
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu3N/Cu2O core–shell nanowires: growth and properties†
Kalliopi
Mavridou
ab,
Maria
Katsikini
 a,
Andreas
Othonos
c,
Nikoletta
Florini
a,
Philomela
Komninou
a and
Matthew
Zervos
*d
a,
Andreas
Othonos
c,
Nikoletta
Florini
a,
Philomela
Komninou
a and
Matthew
Zervos
*d
aSchool of Physics, Aristotle University of Thessaloniki, GR-54124, Thessaloniki, Greece. E-mail: zervos@ucy.ac.cy
bHelmholtz-Zentrum Dresden-Rossendorf, PO Box 01328, Dresden, Germany
cLaboratory of Ultrafast Science, University of Cyprus, Nicosia, PO Box 20537, Nicosia, 1678, Cyprus
dNanostructured Materials and Devices Laboratory, School of Engineering, University of Cyprus, PO Box 20537, Nicosia, 1678, Cyprus
First published on 23rd May 2022
Abstract
CuO nanowires with diameters between 100 and 200 nm, lengths up to ∼10 μm and a uniform distribution have been grown at 600 °C under 100 mL min−1 O2 on 15 mm × 30 mm Cu foils. The CuO nanowires have a monoclinic crystal structure, grow by a vapor–solid mechanism and can be reduced to Cu under H2 at 300 °C but they are shortened, contain residual Cu2O and are eliminated above 400 °C. We develop a strategy to preserve their integrity via the deposition of Cu over the CuO in order to convert them into Cu3N under NH3:H2. The Cu3N nanowires obtained in this way are curly and have a cubic anti-ReO3 crystal structure but are surrounded by a surface shell of Cu2O with a thickness of a few tens of nm as shown by transmission electron microscopy. We find that the CuO NWs coated with Cu having a thickness greater than 200 nm are not fully converted into Cu3N and have an inner core of CuO. The Cu3N nanowires exhibited four maxima in differential transmission at 2.41, 2.17, 1.9 and 1.8 eV, using ultrafast absorption-transmission spectroscopy, corresponding to the M and R direct energy band gaps of Cu3N in good agreement with theory but we find no evidence for quantization. In addition, we observed two minor peaks at 1.69 and 1.67 eV that may be related to transitions between states in the Cu2O shell or Cu3N under compression. Despite the fact that Cu3N has no mid-gap states the photogenerated carriers have lifetimes less than 100 ps, so its potential as a defect tolerant semiconductor for energy conversion is discussed along with its perspective for energy storage.
1. Introduction
Cu3N1 is a binary semiconductor with an indirect energy band gap of 1.0 eV that has a cubic a-ReO3 crystal structure with a lattice constant of 3.8 Å (space group PM![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Number 221).2 It has been described as a defect tolerant semiconductor which is the tendency of a semiconductor to keep its properties despite the presence of crystallographic defects as suggested by Zakutayev et al.3 As such it has been proposed to be used as a solar cell absorber considering that bipolar doping is possible3 but also for energy storage and the realization of batteries4 due to the large voids in the center of its cubic structure that can accommodate foreign atoms, such as lithium.5 It has been found that Li3−xCuxN has a high reversible capacity of 650 mA h g−1 so Cu3N is indeed an attractive material for energy storage and Lithium-ion batteries (LIB)6,7 but so far no one has fabricated a solar cell device using Cu3N exhibiting photovoltaic action.
m, Number 221).2 It has been described as a defect tolerant semiconductor which is the tendency of a semiconductor to keep its properties despite the presence of crystallographic defects as suggested by Zakutayev et al.3 As such it has been proposed to be used as a solar cell absorber considering that bipolar doping is possible3 but also for energy storage and the realization of batteries4 due to the large voids in the center of its cubic structure that can accommodate foreign atoms, such as lithium.5 It has been found that Li3−xCuxN has a high reversible capacity of 650 mA h g−1 so Cu3N is indeed an attractive material for energy storage and Lithium-ion batteries (LIB)6,7 but so far no one has fabricated a solar cell device using Cu3N exhibiting photovoltaic action.
In the past, Cu3N layers have been prepared by a variety of methods including, sputtering,8 molecular beam epitaxy,9 atomic layer deposition10,11 and pulsed laser deposition12,13 but in most cases Cu3N has been obtained by sputtering under Ar and N2. For instance, Birkett et al.8 prepared Cu3N by sputtering under Ar and N2 and the direct energy band gap was found to be 1.68 eV at 300 K which increased very slightly up to 1.7 eV at 4.2 K consistent with their electronic structure calculations. Recently, Matsuzaki et al.14 obtained Cu3N directly from Cu under NH3 and O2 at temperatures ranging between 500 °C and 800 °C. The conversion of Cu into Cu3N was suggested to occur via a two-step reaction of Cu with O2 and then with NH3 which in a way is similar to the conversion of Ga2O3 nanowires into GaN under NH3 at elevated temperatures.15
In the past only, a few have investigated the growth and properties of Cu3N nanowires (NWs). More specifically Wang et al.16 prepared Cu3N NWs by forming Cu(OH)2 NWs on Cu foils that were subsequently exposed to NH3 at 350 °C for 3 h. In addition, Ma et al.17 observed the formation of a planar network of sub-nm diameter Cu3N NWs on Cu(110) foils by strain relief guided growth under N2 at 10−7 mbar and 427 °C. Moreover, Zhang et al.18 obtained Cu3N rods by metal organic chemical vapor deposition (MOCVD) using copper(II) acetylacetonate with H2:NH3 while Hou et al.19 investigated the electrical properties of Ni–Cu3N NWs and used them for water splitting. More recently, Cu3N NWs were prepared by the formation of Cu(OH)2 NWs on Cu foils after which they were converted into Cu3N under a flow of NH3. Following this, Li3N/Cu NWs were obtained by bringing the Cu3N NWs into contact with Li.20 Lastly, Cu3N NWs have also been obtained via an ammonolysis reaction of copper oxide films.21
Here, we have investigated the growth and conversion of CuO into Cu3N NWs. CuO NWs have been obtained in the past mainly via the thermal oxidation of Cu in air or under O2 due to the inherent simplicity of this method.22 Interestingly, a detailed investigation into the growth of CuO NWs on different Cu surfaces was carried out recently by Popovski et al.23 confirming that CuO NWs may be obtained on Cu foils at temperatures as low as 250 °C. The growth of CuO NWs on Cu foils has also been achieved at low temperatures between 250 °C and 450 °C by Gulbransen et al.24 On the other hand, it has been shown by Liang et al.25 that the growth of CuO NWs depends on the annealing temperature and the grain size of the Cu foil that is used as substrate. A smaller grain size and single grain orientation can produce longer, denser and a more uniform distribution of CuO NWs. However, a significant disadvantage of the CuO NWs obtained via the thermal oxidation of Cu foils is that intermediate layers of CuO and Cu2O are formed so a significant lattice mismatch exists between the Cu, CuO and Cu2O which in turn makes the CuO NW network particularly fragile. Nevertheless, it has been shown that the integrity of the CuO NWs can be improved by texturing the surface of the Cu foil.26 It is interesting to point out here that CuO NWs have been used in dye sensitized solar cells,27 sensors,28–31 as well as for catalysis32 so the growth, properties and applications of CuO NWs is an ongoing topic of active investigation which is interesting from a fundamental and technological point of view.33
To the best of our knowledge, no one has investigated previously the conversion of CuO into Cu3N NWs under NH3: H2 which is similar to our earlier investigations on the conversion of Ga2O3 into GaN NWs under NH3 at elevated temperatures.15 We have investigated the structural properties of the CuO and Cu3N NWs by X-ray diffraction, transmission and scanning electron microscopy as well as Raman spectroscopy. In addition, we have carried out ultrafast pump-probe spectroscopy (UPPS) which is a versatile tool for understanding the generation-recombination, mechanisms and pathways, of photo generated electron–holes that depend on the electronic band structure and energetic position of sub-band gap states related to crystal imperfections and impurities that can’t be determined by steady state UV-vis absorption-transmission spectroscopy.
2. Materials and methods
Initially square foils of 10 mm × 10 mm Cu (99.9%) with a thickness of ∼0.15 mm or 150 μm were immersed in a solution of HCl and De-ionized (DI) H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to remove the native oxide. Subsequently the foils were rinsed with DI water, dried with nitrogen and pressed flat. The growth of CuO NWs on the Cu foils was carried out in a 1′′ hot wall, horizontal, chemical vapor deposition (CVD) reactor, capable of reaching 1100 °C that was fed on the upstream side by a manifold consisting of four mass flow controllers connected to Ar, NH3, O2 and H2. The Cu foil was loaded in a quartz boat that was inserted in the reactor after which it was purged with 500 mL min−1 of Ar for 10 min at 1 bar. Then the temperature was ramped at 10 °C min−1 under a flow of 300 mL min−1 O2 up to 600 °C. Upon reaching 600 °C the same flow of O2 was maintained for 120 min. In order to prevent the development of thermal stress in the Cu foil, the cooling rate was adjusted to 5 °C min−1. The conversion of CuO into Cu3N NWs was carried out in the temperature range of 300–400 °C for 60 min under a flow of (a) NH3 (b) NH3:H2 and (c) NH3:O2 after purging with 500 mL min−1 of Ar for 10 min. In all cases the samples were removed from the reactor at room temperature.
:1) to remove the native oxide. Subsequently the foils were rinsed with DI water, dried with nitrogen and pressed flat. The growth of CuO NWs on the Cu foils was carried out in a 1′′ hot wall, horizontal, chemical vapor deposition (CVD) reactor, capable of reaching 1100 °C that was fed on the upstream side by a manifold consisting of four mass flow controllers connected to Ar, NH3, O2 and H2. The Cu foil was loaded in a quartz boat that was inserted in the reactor after which it was purged with 500 mL min−1 of Ar for 10 min at 1 bar. Then the temperature was ramped at 10 °C min−1 under a flow of 300 mL min−1 O2 up to 600 °C. Upon reaching 600 °C the same flow of O2 was maintained for 120 min. In order to prevent the development of thermal stress in the Cu foil, the cooling rate was adjusted to 5 °C min−1. The conversion of CuO into Cu3N NWs was carried out in the temperature range of 300–400 °C for 60 min under a flow of (a) NH3 (b) NH3:H2 and (c) NH3:O2 after purging with 500 mL min−1 of Ar for 10 min. In all cases the samples were removed from the reactor at room temperature.
The morphology and crystal structure of the Cu3N NWs were determined by Scanning Electron Microscopy (SEM, TESCAN) operated at an accelerated voltage of 30 kV and X-ray diffraction (XRD, Rigaku Co) using Cu Kα radiation (λ = 1.54056 Å) with θ–2θ mode ranging from 10 to 80°. Transmission Electron Microscopy (TEM) and high-resolution TEM (HRTEM) observations were carried out in a JEOL JEM F200 CFEG TEM/STEM electron microscope operated at 200 kV (point resolution 0.19 nm). TEM samples were prepared by mechanically rubbing the CuO and Cu3N/Cu2O NWs onto TEM carbon coated copper grids. Raman Spectroscopy (RS) was carried out using a DILOR-XY spectrometer equipped with an optical microscope and a 100× objective; the Raman measurements were performed in the backscattering geometry. The red line (λ = 647.1 nm) of a Kr+ laser was used for excitation and the spectra were calibrated with a Ne lamp. The power on the sample was less than 2 mW.
Finally, the optical properties of the Cu3N NWs were investigated by measuring the time evolution of the differential transmission (dT/T) on a ps time scale by UPPS using a pump with λPU = 400 or 260 nm and diameter of 1 mm on the sample and a probe that was varied between λPR = 450 nm and 750 nm. In this case the CuO NWs were removed from the Cu foil by sonication in isopropanol and dispersed gradually on fused silica (f-SiO2) after which they were converted into Cu3N under NH3:H2 at 400 °C. UPPS was carried out using a Ti: Sapphire ultrafast amplifier system generating 100 fs pulses at 800 nm and running at a repetition rate of 1 kHz. A nonlinear crystal was used to generate 400 nm for the purpose of exciting the sample, whereas part of the fundamental was used to generate a super continuum light for probing different energy states. The measurements were carried out using a typical pump-probe optical setup in a non-collinear configuration, where the probe beam was directed into a spectrometer with 0.5 nm spectra resolution equipped with a fast CCD array.
3. Results and discussion
3.1 Growth of CuO nanowires on Cu foil
In the past, we have obtained CuO NWs by the thermal oxidation of a Cu foil with a thickness of ∼0.15 mm.34 More specifically, the CuO NWs were obtained in a 1′′ hot wall reactor that was purged with Ar for 10 min after which the temperature was ramped to 600 °C using a slow ramp rate of 5 °C min−1 and a flow of 100 mL min−1 O2 that was maintained up to 4 h at 600 °C. The CuO NWs obtained in this way were straight with diameters of ∼100 nm, had lengths of up to ∼10 μm and exhibited clear peaks in the XRD corresponding to the monoclinic crystal structure of CuO but additional peaks were also observed corresponding to the underlying Cu2O and Cu foil.Here, we have grown again CuO NWs on Cu foils at 600 °C for 120 min in order to convert them into Cu3N NWs by thermal nitridation. A typical SEM image of the Cu foils used for the growth of the CuO NWs is shown in Fig. 1(a). This exhibited well resolved peaks in the XRD as shown in Fig. 1(b) corresponding to the face centered cubic (fcc) crystal structure of Cu with a lattice constant of 3.597 Å. A cross-section of the Cu foil is also shown as an inset in Fig. 1(a), from which we find that it has a thickness of about 120 μm. The CuO NWs that were grown on the Cu foil have diameters of ∼100–200 nm, lengths of ∼3–10 μm and form a dark black network with excellent uniformity across areas up to 15 × 30 mm. A typical SEM image of the CuO NWs is shown in Fig. 1(c). In fact, the CuO NWs grow on top of a bilayer of bulk CuO and Cu2O over the Cu foil. A reduction of about 40 μm in the thickness of the Cu foil occurred after growth as shown in Fig. 1(d). The CuO NWs exhibited well resolved peaks in the XRD pattern as shown in Fig. 1(e) corresponding to the monoclinic crystal structure of CuO and also a few peaks due to the Cu2O.
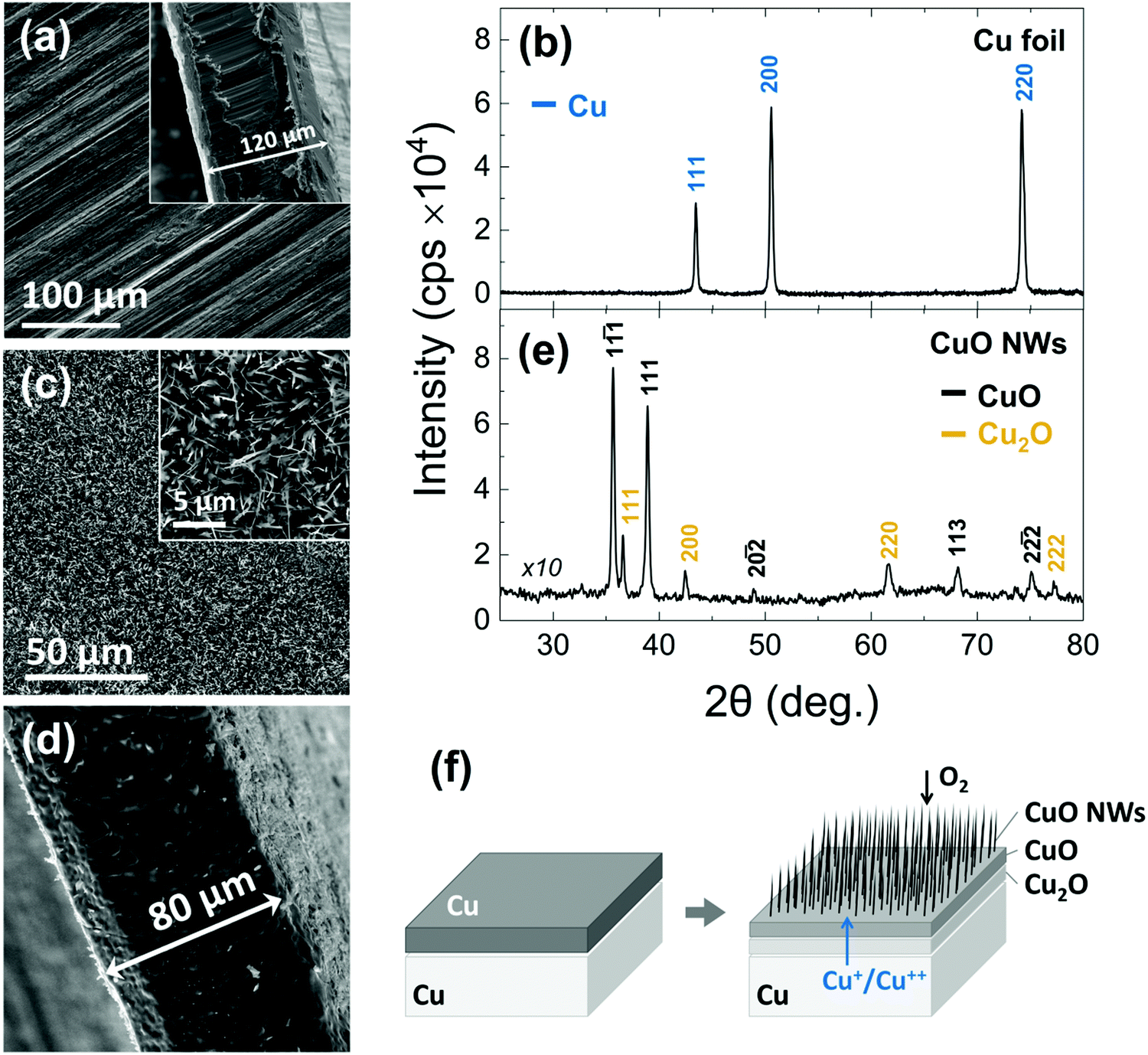 | ||
| Fig. 1 Growth of CuO NWs. (a) SEM image and (b) XRD pattern of the Cu foil that was used for the growth of the CuO NWs. (c) SEM image of the CuO NWs that grow on top of the Cu foil (d) Cross-section image of the Cu foil after removing the bulk CuO layer and CuO NWs (e) XRD pattern of the CuO NWs after being removed from the underlying Cu foil (f) schematic illustration of the growth of CuO NWs. | ||
A typical TEM image of the CuO NWs is illustrated in Fig. 2(a). They have a straight morphology with averaged projected diameters measured as d = 150 ± 30 nm. The corresponding selected area electron diffraction (SAED) pattern recorded along the [111] zone axis (z.a), shown in Fig. 2(b), confirms the base-centered monoclinic crystal structure of the CuO NWs. The 11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) , 11
, 11![[0 with combining macron]](https://www.rsc.org/images/entities/char_0030_0304.gif) and 20 reflections corresponding to the interplanar spacing (d-spacing) of the lattice planes are indicated. A HRTEM image from the surface of a CuO NW is presented in Fig. 2(c). A magnified image of the area bounded by the white frame is given as inset. The lattice fringe spacing, equal to 0.187 nm, corresponds to the d-spacing of the (20) lattice planes of the CuO monoclinic structure.
and 20 reflections corresponding to the interplanar spacing (d-spacing) of the lattice planes are indicated. A HRTEM image from the surface of a CuO NW is presented in Fig. 2(c). A magnified image of the area bounded by the white frame is given as inset. The lattice fringe spacing, equal to 0.187 nm, corresponds to the d-spacing of the (20) lattice planes of the CuO monoclinic structure.
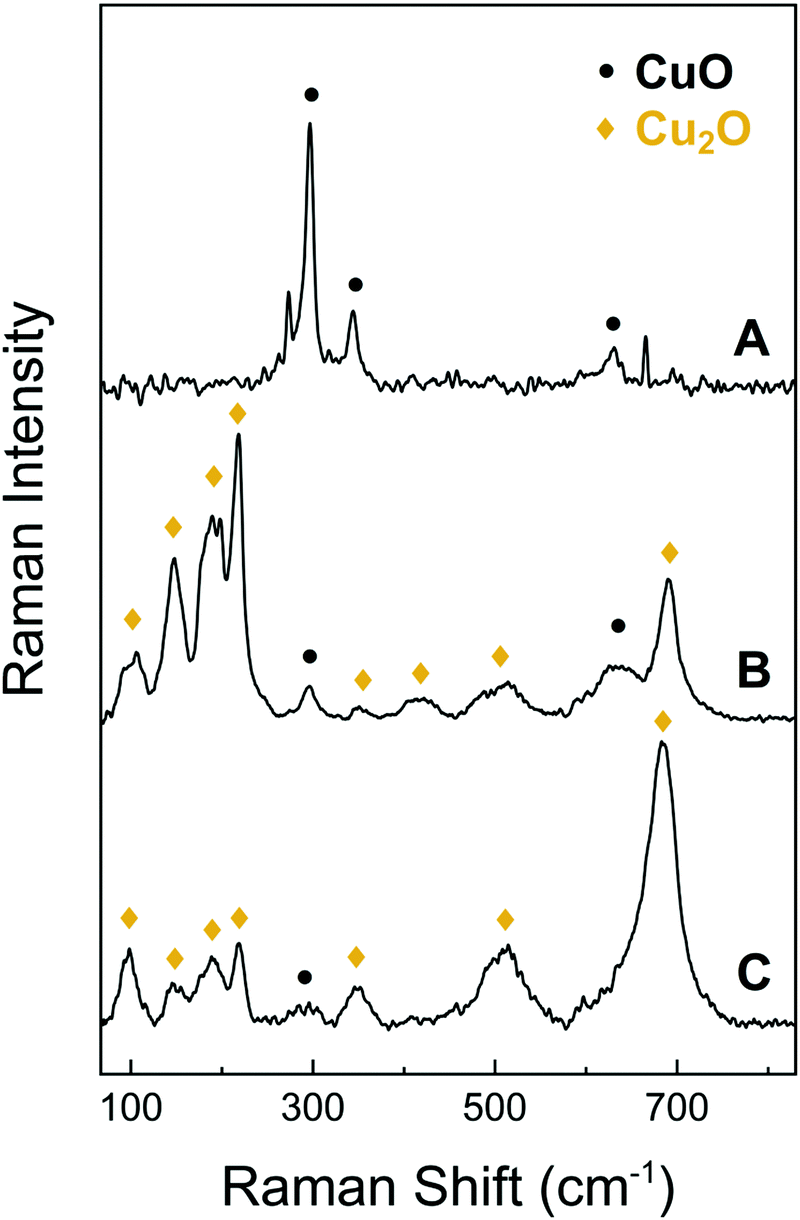 | ||
| Fig. 2 Raman spectra of the CuO NWs (A), the bulk CuO layer that supports the CuO NWs after being detached from the underlying foil (B) and the surface of the bare Cu foil (C). | ||
For completeness it should be mentioned that the CuO NWs grow by a vapor–solid (VS) mechanism whereby Cu diffuses out of the underlying foil and oxygen diffuses inwards along grain boundaries, as illustrated in Fig. 1(f). An extensive description of the growth conditions and their optimization as well as issues pertaining to the integrity of the CuO NW network is provided in the ESI,† S1.
In addition to the above, we investigated the structural properties of the CuO NWs by Raman spectroscopy and found that the conversion of the Cu to CuO NWs occurs gradually, from Cu to Cu2O and then to CuO. As expected, it is found that the CuO NWs consist only of CuO phase, as shown by trace A of Fig. 3. The Raman line observed at 290 cm−1 can be attributed to the Ag mode and the 345 and 630 cm−1 peaks to the Bg modes of the CuO phase.35 The bulk layer supporting the CuO NWs contains both the CuO and Cu2O phases as indicated by trace B in Fig. 3. The peak observed at approximately 515 cm−1 can be assigned to the only Raman active mode T2g of Cu2O. The peaks at 110 cm−1 (Eu), 150 cm−1 (T1u) and 350 cm−1 (A2u) are observed as well and again these are attributed to the Cu2O phase. Additional features at 220 cm−1 (2Eu), in the vicinity of 400–450 cm−1 and at 695 cm−1 are due to multiphonon Raman scattering. Moreover, the signal observed at 190 cm−1 is attributed to local vibrations of Cu on O-sites.36,37 The surface of the Cu foil after the exfoliation of the CuO NWs had a dark red color due to the presence of the Cu2O phase as shown by trace C in Fig. 3; a minor contribution of CuO was also detected.
 | ||
| Fig. 3 (a) TEM image of a CuO NW along with (b) the corresponding SAED pattern recorded along the [111] zone axis of monoclinic CuO. (c) HRTEM image showing the edge side of a CuO NW and as inset, magnification of the area in the rectangle frame indicating the spacing of the (20) lattice fringes of CuO. | ||
In short, a uniform distribution of crystalline CuO NWs over 15 × 30 mm Cu foils was obtained. The CuO NWs are straight, have diameters of 150 ± 30 nm, lengths of ∼3–10 μm, a monoclinic crystal structure and do not contain any Cu2O or Cu.
3.2 Nitridation of CuO into Cu3N Nanowires
Initially we tried to reduce the CuO into Cu NWs under H2 in order to subsequently convert them into Cu3N NWs under NH3: O2 similar to the Cu3N layers that were obtained previously from Cu under NH3: O2.38 We find that the CuO NWs can be reduced to Cu under H2 at 300 °C but contain residual Cu2O and are shortened. The CuO NWs were eliminated above 400 °C, see ESI,† S2. We developed a strategy to preserve their integrity of the CuO NWs under reducing conditions via the deposition of a Cu shell over the CuO NWs in order to convert them into Cu3N under NH3:H2. More specifically Cu with a thickness of 240 nm was deposited on the CuO NWs and the CuO/Cu core–shell NWs were annealed at 400 °C for 60 min under 300:10 mL min−1 NH3:H2, as illustrated in Fig. 4(a). The CuO/Cu core–shell NWs maintained their integrity and were converted into Cu3N NWs that acquired a curly shape as shown in Fig. 4(b) and exhibited well resolved peaks in the XRD pattern belonging to Cu3N as well as a single peak of low intensity at 2θ = 77.4° which may be indexed to the (222) crystallographic plane of the monoclinic crystal structure of Cu2O as shown in Fig. 4(c). The Cu3N phase was also identified by the major peaks of the optical modes T2u (170 cm−1) and T1u (655 cm−1),39 in the Raman spectra shown in Fig. 4(d). A smaller contribution of the Cu2O phase, with peaks at ca. 150 and 220 cm−1 is also detected. For comparison, the Raman spectrum recorded from a Cu3N thin film obtained from annealing of a Cu foil under NH3:H2 flow is shown at the bottom of Fig. 4(d). The Cu2O phase is also detected in this case and can be attributed to the surface native oxide of Cu3N. It is worth noting that the CuO NWs covered with a 480 nm Cu shell that were converted into Cu3N still contained CuO as shown in Fig. 4(d). This can be attributed to the incomplete conversion of the CuO NWs to Cu3N due to the fact that the oxygen atoms had to travel a longer distance in order to out-diffuse from the core during the nitridation process. Moreover, the peak at ca. 655 cm−1 for the Cu3N NWs obtained from CuO NWs covered with a Cu shell of 480 nm is broader compared to that of the Cu3N NWs, obtained from CuO NWs covered with a thin shell of 240 nm, indicating a decrease in crystallinity.
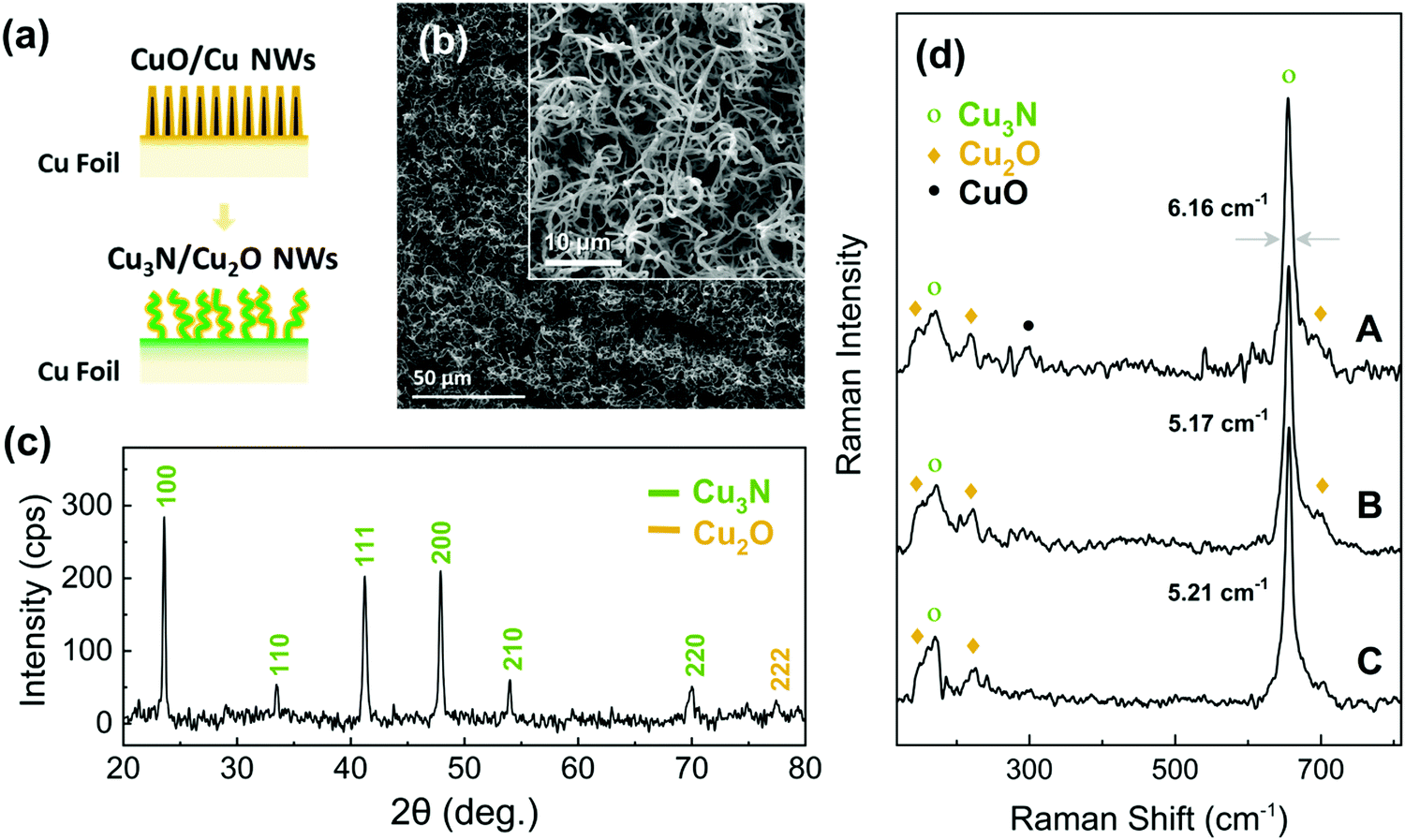 | ||
| Fig. 4 (a) Schematic illustration of the conversion of the CuO into Cu3N NWs (b), (c) SEM image and XRD pattern of the Cu3N/Cu2O NWs obtained from the CuO NWs that were coated with 240 nm Cu (d) Raman spectra of the Cu3N/Cu2O NWs obtained from CuO NWs that were coated with 480 nm Cu (trace A) and 240 nm Cu (trace B) and treated under NH3: H2; also shown the case of a Cu3N thin film obtained from a 240 nm Cu layer that was deposited on a Cu foil and treated under NH3: H2 (trace C). | ||
In order to gain a deeper insight, the structural properties of the Cu3N NWs were investigated by TEM. A curly like morphology with rough side surfaces was observed, as shown in Fig. 5(a). The Cu3N NWs had a diameter d = 320 ± 35 nm. The strong diffraction contrast of the image revealed that the Cu3N NWs actually consist of a CuO core with an averaged projected diameter of 40 ± 5 nm. Detailed SAED analysis demonstrated that the Cu3N NWs are polycrystalline as confirmed by the ring SAED pattern shown in Fig. 5(b). The indexed rings, originating from misoriented crystallites, correspond to the interplanar spacing of 110, 111, 200, 210, 220 and 311 lattice planes of the cubic anti-ReO3 structure of Cu3N. In the SAED pattern of Fig. 5(b) two additional rings were detected assigned to the interplanar spacing of 111 and 211 planes of the cubic Cu2O. The presence of Cu2O has been also confirmed by HRTEM observations. An HRTEM image recorded near the side surface of the Cu3N/Cu2O NW is presented in Fig. 5(c), while a magnification of the area in the rectangular frame (given as inset) illustrates its atomic structure, projected along the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] z.a. of the cubic Cu2O. The viewing direction is also confirmed by the corresponding FFT (2nd inset), calculated from the HRTEM image, where the 111, 11, 00, 220 spatial frequencies of the [10] z.a. are indicated. Two families of {111} lattice fringes of the cubic Cu2O are indicated. It is estimated that a 30 nm Cu2O oxide layer covers the surface of the Cu3N NWs.
10] z.a. of the cubic Cu2O. The viewing direction is also confirmed by the corresponding FFT (2nd inset), calculated from the HRTEM image, where the 111, 11, 00, 220 spatial frequencies of the [10] z.a. are indicated. Two families of {111} lattice fringes of the cubic Cu2O are indicated. It is estimated that a 30 nm Cu2O oxide layer covers the surface of the Cu3N NWs.
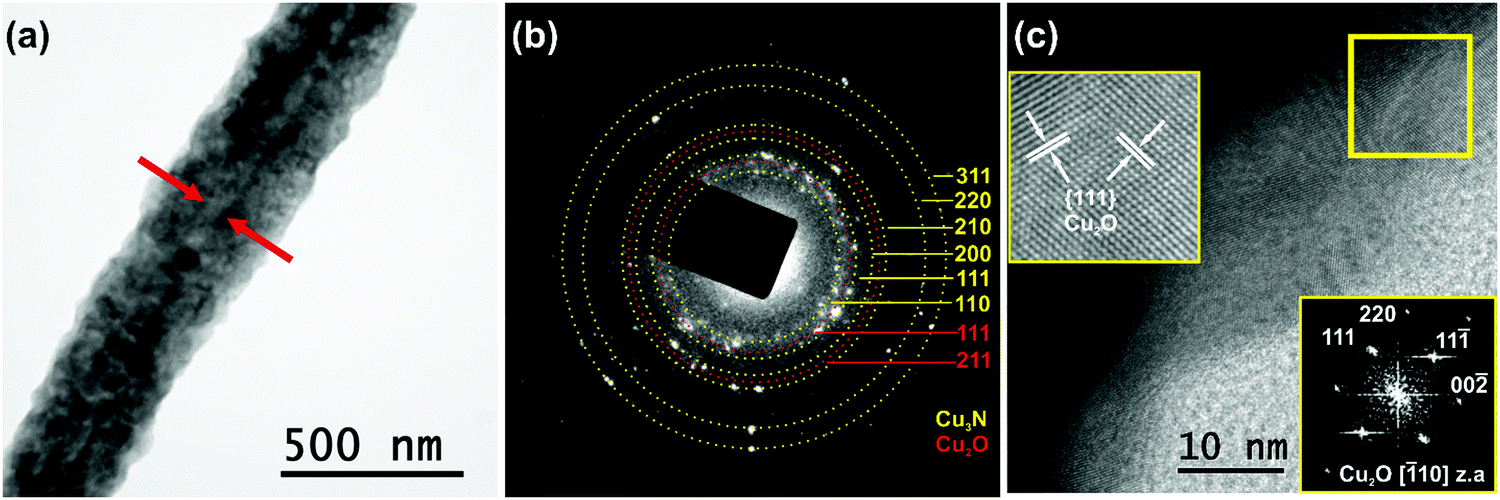 | ||
| Fig. 5 (a) TEM image of a Cu2O/Cu3N core–shell NW; the CuO core is denoted by arrows. (b) Corresponding SAED pattern comprising diffraction rings that correspond to interplanar spacings of the cubic Cu2O and Cu3N structures. (c) HRTEM image of the side surface of a Cu3N/Cu2O NW; the two insets illustrate the atomic structure of the area in the rectangular frame viewed along the [10] Cu2O z.a. and the corresponding FFT. | ||
3.3 Ultrafast pump-probe spectroscopy of Cu3N/Cu2O NWs
In order to measure the optical properties of the Cu3N NWs by UPPS we removed CuO NWs from the Cu foil by sonication in isopropanol and dispersed them gradually on transparent fused silica (f-SiO2) after which they were converted into Cu3N under NH3:H2 at 400 °C as shown in Fig. 6(a). The time evolution of the differential transmission (dT/T) on a ps time scale is shown in Fig. 6(b and c) for different probe wavelengths λPR that vary between 450 and 750 nm after ‘pumping’ the Cu3N NWs with light having a wavelength of λPU = 260 nm and incident energy of 1 and 5 μJ respectively. The incident energy was varied between 1 and 5 μJ to avoid the generation of an overtly high carrier density which leads to a smearing of the spectral structure as we have shown previously.38 In essence the photoexcited electrons will occupy states in the conduction band and recombine over a few tens of ps by moving to states with lower energy, during which the transmission of light with wavelengths that vary between 450 and 750 nm is measured, to find out which states are occupied thereby revealing their energetic position and giving insight into the actual electronic band structure. The Cu3N NWs exhibited maxima at 515 nm (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 2.41 eV), 570 nm (2.17 eV), 650 nm (1.91 eV) and 690 nm (1.8 eV) as measured by UPPS that are shown in Fig. 6(b and c). The lower energy duo of maxima at λPR = 650 nm (1.91 eV) and 690 nm (1.8 eV) correspond to the M and R direct energy band gaps of bulk-Cu3N in good agreement with the electronic structure depicted in Fig. 7(a) while the higher energy maxima at 515 nm (2.41 eV) and 570 nm (2.17 eV) correspond to the M* and R* direct gaps of Cu3N under tensile strain that arise due to the formation of Cu2O on Cu3N as shown in Fig. 7(b). The spectral structure shown in Fig. 6(b and c) is very similar to that obtained from Cu3N layers obtained previously from Cu under a flow of NH3:O2 between 400 and 600 °C.38 The Cu3N layers obtained previously exhibited distinct maxima in differential transmission (dT/T) on a ps time scale at 500 nm (2.48 eV), 550 nm (2.25 eV), 630 nm (1.97 eV) and 670 nm (1.85 eV). We showed that the maxima at 1.97 eV and 1.85 eV correspond to the M and R direct energy band gaps of bulk Cu3N in excellent agreement with density functional theory (DFT) calculations of the electronic band structure. This observation of the M and R direct energy band gaps of Cu3N was possible by virtue of the fact that crystal imperfections, such as N vacancies (VN), Cu interstitials (Cui) etc. give rise to states that are energetically located very close or inside the conduction and valence band edges as shown by Yee et al. who investigated the properties of N-rich Cu3N and confirmed the existence of Cu interstitials (Cui) by photo thermal deflection spectroscopy (PDS). The latter is an indirect method with dramatically higher sensitivity than steady state UV-vis spectroscopy that can be used for the detection of sub-band-gap states that are related to crystal imperfections and/or impurities. In addition, we showed that the maxima at 2.48 eV and 2.25 eV correspond to the M and R direct gaps of certain regions of Cu3N layers under tensile strain that arise due to the formation of CuO and Cu2O, via the out-diffusion of oxygen from the underlying fused silica (f-SiO2) and/or surface oxidation.40 The spectral structure of the Cu3N/Cu2O NWs shown in Fig. 6(b and c) is similar to that obtained previously from Cu3N layers38 but we do not observe any quantization effects, as the peaks corresponding to bulk Cu3N are not blue-shifted to higher energies. However the Cu3N NWs exhibited peaks at 730 nm (1.69 eV) and 740 nm (1.67 eV) shown in Fig. 6(b and c) that were not observed in our previous investigations of Cu3N layers. The maxima at 730 nm (1.69 eV) and 740 nm (1.67 eV) may correspond to the M* and R* direct gaps of the Cu3N closer to the core that could be under compressive strain. These values are close to those found by Birket et al.8 who investigated the variation of the energy band-gap with temperature using Fourier transform infra-red spectroscopy. They prepared Cu3N by sputtering under Ar and N2 and the direct energy band gap was found to be 1.68 eV at 300 K but changed only very slightly up to 1.7 eV at 4.2 K. However, Birket et al.8 showed that the direct and indirect band gap changes from 1.4 eV to 2.0 eV and 0.6 eV to 1.8 eV respectively upon increasing the lattice constant from 3.8 Å to 3.9 Å. Consequently, structural distortion has a pronounced effect on the values of the direct and indirect band-gap and by extension on the optical band gap. This is most likely why there is such a broad variation in the optical band gaps of Cu3N determined previously by others using steady state optical spectroscopy, i.e. UV-vis absorption-transmission, reflectance-transmission spectroscopy, ellipsometry that vary between 1.2 to 2.4 eV.41–51 Alternatively, the maxima observed at 730 nm (1.69 eV) and 740 nm (1.67 eV) in Fig. 6(b and c) may be related to transitions between states in the Cu2O shell. It is useful to mention at this point that Cu2O is a p-type metal-oxide semiconductor that has a direct energy band-gap of 2.1 eV and the Fermi level at the surface is pinned at surface states residing ∼0.4eV below the conduction band edge leading to a depletion.52 It should also be pointed out that Cu2O has acceptor like states located ∼0.16eV above the valence band maximum that are related to copper vacancies (VCu) and give rise to its native p-type conductivity.40,53 Consequently the maxima observed at 730 nm (1.69 eV) and 740 nm (1.67 eV) in Fig. 6(b and c) may be related to transitions between these states that are shown schematically in Fig. 7(d).
2.41 eV), 570 nm (2.17 eV), 650 nm (1.91 eV) and 690 nm (1.8 eV) as measured by UPPS that are shown in Fig. 6(b and c). The lower energy duo of maxima at λPR = 650 nm (1.91 eV) and 690 nm (1.8 eV) correspond to the M and R direct energy band gaps of bulk-Cu3N in good agreement with the electronic structure depicted in Fig. 7(a) while the higher energy maxima at 515 nm (2.41 eV) and 570 nm (2.17 eV) correspond to the M* and R* direct gaps of Cu3N under tensile strain that arise due to the formation of Cu2O on Cu3N as shown in Fig. 7(b). The spectral structure shown in Fig. 6(b and c) is very similar to that obtained from Cu3N layers obtained previously from Cu under a flow of NH3:O2 between 400 and 600 °C.38 The Cu3N layers obtained previously exhibited distinct maxima in differential transmission (dT/T) on a ps time scale at 500 nm (2.48 eV), 550 nm (2.25 eV), 630 nm (1.97 eV) and 670 nm (1.85 eV). We showed that the maxima at 1.97 eV and 1.85 eV correspond to the M and R direct energy band gaps of bulk Cu3N in excellent agreement with density functional theory (DFT) calculations of the electronic band structure. This observation of the M and R direct energy band gaps of Cu3N was possible by virtue of the fact that crystal imperfections, such as N vacancies (VN), Cu interstitials (Cui) etc. give rise to states that are energetically located very close or inside the conduction and valence band edges as shown by Yee et al. who investigated the properties of N-rich Cu3N and confirmed the existence of Cu interstitials (Cui) by photo thermal deflection spectroscopy (PDS). The latter is an indirect method with dramatically higher sensitivity than steady state UV-vis spectroscopy that can be used for the detection of sub-band-gap states that are related to crystal imperfections and/or impurities. In addition, we showed that the maxima at 2.48 eV and 2.25 eV correspond to the M and R direct gaps of certain regions of Cu3N layers under tensile strain that arise due to the formation of CuO and Cu2O, via the out-diffusion of oxygen from the underlying fused silica (f-SiO2) and/or surface oxidation.40 The spectral structure of the Cu3N/Cu2O NWs shown in Fig. 6(b and c) is similar to that obtained previously from Cu3N layers38 but we do not observe any quantization effects, as the peaks corresponding to bulk Cu3N are not blue-shifted to higher energies. However the Cu3N NWs exhibited peaks at 730 nm (1.69 eV) and 740 nm (1.67 eV) shown in Fig. 6(b and c) that were not observed in our previous investigations of Cu3N layers. The maxima at 730 nm (1.69 eV) and 740 nm (1.67 eV) may correspond to the M* and R* direct gaps of the Cu3N closer to the core that could be under compressive strain. These values are close to those found by Birket et al.8 who investigated the variation of the energy band-gap with temperature using Fourier transform infra-red spectroscopy. They prepared Cu3N by sputtering under Ar and N2 and the direct energy band gap was found to be 1.68 eV at 300 K but changed only very slightly up to 1.7 eV at 4.2 K. However, Birket et al.8 showed that the direct and indirect band gap changes from 1.4 eV to 2.0 eV and 0.6 eV to 1.8 eV respectively upon increasing the lattice constant from 3.8 Å to 3.9 Å. Consequently, structural distortion has a pronounced effect on the values of the direct and indirect band-gap and by extension on the optical band gap. This is most likely why there is such a broad variation in the optical band gaps of Cu3N determined previously by others using steady state optical spectroscopy, i.e. UV-vis absorption-transmission, reflectance-transmission spectroscopy, ellipsometry that vary between 1.2 to 2.4 eV.41–51 Alternatively, the maxima observed at 730 nm (1.69 eV) and 740 nm (1.67 eV) in Fig. 6(b and c) may be related to transitions between states in the Cu2O shell. It is useful to mention at this point that Cu2O is a p-type metal-oxide semiconductor that has a direct energy band-gap of 2.1 eV and the Fermi level at the surface is pinned at surface states residing ∼0.4eV below the conduction band edge leading to a depletion.52 It should also be pointed out that Cu2O has acceptor like states located ∼0.16eV above the valence band maximum that are related to copper vacancies (VCu) and give rise to its native p-type conductivity.40,53 Consequently the maxima observed at 730 nm (1.69 eV) and 740 nm (1.67 eV) in Fig. 6(b and c) may be related to transitions between these states that are shown schematically in Fig. 7(d).
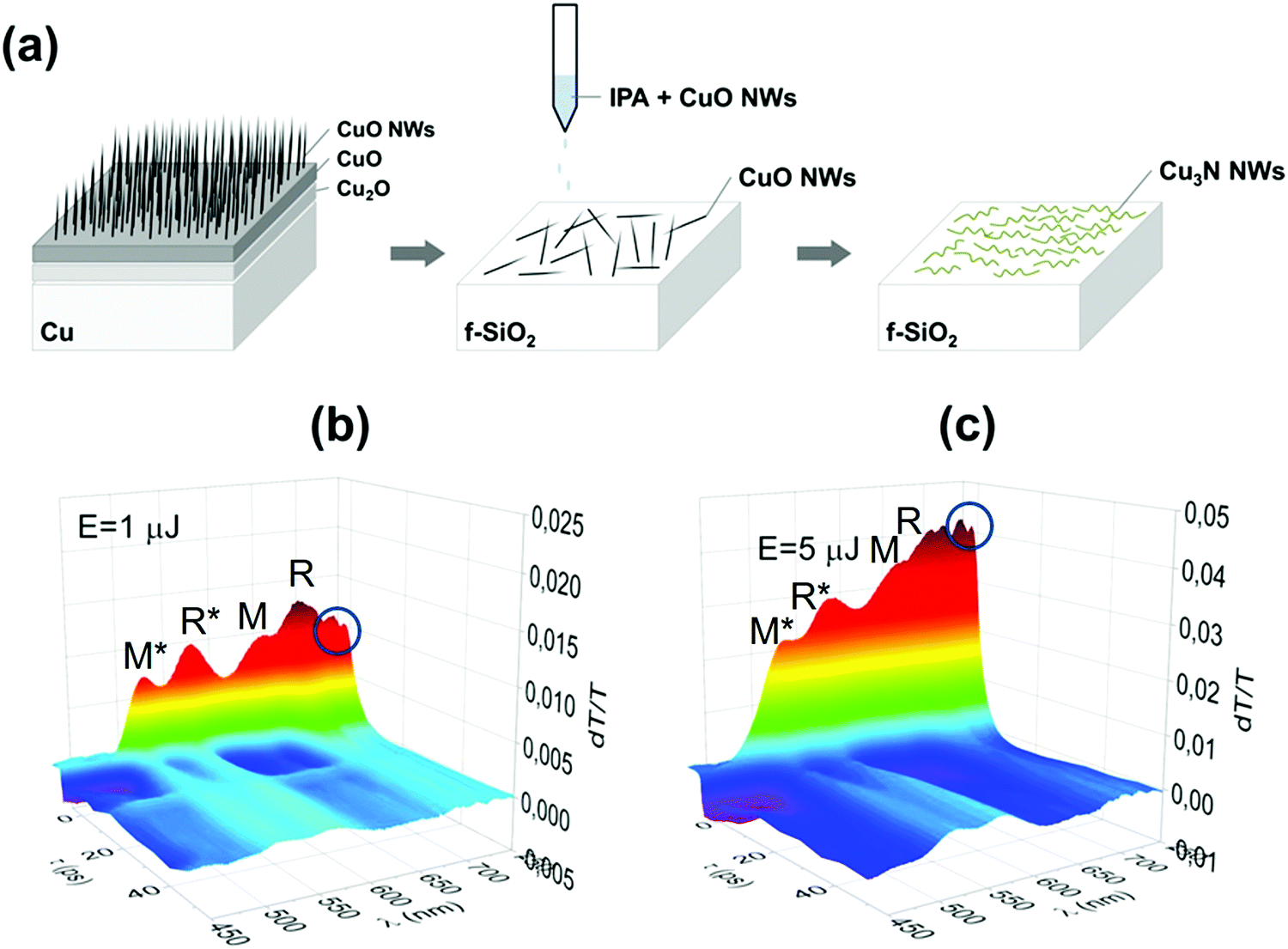 | ||
| Fig. 6 (a) Schematic illustration of the isolation and conversion of CuO into Cu3N/Cu2O NWs. UPPS of Cu3N/Cu2O NWs on f-SiO2 showing the differential transmission dT/T on the vertical axis (no units) versus time delay τ (ps) and probe wavelength λ (nm) for incident energies of (b) E = 1 μJ and (c) E = 5 μJ. The maxima at 515 nm (2.41 eV), 570 nm (2.17 eV) correspond to the M* and R* direct band gaps of Cu3N under tensile strain while those at 650 nm (1.91 eV) and 690 nm (1.8 eV) correspond to the M and R bulk band gaps of Cu3N. The encircled, minor peaks at 730 nm (1.69 eV) and 740 nm (1.67 eV) may be attributed to states in Cu2O or to the M* and R* direct band gaps of Cu3N under compressive strain. | ||
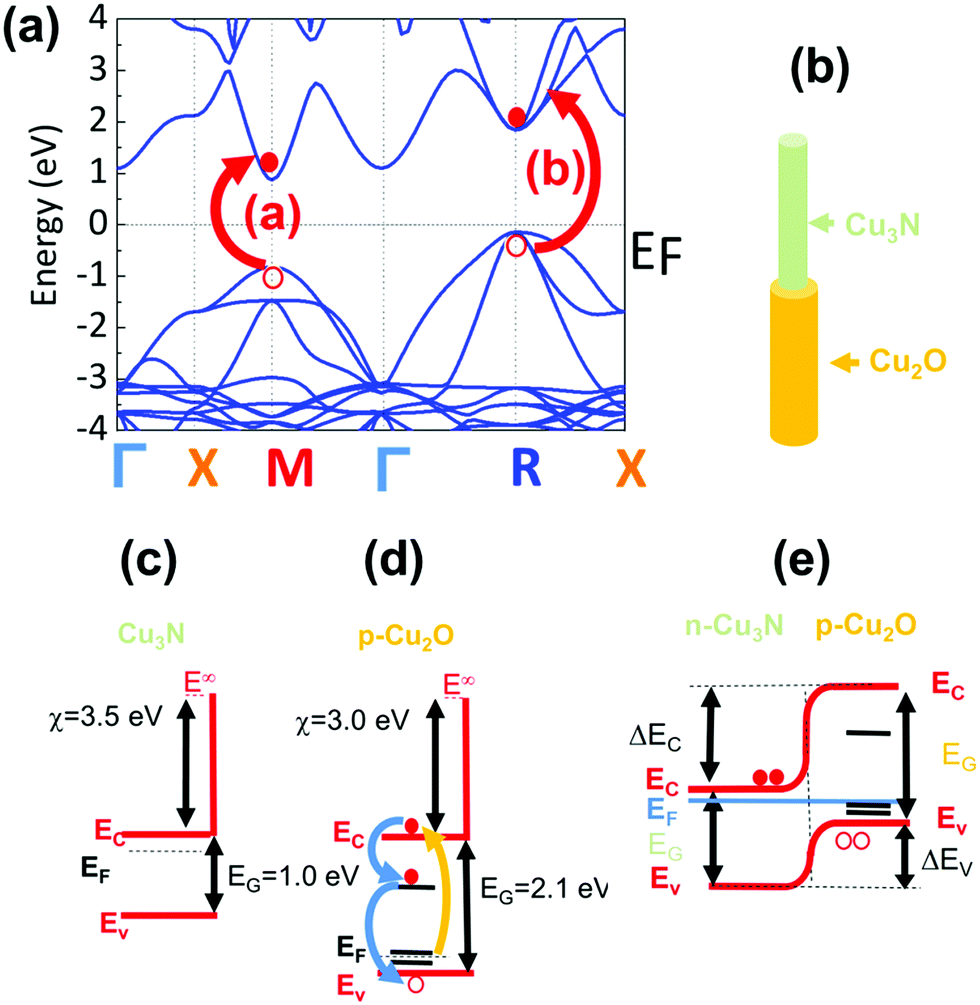 | ||
| Fig. 7 (a) Electronic structure of Cu3N showing the excitation of an electron and its transition across the M and R direct energy bandgaps in good agreement with the energetic position of the maxima in differential transmission observed at 1.91 eV and 1.8 eV corresponding to bulk Cu3N (b) schematic of a Cu3N/Cu2O core–shell NW (c), (d) energy band diagrams of Cu3N and Cu2O showing the states at ∼0.16 eV above EV and ∼0.4 eV below EC (e) energy band diagram of a p–n heterojunction Cu3N/Cu2O core–shell NW. | ||
The energy band diagram of the Cu3N/Cu2O core–shell NW is shown in Fig. 7(e). We have shown previously that oxygen on nitrogen sites (ON) have the lowest formation energy and oxygen acts as a donor in Cu3N.40 Consequently, the Cu3N is expected to be n-type in contrast to CuO and Cu2O that are p-type, hence the Cu3N/Cu2O core–shell NW is a p–n heterojunction. It should be mentioned that Cu2O which is the native oxide of Cu3N was suggested3 that could act in a beneficial way to Cu3N as a surface passivation layer similar to SiO2 in Si solar cells. However, the lifetime of the photogenerated electron–hole pairs is of the order of a few tens of ps as shown by the time evolution of the differential transmission in Fig. 6(b and c) which implies that the photoexcited carriers quickly recombine so the idea that Cu2O may act as a passivation layer for Cu3N may not be so valid in reality. Finally it should be pointed out that despite the fact that Cu3N has a “clean bandgap” with no mid gap states and has been described to be a defect tolerant semiconductor that is attractive as a solar cell absorber, no one has so far fabricated a p–n junction solar cell using Cu3N that exhibits photovoltaic action. In the past, Chen et al.54 fabricated a p–n Cu3N homo junction on indium tin oxide (ITO) and Yee et al.55 fabricated an Al: ZnO/ZnS/Cu3N p–n heterojunction both of which showed rectifying behavior, but no photo generated current, attributed to a high density of N vacancies (VN) and Cu interstitials (Cui) that capture electrons and result into significant Shockley–Read–Hall recombination and suppression of the photogenerated electron–hole pairs. So, while crystal defects such as VN and Cui, do not give rise to mid-gap states they still act in a detrimental fashion that hinders the use of Cu3N as a defect tolerant semiconductor for the fabrication of solar cells. One way of overcoming these difficulties is to grow Cu3N nanostructures on epitaxial, high crystal quality, transparent conducting oxides e.g. Sb: SnO2 NWs which will permit the extraction of the minority carriers over shorter lengths so that Cu3N may be used for energy conversion as a solar cell absorber. Nevertheless, the Cu3N NWs obtained here that have diameters between 100 and 200 nm and lengths up to ∼10 μm can be used directly for energy storage as they have a large surface area and can be easily removed and transferred onto a flexible substrate for the purpose of making devices such as supercapacitors or batteries.
4. Conclusions
CuO NWs with diameters of 150 ± 30 nm, lengths up to 3–10 μm and a uniform distribution have been grown via the thermal oxidation of Cu foils at 600 °C under O2. The CuO NWs have a monoclinic crystal structure and grow by a vapor–solid mechanism on top of bulk CuO and Cu2O that forms first on top of the Cu foil. We developed a strategy to convert the CuO into Cu3N NWs under NH3:H2via the deposition of Cu over the CuO NWs in order to maintain their integrity under reducing conditions. The Cu3N NWs obtained in this way are curly, have a cubic anti-ReO3 crystal structure but are surrounded by a surface shell of Cu2O. The CuO NWs with a 480 nm shell of Cu are not converted completely into Cu3N and consist of a CuO core with thicknesses of a few tens of nm i.e. the CuO/Cu3N core–shell NWs have an averaged projected diameter d = 320 ± 35 nm; 40 ± 5 nm for the core.The Cu3N NWs exhibited maxima in differential transmission at 2.41, 2.17, 1.9 and 1.8 eV as measured by UPPS that correspond to the M and R direct energy band gaps of Cu3N but we find no evidence for quantization. In addition, we observed two minor, but well resolved peaks close to the infra-red at 1.69 and 1.67 eV that may be attributed to the M and R direct energy band gaps of Cu3N under compression or due to recombination via states in the Cu2O. The photogenerated carriers have lifetimes less than 100 ps, so the potential of Cu3N as a defect tolerant semiconductor-solar cell absorber for energy conversion is currently limited in contrast to that for energy storage.
Conflicts of interest
The author declares no competing interest.Acknowledgements
P. K. and N. F. acknowledge support by project “INNOVATION-EL” (MIS 5002772), funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020), co-financed by Greece and the EU (European Regional Development Fund).References
- A. Jiang, M. Qi and J. Xiao, J. Mater. Sci. Technol., 2018, 34, 1467–1473 CrossRef CAS.
- R. Juza and H. Hahn, Z. Anorg. Chem., 1938, 239, 282–287 CrossRef CAS.
- A. Zakutayev, C. M. Caskey, A. N. Fioretti, D. S. Ginley, J. Vidal, V. Stevanovic, E. Tea and S. Lany, J. Phys. Chem. Lett., 2014, 5, 1117–1125 CrossRef CAS PubMed.
- S. Suwannatus, B. Duangsawat and U. Pakdee, Mater. Today, 2018, 5, 15208–15212 CAS.
- J. Wang, F. Li, X. Liu, H. Zhou, X. Shao, Y. Qu and M. Zhao, J. Mater. Chem. A, 2017, 5, 8762–8768 RSC.
- Y. Yue, P. Han, S. Dong, K. Zhang, C. Zhang, C. Shang and G. Cui, Chin. Sci. Bull., 2012, 57, 4111–4118 CrossRef CAS.
- S. Ge, Y. Leng, T. Liu, R. S. Longchamps, X. G. Yang, Y. Gao, D. Wang, D. Wang and C. Y. Wang, Sci. Adv., 2020, 6, 7633 CrossRef PubMed.
- M. Birkett, C. N. Savory, A. N. Fioretti, P. Thompson, C. A. Muryn, A. D. Weerakkody, I. Z. Mitrovic, S. Hall, R. Treharne, V. R. Dhanak, D. O. Scanlon, A. Zakutayev and T. D. Veal, Phys. Rev. B, 2017, 95, 115201 CrossRef.
- K. Matsuzaki, T. Okazaki, Y. S. Lee, H. Hosono and T. Susaki, Appl. Phys. Lett., 2014, 105, 222102 CrossRef.
- Z. Li and R. G. Gordon, Chem. Vap. Deposition, 2006, 12, 435–441 CrossRef CAS.
- J. M. Park, K. Jin, B. Han, M. J. Kim, J. Jung, J. J. Kim and W. J. Lee, Thin Solid Films, 2014, 556, 434–439 CrossRef CAS.
- C. Gallardo-Vega and W. de la Cruz, Appl. Surf. Sci., 2006, 252, 8001–8004 CrossRef CAS.
- G. Soto, J. A. Díaz and W. de la Cruz, Mater. Lett., 2003, 57, 4130–4133 CrossRef CAS.
- K. Matsuzaki, K. Harada, Y. Kumagai, S. Koshiya, K. Kimoto, S. Ueda, M. Sasase, A. Maeda, T. Susaki, M. Kitano, F. Oba and H. Hosono, Adv. Mater., 2018, 30, 1801968 CrossRef PubMed.
- A. Othonos, M. Zervos and C. Christofides, J. Appl. Phys., 2010, 108, 124319 CrossRef.
- Z. Wang, X. Cao, D. Liu, S. Hao, R. Kong, G. Du, A. M. Asiri and X. Sun, Chem. – Eur. J., 2017, 23, 4986–4989 CrossRef CAS PubMed.
- X.-D. Ma, D. I. Bazhanov, O. Fruchart, F. Yildiz, T. Yokoyama, M. Przybylski, V. S. Stepanyuk, W. Hergert and J. Kirschner, Phys. Rev. Lett., 2009, 102, 205503 CrossRef PubMed.
- Y. Zhang, L. Frank Leung-Yuk, Z. Yan and X. Hu, Sci. China, Ser. E: Technol. Sci., 2009, 52, 352–356 CrossRef CAS.
- J. Hou, Y. Sun, Z. Li, B. Zhang, S. Cao, Y. Wu, Z. Gao and L. Sun, Adv. Funct. Mater., 2018, 28, 1803278 CrossRef.
- D. Lee, S. Sun, J. Kwon, H. Park, M. Jang, E. Park, B. Son, Y. Jung, T. Song and U. Paik, Adv. Mater., 2020, 32, 1905573 CrossRef CAS PubMed.
- A. Scigala, E. Szłyk, T. Rerek, M. Wiśniewski, L. Skowronski, M. Trzcinski and R. Szczesny, Materials, 2021, 14, 603 CrossRef CAS PubMed.
- X. Jiang, T. Herricks and Y. Xia, Nano Lett., 2002, 2, 1333–1338 CrossRef CAS.
- G. Fritz-Popovski, F. Sosada-Ludwikowska, A. Köck, J. Keckes and G. A. Maier, Sci. Rep., 2019, 9, 807 CrossRef PubMed.
- E. A. Gulbransen, T. P. Copan and K. F. Andrew, J. Electrochem. Soc., 1961, 108, 119 CrossRef CAS.
- J. Liang, N. Kishi, T. Soga and T. Jimbo, J. Nanomater., 2011, 2011, 1–8 Search PubMed.
- H. Yan, X. Xiao, Z. Chen, Y. Chen, R. Zhou, Z. Wang and M. Hong, Opt. Laser Technol., 2019, 119, 105612 CrossRef CAS.
- S. Anandan, X. Wen and S. Yang, Mater. Chem. Phys., 2005, 93, 35–40 CrossRef CAS.
- D. Li, J. Hu, R. Wu and J. G. Lu, Nanotechnology, 2010, 21, 485502 CrossRef PubMed.
- L. Liao, Z. Zhang, B. Yan, Z. Zheng, Q. L. Bao, T. Wu, C. M. Li, Z. X. Shen, J. X. Zhang, H. Gong, J. C. Li and T. Yu, Nanotechnology, 2009, 20, 085203 CrossRef CAS PubMed.
- O. Lupan, V. Postica, V. Cretu, N. Wolff, V. Duppel, L. Kienle and R. Adelung, Phys. Status Solidi RRL, 2016, 10, 260–266 CrossRef CAS.
- S. Steinhauer, E. Brunet, T. Maier, G. C. Mutinati, A. Köck, O. Freudenberg, C. Gspan, W. Grogger, A. Neuhold and R. Resel, Sens. Actuators, B, 2013, 187, 50–57 CrossRef CAS.
- Y. Feng and X. Zheng, Nano Lett., 2010, 10, 4762–4766 CrossRef CAS PubMed.
- G. Filipič and U. Cvelbar, Nanotechnology, 2012, 23, 194001 CrossRef PubMed.
- A. Othonos and M. Zervos, Nanoscale Res. Lett., 2011, 6, 622 CrossRef PubMed.
- N. A. Mohemmed Shanid, M. Abdul Khadar and V. G. Sathe, J. Raman Spectrosc., 2011, 42, 1769–1773 CrossRef CAS.
- B. K. Meyer, A. Polity, D. Reppin, M. Becker, P. Hering, B. Kramm, P. J. Klar, T. Sander, C. Reindl, C. Heiliger, M. Heinemann, C. Müller and C. Ronning, Semicond. Semimetals, 2013, 201–226 CAS.
- J. Reydellet, M. Balkanski and D. Trivich, Phys. Status Solidi B, 1972, 52, 175–185 CrossRef CAS.
- M. Zervos, A. Othonos, M. Sergides, T. Pavloudis and J. Kioseoglou, J. Phys. Chem. C, 2020, 124, 3459–3469 CrossRef CAS.
- W. Yu, J. Zhao and C. Jin, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 214116 CrossRef.
- M. Zervos, A. Othonos, T. Pavloudis, S. Giaremis, J. Kioseoglou, K. Mavridou, M. Katsikini, F. Pinakidou and E. C. Paloura, J. Phys. Chem. C, 2021, 125, 3680–3688 CrossRef CAS.
- A. Yu, Y. Ma, A. Chen, Y. Li, Y. Zhou, Z. Wang, J. Zhang, L. Chu, L. Yang and X. Li, Vacuum, 2017, 141, 243–248 CrossRef CAS.
- K. V. S. Reddy, A. S. Reddy, P. S. Reddy and S. Uthanna, J. Mater. Sci.: Mater. Electron., 2007, 18, 1003–1008 CrossRef CAS.
- F. Hadian, A. Rahmati, H. Movla and M. Khaksar, Vacuum, 2012, 86, 1067–1072 CrossRef CAS.
- T. Maruyama and T. Morishita, J. Appl. Phys., 1995, 78, 4104–4107 CrossRef CAS.
- X. M. Yuan, P. X. Yan and J. Z. Liu, Mater. Lett., 2016, 60, 1809–1812 CrossRef.
- Y. Hayashi, T. Ishikawa and D. Shimokawa, J. Alloys Compd., 2002, 330–332, 348–351 CrossRef CAS.
- G. Sahoo, S. R. Meher and M. K. Jain, Mater. Sci. Eng., B, 2015, 191, 7–14 CrossRef CAS.
- D. Dorranian, L. Dejam, A. H. Sari and A. Hojabri, J. Theor. Appl. Phys., 2009, 3, 37–41 Search PubMed.
- Y. Du, A. Ji, L. Ma, Y. Wang and Z. Cao, J. Cryst. Growth, 2005, 280, 490–494 CrossRef CAS.
- D. M. Borsa and D. O. Boerma, Surf. Sci., 2004, 548, 95–105 CrossRef CAS.
- K. J. Kim, J. H. Kim and G. H. Kang, J. Cryst. Growth, 2001, 222, 767–772 CrossRef.
- J. W. Hodby, T. E. Jenkins, C. Schwab, H. Tamura and D. Trivich, J. Phys. C: Solid State Phys., 1976, 9, 1429 CrossRef CAS.
- D. O. Scanlon and G. W. Watson, J. Phys. Chem. Lett., 2010, 1, 2582 CrossRef CAS.
- S. C. Chen, S. Y. Huang, S. Sakalley, A. Paliwal, Y. H. Chen, M. H. Liao, H. Sun and S. Biring, J. Alloys Compd., 2019, 789, 428 CrossRef CAS.
- Y. S. Yee, H. Inoue, A. Hultqvist, D. Hanifi, A. Salleo, B. Magyari-Köpe, Y. Nishi, S. F. Bent and B. M. Clemens, Phys. Rev. B, 2018, 97, 245201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00140c |
| This journal is © The Royal Society of Chemistry 2022 |