
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multi-functional porous cobalt catalyst for the selective hydrogenative ring-opening and rearrangement of furfural to cyclopentanol†
Xing-Long
Li
ab and
Rui
Zhu
 *a
*a
aAnhui Province Key Laboratory of Biomass Clean Energy, Department of Chemistry, University of Science and Technology of China, Hefei 230026, China. E-mail: zhr520@mail.ustc.edu.cn
bInstitute of Energy, Hefei Comprehensive National Science Center, Hefei 230031, China
First published on 19th March 2022
Abstract
Developing an economic catalyst to upgrade furfural to cyclopentanol is highly significant for fine chemical synthesis and biomass utilization. In this manuscript, an efficient and economical multi-functional porous Co-400 catalyst was developed. This catalyst obtained porosity, magnetism, Lewis acidity and hydrogenation activity via simple reduction of purchased Co3O4. Various factors were investigated in detail, and 87% yield of CPL could be obtained when Co-400 was used as the catalyst. The active sites of the catalyst were identified according to XRD, IR, XPS, SEM and Raman analysis. Moreover, the keys to the high activity and chemoselectivity of the Co-400 catalyst were mainly attributed to highly dispersed Co0 species and amorphous porous Co3O4 species, which were precisely controlled by the reduction temperature. The catalyst can be easily separated by magnetism. Furthermore, a possible reaction mechanism was proposed based on a series of controlled experiments and catalyst characterizations.
Introduction
To improve the competitiveness of biomass resources against fossil resources, the development of simple and low-cost platform molecule preparation processes and new methods for efficient conversion of platform molecules into fuels and chemicals are important means.1 Biomass-derived furfural (FFA), easily obtained from the dehydration of pentose sugars, is already commercially available at low prices.2 It is considered one of the most important platform compounds that can be subsequently converted into a wide range of high-value chemicals such as furfuryl alcohol (FOL), tetrahydrofuran alcohol (THFOL), furoic acid, 2-methylfuran (2-MF), 2-methyltetrahydrofuran (2-MTHF), levulinic acid, valerolactone, cyclopentanone (CPO) and cyclopentanol (CPL).3CPL and CPO are important fine chemical raw materials that can be used in fields such as solvents, fragrances, cosmetics and agrochemicals (Scheme 1).4 New ether solvents, such as methyl cyclopentyl ether and ethyl cyclopentyl ether, prepared from CPL and CPO have high hydrophobicity, low latent heat of evaporation, difficulty in generating peroxides, easy drying, and acid–base stability; they have been used in important chemical reactions, such as the Grignard reaction and coupling reactions.5 CPL has potential applications in materials, pharmaceutical products, fragrances and solvents.6 Furthermore, CPL is a promising feedstock for various biofuels, including jet fuel and aviation fuel. The world's annual demand for CPL and CPO is more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons. The traditional preparation methods of CPL and CPO are intramolecular decarboxylation ketoneization of adipic acid, hydration of pentene (by steam cracking of naphtha), addition esterification of cyclopentene and acetic acid followed by transesterification with methanol to generate CPL, etc.7 However, these processes require expensive catalysts and harsh reaction conditions (280–300 °C, 25–40 MPa), generate large amounts of waste, and are detrimental to sustainability and process economics.8 Furthermore, the abovementioned processes often use starting materials from fossil resources, and their sustainability has been questioned. Therefore, the preparation of CPL and CPO from biomass-based furfural has high application prospects and industrial value.
000 tons. The traditional preparation methods of CPL and CPO are intramolecular decarboxylation ketoneization of adipic acid, hydration of pentene (by steam cracking of naphtha), addition esterification of cyclopentene and acetic acid followed by transesterification with methanol to generate CPL, etc.7 However, these processes require expensive catalysts and harsh reaction conditions (280–300 °C, 25–40 MPa), generate large amounts of waste, and are detrimental to sustainability and process economics.8 Furthermore, the abovementioned processes often use starting materials from fossil resources, and their sustainability has been questioned. Therefore, the preparation of CPL and CPO from biomass-based furfural has high application prospects and industrial value.
 | ||
| Scheme 1 Some products and applications obtained from conversion of CPO and CPL. | ||
Numerous research groups have reported various supported reducing metals (such as Pd, Pt, Ru, Au, Ni, Cu, and Co) on various acidic supports (such as metal oxides, zeolites, double metal cyanides, metal organic frameworks, and carbon materials) as bifunctional catalysts for the ring-opening rearrangement of furfural to CPL and CPO.9 Noble metal-based catalysts exhibit high activity due to their efficiency in activating H2. The in situ generated intermediates and humins can strongly attach to the metal surface, hindering the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation of CPO. CPO is the main product obtained by using noble metal-based catalysts. In comparison, non-precious metal-based catalysts generally afford CPL, but the catalytic activity is relatively weak and the reaction conditions are relatively harsh. Acidic sites on the supports (eg. Lewis, Brønsted) facilitate the hydrolysis and dehydration steps. Lewis acid sites on the supports were found to be responsible for their selectivity for CPL in aqueous media. Use of Brønsted acid additives or supports containing Brønsted acid centers results in oligomerization (or resinization) of FFA and FOL.10
O hydrogenation of CPO. CPO is the main product obtained by using noble metal-based catalysts. In comparison, non-precious metal-based catalysts generally afford CPL, but the catalytic activity is relatively weak and the reaction conditions are relatively harsh. Acidic sites on the supports (eg. Lewis, Brønsted) facilitate the hydrolysis and dehydration steps. Lewis acid sites on the supports were found to be responsible for their selectivity for CPL in aqueous media. Use of Brønsted acid additives or supports containing Brønsted acid centers results in oligomerization (or resinization) of FFA and FOL.10
Cobalt-based catalysts have numerous important applications; they can be used in oxidation, hydrogenation, isomerization reactions, etc., and are currently widely studied in electrode materials.11 Metal Co has higher hydrogenation selectivity to CO double bonds and repulsion to the furan ring, which is favourable for the highly selective hydrogenation of FFA to FOL. FOL is a key intermediate in the rearrangement reaction, and its yield has a direct effect on the yields of CPL and CPO. In our previous work, it was also found that Cu–Co catalysts prepared by different methods could achieve the selective conversion of FFA to CPO and CPL, respectively.12
Herein, an efficient and economical multi-functional porous Co-400 catalyst was developed for the rearrangement of FFA to CPL. This catalyst obtained porosity, magnetism, Lewis acidity and hydrogenation activity via simple reduction of purchased Co3O4. The effects of the catalyst reduction temperature, reaction temperature, reaction time, hydrogen pressure and catalyst recirculation on the distribution of rearrangement products were investigated. The catalyst structure was characterized and discussed in detail by XRD, XPS, FT-IR spectroscopy, Raman spectroscopy, SEM, etc. Furthermore, a possible reaction mechanism was proposed based on a series of controlled experiments and catalyst characterizations.
Experimental section
Materials and chemicals
Co3O4 was purchased from Sinopharm Holding Chemical Reagent Co., Ltd. Furfural (AR, >99%), furfuryl alcohol (AR, >98%), tetrahydrofurfuryl alcohol (AR, >98%), cyclopentanol (99%), cyclopentanone (99%), and 2-cyclopentenone (97%) were purchased from Aladdin Reagent (Shanghai) Co., Ltd. Purified water was purchased from Hangzhou Wahaha Group Co., Ltd. FFA was used after distillation under reduced pressure. Other reagents not specially emphasized were purchased from Sinopharm Holding Chemical Reagent Co., Ltd.Catalyst preparation
Co-400 Catalyst: A schematic of the catalyst preparation is shown in Fig. S1 (ESI†). An appropriate amount of commercially available Co3O4 was placed in a quartz boat, and the quartz boat was placed in a tube furnace. Catalyst reduction was performed using a gas mixture (100 mL min−1) with a gas composition of H2:N2 = 10:90. The temperature of the tubular furnace was increased from 20 °C to 400 °C at a heating rate of 2 °C min−1, and the temperature was maintained at 400 °C for 3 h. Then, the hydrogen was turned off; the catalyst was cooled to room temperature under a nitrogen atmosphere and taken out and bottled, then labelled as Co-400 catalyst. Co-200 and Co-300 catalysts were prepared with similar methods at reduction temperatures of 200 °C and 300 °C, respectively.
Catalyst characterization
The catalysts were characterized by X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), scanning electron microscopy (SEM), Fourier transform infrared (FT-IR) spectra and Raman spectra.The powder X-ray diffraction (XRD) patterns of the catalysts were measured by an X’pert (PANalytical) diffractometer using Cu Kα radiation at 40 kV and 40 mA; the 2θ ranges were 20–80°.
X-Ray photoelectron spectroscopy (XPS) data were obtained on a Thermo Scientific Escalab 250-X-ray photoelectron spectrometer equipped with a hemispherical electron analyser and Al Kα X-ray source. All binding energy data in the spectra were determined with reference to the C 1s line at 284.8 eV.
Scanning electron microscopy (SEM, Sirion 200, FEI Electron Optics Company, USA) was used to observe the microstructures of the Co catalysts and collect the corresponding data. The tested samples were prepared by ultrasonic suspension in ethanol.
Fourier transform infrared (FT-IR) spectra were recorded with a Nicolet 8700 FT-IR spectrometer at room temperature. The samples were thoroughly ground with potassium bromide and detected after tablet forming.
Raman spectra were collected with a LabRamHR spectrometer with a 532 nm grating at room temperature.
The leaching of Co in the reaction solutions was measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES, Thermo-Jarrell ASH-Atom Scan Advantage). ICP-AES tests: after the reaction, magnets were used to separate out the catalyst. The reaction solution was centrifuged and evaporated to dryness under reduced pressure. The residue was dissolved with concentrated nitric acid and diluted with pure water.
Typical experiments and product analysis
The catalytic conversion of FFA was carried out in a 25 mL stainless steel Parr autoclave equipped with magnetic stirring, a temperature probe and a temperature programmed controller. A typical experiment was to add 0.1 g of FFA, 30 mg of catalyst, and 10 mL of water to a Parr autoclave. After replacing the gas with hydrogen several times, it was charged with hydrogen to the desired pressure and then heated to the desired temperature under magnetic stirring. After holding the reaction for a certain period of time, the temperature was lowered and the pressure was released, and the reactants in the autoclave were transferred out with 20 mL of methanol; N,N-dimethylformamide (DMF) was added as the internal standard. After sampling and centrifugation, a GC chromatograph (Shimadzu GC-2-14C, FID) equipped with a DM-WAX capillary column (30 m × 0.32 mm × 0.25 μm) was used for detection, and the conversion of FFA and yield of the product were calculated according to the following formula:| FFA Conversion/% = (nFFA − mFFA)/nFFA × 100% |
| Yield/% = n(moleofproduct)/nFFA × 100% |
ofproduct): mol of products detected in the reaction solution by GC analysis.
Results and discussion
Catalyst characterization
The catalysts obtained with different reduction temperatures were characterized and discussed firstly to better understand the effect of the catalyst reduction temperature on product selectivity.The XRD patterns of the various Co catalysts were investigated, and the results are listed in Fig. 1. It was found that the diffraction peaks at 31.4°, 37.0°, 38.7°, 45.0°, 55.9°, 59.6°, 65.5° and 77.7° of the Co-200 catalyst could be assigned to the Co3O4 species (JCPDS database PDF# 43-1003).13 The weaker diffraction peaks at 41.7°, 44.7°, 47.6°, and 75.9° were assigned to the Co0 species (JCPDS database PDF# 05-0727).14 This showed that the Co-200 catalyst underwent partial reduction during the low-temperature reduction process, and the observed Co3O4 diffraction peaks indicated that there were still more crystalline Co3O4 species in the catalyst. With the increase of the reduction temperature to 300 °C and 400 °C, the diffraction peaks attributed to Co3O4 disappeared, while the diffraction peaks attributed to Co0 remained weak. The decreased peaks of Co3O4 and the disappearance of the peaks of Co0 suggested that the crystallinity of Co3O4 was largely reduced. This suggested that the Co0 species were highly dispersed in the Co-300 and Co-400 catalysts, while the Co3O4 species changed from a crystalline to an amorphous state.15 Highly dispersed metallic Co0 species provided the catalyst with hydrogenation activity. The amorphous Co3O4 species of the reduction catalyst served as a Lewis acid, promoted the polarization of CO groups and thus facilitated their reduction.16 The amorphous Co3O4 species contain more acidic sites than the crystalline Co3O4 species, which is more conducive to the subsequent ring-opening rearrangement reaction. Meanwhile, the coexistence of Co0 and Co2+/3+ species on the catalyst surface is more conducive to electron transfer, which is beneficial to product selectivity.17
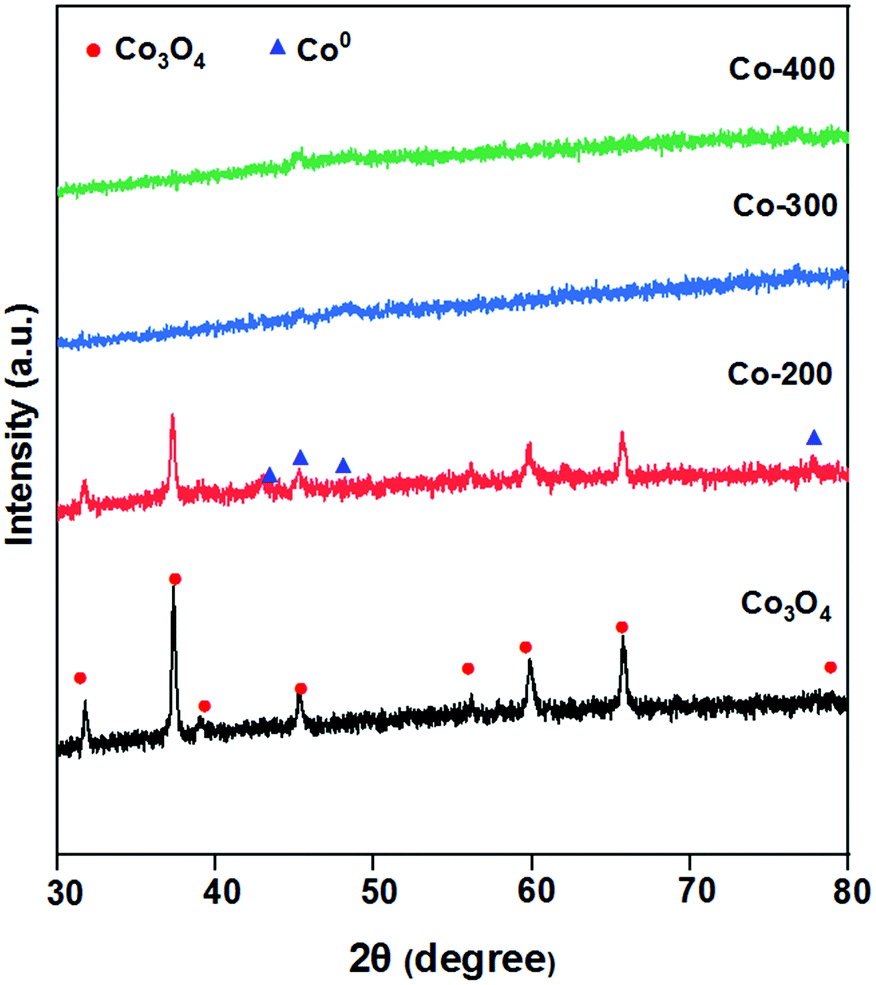 | ||
| Fig. 1 The XRD patterns of the Co-200 catalyst, Co-300 catalyst, Co-400 catalyst and commercial Co3O4. | ||
The FTIR spectra of the catalysts obtained at different reduction temperatures in the range of interest between 400 and 1200 cm−1 are shown in Fig. 2. Two absorption bands were observed in the wavenumber range of 667–575 cm−1, confirming the spinel structure of Co3O4. The peak at 667 cm−1 was attributed to the stretching vibration of Co–O, where Co was in the +2 valence state and was tetrahedrally coordinated. The peak at 575 cm−1 can be assigned to the stretching vibration of Co–O, where Co was in the +3 valence state and thus was in an octahedral coordination.18 The presence of the fingerprint absorption bands confirmed the presence of crystalline Co3O4 species in the commercial Co3O4 and Co-200 catalyst. The wavenumber ranges of these absorption bands were also similar to those reported in the literature.19 It was found that no obvious absorption peaks belonging to crystalline Co3O4 species were observed in the IR spectra of the Co-300 and Co-400 catalysts. This may be due to the possible low-intensity absorption of amorphous Co3O4 in visible light.20 This result was consistent with the XRD pattern (Fig. 1).
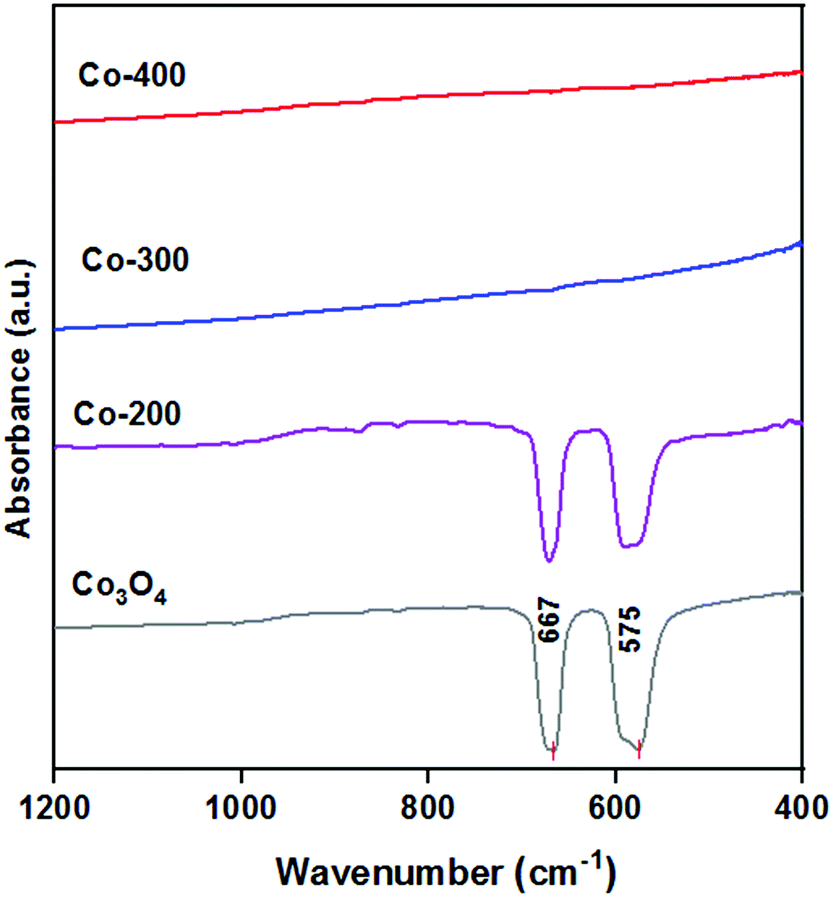 | ||
| Fig. 2 The FTIR spectra of the Co-200 catalyst, Co-300 catalyst, Co-400 catalyst and commercial Co3O4. | ||
The XPS spectra of Co catalysts with different reduction temperatures are shown in Fig. 3. All spectra were calibrated with the C 1s peak at 284.8 eV (Fig. S6, ESI†). According to the XPS analysis of the Co-200 catalyst, the peaks at 782.0 eV and 780.7 eV were assigned to Co 2p3/2, while those at the binding energies of 797.6 eV and 796.1 eV were attributed to Co 2p1/2, indicating that the oxidation states of Co were Co3+ and Co2+, respectively (Fig. 3a). The shakeup satellites located at 785.6 eV and 789.2 eV were assigned to Co 2p3/2, while those at the binding energies of 802.7 eV and 805.3 eV were attributed to Co 2p1/2, indicating that the oxidation states of Co were Co2+ and Co3+, respectively.21 The existence of metallic Co0 species in the Co-200 catalyst was seen from the shoulder peaks around Co 2p1/2 at 794.9 eV and Co 2p3/2 at 779.7 eV.22 These results confirmed that the Co-200 catalyst contained both Co0 and Co3O4 species. The XPS analysis results of the catalysts corroborate the previously obtained XRD results (Fig. 1). As the reduction temperature was increased from 200 °C to 300 °C and 400 °C, it is shown that the catalyst surface of the Co-300 and Co-400 catalysts still contained Co0 and Co3O4 species (Fig. 3b and c). Through the XPS analysis of the catalyst composition ratio, it was found that the proportion of Co0 species decreased and the proportion of Co3O4 (Co2+/Co3+) species increased as the reduction temperature increased from 200 °C to 400 °C (Table S1, ESI†). The observed results show that the proportion of surface Co0 species decreases with increasing reduction temperature. On the one hand, this may be due to the re-oxidation of metallic Co0 species in the catalyst under the condition of exposure to air (during sampling, processing, characterization, etc). On the other hand, this may also be related to the reduction products of Co3O4 at different reduction temperatures. The reduction of Co3O4 generally proceeds through two steps of Co3O4 → CoO → Co0 under a hydrogen atmosphere.23a Kuznetsov et al. have reported that Co3O4 is reduced directly to metallic Co0 species below 291 °C due to the thermodynamic instability of CoO at this temperature.23b However, the reduction of Co3O4 at temperatures above 291 °C proceeded through two steps of Co3O4 → CoO → Co0, with stabilized CoO as an intermediate. The combined effect of the above two factors may be the reason for the gradual decrease of the surface Co0 species content measured with the increase of the reduction temperature. It has been reported in the literature that partially reduced CoOx, in which the metal cations were coordinately unsaturated, can act as a Lewis acid and facilitate preferential adsorption and polarization of carbonyl groups.23c The coexistence of Co0 and Co3O4 species on the catalyst surface suggested that the active sites should have both hydrogenation and Lewis acidity, which both contribute to chemoselectivity.
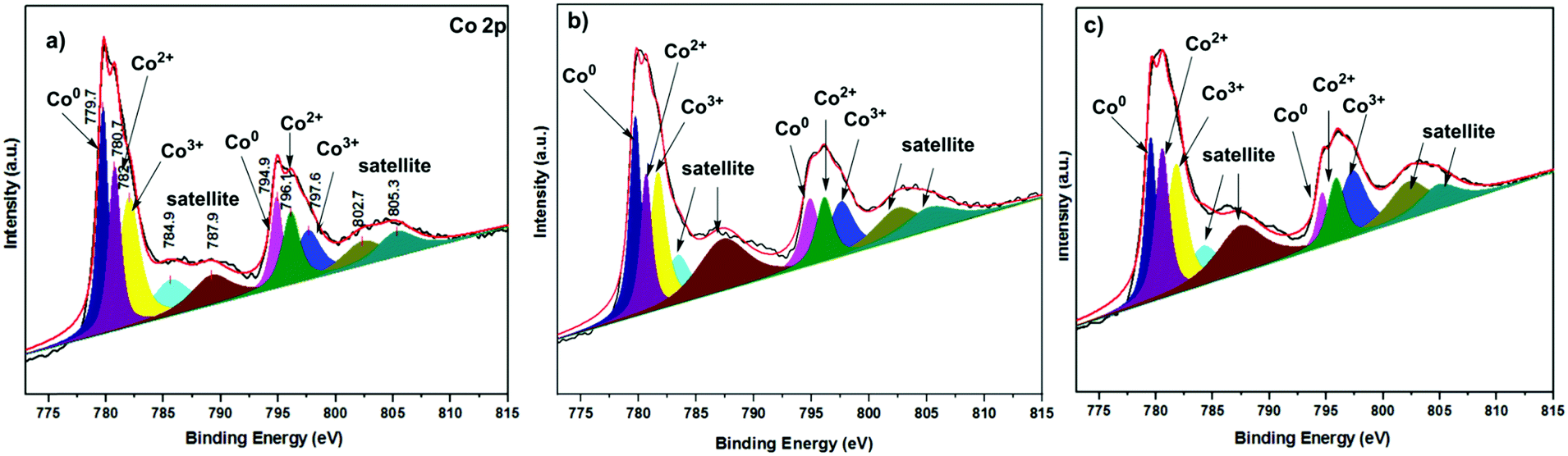 | ||
| Fig. 3 The Co 2p XPS spectra of Co catalysts with different reduction temperatures. (a) Co-200 catalyst, (b) Co-300 catalyst and (c) Co-400 catalyst. | ||
The O 1s XPS spectra of the catalysts obtained at different reduction temperatures were also analysed, and the results are shown in Fig. 4. According to the O 1s XPS analysis of the Co-200 catalyst, the two peaks near 529.6 eV and 530.2 eV can be attributed to the lattice oxygen and metal–oxygen bonds in Co3O4, and one broad peak near 531.5 eV can be attributed to the hydroxyl groups adsorbed on the catalyst surface (Fig. 4a).24 As the reduction temperature increased, the metal–oxygen bond gradually disappeared, and the peaks at 529.6 eV and 531.5 eV represented lattice oxygen and the hydroxyl groups adsorbed on the catalyst surface, respectively (Fig. 4b and c).
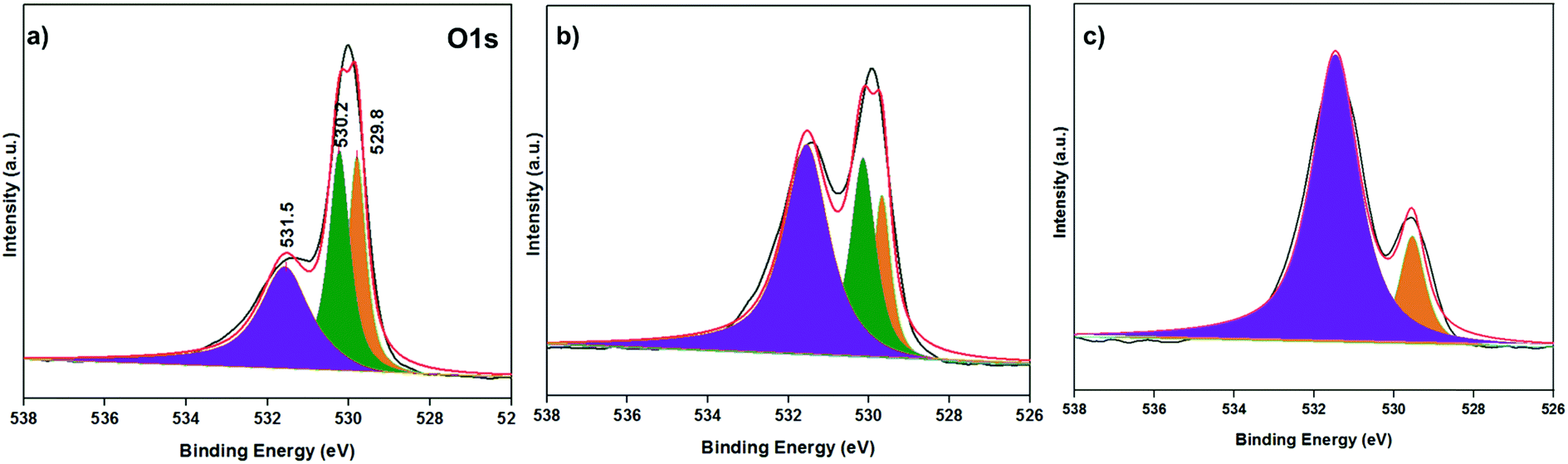 | ||
| Fig. 4 The O 1s XPS spectra of Co catalysts with different reduction temperatures. (a) Co-200 catalyst, (b) Co-300 catalyst and (c) Co-400 catalyst. | ||
The change of the lattice oxygen binding energy may be related to its bonding environment. Lattice oxygen exists in two different bonding environments in Co3O4, namely bonding with Co2+ and Co3+, respectively (Co3O4 = CoO + Co2O3). These two bonding environments merged into one amorphous Co3O4 species of the reduced Co catalysts. This may be related to the obvious transformation of the lattice structure of the catalyst framework from crystalline phase to amorphous phase. Meanwhile, the hydroxyl intensity increased sharply around 531.5 eV, indicating that more hydroxyl groups and oxygen vacancies were created on the amorphous Co3O4 surface layer.25 The hydroxyl groups and oxygen vacancies existing on the catalyst surface could generate Lewis acid–base interactions, which further promoted the ring-opening rearrangement reaction.26
The optical properties of the Co catalysts obtained at different reduction temperatures were characterized by Raman spectroscopy, and the results are listed in Fig. 5. The peaks at 475.78 and 678.54 cm−1 could be assigned to Eg and A1g, while the peaks at 192.91, 517.97 and 612.16 cm−1 could be assigned to the F2g modes of crystalline Co3O4.27 The Raman peaks were caused by the lattice vibrations of the structure, in which the Co2+ and Co3+ cations were located at tetrahedral and octahedral positions in the cubic lattice, respectively. Compared with the unreduced Co3O4 sample, the Raman spectrum of the reduced catalyst showed a negative shift and broadening of the peaks. This may be due to the increased number of oxygen vacancies on the catalyst surface and partial oxide reduction. This result was consistent with that of the O 1s XPS analysis. The above results showed that the reduced catalyst consisted of amorphous Co3O4 species and Co0 species.28 This is consistent with the results of the XPS and XRD analysis.
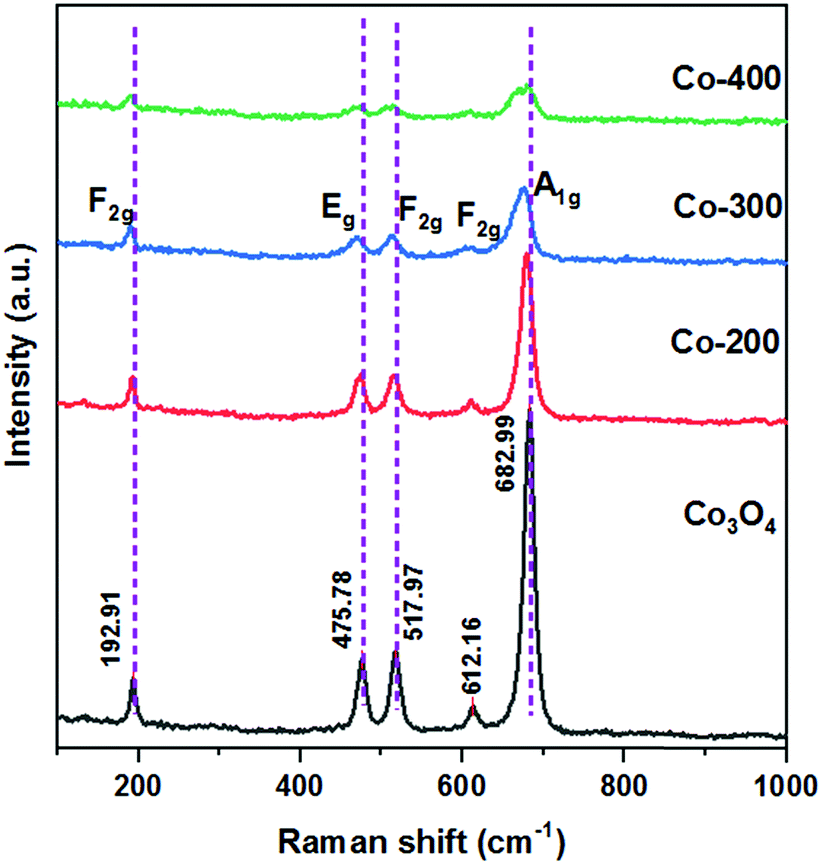 | ||
| Fig. 5 The Raman spectra of the Co-200 catalyst, Co-300 catalyst, Co-400 catalyst and commercial Co3O4. | ||
It can be seen from the SEM images that after increasing the reduction temperature from 200 °C to 400 °C, the surface of the catalyst gradually changed from a compact and flat structure to a rough and porous structure (Fig. 6). Zhou et al. reported that the porous surface of Co3O4 contained a large number of hydroxyl groups and oxygen vacancies, which may be more conducive to the improvement of the reaction carbon yield and the mass transfer.29
 | ||
| Fig. 6 The SEM images of Co catalysts obtained with different reduction temperatures. (a) Co-200 catalyst, (b) Co-300 catalyst, (c) Co-400 catalyst. | ||
Optimization of reaction conditions
Using FFA as the substrate and water as the solvent, the effects of the catalyst species on the product distribution for the conversion of FFA to CPO and CPL were investigated. The results are listed in Table 1.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conversion/% | Yield/% | ||||
| FOL | CPEO | CPO | CPL | Others | |||
| Reaction conditions: FFA 0.1 g, catalyst 30 mg, solvent 10 mL H2O, 160 °C, 5 h, 4 MPa H2, mole yield.a Commercial Co3O4 was used directly without reduction. [n.d]: not detected. Others may be include of humins, THFOL and levulinic acid, etc. | |||||||
| 1 | Co-200 | 100 | 58 | 1 | 27 | 5 | 9 |
| 2 | Co-300 | 100 | 6 | 1 | 36 | 47 | 10 |
| 3 | Co-400 | 100 | 6 | 2 | 8 | 74 | 10 |
| 4 | RANEY® Co | 100 | 88 | 0 | 0 | 0 | 12 |
| 5a | Co3O4 | 6 | 1 | 0 | 0 | 0 | 5 |
| 6 | — | 3 | 0 | 0 | 0 | 0 | 3 |
| 7 | RANEY® Ni | 100 | 0 | 0 | 17 | 28 | 55 |
The catalytic effects of the Co-200, Co-300 and Co-400 catalysts on the ring-opening rearrangement of FFA at the reduction temperatures of 200 °C, 300 °C and 400 °C were compared initially. It was found that FFA was completely converted by using the Co-200 catalyst, while the main products were 58% yield of FOL and 27% yield of CPO (Table 1, entry 1). This indicated that the Co-200 catalyst had hydrogenation activity and its Lewis acidity was weak at low reduced temperature. The main products obtained by using the Co-300 catalyst were CPO and CPL, but the selectivity of the products was poor (Table 1, entry 2). FFA could be completely converted by using the Co-400 catalyst, and the obtained products were mainly isomerized products in which the yield of CPL was 74% and the yield of CPO was 8% (Table 1, entry 3). This showed that Co-400 catalyst has higher hydrogenation activity and Lewis acidity. As widely accepted, the conversion of FFA to CPO and CPL must undergo hydrogenation, ring-rearrangement, hydrogenation, and dehydration steps over catalysts which possess hydrogenation activity and Lewis acidity in water. The prepared Co-400 catalyst not only had a porous structure, but also contained a suitable proportion of amorphous Co3O4 and Co0 on the catalyst surface according to the catalyst characterization. More hydroxyl groups and oxygen vacancies were created on the amorphous Co3O4 surface layer, which not only facilitated the mass transfer of the reaction, but also improved the chemical selectivity of the reaction.
Meanwhile, using RANEY® Co as a catalyst, FFA was completely converted, and the main product was FOL with a yield of 88% (Table 1, entry 4). No obvious ring-opening rearrangement products of CPO and CPL were observed during the reaction, indicating that the RANEY® Co catalyst had high selectivity to FOL. In fact, reduction of FFA with a Co-based catalyst usually only converts the aldehyde group of FFA to FOL or 2-MF.30a It was found that only 6% of FFA was converted by using unreduced Co3O4 as the catalyst (Table 1, entry 5). FFA was basically not converted when no catalyst was present (Table 1, entry 6). The yields of CPO and CPL obtained by using RANEY® Ni as the catalyst were 17% and 28%, respectively. Moreover, the by-products were mainly overhydrogenation products of tetrahydrofuran and THFOL due to the strong hydrogenation activity of RANEY® Ni (Table 1, entry 7).30b Compared with the reaction carbon yield obtained by a previously reported Cu–Co3O4 catalyst, the carbon yield obtained by using the Co-400 catalyst was relatively high.12 This may be due to the fact that the surface of Co catalyst had weak affinity for CC bonds and strong adsorption capacity for the oxygen atoms of CO bonds.31 After the aldehyde groups adsorbed on the surface of the Co catalyst were hydrogenated to hydroxyl groups, the substrate would be detached from the surface of the catalyst.32 FOL is an important intermediate in the rearrangement reaction, and the improvement selectivity of FOL is beneficial to the improvement yield of the rearrangement product. In fact, the structural instability of FOL is prone to intermolecular aldol condensation, dehydration or hydrolysis side reactions, which can lead to the formation of complex oligomers.33 This is the main reason for the unbalanced carbon yield of the reaction.
Then, other reaction conditions were screened and optimized in detail as follows.
Reaction temperature
The reaction temperature has a great effect on the selectivity and yield of CPO and CPL. The detailed temperature effects were investigated in the range of 130 °C to 170 °C, and the product distribution results are shown in Fig. 7. FFA was almost completely converted and 16% yield of FOL remained, while the main products obtained were 27% CPO and 50% CPL at 130 °C. This showed that the rearrangement reaction of FFA could take place at a relatively low temperature. The carbon yield of the reaction was high, and only about 6% yield of by-products was observed. The yields of FOL and CPO gradually decreased while the yield of CPL gradually increased as the reaction temperature increased from 130 °C to 160 °C. The highest yield of CPL was 81% at 160 °C. CPO was gradually converted to CPL at high temperature. The yield of the over-hydrogenation product THFOL was 5% and the carbon yield of the reaction was obviously decreased at 170 °C. It was shown that under high temperature conditions, the side reactions of the raw materials and intermediates were increased. This may be due to the fact that the intermediates and products were more prone to side reactions, which hinders the conversion of intermediates to products at high temperatures.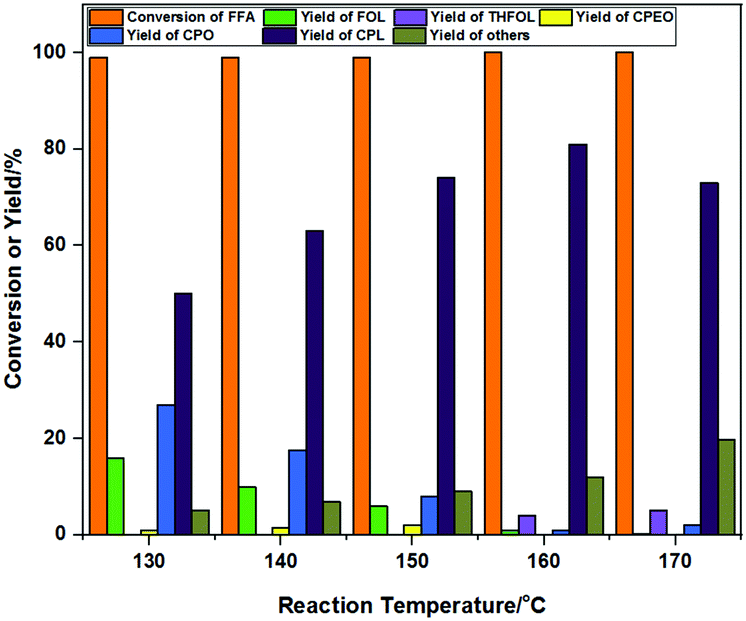 | ||
| Fig. 7 The effects of the reaction temperature on the product distribution. Reaction conditions: FFA 0.1 g, Co-400 catalyst 30 mg, solvent 10 mL H2O, 5 h, 4 MPa H2, mole yield. | ||
Hydrogen pressure
The effects of hydrogen pressure on the product distribution when using Co-400 as a catalyst are shown in Fig. 8. It can be seen that the conversion of FFA was 47%, and the main products were FOL (yield of 36%), CPO (yield of 5%) and CPL (yield of 2%) under 0.1 MPa H2. This showed that the hydrogenation activity of the Co-400 catalyst was relatively weak at lower hydrogen pressure, which is not conducive to the acquisition of CPL. FFA was completely converted, and only about 3% yield of FOL remained, while the main products were a mixture of CPO (yield of 46%) and CPL (yield of 43%) as the reaction hydrogen pressure increased to 1 MPa. This showed that increasing the hydrogen pressure could significantly promote the isomerization of FFA to CPO and CPL, which was similar to results reported in the literature.34 The yield of CPO decreased continuously, and the yield of CPL increased initially and decreased subsequently as the pressure increased from 1 MPa to 4 MPa. The highest yield of CPL was 86% at a hydrogen pressure of 3 MPa. With increasing hydrogen pressure, the total yield of CPO and CPL was basically unchanged although the yield of CPL increased. The improved yield of CPL was due to the further hydrogenation of CPO, and there were few side reactions in the process. This result was also confirmed in controlled experiments of the reaction mechanism when CPO was used as the substrate (Scheme 3d). The selectivity of CPL decreased with the increase of hydrogen pressure due to the increased formation of THFOL resulting from overhydrogenation.35 It can be seen from the characterization (XRD, XPS, IR and Raman) that the presence of Co0 and amorphous Co3O4 species in the Co-400 catalyst endowed the catalyst with both hydrogenation activity and Lewis acidity. The rough and porous structure and suitable Co0/Co2+/3+ ratio were beneficial to improve the product selectivity. The optimum reaction pressure of the catalyst was 3 MPa H2.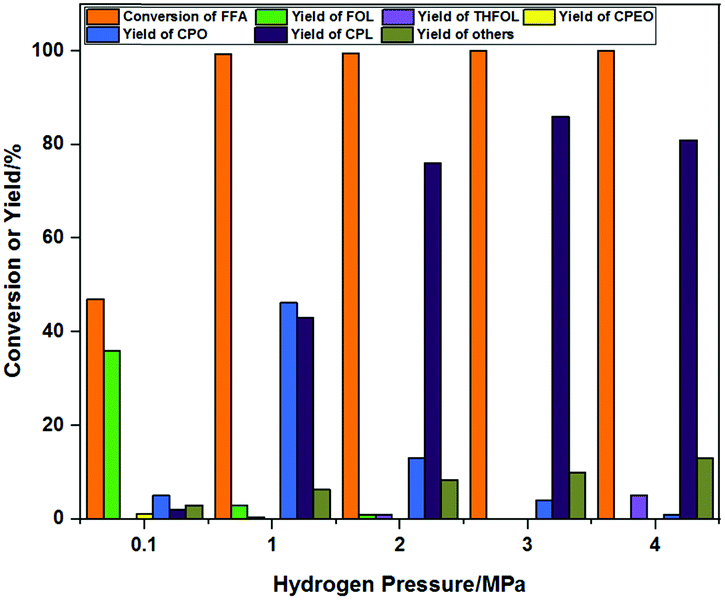 | ||
| Fig. 8 The effects of hydrogen pressure on the product distribution. Reaction conditions: FFA 0.1 g, Co-400 catalyst 30 mg, H2O 10 mL, 160 °C, 5 h, mole yield. | ||
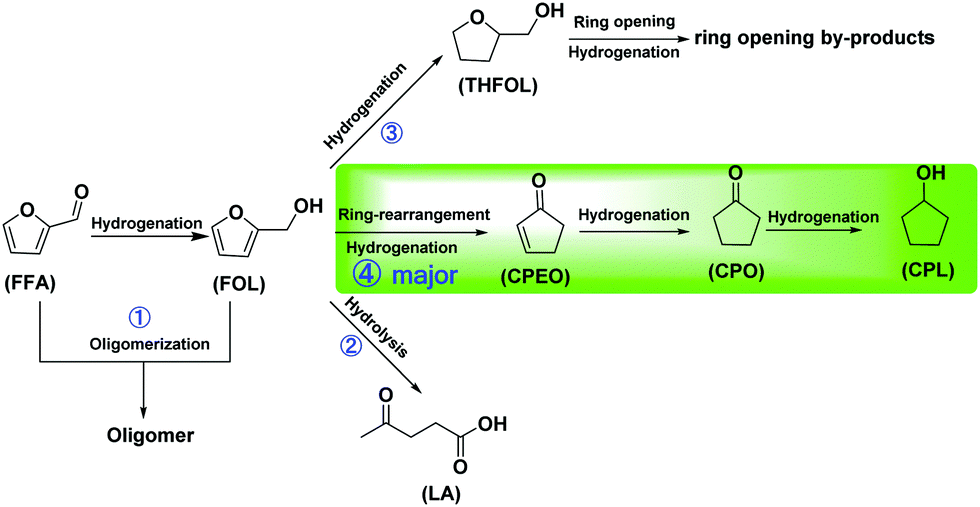 | ||
| Scheme 2 Proposed reaction pathway for the conversion of FFA to CPL with the multi-functional porous Co-400 catalyst. | ||
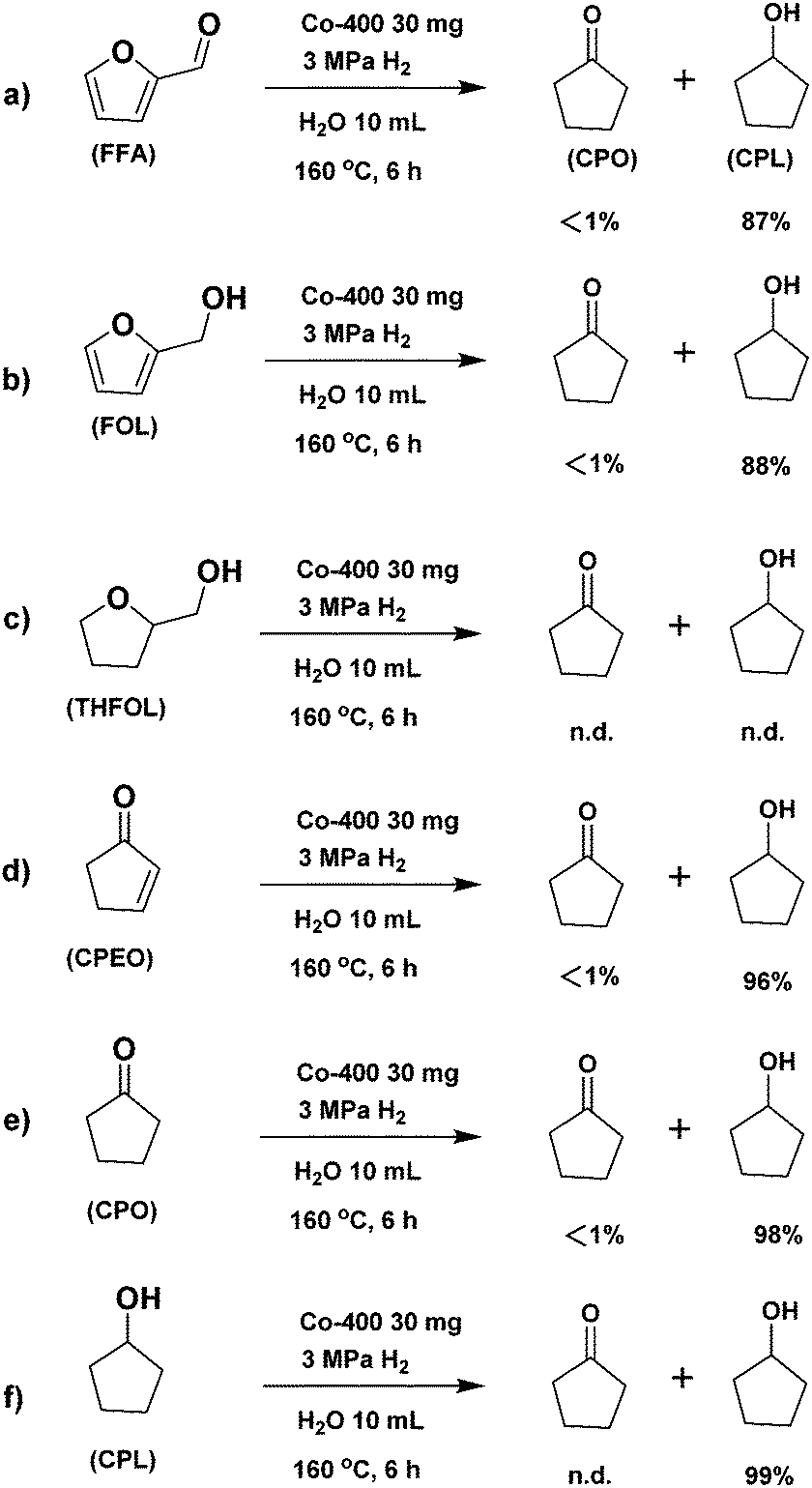 | ||
| Scheme 3 Control experiments. n.d.: not detected. | ||
Catalyst amount
Since the catalyst possessed both hydrogenation activity and Lewis acidity, the ratio of catalyst to substrate had a great effect on the product distribution. The effects of the catalyst amount on product selectivity were investigated, and the results are shown in Fig. 9. The conversion of FFA was 87%, and the main products were 54% yield of FOL and 20% yield of isomerization product CPO when using 10% Co-400 catalyst. The conversion of FFA was completed, and the obtained main products were CPO (yield of 34%) and CPL (yield of 55%) as the catalyst amount increased to 20%. The increased yield of CPL indicated that the overall hydrogenation activity was greatly improved in the presence of 20% Co-400 catalyst. The yield of CPL increased initially to 86% and decreased subsequently to 65% with further increase of the catalyst amount from 20% to 50%. The amount of the over-hydrogenation product THFOL increased to 16%, while the reaction carbon loss increased to 18% when the catalyst amount was increased to 50%. This suggested that the increase in the catalyst amount provided more hydrogenation active sites and Lewis acid sites, resulting in a decrease in the selectivity of ring-opening isomerization. The by-products mainly included levulinic acid and undetectable oligohumins.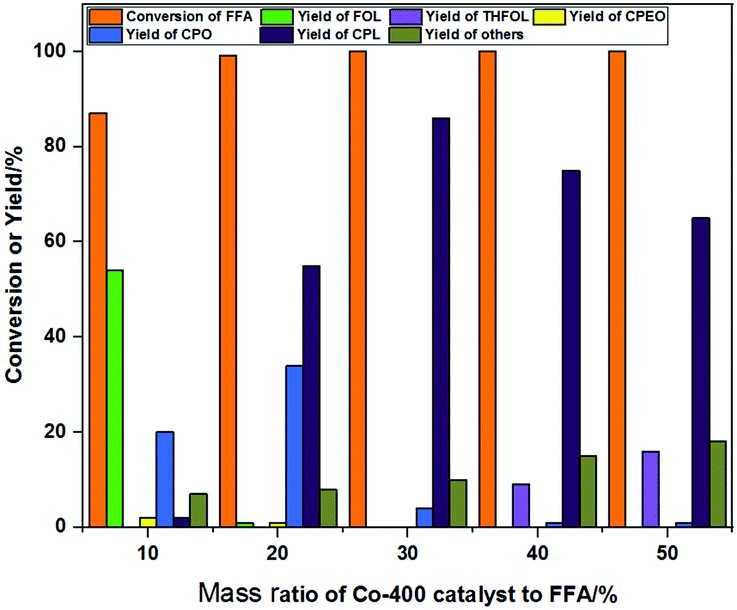 | ||
| Fig. 9 The effects of the catalyst amount on the product distribution. Reaction conditions: FFA 0.1 g, Co-400 catalyst, H2O 10 mL, 160 °C, 5 h, 3 MPa H2, mole yield. | ||
Reaction time
The investigation of the product distribution with reaction time helped us to better judge the reaction mechanism, and the relevant results are listed in Fig. 10. FFA was converted completely, and the main products were FOL and CPO with yields of 40% and 34% after reaction for 1 h, respectively. The yield of CPO gradually increased and then decreased with the increase of the reaction time from 1 h to 6 h. The yield of CPL gradually increased (from 17% to 87%) with the increase of the reaction time and was accompanied by decreased yields of FOL and CPO. This confirmed that FOL was the initial intermediate of the reaction.36 Furthermore, the formation of CPEO was observable during the initial reaction (<2 h) and was not observed until the reaction proceeded to 3 h, where the yields of CPO and CPL increased to 36% and 53%, respectively. This suggested that CPEO is another key intermediate for the reaction, which was consistent with results reported in the literature.37 The increase of by-products was not obvious with the extension of time. The optimal reaction time was 6 h, and the highest yield of CPL was 87%.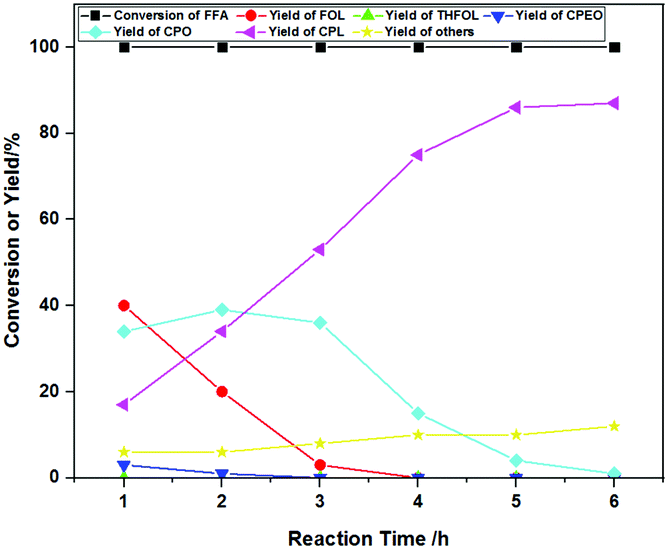 | ||
| Fig. 10 The effects of the reaction time on the product distribution. Reaction conditions: FFA 0.1 g, Co-400 catalyst 30 mg, H2O 10 mL, 160 °C, 3 MPa H2, mole yield. | ||
Catalyst recirculation
Reusability, an important indicator of catalyst performance, was evaluated, and the results are shown in Fig. 11. Because the catalyst was magnetic, it could be easily separated from the system (Fig. S3–S5, ESI†). The operation of the catalyst circulation is shown in the supplementary information (SI, Experimental, “Catalyst Cycle Experiment”). The yield of CPL decreased from 86% to 78%, and FFA was almost completely converted in each cycle after the catalyst was reused 4 times. With the increase of cycle time, the yield of CPO continued to increase, but the total yield of CPO and CPL remained basically unchanged. This indicated that the Lewis acidity of the reused Co-400 catalyst was not significantly weakened. Through the previous investigation of the catalyst amount (Fig. 9), it was found that reducing the catalyst amount (from 30% to 20%) may lead to weakening of the hydrogenation activity without affecting the overall yield of CPO and CPL.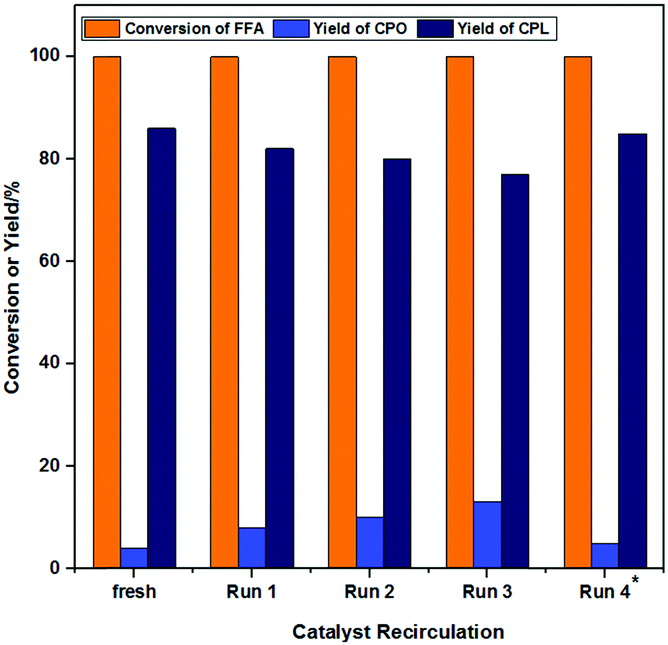 | ||
| Fig. 11 The effects of cycle times on the product distribution. Reaction conditions: FFA 0.1 g, Co-400 catalyst 30 mg, H2O 10 mL, 160 °C, 5 h, 3 MPa H2, mole yield. Run 4* means adding 10 mg of fresh catalyst for the cyclic reaction. | ||
The catalysts were characterized by XRD (Fig. 12a), XPS (Fig. 12b–e), IR (Fig. S7, ESI†), SEM (Fig. S8, ESI†) and Raman (Fig. S9, ESI†) characterizations in order to investigate the structure of the reused Co-400 catalyst. It could be seen from the above characterization that the structure and composition of the Co-400 catalyst did not change significantly after recirculation, which indicated that the catalyst structure remained stable after the reaction. The leaching of metal Co in the reaction solution during the cycle reaction was investigated, and the results are listed in Table S2 (ESI†). As the number of cycles increased, the loss of Co in the solution after the reaction was 0.4%, 0.3%, 0.2% and 0.2%, respectively. This indicated that the leaching of Co in the reaction solution was not obvious during the reaction process. The weakening of the catalyst activity should be attributed to catalyst loss during the cycle operation. To test this speculation, 10 mg of fresh Co-400 catalyst was supplemented in Run 4, and the yield of CPL encouragingly increased to 85%. Therefore, the maintenance of the catalyst structure and the improvement of the product selectivity after the supplementation of additional catalyst further confirmed that the weakening of the catalyst activity is due to catalyst loss during the cycle operation.
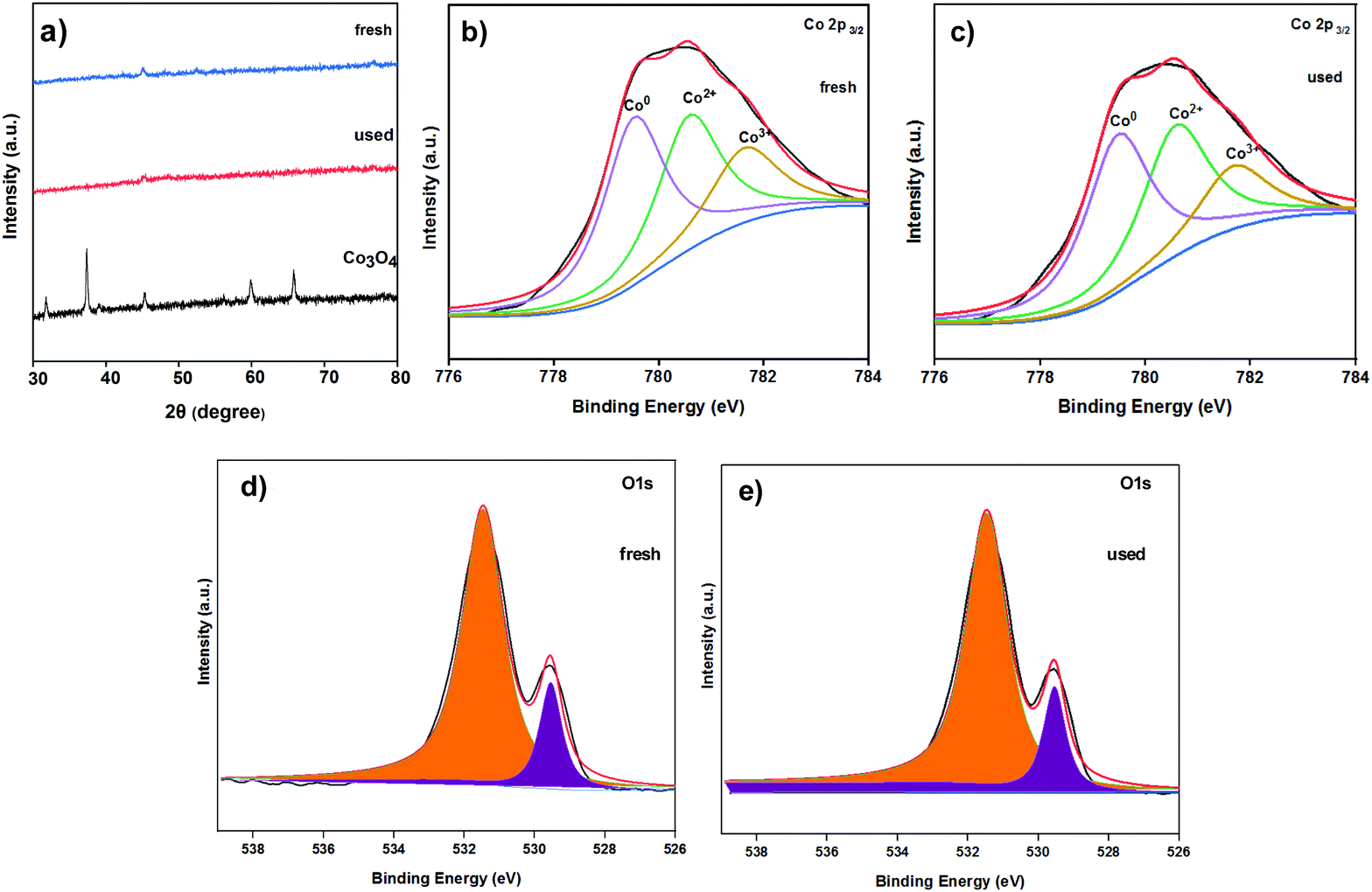 | ||
| Fig. 12 The XRD pattern and XPS spectra of the Co catalysts. (a) The XRD pattern of fresh Co-400 catalyst, Co-400 catalyst after reuse 4 times, and commercial Co3O4; (b) Co 2P3/2 XPS spectra of fresh Co-400 catalyst; (c) Co 2P3/2 XPS spectra of Co-400 catalyst after reuse 4 times; (d) O 1s XPS spectra of fresh Co-400 catalyst; (e) O 1s XPS spectra of Co-400 catalyst after reuse 4 times. | ||
Reaction pathway
The conversion of FFA in water may include the following four pathways in the presence of a catalyst (Scheme 2): (1) FFA is hydrogenated at the hydrogenation site of the catalyst to obtain FOL. Due to the existence of water and acid sites on the catalyst, FFA and FOL may undergo oligomerization to obtain oligomers that are difficult to detect in the GC analysis. The further competitive reaction of FOL in the presence of the catalyst directly affects the product distribution. (2) FOL is hydrolysed to levulinic acid (LA) at the catalyst acid sites. (3) After excessive hydrogenation of FOL to THFOL under severe conditions, there may be further ring-opening hydrogenation to by-products such as amyl alcohol. (4) FOL undergoes ring-opening rearrangement and hydrogenation to afford cyclopentenone (CPEO) in the presence of the catalyst.38In order to investigate the specific conversion pathway of FFA in water in the presence of the catalysts, control experiments were performed (Scheme 3). The yield of CPL was 87% when using FFA as the substrate, according to the results of the aforementioned optimization of the reaction conditions (Scheme 3a). The yield of CPL was 88% when using FOL, which indicated that the conversion effect of FOL to CPL is similar to that of FFA (Scheme 3b). No CPO and CPL were observed when using THFOL as the raw material, indicating that THFOL was not an intermediate of the ring-opening rearrangement (Scheme 3c). The high yield of CPL was obtained by using CPEO and CPO as raw materials, indicating that the further hydrogenation of CPEO and CPO had fewer side reactions, and the carbon yield loss basically does not occur in this step (Schemes 3d–e). 99% yield of CPL was recovered, indicating that the further conversion of CPL under the catalytic system was inhibited by using CPL as the raw material (Scheme 3f). Meanwhile, the reaction under the additional addition of acid and alkali was investigated (Scheme S1, ESI†). Adding alkali to the standard reaction system significantly inhibited the ring-opening isomerization reaction of FFA, and no isomerization product was observed (Scheme S1a, ESI†). Adding Brønsted acid to the standard reaction system significantly inhibited the ring-opening isomerization reaction. Meanwhile, the formation of levulinic acid was observed. The presence of Brønsted acid increased the formation of humins, and the carbon yield decreased significantly (Scheme S1b, ESI†). It can be seen from the control experiments that the ring-opening rearrangement reaction of FFA in the presence of the Co-400 catalyst mainly proceeds by path 4 (Fig. S2, ESI†).
No over-hydrogenation product (THFOL) or hydrolysis product (levulinic acid) was detected under the optimal conditions, indicating that FFA was less converted through route 2 and route 3 in water. The non-conservation of the reaction carbon yield was mainly caused by the oligomerization side reaction of raw materials and intermediate products in the presence of the acid sites on the catalyst. It was found that the amount of catalyst, catalyst reduction temperature and hydrogen pressure had obvious effects on the product distribution. The appropriate proportion of Co0 species and amorphous Co3O4 species in the catalyst provided hydrogenation activity and Lewis acid activity, respectively. Meanwhile, the rough and porous structure of the Co-400 catalyst improved the chemoselectivity of the catalyst and afforded CPL as the main product in water.
Conclusion
In summary, an efficient and economical multi-functional porous Co-400 catalyst that obtained porosity, magnetism, Lewis acidity and hydrogenation activity via simple reduction of commercially available Co3O4 for the conversion of FFA to CPL was developed. The highest yield of CPL was 87% by using the Co-400 catalyst at 160 °C, 3 MPa H2, and 6 h. Various characterization methods were used to analyse the catalyst structure. It was found that with the increase of reduction temperature, the crystalline Co3O4 species in the catalyst transformed to the amorphous Co3O4 species while dispersed Co0 species were generated in the catalyst surface, according to the XRD, IR, Co 2p XPS and Raman analysis. More hydroxyl groups and oxygen vacancies were created on the amorphous Co3O4 surface layer in the Co-400 catalyst, which can generate Lewis acid–base interactions and facilitate the ring-opening rearrangement reaction according to the O 1s XPS and Raman analysis. The surface of catalyst gradually changed from a compact and flat structure to a rough and porous structure with increasing reduction temperature from 200 °C to 400 °C, as observed by SEM analysis. The porous surface of Co3O4 contained a large number of hydroxyl groups and oxygen vacancies, which were more conducive to the improvement of the reaction carbon yield and the mass transfer. The reaction factors were investigated in detail, and it was found that the amount of catalyst, catalyst reduction temperature and hydrogen pressure had obvious effects on the product distribution. The structure and composition of the catalyst did not change significantly after cycling, indicating that the catalyst structure remained stable after the recycle reaction. The catalysts can be easily separated by magnetism. The slight decrease in the catalyst activity after 4 cycles was mainly attributed to loss of the catalyst during the cycle operation. Meanwhile, the possible reaction pathways of FFA in water were discussed, and the possible reaction mechanism was given according to control experiments. This work provided an effective strategy to regulate catalyst structure and product selectivity by changing the catalyst reduction conditions, and it revealed the synergistic effect of catalyst hydrogenation and acid catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the Anhui Natural Science Foundation Project (2008085QB63), and the University Synergy Innovation Program of Anhui Province [GXXT-2021-023]. The authors thank Anhui Kemi Machinery Technology Co., Ltd. for equipment that benefited our ability to conduct this study.References
- (a) V. G. Yadav, G. D. Yadav and S. C. Patankar, Clean Technol. Environ. Policy, 2020, 22, 1757–1774 CrossRef PubMed; (b) W. Dwi Prasetyo, Z. A. Putra, M. R. Bilad, T. M. I. Mahlia, Y. Wibisono, N. A. H. Nordin and M. D. H. Wirzal, Polymers, 2020, 12, 1091 CrossRef PubMed.
- (a) B. R. Caes, R. E. Teixeira, K. G. Knapp and R. T. Raines, ACS Sustainable Chem. Eng., 2015, 3, 2591–2605 CrossRef CAS; (b) P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- (a) P. Khemthong, C. Yimsukanan, T. Narkkun, A. Srifa, T. Witoon, S. Pongchaiphol, S. Kiatphuengporn and K. Faungnawakij, Biomass Bioenergy, 2021, 148, 106033 CrossRef CAS; (b) X. Zhang, S. Xu, Q. Li, G. Zhou and H. Xia, RSC Adv., 2021, 11, 27042–27058 RSC; (c) J. Zhu and G. Yin, ACS Catal., 2021, 11, 10058–10083 CrossRef CAS; (d) S. Karnjanakom, A. Bayu, P. Maneechakr, C. Samart, S. Kongparakul and G. Q. Guan, ACS Sustainable Chem. Eng., 2021, 9, 14170–14179 CrossRef CAS.
- J. Scognamiglio, L. Jones, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2012, 50, S608–S612 CrossRef CAS PubMed.
- G. de Gonzalo, A. R. Alcántara and P. Domínguez de María, ChemSusChem, 2019, 12, 2083–2097 CrossRef CAS PubMed.
- J. Kijeński, P. Winiarek, T. Paryjczak, A. Lewicki and A. Mikołajska, Appl. Catal., A, 2002, 233, 171–182 CrossRef.
- (a) M. Renz, Eur. J. Org. Chem., 2005, 979–988 CrossRef CAS; (b) K. A. Dubkov, G. I. Panov, E. V. Starokon and V. N. Parmon, Catal. Lett., 2002, 77, 197–205 CrossRef CAS; (c) J. Marquié, A. Laporterie, J. Dubac and N. Roques, Synlett, 2001, 0493–0496 CrossRef.
- S. Van de Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465–1479 RSC.
- (a) M. Hronec and K. Fulajtarová, Catal. Commun., 2012, 24, 100–104 CrossRef CAS; (b) G. S. Zhang, M. M. Zhu, Q. Zhang, Y. M. Liu, H. Y. He and Y. Cao, Green Chem., 2016, 18, 2155–2164 RSC; (c) J. H. Guo, G. Y. Xu, Z. Han, Y. Zhang, Y. Fu and Q. Guo, ACS Sustainable Chem. Eng., 2014, 2, 2259–2266 CrossRef CAS; (d) Y. Wang, S. Y. Sang, W. Zhu, L. J. Gao and G. M. Xiao, Chem. Eng. J., 2016, 299, 104–111 CrossRef CAS; (e) Q. Deng, X. H. Wen and P. Zhang, Catal. Commun., 2019, 126, 5–9 CrossRef CAS; (f) X. Li, Q. Deng, L. K. Zhang, J. Wang, R. Wang, Z. L. Zeng and S. G. Deng, Appl. Catal., A, 2019, 575, 152–158 CrossRef CAS.
- (a) T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS; (b) X. Li, Q. Deng, L. Yu, R. Gao, Z. Tong, C. Lu, J. Wang, Z. Zeng, J. J. Zou and S. Deng, Green Chem., 2020, 22, 2549–2557 RSC; (c) R. M. Mironenko, V. P. Talsi, T. I. Gulyaeva, M. V. Trenikhin and O. B. Belskaya, React. Kinet., Mech. Catal., 2019, 126, 811–827 CrossRef CAS.
- (a) G. H. Wang, X. H. Deng, D. Gu, K. Chen, H. R. Tüysüz, B. Spliethoff, H. J. Bongard, C. Weidenthaler, W. Schmidt and F. Schüth, Angew. Chem., 2016, 128, 11267–11271 CrossRef; (b) Z. Gholami, Z. Tišler and V. Rubáš, Catal. Rev., 2021, 63, 512–595 CrossRef CAS; (c) X. R. Hu, Y. Wang, Q. S. Wu and J. F. Li, Ionics, 2021, 1–27 Search PubMed; (d) O. K. Mmelesi, N. Masunga, A. Kuvarega, T. T. Nkambule, B. B. Mamba and K. K. Kefeni, Mater. Sci. Semicond. Process., 2021, 123, 105523 CrossRef CAS; (e) C. Li, X. Tong, P. Yu, W. Du, J. Wu, H. Rao and Z. M. Wang, J. Mater. Chem., 2019, 7, 16622–16642 RSC; (f) I. Borthakur, A. Sau and S. Kundu, Coord. Chem. Rev., 2022, 451, 214257 CrossRef CAS; (g) L. Lukasevics and L. Grigorjeva, Org. Biomol. Chem., 2020, 18, 7460–7466 RSC; (h) S. Iravani and R. S. Varma, Green Chem., 2020, 22, 2643–2661 RSC; (i) A. Badruzzaman, A. Yuda, A. Ashok and A. Kumar, Inorg. Chim. Acta, 2020, 511, 119854 CrossRef CAS; (j) Z. Ruan and Z. Li, Polym. Chem., 2020, 11, 764–778 RSC; (k) M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 1–8 Search PubMed.
- X. L. Li, J. Deng, J. Shi, T. Pan, C. G. Yu, H. J. Xu and Y. Fu, Green Chem., 2015, 17, 1038–1046 RSC.
- C. H. Huang, S. J. Liu and W. S. Hwang, Energy, 2011, 36, 4410–4414 CrossRef CAS.
- S. A. Lee, T. H. Lee, C. Kim, M. J. Choi, H. Park, S. Choi, J. Lee, J. Oh, S. Y. Kim and H. W. Jang, ACS Catal., 2019, 10, 420–429 CrossRef.
- W. Q. Qin, C. R. Yang, X. H. Ma and S. S. Lai, J. Alloys Compd., 2011, 509, 338–342 CrossRef CAS.
- L. L. Zhang, M. X. Zhou, A. Q. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 CrossRef CAS PubMed.
- (a) M. Salavati-Niasari and F. Davar, Int. J. Nanosci., 2009, 8, 273–276 CrossRef CAS; (b) M. Herrero, P. Benito, F. M. Labajos and V. Rives, Catal. Today, 2007, 128, 129–137 CrossRef CAS.
- (a) A. D. Jagadale, V. S. Kumbhar, R. N. Bulakhe and C. D. Lokhande, Energy, 2014, 64, 234–241 CrossRef CAS; (b) M. K. Lima-Tenorio, C. S. Ferreira, Q. H. F. Rebelo, R. F. B. D. Souza, R. R. Passos, E. A. G. Pineda and L. A. Pocrifka, Mater. Res., 2018, 21, e20170521 CAS; (c) H. Cai, Q. Wei, H. Xiao, H. Liu and J. Wang, J. Mater. Sci.: Mater. Electron., 2020, 31, 7606–7615 CrossRef CAS; (d) F. Liu, H. Su, L. Jin, H. Zhang, X. Chu and W. Yang, J. Colloid Interface Sci., 2017, 505, 796–804 CrossRef CAS PubMed; (e) M. Aadil, S. Zulfiqar, M. Shahid, S. Haider, I. Shakir and M. F. Warsi, J. Alloys Compd., 2020, 844, 156062 CrossRef CAS; (f) R. A. Nuamah, S. Noormohammed and D. K. Sarkar, Int. J. Energy Res., 2021, 1–11 Search PubMed.
- M. Tiwari, M. A. Arya, P. V. More, S. Parmar, S. Datar and P. K. Khanna, J. Nanosci. Nanotechnol., 2020, 20, 2847–2857 CrossRef CAS PubMed.
- (a) J. Yu, Y. Zhong, X. Wu, J. Sunaro, W. Zhou and Z. Shao, Adv. Sci., 2018, 5, 1800514 CrossRef PubMed; (b) J. Yang, J. K. Cooper, F. M. Toma, K. A. Walczak, M. Favaro, J. W. Beeman, L. H. Hess, C. Wang, C. Zhu, S. Gul, J. Yano, C. Kisielowski, A. Schwartzberg and I. D. Sharp, Nat. Mater., 2017, 16, 335–341 CrossRef CAS PubMed; (c) H. F. Peng, W. L. Zhang, Y. Song, F. X. Yin, C. W. Zhang and L. Zhang, Catal. Today, 2020, 355, 286–294 CrossRef CAS.
- A. B. Mandale, S. Badrinarayanan, S. K. Date and A. P. B. Sinha, J. Electron Spectrosc. Relat. Phenom., 1984, 33, 61–72 CrossRef CAS.
- (a) B. Tomić-Tucaković, D. Majstorović, D. Jelić and S. Mentus, Thermochim. Acta, 2012, 541, 15–24 CrossRef; (b) A. N. Kuznetsov and N. F. Kulish, Izv. Akad. Nauk SSSR, Otd. Tekh. Nauk, Metall. Topl., 1959, 4, 52–58 Search PubMed; (c) J. Heveling, W. G. Augustyn and L. M. Cele, Ind. Eng. Chem. Res., 2014, 53, 13910–13919 CrossRef.
- (a) C. D. Wagner, D. A. Zatko and R. H. Raymond, Anal. Chem., 1980, 52, 1445–1451 CrossRef CAS; (b) G. Tyuliev and S. Angelov, Appl. Surf. Sci., 1988, 32, 381–391 CrossRef CAS.
- (a) X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed; (b) T. Xia, C. Zhang, N. A. Oyler and X. Chen, Adv. Mater., 2013, 25, 6905–6910 CrossRef CAS PubMed; (c) T. Xia, P. Wallenmeyer, A. Anderson, J. Murowchick, L. Liu and X. Chen, RSC Adv., 2014, 4, 41654–41658 RSC.
- P. A. R. D. Jayathilaka, M. A. K. L. Dissanayake, I. Albinsson and B. E. Mellander, Electrochim. Acta, 2002, 47, 3257–3268 CrossRef CAS.
- (a) Y. C. Liu, J. A. Koza and J. A. Switzer, Electrochim. Acta, 2014, 140, 359–365 CrossRef CAS; (b) Y. Zhao, S. Chen, B. Sun, D. Su, X. Huang, H. Liu, Y. Yan, K. Sun and G. Wang, Sci. Rep., 2015, 5, 7629 CrossRef CAS PubMed; (c) J. Zhang, W. Gao, M. Dou, F. Wang, J. Liu, Z. Li and J. Ji, Analyst, 2015, 140, 1686–1692 RSC; (d) L. M. Alrehaily, J. M. Joseph and J. C. Wren, Phys. Chem. Chem. Phys., 2015, 17, 24138–24150 RSC; (e) V. Khadzhiev, M. Iliev and I. Vergilov, J. Phys. C: Solid State Phys., 1988, 21, L199–L201 CrossRef CAS; (f) G. Wang, X. Shen, J. Horvat, B. Wang, H. Liu, D. Wexler and J. Yao, J. Phys. Chem. C, 2009, 113, 4357–4361 CrossRef CAS.
- (a) L. Zhang, X. Zhao, W. Ma, M. Wu, N. Qian and W. Lu, CrystEngComm, 2013, 15, 1389–1396 RSC; (b) J. Zhu, L. Huang, Y. Xiao, L. Shen, Q. Chen and W. Shi, Nanoscale, 2014, 6, 6772–6781 RSC; (c) J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- Q. Zhou, S. Liu, S. Zhang, Y. Che and L. H. Gan, Front. Mater., 2021, 8, 206 Search PubMed.
- (a) C. Y. Li, C. X. Ke, R. R. Han, G. L. Fan, L. Yang and F. Li, Mol. Catal., 2018, 455, 78–87 CrossRef CAS; (b) Y. Xu, S. B. Qiu, J. X. Long, C. G. Wang, J. M. Chang, J. Tan, Q. Y. Liu, L. L. Ma, T. J. Wang and Q. Zhang, RSC Adv., 2015, 5, 91190–91195 RSC.
- (a) S. Sitthisa, T. Sooknoi, Y. Ma, P. B. Balbuena and D. E. Resasco, J. Catal., 2011, 277, 1–13 CrossRef CAS; (b) Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- (a) S. Srivastava, P. Mohanty, J. K. Parikh, A. K. Dalai, S. S. Amritphale and A. K. Khare, Chin. J. Catal., 2015, 36, 933–942 CrossRef CAS; (b) H. C. Zhou, J. L. Song, H. L. Fan, B. B. Zhang, Y. Y. Yang, J. Y. Hu, Q. G. Zhu and B. X. Han, Green Chem., 2014, 16, 3870–3875 RSC; (c) L. Xu, R. Nie, X. Lyu, J. Wang and X. Lu, Fuel Process. Technol., 2020, 197, 106205 CrossRef CAS.
- (a) M. Hronec, K. Fulajtarová and T. Liptaj, Appl. Catal., A, 2012, 437, 104 CrossRef; (b) Q. Deng, R. Gao, X. Li, J. Wang, Z. Zeng, J. J. Zou and S. Deng, ACS Catal., 2020, 10, 7355–7366 CrossRef CAS; (c) M. Hronec, K. Fulajtarová and M. Micušik, Appl. Catal., A, 2013, 468, 426 CrossRef CAS; (d) H. Y. Zhu, M. H. Zhou, Z. Zeng, G. M. Xiao and R. Xiao, Korean J. Chem. Eng., 2014, 31, 593 CrossRef CAS; (e) R. Q. Fang, H. L. Liu, R. Luque and Y. W. Li, Green Chem., 2015, 17, 4183 RSC; (f) M. Hronec, K. Fulajtárová, I. Vávra, T. Soták and E. Dobročka, Appl. Catal., B, 2016, 181, 210 CrossRef CAS.
- S. Dutta and N. S. Bhat, ACS Omega, 2021, 6, 35145–35172 CrossRef CAS PubMed.
- C. Herrera, D. Fuentealba, I. T. Ghampson, C. Sepulveda, J. L. García-Fierro, R. I. Canales and N. Escalona, Catal. Commun., 2020, 144, 106092 CrossRef CAS.
- (a) X. Li, Q. Deng, S. H. Zhou, J. D. Zou, J. Wang, R. Wang, Z. L. Zeng and S. G. Deng, J. Catal., 2019, 378, 201–208 CrossRef CAS; (b) P. Jia, X. C. Lan, X. D. Li and T. F. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15221–15229 CrossRef CAS.
- (a) C. Y. Liu, R. P. Wei, G. L. Geng, M. H. Zhou, L. J. Gao and G. M. Xiao, Fuel Process. Technol., 2015, 134, 168–174 CrossRef CAS; (b) Y. L. Yang, Z. T. Du, Y. Z. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC; (c) S. J. Zhang, H. Ma, Y. X. Sun, Y. Luo, X. Liu, M. Y. Zhang, J. Gao and J. Xu, Green Chem., 2019, 21, 1702–1709 RSC; (d) D. D. Wang, M. Al-Mamun, W. B. Gong, Y. Lv, C. Chen, Y. Lin, G. Z. Wang, H. M. Zhang and H. J. Zhao, Nano Res., 2021, 14, 2846–2852 CrossRef CAS.
- (a) R. M. Mironenko, O. B. Belskaya, V. P. Talsi and V. A. Likholobov, J. Catal., 2020, 389, 721–734 CrossRef CAS; (b) Y. Zhang, G. Fan, L. Yang and F. Li, Appl. Catal., A, 2018, 561, 117–126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ma00127f |
| This journal is © The Royal Society of Chemistry 2022 |