
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMononuclear nickel(II) complexes as electrocatalysts in hydrogen evolution reactions: effects of alkyl side chain lengths†
Arpita
Barma
a,
Malay
Chakraborty
a,
Swapan Kumar
Bhattacharya
 *a,
Pritam
Ghosh
*b and
Partha
Roy
*a
*a,
Pritam
Ghosh
*b and
Partha
Roy
*a
aDepartment of Chemistry, Jadavpur University, Kolkata 700 032, India. E-mail: partha.roy@jadavpuruniversity.in; skbhatt7@yahoo.co.in
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: ppritamghosh@gmail.com
First published on 1st August 2022
Abstract
We report three mononuclear Ni(II) complexes, namely, [Ni(L1)2] (1), [Ni(L2)2] (2) and [Ni(L3)2] (3), where HL1 = 1-((4-hydroxybutylimino)methyl)naphthalen-2-ol, HL2 = 1-((5-hydroxypentylimino)methyl)naphthalen-2-ol and HL3 = 1-((6-hydroxyhexylimino)methyl)naphthalen-2-ol, as electrocatalysts for hydrogen evolution reactions (HERs). Complexes 1, 2 and 3 were characterized by various standard analytical methods. Single-crystal X-ray structure analysis reveals that nickel is in square planar geometry in all the complexes. These complexes act as efficient electrocatalysts in HERs using acetic acid (AA) and trifluoroacetic acid (TFA) as the proton source in DMF. Controlled-potential electrolysis experiments show that these complexes are capable of reducing protons of AA and TFA to produce H2. Control experiments show that the complexes are essential for improved production of hydrogen. Theoretical calculations were performed to support the mechanism of HER and to check the effect of chain lengths on the catalytic activity. The catalytic activity runs in the order of complex 1 > 2 > 3.
Introduction
The need for the development and improvement of secure, sustainable and eco-friendly energy resources is one of the most vital scientific and technical challenges in the present century due to the finite resources of fossil fuels. It is common to use fossil fuel that releases the greenhouse gas, carbon dioxide, into the environment resulting in global warming. In this respect, hydrogen may be considered as a potential candidate for use as an apt energy carrier. The reaction by which molecular hydrogen is produced by two protons via the two-electron reduction is known as the hydrogen evolution reaction (HER).1To design an efficient and suitable catalyst for HERs few things such as low price of the metal, metal with labile oxidation states, and coordination sites are to be kept in mind. Platinum has been established as an efficient catalyst for the production of hydrogen.2 However, it is available in limited quantity and highly expensive. Thus, its extensive use as the catalyst is restricted. Therefore, alternatives are being investigated with much cheaper earth-abundant metals as the catalyst. There are reports of different transition metal complexes3 with Fe,4 Mn,5 Cu,6 Co7 and Ni8 as electrocatalysts for HERs. Various compounds of nickel and cobalt have also been used as the electrocatalysts for water splitting, hydrogen generation, and carbon dioxide reduction, showing their efficient role as electrocatalysts.9 Due to their inherent better ability towards hydrogen evolution, complexes of cobalt and nickel gain intense interest of researchers as high-performance catalysts.10 Moreover, Ni(II) systems are abundant in earth,11 and have relevance to biology.12 Typically, the hydrogen evolution reaction is operated via a metal-centered route, and involves a metal hydride. However, the electrocatalysts consisting of earth-abundant elements are rarely prevailing for HERs under pure aqueous conditions. In most of the cases, organic acids have been used as the substrate in organic media with metal complexes as electrocatalysts.
Nickel(II) complexes with a square planar geometry have been used as catalysts for HERs. Ligands with different donor atoms were designed to get various types of Ni(II) complexes. Generally, NiN4, NiS4, NiP4, NiN2S2 and NiS2O2 cores have been employed in such catalytic reactions (Scheme 1). Fisher and Eisenberg reported a nickel(II) complex involving a macrocycle with a NiN4 core as the ligand, as an electrocatalyst for the reduction of carbon dioxide into carbon monoxide and hydrogen as major products.13 After that, a number of complexes with N4-donor atoms have been reported in the literature for the same. Mitsopoulou et al. reported three Ni(II) diphenyl-1,2-dithiolene complexes with different substituents on the benzene ring having a NiS4 core for this purpose and showed the effects of the substituents on the results.14 Helm and coworkers used a mononuclear nickel(II) complex with two seven-membered cyclic diphosphine ligands having P donor atoms as an highly efficient electrocatalyst for the hydrogen generation.15 Bergamini and Natali reported a nickel(II) bis(diphosphine) complex as a catalyst for the HER in homogeneous media as well as in heterogeneous media.16 Eisenberg et al. reported a series of nickel complexes with two ligands of N,S or O,S donor sites having square planar coordination for hydrogen evolution with a high turnover number and high stability under conditions of hydrogen production.17 In comparison to these square planar Ni(II) complexes, very limited number of articles describe the NiN2O2 core as the electrocatalyst for hydrogen evolution. Soo et al. reported a NiN2O2 core as an electrocatalyst for the HER. The presence of alkali metals in the second sphere ether appendages actually increased the hydrogen evolution.18 The same group has recently reported another nickel complex with a NiO2N2 core with different ligand backbones for hydrogen evolution with TONs up to 3880.19
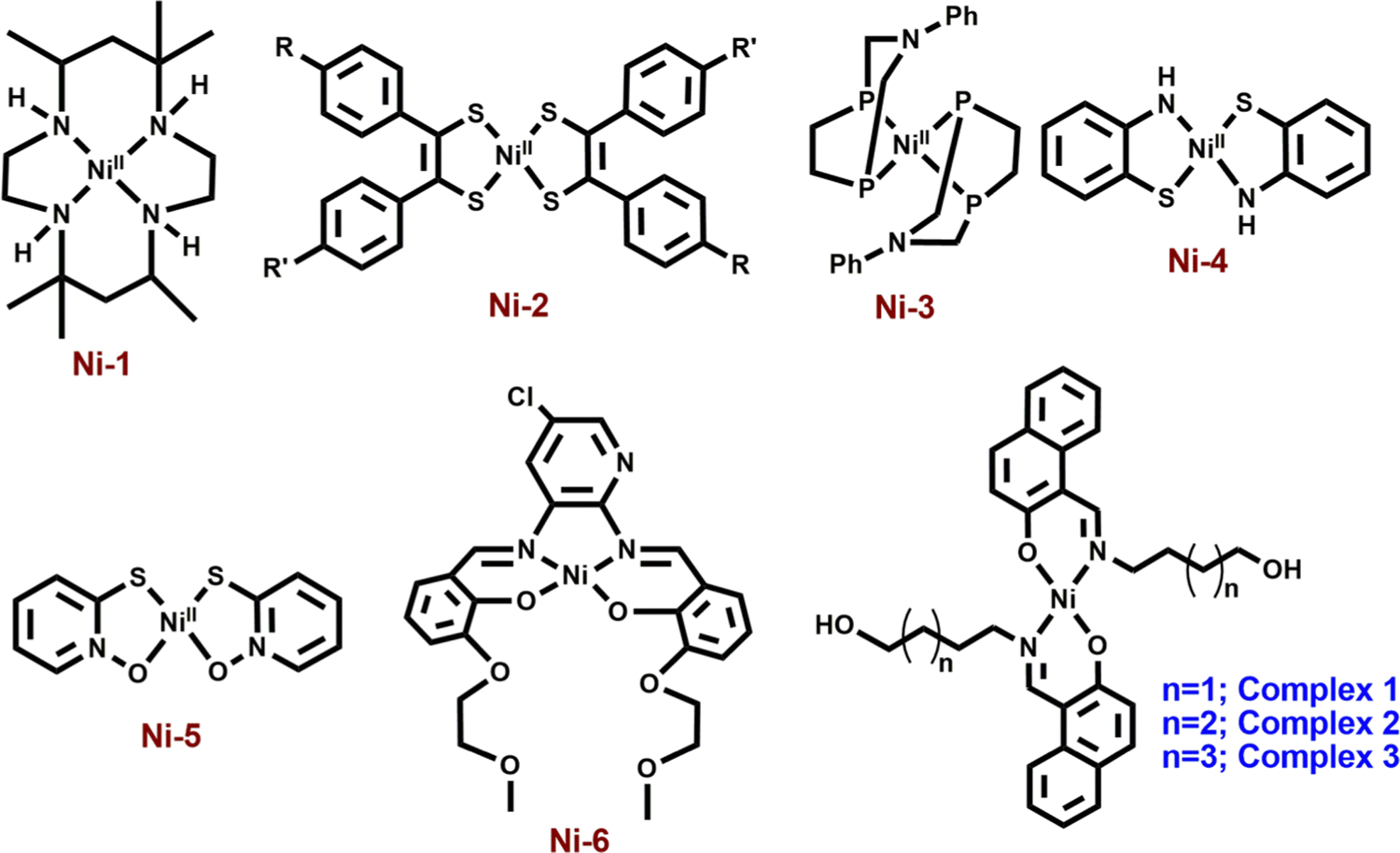 | ||
| Scheme 1 Different types of mononuclear nickel complexes with NiN4 (Ni-1),12 NiS4 (Ni-2),13 NiP4 (Ni-3),14 NiS2N2 (Ni-4),16 NiS2O2 (Ni-5)16 and NiN2O2 (Ni-6)17 cores for hydrogen evolution reactions. | ||
Herein, we report the electro-catalytic behavior of three neutral, monomeric Ni(II) complexes, [Ni(L1)2] (1), [Ni(L2)2] (2) and [Ni(L3)2] (3), where HL1 = 1-((4-hydroxybutylimino)methyl)naphthalen-2-ol, HL2 = 1-((5-hydroxypentylimino)methyl)naphthalen-2-ol and HL3 = 1-((6-hydroxyhexylimino)methyl)naphthalen-2-ol, for HERs. Complexes 1, 2 and 3 have been synthesized easily and characterized by several standard methods including single-crystal X-ray diffraction analysis. All of these complexes have been used as electrocatalysts for the reduction of proton in DMF with acetic acid (AA) and trifluoroacetic acid (TFA) as the proton source. Here, we aim to make variation in the ligand chain length of the complexes and to compare the electro-catalytic results obtained under identical conditions. Some theoretical calculations have been performed with insights into the hydrogen evolution mechanism and the effect of chain lengths of ligands on the results.
Experimental section
Materials and physical methods
2-Hydroxy-1-naphthaldehyde, 4-amino-1-butanol, 5-amino-1-pentanol, 6-amino-1-hexanol and nickel(II) perchlorate hexahydrate were purchased from Sigma Aldrich and were used without further purification. Other reagents and solvents were obtained from different commercial sources and used without further purification. Elemental analyses of complexes 1, 2 and 3 were performed using a Perkin Elmer 2400C elemental analyzer. 1H NMR spectra of three Schiff-base ligands, complexes 1, 2 and 3, were recorded using a Bruker 400 MHz spectrometer. FT-IR spectra of complexes 1, 2 and 3 were recorded using a Perkin Elmer spectrometer (Spectrum Two) with the samples by the ATR method. The UV-visible spectral measurements of complexes 1, 2 and 3 were performed using an Agilent 8453 diode array spectrophotometer. Cyclic voltammograms were obtained using an electrochemical analyzer (CHI 600C, CH Instruments,) under air-free conditions using a conventional three-electrode cell in which a glassy carbon electrode was used as the working electrode, a saturated Ag/AgCl electrode as the reference electrode, and a platinum wire as the auxiliary electrode. The surface area of the glassy carbon working electrode is 0.07 cm2. The gas evolved during bulk electrolysis was detected using a GC instrument of model no. 7890B (G3440B), serial no. CN14333203, fitted with TCD. For this, 500 μL gas was taken out in a gas tight syringe from the head space and was injected into the inlet of the GC.Synthesis of complexes 1, 2 and 3
Data for HL1: yield = 0.068 g, 92%; anal. calc. (%) for C15H17NO2: C, 74.05; H, 7.04; N, 5.76. Found: C, 73.97; H, 6.96; N, 5.79; 1H NMR (400 MHz, DMSO-d6, δ ppm, TMS) 14.13 (1H, s), 9.08 (1H, s), 8.07 (1H, d, J = 8.4 Hz), 7.71 (1H, d, J = 9.2 Hz), 7.62 (1H, d, J = 7.6 Hz), 7.42 (1H, m), 7.19 (1H, t, J = 7.2 Hz), 6.72 (1H, d, J = 9.2 Hz), 4.51 (1H, s), 3.66 (2H, t, J = 4.4 Hz), 3.45 (2H, t, J = 4.4 Hz), 1.72 (2H, m), 1.54 (2H, m); ESI-MS+ (m/z): 244.23 [(HL1 + H+)].
Data for HL2: yield = 0.070 g, 90%; anal. calc. (%) for C16H19NO2: C, 74.68; H, 7.44; N, 5.44. Found: C, 74.57; H, 7.36; N, 5.39; 1H NMR (400 MHz, DMSO-d6, δ ppm, TMS) 14.13 (1H, s), 9.09 (1H, s), 8.06 (1H, d, J = 8.0 Hz), 7.71 (1H, d, J = 9.2 Hz), 7.62 (1H, d, J = 7.6 Hz), 7.42 (1H, t, J = 7.6 Hz), 7.18 (1H, t, J = 7.2 Hz), 6.72 (1H, d, J = 9.2 Hz), 4.33 (1H, s), 3.65 (2H, t, J = 6.8 Hz), 3.42 (2H, t, J = 6.0 Hz), 1.49 (2H, m), 1.29 (2H, m); ESI-MS+ (m/z): 258.11 [(HL2 + H+)].
Data for HL3: yield = 0.069 g, 85%; anal. calc. (%) for C17H21NO2: C, 75.25; H, 7.80; N, 5.16. Found: C, 75.17; H, 7.76; N, 5.29; 1H NMR (400 MHz, DMSO-d6, δ ppm, TMS) 14.12 (1H, s), 9.08 (1H, s), 8.06 (1H, d, J = 8.4 Hz), 7.70 (1H, d, J = 9.2 Hz), 7.61 (1H, d, J = 8.0 Hz), 7.41 (1H, t, J = 7.2 Hz), 7.18 (1H, t, J = 7.6 Hz), 6.71 (1H, d, J = 9.2 Hz), 4.36 (1H, s), 3.63 (2H, t, J = 7.2 Hz), 3.39 (2H, t, J = 6.0 Hz), 1.67 (2H, m), 1.33 (2H, m); ESI-MS+ (m/z): 272.13 [(HL3 + H+)].
Data for 1: yield, 0.094 g, 58%; anal. calc. for C30H31NiN2O4: C, 66.45; H, 5.76; N, 5.17; found: C, 66.33; H, 5.80; N, 5.06%. 1H NMR (400 MHz, DMSO-d6, δ ppm, TMS) 9.11 (1H, s), 8.12 (1H, d, J = 8.4 Hz), 7.83 (1H, d, J = 8.8 Hz), 7.74 (1H, d, J = 8.0 Hz), 7.50 (1H, t, J = 7.6 Hz), 7.25 (1H, t, J = 7.2 Hz), 6.82 (1H, d, J = 8.8 Hz), 4.45 (1H, s), 4.17 (2H, t, J = 5.2 Hz), 3.50 (2H, t, J = 5.6 Hz), 1.97 (2H, m), 1.65 (2H, m).
Data for 2: yield, 0.106 g, 62%; anal. calc. for C32H36NiN2O4: C, 67.27; H, 6.35; N, 4.90; found: C, 67.20; H, 6.26; N, 5.05%. 1H NMR (400 MHz, DMSO-d6, δ ppm, TMS) 9.15 (1H, s), 8.12 (1H, d, J = 8.8 Hz), 7.85 (1H, d, J = 8.8 Hz), 7.74 (1H, d, J = 8.0 Hz), 7.48 (1H, t, J = 7.6 Hz), 7.27 (1H, t, J = 7.2 Hz), 6.80 (1H, d, J = 8.8 Hz), 4.35 (1H, s), 4.24 (2H, t, J = 5.2 Hz), 3.43 (2H, t, J = 5.2 Hz), 1.94 (2H, m), 1.52 (2H, m).
Data for 3: yield, 0.104 g, 58%; anal. calc. for C34H40NiN2O4: C, 68.13; H, 6.73; N, 4.67; found: C, 68.05; H, 6.80; N, 4.58%. 1H NMR (400 MHz, DMSO-d6, δ ppm, TMS) 9.12 (1H, s), 8.11 (1H, d, J = 8.0 Hz), 7.89 (1H, d, J = 8.8 Hz), 7.74 (1H, d, J = 7.2 Hz), 7.50 (1H, t, J = 7.2 Hz), 7.25 (1H, t, J = 8.0 Hz), 6.79 (1H, d, J = 8.8 Hz), 4.32 (1H, s), 3.38 (2H, t, J = 5.2 Hz), 3.33 (2H, t, J = 6.4 Hz), 2.29 (2H, m), 1.48 (2H, m).
CAUTION: nickel(II) perchlorate hexahydrate can be explosive on heating. Therefore, the perchlorate salts should be handled with care.
X-ray data collection and structure determination
Data collection and refinement parameters for complexes 1, 2 and 3 are summarized in Table 1. The X-ray diffraction experiments were performed using a BRUKER D8 QUEST CCD diffractometer for 1 and a Bruker APEX-II CCD diffractometer for 2 and 3 with graphite-monochromated Mo Kα radiation at 294(2) K. Data processing was done using the Bruker APEX2 and SAINT packages.20 Absorption corrections, which are based on multi-scans using the SADABS software,20 were applied to intensity data. The structures of complexes 1, 2 and 3 were solved by direct methods utilizing SHELXT21 and refined with full-matrix least-squares on F2 on all unique reflections employing SHELXL-2014/7.22 All the non-hydrogen atoms of all of three complexes were refined anisotropically. The crystals selected for the single-crystal X-ray diffraction analysis of complexes 1 and 3 were refined as merohedral twins in both the complexes with a fractional contribution of minor components of 0.18(3) and 0.03(2), respectively. In complex 1, the quality of crystal was poor. The poor quality as well as the presence of twinning in 1 may account for the limited overall precision of its structure, high residual peaks and the relatively high values of R and wR2 parameters.| Complex | 1 | 2 | 3 |
|---|---|---|---|
| Formula | C30H31N2Ni1O4 | C32H36N2 Ni1O4 | C34H40N2 Ni1O4 |
| Formula weight | 542.26 | 571.32 | 599.39 |
| T (K) | 298(2) | 298(2) | 298(2) |
| Crystal color | Green | Green | Green |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) | 10.7840(8) | 4.9186(4) | 5.1838(7) |
| b (Å) | 4.9890(4) | 16.7080(14) | 16.905(2) |
| c (Å) | 23.8099(18) | 16.9064(14) | 17.141(2) |
| α (°) | 90 | 86.436(3) | 84.192(5) |
| β (°) | 90.846(2) | 84.966(2) | 87.843(4) |
| γ (°) | 90 | 89.509(2) | 89.703(4) |
| V (Å3) | 1280.87(17) | 1381.3(2) | 1493.3(3) |
| Z | 2 | 2 | 2 |
| Crystal dimensions (mm) | 0.40 × 0.26 × 0.13 | 0.41 × 0.23 × 0.12 | 0.46 × 0.29 × 0.16 |
| F(0 0 0) | 570.0 | 604.0 | 636.0 |
| D c (g cm−3) | 1.406 | 1.374 | 1.333 |
| λ(Mo Kα) (Å) | 0.71073 | 0.71073 | 0.71073 |
| θ range (°) | 2.53–27.11 | 2.65–27.25 | 2.39–27.00 |
| Reflection collected/unique/observed | 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 207, 2837, 2507 207, 2837, 2507 |
70198, 6202, 5565 |
78729, 6626, 5259 |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan |
| R int | 0.0306 | 0.0320 | 0.0350 |
| Final R1 index [I > 2σ(I)] | 0.0352 | 0.0348 | 0.0498 |
| Final wR2 index (all reflections) | 0.1324 | 0.1024 | 0.1397 |
| Goodness-of-fit | 1.166 | 1.022 | 0.925 |
CCDC 2062677, 2062678 and 2062679 contain the supplementary crystallographic data for complexes 1, 2 and 3, respectively.†
Results and discussion
Synthesis of ligands and their characterization by ESI mass and 1H spectral analysis
1-((4-Hydroxybutylimino)methyl)naphthalen-2-ol (HL1), 1-((5-hydroxypentylimino)methyl)naphthalen-2-ol (HL2) and 1-((6-hydroxyhexylimino)methyl)naphthalen-2-ol (HL3) were synthesized by a Schiff-base condensation reaction between 2-hydroxy-1-naphthaldehyde and the respective amine in 1:1 proportion in acetonitrile with high yields (Scheme S1, ESI†). Complexes 1, 2 and 3 were obtained by the reactions between the ligands and nickel(II) perchlorate hexahydrate in 2:1 proportion without the addition of any external base.
ESI-mass spectrometric measurements of HL1, HL2 and HL3 were performed with their methanolic solutions (Fig. S1–S3, ESI†). Mass spectrum of HL1 shows an m/z peak at 244.23, which may be attributed to the [HL1 + H+] species (calculated value 244.31). The m/z peak at 258.11 may be assigned to the presence of [HL2 + H+] species (calculated value 258.15). The mass spectrum of HL3 shows an m/z peak at 272.13, which may be due to the presence of [HL3 + H+] (calculated value 272.17). These ligands were further characterized by 1H NMR spectral studies (Fig. S4–S6, ESI†). Spectra were obtained in DMSO-d6. NMR spectral studies support the formation of Schiff-base ligands. All of these compounds show a peak at around 14.1 ppm, which indicates the presence of a phenolic OH group. The peak at around 9.0 ppm in all the ligands may be due to the presence of an imine proton, indicating the formation of Schiff-base compounds. Signals for aromatic protons for all of the ligands appear in the appropriate positions. Signals for methylene protons also appear in their usual positions. All of these analyses support formation of the Schiff-base ligands.
Characterization of complexes 1, 2 and 3
space group. The asymmetric unit consists of one nickel centre and a pair of ligands for all of the complexes (Fig. 1). All of 1, 2 and 3 are mononuclear complexes (Fig. S7–S9, ESI†). The selected bond lengths and bond angles are listed in Table S1 (ESI†). All of these complexes may be summarized as the NiO2N2 core. For all of the complexes, the Ni atom is coordinated to N and O donor atoms from a ligand and another set of N and O donor atoms from a different ligand. Ni atom is in a square planar geometry. Apart from the hydroxyl alkyl chain, the rest of the molecule is planar. For complex 1, angles O1–Ni1–O1a and N1–Ni1–N1a are 180°, whereas other donor–metal–donor angles range from 87.86 to 92.14°. While other donor–metal–donor angles vary from 87.82 to 92.18° for complex 2, they vary from 88.44 to 91.56° for complex 3.
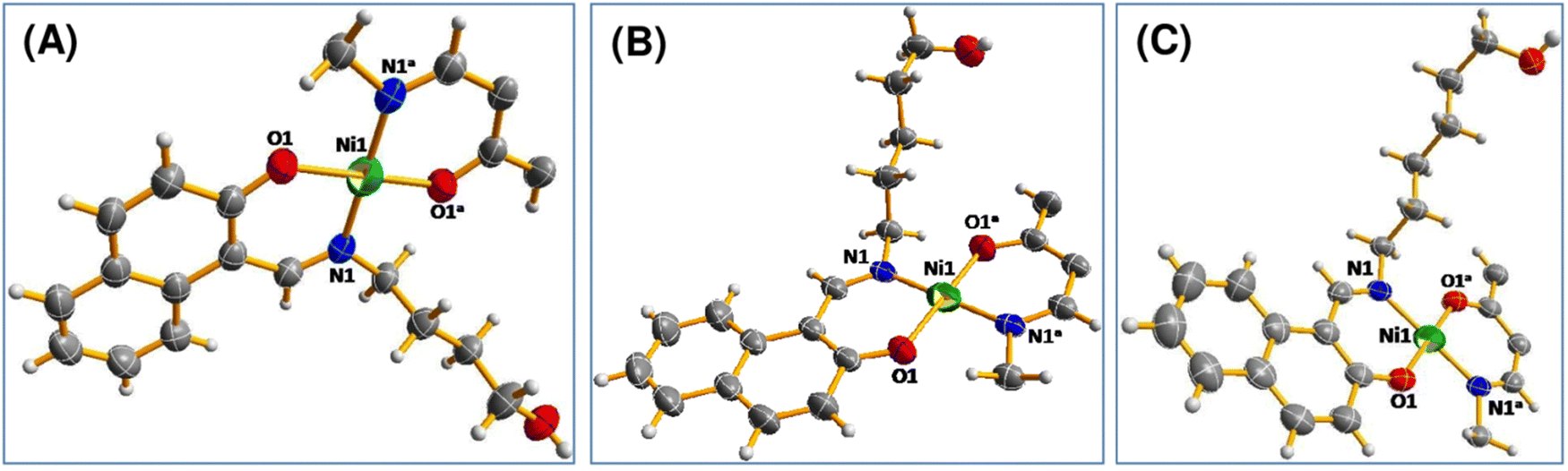 | ||
| Fig. 1 Perspective view of (A) complex 1, (B) complex 2 and (C) complex 3 with displacement ellipsoids drawn at the 50% probability level. | ||
For other two complexes, all of the large donor–metal–donor angles are 180°. For a four-coordinate complex, Houser and co-workers proposed the four-coordinate τ4 index to ascertain the geometry around the metal center. The four-coordinate τ4 index value was obtained using the following formula:23

ESI-mass, FT-IR, UV-vis and 1H NMR spectral characterization of the complexes
ESI-mass spectra of complexes 1, 2 and 3 were obtained with their methanolic solutions (Fig. S10–S12, ESI†). All of them behave similarly. The mass spectrum of 1 exhibits an m/z peak at 565.0727, which may be attributed to the [Ni(L1)2 + Na+] species (calculated value = 565.16). This indicates that the complex exists mainly as a mononuclear nickel(II) species in solution. For complex 2, an m/z peak appears at 593.10, which may be assigned to [Ni(L2)2 + Na+] (calculated value = 565.19). Similarly, the m/z peak at 621.14 of complex 3 appears due to the presence of [Ni(L3)2 + Na+] species (calculated value = 621.22).25FT-IR spectra of complexes 1, 2 and 3 were recorded with a solid sample by the ATR technique (Fig. S13–S15, ESI†). In the IR spectra, broad bands appear at 3536 cm−1, 3316 cm−1 and 3272 cm−1 for complexes 1, 2 and 3, respectively. The peaks were assigned to O–H stretching for the presence of a hydroxyl group in the ligand part of the complexes. The presence of a methylene group in the complexes has been evidenced by the appearance of unsymmetrical and symmetrical frequencies of νC–H in the range of 2800–3000 cm−1. In the IR spectra of complexes 1, 2 and 3, intense bands appeared at ∼1657 cm−1, 1658 cm−1 and 1642 cm−1 respectively. These may be attributed to the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety in the complexes. The conclusive evidence for the formation of Ni–N and Ni–O bonds in the IR spectra of these complexes is also observed with characteristic bands. These bands support the fact that the metal ion has been effectively coordinated to the four coordinating heteroatoms (NONO). These data are also supported by the literature (Table S2, ESI†).26
N moiety in the complexes. The conclusive evidence for the formation of Ni–N and Ni–O bonds in the IR spectra of these complexes is also observed with characteristic bands. These bands support the fact that the metal ion has been effectively coordinated to the four coordinating heteroatoms (NONO). These data are also supported by the literature (Table S2, ESI†).26
The UV-vis spectra of complexes 1, 2 and 3 were recorded in the range of 200–800 nm in DMF at room temperature (Fig. S16–S18, ESI†). The electronic spectra of these complexes are grossly similar in nature. A broad band at around 600 nm was observed for each of the complexes, which may be attributed to the d–d transition. These are weak in intensity as they are Laporte forbidden.27 The bands at 275 nm for complex 1, 279 nm for complex 2 and 274 nm for complex 3 may be assigned to the π–π* transitions of the phenolic chromophore. The bands between 300 and 500 nm may be assigned to the π–π* transition of the azomethine chromophore and the benzene ring and the n–π* transition of the azomethine chromophore.28 The higher intensity charge-transfer transition has been observed at a wavelength near 420 nm for all three complexes. These are attributed to O− (of naphthalen-1-olate) → Ni(II), N(amino) → Ni(II) LMCT and intra-ligand charge transfer transitions.29 All of these bands corroborate the structure of the complexes (Table S3, ESI†).
1H NMR spectra of complexes 1, 2 and 3 were obtained in DMSO-d6 (Fig. S19–S21, ESI†). The behavior of all the ligands in the presence of Ni2+ is grossly similar. All of these complexes are square planar in geometry and the metal ion in the complex does not possess any unpaired electron. The peak for phenolic OH at ∼14 ppm is absent in the NMR spectra of all the complexes. This indicates that the phenolic OH group is deprotonated during complex formation and then coordinated to the metal center. The signal for the imine proton at ∼8 ppm in the ligands undergoes a shift to ∼9 ppm for all the complexes, confirming complex formation via azomethine nitrogen atoms. Signals for aromatic protons shift towards higher δ values. All other peaks appear in their usual positions. The fact indicates that all of the complexes remain the same in the DMSO solution.25
Electrochemistry
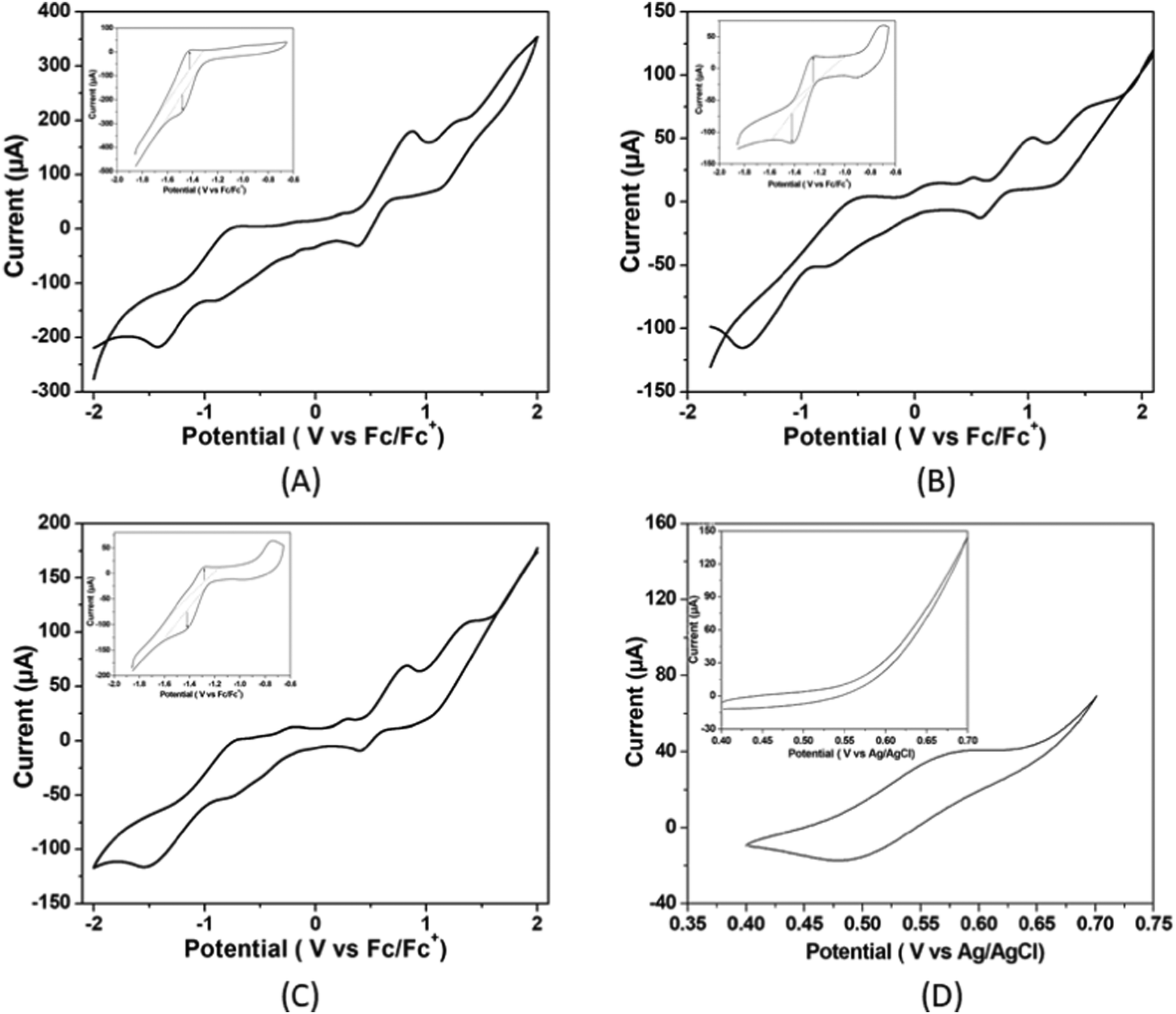 | ||
| Fig. 2 Cyclic voltammograms of 5.36 μM of (A) complex 1, (B) complex 2 (C) complex 3 and (D) ferrocene (inset: cyclic voltammograms scanned over the presently important region of potential of reversible Ni(II/I) couple) in air free DMF solutions with 0.1 M of [n-Bu4N]Br as the supporting electrolyte at a scan rate of 50 mV s−1. Here, ferrocene is the internal standard. | ||
The current responses of the redox events at −1.48, −1.51, and −1.52 V versus Fc/Fc+ for complexes 1, 2 and 3 respectively at multiple scan rates from 0.02 to 0.15 V s−1 were used to construct the Cottrell plot. The plots show linear dependence of the current response on the square root of the scan rate (Fig. S22, ESI†). This indicates that for the three complexes, the reduction is diffusion limited with a diffusion coefficient of 1.07 × 10−5 for complex 1, 0.77 × 10−5 for complex 2 and 0.72 × 10−5 for complex 3 (please see ESI†). It has been observed that the catalytic-to-peak current ratio (icat/ip) increases for all the three complexes with the increase in acid concentration (Fig. S23, ESI†).
The electrochemical properties of three nickel(II) complexes with salen-type ligands were influenced by the presence of different substituents of the imine linkage.33 Ren and co-workers showed that the electrochemical reduction ability of nickel(II) complexes with cyclam ligands from H+ to H2 is dependent on the substituents of the macrocyclic ligands.34 Thus, it is expected that changes in the chain length of ligands in the Ni(II) complexes in the present study could influence Ni(II)/Ni(I) potentials of the complexes.
To determine the activity of complexes 1, 2 and 3 as electrocatalysts, first, cyclic voltammograms of the complexes were recorded in the presence of acetic acid (pKa = 13.5) in DMF.35 The addition of CH3COOH aliquots at a concentration ranging from 0.00 to 24.00 mM to the solutions of complexes 1, 2 and 3 in DMF triggers the systematic increase in catalytic current (icat) at −1.48, −1.51, and −1.52 V respectively versus Fc/Fc+ (Fig. 3).
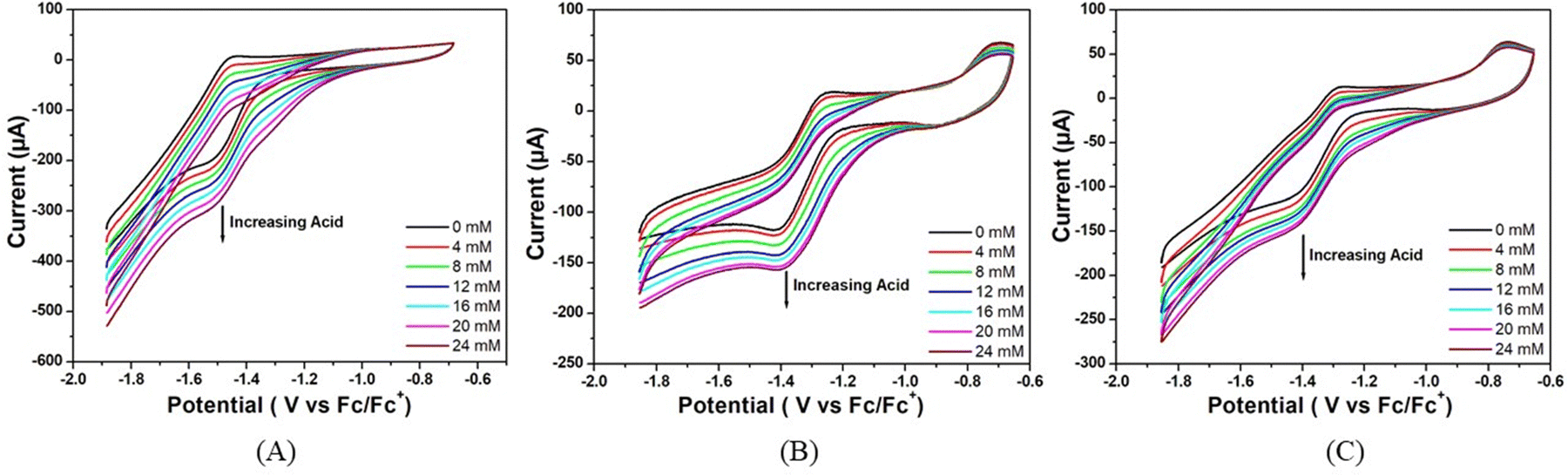 | ||
| Fig. 3 Cyclic voltammograms of (A) complex 1, (B) complex 2 and (C) complex 3 (5.36 μM) in the absence of acetic acid (black trace) and in the presence of different concentrations of acetic acid in air-free DMF. Conditions: 25 °C, 0.1 M [n-Bu4N]Br as the supporting electrolyte, scan rate = 50 mV s−1, glassy carbon working electrode, Pt wire counter electrode and the potential are referenced against Fc/Fc+. | ||
The onset potentials for the reduction of hydrogen ions in the presence of the three complexes studied are −1.13 V, −1.15 V and −1.20 V versus Fc/Fc+, respectively, indicating that the catalytic activity is in the order: complex 1 > complex 2 > complex 3.
The electrocatalytic activity of these complexes was also accessed using trifluoroacetic acid (pKa = 6.0) in DMF as a proton source.36 As shown in Fig. 4, there is an increase in the catalytic current (icat) at −1.48, −1.51, and −1.52 V versus Fc/Fc+ for complexes 1, 2 and 3 respectively upon successive addition of CF3COOH (0.00 to 24.00 mM). For all the three complexes, the peak current increases and the peak potential is shifted towards less negative potential (anodically) with the increase in acid concentration in the solution. This signifies that the greater diffusion of proton from bulk solution to the electrode surface makes the reduction process much easier at high concentrations of acid. Notably, for all the three complexes, the sequence of peak currents that also represents their catalytic activity at any particular acid concentration is the same as the order of post peak current values, which decrease sharply with the decrease in potential due to increased hydrogen evolution.
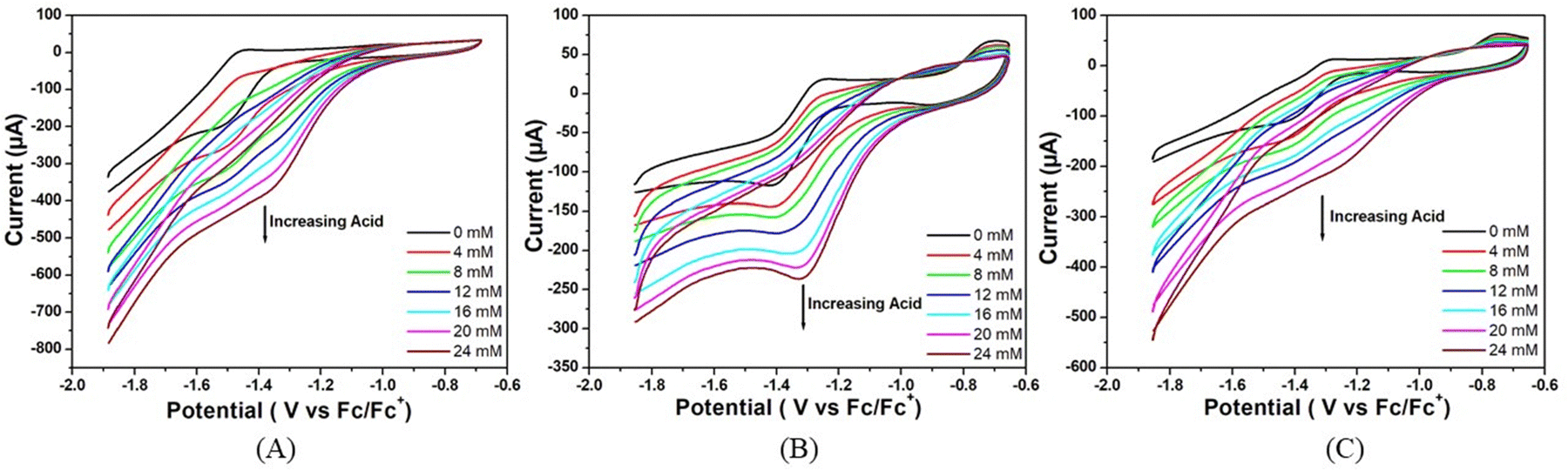 | ||
| Fig. 4 Cyclic voltammograms of (A) complex 1, (B) complex 2 and (C) complex 3 (5.36 μM) in the absence of trifluoroacetic acid (black trace) and in the presence of different concentrations of trifluoroacetic acid in air-free DMF. Conditions: 25 °C, 0.1 M [n-Bu4N]Br as supporting electrolyte, scan rate = 50 mV s−1, glassy carbon working electrode, Pt wire counter electrode and the potential is referenced against Fc/Fc+. | ||
This rise in current in both the cases could be attributed to the generation of H2 from catalytic reduction of protons. The reduction potentials of these complexes slightly change towards more anodic values with the sequential enhancements in the concentration of the acid. The overpotential for hydrogen evolution was calculated following the methods reported by Fourmond et al.35 from the theoretical half wave potential, ET1/2, based on the expression found in the supporting document and the experimental potential Ecat/2 (eqn (1)):
| Over potential (η) = |ET1/2 − Ecat/2| | (1) |
Based on eqn (1), this reduction occurs at overpotentials of −0.52 V, −0.55 V and −0.56 V for complexes 1, 2 and 3 respectively for the former case, where acetic acid is the proton source. The corresponding values are −0.22 V, −0.23 V and −0.24 V for complexes 1, 2 and 3 respectively for the latter case, where trifluoroacetic acid is the proton source.
In both the cases, the catalytic activity of the three homogeneous catalysts as obtained from the overpotential value is in the following order: complex 1 > complex 2 > complex 3. Moreover, the large difference in overpotential for each complex for the two proton sources is expected for the increased diffusion of protons in the latter case.
A table is given in ESI† (Table S4), where few of recently reported Ni(II) complexes8b,c,37 are listed with their overpotential values. In most of the cases, the medium of experiment was organic. The proton source varies as acetic acid, trifluoroacetic acid, perchloric acid or buffer solution. If we look into the values of overpotential, they are in the range of few millivolts. While comparing with the other reported overpotential values for the electrocatalytic hydrogen evolution catalysed by nickel complexes as enlisted in the table, it can be concluded that these three nickel(II) complexes are more effective in this field.
The stability of all three complexes have been checked by repeated cyclic voltammetry (CV) studies up to 65 cycles using TFA as the proton source (Fig. 5). It has been observed that no new peaks are generated in the CV and also the colour of the solution remains the same during the study. In all cases, the reduction peak current increases continuously and reaches almost a steady value at the 55th cycle. The peak current with respect to the lowest amount is increased by 12% for complex 1, 14% for complex 2, and 28% for complex 3. The current of complex 1 is always greater than the remaining two complexes. The increase in current during repeated cyclic voltammetric operation might be due to the cleansing of the fine surface of the electrode and opening up of new channels by the initial H+ ion penetration at the metal centre and the associated H2 evolution. Thus, more active sites of the electrode surface are created for further reaction. Greater the size of the alkyl group, the greater is the initial prevention of H+ for approaching towards the metal centre for reduction. On application of the negative potential, the proton penetrates through the barrier and creates new channels for further reaction. Since the initial barrier varies in the order of complex 3 > complex 2 > complex 1, the increase in the peak current on cycling is in the reverse order.
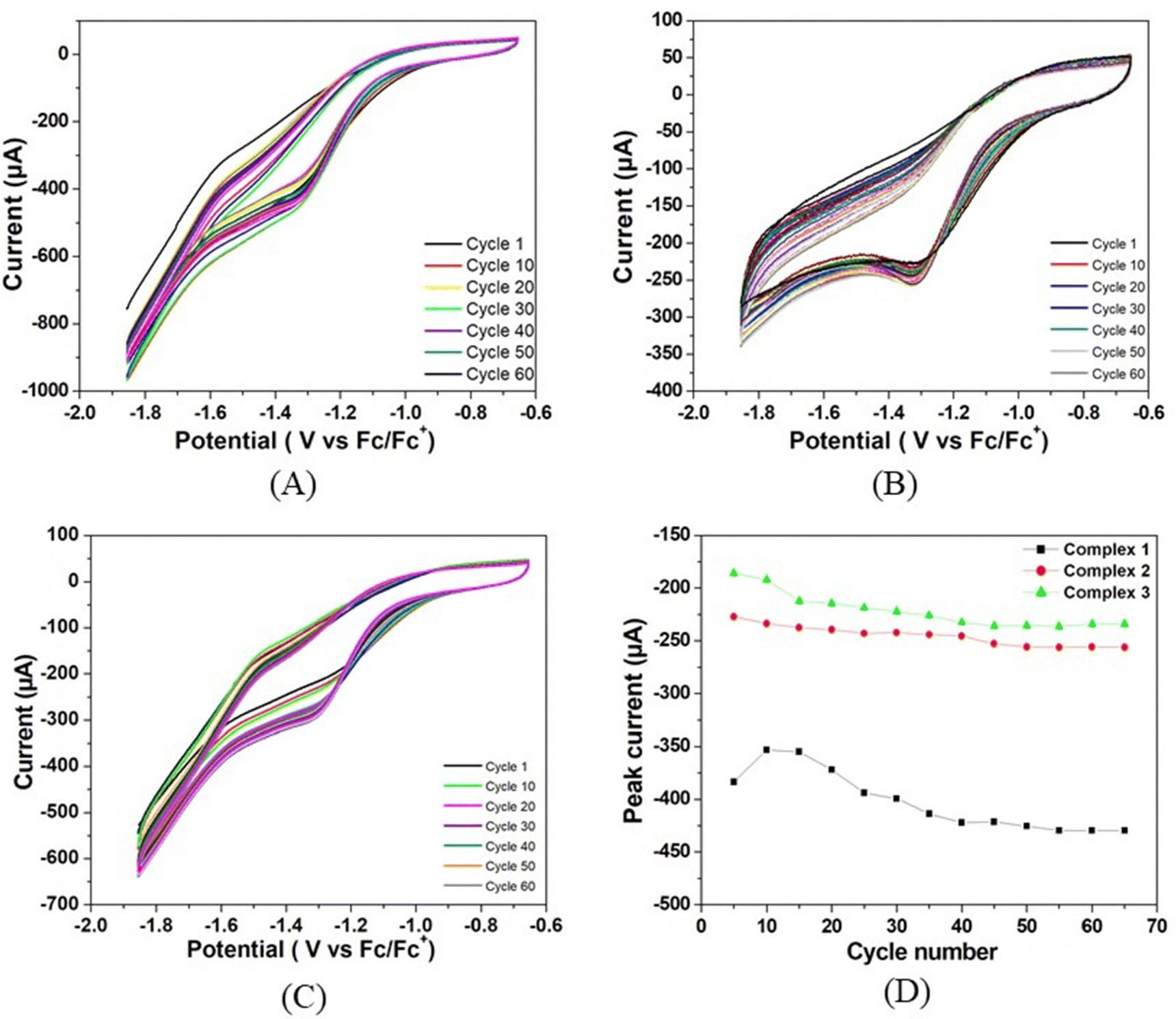 | ||
| Fig. 5 Repeated cyclic voltammetry studies of (A) complex 1, (B) complex 2, and (C) complex 3. (D) Peak current vs. cycle number profiles for all the three complexes. | ||
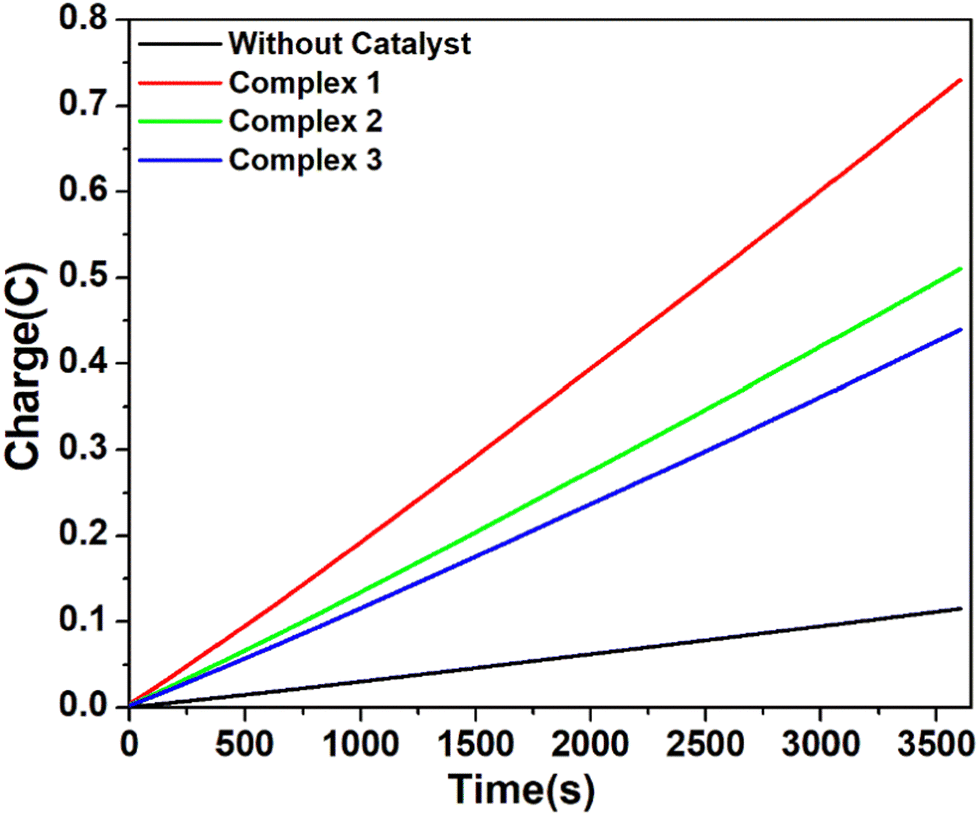 | ||
| Fig. 6 Charge build up vs. time plots in the CPE (controlled potential electrolysis) experiment of complex 1 (red), complex 2 (green), and complex 3 (blue) and without catalyst (black) at potential −1.5 V versus Fc/Fc+. Conditions: 5.36 μM complex in air-free DMF with 0.1 M [n-Bu4N]Br as the supporting electrolyte and 24 mM CH3COOH as the proton source. | ||
| Complex | Solvent | Proton source | q (C) | n (× 10−6) | TON | TOF (s−1) | Faradaic efficiency (%) |
|---|---|---|---|---|---|---|---|
| 1 | DMF | Acetic acid | 0.62 | 3.21 | 23.95 | 251.25 | 67.56 |
| 2 | 0.40 | 2.07 | 15.45 | 133.75 | 48.64 | ||
| 3 | 0.33 | 1.71 | 12.76 | 92.92 | 41.35 | ||
| 1 | Trifluoroacetic acid | 1.30 | 6.74 | 50.30 | 426.67 | 81.78 | |
| 2 | 0.50 | 2.59 | 19.33 | 188.75 | 61.41 | ||
| 3 | 0.38 | 1.97 | 14.70 | 126.67 | 59.58 |
However, when the CPE experiments were done with 5.36 μM of complexes 1, 2 and 3 separately in DMF solutions in the presence of trifluoroacetic acid (24 mM), the same trend was obtained (Fig. 7). At an applied potential of −1.50 V versus Fc/Fc+, the maximum charge reached 1.41 C for complex 1, 0.61 C for complex 2 and 0.49 C for complex 3 during 1 hour of electrolysis. CPE experiments under the same potential with a catalyst-free solution gave only a charge of 0.11 C, showing that complex 1 is again more effective than the remaining two complexes in hydrogen production under such conditions. This study suggests that all these three complexes are capable of catalysing the reduction of protons from acid to H2. The evolution of H2 gas has been confirmed by the use of gas chromatography (Fig. S25, ESI†). The turn over number (TON) and faradaic efficiency were calculated for all the complexes (Tables S5 and S6, ESI†), and the result of CPE experiments is enlisted in Table 2.
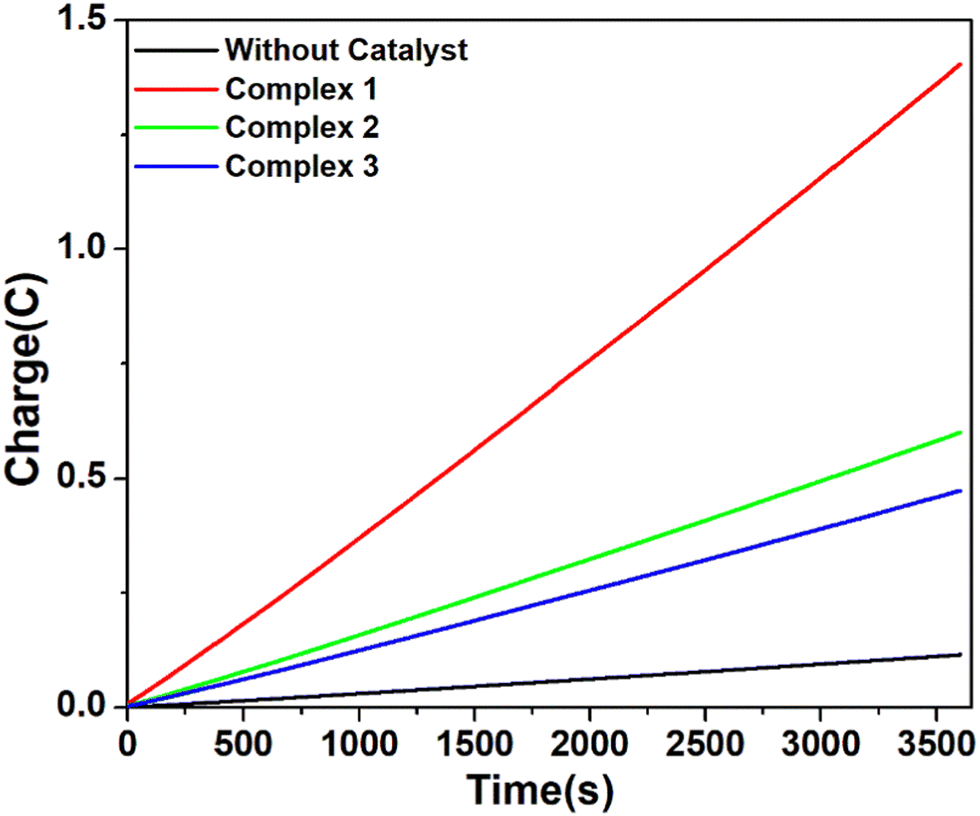 | ||
| Fig. 7 Charge build up vs. time plots in the CPE (controlled potential electrolysis) experiment of complex 1 (red), complex 2 (green), and complex 3 (blue) and without catalyst (black) at potential −1.5 V versus Fc/Fc+. Conditions: 5.36 μM complex in air-free DMF with 0.1 M[n-Bu4N]Br as the supporting electrolyte and 24 mM CF3COOH as the proton source. | ||
The turnover frequency (TOF) for hydrogen evolution using all the three complexes 1, 2 and 3 as electrocatalysts was estimated by the foot-of-the-wave analysis (FOWA).38 The FOWA has been considered near the foot of the catalytic wave, where the catalytic wave does not get affected much by phenomena such as substrate consumption, diffusion and shape of CV dominated mainly by catalytic phenomenon.39 The icat/ip was plotted against 1/(1 + exp[(F/RT)(E − E1/2)]) for both the complexes, as given in Fig. S26 and S27 (ESI†), and the obtained TOF values are presented in Table S5 (ESI†).
η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logi logi | (2) |
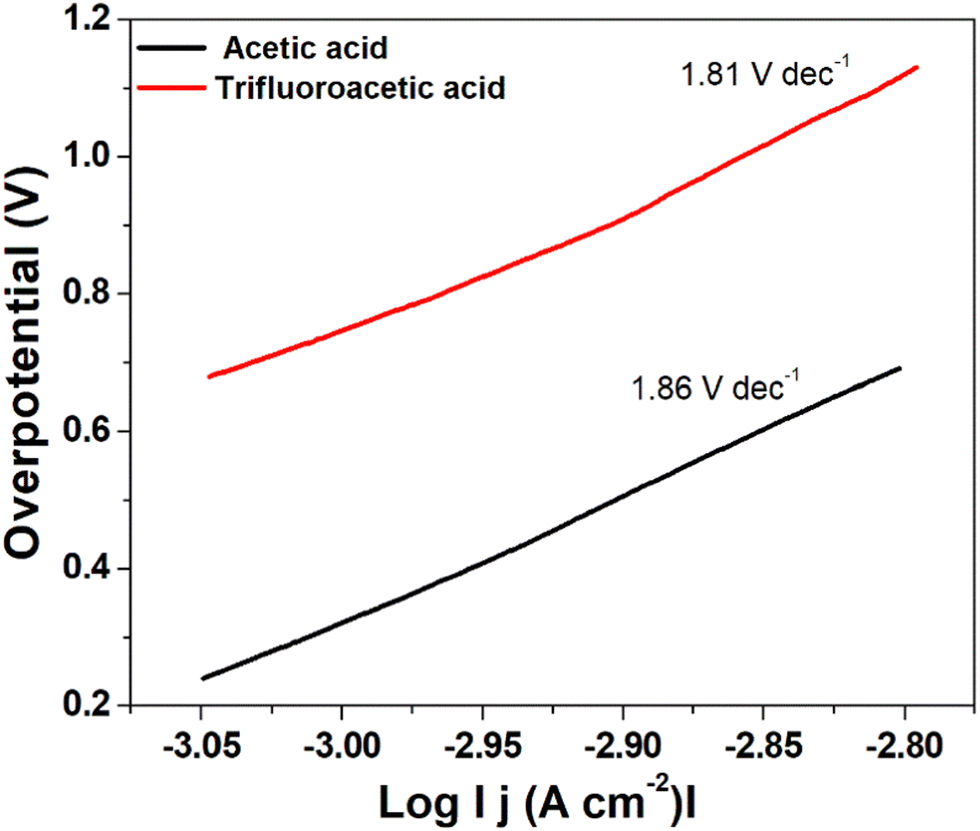 | ||
| Fig. 8 Tafel plot for hydrogen evolution catalysed by complex 1 using trifluoroacetic acid (red line) and acetic acid (black line) as the acid source. | ||
A low η value corresponds to a large exchange current density (i0, current density at η = 0), whereas a low b value indicates better hydrogen evolution and hence a better catalytic activity.40 In this case, the determined Tafel slopes for complex 1 are 1.81 V dec−1 and 1.86 V dec−1 for the acid sources trifluoroacetic acid and acetic acid, respectively indicating that trifluoroacetic acid will lead to a faster increase in reaction rate (fast proton discharge kinetics on the working electrode) with the increase in potential.
Mechanistic pathways are reportedly known in the literature, based on different reports we propose the plausible mechanism, as depicted in Fig. S32a (ESI†). The pristine catalyst with the Ni(II) center reduced first to Ni(I), which then undergoes oxidative protonation to form a hydride complex {Ni(III)–H} (Tables S9–S20 for geometry optimized coordinates and summary of natural population analysis for complexes and intermediates, ESI†). As per the report of Cao et al.,42 this Ni(III)–H hydride complex either reacts with a proton to produce H2 and Ni(III) complex (heterolytic route), or is further reduced to Ni(II)–H. This Ni(II)–H can react with a proton to give Ni(II) and H2, or react with another Ni(II)–H to produce H2 and 2 molecules of Ni(I) complex. Another way is that Ni(III)–H underwent bimolecular hydrolysis to produce H2 and the original starting Ni(II) complex. It can be concluded that once the hydride complex is formed, it can liberate hydrogen, and by simultaneous reduction, it gets back to the initial catalyst to continue the cycle.42,43 This suggests that the key intermediate is Ni(III)–H complexes in a mechanistic cycle of HER. In this work, we wanted to model those complexes and understand their geometries, which, on the one hand, helps to understand the present outcome and, on the other hand, can suggest better geometries for future development. Therefore, we took aid of DFT calculation that helps to understand an interesting trend in dipole moment (Table S21, ESI†), where the highest value was obtained for complex 1. Indeed, we hypothesize that one reason of showing better catalytic activity for complex 1 can be associated with a higher dipole moment that possibly helps in forming the hydride complex leading to catalysis.42–44 To understand the geometry of the hydride intermediates, it was calculated at the level of B3LYP + COSMO(H2O) + def2TZVP(Ni) + def2-SVP(C,H,N,O) which reveals a distorted penta-coordination network for the Ni(III) centre in the complexes. Metal centres of catalysts (complexes 1, 2 and 3) comprise the HOMO density, whereas for hydride complexes the metal centres in LUMO comprise the density as obtained from the examined frontier molecular orbitals (Fig. S32b–d and ESI† for population analysis). Thus, from DFT calculations, we understood that shorter aliphatic substitution gives a higher dipole moment that could have an influence on higher catalytic activity. Another aspect that originates from the DFT calculation regarding future development is to keep an aromatic group at the end of the aliphatic chain for aromatic interaction with adjacent aromatic groups for complex 2. As we observed for complex 2 and its hydride derivative, the aliphatic group is finally inclined towards the aromatic groups. However, for complexes 1 and 3, such observation was not seen. This possibly suggests an optimum chain length for inclination, and here substitution with an aromatic group could give higher stability of the complex, eventually better catalytic activity.
Conclusions
In summary, we have successfully synthesized three mononuclear nickel(II) complexes with similar N and O donor ligands and characterized them by various standard methods. These complexes have been found to be active electrocatalysts for hydrogen evolution reactions using acetic acid and trifluoroacetic acid as the substrates in DMF. The TOF values of these catalysts decrease with the increase in the chain length of the hydroxyalkyl group. The Ni(II) centre in these complexes is reduced to Ni(I) species and then converted into Ni(III)-hydride, which ultimately generates hydrogen and returns to the Ni(II) state. This possible mechanism has been supported by theoretical calculations. Thus, in this study, it has been demonstrated that the length of the alkyl side chain has significant effects on the catalytic ability in HERs and can be judiciously designed to get optimized efficiency.Author contributions
Arpita Barma: conceptualization, formal analysis, investigation, methodology; Malay Chakraborty: formal analysis, investigation; Swapan Kumar Bhattacharya: formal analysis, supervision, validation, writing – original draft; Pritam Ghosh: methodology, software, validation, writing – original draft; Partha Roy: conceptualization, resources, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AB wishes to thank CSIR, New Delhi for providing her a fellowship.References
- J.-W. Wang, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2019, 378, 237–261 CrossRef CAS.
- (a) M. G. Pfeffer, T. Kowacs, M. Wächtler, J. Guthmuller, B. Dietzek, J. G. Vos and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 6627–6631 CrossRef CAS PubMed; (b) H. Ozawa, M.-A. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926–4927 CrossRef CAS PubMed; (c) D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS; (d) K. Zeng and D. K. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS; (e) A. Lasia, Hydrogen evolution reaction, InHandbook of Fuel Cells, John Wiley & Sons, New York, 2010 Search PubMed; (f) J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef; (g) J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- (a) K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed; (b) L. Tong, L. Duan, A. Zhou and R. P. Thummel, Coord. Chem. Rev., 2020, 402, 213079 CrossRef CAS.
- T. Agarwal and S. Kaur-Ghumaan, Coord. Chem. Rev., 2019, 39, 88–219 Search PubMed.
- M. D. Sampson and C. P. Kubiak, Inorg. Chem., 2015, 54, 6674–6676 CrossRef CAS PubMed.
- (a) A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS PubMed; (b) P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- (a) N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2020, 142, 274–282 CrossRef CAS PubMed; (b) T. Straistari, R. Hardré, J. Fize, S. Shova, M. Giorgi, M. Réglier, V. Artero and M. Orio, Chem. Eur. J., 2018, 24, 8779–8786 CrossRef CAS PubMed; (c) N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed.
- (a) T. Fogeron, T. K. Todorova, J.-P. Porcher, M. Gomez-Mingot, L.-M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, Dalton Trans., 2019, 48, 14653–14661 RSC; (b) Y.-P. Zhang, M. Zhang, X.-R. Chen, C. Lu, D. J. Young, Z.-G. Ren and J.-P. Lang, Inorg. Chem., 2020, 59, 1038–1045 CrossRef CAS PubMed; (c) R. Shen, J. Xie, Q. Xiang, X. Chen, J. Jiang and X. Li, Chin. J. Catal., 2019, 40, 240–288 CrossRef CAS; (d) D. Jana, H. K. Kolli, S. Sabnam and S. K. Das, Chem. Commun., 2021, 57, 9910–9913 RSC; (e) S. Sinha, G. N. Tran, H. Na and L. M. Mirica, Chem. Commun., 2022, 58, 1143–1146 RSC; (f) A. Cabrera-García, V. Blay, R. Blay-Roger, Á. G. Ravelo, J. González-Platas, M. C. Arévalo, J. Sanchiz and P. Martín-Zarza, Chem. Eng. J., 2021, 420, 130342 CrossRef.
- (a) P. Bhanja, B. Mohanty, S. Chongdar, A. Bhaumik, B. K. Jena and S. Basu, ACS Appl. Energy Mater., 2022, 5, 3558–3567 CrossRef CAS; (b) S. Bhattacharjee, S. Bera, R. Das, D. Chakraborty, A. Basu, P. Banerjee, S. Ghosh and A. Bhaumik, ACS Appl. Mater. Interfaces, 2022, 14, 20907–20918 CrossRef CAS PubMed; (c) H. Xu, H. Shang, C. Wang and Y. Du, Coord. Chem. Rev., 2020, 418, 213374 CrossRef CAS; (d) J. Mohammed-Ibrahim and X. Sun, J. Energy Chem., 2019, 34, 111–160 CrossRef.
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC.
- J. W. Morgan and E. Anders, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6973–6977 CrossRef CAS PubMed.
- J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560–8564 CrossRef CAS PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- A. Zarkadoulas, M. J. Field, C. Papatriantafyllopoulou, J. Fize, V. Artero and C. A. Mitsopoulou, Inorg. Chem., 2016, 55, 432–444 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- G. Bergamini and M. Natali, Dalton Trans., 2019, 48, 14653–14661 RSC.
- A. Das, Z. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, ACS Catal., 2015, 5, 1397–1406 CrossRef CAS.
- H. Shao, S. K. Muduli, P. D. Tran and H. S. Soo, Chem. Commun., 2016, 52, 2948–2951 RSC.
- X. Li, Ho, H. Shao, Y. Y. Ng, R. Ganguly, Y. Lu and H. S. Soo, Inorg. Chem., 2019, 58, 1469–1480 CrossRef PubMed.
- APEX-II, SAINT and SADABS, Bruker AXS Inc., Madison, WI, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) S. Halder, J. Mondal, J. Ortega-Castro, A. Frontera and P. Roy, Dalton Trans., 2017, 46, 1943–1950 RSC; (b) A. Bhattacharjee, S. Dey and P. Roy, Inorg. Chim. Acta, 2019, 490, 93–103 CrossRef CAS.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introduction to spectroscopy, Cengage Learning, Stamford, USA, 5th edn, 2015 Search PubMed.
- (a) M. F. Mehrjardi, H. Kargar, R. B.-Ardakani, M. Ashfaqc, K. S. Munawar and M. N. Tahir, J. Mol. Struct., 2022, 1251, 132037 CrossRef; (b) G. R. Reddy, S. Balasubramanian and K. Chennakesavulu, J. Mater. Chem. A, 2014, 2, 15598–15610 RSC; (c) A. D. Khalaji, M. Nikookar and D. Das, J. Therm. Anal. Calorim., 2014, 115, 409–417 CrossRef CAS; (d) S. N. Shukla, P. Gaur, M. L. Raidas and B. Chaurasia, J. Mol. Struct., 2020, 1202, 127362 CrossRef CAS; (e) H. Kargr, A. A. Ardakani, M. N. Tahir, M. Ashfaqc and K. S. Munawar, J. Mol. Struct., 2021, 129842 CrossRef.
- (a) S. Chattopadhyay, M. S. Ray, S. Chaudhuri, G. Mukhopadhyay, G. Bocelli, A. Cantoni and A. Ghosh, Inorg. Chem. Acta, 2006, 359, 1367–1375 CrossRef CAS; (b) M. S. Ray, S. Chaudhuri, L. Right, G. Bocelli, G. Mukhopadhyay and A. Ghosh, Polyhedron, 2003, 22, 617–624 CrossRef CAS.
- (a) B. S. Garg and D. N. Kumar, Spectrochim. Acta, Part A, 2003, 59, 229–234 CrossRef CAS; (b) A. Anthonysamy and S. Balasubramanian, Inorg. Chem. Commun., 2005, 8, 908–911 CrossRef CAS.
- (a) M. Salehi, F. Rahimifar, M. Kubicki and A. Asadi, Inorg. Chim. Acta, 2016, 443, 28–35 CrossRef CAS; (b) A. Barma, D. Ghosh, P. Karmakar and P. Roy, J. Mol. Struct., 2022, 1250, 131687 CrossRef CAS.
- A. Ourari, Y. Ouennoughi, D. Aggoun, M. S. Mubarak, E. M. Pasciak and D. G. Peters, Polyhedron, 2014, 67, 59–64 CrossRef CAS.
- L.-Q. Chai, H.-S. Zhang, J.-J. Huang and Y.-L. Zhang, Spectrochim. Acta, Part A, 2015, 137, 661–669 CrossRef CAS PubMed.
- J. Datta, S. Bhattacharya and K. K. Kundu, Aust. J. Chem., 1983, 36, 1779–1784 CrossRef CAS.
- C. Chen, X. Li, F. Deng and J. Li, RSC Adv., 2016, 6, 79894–79899 RSC.
- T. D. Cook, S. F. Tyler, C. M. McGuire, M. Zeller, P. E. Fanwick, D. H. Evans, D. G. Peters and T. Ren, ACS Omega, 2017, 2, 3966–3976 CrossRef CAS PubMed.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS PubMed.
- (a) V. Fourmond, S. Canaguier, B. Golly, M. J. Field, M. Fontecave and V. Artero, Energy Environ. Sci., 2011, 4, 2417–2427 RSC; (b) G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- (a) J.-P. Cao, T. Fang, L.-Z. Fu, L.-L. Zhou and S. Zhan, Int. J. Hydrogen Energy, 2014, 39, 10980–10986 CrossRef CAS; (b) R. Jain, A. A. Mamun, R. M. Buchanan, P. M. Kozlowski and C. A. Grapperhaus, Inorg. Chem., 2018, 57, 13486–13493 CrossRef CAS PubMed; (c) G. Bergamini and M. Natali, Dalton Trans., 2019, 48, 14653–14661 RSC; (d) Y.-P. Zhang, M. Zhang, X.-R. Chen, C. Lu, D. J. Young, Z.-G. Ren and J.-P. Lang, Inorg. Chem., 2020, 59, 1038–1045 CrossRef CAS PubMed; (e) J.-M. Lei, S.-P. Luo and S.-Z. Zhan, Int. J. Hydrogen Energy, 2018, 43, 19047–19056 CrossRef CAS; (f) Q.-X. Peng, D. Xue, S.-Z. Zhan and C.-L. Ni, Appl. Catal. B, 2017, 219, 353–361 CrossRef CAS; (g) L. Chen, G. Chen, C.-F. Leung, S.-M. Yiu, C.-C. Ko, E. A. Mallart, M. Robert and T.-C. Lau, ACS Catal., 2015, 5, 356–364 CrossRef CAS.
- (a) C. Costentin, S. Drouet, M. Robert and J.-M. Saveant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed; (b) C. Costentin, M. Robert and J.-M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC; (c) C. Costentin and J.-M. Saveant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS; (d) R. J. DiRisio, J. E. Armstrong, M. A. Frank, W. R. Lake and W. R. McNamara, Dalton Trans., 2017, 46, 10418–10425 RSC.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- (a) V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC; (b) M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 RSC; (c) X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- (a) TURBOMOLE, Version 7.3, a development, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007 Search PubMed; (b) E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- Y. Han, H. Fang, H. Jing, H. Sun, H. Lei, W. Lai and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 5457–5462 CrossRef CAS PubMed.
- K. Majee, J. Patel, S. Rai, B. Das, B. Panda and S. K. Padhi, Phys. Chem. Chem. Phys., 2016, 18, 21640–21650 RSC.
- N. Villegas-Escobar, D. E. Ortega, D. Cortes-Arriagada, R. Duran, D. Yepes, S. Gutierrez-Oliva and A. Toro-Labbe, J. Phys. Chem. C, 2017, 121, 12127–12135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2062677 (1), 2062678 (2) and 2062679 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ma00462c |
| This journal is © The Royal Society of Chemistry 2022 |