
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver modified copper foam electrodes for enhanced reduction of CO2 to C2+ products†
Chong
Wang
a,
Chengdeng
Wang
a,
Zhihao
Xiong
a,
Jiashuai
Wang
a,
Wenyuan
Zhang
a,
Haofeng
Shi
a,
Donghua
Wang
a,
Yousong
Gu
 a,
Zhiming
Bai
*c,
Yan
Gao
*b and
Xiaoqin
Yan
*a
a,
Zhiming
Bai
*c,
Yan
Gao
*b and
Xiaoqin
Yan
*a
aSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, People's Republic of China. E-mail: xqyan@mater.ustb.edu.cn
bLaboratory of Nanomaterials, National Center for Nanoscience and Technology, Beijing, 100190, People's Republic of China. E-mail: gaoyan@nanoctr.cn
cSchool of Civil and Resource Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China. E-mail: baizhiming2008@126.com
First published on 20th May 2022
Abstract
Electrochemical CO2 reduction reactions (CO2RRs) have been recognized as a promising solution to environmental and energy problems due to their ability to convert intermittent renewable electricity into hydrocarbon fuels and value-added chemicals. Copper has a unique ability to electrochemically reduce CO2 to produce C2+ hydrocarbon products compared to other metals. However, it is still suffering from low Faraday efficiency (FE) limitations for specific products. Herein, we have prepared an Ag-modified Cu/CuO–Ag catalyst with a porous structure via a galvanic replacement reaction method, which reduced CO2 to C2+ products with the FE of 52.5% at −1.1 V (vs. RHE). The experimental results and density functional theory (DFT) calculations show that the addition of Ag in Cu is beneficial to increasing the coverage of *CO on the Cu surface, which can decrease the potential barrier energy of the C–C coupling reaction and favor the generation of C2+ products. The results of this study may be helpful to the design of efficient tandem catalysts for electrochemical CO2 reduction.
Introduction
The rising of CO2 emissions in the atmosphere due to the combustion of fossil fuels has caused a number of environmental problems, such as the greenhouse effect and sea level rise, which has induced great concern about global climate change.1,2 Therefore, converting CO2 into chemicals and fuels not only reduces the carbon dioxide content in the atmosphere but also recovers carbon resources and alleviates the energy crisis.3–5 Among various CO2 reduction methods, the electrochemical CO2 reduction reaction (CO2RR), especially in aqueous media, is attracting a lot of attention. The electrochemical reduction of CO2 to small organic molecules with high energy density by using renewable energy has the advantages of high efficiency, controllable selectivity, simple reaction unit and great potential for industrial applications.6–9Nowadays, the common catalysts for the CO2RR are based on transition metals or their compounds, which have been reported to be used for electrochemical CO2 reduction to produce different products.10,11 For example, Au, Ag and Zn are capable of reducing CO2 to carbon monoxide, while Sn, In and Bi are more inclined to reduce CO2 to formate.12–18 Copper has attracted great interest as the only metal material capable of electrochemically converting carbon dioxide to hydrocarbons and alcohols in aqueous solution.19–21 Unfortunately, Cu-based catalysts still face a series of issues in the reduction of CO2, such as high overpotential, low current density and low product selectivity.22 Several strategies have been proposed to improve the properties of Cu-based catalysts, including engineering low-coordinated sites and defects,23–25 modifying the size and shape of nanostructures,26,27 and tuning the oxidation state.22,28,29 For example, it has been reported that a copper catalyst synthesized from the reduction of oxidized Cu showed higher activity and better C2+ product selectivity in the electrochemical reduction of carbon dioxide compared with unoxidized copper foil.30 The results show that the reduced copper oxide electrode can expose more optimized surfaces with edges and steps for the initial activation of carbon dioxide molecules and the dimerization of related intermediates.
Currently, a promising method is to combine copper-based catalysts with second catalysts (such as gold, silver and zinc) to further increase the surface coverage of *CO, which contributes to the formation of C2+ products.31–33 For example, Du et al. constructed silver–copper bimetallic catalysts with sharp interfaces via sequential precipitation for the efficient production of ethylene. The Faraday efficiency of ethylene production was more than twice that of the pure copper catalyst. DFT calculation shows that the adsorption energy of CO on Cu is three times that of Ag. The CO molecules are first formed on Ag atoms and then trapped by Cu atoms through the Ag/Cu interface, resulting in a large number of CO molecules aggregated on the Cu side of the Ag/Cu interface. Therefore, the increase of the FE for the C2+ products is due to the tandem effect of the silver–copper metal sites near the interface.34 Although CuAg bimetallic catalysts can improve the C2+ product selectivity, the activity is still low and the current density is not sufficient for commercialization.35 Moreover, the reaction mechanism of converting CO2 to C2+ products over Ag-modified Cu-based catalysts should be further elucidated.
Here, Ag-modified Cu/CuO (Cu/CuO–Ag) catalysts with a porous 3D structure were synthesized by a galvanic replacement reaction (GRR) between Ag+ and Cu. This special mesoscopic architecture provides the best combination between copper and silver, which dramatically improves the Faraday efficiency and current density of ethylene formation. The electrochemical studies show that Cu/CuO–Ag has good catalytic activity toward the reduction of CO2, and the FE for C2+ reaches 52.5% at −1.1 V (vs. RHE), much greater than that of the Cu/CuO catalyst (32.2%). DFT calculation shows that increasing the coverage of *CO on the Cu surface facilitates the C–C dimerization reaction and improves the selectivity of C2+ products.
Experimental
Chemicals
All chemicals were used as received unless otherwise stated. Copper foil (Cu, 99.99%, thickness 0.25 mm) and silver nitrate (AgNO3, 99.99%) were purchased from Innochem. Potassium hydrogen carbonate (KHCO3, 99.99%) was purchased from J & K chemical. Sodium chloride (NaCl, 99.9%) was bought from HWRK Chem. Copper(II) sulfate pentahydrate (CuSO4·5H2O) was bought from Aladdin. Nitric acid (HNO3, 65%), phosphoric acid (H3PO4, 85%) and sulfuric acid (H2SO4, 98%) were from Sinopharm Chemical Reagent. Nitrogen (N2, 99.9999%) and carbon dioxide (CO2, 99.999%) were provided by Beijing Beiwen Gases Company.Synthesis of electrocatalysts
Characterization of the catalysts
The surface morphology and elemental composition of the catalysts were characterized by scanning electron microscope (Gemini-500) coupled with energy-dispersive X-ray spectroscopy (EDX). The microscopic morphology of the catalysts was further studied by transmission electron microscopy (TEM, FEI TALOS 200X). The crystalline structures and phases were characterized by an X-ray diffractometer (XRD UltimaIV, SmartLab). X-ray photoelectron spectroscopy (XPS, Kratos Axis Ultra spectrometer instrument) was used for analyzing the chemical compositions of the samples. The electrochemically active surface areas (ECSAs) of the Cu foam, Cu/CuO and Cu/CuO–Ag were determined by double layer capacitance measurements. Cyclic voltammetry (CV) measurements were performed from −0.11 V to −0.01 V (vs. RHE) at various scan rates in the N2 saturated state. The non-Faraday current density (Δj) was calculated from the equation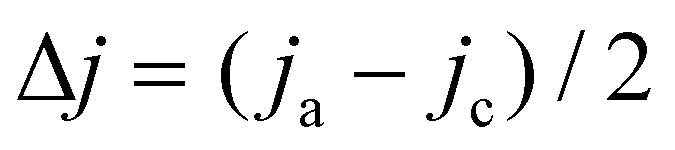 , whereby jc and ja are the cathode and anode current densities at −0.06 V vs. RHE. Plotting Δj against the scan rate, the slope was determined as the double layer capacitance (CDL) value. The roughness factors were evaluated by the equation RF = CDL/Cs, with CDL of Cu foil as the general Cs.
, whereby jc and ja are the cathode and anode current densities at −0.06 V vs. RHE. Plotting Δj against the scan rate, the slope was determined as the double layer capacitance (CDL) value. The roughness factors were evaluated by the equation RF = CDL/Cs, with CDL of Cu foil as the general Cs.
Electrochemical CO2 reduction measurements
A CHI660E potentiostat was used for all CO2 reduction experiments. A specially designed airtight glass electrolytic cell (H-type) was used to conduct the CO2 electrolysis test. During potentiostatic CO2 electrolysis, carbon dioxide is continuously injected into the cathode solution at a speed of 30 mL min−1 by a Mass Flow Controller (MFC) for gases. Electrodes were pre-reduced for more than 30 min to reduce CuO to Cu at −2.0 V (vs. Ag/AgCl). The electrochemical reduction potential of CO2 was investigated by performing linear sweep voltammetry (LSV) at a scan rate of 50 mV s−1 from 0 V to −2.0 V (vs. Ag/AgCl). All potentials are reported relative to a reversible hydrogen electrode (RHE) through the below equation: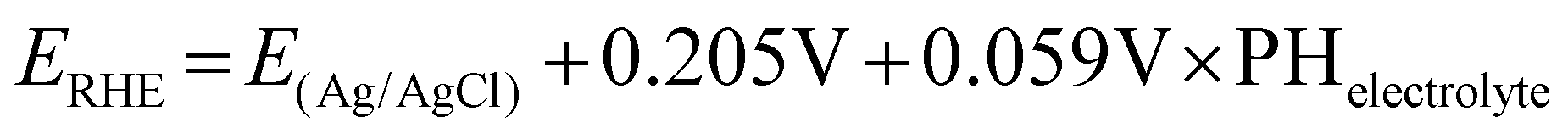 | (1) |
To evaluate the selectivity of CO2 reduction, we used online electrochemical gas chromatography (GC, Shimadzu GC-2014c) to detect the gas product during the CO2RR through the continuous flow of an electrolytic cell. The GC was calibrated regularly using a standard gas combination under standard circumstances. The faradaic efficiency (FE) data of the gas product was computed from the following equation:
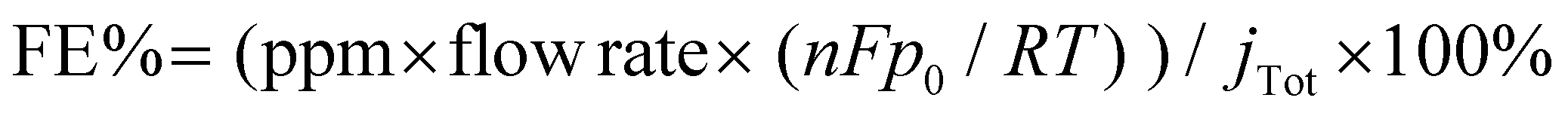 | (2) |
The electrolytes were collected separately after 1 h of electrolysis at each potential and the liquid products were analyzed quantitatively using the Ascend-400 MHz nuclear magnetic resonance (NMR) spectrometer. After 1 h of electrolysis, 0.5 mL of electrolyte was removed, and 0.1 mL of D2O and 1.67 ppm of dimethyl sulfoxide (DMSO) were added to it as an internal standard. A pre-saturation technique is used to suppress the water signal. The FE data for liquid products are computed using the following formula:
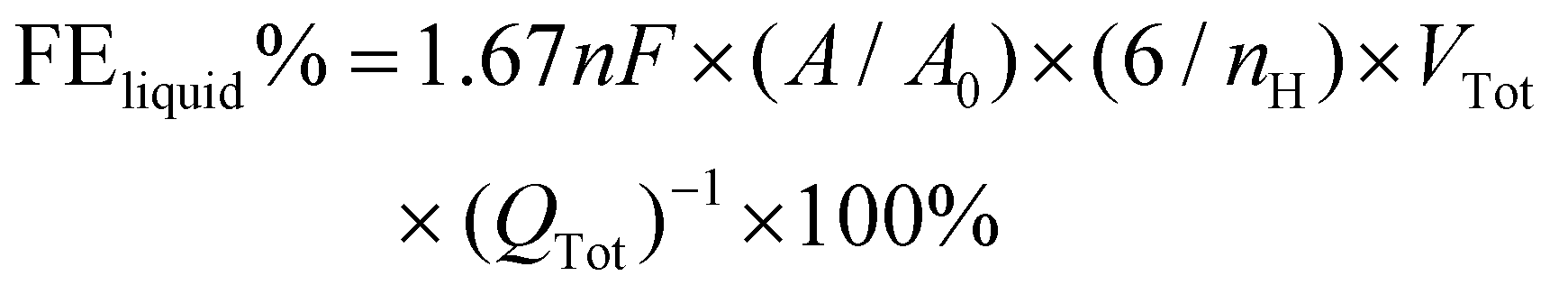 | (3) |
Density functional theory calculations
All DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP).36–38 We used the generalized gradient approximation and the Perdew–Burke–Ernzerhof exchange–correlation functional.39 The projection enhanced wave (PAW) method was used to deal with the interaction between electrons and ions, and the energy cutoff of the plane wave base was set as 450 eV.40 A (3 × 3 × 1) Monkhorst–Pack k-point mesh was used to optimize the surface structures. The convergence criteria for residual forces and energies were set to 0.01 eV Å−1 and 10−4 eV for vacuum spaces greater than 20 Å in the z-direction to avoid interactions between periodic cells during structural relaxation.We first optimized the crystal structure and found the equilibrium lattice constant to be αCu = 3.631. Then a 4-layer model with a p (3 × 3) Cu(111) supercell was used, with the top two layers relaxed and the bottom two layers fixed. The Nudged Elastic Band (NEB)41 and Dimer's methods42 were used to locate the transition state structures, and to further optimize the transition state using a residual force convergence criterion of less than 0.05 eV Å−1.
Results and discussion
The method of preparing Cu/CuO and decorating it with Ag-islands (Cu/CuO–Ag) is shown schematically in Fig. 1. Firstly, the Cu foam was prepared by the hydrogen bubble template method. When high current densities of j = −2 A cm−2 are applied in the high acid environment (1.5 M H2SO4), the hydrogen evolution reaction (HER) is superimposed on the corresponding metal deposition and gas bubbles temporarily appear on the electrode as geometric templates for Cu deposition. The main source of HER in an acidified aqueous solution is proton reduction (eqn (4)) and water splitting (eqn (5)).| HER (proton reduction): 2H+ + 2e− → H2 | (4) |
| HER (water splitting): 2H2O + 2e− → H2 + 2OH− | (5) |
| Metal deposition: Cu2+ + 2e− → Cumet | (6) |
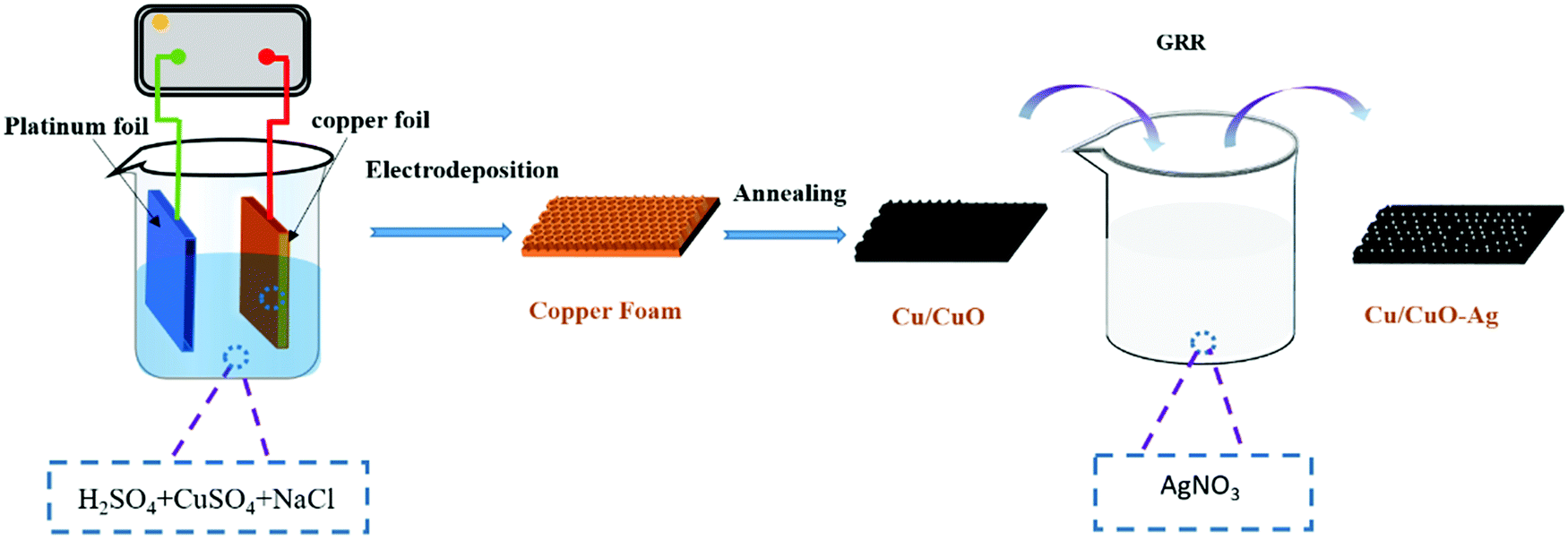 | ||
| Fig. 1 Schematic route for the preparation of Cu/CuO and Cu/CuO–Ag. | ||
The morphological characteristics of copper foams after deposition for 20 s are shown in Fig. S1 (ESI†). The original Cu foam shows an obvious pore size gradient along the surface normal. The smallest pore is close to the supporting electrode (copper foil), and the largest pore is located on the outermost surface of the Cu foam. The interconnected pores are visible with surface pore sizes varying from 50 to 80 μm, which provide adequate space for the electrolyte to circulate freely. The sidewalls of the pores are composed of loosely packed copper dendrites, thus forming “secondary” nanopores in the Cu foam. Then, the Cu foam was further thermally annealed in air at 250 °C for 3 h, thereby converting the catalyst materials into Cu/CuO foams. The overall morphology of the copper foam remains essentially unchanged following thermal annealing, which is proved by SEM (Fig. 2a and b). The Cu/Cu2O–Ag was prepared by immersing Cu/CuO into an aqueous AgNO3 solution for 1–5 min. Since the standard reduction potential of the Cu2+/Cu redox couple (0.34 V vs. SHE) is lower than that of Ag+/Ag (0.80 V vs. SHE), the following primary galvanic replacement reaction (GRR) occurs spontaneously:
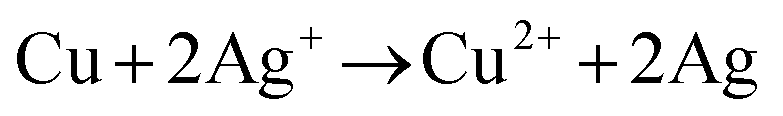 | (7) |
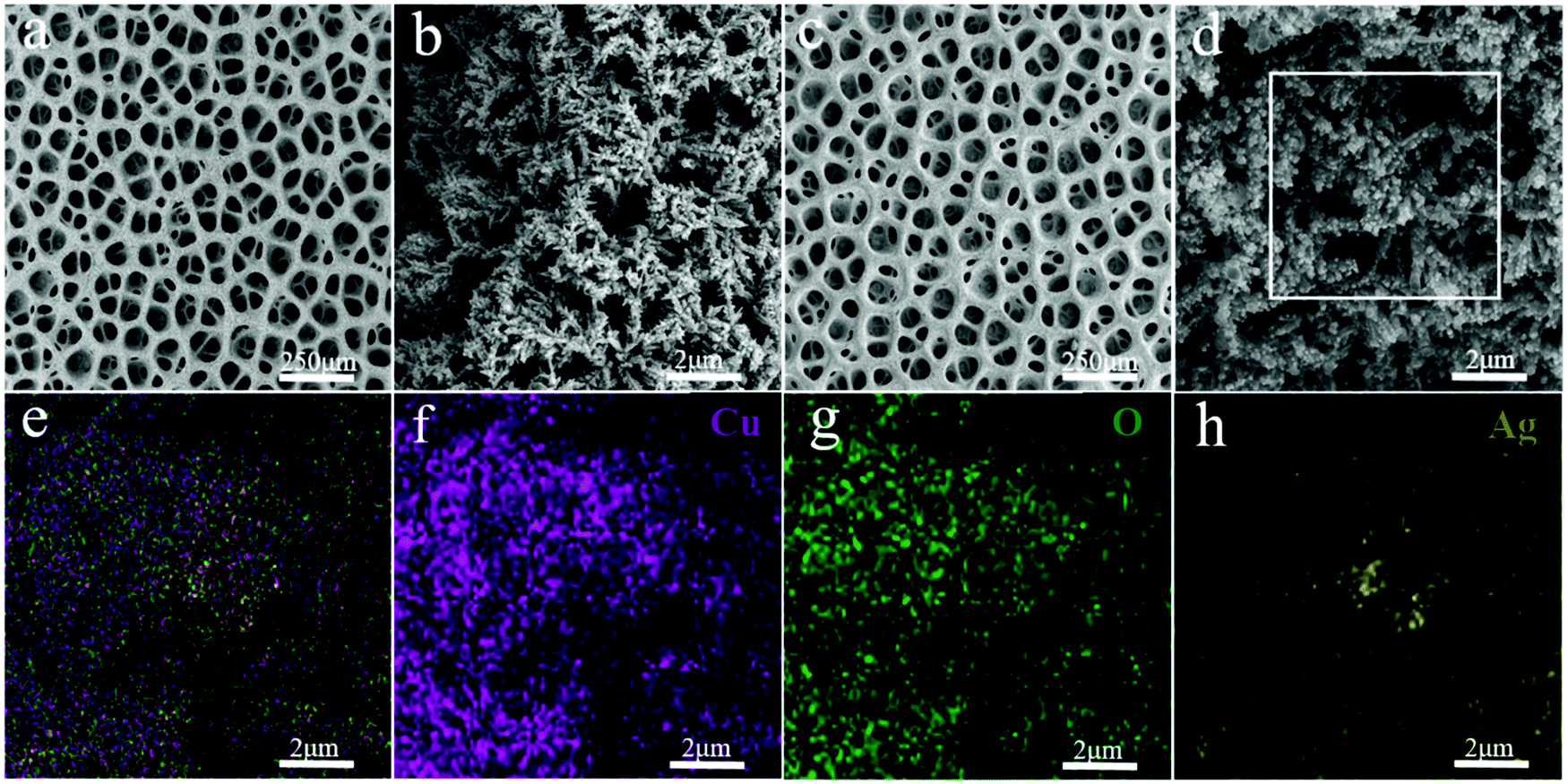 | ||
| Fig. 2 (a) and (b) SEM images of Cu/CuO. (c) and (d) SEM images of Cu/CuO–Ag. (e–h) EDX images of overlap, Cu, O and Ag of Cu/CuO–Ag. | ||
After 3 min of GRR in 10 mM AgNO3 solution, silver nanoparticles (NPs) with the size of 50–100 nm were uniformly distributed on the surface of Cu/CuO foams (Fig. 2c and d). The EDX shows that Cu, O and Ag are uniformly distributed in the Cu/CuO–Ag nanocomposites (Fig. 1e–h), which matches well with the results of the above SEM characterizations. The TEM image (Fig. S2, ESI†) shows that silver nanoparticles are attached to copper dendrites. In addition, we also characterized the distribution of Ag nanoparticles in the Cu/CuO–Ag1 and Cu/CuO–Ag5 samples. The images in Fig. S3 (ESI†) show a gradual increase in the average size of the Ag nanoparticles from the Cu/CuO–Ag1 to Cu/CuO–Ag5 samples. The roughness factor of the foam structured catalyst was estimated using the double layer capacitance in the nonfaradaic regions of the CV method. The electrodeposited catalysts have a larger roughness factor than that of the Cu foil catalysts. The roughness of the catalysts follows the order as Cu foam < Cu/CuO–Ag < Cu/CuO (Fig. S4 and Table S1, ESI†).
The XRD of Cu/CuO and Cu/CuO–Ag show peaks that belong to the diffraction of CuO(PDF NO. 45-0793) and Cu(PDF No. 04-0836) (Fig. 3a). The peaks at 38.3°, 44.4°, 64.4° and 77.3° of Cu/CuO–Ag could be ascribed to Ag(111), Ag (200), Ag(220) and Ag(310), respectively. XPS was used to study the chemistry states and surface composition of the Cu/CuO–Ag catalysts. The catalyst exhibits a pair of spin–orbiting double Cu 2p peaks, which may be attributed to Cu 2p3/2 and Cu 2p1/2, respectively (Fig. 3b). The fitting peaks at 933.8eV and 953.2 eV are ascribed to the Cu 2p3/2 and Cu 2p1/2 peaks of Cu2+, respectively. Furthermore, the presence of CuO is supported by the presence of two discernible shake-up satellite peaks.43 The fitted peak at 932.1 eV makes it hard to distinguish the Cu+ and Cu0 directly by XPS since the difference in binding energy between Cu+ and Cu0 is only 0.1 eV.44 The chemical state of Cu was further verified using Auger electron spectroscopy (Fig. 3c). Cu LMM XAES spectra indicate the presence of two surface Cu species near 914 eV (Cu+) and 918 eV (Cu0). The silver 3d3/5 and 3d3/2 peaks of the Cu/CuO–Ag are located at 368.6 and 374.6 eV (Fig. 3d), which also confirms the existence of Ag on Cu/CuO–Ag.45 The Cu/CuO and Cu/CuO–Ag catalysts were also characterized after pre-reduction (Fig. S5, ESI†). It can be confirmed by XRD and XPS that the CuO components have been reduced to Cu in both catalysts.
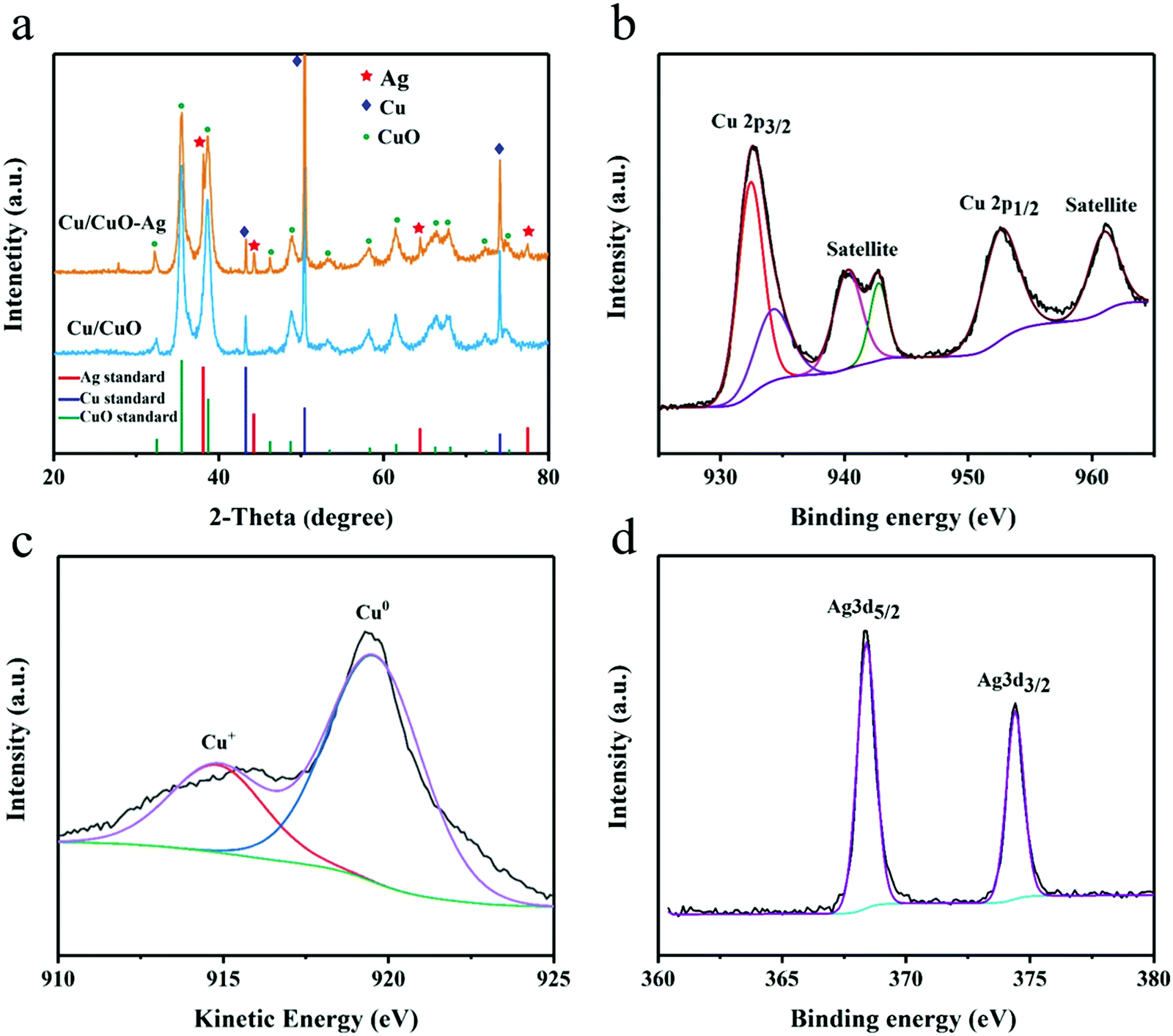 | ||
| Fig. 3 (a) XRD patterns of Cu/CuO and Cu/CuO–Ag. (b) High-resolution Cu 2p XPS spectra, (c) Auger spectra of Cu LMM, and (d) Ag 3d XPS spectra of Cu/CuO–Ag. | ||
The selectivity and activity of the CO2RR were evaluated in a three-electrode H-type cell at a constant external electrode potential (Fig. S6, ESI†). The catalytic activity of the prepared catalysts was first investigated by linear voltammetry. As shown in Fig. 4a, both catalysts exhibit higher current density to reduce CO2 compared to the copper foil. The total current density of Cu/CuO and Cu/CuO–Ag increases from 1.05 mA cm−2 to 18.42 mA cm−2 and from 1.45 mA cm−2 to 22.76 mA cm−2 when the potential decreased from −0.47 V to −1.20 V (vs. RHE). Therefore, the total current density of the Cu/CuO and Cu/CuO–Ag catalysts shows similar trends, but the current density of the Cu/CuO–Ag catalyst is greater than that of the Cu2O catalyst, which indicates that the introduction of Ag improved the catalytic activity of the catalysts. In addition, the total current density of Cu/CuO is greatly improved compared with the original Cu foam at different potentials (Fig. S8a, ESI†), which may be caused by the significant increase in surface roughness of the Cu foam after annealing reduction (Table S1, ESI†).
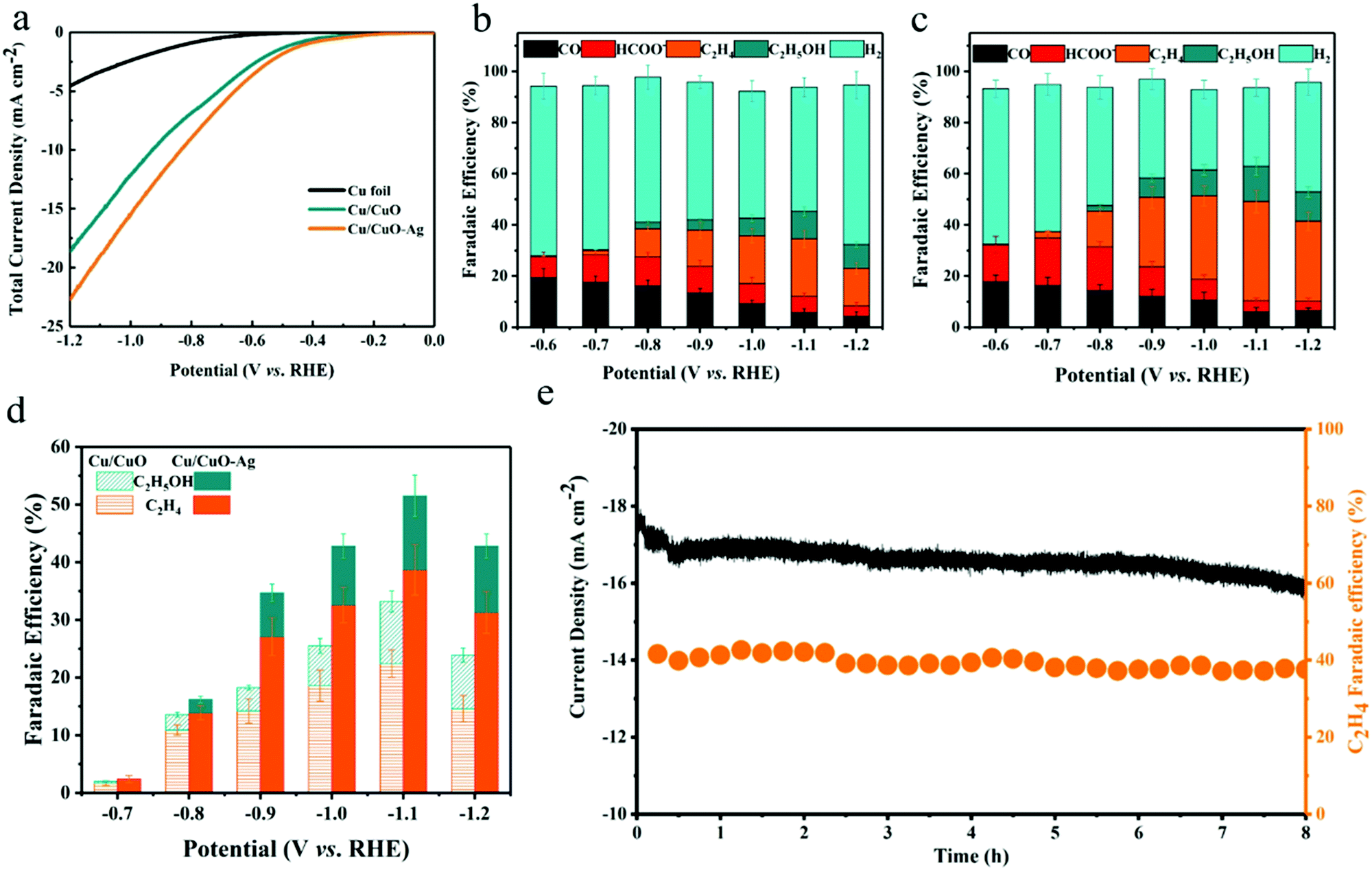 | ||
| Fig. 4 Electrocatalytic carbon dioxide reduction performance on Cu/CuO, Cu/CuO–Ag and Cu foil catalysts. (a) LSV curves of Cu foil, Cu/CuO and Cu/CuO–Ag. Faraday efficiency for each gaseous product (H2, CO, C2H4) and the main liquid product (HCOO−, C2H5OH) over (b) Cu/CuO, and (c) Cu/CuO–Ag in the potential range of −0.8 to −1.2 V (vs. RHE). (d) Faraday efficiency for the C2+ product of Cu/CuO and Cu/CuO–Ag in the potential range of −0.7 to −1.2 V (vs. RHE). (e) The FE of C2H4 and total current density of the Cu/CuO–Ag catalyst over 8 hours at −1.1 V (vs. RHE). | ||
The selectivity of the products was obtained by GC and 1H NMR analysis after applying different potentials in a 0.1 M KHCO3 solution with saturated CO2 and electrolysis at each potential for 1 h. The geometric current density of Cu/CuO–Ag remained stable during 60 min of electrolysis (Fig. S7, ESI†). The FE values of the carbon dioxide reduction products on the Cu/CuO and Cu/CuO–Ag catalysts are shown in Fig. 4b and c. For the Cu/CuO–Ag catalysts, the reduction of CO2 produced almost no C2+ product, and the main product is H2 with FE of 62.01–56.7% at overpotentials from −0.6 V to −0.7 V (vs. RHE). Significantly, the FE of C2H4 increases from 13.9% at −0.8 V to 38.7% at −1.1 V (vs. RHE). The FE of C2H4 for the Cu/CuO–Ag catalyst was 38.7% at −1.2 V (vs. RHE), which is greater than the Faraday efficiency of Cu/CuO (22.4%) at the identical potential. By contrast, copper foil and copper foam catalysts produce much fewer C2H4 (Fig. S8b, ESI†). The copper foam catalyst generates few hydrocarbons and the major formic acid product exceeds 26% at −1.0 V (vs. RHE), which is consistent with the literature.46 Quite intriguingly, the Faraday efficiency of the formate for the Cu/CuO catalyst has remained at a relatively low level and would not exceed 11% at any electrolytic potential. In contrast, the Faraday efficiencies for ethylene and ethanol products reach 22.4% and 12.8%, respectively.
The key to reducing CO2 to HCOOH or CO is whether the CO2 is adsorbed by its oxygen atoms or carbon atoms. There is no breaking of the CO bond during the formation of HCOOH. The CO2 molecules form *CO2˙− free radical anions on the surface of the catalysts and extract a proton from the adjacent water, which then results in the production of formate.47 The different distribution of CO2RR products between Cu foams and Cu/CuO catalysts is mainly due to the weakly coordinated surface sites caused by electrochemical reduction of Cu/CuO foams under CO2RR conditions, which are particularly active for carbon–carbon coupling reactions.48
Fig. 4d summarizes the C2+ product FEs for Cu/CuO and Cu/CuO–Ag catalysts. The Ag/Cu catalysts show the highest FEs for the C2+ products (52.5% at −1.1 V vs. RHE) as compared to Cu/CuO (32.2%), which indicates that the addition of Ag nanoparticles can efficiently improve the selectivity of the C2+ products. We also test the electrochemical reduction characteristics of the Cu/CuO–Ag catalysts by varying the Ag content. FE of the C2+ product shows a volcanic variation with increasing Ag loading, which indicates that there is an optimal amount of Ag loading (Fig. S9, ESI†). When the content of Ag is low, it does not provide enough CO intermediates for the copper atoms to promote the C–C coupling reaction. While the higher content of Ag leads to severe aggregation of silver nanoparticles, resulting in elevated FE of CO and lower FE of C2+. Ag foam and Cu–Ag foam were also prepared in a similar way (Fig. S10, ESI†). XRD patterns show that separated pure metal Cu and Ag phases are also formed in the Cu–Ag foam (Fig. S11, ESI†). Their performance for CO2 reduction was tested under the same conditions (Fig. S12, ESI†), and the CO2 reduction product on the Ag foam is dominated by CO, yielding a very small amount of C2H4 (FE < 3%). In contrast, the selectivity of the Cu–Ag foam for the C2+ product is significantly improved, reaching a Faraday efficiency of 27.5% for the C2+ product at −1.1 V (vs. RHE). However, it is lower than that of Cu/CuO–Ag (52.5%), indicating that the synergistic effect between the oxidation-derived Cu and Ag modifications leads to the high selectivity of the Cu/CuO–Ag catalyst for the C2+ product.
Cu/CuO–Ag shows good stability with continuous reduction of CO2 to C2H4 at −1.1 V (vs. RHE) for 8 h (Fig. 4e). The FE 39% of the C2H4 products remains almost unchanged. A longer test was conducted for 20 h, and the catalytic activity of Cu/CuO–Ag and the faradaic efficiency for ethylene hardly changes (Fig. S13, ESI†). In addition, the Cu/CuO–Ag catalyst remains in its original porous morphology (Fig. S14, ESI†), which indicates that the Cu/CuO–Ag catalyst has excellent stability.
The FE of C2H4 for the Cu/CuO–Ag catalyst is obviously higher than that of the Cu/CuO catalyst, which indicates that the introduction of Ag contributes to the production of C2+ products. Furthermore, we discuss the possible catalytic mechanism of reducing CO2 to C2H4 in this catalyst (Fig. 5a). At present, *CO is recognized as a critical intermediary in the electrochemical reduction of carbon dioxide to hydrocarbons.49 The formation of *CO intermediates from CO2 molecules usually undergoes two processes. First, the CO2 molecule is caught by Cu and Ag atoms, and both a proton and an electron are transferred.50
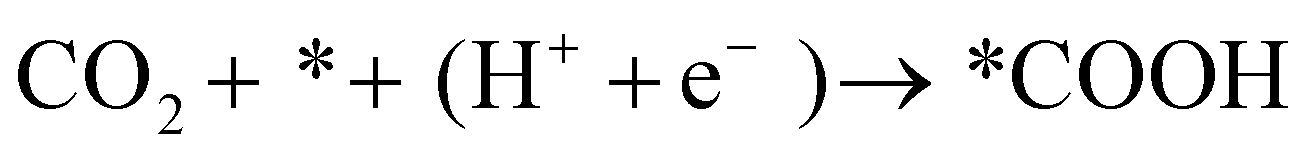 | (11) |
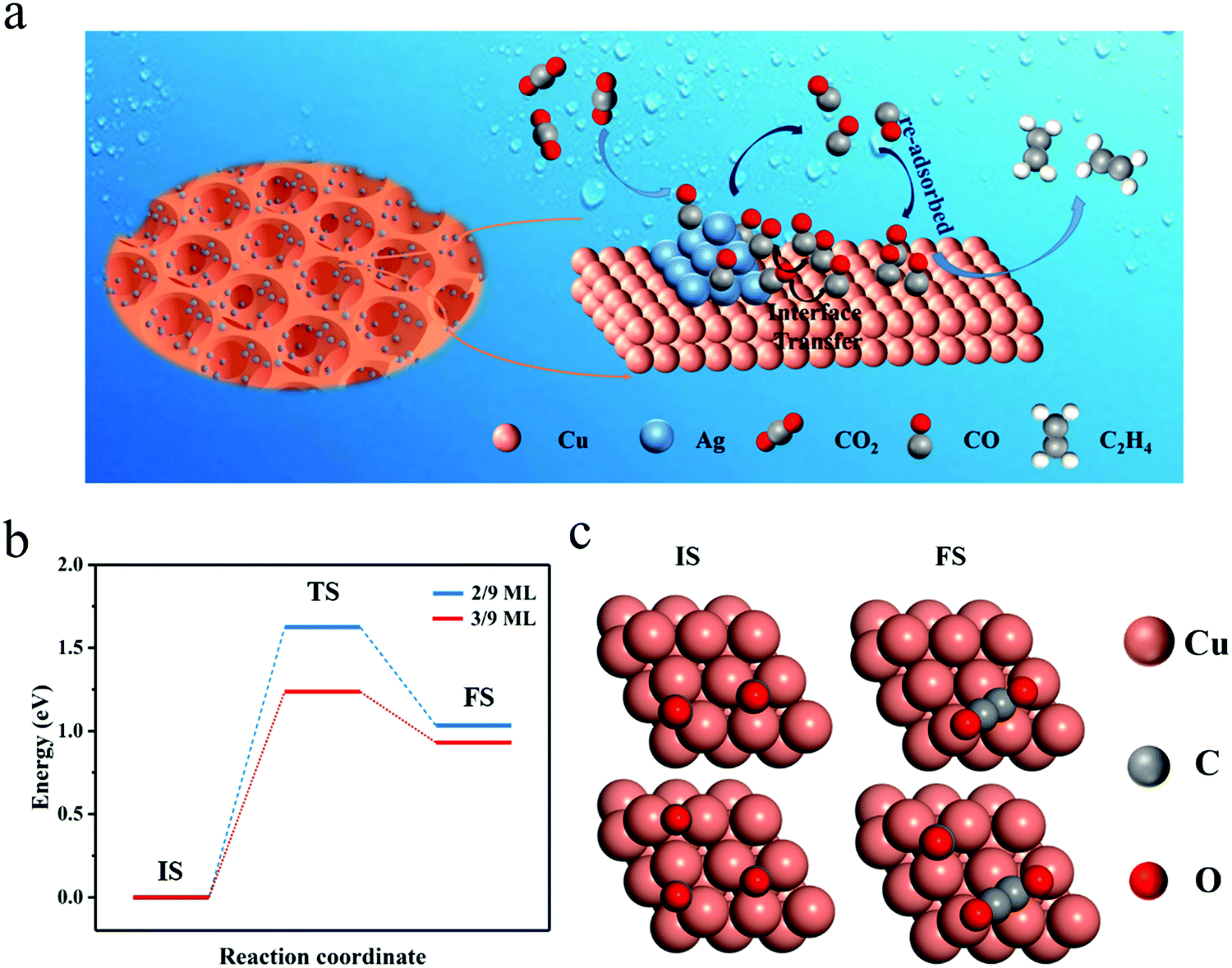 | ||
| Fig. 5 (a) Mechanism for the electrochemical reduction of CO2 to ethylene on the Cu/CuO–Ag catalyst. (b) The activation energy barrier of carbon CO at different *CO coverages. (c) The corresponding configurations of the initial state and final state on the CO coverage of 2/9 ML and 3/9 ML. IS, initial state; TS, transition state; FS, final state. | ||
Next, the *COOH intermediate is further reduced to *CO.
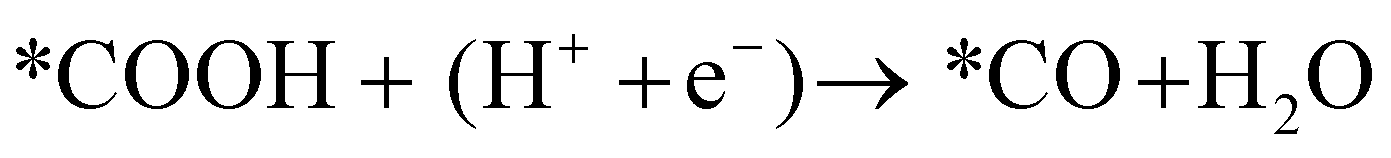 | (12) |
Ag atoms are more active than Cu atoms in the production of carbon monoxide molecules.51 The *CO generated on Ag is more easily desorbed as *CO is weakly bound to Ag, increasing the local CO concentration around the electrode surface. The CO will be temporarily trapped in the pores of the foam catalyst and re-adsorbed by the Cu sites. Additionally, the *CO intermediate produced on the Ag atoms can be captured by the Cu atoms through the Cu–Ag interface, leading to a high surface coverage of *CO. In the step of further reduction of the CO intermediate to ethylene on the Cu surface, the rate-determining step (RDS) is the dimerization of CO leading to the formation of a C–C bond.52 We consider that the coverage of CO intermediates on the catalyst surface has an important effect on the CO dimerization reaction and high surface coverage of *CO may facilitate the synthesis of the C2+ product, which is further proved by DFT calculation below.
Computationally, a (3 × 3 × 4) Cu(111) supercell was built to simulate the catalyst. With DFT, we calculated the C–C coupling barrier energy for *CO coverage of 2/9 ML and 3/9 ML to simulate the effect of high and low* CO coverage on the Cu surface for the synthesis of C2+ products (Fig. S15, ESI†), respectively. Although solvent molecules have a certain effect on the CO2RR,53 in order to simplify the model, we ignore the role of solvent molecules, and here the geometry of *OCCO is stable, the same as that under no charged water layer.52 As shown in Fig. 5b and c, increasing the CO coverage from 2/9 ML to 3/9 ML decreases the barrier from 1.62 eV to 1.13 eV, a reduction of 0.49 eV. The lowering of the barrier energy of the C–C coupling reaction is due to the interaction between the adsorbates (CO), which weakens the carbon bonds on the surface and makes the carbon–carbon bonds more easily formed (Table S2, ESI†).
Conclusion
In summary, we have successfully prepared Cu/CuO–Ag catalysts with porous structures using a galvanic replacement method and investigated their catalytic activity for the electrochemical reduction of CO2. The selectivity of CO2 reduction to ethylene was significantly improved by the synergistic effect of oxide-derived copper and Ag metals modified on the copper surface. The Faraday efficiency of ethylene reaches 38.6% at −1.1 V (vs. RHE) and remains stable over 8 h. Compared to Cu/CuO catalysts, the Cu/CuO-Ag displays better electrochemical performance for the reduction of CO2 to ethylene, which is due to the efficient spillover of CO from Ag to the Cu surface. DFT calculations indicate that the high CO coverage on the Cu surface decreases the C–C coupling barrier energy and facilitates the formation of C2+ products. In addition, the porous structure of the Cu/CuO–Ag catalyst facilitates readsorption of the intermediate CO by the Cu surface for further reduction to C2+ products. This work shows strategies for developing efficient Cu-based bimetallic catalysts for the selective formation of C2+ products in CO2RR processes and provides greater insight into the catalyst design for the electrochemical reduction of CO2.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (NSFC) (No. 51772024, X. Q. Y.; 51772057, Y. G.).References
- Y. Wang, J. Liu, Y. Wang, A. M. Al-Enizi and G. Zheng, Tuning of CO2 Reduction Selectivity on Metal Electrocatalysts, Small, 2017, 13, 1701809 CrossRef
.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, What Should We Make with CO2 and How Can We Make It?, Joule, 2018, 2, 825–832 CrossRef CAS
- D. T. Whipple and P. J.-A. Kenis, Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T.-M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Enzymatic conversion of carbon dioxide, Chem. Soc. Rev., 2015, 44, 5981–6000 RSC
- J. Qiao, Y. Liu, F. Hong and J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC
- S. Chu, Y. Cui and N. Liu, The path towards sustainable energy, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed
- L. Zhang, Z. J. Zhao and J. Gong, Nanostructured Materials for Heterogeneous Electrocatalytic CO2 Reduction and their Related Reaction Mechanisms, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed
- B. Kumar, J. P. Brian, V. Atla, S. Kumari, K. A. Bertram, R. T. White and J. M. Spurgeon, New trends in the development of heterogeneous catalysts for electrochemical CO2 reduction, Catal. Today, 2016, 270, 19–30 CrossRef CAS
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 Reduction: A Classification Problem, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed
- L. Hou, J. Yan, L. Takele, Y. Wang, X. Yan and Y. Gao, Current progress of metallic and carbon-based nanostructure catalysts towards the electrochemical reduction of CO2, Inorg. Chem. Front., 2019, 6, 3363–3380 RSC
- A. J. Welch, J. S. DuChene, G. Tagliabue, A. Davoyan, W.-H. Cheng and H. A. Atwater, Nanoporous Gold as a Highly Selective and Active Carbon Dioxide Reduction Catalyst, ACS Appl. Energy Mater., 2018, 2, 164–170 CrossRef
- S.-Q. Liu, S.-W. Wu, M.-R. Gao, M.-S. Li, X.-Z. Fu and J.-L. Luo, Hollow Porous Ag Spherical Catalysts for Highly Efficient and Selective Electrocatalytic Reduction of CO2 to CO, ACS Sustainable Chem. Eng., 2019, 7, 14443–14450 CrossRef CAS
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, Electrodeposited Zn Dendrites with Enhanced CO Selectivity for Electrocatalytic CO2 Reduction, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Promises of Main Group Metal–Based Nanostructured Materials for Electrochemical CO2 Reduction to Formate, Adv. Energy Mater., 2019, 10, 1902338 CrossRef
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate, Nat. Commun., 2018, 9, 1320 CrossRef PubMed
- Z. B. Hoffman, T. S. Gray, K. B. Moraveck, T. B. Gunnoe and G. Zangari, Electrochemical Reduction of Carbon Dioxide to Syngas and Formate at Dendritic Copper–Indium Electrocatalysts, ACS Catal., 2017, 7, 5381–5390 CrossRef CAS
- W. Ma, S. Xie, X. G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D. Y. Wu and Y. Wang, Promoting electrocatalytic CO2 reduction to formate via sulfur-boosting water activation on indium surfaces, Nat. Commun., 2019, 10, 892 CrossRef PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E.-L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- Z. Ni, H. Liang, Z. Yi, R. Guo, C. Liu, Y. Liu, H. Sun and X. Liu, Research progress of electrochemical CO2 reduction for copper-based catalysts to multicarbon products, Coord. Chem. Rev., 2021, 441, 213983 CrossRef CAS
- A. H. Shah, Y. Wang, S. Hussain, M. B. Akbar, A. R. Woldu, X. Zhang and T. He, New aspects of C2 selectivity in electrochemical CO2 reduction over oxide-derived copper, Phys. Chem. Chem. Phys., 2020, 22, 2046–2053 RSC
- Z. Wang, G. Yang, Z. Zhang, M. Jin and Y. Yin, Selectivity on Etching: Creation of High-Energy Facets on Copper Nanocrystals for CO2 Electrochemical Reduction, ACS Nano, 2016, 10, 4559–4564 CrossRef CAS PubMed
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, Highly Electrocatalytic Ethylene Production from CO2 on Nanodefective Cu Nanosheets, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed
- F. Pan and Y. Yang, Designing CO2 reduction electrode materials by morphology and interface engineering, Energy Environ. Sci., 2020, 13, 2275–2309 RSC
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction, Adv. Mater., 2018, 30, 1803111 CrossRef
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard, 3rd, J. Yano and E. J. Crumlin, Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CrossRef CAS
- S. Wang, T. Kou, S. E. Baker, E. B. Duoss and Y. Li, Recent progress in electrochemical reduction of CO2 by oxide-derived copper catalysts, Mater. Today Nano, 2020, 12, 100096 CrossRef
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, Structural Sensitivities in Bimetallic Catalysts for Electrochemical CO2 Reduction Revealed by Ag–Cu Nanodimers, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS
- Y. Zhu, X. Cui, H. Liu, Z. Guo, Y. Dang, Z. Fan, Z. Zhang and W. Hu, Tandem catalysis in electrochemical CO2 reduction reaction, Nano Res., 2021, 14, 4471–4486 CrossRef CAS
- L. Hou, J. Han, C. Wang, Y. Zhang, Y. Wang, Z. Bai, Y. Gu, Y. Gao and X. Yan, Ag nanoparticle embedded Cu nanoporous hybrid arrays for the selective electrocatalytic reduction of CO2 towards ethylene, Inorg. Chem. Front., 2020, 7, 2097–2106 RSC
- J. Wang, Z. Li, C. Dong, Y. Feng, J. Yang, H. Liu and X. Du, Silver/Copper Interface for Relay Electroreduction of Carbon Dioxide to Ethylene, ACS Appl. Mater. Interfaces, 2019, 11, 2763–2767 CrossRef CAS
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. G. Kresse and J. J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed
- J. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev., 1996, 77, 3856 Search PubMed
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef
- S. A. Trygubenko and D. J. Wales, A doubly nudged elastic band method for finding transition states, J. Chem. Phys., 2004, 120, 2082–2094 CrossRef CAS
- D. Sheppard, R. Terrell and G. Henkelman, A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives, J. Chem. Phys., 2008, 111, 7010 Search PubMed
- F. Du, Q.-Y. Chen and Y.-H. Wang, Effect of annealing process on the heterostructure CuO/Cu2O as a highly efficient photocathode for photoelectrochemical water reduction, J. Phys. Chem. Solids, 2017, 104, 139–144 CrossRef CAS
- T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, XPS study of the Cu@Cu2O core-shell nanoparticles, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS
- Y. Mou, H. Wang, Y. Peng, J. Liu, H. Cheng, Q. Sun and M. Chen, Low temperature enhanced flexible conductive film by Ag flake/ion composite ink, Mater. Des., 2020, 186, 108339 CrossRef CAS
- S. Sen, D. Liu and G. T.-R. Palmore, Electrochemical Reduction of CO2 at Copper Nanofoams, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS
- Y. Zhong, S. Wang, M. Li, J. Ma, S. Song, A. Kumar, H. Duan, Y. Kuang and X. Sun, Rational design of copper-based electrocatalysts and electrochemical systems for CO2 reduction: From active sites engineering to mass transfer dynamics, Mater. Today Phys., 2021, 18, 100354 CrossRef CAS
- A. Dutta, M. Rahaman, N. C. Luedi and P. Broekmann, Morphology Matters: Tuning the Product Distribution of CO2 Electroreduction on Oxide-Derived Cu Foam Catalysts, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kuhl, R. Garcia-Muelas, N. Lopez and B. R. Cuenya, Revealing the CO Coverage-Driven C–C Coupling Mechanism for Electrochemical CO2 Reduction on Cu2O Nanocubes via Operando Raman Spectroscopy, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS
- F. Calle-Vallejo and M. T. Koper, Theoretical considerations on the electroreduction of CO to C2 species on Cu(100) electrodes, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed
- X. Hu, S. Yao, L. Chen, X. Zhang, M. Jiao, Z. Lu and Z. Zhou, Understanding the role of axial O in CO2 electroreduction on NiN4 single-atom catalysts via simulations in realistic electrochemical environment, J. Mater. Chem. A, 2021, 9, 23515–23521 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00188h |
| This journal is © The Royal Society of Chemistry 2022 |