phys. Chem. A, 2015, 119, 8106–8116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis, spectroscopic characterization and data, additional photophysical studies, and FESEM data. See DOI: https://doi.org/10.1039/d1ma01179k |
| This journal is © The Royal Society of Chemistry 2022 |