
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining the most suitable energetic tetrazole and triazole moieties: synthesis and characterization of 5-(1-hydroxy-3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole and its nitrogen-rich ionic derivatives†
Lukas
Bauer
,
Maximilian
Benz‡
,
Thomas M.
Klapötke
 *,
Cedric
Pignot
and
Jörg
Stierstorfer
*,
Cedric
Pignot
and
Jörg
Stierstorfer
Department of Chemistry, Ludwig-Maximilian University Munich, Butenandtstr. 5-13, 81377 Munich, Germany. Web: http://www.hedm.cup.uni-muenchen.deE-mail: tmk@cup.uni-muenchen.de
First published on 19th March 2022
Abstract
With the synthesis of 5-(1-hydroxy-3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole (1) we can report the first successful production of a combined bis-heterocyclic system composed of 1-hydroxy-tetrazole and 1-hydroxy-1,2,4-triazole. The straightforward synthesis modifies the cyano group of the nitro-triazole starting material within four steps to the tetrazol-1-ol and finally oxidizes the triazole selectively to the 1-hydroxy-triazole, resulting in compound 1, which is a hybrid molecule of the two in energetic materials chemistry established bis-heterocyclic motifs 1,1′-dihydroxy-3,3′-dinitro-bis-1,2,4-triazole and 1,1′-dihydroxy-5,5′-bistetrazole. In order to tune and improve the performance parameters, several nitrogen rich bases were used to obtain the respective ionic derivatives. All compounds were characterized using multinuclear NMR and IR spectroscopy, differential thermal analysis (DTA) and elemental analysis. Several compounds could be further analyzed using X-ray diffraction measurements. The heats of formation for all investigated compounds were determined and the detonation properties (EXPLO5 V6.05.02) were calculated. These data, as well as the sensitivity values, were compared to related substances.
Introduction
Since the start of large-scale production of RDX during World War II, the substance, and its eight-membered ring homolog HMX, has been included in almost all energetic military charges as a high explosive (HE) component.1–3 Despite the good performance data and the simple synthesis, alternative substances are currently being explored, because the main disadvantage of RDX and HMX is their decomposition product's toxicity and environmental incompatibility.4–6 Therefore, new generation molecules should avoid this drawback and be as environmentally friendly as possible. Another criterion for a possible substitute is, above all, at least comparable performance values to that of RDX or even HMX for the final substance. The best case would be an insensitive compound outperforming RDX/HMX.7–10Of high potential for replacing the currently common secondary explosives are triazole-, tetrazole- and furazan-based compounds, especially in the form of bis-heterocycles. These scaffolds exhibit high densities, highly endothermic heats of formation and therefore good performance compared to several single ring systems.7,11–14
Since, in addition to a high heat of formation, density plays the major role in the calculation of detonation parameters, some recurring strategies are widely used to increase it, such as nitration of the molecular backbone, formation of a system consisting of π-stacking parts or the synthesis of ionic derivatives to maximize the packing coefficient.8,15,16
Another approach is to utilize N-oxidation, which not only has the advantage of increasing the oxygen balance but also boosting the density through the addition of a further heteroatom. Fig. 1 depicts some prominent molecular moieties containing bis-heterocyclic systems and N-oxides at the respective position 1.17–23 Among the most promising molecules developed with the aim of replacing RDX are the bis-hydroxylammonium derivatives of 3,3′-dinitro-bis-1,2,4-triazol-1,1′-diolate (MAD-X1)19 and 5,5′-bistetrazol-1,1′-diolate (TKX-50),18 which are attracting great attention in the international community.24–30
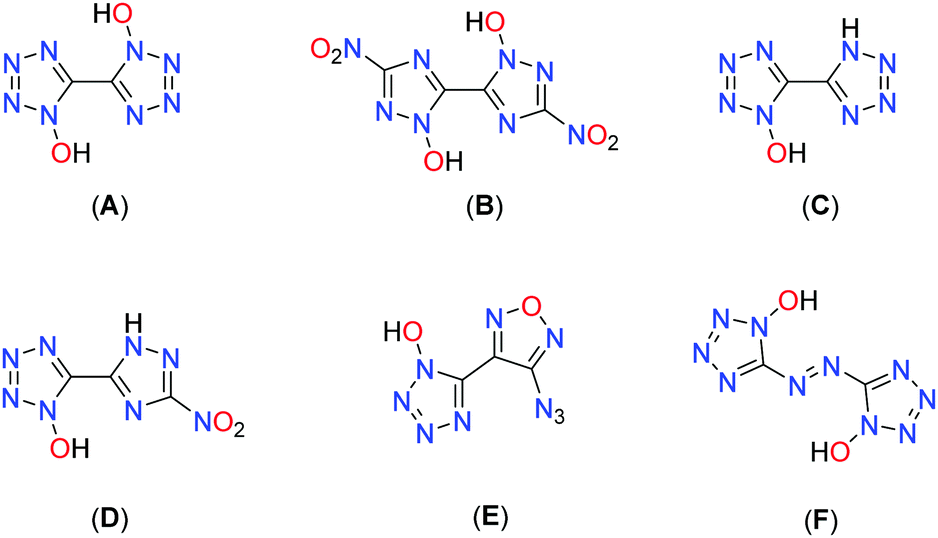 | ||
| Fig. 1 Structures of literature known N1-oxidized bicyclic triazole and tetrazole motifs: 1,1′-dihydroxy-5,5′-bistetrazole (A), 1,1′-dihydroxy-3,3′-dinitro-bis-1,2,4-triazole (B), 1-hydroxy-5,5′-bistetrazole (C), 3-nitro-(1,2,4-triazol-5-yl)-1-hydroxy-tetrazole (D), 3-azido-(furazan-4-yl)-1-hydroxy-tetrazole (E), and 1,1′-dihydroxy-5,5′-azotetrazole (F). | ||
Combination of 1-hydroxy-tetrazole and 1-hydroxy-3-nitro-1,2,4-triazole yields the hybrid molecule 1-hydroxy-3-nitro-(1,2,4-triazol-5-yl)-1-hydroxy-tetrazole (1) with the aim to combine the positive properties of the fundamental symmetric bis-heterocycles in a new asymmetric molecule: preferrable the high heat of formation of 1,1′-dihydroxy-5,5′-bistetrazole (A) with the high density and insensitivity toward external stimuli of 1,1′-dihydroxy-3,3′-dinitro-bis-1,2,4-triazole (Fig. 2).
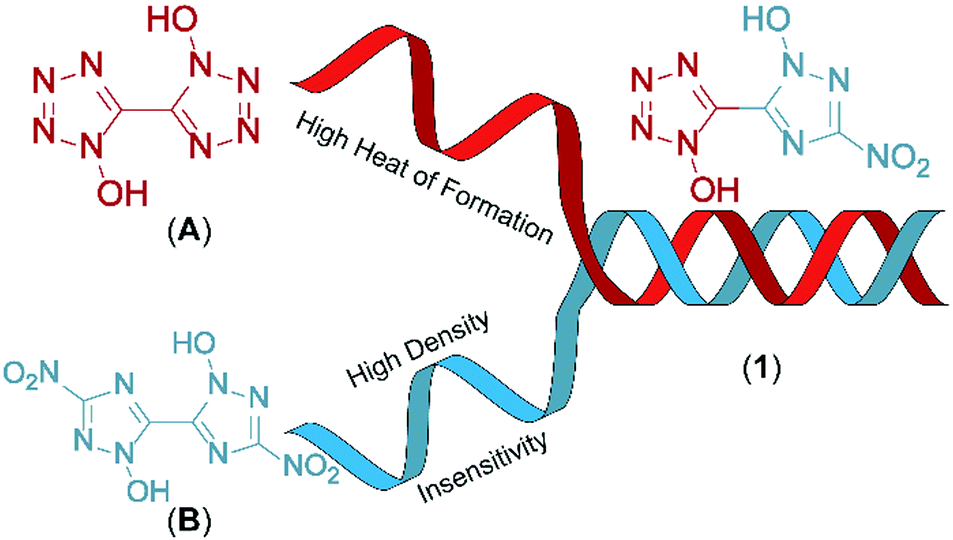 | ||
| Fig. 2 Graphical illustration of the combination of 1,1′-dihydroxy-5,5′-bistetrazole (A) and 1,1′-dihydroxy-3,3′-dinitro-1,2,4-bistriazole (B) resulting in the hybrid molecule 5-(1-hydroxy-3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole (1). | ||
Results and discussion
Synthesis
The synthesis starts from 3-amino-1,2,4-triazol-5-carbonitrile, which was synthesized according to a literature known procedure starting from oxalic acid and aminoguanidinium hydrogencarbonate.31 Diazotization in 1 M sulfuric acid and excess of sodium nitrite led to the nitro-triazole derivative. Therefore, a modified procedure was established, which prevents the long presence of the extremely sensitive diazonium intermediate (for more information see the ESI†). Afterwards, the nitrile function was converted into a 1-hydroxy-tetrazole moiety by a proven procedure within four reaction steps (Scheme 1). This reaction sequence includes the addition of hydroxylamine, the diazotization of the aminoxime in hydrochloric acid precipitating the chloroxime, chlorine-azide exchange and finally a proton promoted ring closing reaction to end up with 5-(3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole (D) in 94% yield.21 The overall yield of D starting from 3-amino-1,2,4-trazole-5-carbonitrile is 61%.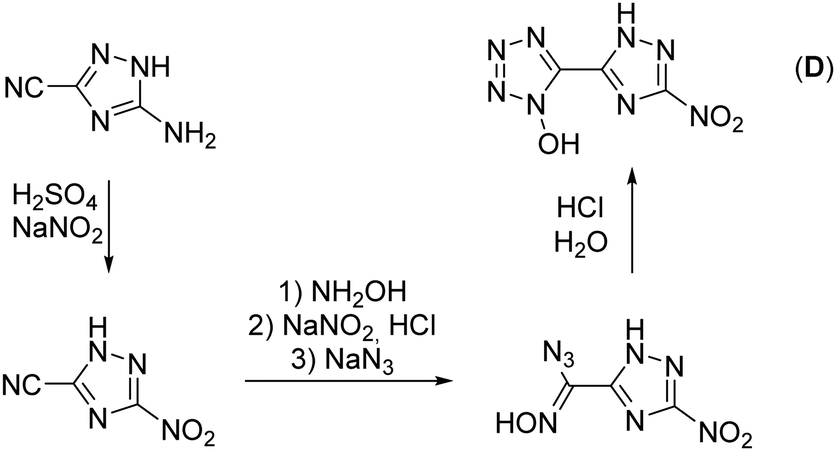 | ||
| Scheme 1 Literature known synthetic pathway for the precursor compound for the synthesis of 1. | ||
Oxidation of 5-(3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole at the triazole yielded 5-(1-hydroxy-3-nitro-1,2,4-triazol-5-yl)-1-hydroxy-tetrazole trihydrate (1) and was performed with Oxone® as an oxidant (Scheme 2). This has already been established as a suitable reagent in similar reactions.16,32,33 The reaction was buffered to pH = 7 in an aqueous medium using tri-sodium phosphate dodecahydrate. Other buffer reagents, such as sodium and potassium acetate and sodium hydrogen carbonate, as well as non-buffered conditions were examined but did not yield 1 at all. Maximal yields of 70% were achieved with a reaction time of 16 h at room temperature (25 °C). Due to the electron withdrawing effect of the nitro group of the triazole, the oxidation occurs selectively at the meta position to the nitro function. All attempts to dehydrate 1 failed, as it kept drawing and absorbing water from the air.
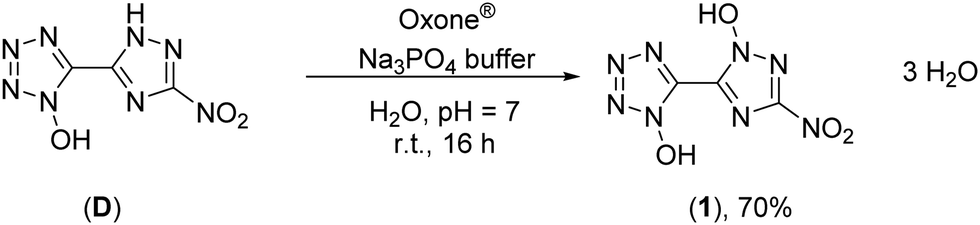 | ||
| Scheme 2 Synthesis of 1 with Oxone® as an oxidation agent. | ||
Ionic derivatives 2–7 were obtained by dissolving 1 in ethanol or ethanol/water mixtures and adding two equivalents of the respective base to the solution. For the ammonium salt 2, gaseous ammonia was bubbled through the solution for about 1 minute. Compounds 2–5 precipitated immediately as water free salts and were used for characterization without further purification after washing with a small amount of the respective solvent. For compounds 6 and 7 the solution had to be reduced to precipitate the solid. In the case of compounds 2–6, the double salts were obtained. Despite the use of two equivalents of the base, only the mono triaminoguanidinium derivative 7 could be obtained, which is known to happen from similar systems (Scheme 3).34
 | ||
| Scheme 3 Synthetic pathway for the formation of ionic derivatives 2–7. | ||
Crystal structures
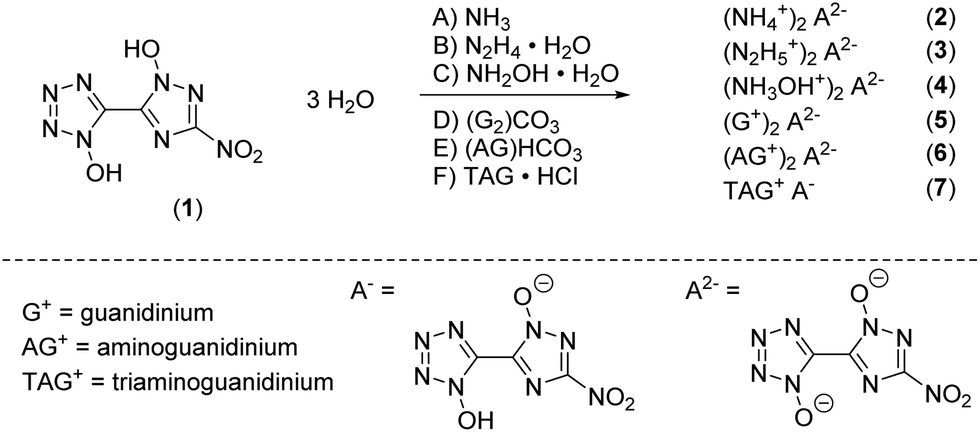
Single crystals suitable for X-ray diffraction experiments could be obtained by recrystallizing from common organic solvents for all compounds except 5 due to its bad solubility and crystallization behavior. Even though bis-hydroxylammonium derivative 4 was obtained solvent free, only a crystal structure of the hydrate adduct could be obtained. Details on the measurement and refinement data for every crystallographically investigated compound are listed in the ESI† (Tables S1–S3). The crystal data sets were uploaded to the CSD database with the CCDC numbers 2144424–2144432.†Compound 1 crystallizes as a trihydrate in the orthorhombic space group Cmca with a cell volume of 1969.5(7) Å3 and eight molecular units per unit cell. The cell constants are a = 6.239(12) Å, b = 7.842(16) Å and c = 40.259(8) Å while the density is 1.809 g cm−3 at 293 K. Compared to the room temperature densities of 1,1′-dihydroxy-5,5′-bistetrazole·2 H2O (1.778 g cm−3) and 1,1′-dihydroxy-3,3′-dinitro-bis-1,2,4-triazole·2 H2O (1.829 g cm−3), 1 lies in the middle of the two values as it is a chemical mixture of both of them.18,19 The crystal structure is depicted in Fig. 1a. The bond lengths in both azole rings are in the range of N–N (N5–N6, 1.332(3) Å; N2–N3, 1.309(4) Å) and N–C (N1–C1, 1.339(4) Å; N7–C2, 1.341(4) Å) single and double bonds, respectively. The bond lengths for the N-oxides are 1.339(3) Å (O1–N1) and 1.337(3) Å (O2–N5) which is a normal value for N-bonded hydroxides donating electron density into the aromatic system. The angles N1–O1–H1 (107(3)°) and N5–O2–H2 (102(3)°) are close to the ideal tetrahedral angle, yet deviate slightly from it, which can be explained by the hydrogen bridges that have formed. Due to the orthorhombic symmetry, the molecule itself is completely planar (N1–C1–C2–N5, 180.00°; O3–N8–C3–N7, 180.00°). Each bis-heterocycle is surrounded by eight water molecules. All heteroatoms with the exception of N2 and N8 are involved in the formation of a network consisting of strong to moderately strong hydrogen bonds, which cover a distance range of 1.57(4) Å (O2–H2_a⋯O6) to 2.63(6) Å (O6–H2_b⋯O4). This results in the layered stacking of 1 shown in Fig. 3b with a layer distance of 3.12 Å.
 | ||
| Fig. 3 (a) Molecular structure of 1 as determined by X-ray diffraction with thermal ellipsoids drawn at the 50% probability level; (b) 2D layer structure of 1 viewed along the b axis. | ||
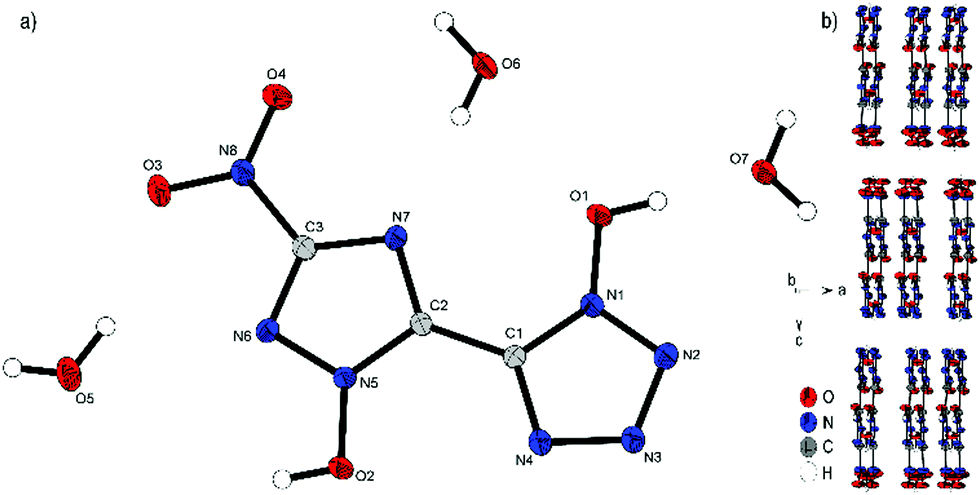
Bis-ammonium derivate 2 crystallizes in the monoclinic space group P21/n with a cell volume of 914.2 Å3 and four molecular units per unit cell. The cell constants are a = 4.3259(1) Å, b = 26.9849(8) Å and c = 7.9243(2) Å, while the density is 1.803 g cm−3 at 106 K. The two aromatic rings with their N-oxides form an almost planar configuration (O1–N1–C1–C2, 0.5(3)°; N4–C1–C2–N7, 170.00(5)°; O2–N5–C2–C1, 175.6(2)°). Only the nitro group of the triazole is twisted out of this plane 13.2(2)°(O3–N8–C3–N6). Every anionic bis-heterocycle is surrounded by nine ammonium cations, forming a large number of hydrogen bonds. 2 forms a wave like structure along the b axis (Fig. 4b). The change of orientation takes place after two formula units of the anion. The ammonium cations are located between the anion layers.
 | ||
| Fig. 4 (a) Molecular structure of 2 as determined by low temperature X-ray diffraction with thermal ellipsoids drawn at the 50% probability level; (b) zig-zig pattern of 2 viewed along the ac plane. | ||
Compound 3 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a cell volume of 528.11(19) Å3 and two molecular units per cell. The constants are a = 3.6755(4) Å, b = 8.0751(9) Å and c = 17.8693(18) Å while the density is 1.750 g cm−3 at 173 K. The nearly planar anion (N5–C2–C1–N1, 175.4(3)°; O4–N8–C3–N6, 173.1(3)°; O1–N1–N2–N3, 179.3(3)°) forms a layer like structure along the bc plane, as depicted in Fig. 5b. The free spaces between the respective layers are filled by hydrazinium cations, which form a large number of hydrogen bonds to the double anion.
with a cell volume of 528.11(19) Å3 and two molecular units per cell. The constants are a = 3.6755(4) Å, b = 8.0751(9) Å and c = 17.8693(18) Å while the density is 1.750 g cm−3 at 173 K. The nearly planar anion (N5–C2–C1–N1, 175.4(3)°; O4–N8–C3–N6, 173.1(3)°; O1–N1–N2–N3, 179.3(3)°) forms a layer like structure along the bc plane, as depicted in Fig. 5b. The free spaces between the respective layers are filled by hydrazinium cations, which form a large number of hydrogen bonds to the double anion.
 | ||
| Fig. 5 (a) Molecular structure of 3 as determined by low temperature X-ray diffraction with thermal ellipsoids drawn at the 50% probability level; (b) layer structure of 3 viewed along the a axis. | ||
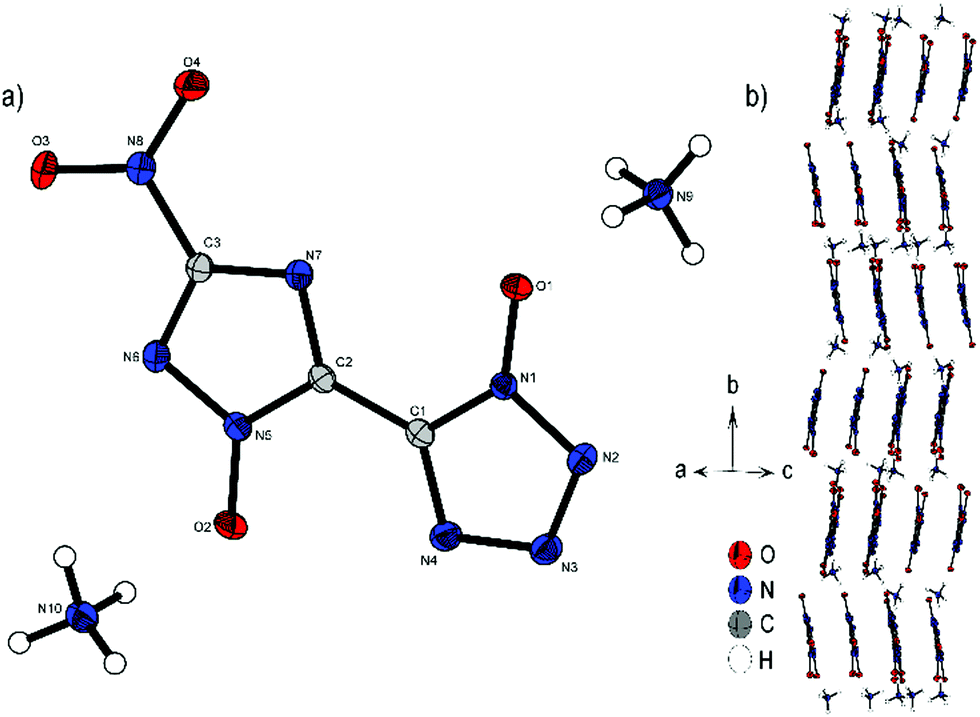
Compound 4 crystallizes with the inclusion of one water molecule in the monoclinic space group P21/c with a cell volume of 1100.2(3) Å3 and four molecular units per cell. The cell constants are a = 7.3751(11) Å, b = 15.0961(19) Å and c = 10.4402(14) Å while the density is 1.800 g cm−3 at 298 K. The anionic moiety is presented in almost planar conformation (O1–N1–C1–C2, −1.0(4)°; O2–N5–C2–C1, 0.3(4)°; O3–N8–C3–N6, 0.0(4)°). No other anion structure features this kind of coplanar arrangement of the aromatic rings. Along the ac plane, the bis-heterocycle forms a zig-zag-like structure, which is due to the same space group very similar to the three-dimensional structure of 2. The cations and the crystal water form layers between the respective linear chains of anions (Fig. 6b).
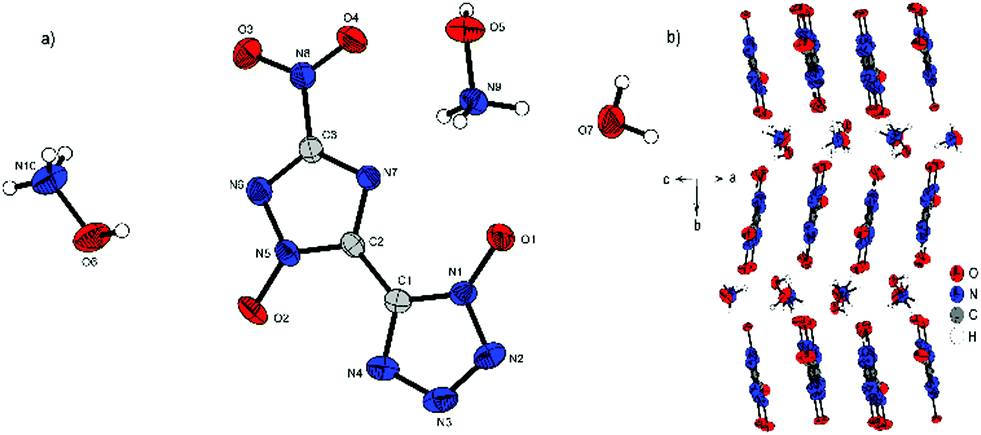 | ||
| Fig. 6 (a) Molecular structure of 4·H2O as determined by X-ray diffraction with thermal ellipsoids drawn at the 50% probability level; (b) zig-zag pattern of 4 along the b axis. | ||
Bis-aminoguanidinium derivative 6 crystallizes in the monoclinic space group I2/a with a cell volume of 2835.0(1) Å3 and eight molecules per unit cell. The cell constants are a = 11.245(2) Å, b = 6.7268(15) Å and c = 37.486(8) Å while the density is 1.698 g cm−3 at 100 K. Compound 6 is the most twisted anion structure for double deprotonated anions compared in this study. The two azole rings are twisted by 148.9(4)°(N4–C1–C2–N7) toward each other. As shown in Fig. 7b, the maximum number and strength of hydrogen bonds with the aminoguanidinium cation is achieved by this twist of the anion. Along b, the formation of channels of anions and cations occurs. It can also be observed that the aminoguanidinium units are twisted in relation to each other. This also results from the maximization of the intermolecular bonds.
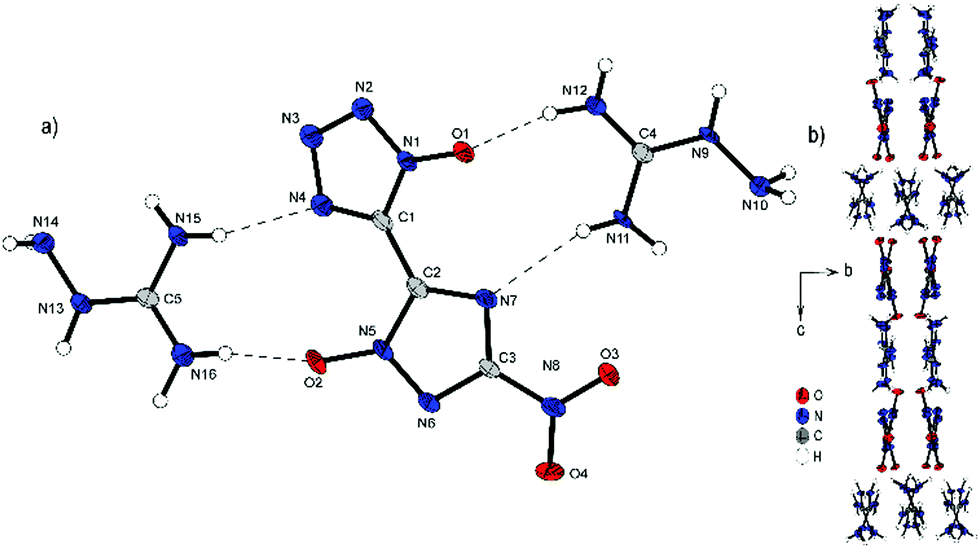 | ||
| Fig. 7 (a) Molecular structure of 6 as determined by low temperature X-ray diffraction with thermal ellipsoids drawn at the 50% probability level; (b) pattern of 6 viewed along the a axis. | ||
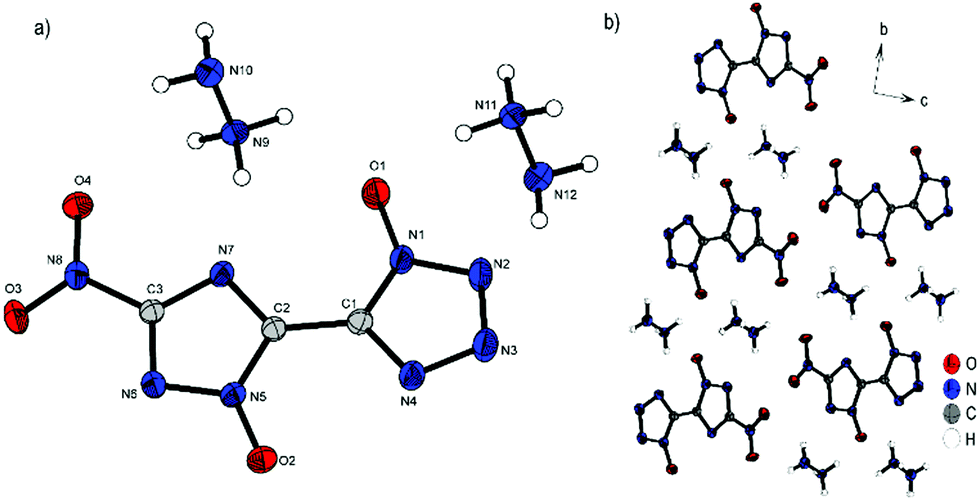
Compound 7 crystallizes in the triclinic space group P with a cell volume of 611.07(13) Å3 and two molecular units per cell. The constants are a = 7.5556(8) Å, b = 8.1499(8) Å and c = 11.4798(12) Å while the density is 1.730 g cm−3 at 102 K. It is the only compound that is preserved only as a single salt. Compared to the structures described above, this also results in the unique assembly of 7. The single deprotonation occurs at the triazole OH group, since the proton there has a lower pKa value than the proton at the tetrazole as a result of the nitro substituent. This deprotonation also leads to the rotation of the rings relative to each other into a sterically unfavorable position, so that both hydroxy units are on the same side (N7–C2–C1–N4, 15.3(2)°). This ring formation is stabilized by a strong intramolecular interaction formed between the two hydroxy groups and the remaining proton. H1 can be classified as a divided proton, since it is more likely to be bound to O1, but also has a very strong interaction with O2 (O1–H1, 1.05(4) Å; O2–H1, 1.47(4) Å; O1–H1–O2, 171(2)°). Compared to the neutral compound 1, the bonds O1–H1 and O2–H1 (O1–H1, 0.86(4) Å; O2–H2, 0.85(4) Å) are significantly prolonged. Viewed along the ab plane (Fig. 8b), the mono-anion forms an oblique cylindrical structure in which the heterocycles are alternately superimposed. This pattern is embedded by triaminoguanidinium cations. Along the space diagonal (Fig. 8c), anions and cations are arranged in pairs in a chessboard-like structure, with the respective orientation flipped by 90° toward each other.
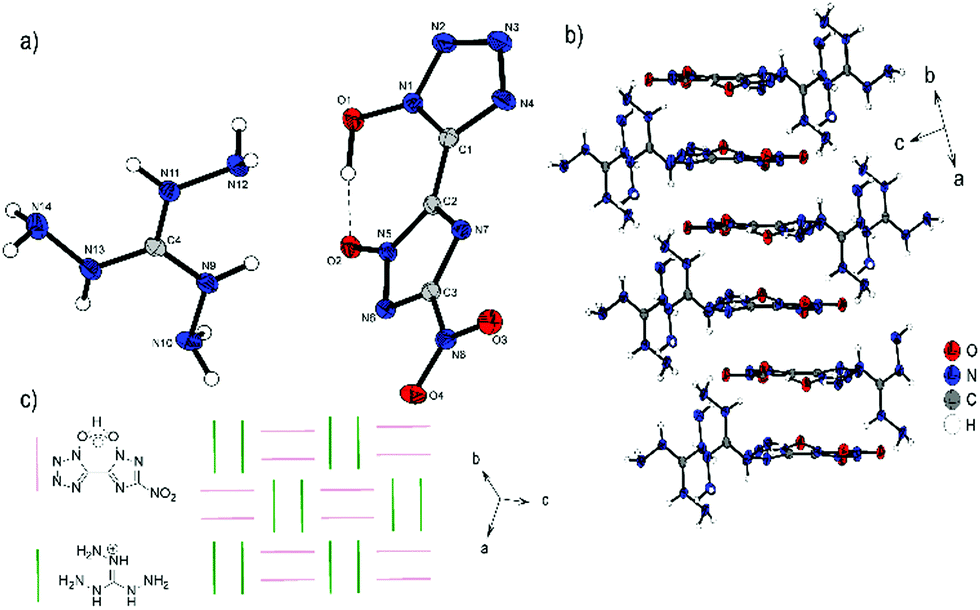 | ||
| Fig. 8 (a) Molecular structure of 7 as determined by low temperature X-ray diffraction with thermal ellipsoids drawn at the 50% probability level; (b) 2D layer structure of 7 viewed along the ab plane; (c) schematic arrangement of the anions and cations viewed along the space diagonal. | ||
NMR Spectroscopy
All investigated compounds were characterized using 1H, 13C and 14N NMR spectroscopy. Additionally, compounds 1 and 2 were analyzed using proton coupled 15N NMR spectroscopy. The proton NMR spectrum of 1 shows one broad signal at around 7.80 ppm representing the acidic OH protons. The proton signals for the cations in 2 (7.25 ppm), 3 (7.13 ppm) and 4 (9.66 ppm) can be found as broad singlets in the classical region for the respective cations.35,36 Derivative 5 shows one singlet resonance at 7.13 ppm for the guanidinium cation. For the aminoguanidinium protons in compound 6 three different signals could be obtained. Broadened singlets at 8.93 ppm (NH-NH2) and 7.21 ppm (C-NH2) and a sharp resonance at 4.65 ppm (N-NH2) are characteristic for aminoguanidinium cations.37 TAG protons for 7 can be assigned to two broad singlets at 8.61 ppm (NH-NH2) and 5.68 ppm (N-NH2). All compounds show three signals for the bis-heterocyclic moiety. The resonance, with the greatest shift to lower fields always belongs to the triazole carbon C-NO2 appearing in the range of 155.3 to 151.2 ppm. The two remaining bridging carbon signals emerge in the region of 137.0 to 134.9 ppm for the tetrazole and 133.3 to 132.0 ppm for the triazole. Compounds 5, 6, and 7 exhibit one additional signal for the cations at 158.1, 159.0 and 159.0 ppm, respectively. 14N measurements show resonances in the area of around −28 ppm for the nitro groups of the triazole moieties. Additionally, spectra of compounds 2–4 show sharp signals for the respective cations at around −360 ppm.Proton coupled 15N NMR measurements were measured for neutral compound 1 and bis-ammonium salt 2 as a representative for the anion moiety. For the two bis-heterocycles, eight different resonances were obtained. An additional signal is detectable in the spectrum of 2. This belongs to the resonance of the ammonium cation. By comparison with the spectra of the respective symmetric molecules 1,1-dihydroxy-5,5′-bistetrazole (A)17 and 1,1′-dihydroxy-3,3′-dinitro-bis-1,2,4-triazole (B)19 all signals could be assigned, as shown in Fig. 9. The double deprotonation results in a shift of some signals to lower fields. That is the case for the two oxygen-substituted nitrogen atoms N1 and N5 as well as N3. The nitrogen atoms N2, N4, N6, N7 and N8 remain almost unchanged by the deprotonation, with respect to the chemical shift.
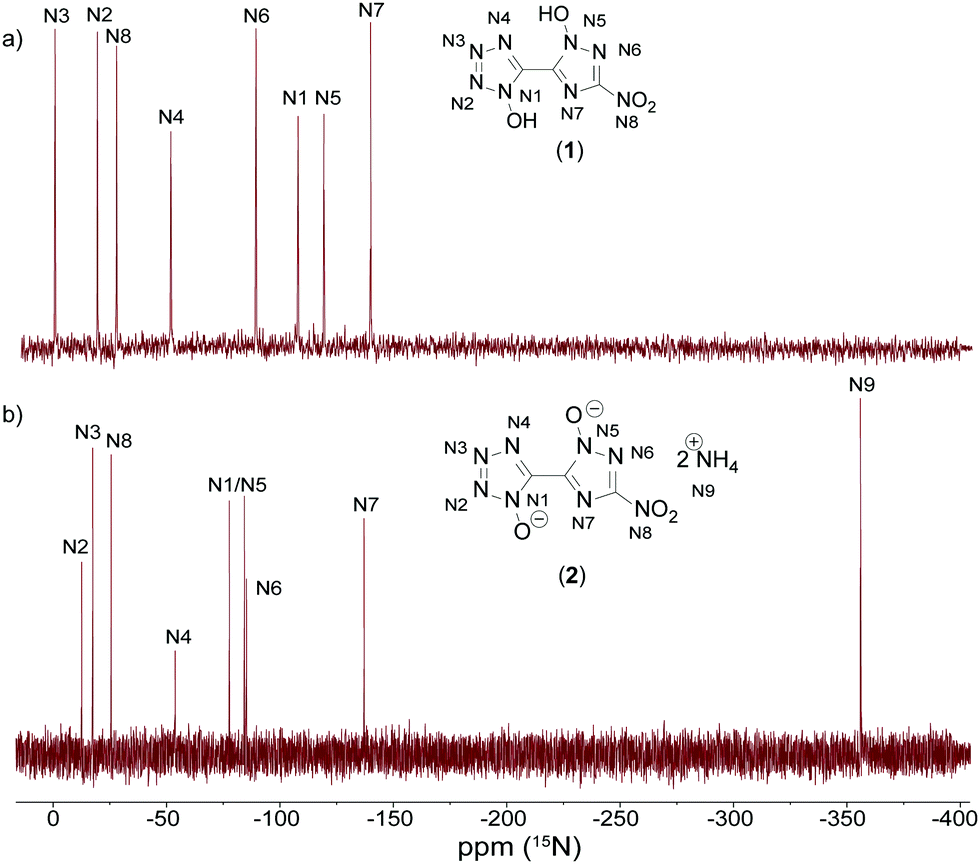 | ||
| Fig. 9 Proton coupled 15N NMR spectra of 1 (top, (a)) and 2 (bottom, (b)) measured in DMSO-D6. Chemical shifts are given with respect to MeNO2. | ||
Physicochemical properties
Experiments for the thermal behavior were determined through DTA measurements with a heating rate of 5 °C min−1. All respective values for compounds 1–7 are listed in Table 1. Neutral compound 1 decomposes at 167 °C and starts losing its crystal waters from around 71 °C indicated by an endothermal signal, which was further confirmed by a TGA measurement (for more details see the ESI†). Compounds 2–4 decompose sharply without any other measurable signal before the exothermic event at 265 °C, 206 °C and 205 °C, respectively. Compounds 5–7, all consisting of a guanidinium-based cation, feature endothermic signals prior to their decomposition points. Guanidinium salt 5 shows a melting point at 200 °C (Tdec = 270 °C) and 6–7 melt close to their decomposition (Tmelt = 217 °C (6); 165 °C (7); Tdec = 219 °C (6); 204 °C (7)). Derivatives 1–2 as well as 5–6 are insensitive toward friction and impact, whereas 3, 4 and 7 are moderately sensitive with values of IS = 7 J and FS = 192 N for 7 and IS = 20 J and FS = 324 N for 4. The densities recalculated to room temperature are in the range of 1.649 g cm−3 (6) to 1.885 g cm−3 (4) for ionic derivatives 2–7 and 1.808 g cm−3 for neutral compound 1. The detonation velocities for all synthesized compounds are above 8200 m s−1, and for 3, 4 and 7 are even over 9000 m s−1. Bis-hydroxylammonium derivative 4, which is particularly interesting because of its energetic properties, is the overall best compound and has a calculated detonation velocity of 9490 m s−1 and detonation pressure of 386 kbar. These values fit very well with the calculated values for MAD-X1 and TKX-50, since they are approximately in the middle of both and thus support the theory that 4 is also a hybrid molecule in terms of its properties.18,19| 1 | 2 | 3 | 4 | 5 | 6 | 7 | TKX-50 | MAD-X1 | β-HMX | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Impact sensitivity (BAM drophammer (1 of 6)). b Friction sensitivity (BAM friction tester (1 of 6)). c Electrostatic discharge device (OZM research). d From X-ray diffraction analysis recalculated to 298 K; * Density is based on gas pycnometer measurement at 25 °C. e Nitrogen content. f Oxygen balance with respect to the formation of CO2. g Decomposition temperature (DTA; β = 5 °C min−1). h Calculated enthalpy of formation. i Calculated energy of formation. j Energy of explosion. k Detonation temperature. l Volume of detonation products (assuming only gaseous products). m Detonation pressure at Chapman-Jouguet point. n Detonation velocity. | ||||||||||
| Formula | C3H8N8O7 | C3H8N10O4 | C3H10N12O4 | C3H8N10O6 | C5H12N14O4 | C5H14N16O4 | C4H10N14O4 | C2H8N10O4 | C4H8N10O8 | C4H8N8O8 |
| M [g mol−1] | 268.13 | 248.16 | 278.19 | 280.16 | 332.24 | 362.28 | 318.21 | 236.15 | 324.17 | 296.16 |
| ISa [J] | >40 | >40 | 10 | 20 | >40 | 40 | 7 | 20 | >40 | 7 |
| FSb [N] | >360 | >360 | 216 | 324 | >360 | >360 | 192 | 120 | >360 | 112 |
| ESD [J]c | 0.50 | 0.37 | 0.10 | 0.20 | 0.25 | 0.25 | 0.10 | 0.10 | 0.50 | 0.20 |
| P [g cm−3] | 1.808 | 1.753 | 1.718 | 1.885* | 1.707* | 1.649 | 1.681 | 1.877 | 1.90 | 1.904 |
| N [%] | 44.8 | 56.4 | 60.4 | 50.0 | 59.0 | 61.9 | 61.6 | 59.3 | 43.2 | 37.84 |
| Ω [%] | −19.2 | −38.7 | −40.3 | −22.8 | −57.8 | −57.4 | −45.3 | −27.1 | −19.7 | −21.6 |
| T dec [°C] | 169 | 265 | 206 | 205 | 270 | 219 | 204 | 221 | 217 | 279 |
ΔfH0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h [kJ mol−1] h [kJ mol−1] |
−261.4 | 200.6 | 547.3 | 316.4 | 261.9 | 490.6 | 615.5 | 447.0 | 222.3 | 74.8 |
| ΔfU0i [kJ mol−1] |
−233.0 | 227.8 | 579.6 | 346.2 | 299.1 | 532.7 | 650.2 | 474.3 | 254.5 | 109.5 |
| Explo5 V6.05.02 | ||||||||||
| −ΔExU0j [kJ kg−1] |
4587 | 4378 | 5825 | 5631 | 3557 | 3995 | 6383 | 5758 | 5670 | 5700 |
|
T
detk [K] |
3126 | 2995 | 3353 | 3573 | 2518 | 2709 | 3978 | 3513 | 3670 | 3623 |
|
V
0l [L kg−1] |
820 | 873 | 907 | 831 | 865 | 889 | 875 | 910 | 778 | 763 |
|
P
CJm [kbar] |
313 | 284 | 311 | 386 | 244 | 249 | 318 | 401 | 381 | 378 |
|
V
detn [m s−1] |
8666 | 8650 | 9052 | 9490 | 8271 | 8358 | 9058 | 9940 | 9270 | 9193 |
At least since the synthesis of TKX-50 and MAD-X1 and their establishment as green secondary explosive replacements, the benefit of introducing N-oxidation to nitrogen-rich azoles is undeniable.18,38–41 The advantages are not only the apparent increase of the oxygen balance, but also the increase of the respective densities as well as higher thermal robustness, since the introduction of an oxygen adds another site for the hydrogen bridge acceptor.
Hydroxylammonium, formally oxidized ammonium, is for several reasons of great interest in energetic materials chemistry. As for the oxidation of the heterocycles, the main advantages are the increased oxidation balance and the elevated density of the resulting salt. With the increasing numbers of N-oxides, a clear trend is visible of better performance parameters for a growing number of introduced oxygen atoms. Fig. 10 depicts the investigation on the effect of rising numbers of N-oxidations based on the heterocyclic motif 5-(3-nitro-1,2,4-triazol-5-yl)-tetrazole.42 Hydroxylammonium was chosen as the cation for this comparison, but similar trends can be seen for other salts. For the decomposition point, an increase of about 25 °C compared to the minor oxidized compounds G and H is observed for 4, even if this trend is not completely unambiguous. The densities were all determined through pycnometric measurements and are therefore easily comparable among themselves. The density of 4 is more than 0.1 g cm−3 higher than that of G, which is due to the double N-oxidation. The closely related detonation rate also increases with increasing N-oxidation. In general, it can be noted that the rise in density and detonation velocity from G to H is not as large as from H to 4. However, it can be stated that there is a clear improvement in all energetic parameters with increasing numbers of N-oxides.
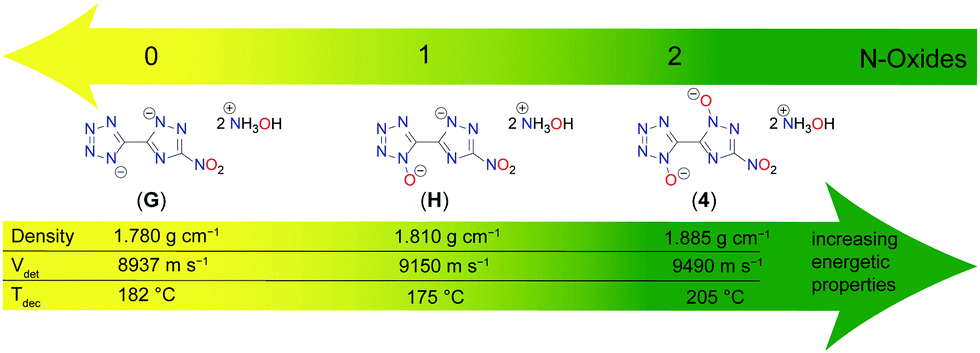 | ||
| Fig. 10 Comparison of the density, detonation velocity and decomposition temperature of bis-hydroxylammonium salts 4 and the respective 5-(3-nitro-1,2,4-triazol-5-yl)-tetrazole homologues without (G) and with one (H) N-oxide. | ||
Conclusions
The skeleton based on 1-hydroxy-3-nitro-1,2,4-triazole combined with 1-hydroxy-tetrazole has proved to be an interesting platform for new energetic materials. The neutral compound 1 could finally be synthesized through selective oxidation of the triazole moiety using Oxone® under phosphate buffered conditions. The six ionic derivatives 2–7 were obtained by reaction of the respective bases with 1 in an alcoholic medium in good yields and were intensively characterized. While insensitive bis-ammonium and bis-guanidinium derivatives 2 and 5 are the thermally most stable ones (265 °C for 2 and 270 °C for 5), the bis-hydroxylammonium derivative 4 is, as expected, the best performing compound with a detonation velocity of 9490 m s−1 and a moderate sensitivity of IS = 20 J and FS = 324 N. As it is the hybrid-molecule of TKX-50 and MAD-X1, an overlap of some properties and characteristics was expected. 4 exhibits moderate sensitivities, a high density and high heat of formation, which results in high calculated detonation properties and thus confirms the concept under investigation. The method of N-oxidation on nitrogen-rich azoles again showed enormous improvement compared to the minor oxidized molecules (G and H) and an increase in all energetically relevant properties. Generally, all compounds investigated show consistently convincing performance data, even if synthesis is more demanding than for the symmetric 1,1′-dihydroxy-5,5′-bistetrazole (A) and 1,1′-dihydroxy-3,3′-dinitro-bis-1,2,4-triazole (B).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of this work by Ludwig-Maximilian University (LMU), the Office of Naval Research (ONR) under grant no. ONR N00014-19-1-2078 and the Strategic Environmental Research and Development Program (SERDP) under contract no. W912HQ19C0033 is gratefully acknowledged. We thank Prof. Konstantin Karaghiosoff for NMR measurements, Jasmin Lechner for TGA measurement, Alexander Harter for pycnometric measurements and Stefan Huber for assistance in sensitivity measurements.Notes and references
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- D. Kumar, Y. Tang, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2018, 24, 17220–17224 CrossRef CAS PubMed.
- M. C. Schulze, B. L. Scott and D. E. Chavez, J. Mater. Chem. A, 2015, 3, 17963–17965 RSC.
- H. Gao, Q. Zhang and J. M. Shreeve, J. Mater. Chem. A, 2020, 8, 4193–4216 RSC.
- M. Deng, Y. Feng, W. Zhang, X. Qi and Q. Zhang, Nat. Commun., 2019, 10, 1339 CrossRef PubMed.
- S. Li, Y. Wang, C. Qi, X. Zhao, J. Zhang, S. Zhang and S. Pang, Angew. Chem., Int. Ed., 2013, 52, 14031–14035 CrossRef CAS PubMed.
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, S. Song, Z. Yang, X. Qi, K. Wang, Y. Liu, Q. Zhang and Y. Tian, Nat. Commun., 2018, 9, 2444 CrossRef PubMed.
- J. Zhang, Y. Feng, Y. Bo, R. J. Staples, J. Zhang and J. M. Shreeve, J. Am. Chem. Soc., 2021, 143, 12665–12674 CrossRef CAS PubMed.
- A. K. Chinnam, Q. Yu, G. H. Imler, D. A. Parrish and J. M. Shreeve, Dalton Trans., 2020, 49, 11498–11503 RSC.
- O. T. O’Sullivan and M. J. Zdilla, Chem. Rev., 2020, 120, 5682–5744 CrossRef PubMed.
- A. A. Larin, A. V. Shaferov, M. A. Epishina, I. N. Melnikov, N. V. Muravyev, I. V. Ananyev, L. L. Fershtat and N. N. Makhova, ACS Appl. Energy Mater., 2020, 3, 7764–7771 CrossRef CAS.
- J. R. Yount, M. Zeller, E. F. C. Byrd and D. G. Piercey, J. Mater. Chem. A, 2020, 8, 19337–19347 RSC.
- M. L. Gettings, M. T. Thoenen, E. F. C. Byrd, J. J. Sabatini, M. Zeller and D. G. Piercey, Chem. – Eur. J., 2020, 26, 14530–14535 CrossRef CAS PubMed.
- Y. Tang, W. Huang, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2020, 142, 7153–7160 CrossRef CAS PubMed.
- S. E. Creegan, M. Zeller, E. F. C. Byrd and D. G. Piercey, Cryst. Growth Des., 2021, 21, 3922–3927 CrossRef CAS.
- N. Fischer, T. M. Klapötke, M. Reymann and J. Stierstorfer, Eur. J. Inorg. Chem., 2013, 2167–2180 CrossRef CAS.
- N. Fischer, D. Fischer, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Mater. Chem., 2012, 22, 20418–20422 RSC.
- A. A. Dippold and T. M. Klapötke, J. Am. Chem. Soc., 2013, 135, 9931–9938 CrossRef CAS PubMed.
- D. Fischer, T. M. Klapötke, M. Reymann, P. C. Schmid, J. Stierstorfer and M. Sućeska, Propellants, Explos., Pyrotech., 2014, 39, 550–557 CrossRef CAS.
- A. A. Dippold, D. Izsák and T. M. Klapötke, Chem. – Eur. J., 2013, 19, 12042–12051 CrossRef CAS PubMed.
- J. Zhang, P. Yin, G. Pan, Z. Wang, J. Zhang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, New J. Chem., 2019, 43, 12684–12689 RSC.
- D. Fischer, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Chem. – Eur. J., 2013, 19, 4602–4613 CrossRef CAS PubMed.
- B. Yuan, Z. Yu and E. R. Bernstein, J. Phys. Chem. A, 2015, 119, 2965–2981 CrossRef CAS PubMed.
- J. C. Bennion, J. A. Ciezak-Jenkins and T. A. Jenkins, Propellants, Explos., Pyrotech., 2019, 44, 1263–1269 CrossRef CAS.
- M. Herrmann and U. Förter-Barth, Propellants, Explos., Pyrotech., 2021, 46, 262–266 CrossRef CAS.
- X. Xu, D. Chen, H. Li, R. Xu and H. Zhao, ChemistrySelect, 2020, 5, 1919–1924 CrossRef CAS.
- X. Xu, D. Chen, H. Li, M. Yan, Y. Xiong, H. Zhao and R. Xu, RSC Adv., 2020, 10, 11939–11944 RSC.
- Y. Xu, Y. Tan, W. Cao, Y. Zhao and B. Tian, J. Phys. Chem. C, 2020, 124, 5987–5998 CrossRef CAS.
- J. L. Gottfried, T. M. Klapötke and T. G. Witkowski, Propellants, Explos., Pyrotech., 2017, 42, 353–359 CrossRef CAS.
- L. Kukuljan and K. Kranjc, Tetrahedron Lett., 2019, 60, 207–209 CrossRef CAS.
- M. Göbel, K. Karaghiosoff, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Am. Chem. Soc., 2010, 132, 17216–17226 CrossRef PubMed.
- T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Chem. – Eur. J., 2011, 17, 13068–13077 CrossRef PubMed.
- K. Hafner, T. M. Klapötke, P. C. Schmid and J. Stierstorfer, Eur. J. Inorg. Chem., 2015, 2794–2803 CrossRef CAS.
- L. Hu, R. J. Staples and J. M. Shreeve, Chem. Commun., 2021, 57, 603–606 RSC.
- J. Ma, A. K. Chinnam, G. Cheng, H. Yang, J. Zhang and J. M. Shreeve, Angew. Chem., Int. Ed., 2021, 60, 5497–5504 CrossRef CAS PubMed.
- Y. Tang, W. Huang, A. K. Chinnam, J. Singh, R. J. Staples and J. M. Shreeve, Inorg. Chem., 2021, 60, 8339–8345 CrossRef CAS PubMed.
- D. G. Piercey, D. E. Chavez, B. L. Scott, G. H. Imler and D. A. Parrish, Angew. Chem., Int. Ed., 2016, 55, 15315–15318 CrossRef CAS PubMed.
- D. G. Piercey, D. E. Chavez, S. Heimsch, C. Kirst, T. M. Klapötke and J. Stierstorfer, Propellants, Explos., Pyrotech., 2015, 40, 491–497 CrossRef CAS.
- Y. Tang, K. Li, A. K. Chinnam, R. J. Staples and J. M. Shreeve, Dalton Trans., 2021, 50, 2143–2148 RSC.
- J. Yuan, X. Long and C. Zhang, J. Phys. Chem. A, 2016, 120, 9446–9457 CrossRef CAS PubMed.
- M. Dachs, A. A. Dippold, J. Gaar, M. Holler and T. M. Klapötke, Z. Anorg. Allg. Chem., 2013, 639, 2171–2180 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: A detailed Experimental and X-ray diffraction section, details and graphic illustrations of the thermal analysis and the computational methods. CCDC 2144424–2144432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ma00135g |
| ‡ First author. |
| This journal is © The Royal Society of Chemistry 2022 |