
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColor-tunable persistent luminescence hybrid materials via radiative energy transfer†
Dan
Zhao
abcde,
Lijun
Song
 *abde,
Jinlei
Li
abde,
Fenglong
Lin
abde,
Shenglong
Wang
abde,
Yincai
Wu
abde and
Fulin
Lin
abde
*abde,
Jinlei
Li
abde,
Fenglong
Lin
abde,
Shenglong
Wang
abde,
Yincai
Wu
abde and
Fulin
Lin
abde
aFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, 350002, China. E-mail: slj@fjirsm.ac.cn
bXiamen Institute of Rare Earth Materials, Haixi Institute, Chinese Academy of Sciences, Xiamen, 361021, China
cCollege of chemical Engineering, Fuzhou University, Fuzhou, Fujian 350108, China
dKey Laboratory of Design and Assembly of Functional Nanostructures, Fujian Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
eXiamen Key Laboratory of Rare Earth Photoelectric Functional Materials, Chinese Academy of Sciences, Xiamen, 361021, China
First published on 23rd June 2022
Abstract
Colorful, persistent luminescence materials are in great demand in many technological fields but their synthesis remains a challenge. Herein, a unique strategy is reported, that is, a full spectrum adjustment of the persistent luminescence was achieved by using an organic polymer fluorescent dye (PFD) as an efficient light-conversion material. The observed emission spectra upon photoexcitation of these polymer films were assigned to dual emission bands: the fluorescence emission of the PFD and the phosphorescence emission of the phosphor. There is a balance point between the two emission bands, and thus we can precisely control the luminescence colors by adjusting the left and right luminous intensity. Moreover, phosphorescence emission colors of films can be tuned from blue, purple and green to red by varying the concentration of PFD and the type of phosphor. These emission lights were achieved through a radiative energy transfer pathway between light-conversion molecules and the phosphor. A possible mechanism of the light conversion process was proposed. The results presented here would offer a promising way to effectively and conveniently broaden the color of persistent luminescence, which may open up new opportunities for individual customization, colorful surface coating and anti-counterfeiting of polymers.
Introduction
Persistent luminescence, also known as long-lasting phosphorescence or an afterglow, is a special optical phenomenon characterized by the ability of some materials to emit light even after the cessation of external light stimulations.1 In 1996, Matsuzawa et al. first reported an ultralong green phosphor, rare earth-doped strontium aluminate with 30 hours of afterglow lifetime.2 Currently, blue and green persistent luminescence phosphors have been well-developed and commercialized due to their excellent performance,3–8 but persistent luminescence materials of other colors are still being developed in the laboratory due to their difficult synthesis and poor performance. Colorful spectra will open up opportunities for persistent luminescence materials for applications, especially in new types of light sources,8,9 optoelectronic devices,10–12 individual customization,13,14 and advanced information security/information anticounterfeiting.15–17 Therefore, the development of color-tunable photoluminescence materials has important scientific significance and practical value but remains challenging.Recently, more efforts have been devoted to preparing tunable multicolor materials.18–21 A straightforward and effective way to achieve a considerable variation in the emission color is to combine materials with different luminescence bands.22–25 Gong et al. reported full-spectrum persistent luminescence tuning by spinning the CsPbX3 perovskite quantum dots as an efficient light conversion layer on the surface of a CaAl2O4:Eu2+,Nd3+ afterglow phosphor.26 Shintaro Furukawa et al. prepared a polymer film with tunable luminescence color by adding a 2-(2′-hydroxyphenyl) imidazo [1,2-α] pyridine derivative.27 The polymer films exhibit a wide range of colors, from purple to orange, by changing the concentration and the type of the polymer matrix. Since their preparation methods are all spin coating, the application in many scenarios is restricted. Zhao dan et al. used the inclusion complex of rhodamine 6G with (2-hydrosypropyl)-β-cyclodextrin as light conversion materials to fine-tune the persistent luminescence from green to orange.28 However, small-molecule organic dyes undergo fast dye photodegradation, which hinders their practical application. Polymer fluorescent dyes (PFDs) are functional polymers which have a chromophoric center within their structure.29–33 Owing to their inherent non-leaching behavior and light aging combined with the special properties of polymers, polymer bonded dyes or reactive dyes may play more important roles.34–39
Herein, we prepared a series of fascinating multicolored photoluminescent composite films by simple mixing PFDs with three kinds of persistent luminescence phosphors in silica gel medium at room temperature (Fig. 1a). The spectroscopic and photophysical properties of composite films were characterized and the possible mechanism of the light conversion process were investigated. Remarkably, the corresponding films show various emission colors as precisely measured from the CIE coordinates, which demonstrate that a full spectrum adjustment can be achieved through radiative energy transfer.40–42
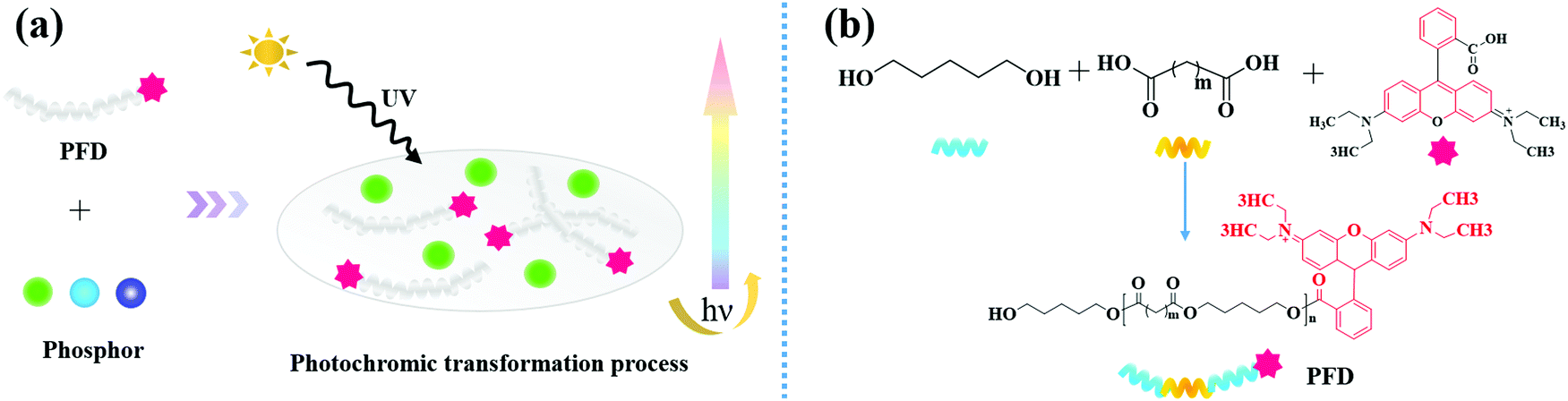 | ||
| Fig. 1 (a) Illustration for full-spectrum persistent luminescence tuning using PFD; (b) schematic diagram of synthesis of PFD by covalently attaching the rhodamine fluorescent dye onto PEDO-based aliphatic polyester chains. | ||
Experimental
Chemicals
Aluminum oxide (Al2O3, 99.99%), strontium carbonate (SrCO3, 99.99%), europium nitrate hexahydrate (Eu(NO3)3·6H2O, 99.99%), dysprosium nitrate hexahydrate (Dy(NO3)3·6H2O, 99.99%), calcium carbonate (CaCO3, AR), europium(III) oxide (Eu2O3, 99.99%), neodymium(III) oxide (Nd2O3, 99.99%), lanthanum(III) oxide (La2O3, 99.99%) and boric acid(H3BO3, AR) were purchased from Aladdin Chemistry Co., Ltd (Shanghai, China). A silicone elastomer and the curing agent were purchased from Beijing Sanjing Xinde Technology Co., Ltd (Beijing, China). The PFD was kindly provided by Zhejiang Wanlong Chemical CO., Ltd. (Zhejiang, China). All the agents were used as received without any further purification.Synthesis of SrAl2O4: Eu2+, Dy3+ (SA2O4) phosphors
The powders of SA2O4 phosphors were synthesized via the solid-state reactions at 1350 °C according to the reference method.43 The molar ratio of SrCO3, Al2O3, Eu(NO3)3·6H2O, Dy(NO3)3·6H2O, and H3BO3 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.02:0.04:0.1. The starting materials were mixed thoroughly in a ball mill for 4 h and subsequently heated to 1350 °C under a mild reducing atmosphere of activated carbon for 4 h. After cooling, the sintered products were re-milled and sieved to get the phosphors.
:1:0.02:0.04:0.1. The starting materials were mixed thoroughly in a ball mill for 4 h and subsequently heated to 1350 °C under a mild reducing atmosphere of activated carbon for 4 h. After cooling, the sintered products were re-milled and sieved to get the phosphors.
Synthesis of Sr4Al14O25: Eu2+, Dy3+ (S4A14O25) phosphors
The powders of S4A14O25 phosphors were prepared using the solid-state reaction method.44 The molar ratio of SrCO3, Al2O3, Eu(NO3)3·6H2O, Dy(NO3)3·6H2O, and H3BO3 is 4:7:0.02:0.04:0.1. The raw materials were thoroughly mixed by using a ball mill for 4 h. After being dried at 120 °C for 24 h, the final luminescent powders were obtained by calcining the raw mixture at 1350 °C for 4 h in a reducing environment.
Synthesis of CaAl2O4: Eu2+, Nd3+, La3+ (CA2O4) phosphors
The powder of CA2O4 were prepared by the solid-state reactions method.45 The molar ratio of CaCO3, Al2O3, Eu2O3, Nd2O3, La2O3 and H3BO3 is 1:1:0.02:0.02:0.01:0.1. The starting materials were mixed in a ball mill for 4 h and subsequently heated to 1320 °C under a mild reducing atmosphere of activated carbon for 2 h. After cooling, the sintered products were re-milled and sieved to get the phosphors.
Preparation of SA2O4/PFD, S4A14O25/PFD and CA2O4/PFD composite films
The SA2O4/PFD, S4A14O25/PFD and CA2O4/PFD composite were fabricated through addition of PFD and phosphors onto the flexible silicone substrates, respectively. The mixture of a silicone elastomer, the curing agent, phosphors and PFD with a weight ratio of 5:5: 2: x (0#: x = 0 wt%, 1#: x = 0.01 wt%, 2#: x = 0.025 wt%, 3#: x = 0.05 wt%, 4# x = 0.1 wt%, 5# x = 0.15 wt%, 6#: x = 0.2 wt%, 7#: x = 0.25 wt%, 8#: x = 0.5 wt%, 9#: x = 0.75 wt%, 10#: x = 1 wt%, 11#: x = 1.25 wt%, 12#: x = 1.5 wt%, 13#: x = 1.75 wt%, 14#: x = 2 wt%, x were based on the percentage of phosphor mass) were stirred at room temperature. Then, the mixture was poured into a round mold with a diameter of ∼3 cm and a thickness of 1 mm. The mixture formed a thin film and was aged at 60 °C for 1 h.
Instrumentation
The samples were tested using a Fourier-transform infrared (FT-IR) spectrometer (Thermo Fisher Nicolet iS 50) in the range of 500–4000 cm−1 with a resolution of 4 cm−1. Ultraviolet-visible (UV-vis) absorption spectra were recorded on a UV-vis-NIR spectrophotometer (Agilent Cary 5000). The surface morphologies of the phosphor particles and phosphor/PFD composites were inspected using a Field Emission Scanning Electron Microscope (FESEM, Thermo Fisher-Apreo S LoVac). The X-ray diffraction (XRD) measurements were carried out on an X-ray powder diffractometer (Rigaku Miniflex 600, Cu Kα radiation, λ = 1.5418 Å) with an angle (2θ) range of 3° to 70°. The photoluminescence performances (excitation and emission spectra), steady state photoluminescence quantum yield and CIE chromaticity coordinates were measured using a Fluorescence Spectrometer (Edinburgh Instruments FLS 980, under 365 nm excitation). Afterglow decay curves were tested using a PR-305 afterglow Brightness tester (SENSING Instruments Co., Ltd. China) where the samples were excited using a 150 W xenon arc lamp, and the excitation wavelength is 365 nm. The excitation time is 15 min with excitation illumination of 1000 lx to ensure that our samples fully absorb energy. The detector is HAMAMATSU CR114. The data were recorded 10 s later. All of the measurements were carried out at room temperature.Results and discussion
In this work, we employed organic polymer fluorescent dyes as high-efficiency light conversion materials to realize the full spectrum controllable adjustment of the persistent luminescence.As outlined in Fig. 1b, the PFD was produced by covalently attaching the rhodamine B (RhB) fluorescent dye onto 1,5-pentanediol (PEDO)-based aliphatic polyester chains via esterification followed by polycondensation. Fig. 2a shows the FT-IR spectrum of PFD, RhB and PEDO. Comparing the FT-IR spectra of PFD to PEDO, the absorption peaks at 2936 cm−1 and 2853 cm−1 of the methylene group on PEDO can be observed from the FT-IR spectra of PFD. Due to the esterification reaction, the absorption peak of the hydroxyl group on PEDO at 3300 cm−1 is weakened in PFD. Meanwhile, it can be seen from the FT-IR spectra of PFD and RhB that there is no obvious difference between the two spectra at the main characteristic absorption peaks. Typical absorption peaks of RhB can be observed from the infrared spectrum of the PFD.46 1601 cm−1, 1503 cm−1 and 1450 cm−1 were assigned to the stretching resonances of the benzene ring on RhB; 1254 cm−1 was assigned to the characteristic absorption peak of the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O on RhB; 1068 cm−1 was assigned to the stretching resonances of –C–O–C– on RhB; 3502 cm−1 was assigned to the stretching resonances of –OH on RhB. The enhancement of the stretching vibration peak of CO at 1254 cm−1 and the weakening of the stretching vibration peak of the terminal hydroxyl at 3502 cm−1 on the PFD infrared spectrum can explain the higher molecular weight of polyester to a certain extent.47,48 The peak of the terminal hydroxyl group on the PFD infrared spectrum shifted from 3340 cm−1 to 3502 cm−1, indicating that RhB formed a strong hydrogen bond with the polyester after being grafted onto the polyester segment.
O on RhB; 1068 cm−1 was assigned to the stretching resonances of –C–O–C– on RhB; 3502 cm−1 was assigned to the stretching resonances of –OH on RhB. The enhancement of the stretching vibration peak of CO at 1254 cm−1 and the weakening of the stretching vibration peak of the terminal hydroxyl at 3502 cm−1 on the PFD infrared spectrum can explain the higher molecular weight of polyester to a certain extent.47,48 The peak of the terminal hydroxyl group on the PFD infrared spectrum shifted from 3340 cm−1 to 3502 cm−1, indicating that RhB formed a strong hydrogen bond with the polyester after being grafted onto the polyester segment.
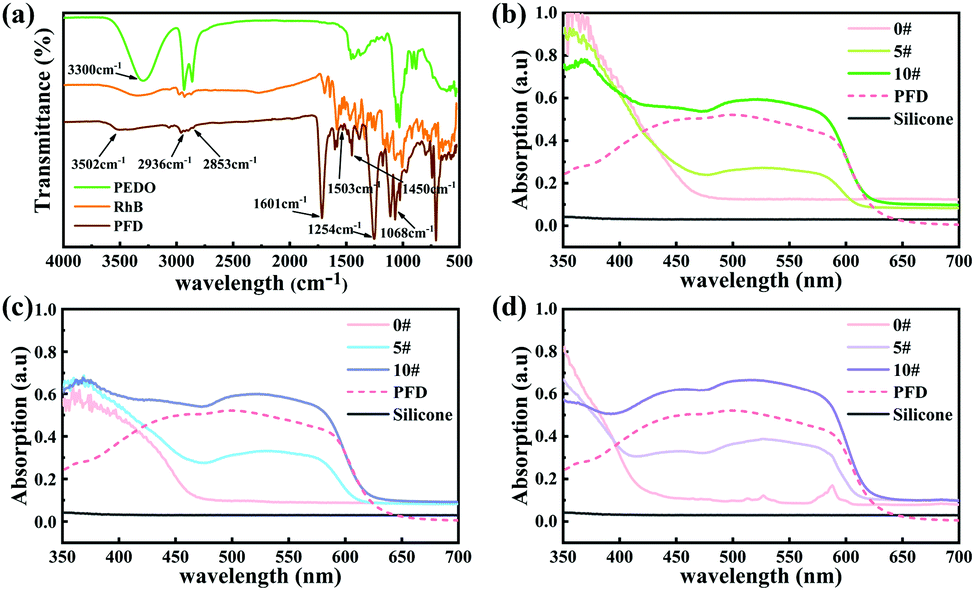 | ||
| Fig. 2 (a) FT-IR spectra of PFD, RhB and PEDO. (b) UV-Vis absorption spectra of SA2O4/PFD composite films (0#, 5#, 10#) and PFD. (c) UV-Vis absorption spectra of S4A14O25/PFD composite films (0#, 5#, 10#) and PFD. (d) UV-Vis absorption spectra of CA2O4/PFD composite films (0#, 5#, 10#) and PFD. | ||
The UV-vis absorption spectra of SA2O4/PFD composite films (0#, 5#, 10#), S4A14O25/PFD composite films (0#, 5#, 10#), CA2O4/PFD composite films and PFD were recorded in Fig. 2b–d. Generally, the flexible silicone substrates had no absorption in the range of 350–700 nm. The composite film 0# has strong absorption in the range of 350–470 nm, 350–460 nm and 350–425 nm, which is derived from the absorption of SA2O4, S4A14O25 and CA2O4 phosphors, respectively. The PFD had a wide visible-light absorption band in the range of 410–590 nm. Therefore, with the gradual increase of PFD content from sample 0# to sample 10#, the absorption of SA2O4/PFD composite films, S4A14O25/PFD composite films and CA2O4/PFD composite films in the range of 430–590 nm gradually increased.
The XRD patterns of PFD, silicone, SA2O4, S4A14O25, CA2O4, and the related PFD composite films are shown in Fig. 3a–d. Fig. 3a shows the XRD patterns of PFD and silicone substrates. The broad peak indicates these two compounds are amorphous structures (Fig. 3a). The prepared SA2O4, S4A14O25 and CA2O4 long afterglow powders were analyzed using MDI Jade5.0 software and compared with standard PDF cards. It can be seen from Fig. 3b that the characteristic diffraction peaks of the SA2O4 long afterglow powder are completely consistent with those of monoclinic SrAl2O4 (JPCDS No. 34-0379), and the relevant lattice parameters are a = 8.4427 Å, b = 8.8220 Å, c = 5.1603 Å. It can be seen from Fig. 3c that the characteristic diffraction peaks of the S4A14O25 long afterglow powder are completely consistent with those of the monoclinic S4A14O25 (JPCDS No. 52-1876), and the relevant lattice parameters are a = 24.791 Å, b = 8.4858 Å, c = 4.8865 Å. It can be seen from Fig. 3d that the characteristic diffraction peaks of CA2O4 long afterglow powder are completely consistent with the orthorhombic CA2O4 (JPCDS No. 53-0191), and the relevant lattice parameters are a = 8.68683 Å, b = 8.08354 Å, and c = 15.19239 Å. This means that we have successfully prepared SA2O4, S4A14O25 and CA2O4 long afterglow powders, ensuring the luminescence properties of the long afterglow powders. Meanwhile, from the Fig. 3b–d, we can see that the SA2O4, S4A14O25 and CA2O4 phosphors in the composite films still maintain their crystal structure, which can ensure the luminescence properties of phosphor.
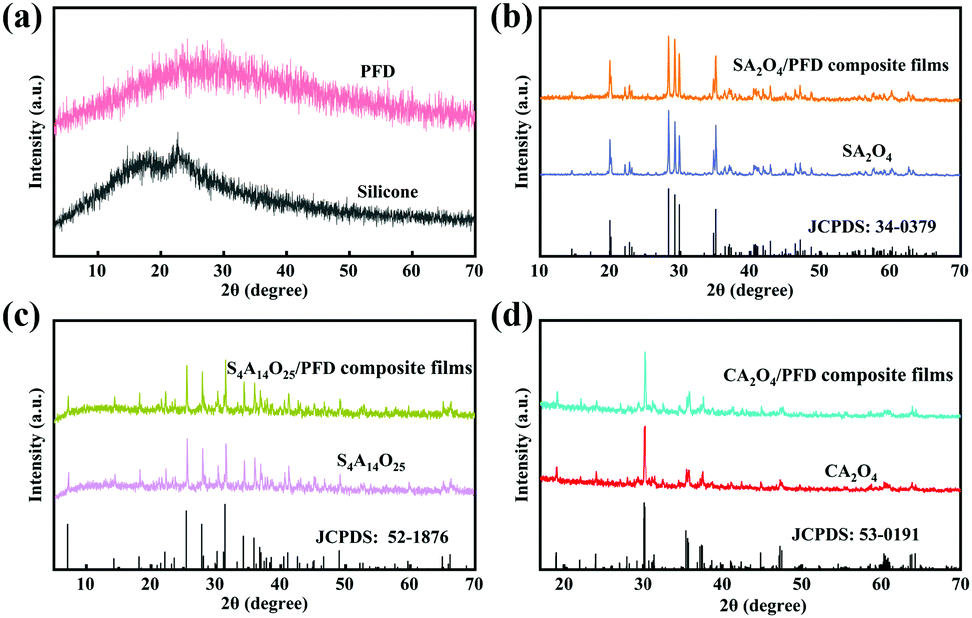 | ||
| Fig. 3 (a) XRD patterns of PFD and silicone, respectively. (b) XRD patterns of SA2O4 and SA2O4/PFD composite films. (c) XRD patterns of S4A14O25 and S4A14O25/PFD composite films. (d) XRD patterns of CA2O4 and CA2O4/PFD composite films. | ||
The morphologies of SA2O4, S4A14O25, CA2O4 phosphors and PFD are shown in Fig. 4a–d, respectively. The as-synthesized SA2O4, S4A14O25, C2A4O phosphors are neat and tidy, with some debris from grinding, and the particle size is between 10 and 20 μm. This particle size does not affect its dispersibility in applications, and can also ensure its luminous intensity, which was confirmed in subsequent applications. The PFD displays a loose lamellae-shaped assembly with some large agglomerates of nanoparticles (Fig. 4d). SEM characterization of the hybrid films (Fig. 4e) clearly illustrates a homogeneous distribution of the phosphor and PFD within the film. The cross-section of the film shows that the phosphor and PFD are well embedded and dispersed in the flexible silicone substrates (Fig. 4f).
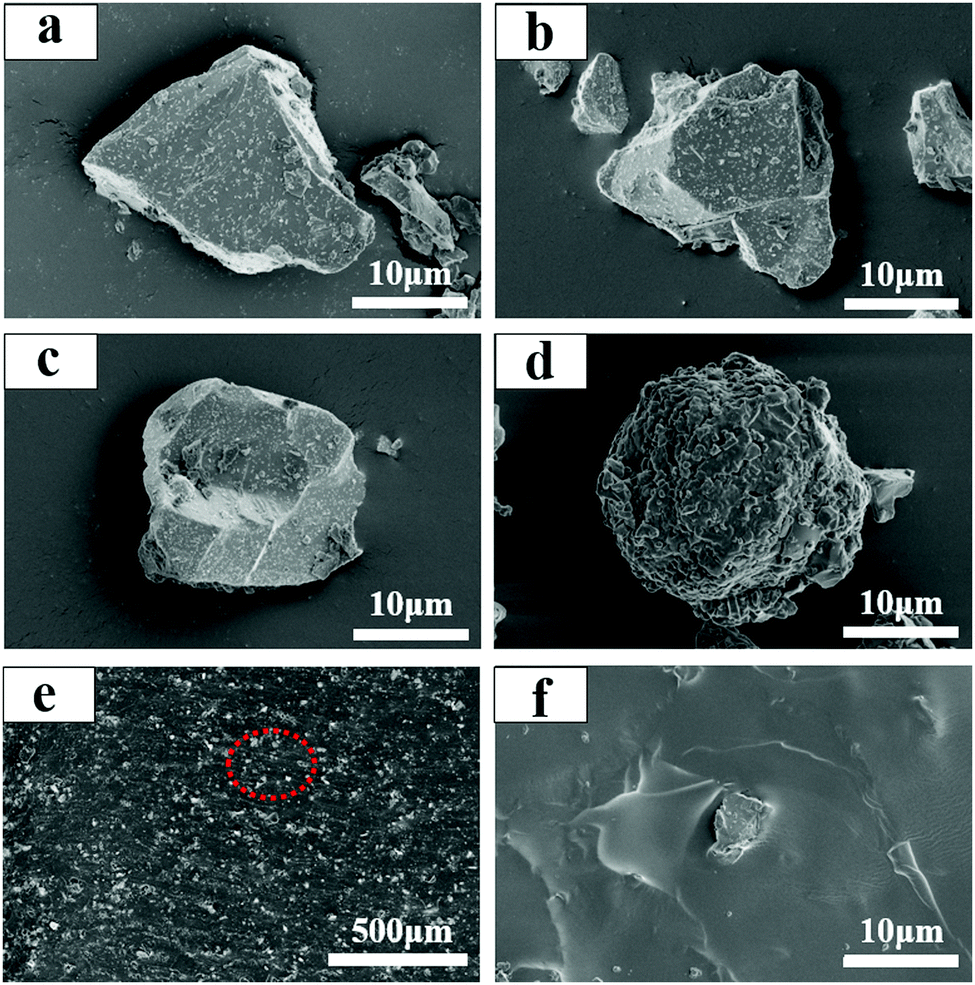 | ||
| Fig. 4 (a) SEM micrograph of SA2O4. (b) SEM micrograph of S4A14O25. (c) SEM micrograph of C2A4O. (d) SEM micrograph of PFD. (e and f) SEM micrograph of CA2O4/PFD composite films with different scales. | ||
Fig. 5a shows the PL excitation spectrum of the SA2O4, S4A14O25, and CA2O4 phosphors monitored at the maximum emission wavelengths 450 nm, 490 nm and 525 nm, respectively. Fig. 5b shows the PL excitation spectrum monitored at the emission wavelength 616 nm and emission spectrum of the PFD. The excitation spectrum of the PFD overlapped with the emission spectrum of phosphor (Fig. 5c). Therefore, theoretically, the phosphor can be used as an excitation light source to excite the PFD effectively, to produce color converted long afterglow films. Furthermore, as shown in Fig. 5d–f, the phosphorescence emission spectra of S2A4O/PFD composite films, S4A14O25/PFD composite films and CA2O4/PFD composite films were recorded, respectively. Since the silicon substrate is transparent, there are no fluorescence absorption and emission signals. Thus, the observed emission spectra upon photoexcitation of these polymer films were assigned to dual emission bands: the fluorescence emission of the PFD and the phosphorescence emission of phosphor. After further observation and research, there is an equal strength point between the two emission bands (Fig. 5d–f). Since the luminescence color of the composite films was a mixed fluorescence color of the phosphor and PFD, around this point, by adjusting the left and right luminous intensities, in other words, by changing the mass ratio of the phosphor and PFD, a precise control of the luminescence color was achieved. In order to further study the mechanism of light conversion, we selected sample 10# in the three groups of composite films to perform peak fitting. As shown in Fig. 5d–f, the left peaks between 450 nm to 575 nm, 430 nm to 570 nm and 400 nm to 530 nm were assigned to the typical afterglow phosphorescence emission of S2A4, S4A14O25 and CA2O4, respectively. The right peaks were assigned to the photoluminescence fluorescence emission of PFD. When the mass ratio of phosphor/PFD composite films increased from 0 wt% to 2 wt%, the emission intensity of left peaks at the wavelength of 450 nm, 490 nm and 525 nm decreased respectively. The reason for the decrease in the intensity of the left peak is that the PFD partially absorbs the photons emitted by the phosphor. In contrast, the maximum emission peak intensity of right peaks gradually increased and shifted from 595 nm to 612 nm, which is assigned to the concentration effect. The intensity of the right peak is much higher than that of the left peak, indicating that the phosphor successfully transfers energy to the PFD. The emission peaks of the SA2O4/PFD, S4A14O25/PFD, CA2O4/PFD composite films 10# (Fig. 5g–i) were well fitted, in which fitting curves 10-1 could be divided into 10-2, 10-3 and 10-4 sub-peaks, respectively. The ratio of the peak area is 0.18:0.26:0.56 for the fitting peaks 10-2, 10-3, and 10-4 of the SA2O4/PFD composite films 10#. The ratio of peak area is 0.13:0.32:0.55 for fitting peaks 10-2, 10-3, and 10-4 of the S4A14O25/PFD composite films 10#. The ratio of peak area is 0.1:0.35:0.55 for fitting peaks 10-2, 10-3, and 10-4 of the CA2O4/PFD composite films 10#, which is consistent with the concentration ratio of adding PFD. Although the physical meaning of three-component peak was not clear, it is significant that this method can be used to make a semi-quantitative interpretation.
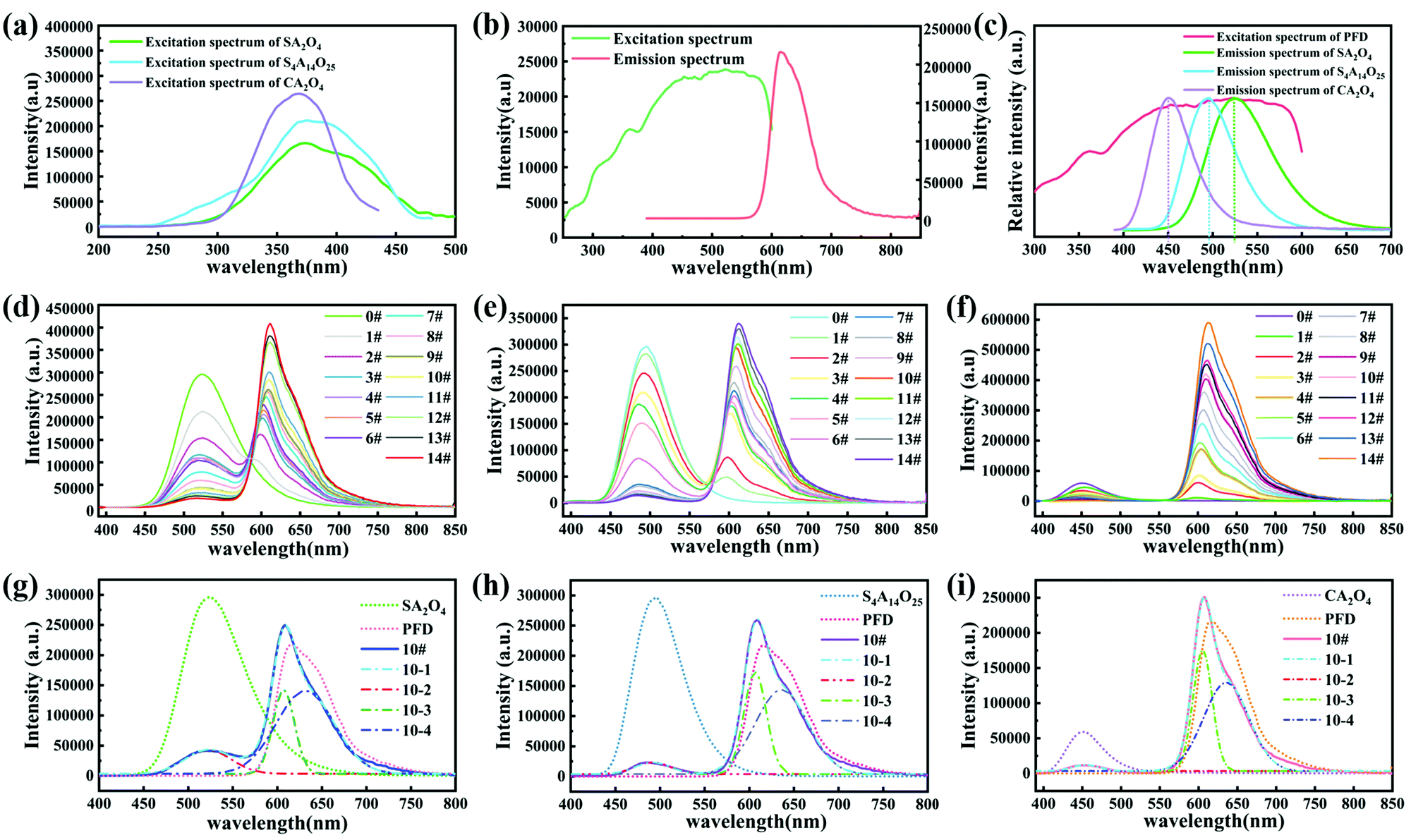 | ||
| Fig. 5 (a) Excitation of SA2O4, S4A14O25, and CA2O4 phosphors. (b) Excitation and emission spectrum of PFD. (c) Emission spectrum of SA2O4, S4A14O25, and CA2O4 phosphors (λEX = 365 nm) and excitation spectrum of PFD (λEM = 525 nm). (d) Emission spectrum (λEX = 365 nm) of SA2O4/PFD composite films with various contents of PFD. (e) Emission spectrum (λEX = 365 nm) of S4A14O25/PFD composite films with various contents of PFD. (f) Emission spectrum (λEX = 365 nm) of CA2O4/PFD composite films with various contents of PFD. Multi-peak fitting curve of prepared SA2O4/PFD composite films 10# (g), S4A14O25/PFD composite films 10# (h), and CA2O4/PFD composite films 10# (i). | ||
The CIE 1931 chromaticity diagram of the luminescing films is shown in Fig. 6a. When changing the molar ratio and the type of excitation light source, we observed that the film colours varied from green, blue and purple to red, respectively. The phosphor in the luminescent film acts as an energy storage light source to harvest the photons and store the excitation energy, and then slowly releases photons. The persistently emitted photons from phosphors are captured by PFD molecules, followed by photon emission through a down-conversion process. The emitted light color of these composites is a linear combined color of the PFD and phosphor.49 Therefore, the colors of luminescent films can be facilely fine-tuned by changing various the concentration of PFD and the type of the phosphor light sources, which is consistent with the change of the fluorescence spectrum. In fact, from the CIE chromaticity diagram, we have successfully achieved tuneable persistent luminescence through three directions.
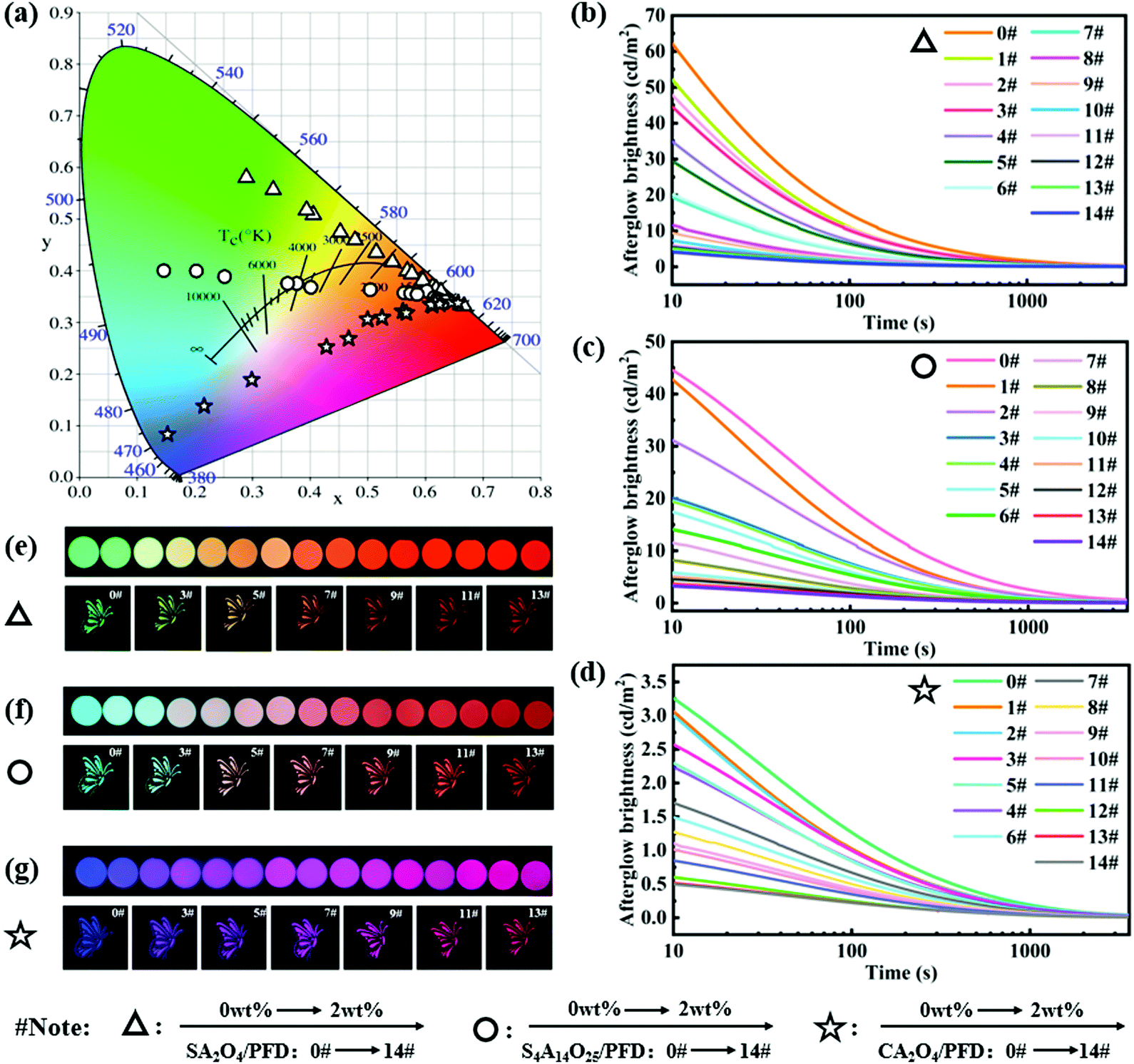 | ||
| Fig. 6 (a) CIE 1931 chromaticity diagram for SA2O4/PFD composite films, S4A14O25/PFD composite films, and CA2O4/PFD composite films with various contents of the PFD. (b–d) Afterglow decay curves (0–3600 s) of SA2O4/PFD composite films, S4A14O25/PFD composite films, and CA2O4/PFD composite films with various contents of the PFD. (e–g) Optical images photographed in the darkroom of SA2O4/PFD composite films, S4A14O25/PFD composite films, and CA2O4/PFD composite films with various contents of the PFD (afterglow images recorded at a delay time of 5 min after saturated excitation with a 365 nm UV lamp). | ||
To gain more insights into the energy transfer process in composite films, we recorded the afterglow decay curves of the SA2O4/PFD composite films, S4A14O25/PFD composite films, and CA2O4/PFD composite films, respectively. As shown in Fig. 6b–d, the afterglow intensity of the film slowly decreased over a period of time, and the intensity remained at a relatively high level after attenuation for 2 h, confirming that the long afterglow composite films were fabricated. The afterglow decay process shows the process of phosphorescence. The afterglow decay process shows a similar tendency: rapid decay and slow decay process, verifying the similar radiant energy transfer mechanism.26,50 Meanwhile, as the PFD content increases, the initial brightness of afterglow gradually decreases. This is because that the three phosphors are energy storage light sources,51 and the photons emitted by the phosphors were partially absorbed by the PFD.26 Through calculations, we obtain the energy transfer efficiency (ηET) of the composite films.52,53 The specific ηET date of the composite film are summarized in Table S1 (ESI†) and the efficiency curves are shown in Fig. S2 (ESI†). It can be concluded from the calculation that the energy transfer efficiency of the composite film decreases with the increase of the PFD content. The highest energy transfer efficiencies of the SA2O4/PFD, S4A14O25/PFD and CA2O4/PFD composite films are 16.5%, 27.7% and 15.5%, respectively. From the afterglow optical images (Fig. 6e–g), we can clearly observe that the colors of luminescent films gradually changed from green, blue and purple to red, which demonstrates that full spectrum adjustment can be achieved through radiative energy transfer.
Conclusions
In summary, full spectrum adjustment has been successfully achieved through a simple, fast and low-cost procedure. By changing the concentrations of PFD and the type of the phosphor light source, light intensity of the two types of emissions can be tuned, respectively, resulting in a tunable luminescence color of the PFD-doped polymer films. We demonstrated how to make full spectrum luminescence in a wide range of colors from blue, purple and green to red, respectively. The composite films have various uses, such as in optical materials and surface coating. Our strategy provides a promising system for fine-tuning the persistent luminescence, which may open up new opportunities for persistent luminescence materials toward individual customization, colorful surface coating and anti-counterfeiting of polymer, etc.Author contributions
Zhao dan: conceptualization, validation, formal analysis, investigation, writing – original draft, writing – review & editing. Lijun Song: resources, supervision, project administration, funding acquisition, writing – review & editing. Jinlei Li: software. Fenglong Lin: methodology. Shenglong Wang: data curation. Yincai Wu: visualization. Fulin Lin: test, analysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZR122), the project “The key technology of Rare earth long glow materials” and the joint luminescent fibre lab by Xinsiyuan New materials Co. Ltd and Xiamen Institute of Rare Earth Materials.References
- N. Siraj, B. El-Zahab, S. Hamdan, T. E. Karam, L. H. Haber, M. Li, S. O. Fakayode, S. Das, B. Valle, R. M. Strongin, G. Patonay, H. O. Sintim, G. A. Baker, A. Powe, M. Lowry, J. O. Karolin, C. D. Geddes and I. M. Warner, Anal. Chem., 2015, 88, 170–202 CrossRef PubMed.
- T. Matsuzawa, Y. Aoki, N. Takeuchi and Y. Murayama, J. Electrochem. Soc., 1996, 143, 2670–2673 CrossRef CAS.
- E. Finley, A. Cobb, A. Duke, A. Paterson and J. Brgoch, ACS Appl. Mater. Interfaces, 2016, 8, 26956–26963 CrossRef CAS PubMed.
- S. Liao, X. Ji, Y. Liu and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 39064–39073 CrossRef CAS PubMed.
- C. Wang, T. Takeda, O. Melvin Ten Kate, S. Funahashi, R. Xie, K. Takahashi and N. Hirosaki, ACS Appl. Mater. Interfaces, 2019, 11, 29047–29055 CrossRef CAS PubMed.
- Y. Zu, J. Xi, L. Li, J. Dai, S. Wang, F. Yun, B. Jiao, H. Dong, X. Hou and Z. Wu, ACS Appl. Mater. Interfaces, 2019, 12, 2835–2841 CrossRef PubMed.
- S. Ghose, S. Singh and T. S. Bhattacharya, ACS Appl. Mater. Interfaces, 2020, 12, 7727–7735 CrossRef CAS PubMed.
- L. Wang, R. Xie, T. Suehiro, T. Takeda and N. Hirosaki, Chem. Rev., 2018, 118, 1951–2009 CrossRef CAS PubMed.
- X. Zhang, W. Liu, G. Z. Wei, D. Banerjee, Z. Hu and J. Li, J. Am. Chem. Soc., 2014, 136, 14230–14236 CrossRef CAS PubMed.
- V. K. Praveen, S. J. George, R. Varghese, C. Vijayakumar and A. Ajayaghosh, J. Am. Chem. Soc., 2006, 128, 7542–7550 CrossRef CAS PubMed.
- A. Feinle, F. Lavoie-Cardinal, J. Akbarzadeh, H. Peterlik, M. Adlung, C. Wickleder and N. Hüsing, Chem. Mater., 2012, 24, 3674–3683 CrossRef CAS PubMed.
- Y. Liu, H. Wang, P. Liu, H. Zhu, B. Shi, X. Hong and F. Huang, Angew. Chem., Int. Ed., 2021, 60, 5766–5770 CrossRef CAS PubMed.
- L. Jia, S. Zeng, H. Ding, A. T. Smith, A. M. Lachance, M. M. Farooqui, D. Gao, J. Ma and L. Sun, Adv. Funct. Mater., 2021, 2104427 CrossRef CAS.
- W. Tian, J. Zhang, J. Yu, J. Wu, J. Zhang, J. He and F. Wang, Adv. Funct. Mater., 2018, 28, 1703548 CrossRef.
- M. Zuo, W. Qian, T. Li, X. Hu, J. Jiang and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 39214–39221 CrossRef CAS PubMed.
- T. Si, Q. Zhu, J. Xiahou, X. Sun and J. Li, ACS Appl. Electron. Mater., 2021, 3, 2005–2016 CrossRef CAS.
- C. Shi, X. Shen, Y. Zhu, X. Li, Z. Pang and M. Ge, ACS Appl. Mater. Interfaces, 2019, 11, 18548–18554 CrossRef CAS PubMed.
- M. Aghajamali, M. Iqbal, T. K. Purkait, L. Hadidi, R. Sinelnikov and J. G. C. Veinot, Chem. Mater., 2016, 28, 3877–3886 CrossRef CAS.
- J. Gil-Rostra, F. J. Ferrer, J. P. Espinós, A. R. González-Elipe and F. Yubero, ACS Appl. Mater. Interfaces, 2017, 9, 16313–16320 CrossRef CAS PubMed.
- S. Kamal, K. P. Bera, M. Usman, B. Sainbileg, S. Mendiratta, A. Pathak, A. I. Inamdar, C. Hung, M. Hayashi, Y. Chen and K. Lu, ACS Appl. Nano Mater., 2021, 4, 2395–2403 CrossRef CAS.
- T. Huang, Y. Niu, F. Zhang, L. Zhang, S. Chen and Q. J. Yu, Appl. Mater. Today, 2017, 9, 176–183 CrossRef.
- T. Wang, P. Li and H. Li, ACS Appl. Mater. Interfaces, 2014, 6, 12915–12921 CrossRef CAS PubMed.
- S. Das, J. Manam and S. K. Sharma, New J. Chem., 2017, 41, 5934–5941 RSC.
- S. Das, J. Manam and S. K. Sharma, ECS J. Solid State Sci. Technol., 2016, 5, R98–R103 CrossRef CAS.
- Q. Tang, M. Wang, Z. Wang, W. Sun and R. Shang, Chem. Commun., 2017, 53, 4815–4817 RSC.
- Z. Gong, W. Zheng, Y. Gao, P. Huang, D. Tu, R. Li, J. Wei, W. Zhang, Y. Zhang and X. Chen, Angew. Chem., Int. Ed., 2019, 58, 6943–6947 CrossRef CAS PubMed.
- S. Furukawa, H. Shono, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2014, 6, 16065–16070 CrossRef CAS PubMed.
- L. S. L. H. Dan Zhao, ACS Omega, 2021, 39, 25585–25593 CrossRef PubMed.
- Y. Akae, H. Sogawa and T. Takata, Angew. Chem., Int. Ed., 2018, 57, 14832–14836 CrossRef CAS PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- J. Yan, Z. Wang, X. Lin, C. Hong, H. Liang, C. Pan and Y. You, Adv. Mater., 2012, 24, 5617–5624 CrossRef CAS PubMed.
- C. Calvino, A. Guha, C. Weder and S. Schrettl, Adv. Mater., 2018, 30, 1704603 CrossRef PubMed.
- H. Li, X. Zhang, X. Zhang, K. Wang, H. Liu and Y. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 4241–4246 CrossRef CAS PubMed.
- M. Sun, C. Hong and C. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS PubMed.
- O. Lavastre, I. Illitchev, G. Jegou and P. H. Dixneuf, J. Am. Chem. Soc., 2002, 124, 5278–5279 CrossRef CAS PubMed.
- S. Uchiyama, N. Kawai, A. P. de Silva and K. Iwai, J. Am. Chem. Soc., 2004, 126, 3032–3033 CrossRef CAS PubMed.
- L. Xia, Y. Xue, K. Xiong, C. Cai, Z. Peng, Y. Wu, Y. Li, J. Miao, D. Chen, Z. Hu, J. Wang, X. Peng, Y. Mo and L. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 26405–26413 CrossRef CAS PubMed.
- L. Xu, X. Shen, Z. Zhou, T. He, J. Zhang, H. Qiu, M. L. Saha, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 16920–16924 CrossRef CAS PubMed.
- H. Liu, P. Wu, S. Kuo, C. Chen, E. Chang, C. Wu and Y. Chan, J. Am. Chem. Soc., 2015, 137, 10420–10429 CrossRef CAS PubMed.
- M. Ganschow, C. Hellriegel, E. Kneuper, M. Wark, C. Thiel, G. Schulz-Ekloff, C. Bräuchle and D. Wöhrle, Adv. Funct. Mater., 2004, 14, 269–276 CrossRef CAS.
- X. Zhong, T. Chervy, L. Zhang, A. Thomas, J. George, C. Genet, J. A. Hutchison and T. W. Ebbesen, Angew. Chem., Int. Ed., 2017, 56, 9034–9038 CrossRef CAS PubMed.
- H. M. Nguyen, O. Seitz, W. Peng, Y. N. Gartstein, Y. J. Chabal and A. V. Malko, ACS Nano, 2012, 6, 5574–5582 CrossRef CAS PubMed.
- S. Han, K. C. Singh, T. Cho, H. Lee, D. Jakhar, J. P. Hulme, C. Han, J. Kim, I. Chun and J. Gwak, J. Lumin., 2008, 128, 301–305 CrossRef CAS.
- Z. Yuan, C. Chang, D. Mao and W. Ying, J. Alloys Compd., 2004, 377, 268–271 CrossRef CAS.
- W. W. G. Z. Yang Zhiping, J. Chin. Ceram. Soc., 2004, 32, 118–122 Search PubMed.
- Z. Xu, X. Sun, P. Ma, Y. Chen, W. Pan and J. Wang, J. Mater. Chem. C, 2020, 8, 4557–4563 RSC.
- J. Lu, L. Wu and B. Li, J. Appl. Polym. Sci., 2017, 134, 44544 Search PubMed.
- X. H. L. M. Zhang Li, J. Funct. Polym., 2017, 4, 437–443 Search PubMed.
- L. Wang, R. Xie, T. Suehiro, T. Takeda and N. Hirosaki, Chem. Rev., 2018, 118, 1951–2009 CrossRef CAS PubMed.
- Y. Zheng, H. Wei, P. Liang, X. Xu, X. Zhang, H. Li, C. Zhang, C. Hu, X. Zhang, B. Lei, W. Y. Wong, Y. Liu and J. Zhuang, Angew. Chem., Int. Ed., 2021, 60, 22253–22259 CrossRef CAS PubMed.
- F. Clabau, X. Rocquefelte, S. Jobic, P. Deniard, M. H. Whangbo, A. Garcia and T. Le Mercier, Chem. Mater., 2005, 17, 3904–3912 CrossRef CAS.
- Z. Chen, L. Luo, Y. Li, J. Li and Q. Wei, J. Lumin., 2019, 216, 116756 CrossRef CAS.
- Y. Luo and Z. Xia, J. Phys. Chem. C, 2014, 118, 23297–23305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00327a |
| This journal is © The Royal Society of Chemistry 2022 |