DOI:
10.1039/D2MA00378C
(Paper)
Mater. Adv., 2022,
3, 4899-4907
Non-ammonolysis synthesis and characterisation of environmentally benign yellow pigments based on calcium–tantalum perovskite oxynitrides†
Received
1st April 2022
, Accepted 9th May 2022
First published on 19th May 2022
Abstract
Calcium–tantalum-based perovskite oxynitrides were synthesised as candidates for environmentally benign inorganic yellow pigments. The compounds were prepared via a urea-nitriding method, which does not involve a harmful ammonolysis process. Rapid single-phase synthesis was achieved under the optimum conditions, and only removable by-products were generated. Although the colours and bandgap energies of the obtained calcium–tantalum oxynitrides, CaTa(O,N)3, fell short of those of CaTaO2N synthesised by ammonolysis, they were made comparable by doping with La3+. Among the samples synthesised in this study, the most brilliant yellow hue was achieved with Ca0.8La0.2Ta(O,N)3, which has lightness (L*) and yellowness (b*) values comparable to those of conventional inorganic yellow pigments. In this synthetic method, harmful substances are eliminated both from the products and the synthesis process. In addition, the short heat-treatment duration enables the products to be obtained as fine powders suitable for pigment applications. Calcium–tantalum-based perovskite oxynitrides prepared by the strategy developed in this study have potential applications as environmentally benign yellow pigments.
Introduction
Our world contains a variety of colours. Colour has psychological and aesthetic importance in daily life.1,2 Inorganic pigments have been used in a wide variety of applications such as ceramics, glasses, plastics, inks and others.3 However, inorganic pigments are not used only because of their colouristic properties. In a wide range of applications, their superior thermal and chemical stabilities, and resistance to climatic conditions give inorganic pigments advantages over their organic counterparts.3 Though various colours exist in inorganic pigment, yellow is in particularly high demand because of its high visibility and use in many applications, e.g., traffic signs, road markers, construction and others.4–6 Chromium yellow (PbCrO4),7 cadmium yellow (CdS·ZnS),8 and nickel–antimony–titanium yellow (NiO–Sb2O3–TiO2)9 have been popularly employed as inorganic yellow pigments for such applications. However, there is growing concern that the toxic elements (Pb, Cr, Cd and Sb) in these pigments critically affect human health as well as the environment.10–13 Recently, the Institute for Health Metrics and Evaluation estimated that in 2019, Pb exposure accounted for 0.90 million deaths and 21.7 million years lost of healthy life (disability-adjusted life years) world-wide owing to long-term health effects.14 This situation has led to governmental authorities strictly limiting the use of inorganic yellow pigments that contain hazardous elements. The development of alternatives that are applicable as environmentally-friendly yellow pigments is therefore strongly desired, and considerable efforts have been devoted to developing such alternative materials.15–24 For example, praseodymium yellow (ZrSiO4:Pr)21,22 and bismuth vanadate (BiVO4)23,24 have been developed as metal-oxide-based yellow pigments. However, the preparation of ZrSiO4:Pr is time consuming and its particles are poorly dispersible, which limits its use in paints and inks. Among several potential candidates, metal oxynitrides are also drawing much attentions as alternatives because they exhibit brilliant colours but do not contain toxic elements.25,26 Jansen and Letschert developed pigments based on CaTaO2N–LaTaON2 solid solutions, which are able to substitute conventional inorganic yellow-red pigments.25 Kim also reported that the preparation of CaTaO2N exhibited a chromatic colour of yellow, close to that of the commercial cadmium yellow pigment.26 However, there are several obstacles in the preparation of metal oxynitrides, and these need to be resolved. Ammonolysis, which involves the use of harmful NH3 gas for long time at high temperature, is usually required for synthesis of metal oxynitride.27–29 The single-phase synthesis of CaTaO2N by ammonolysis usually requires heat treatment at temperatures above 1000 °C for several days.30 Even though the final products are environmentally benign, their use is unfavourable if materials that affect the environment and/or human health are used in their preparation. Prolonged heat treatment at high temperatures is also undesirable because it consumes a large amount of energy. There is also a cost issue because of the continuous flow of expensive NH3 gas for a long period of time. In addition, the products grow into large crystalline grains, which are unsuitable for applications as pigments where a fine morphology is desired. In the context of these circumstances, several approaches for eliminating the use of toxic NH3 gas by using solid nitriding agents for metal oxynitride synthesis have been proposed.31–38 We recently developed a urea-nitriding method for synthesizing metal oxynitrides, namely GaN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnO and LaTiO2N, and the nitriding mechanisms were elucidated.39,40 The shorter heat-treatment duration compared with those in processes that involve ammonolysis achieved a reduction in energy consumption and enabled the formation of finer sample powders. More recently, the strontium–tantalum oxynitride, SrTaO2N, and its solid solutions with Sr1.4Ta0.6O2.9 have also been prepared by the urea-nitriding method using SrCO3 and Ta2O5 gel as precursors.41 The oxygen and nitrogen contents in the solid solutions can be controlled by changing the quantity of urea in the precursor mixture. The solid solutions exhibited various chromatic colours, suggesting their potential applications as secure inorganic pigments. However, tantalum-based oxynitrides that exhibit a brilliant yellow colour comparable to those of PbCrO4, CdS and ZrSiO4:Pr have not been obtained.
:ZnO and LaTiO2N, and the nitriding mechanisms were elucidated.39,40 The shorter heat-treatment duration compared with those in processes that involve ammonolysis achieved a reduction in energy consumption and enabled the formation of finer sample powders. More recently, the strontium–tantalum oxynitride, SrTaO2N, and its solid solutions with Sr1.4Ta0.6O2.9 have also been prepared by the urea-nitriding method using SrCO3 and Ta2O5 gel as precursors.41 The oxygen and nitrogen contents in the solid solutions can be controlled by changing the quantity of urea in the precursor mixture. The solid solutions exhibited various chromatic colours, suggesting their potential applications as secure inorganic pigments. However, tantalum-based oxynitrides that exhibit a brilliant yellow colour comparable to those of PbCrO4, CdS and ZrSiO4:Pr have not been obtained.
The main aims of the current work were the ammonolysis-free synthesis and characterisation of the calcium–tantalum perovskite oxynitride, CaTa(O,N)3, which has potential applications as an environmentally benign yellow pigment. There have been several reports on the synthesis of CaTa(O,N)3 through a non-ammonolysis process, e.g., calcination of CaCO3 and TaON,31 and nitridation of Ca2Ta2O7 with C3N4.36 However, because of the presence of raw material residues and the generation of by-products, single-phase synthesis of calcium–tantalum perovskite oxynitrides has not been achieved. In addition, a detailed colour evaluation of the products was not carried out in either of these reports. In this paper, we determined the precursor composition needed to obtain CaTa(O,N)3 in a single phase by using two different Ca sources, i.e., calcium carbonate (CaCO3) and calcium hydroxide (Ca(OH)2). The optical absorption properties of metal oxynitrides depend on their band structures. When both O2− and N3− ions occupy the anion sites of the perovskite structure, the O 2p and N 2p states are hybridised, and the bandgap energies of the metal oxynitrides are lower than those of the corresponding metal oxides.28,29,42 The ratio of O to N therefore critically affects the metal oxynitride colour. For example, in the case of CaTaO2N, the synthesised products often lack N, and the molar ratio of oxygen to nitrogen is not always 2:1.43 This is why the chemical formula of the calcium–tantalum perovskite oxynitride is represented by CaTa(O,N)3 in this paper. In addition to the colour coordinates, the bandgap energies and the O/N ratios of the obtained products were therefore evaluated in detail. To achieve the synthesis of calcium–tantalum-based oxynitrides with colours equivalent to those of commercially available inorganic yellow pigments such as BiVO4 and ZrSiO4:Pr, partial substitution of La3+ at Ca2+ sites of CaTa(O,N)3 for adjustment of the bandgap energy, i.e., the O/N ratio, was also investigated.
Experimental procedure
Details of the materials employed in this study are provided in the ESI.† As calcium sources, CaCO3 and Ca(OH)2 were selected for the preparation of CaTa(O,N)3 on the basis of cost, safety, and ease of handling. For the preparation of La-doped CaTa(O,N)3, lanthanum hydroxide (La(OH)3) was used as the lanthanum source. Tantalum oxide (Ta2O5) gel, which was synthesised via the sol–gel method, was used as the tantalum source for preparation of calcium–tantalum-based oxynitrides. Tantalum(V) ethoxide (Ta(OC2H5)5; 1 mmol) was added to ethanol (EtOH) (1 mL). Then, deionised (DI) water (5 mL) was added to the EtOH solution and the mixture was stirred vigorously to achieve hydrolysis and polycondensation of Ta(OC2H5)5. The mixture was centrifuged and the Ta2O5 gel was obtained by drying the separated product overnight in an oven at 50 °C. The Ta2O5 gel, Ca source (CaCO3 or Ca(OH)2) and urea (CO(NH2)2) were put in an agate mortar and were mixed well with a pestle for 10 min under dry conditions. The molar ratio of Ca (in the Ca source)/Ta (in the Ta2O5 gel)/urea in the precursor mixture was varied to obtain perovskite oxynitrides without any impurity phases. The synthesis of CaTa(O,N)3 was carried out in a horizontal quartz furnace tube under a flow of N2 gas (purity 99.99%) at 0.3 L min−1. An alumina boat loaded with the precursor mixture was placed at the centre of the furnace. The heating rate was 15 °C min−1. The temperature of heat-treatment was 950 °C. The heat-treatment duration was fixed at 2 h.
X-Ray diffraction (XRD) measurement, scanning electron microscopy (SEM), ultraviolet-visible (UV-vis) spectroscopy, Fourier-transform infrared (FT-IR) spectroscopy, chromatic property determination, oxygen/nitrogen combustion analysis and X-ray fluorescence (XRF) spectroscopy were used to characterise the obtained products. Details of the apparatus and methods used in this study are described in the ESI.†
Results and discussion
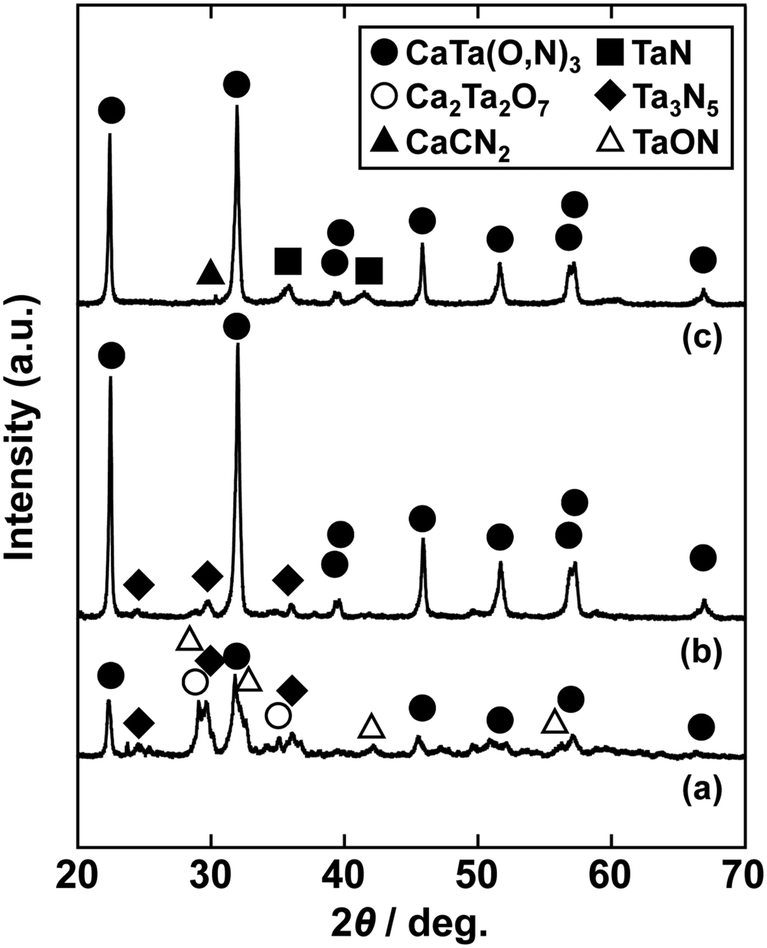
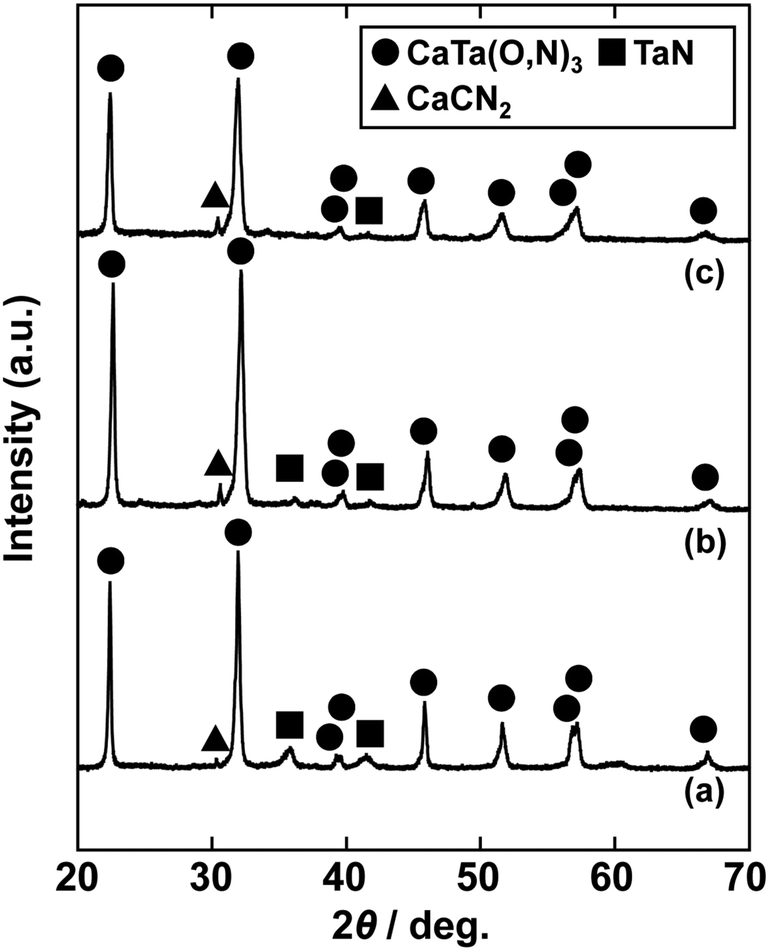
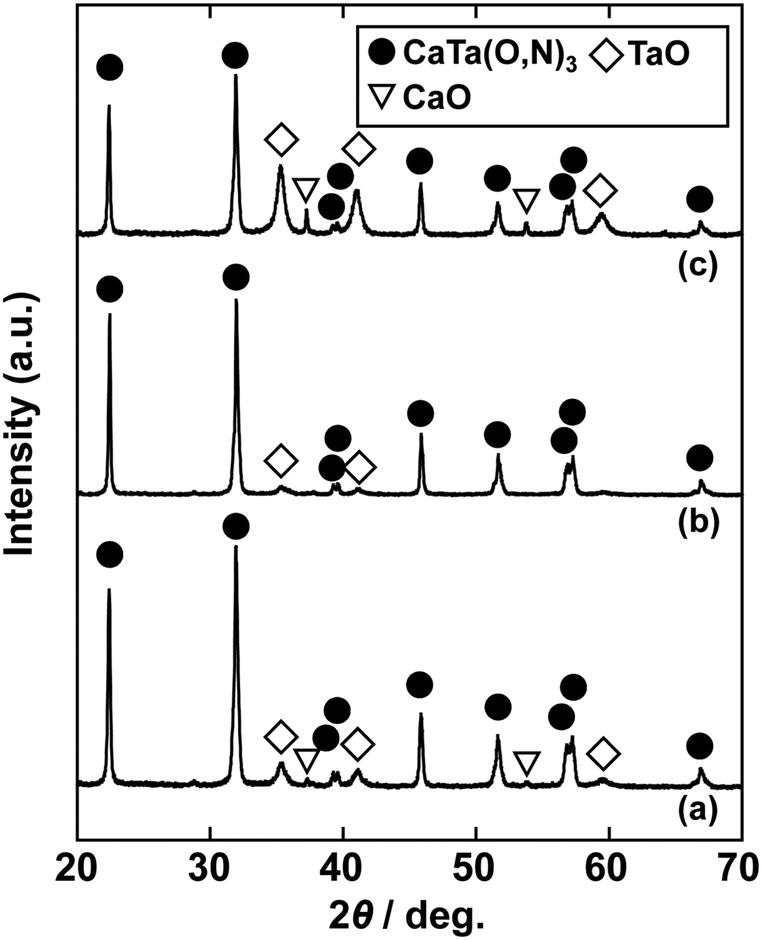
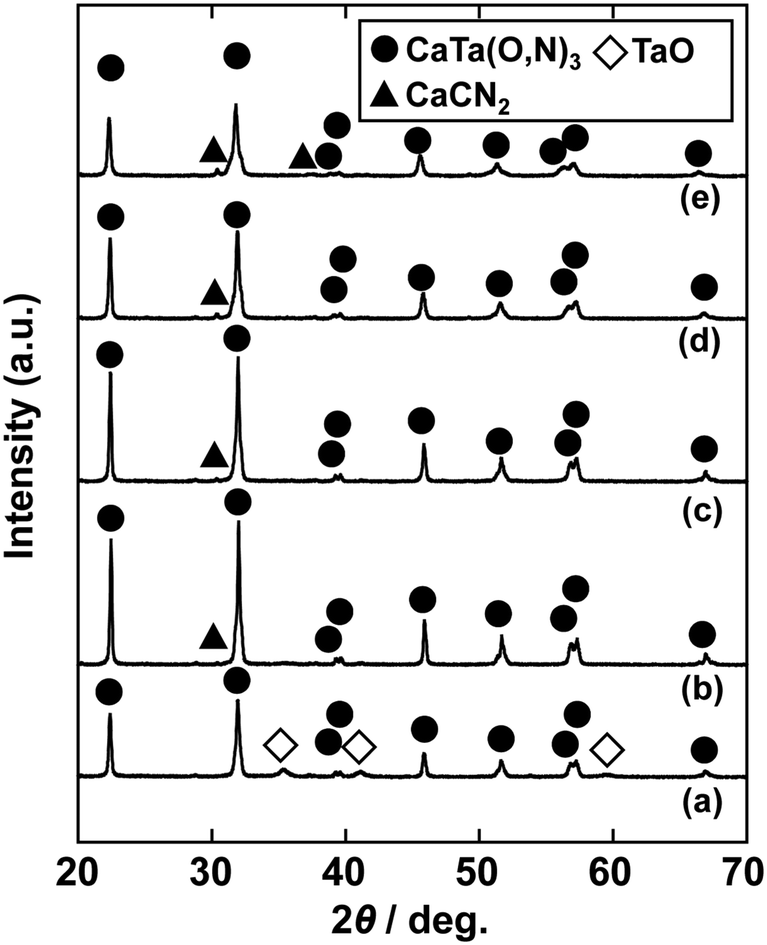
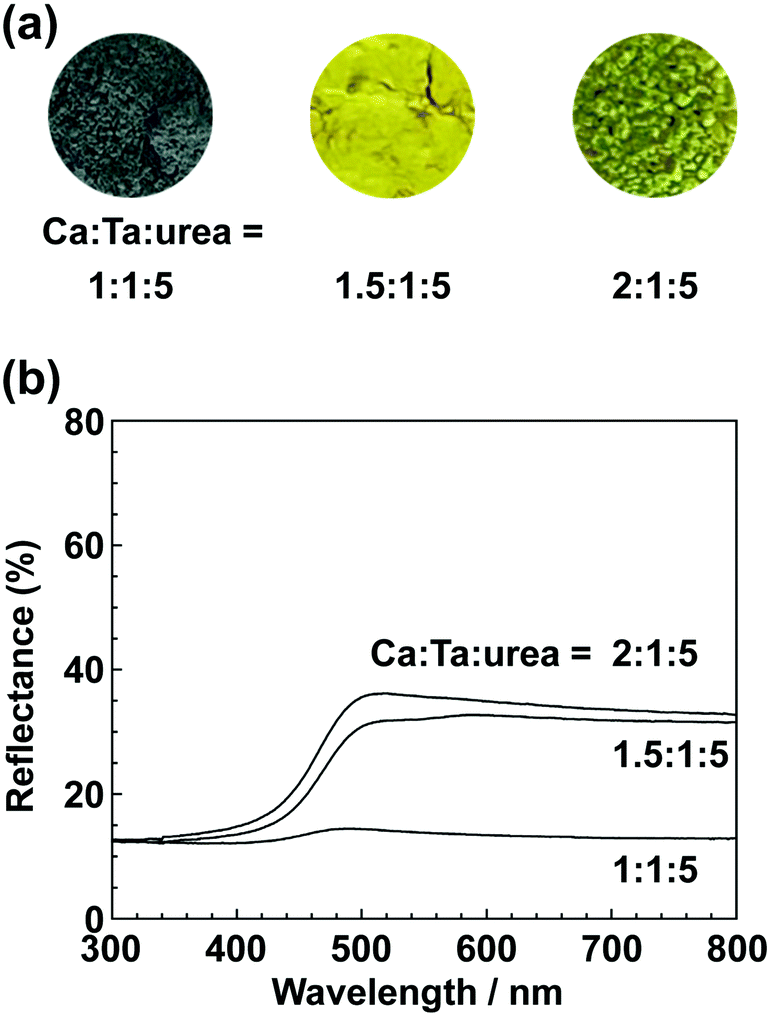
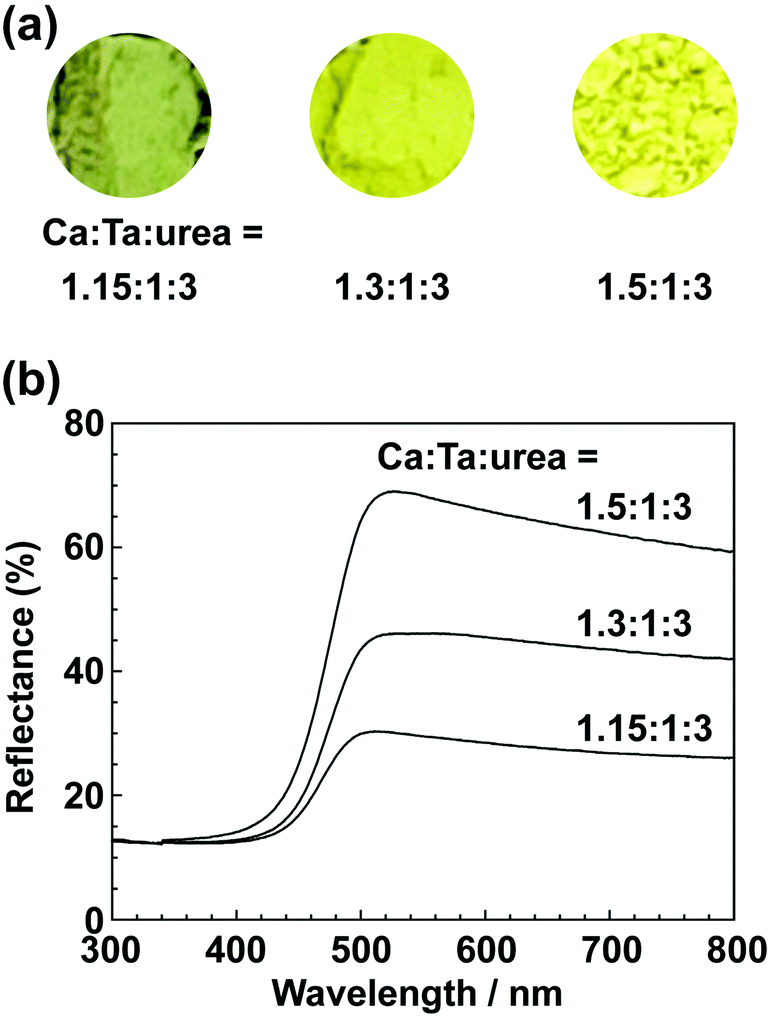
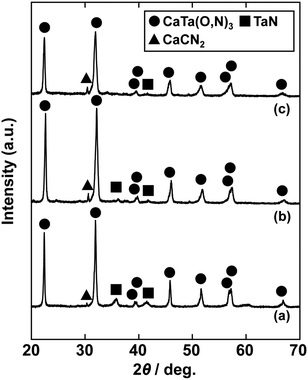
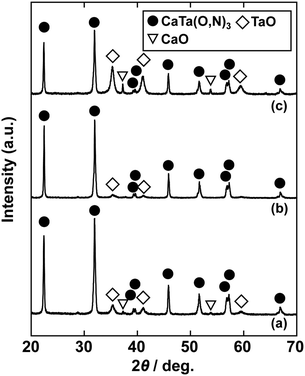
In the previous report on the preparation of SrTaO2N from a Ta2O5 gel and urea, a single-phase synthesis was achieved by using SrCO3 as the Sr source.41 In this study, we thus first examined the use of CaCO3 as the Ca source in the preparation of CaTa(O,N)3. Information obtained by characterisation of products synthesised with various molar ratios of urea can be used to identify the optimum conditions for the formation of perovskite oxynitrides.41 We therefore varied the urea content relative to those of CaCO3 and the Ta2O5 gel in the precursor mixture. The Ca and Ta molar ratio (x:y) in the precursor was fixed at 1:1 and the molar ratio of urea (z) was changed from 3 to 5. The XRD patterns of the products prepared from CaCO3, Ta2O5, and urea (x:y:z = 1:1:3, 1:1:4 and 1:1:5) are shown in Fig. 1. Photographs of the products are provided in the ESI† (Fig. S1). At the lowest urea content (x:y:z = 1:1:3), diffraction peaks assignable to the perovskite phase appeared, but other phases, such as Ca2Ta2O7, Ta3N5 and TaON, were also detected. As shown in Fig. S1a (ESI†), the colour of this product was pale beige, far from the yellow colour of CaTaO2N. In the cases of higher amounts of urea (x:y:z = 1:1:4 and 1:1:5), sharp and strong diffraction peaks corresponding to the perovskite phase were observed. The positions of these peaks correspond well to those of CaTaO2N (orthorhombic perovskite, space group: Pnma).44 Small amounts of tantalum nitrides (Ta3N5 and TaN) were detected as impurities. The grey colour of the product obtained with x:y:z = 1:1:5 (Fig. S1c, ESI†) was probably caused by the presence of TaN. These results indicate that z = 4 or higher is appropriate for obtaining CaTaO2N as the main product. However, post-treatment to remove the tantalum nitride by-products that dull the colour tone of the products is difficult. To suppress the generation of tantalum nitrides, we investigated the synthesis of CaTa(O,N)3 at various ratios of Ca to Ta. The calcium-based by-products can potentially be removed by post-treatment, therefore the molar ratio of Ca to Ta was increased. The Ta and urea molar ratio (y:z) in the precursor mixture was fixed at 1:5 and the molar ratio of Ca (x) was changed from 1 to 2. The XRD patterns of the products prepared from CaCO3, Ta2O5 and urea (x:y:z = 1:1:5, 1.5:1:5 and 2:1:5) are shown in Fig. 2. Photographs of the products are shown in the ESI† (Fig. S2). When the amount of CaCO3 was increased by a factor of 1.5 (Fig. 2(b)), the main product was CaTa(O,N)3, and CaCN2 was generated as a new by-product. It should be noted that the generation of CaCN2 was not observed under any conditions in a previous study of the preparation of CaTa(O,N)3 by using C3N4.36 Both are methods for the preparation of oxynitrides by using solid nitriding agents, but the nitridation processes with C3N4 and urea presumably differ. The colour of this product was pale yellow (Fig. S2b, ESI†). This indicates that the formation of TaN was suppressed, but weak TaN peaks were still present in the XRD pattern. However, when the amount of CaCO3 was doubled, the diffraction peaks of TaN almost disappeared (Fig. 2(c)). This shows that increasing the amount of CaCO3 effectively suppressed the formation of TaN in the product. CaCN2 decomposes to NH3 and CaCO3 when reacted with water, and CaCO3 is soluble in acidic solutions. The CaCN2 by-product can therefore be removed by treatment with water and hydrochloric acid (HCl aq.) The FT-IR spectra and XRD patterns of samples prepared from CaCO3, Ta2O5 and urea before and after washing with DI water and HCl aq. are provided in Fig. S3 and S4, respectively, in the ESI.† As depicted in Fig. S3, ESI† the peak attributed to the stretching vibration of carbodiimide (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN) in CaCN2 appeared at approximately 2000–2200 cm−1 in the FT-IR spectrum of the product before washing,45 but this peak was not present in the spectrum of the product after washing. In the XRD pattern of the products after washing (Fig. S4, ESI†), the peaks assignable to CaCN2 disappeared and only peaks corresponding to CaTa(O,N)3 were observed. These results show that CaTa(O,N)3 could be obtained almost as a single phase by sequential washing with DI water and HCl aq. Next, Ca(OH)2 was examined as a Ca source for the preparation of CaTa(O,N)3. Similarly to the case for CaCO3, the effects of varying the amount of urea relative to those of Ca(OH)2 and Ta2O5 gel in the precursor mixture were investigated. The Ca and Ta molar ratio (x:y) in the precursor mixture was fixed at 1:1 and the urea molar ratio (z) was varied from 3 to 5. Fig. 3 depicts the XRD patterns of the products prepared from Ca(OH)2, Ta2O5 and urea (x:y:z = 1:1:3, 1:1:4 and 1:1:5). Photographs of the products are provided in the ESI† (Fig. S5). For all precursor compositions, a perovskite phase, which corresponded to CaTa(O,N)3, was formed as the main phase of the obtained products. The generated by-products differed from those generated with the products prepared by using CaCO3 as the Ca source. In the case of CaCO3, tantalum nitrides, i.e., TaN and Ta3N5, were formed as by-products, but in the case of Ca(OH)2, the by-products were metal oxides, i.e., TaO and CaO. In addition, the lowest urea amount, z = 3, was enough to achieve nitridation to CaTa(O,N)3 when Ca(OH)2 was used as the Ca source, but z = 5 was required when CaCO3 was used. As in the case of CaCO3, the effect of increasing the molar ratio of Ca to Ta was investigated. The Ta to urea molar ratio (y:z) in the precursor mixture was fixed at 1:3 and the Ca molar ratio (x) was varied from 1 to 2. Fig. 4 shows XRD patterns of the products prepared from Ca(OH)2, Ta2O5 and urea (x:y:z = 1:1:3, 1.15:1:3, 1.3:1:3, 1.5:1:3 and 2:1:3). Photographs of the products are given in the ESI† (Fig. S6). When the Ca ratio (x) was 1.15 or higher, the impurity phases of TaO and CaO disappeared or became negligible. Similarly to the case for CaCO3, CaCN2 was formed as a by-product when the ratio of Ca in the precursor was increased. As shown in Fig. S6, ESI† the colour of the sample was grey when the Ca ratio, x, was 1, but the blackness decreased with increasing value of x, and the colour of the sample with x = 1.5 was bright yellow. However, the yellowish colour faded and the sample became whitish beige at x = 2. In the XRD pattern of this sample, all the peaks attributed to CaTa(O,N)3 broadened, which suggests decreased crystallinity. Molar ratios of Ca/Ta/urea = 1.5:1:3 are therefore considered to be the optimum composition. The CaCN2 by-product can be removed by washing with DI water and HCl aq., as described above, therefore CaTa(O,N)3 can be obtained as a single phase by washing. The high-temperature reaction time needed to synthesise CaTaO2N by ammonolysis of Ca2Ta2O7 is longer than those needed to synthesise other tantalum-based perovskite oxynitrides, e.g., BaTaO2N.30 For example, it has been reported that Ca2Ta2O7 remains after heat treatment under a flow of NH3 at 1100 °C for 240 h. Sun et al. achieved the synthesis of CaTaO2N from the reaction of CaCO3 and TaON by heat treatment for 3 h, but Ca2Ta2O7 remained, and Ta4N5 and TaON were formed as impurities.31 Masubuchi and co-workers recently reported an ammonia-free synthesis of CaTa(O,N)3via the reaction of Ca2Ta2O7 and C3N4.36 The heat-treatment time was decreased to 6 h, but a certain amount of Ca2Ta2O7 remained and a single-phase CaTa(O,N)3 was not obtained. In contrast, in the present method, optimisation of the precursor composition suppresses formation of by-products that are difficult to remove, and only soluble CaCN2 is produced as a by-product. It is therefore possible to obtain a single phase of CaTaO2N with a short calcination time and by using a simple washing process. XRD pattern of the products prepared from either CaCO3 or Ca(OH)2 after washing with DI water and HCl aq. are given in the ESI† (Fig. S7). The morphologies of CaTa(O,N)3 were observed using SEM (Fig. 5). The average particle sizes of the products were ca. 100 nm, which is smaller than those reported in the literature for CaTaO2N prepared by ammonolysis. In addition, the crystalline sizes of the corresponding products, which were estimated from the XRD data by using the Scherrer equation, were 31 nm (Ca source: CaCO3) and 28 nm [Ca source: Ca(OH)2)] This suggests that crystal growth of CaTa(O,N)3 during the urea-nitriding process was effectively suppressed. The grain and crystalline sizes are smaller because of the lower temperature and shorter heat-treatment duration in the urea-nitriding process.
CN) in CaCN2 appeared at approximately 2000–2200 cm−1 in the FT-IR spectrum of the product before washing,45 but this peak was not present in the spectrum of the product after washing. In the XRD pattern of the products after washing (Fig. S4, ESI†), the peaks assignable to CaCN2 disappeared and only peaks corresponding to CaTa(O,N)3 were observed. These results show that CaTa(O,N)3 could be obtained almost as a single phase by sequential washing with DI water and HCl aq. Next, Ca(OH)2 was examined as a Ca source for the preparation of CaTa(O,N)3. Similarly to the case for CaCO3, the effects of varying the amount of urea relative to those of Ca(OH)2 and Ta2O5 gel in the precursor mixture were investigated. The Ca and Ta molar ratio (x:y) in the precursor mixture was fixed at 1:1 and the urea molar ratio (z) was varied from 3 to 5. Fig. 3 depicts the XRD patterns of the products prepared from Ca(OH)2, Ta2O5 and urea (x:y:z = 1:1:3, 1:1:4 and 1:1:5). Photographs of the products are provided in the ESI† (Fig. S5). For all precursor compositions, a perovskite phase, which corresponded to CaTa(O,N)3, was formed as the main phase of the obtained products. The generated by-products differed from those generated with the products prepared by using CaCO3 as the Ca source. In the case of CaCO3, tantalum nitrides, i.e., TaN and Ta3N5, were formed as by-products, but in the case of Ca(OH)2, the by-products were metal oxides, i.e., TaO and CaO. In addition, the lowest urea amount, z = 3, was enough to achieve nitridation to CaTa(O,N)3 when Ca(OH)2 was used as the Ca source, but z = 5 was required when CaCO3 was used. As in the case of CaCO3, the effect of increasing the molar ratio of Ca to Ta was investigated. The Ta to urea molar ratio (y:z) in the precursor mixture was fixed at 1:3 and the Ca molar ratio (x) was varied from 1 to 2. Fig. 4 shows XRD patterns of the products prepared from Ca(OH)2, Ta2O5 and urea (x:y:z = 1:1:3, 1.15:1:3, 1.3:1:3, 1.5:1:3 and 2:1:3). Photographs of the products are given in the ESI† (Fig. S6). When the Ca ratio (x) was 1.15 or higher, the impurity phases of TaO and CaO disappeared or became negligible. Similarly to the case for CaCO3, CaCN2 was formed as a by-product when the ratio of Ca in the precursor was increased. As shown in Fig. S6, ESI† the colour of the sample was grey when the Ca ratio, x, was 1, but the blackness decreased with increasing value of x, and the colour of the sample with x = 1.5 was bright yellow. However, the yellowish colour faded and the sample became whitish beige at x = 2. In the XRD pattern of this sample, all the peaks attributed to CaTa(O,N)3 broadened, which suggests decreased crystallinity. Molar ratios of Ca/Ta/urea = 1.5:1:3 are therefore considered to be the optimum composition. The CaCN2 by-product can be removed by washing with DI water and HCl aq., as described above, therefore CaTa(O,N)3 can be obtained as a single phase by washing. The high-temperature reaction time needed to synthesise CaTaO2N by ammonolysis of Ca2Ta2O7 is longer than those needed to synthesise other tantalum-based perovskite oxynitrides, e.g., BaTaO2N.30 For example, it has been reported that Ca2Ta2O7 remains after heat treatment under a flow of NH3 at 1100 °C for 240 h. Sun et al. achieved the synthesis of CaTaO2N from the reaction of CaCO3 and TaON by heat treatment for 3 h, but Ca2Ta2O7 remained, and Ta4N5 and TaON were formed as impurities.31 Masubuchi and co-workers recently reported an ammonia-free synthesis of CaTa(O,N)3via the reaction of Ca2Ta2O7 and C3N4.36 The heat-treatment time was decreased to 6 h, but a certain amount of Ca2Ta2O7 remained and a single-phase CaTa(O,N)3 was not obtained. In contrast, in the present method, optimisation of the precursor composition suppresses formation of by-products that are difficult to remove, and only soluble CaCN2 is produced as a by-product. It is therefore possible to obtain a single phase of CaTaO2N with a short calcination time and by using a simple washing process. XRD pattern of the products prepared from either CaCO3 or Ca(OH)2 after washing with DI water and HCl aq. are given in the ESI† (Fig. S7). The morphologies of CaTa(O,N)3 were observed using SEM (Fig. 5). The average particle sizes of the products were ca. 100 nm, which is smaller than those reported in the literature for CaTaO2N prepared by ammonolysis. In addition, the crystalline sizes of the corresponding products, which were estimated from the XRD data by using the Scherrer equation, were 31 nm (Ca source: CaCO3) and 28 nm [Ca source: Ca(OH)2)] This suggests that crystal growth of CaTa(O,N)3 during the urea-nitriding process was effectively suppressed. The grain and crystalline sizes are smaller because of the lower temperature and shorter heat-treatment duration in the urea-nitriding process.
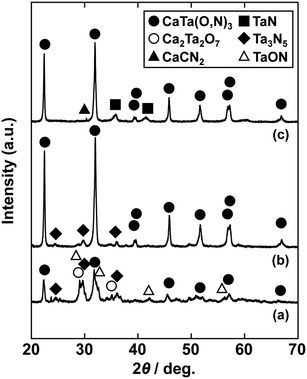 |
| | Fig. 1 XRD patterns of products prepared from CaCO3, Ta2O5 gel and urea after heat-treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were (a) 1:1:3, (b) 1:1:4 and (c) 1:1:5. | |
 |
| | Fig. 2 XRD patterns of products prepared from CaCO3, Ta2O5 gel, and urea after heat treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were (a) 1:1:5, (b) 1.5:1:5 and (c) 2:1:5. | |
 |
| | Fig. 3 XRD patterns of products prepared from Ca(OH)2, Ta2O5 gel and urea after heat treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were (a) 1:1:3, (b) 1:1:4 and (c) 1:1:5. | |
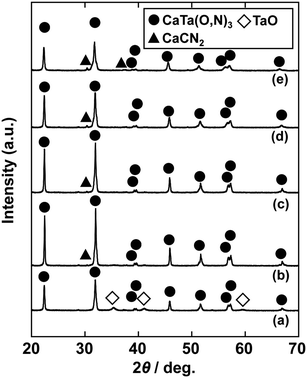 |
| | Fig. 4 XRD patterns of products prepared from Ca(OH)2, Ta2O5 gel and urea after heat treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were (a) 1:1:3, (b) 1.15:1:3, (c) 1.3:1:3, (d) 1.5:1:3 and (e) 2:1:3. | |
 |
| | Fig. 5 SEM images of CaTa(O,N)3 prepared from Ca source ((a) CaCO3, (b) Ca(OH)2), Ta2O5 gel and urea after heat treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were (a) 2:1:5 and (b) 1.5:1:3. | |
The optical properties of the CaTa(O,N)3 samples prepared from both CaCO3 and Ca(OH)2 were evaluated. Photographs and UV-vis diffuse reflectance spectra (DRS) of the products prepared from CaCO3 and Ca(OH)2 are shown in Fig. 6 and 7, respectively. All the samples were washed with DI water, and then with HCl aq. It has been reported that the sample crystallinity affects the absorption-edge steepness of the DRS.46 The absorption-edge profiles of the products obtained using Ca(OH)2 were much steeper than those of the products prepared from CaCO3. This is primarily because of the higher crystallinity of the products obtained from Ca(OH)2. The peaks in the XRD patterns of the products prepared using Ca(OH)2 were also much sharper than those in the XRD patterns of the products prepared using CaCO3 (Fig. S7, ESI†). These results suggest that CaTa(O,N)3 prepared from Ca(OH)2 has a higher crystallinity than that prepared from CaCO3. The thermal decomposition temperature of CaCO3 is 825 °C, which means that CaCO3 is stable even at high calcination temperatures. The thermal decomposition temperature of Ca(OH)2 is 580 °C, which is much lower than that of CaCO3. The reaction of Ca(OH)2 with urea and its derivatives therefore starts at a lower temperature during calcination in the synthesis of CaTa(O,N)3. The difference between the thermal decomposition temperatures of CaCO3 and Ca(OH)2 is therefore an important factor in the difference between the crystallinities of CaTa(O,N)3 obtained from these Ca sources.
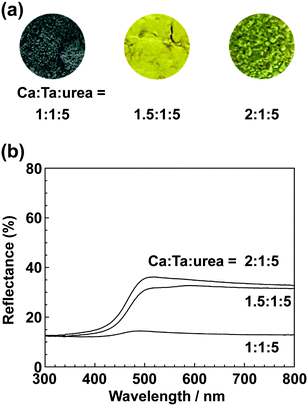 |
| | Fig. 6 (a) Photographs and (b) UV-vis DRS of CaTa(O,N)3 prepared from CaCO3, Ta2O5 gel, and urea after heat treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were 1:1:5, 1.5:1:5 and 2:1:5. | |
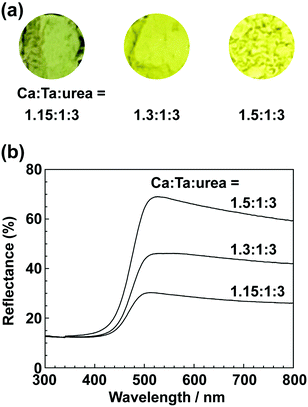 |
| | Fig. 7 (a) Photographs and (b) UV-vis DRS of CaTa(O,N)3 prepared from Ca(OH)2, Ta2O5 gel, and urea after heat treatment at 950 °C for 2 h. Molar ratios of Ca/Ta/urea in precursors were 1.15:1:3, 1.3:1:3 and 1.5:1:3. | |
All the spectra exhibited strong absorption of blue light at wavelengths below 500 nm; blue is a complementary colour to yellow. This absorption originates from electronic transitions between the valence band formed by hybridisation of O 2p/N 2p orbitals and the conduction band consisting of the 5d orbital of Ta.29,30 Regardless of the Ca source, an increase in the proportion of Ca in the precursor resulted in an increase in the maximum reflectance intensity of the products in the long-wavelength range 500–800 nm. As mentioned above, the product prepared using CaCO3 at a lower Ca ratio was contaminated with TaN, and this caused a grey colour. However, even for the products in which formation of TaN as a by-product was sufficiently suppressed, their reflectance was lower and their colour was a duller yellow than those reported in the literature. This is presumably because of reduction of Ta5+ in CaTa(O,N)3 to Ta4+ by urea, which also acts as a reducing agent.40,41 In other words, the dull colour arose from a tantalum species, which was reduced during the urea-nitriding process. The Commission Internationale de l’Eclairage (CIE) L*a*b*Ch° colour coordinate parameters of CaTa(O,N)3 prepared using CaCO3 (Ca:Ta:urea = 2:1:5) and Ca(OH)2 (Ca:Ta:urea = 1.5:1:3) are summarised in Table 1. The bandgap energy (Eg) values, which were estimated from UV-vis DRS, are also given in Table 1. The hue angles (h°) of the products were 104° and 110°, which implies that the colours were in the boundary region between yellow and green. Although the redness (a*) value was comparable to those of conventional yellow pigments (−7.90 for cadmium yellow and −3.28 for ZrSiO4:Pr), the lightness (L*), yellowness (b*) and chroma (C) values of CaTa(O,N)3 prepared from CaCO3 were considerably lower than those of CaTaO2N prepared by ammonolysis.26 In the case of the product prepared from Ca(OH)2, the L* value was close to that of CaTaO2N prepared by ammonolysis, but the b* and C values were still low. The estimated bandgap energies for the products prepared from CaCO3 and Ca(OH)2 (2.57 and 2.59 eV, respectively) were larger than the reported value for CaTaO2N prepared via ammonolysis (2.47 eV).47 It is well known that the O/N ratio strongly affects the band structures of oxynitrides, i.e., a lower nitrogen content leads to widening of the bandgap compared with those of oxynitrides with an ideal O/N ratio.25,47 The CaTa(O,N)3 samples prepared using CaCO3 and Ca(OH)2 were analysed by the O/N combustion method. The data in Table 2 show that the nitrogen contents of oxynitrides prepared by the urea-nitriding method were consistently lower than that of CaTaO2N obtained through ammonolysis.
Table 1 CIE L*a*b*Ch° colour coordinates and bandgap energies (Eg) of CaTa(O,N)3 prepared from Ca source (CaCO3 or Ca(OH)2), Ta2O5 gel and urea after heat treatment at 950 °C for 2 h
| Ca source |
Molar ratios of Ca/Ta/urea in the precursors |
Colour coordinates |
E
g/eV |
|
L* |
a* |
b* |
C
|
h° |
|
# The values of a* and b* for CaTaO2N prepared by ammonolysis were extracted from the positions in the plots in a figure in the literature by using ImageJ software. |
| CaCO3 |
2:1:5 |
57.0 |
−5.89 |
+24.1 |
24.9 |
104 |
2.57 |
| Ca(OH)2 |
1.5:1:3 |
82.3 |
−15.4 |
+41.8 |
44.5 |
110 |
2.59 |
| Reference data for CaTaO2N prepared by ammonolysis26 |
75.5 |
−5.23# |
+61.7# |
62.0 |
94.8 |
— |
Table 2 Oxygen and nitrogen contents (determined by combustion method) of CaTa(O,N)3 prepared from Ca source (CaCO3 or Ca(OH)2), Ta2O5 gel and urea after heat treatment at 950 °C for 2 h
| Ca source |
Molar ratios of Ca/Ta/urea in the precursors |
O/N molar ratio in products (O + N = 3) |
| O |
N |
| CaCO3 |
2:1:5 |
2.54 |
0.46 |
| Ca(OH)2 |
1.5:1:3 |
2.32 |
0.68 |
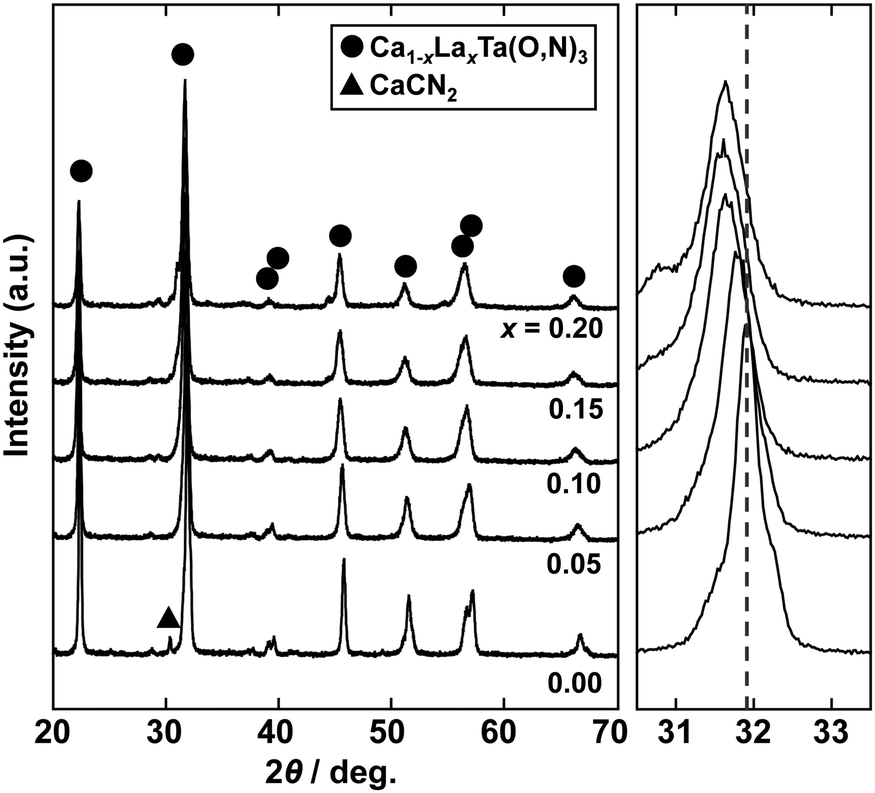
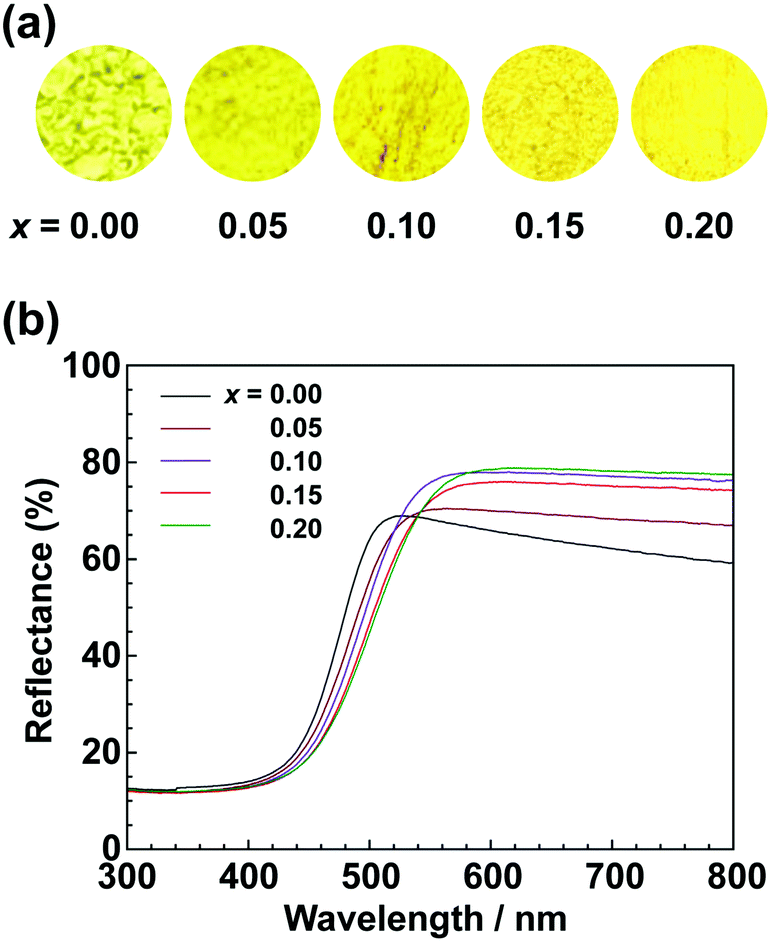
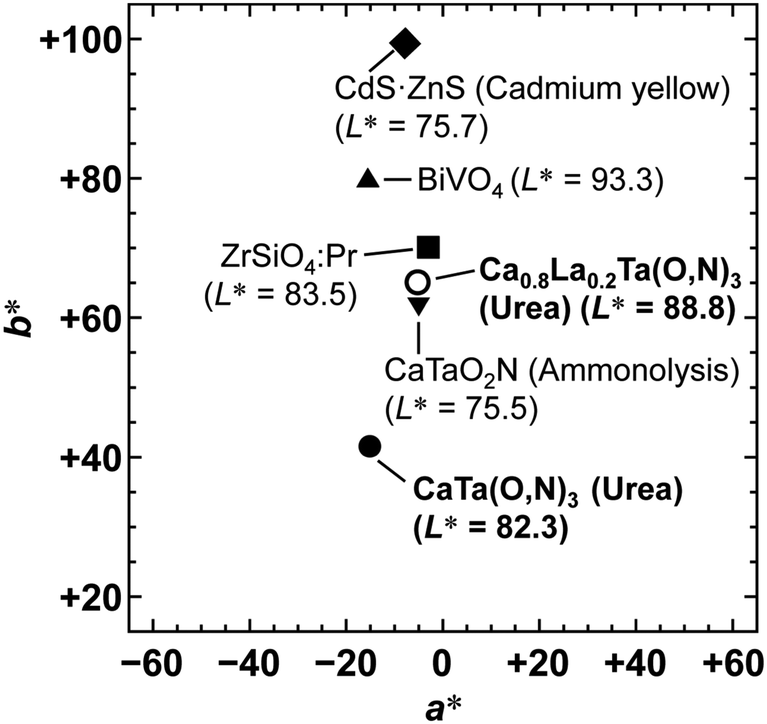
To achieve a colour equivalent to the ideal for CaTaO2N, the lack of nitrogen in the perovskite structure needs to be compensated for. As mentioned above, increasing the amount of urea, i.e., the nitrogen source in the precursor, is undesirable because it leads to increased formation of tantalum nitride by-products such as TaN. However, it is possible to control the O and N contents by changing the A site cation in the metal oxynitrides, AB(O,N)3, in which Ta5+ occupies the B site.25,47 For example, when divalent cations such as Ca2+ are used, the O:N molar ratio is ideally 2:1, as in CaTaO2N, but when trivalent cations such as La3+ are used, the O:N molar ratio is 1:2 and the N content is greater, as in LaTaON2. In other words, it can be speculated that if a certain portion of the Ca2+ in CaTaO2N is substituted by La3+, the N ratio can be increased while maintaining the perovskite structure. Fig. 8 displays XRD patterns of the products obtained from La-doped calcium–tantalum oxynitrides, i.e., Ca1−xLaxTa(O,N)3, with various La contents (x = 0.00, 0.05, 0.10, 0.15 and 0.20). The molar ratios of (Ca + La)/Ta/urea were 1.5:1:3. All the products crystallised with an almost single-phase perovskite structure without any impurity phases. This suggests that solid solutions of CaTa(O,N)3 and LaTa(O,N)3 are formed in the examined Ca/La composition range. When the doping amount of La3+ was further increased (x > 0.20), LaN and Ca2LaTaO6 were generated as impurity phases (Fig. S8 in the ESI†). In addition, the yellowness (b*) value of the products decreased in the colour evaluation. Therefore, a detailed investigation of Ca1−xLaxTa(O,N)3 in the range 0 ≤ x ≤ 0.2 was performed. Fig. 8 shows that all the diffraction peaks consecutively shifted towards lower angles with increasing La3+ uptake. This indicates continuous expansion of the AB(O,N)3 unit cell on substitution of Ca2+ with La3+. The lattice parameters and volumes of the products are listed in Table S1 in the ESI.† The lattice parameters and volumes increased with increasing doping with La3+ ions at the A sites. This reveals that smaller Ca2+ ions (ionic radius: 0.134 nm)48 in CaTa(O,N)3 were replaced with larger La3+ ions (ionic radius: 0.136 nm).48 Photographs and UV-vis DRS of Ca1−xLaxTa(O,N)3 (x = 0.00, 0.05, 0.10, 0.15 and 0.20) are depicted in Fig. 9. The products were washed sequentially with DI water and HCl aq. to remove the CaCN2 by-product. All the products exhibited a bright yellow colour, as shown in Fig. 9(a). Higher reflectances were observed on the longer-wavelength side of the DRS even with increasing La3+ ion concentration. This suggests that substitution of Ca2+ ions with La3+ ions did not induce the reduction of Ta5+ ions during the urea-nitriding process. The spectroscopic curves shifted towards the longer-wavelength side with increasing La3+ ion concentration. The CIE L*a*b*Ch° colour coordinate parameters and bandgap energies of Ca1−xLaxTa(O,N)3 (x = 0.00, 0.05, 0.10, 0.15 and 0.20) are given in Table 3. The hue angle parameters were located in the yellow region (70° < h° < 105°), therefore all the products were yellow. The yellowness (b*) value increased in the positive direction, from +41.8 to +65.3, with increasing doping amount of La3+ from 0 to 0.2. The colour saturation parameter (C) also increased with increasing b* value. Among the Ca1−xLaxTa(O,N)3 samples prepared in this study, Ca0.8La0.2Ta(O,N)3 had the highest b* and C values. This indicates that this oxynitride exhibited the most brilliant yellow colour. The estimated bandgap energy (Eg) of Ca0.8La0.2Ta(O,N)3 was 2.48 eV, which is smaller than that of CaTa(O,N)3 (Eg = 2.56 eV; described above) and comparable to that of CaTaO2N synthesised by ammonolysis (Eg = 2.47 eV).47 The elemental contents of Ca0.8La0.2Ta(O,N)3 were determined by O/N combustion analysis and XRF spectroscopy. The O:N ratio obtained by combustion analysis was 2.28:0.72 and that obtained by XRF spectroscopy was 2.11:0.89. These values are closer to an O/N ratio of 2:1 than is that of CaTa(O,N)3 (Table 4). These results reveal that the partial substitution of Ca2+ sites by La3+ compensates for the deficiency of N3− at the anion sites of a Ca–Ta-based perovskite oxynitride. This results in a bandgap energy and chromaticity that resemble those of CaTaO2N with an ideal O/N ratio. The distortion of Ta–O/N–Ta bonds is also a critical factor in the electronic structures of Ta-based perovskite oxynitrides because hybridisations between the 5d orbital of Ta and the 2p orbitals of O and N contribute significantly to the conduction and valence bands.47,49 The substitution of Ca2+ with La3+ induces changes in the Ta–O/N–Ta bond angles and this may contribute to the variations in the bandgap energies of Ca1−xLaxTa(O,N)3. The colour coordinate data for CaTa(O,N)3 and Ca0.8La0.2Ta(O,N)3 prepared in this study were compared with those reported in the literature for CaTaO2N synthesised by ammonolysis26 and conventional yellow pigments such as cadmium yellow,25 BiVO420 and ZrSiO4:Pr20 (Fig. 10). Although the yellowness (b*) value for CaTa(O,N)3 prepared by using urea fell short of those for CaTaO2N prepared by ammonolysis and conventional yellow pigments, that for Ca1−xLaxTa(O,N)3 was almost comparable to those of CaTaO2N obtained by ammonolysis and ZrSiO4:Pr. The lightness (L*) value of Ca1−xLaxTa(O,N)3 was higher than those of CaTaO2N and cadmium yellow. SEM was used to observe the morphology of Ca0.8La0.2Ta(O,N)3 (Fig. S9 in the ESI†). Similarly to that of CaTa(O,N)3 (Fig. 5), the particle size was ca. 100 nm. The crystalline size of Ca0.8La0.2Ta(O,N)3 estimated from the XRD pattern by using the Scherrer equation was 28 nm. These small grain and crystalline sizes are suitable for pigment applications. These results therefore show that Ca1−xLaxTa(O,N)3 samples prepared via the urea-nitriding method have good potential as environmentally benign alternatives to conventional inorganic yellow pigments that contain harmful elements.
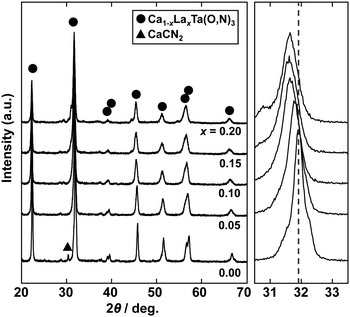 |
| | Fig. 8 XRD patterns of Ca1−xLaxTa(O,N)3 (x = 0.00, 0.05, 0.10, 0.15 and 0.20) prepared from Ca(OH)2, La(OH)3, Ta2O5 gel and urea after heat treatment at 950 °C for 2 h. Enlargements of main reflections at approximately 32° are shown on the right; dashed line provides a guide for the eye. | |
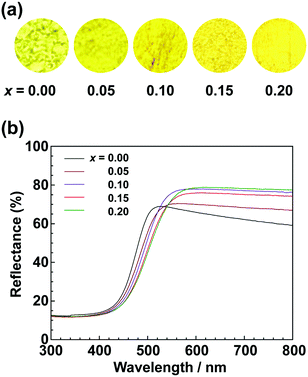 |
| | Fig. 9 (a) Photographs and (b) UV-vis DRS of Ca1−xLaxTa(O,N)3 (x = 0.00, 0.05, 0.10, 0.15 and 0.20) obtained from Ca(OH)2, La(OH)3 Ta2O5 gel and urea after calcination at 950 °C for 2 h. | |
Table 3 CIE L*a*b*Ch° colour coordinates and bandgap energies (Eg) of Ca1−xLaxTa(O,N)3 (x = 0.00, 0.05, 0.10, 0.15 and 0.20) prepared from Ca(OH)2, La(OH)3 Ta2O5 gel and urea after heat treatment at 950 °C for 2 h
|
x in Ca1−xLaxTa(O,N)3 |
Colour coordinates |
E
g/eV |
|
L* |
a* |
b* |
C
|
h° |
| 0.00 |
82.3 |
−15.4 |
+41.8 |
44.5 |
110 |
2.59 |
| 0.05 |
90.0 |
−13.8 |
+55.9 |
57.5 |
104 |
2.56 |
| 0.10 |
91.1 |
−10.9 |
+62.1 |
63.0 |
100 |
2.53 |
| 0.15 |
88.2 |
−7.88 |
+62.8 |
63.3 |
97 |
2.50 |
| 0.20 |
88.8 |
−5.51 |
+65.3 |
65.5 |
95 |
2.48 |
Table 4 Oxygen and nitrogen contents of Ca1−xLaxTa(O,N)3 (x = 0.00 and 0.20) prepared from Ca(OH)2, La(OH)3, Ta2O5 gel and urea after heat treatment at 950 °C for 2 h
| Sample |
O/N molar ratio in the product analysed by combustion method (O + N = 3) |
O/N molar ratio in the product analysed by XRF spectroscopy (O + N = 3) |
| O |
N |
O |
N |
| CaLaTa(O,N)3 |
2.32 |
0.68 |
2.18 |
0.82 |
| Ca0.8La0.2Ta(O,N)3 |
2.28 |
0.72 |
2.11 |
0.89 |
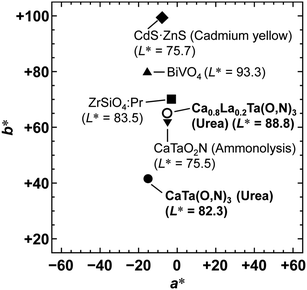 |
| | Fig. 10 CIE L*a*b* colour coordinate plots for Ca1−xLaxTa(O,N)3 (x = 0, 0.2) prepared by urea-nitriding method in this study. Data for CaTaO2N prepared via the ammonolysis method26 and commercially available yellow pigments (CdS–ZnS (cadmium yellow),25 BiVO420 and ZrSiO4:Pr20) are also displayed for comparison. | |
Conclusions
Calcium–tantalum-based perovskite oxynitrides were successfully synthesised without use of an ammonolysis process. As a Ca source, Ca(OH)2 was more suitable than CaCO3 because the CaTa(O,N)3 product had a bright yellow colour and the formation of by-products that are difficult to remove was suppressed. Increasing the amount of the Ca source in the precursor suppressed the formation of tantalum nitride by-products. Because the CaCN2 by-product and excess Ca components remaining after nitridation could be removed by sequential washing with DI water and HCl aq., CaTa(O,N)3 was obtained as a single phase. The CaTa(O,N)3 obtained by this method was not completely nitrided and did not give ideal CaTaO2N. The proportion of N3− at the anion sites of the perovskite structure was adjusted by increasing the total cation charge by partial substitution of Ca2+ with La3+. The bandgap energy of Ca0.8La0.2Ta(O,N)3 was comparable to that of CaTaO2N prepared by ammonolysis and its colour parameters were comparable to those of commercially available inorganic yellow pigments. In this approach, hazardous components are eliminated both from the preparation process and the final products. The urea-nitriding method enables shortening of the high-temperature reaction time. This is advantageous in terms of reduced energy consumption. In addition, because of the shortened reaction time, the sample particles are finer. This is an important advantage in pigment applications. We therefore believe that this developed approach to the preparation of oxynitride pigments will help the inorganic pigment industry to achieve sustainable development goals.
Author contributions
T. Sakata: investigation, writing – original draft; S. Urushidani: investigation; N. Tarutani: investigation, methodology, writing –review & editing; K. Katagiri: conceptualization, funding acquisition, methodology, supervision, writing – review & editing; K. Inumaru: supervision; K. Koyama: investigation; N. Iwata: investigation; Y. Masubuchi: resources.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. JP16H06439, JP18K19132, JP18H01709, JP19H04699, and JP20H02439). We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
References
-
K. Nassau, The Physics and Chemistry of Color: The Fifteen Causes of Color, John Wiley & Sons, Inc., New York, NY, USA, 2nd edn, 2001 Search PubMed.
-
R. G. Kuehni, Color: An Introduction to Practice and Principles, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2nd edn, 2005 Search PubMed.
-
E. B. Faulkner and R. J. Schwartz, High Performance Pigments, Wiley-VCH, Weinheim, 2nd edn, 2009 Search PubMed.
- R. A. Eppler and E. R. Douglas, Ceram. Eng. Sci. Proc., 1994, 15, 281–288 CrossRef CAS.
- J. A. Badenes, M. Llusar, M. A. Tena, J. Calbo and G. Monrós, J. Eur. Ceram. Soc., 2002, 22, 1981–1990 CrossRef CAS.
- A. Mahmoodi, Z. Jiryaei, A. Dadras, M. Khorasani and X. Shi, J. Cleaner Prod., 2021, 319, 128733 CrossRef CAS.
- A. Amat, C. Miliani and S. Fantacci, RSC Adv., 2016, 6, 36336–36344 RSC.
- A. Cesaratto, C. D’Andrea, A. Nevin, G. Valentini, F. Tassone, R. Alberti, T. Frizzi and D. Comelli, Anal. Methods, 2014, 6, 130–138 RSC.
- G. Zeng, J. Yang, R. Hong, Z. Li, Y. Chen, F. Li, Q. Wu, L. Liu and X. Jiang, Ceram. Int., 2018, 44, 8788–8794 CrossRef CAS.
- S. H. Gilani and M. Marano, Environ. Res., 1979, 19, 427–431 CrossRef CAS PubMed.
- L. Järup and A. Åkesson, Toxicol. Appl. Pharmacol., 2009, 238, 201–208 CrossRef PubMed.
- C. J. Boreiko and T. G. Rossman, Toxicol. Appl. Pharmacol., 2020, 403, 115156 CrossRef CAS PubMed.
- A. L. Wani, A. Ara and J. A. Usmani, Interdiscip. Toxicol., 2015, 8, 55–64 CrossRef CAS PubMed.
- World Health Organization, Lead poisoning, https://www.who.int/news-room/fact-sheets/detail/lead-poisoning-and-health (accessed March 2022).
- V. S. Vishnu, S. Jose and M. L. Reddy, J. Am. Ceram. Soc., 2011, 94, 997–1001 CrossRef CAS.
- T. Masui, T. Honda, Wendusu and N. Imanaka, Dyes Pigm., 2013, 99, 636–641 CrossRef CAS.
- S. P. Radhika, K. J. Sreeram and B. U. Nair, ACS Sustainable Chem. Eng., 2014, 2, 1251–1256 CrossRef CAS.
- S. Chen, M. Cai and X. Ma, J. Alloys Compd., 2016, 689, 36–40 CrossRef CAS.
- Wendusu, T. Honda, T. Masui and N. Imanaka, RSC Adv., 2013, 3, 24941–24945 RSC.
- K. Minagawa, Y. Nishiguchi, R. Oka and T. Masui, ACS Omega, 2021, 6, 3411–3417 CrossRef CAS PubMed.
- J. A. Badenes, J. B. Vicent, M. Llusar, M. A. Tena and G. Monr, J. Mater. Sci., 2002, 37, 1413–1420 CrossRef CAS.
- G. D. Nero, G. Cappelletti, S. Ardizzone, P. Fermo and S. Gilardoni, J. Eur. Ceram. Soc., 2004, 24, 3603–3611 CrossRef.
- R. Nayak, A. Suryanarayana and S. B. Rao, J. Sci. Ind. Res., 2000, 59, 833–837 CAS.
- X. Zhang, T. Chen, Y. Xu, W. Jiang, J. Liu and Z. Xie, J. Sol-Gel Sci. Technol., 2019, 91, 127–137 CrossRef CAS.
- M. Jansen and H. P. Letschert, Nature, 2000, 404, 980–982 CrossRef CAS PubMed.
- Y.-I. Kim, Ceram. Int., 2014, 40, 5275–5281 CrossRef CAS.
- P. Maillard, F. Tessier, E. Orhan, F. Cheviré and R. Marchand, Chem. Mater., 2005, 17, 152–156 CrossRef CAS.
- A. Fuertes, J. Mater. Chem., 2012, 22, 3293–3299 RSC.
- F. Tessier, P. Maillard, F. Cheviré, K. Domen and S. Kikkawa, J. Ceram. Soc. Jpn., 2009, 117, 1–5 CrossRef CAS.
- Y.-I. Kim, P. M. Woodward, K. Z. Baba-Kishi and C. W. Tai, Chem. Mater., 2004, 16, 1267–1276 CrossRef CAS.
- S.-K. Sun, Y. Masubuchi, T. Motohashi and S. Kikkawa, J. Eur. Ceram. Soc., 2015, 35, 3289–3294 CrossRef CAS.
- J. Odahara, A. Miura, N. C. Rosero-Navarro and K. Tadanaga, Inorg. Chem., 2018, 57, 24–27 CrossRef CAS PubMed.
- S.-L. Chen, W.-M. Guo, S.-K. Sun, Y. Masubuchi, M. Lv, H.-T. Lin and C.-Y. Wang, Ceram. Int., 2018, 44, 4504–4507 CrossRef CAS.
- A. Hosono, Y. Masubuchi, S. Yasui, M. Takesada, T. Endo, M. Higuchi, M. Itoh and S. Kikkawa, Inorg. Chem., 2019, 58, 16752–16760 CrossRef CAS PubMed.
- Y. Masubuchi, M. Tadaki and S. Kikkawa, Chem. Lett., 2018, 47, 31–33 CrossRef CAS.
- Q. Yang, Y. Masubuchi and M. Higuchi, Ceram. Int., 2020, 46, 13941–13944 CrossRef CAS.
- A. Gomathi, S. Reshma and C. N.-R. Rao, J. Solid State Chem., 2009, 182, 72–76 CrossRef CAS.
- Q. Gao, C. Giordano and M. Antonietti, Small, 2011, 7, 3334–3340 CrossRef CAS PubMed.
- K. Katagiri, Y. Hayashi, R. Yoshiyuki, K. Inumaru, T. Uchiyama, N. Nagata, Y. Uchimoto, A. Miyoshi and K. Maeda, Inorg. Chem., 2018, 57, 13953–13962 CrossRef CAS PubMed.
- R. Okada, K. Katagiri, Y. Masubuchi and K. Inumaru, Eur. J. Inorg. Chem., 2019, 1257–1264 CrossRef CAS.
- T. Sakata, R. Yoshiyuki, R. Okada, S. Urushidani, N. Tarutani, K. Katagiri, K. Inumaru, K. Koyama and Y. Masubuchi, Inorg. Chem., 2021, 60, 4852–4859 CrossRef CAS PubMed.
- R. J. Xie and H. T. Hintzen, J. Am. Ceram. Soc., 2013, 96, 665–687 CrossRef CAS.
- F. Oehler, R. Naumann, R. Köferstein, D. Hesse and S. G. Ebbinghaus, Mater. Res. Bull., 2016, 73, 276–283 CrossRef CAS.
- E. Günther, R. Hagenmayer and M. Jansen, Z. Anorg. Allg. Chem., 2000, 626, 1519–1525 CrossRef.
- U. Berger and W. Schnick, J. Alloys Compd., 1994, 206, 179–184 CrossRef CAS.
- F. Cheviré, F. Tessier and R. Marchand, Eur. J. Inorg. Chem., 2006, 1223–1230 CrossRef.
- Y. Wang, Y. Kang, H. Zhu, G. Liu, J. T.-S. Irvine and X. Xu, Adv. Sci., 2021, 8, 2003343 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- S. Balaz, S. H. Porter, P. M. Woodward and L. J. Brillson, Chem. Mater., 2013, 25, 3337–3343 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kiyofumi
Katagiri
a,
Kiyofumi
Katagiri