
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of cation vacancies at tetrahedral sites in cobalt spinel oxides on oxygen evolution catalysis†
Wei
Liu
 a,
Masao
Kamiko
a,
Ikuya
Yamada
b and
Shunsuke
Yagi
*a
a,
Masao
Kamiko
a,
Ikuya
Yamada
b and
Shunsuke
Yagi
*a
aInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-Ku, Tokyo, 153-8505, Japan. E-mail: syagi@iis.u-tokyo.ac.jp
bDepartment of Materials Science, Graduate School of Engineering, Osaka Metropolitan University, 1-1 Gakuen-cho, Naka-ku, Sakai, Osaka, 599-8531, Japan
First published on 23rd August 2022
Abstract
Cobalt spinel oxides exhibit high catalytic activity for the oxygen evolution reaction (OER). Therefore, accurately identifying the effects of ions from different sites on OER catalysis is essential to improve catalytic activity. Co ions at octahedral sites are usually considered as active sites, whereas those at tetrahedral sites are less active. In this study, the effects of cation vacancies at the tetrahedral sites were investigated for the OER, excluding the contributions from the active cations at the octahedral sites, using a series of Zn-substituted cobalt spinel oxides, ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1), where Co ions occupy only the tetrahedral sites because Al ions tend to occupy the octahedral sites. Among them, Zn0.6Co0.4Al2O4 exhibited the highest OER activity and comparable kinetics to that of Co3O4, which is attributed to the cation vacancies generated by Zn dissolution. The Al ions in the structure enhanced the stability of the cobalt spinel oxides, and the spinel structure of Zn0.6Co0.4Al2O4 was retained after 1200 OER cycles. By excluding the active sites from the octahedral sites, this study demonstrates that a certain degree of cation vacancies at the tetrahedral sites can effectively improve the activity of less active Co ions at the tetrahedral sites, thus providing an important basis for future catalyst design.
Introduction
As a highly efficient and environmentally friendly fuel, hydrogen has received significant research attention. Water electrolysis is an effective technology to produce hydrogen on the cathode, while oxygen evolution reaction (OER) occurs on the anode. However, OER faces significant overpotential and slow kinetics due to its multiple-step reactions.1 Therefore, affordable catalysts are necessary for reducing the energy barrier and increasing the reaction rate of the OER. Most commercial OER catalysts, such as IrO2 and RuO2, are noble metal-based materials used in industrial electrolyzers; however, they are expensive, rare, and cannot meet the increasing demand for hydrogen production.2,3 Therefore, numerous studies have explored cost-effective and catalytically active materials.4–7 Nickel-based materials exhibit high OER catalytic activity in alkaline electrolyzers.8 Besides nickel, it is still necessary to develop other transition metal-based catalysts for further enhancement of the catalytic activity.Cobalt spinel oxide Co3O4 has been reported as a highly active catalyst for OER in alkaline solutions.9–13 In the spinel oxide structure, cations in tetrahedral and octahedral coordination exist in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Because the orbital overlap between the cations of octahedral coordination and the surrounding oxygen ions is larger than that of tetrahedral coordination, adjusting the strength of the M–O bonds of octahedral sites should effectively improve the catalytic activity. According, many researchers have attempted to modify cations at the octahedral sites to improve the OER activity.14–17
:2. Because the orbital overlap between the cations of octahedral coordination and the surrounding oxygen ions is larger than that of tetrahedral coordination, adjusting the strength of the M–O bonds of octahedral sites should effectively improve the catalytic activity. According, many researchers have attempted to modify cations at the octahedral sites to improve the OER activity.14–17
However, cations at the tetrahedral sites also affect OER activity. In our previous study, Co ions at the tetrahedral sites were partially replaced with Zn ions, and the substituted sample Zn0.4Co2.6O4 showed higher activity than pure Co3O4.18 The reason for this behavior is that Zn has a stronger leaching tendency than Co ions, and the resulting structure with cation vacancies at the tetrahedral sites showed a higher OER activity. Similarly, Wang et al. reported that Zn0.4Ni0.6Co2O4 spinel nanoparticles supported on N-doped nanotubes showed a high activity on OER because Zn ions dissolve in an alkaline solution, generating cation vacancies and reinforcing the Co–O covalency.4,19 These studies have contributed to the knowledge about the origin of catalytic activity from different sites in spinel oxide structures. However, all the cations (Co2+, Co3+, and Ni2+ in these studies) are catalytically active except Zn2+, leading to difficulties in distinguishing the contributions of the active ions at the tetrahedral and octahedral sites. In particular, the possible effects of tetrahedral vacancies on tetrahedral active cations and/or octahedral active cations to enhance the OER remain unclear.
To overcome these existing limitations, Zn- and Al-substituted Co3O4, denoted as ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1) were synthesized in the present study, and their activities on OER were evaluated in 0.1 M KOH aqueous solution. Zn2+ and Al3+ ions were selected as substitution ions because they are not active on OER catalysis with non-variable valence states, and they preferentially occupy the tetrahedral and octahedral sites than Co ions, respectively. Detailed spectroscopic and electrochemical analyses were conducted to examine the contribution of cation vacancies at the tetrahedral sites to OER catalysis, sufficiently eliminating the impacts of other factors. Although CoAl2O4 was expected to have the highest catalytic activity because Co2+ ions act as highly active sites compared to Zn2+ ions, Zn0.6Co0.4Al2O4 with a middle Zn substitution amount showed the highest OER activity among all the samples. In addition, Al ions in the structure stabilized the Co2+ ions in the cobalt spinel oxides, and the spinel structure of Zn0.6Co0.4Al2O4 was retained after 1200 OER cycles.
Results and discussion
The crystal structures of the as-synthesized spinel oxides were examined using synchrotron X-ray diffraction (SXRD), as displayed in Fig. 1a. The diffraction peaks of all the samples, except ZnAl2O4, corresponded well with the reported ZnAl2O4 (ICSD No. 163268) and CoAl2O4 (ICSD No. 290133), indicating that the typical spinel oxide structures were obtained.20,21 For the as-synthesized ZnAl2O4, in addition to the spinel oxide peaks with strong intensities, some impurity peaks were consistent with those for ZnO. However, ZnO shows little catalytic activity for OER; therefore, its existence does not affect the evaluation of the OER activity of spinel oxide samples in this study.22 Rietveld refinement of the SXRD patterns was conducted to reveal the atom occupancy in Zn1−xCoxAl2O4, and the results are displayed in Fig. S1 and Table S1 (ESI†). No evident cation vacancy was observed from the fitting. Co and Zn ions primarily occupied the tetrahedral (8a) sites, and Al ions primarily occupied the octahedral (16d) sites, as shown in Fig. S2a (ESI†). In addition, the lattice constant decreased with the increase in the amount of Zn, as shown in Fig. S2b (ESI†). The decrease in lattice constants was found to be against the larger radius of Zn2+ (tetrahedral: 0.6 Å) than that of Co2+ (tetrahedral: 0.58 Å) ion. However, considering the large difference in the radii of Al3+ ions in the two different sites (tetrahedral: 0.39 Å, octahedral: 0.535 Å), mixed occupancy of Al ions accounted for the decrease in lattice constants. The results are also consistent with those of previous studies.20,21 The structures of the as-synthesized Zn- and Al-substituted Co spinel oxides are illustrated in Fig. 1b.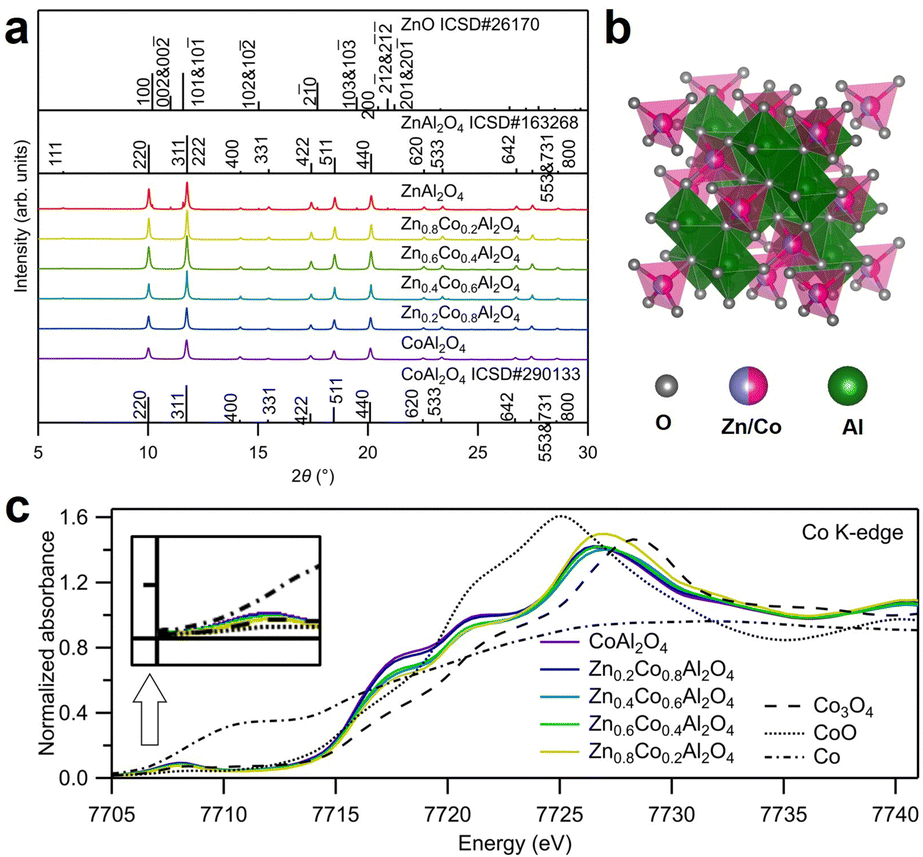 | ||
| Fig. 1 (a) Synchrotron X-ray diffraction (SXRD) patterns for ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1) utilizing an irradiated X-ray wavelength of 0.49995 Å. The wavelength was calibrated using a standard sample of CeO2. (b) Illustration of the crystal structure of ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1). (c) Normalized Co K-edge X-ray absorption near edge structure (XANES) spectra for ZnxCo1−xAl2O4 (x = 0.2, 0.4, 0.6, 0.8, and 1) with Co,CoO and Co3O4 as standard references. | ||
X-Ray absorption spectroscopy (XAS) was conducted to investigate the environment around Co; the obtained X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra near Co K-edge are displayed in Fig. 1c and Fig. S3 (ESI†), respectively. The pre-edge peaks at 7708 eV in Fig. 1c for ZnxCo1−xAl2O4 were more significant than that for Co3O4. The emergence of the pre-edge peaks was contributed by the partly permitted 1s to 3d transition of Co ions at the tetrahedral sites. The values of absorption edge energies E0 are summarized in Table S2 (ESI†). As shown in Fig. 1c and Table S2 (ESI†), E0 for all the samples are close to that of CoO, indicating that Co primarily exists as Co2+ ions. Curve fitting was conducted for the first shell in the EXAFS spectra in Fig. S3 (ESI†), and the fitting results are displayed in Table S2 (ESI†). The coordination numbers (CN) are in the range of 4.32–4.76, which are closer to the theoretical value of tetrahedral coordination (CN: 4) than that of octahedral coordination (CN: 6). The clear pre-edge peaks, similar absorption edge energies, and CN values (close to 4) for the synthesized spinel oxides (ZnxCo1−xAl2O4) indicate that most Co ions with +2 valence state occupied the tetrahedral sites, which is consistent with the results obtained from Rietveld refinement.
Fig. S4 (ESI†) shows the scanning electron microscopy (SEM) images of the synthesized spinel oxides ZnxCo1−xAl2O4. All the samples exhibited similar powder morphologies, with particle size at several micrometers. The elemental dispersive spectroscopy (EDS) spectra and analysis results are shown in Fig. S5 and Table S3 (ESI†), respectively. The ratios of Zn/Co ions in the spinel oxides ZnxCo1−xAl2O4 determined using EDS were consistent with their nominal ratios. The specific surface areas were determined using N2 adsorption/desorption isotherms (Fig. S6a, ESI†) with Brunauer–Emmett–Teller (BET) analysis, and the results are displayed in Table S4 (ESI†). Pore size distributions were also obtained using the Barrett–Joyner–Halenda method and the results (Fig. S6b, ESI†) suggest the existence of mesopores with diameters ranging from 17 to 30 nm in all the samples.
Cyclic voltammetry (CV) was conducted to evaluate the OER catalytic activities of ZnxCo1−xAl2O4. The anodic and cathodic sweeps were averaged to eliminate the influence of double-layer capacitance, and the obtained linear sweep voltammograms of the first cycle are displayed in Fig. 2a. The currents were standardized with the surface area of the as-synthesized catalyst powders determined by BET analysis. As shown in Fig. 2a, Zn0.6Co0.4Al2O4 shows the smallest overpotential to reach 5 μA cm−2 among all the samples. The overpotentials followed volcano relationships with Zn ratios for the OER, as shown in the inset graph. Tafel plots in Fig. 2b provide the Tafel slopes, which are related to the OER kinetics. The Tafel slope for Zn0.6Co0.4Al2O4 was the smallest (71.8 mV dec−1), indicating the fastest OER kinetics, and the second smallest value (81.3 mV dec−1) was for Zn0.4Co0.6Al2O4. The large difference in the Tafel slopes of Zn0.6Co0.4Al2O4 and Zn0.4Co0.6Al2O4 indicates that the OER process for the two catalysts had a different rate-determining step than those for other samples.23 Zn0.6Co0.4Al2O4 shows the smallest overpotential (0.44 V) and Tafel slope (71.8 mV dec−1), indicating the highest OER catalytic activity among all the spinel oxide samples. The large overpotential and Tafel slope of ZnAl2O4 were predictable because no OER active site (Co ions in this spinel oxide) was present. In contrast, the large overpotential of more than 0.7 V and Tafel slope of 265.6 mV dec−1 in CoAl2O4 was unexpected because the Co amount in CoAl2O4 was the largest among all the spinel oxides. The tendency that the Co spinel oxide with a middle Zn substitution amount had the highest OER activity was reported in our previous study;18 the origin of the OER enhancement by Zn substitution may be attributed to the cation vacancies generated by Zn dissolution, which is discussed further in a subsequent paragraph.
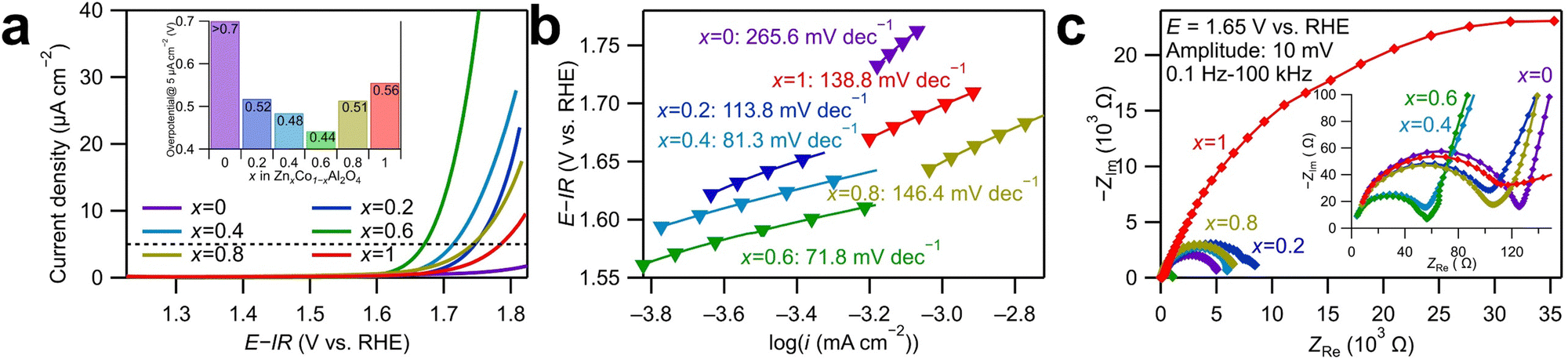 | ||
| Fig. 2 (a) Linear sweep voltammograms for ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1) in 0.1 M KOH aqueous solution with a scan rate of 10 mV s−1. Overpotentials at 5 μA cm−2 are shown in the inset graph. (b) Tafel plots for ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1) derived from the linear sweep voltammograms. (c) Nyquist plots for ZnxCo1−xAl2O4 measured using electrochemical impedance spectroscopy at 1.65 V vs. reversible hydrogen electrode (RHE). | ||
Nyquist plots of electrochemical impedance spectroscopy (EIS) spectra for the catalyst samples are displayed in Fig. 2c. The potential was set to 1.65 V vs. reversible hydrogen electrode (RHE), where OER is the dominant reaction. Two semicircles were observed for all the samples in the Nyquist plots. The EIS spectra were fitted using the equivalent circuit displayed in Fig. S7 (ESI†), and the obtained fitting results are shown in Table S5 (ESI†). Because the semicircles deviated from perfect circles, constant phase elements, indicated by Q, were used in the equivalent circuit instead of capacitors C. R1 is the system resistance, which hardly changes across all the samples. The two semicircles indicate the following two processes with different time constants: the one in the high-frequency range corresponds to the electron transfer between the modified electrode surface and the electrolyte, while the other semicircle in the low-frequency range is assigned to the adsorption/desorption process of charged OER intermediates, such as OH−.24 The fitted R2 values of Zn0.4Co0.6Al2O4 and Zn0.6Co0.4Al2O4 were smaller than other samples by approximately 55 Ω, which is consistent with their smaller Tafel slopes shown in Fig. 2c, suggesting a faster electron transfer process. For the semicircles in the low-frequency range, Zn0.6Co0.4Al2O4 exhibited the smallest R3 and the largest Q3 values, indicating the fastest adsorption/desorption rate and largest reaction area during the OER process. Fig. S8 (ESI†) presents the double-layer capacitances (Cdl) of the spinel oxides, calculated as specified in ESI.†25 The largest Cdl value, 40.1 μF of Zn0.6Co0.4Al2O4 indicates the largest double-layer capacitance, which is consistent with the largest fitted Q3 from the EIS spectra. It is noticed that a scale relationship cannot be observed from the values of double layer capacitance and the BET surface area, suggesting the difference in the adsorption abilities to OH− ions and N2 gas, which may be resulted from the generation of cation vacancies by a possible Zn dissolution of Zn-substituted samples. Therefore, the highest activity of Zn0.6Co0.4Al2O4 might have accounted for the improved kinetics of OER and increased amount of adsorbed OH−.
The structural stability of the most active catalyst Zn0.6Co0.4Al2O4 was investigated using long-term tests as displayed in Fig. 3. The CV curves in Fig. 3a show that the area between cathodic and anodic sweeps in the range of 1–1.5 V vs. RHE increased with cycle numbers, indicating an increase in double-layer capacitance. In the averaged linear sweep voltammograms shown in Fig. 3b, the overpotential at 0.02 mA cm−2 slightly decreased from 1.72 to 1.71 V after 1200 CV cycles. Similarly, as shown in the Tafel plots (Fig. 3c), the Tafel slope increased slightly from 71.8 mV dec−1 to 85.7 mV dec−1 after 1200 cycles. The above results revealed that the catalytic activity of Zn0.6Co0.4Al2O4 was kinetically lowered. The decrease in the size of the larger semicircle in the low-frequency region of the Nyquist plot (Fig. 3d) also provides evidence of the increase in double-layer capacitance and decrease in resistance during the adsorption/desorption process of OER intermediates, which may explain the decrease in the overpotential after 1200 cycles. These electrochemical results demonstrate that a small structural change occurred on the catalyst surface after 1200 CV cycles.
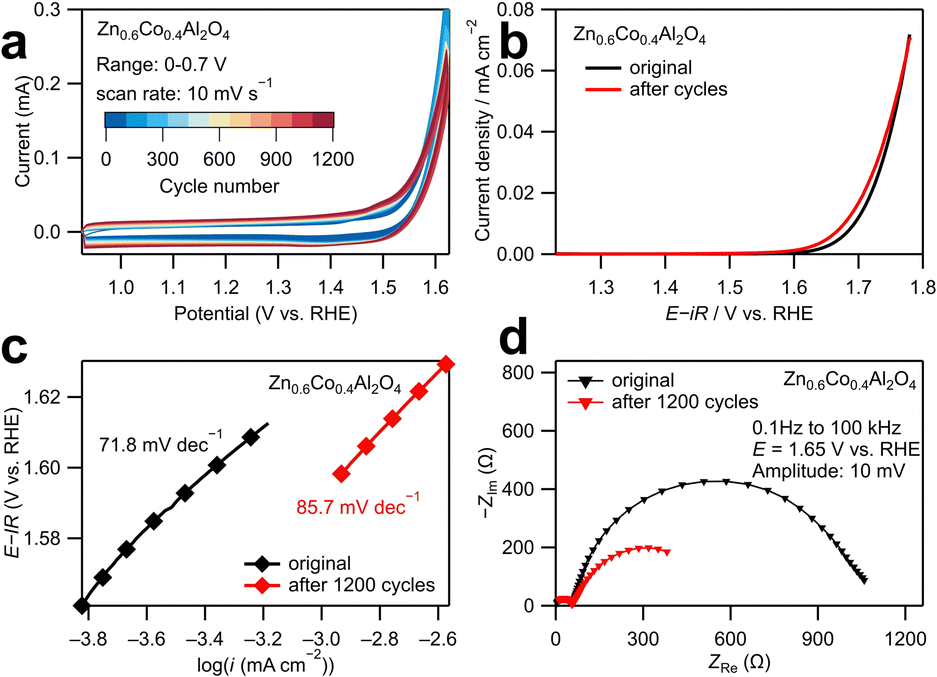 | ||
| Fig. 3 (a) Cyclic voltammograms of 1200 cycles for Zn0.6Co0.4Al2O4 in the range of 0.926–1.626 V vs. RHE with a scan rate of 10 mV s−1. (b) Averaged linear sweep voltammograms. (c) Tafel plots and (d) Nyquist plots for Zn0.6Co0.4Al2O4 before and after the 1200 cycles. | ||
To investigate the change in the crystal structure of the catalysts after OER tests, XAS around Co K-edge and X-ray photoelectron spectroscopy (XPS) analyses were conducted as displayed in Fig. 4a–e. The XANES and EXAFS spectra in Fig. 4a and b showed that the spectral curve after 1200 cycles almost totally overlapped with that before the electrochemical tests, indicating that the spinel structure was maintained, and the atomic occupancy and valence states of Co ions did not change after the electrochemical tests. Fig. 4c and d show the deconvoluted XPS spectral peaks corresponding to Co 2p and O 1s. The wide-scan spectra are shown in Fig. S9 (ESI†), and the fitting results are listed in Table S6 (ESI†). In Fig. 4c, the peaks are only contributed by Co2+ ions and their satellite effect, irrespective of the electrochemical tests; this indicates that only Co2+ exists in the remaining catalysts.26–28 The O 1s peak was deconvoluted into three subpeaks: oxygen from the lattice (OL), oxygen near the defects (OD), and oxygen adsorbed on the surface (OA).28 The significant increase in the OD ratio implies that numerous defects were generated on the surface after the electrochemical test. In addition, the possibility of cation dissolution on the catalyst surface was examined by the elemental ratio analysis from the XPS spectra, as shown in Fig. 4e. The amount of Co ions was set to one to compare the relative amounts of Zn and Al ions. After 1200 cycles, the relative amount of Al ions did not change, indicating a high stability. This high stability of Al ions contradicts their amphoteric property (soluble in both acids and bases). However, according to Wu et al.'s study,16 the high stability of CoAl2O4 during OER in alkaline solution is attributed to the O 2p band center, which is far from the Fermi level. To prove their assumption, they adjusted the O 2p band center close to the Fermi level by partially substituting Al with Fe ions, leading to dissolution of Al ions and surface reconstruction from spinel oxide to oxyhydroxide species.16 In contrast, the relative amount of Zn ions decreased from 1.21 to 0.98, suggesting that Zn ion dissolution evidently occurred during the OER tests. The dissolution of Zn ions into the electrolyte was also confirmed by the cation concentration in the electrolyte after 1200 cycles using ICP-AES, as displayed in Table S7 (ESI†). Because approximately 20% of Zn ions were dissolved, which is considerably larger than the ratio of Zn ions that occupy the octahedral sites (approximately 10%), this implies that Zn ions were primarily dissolved from the tetrahedral sites. The dissolution of Zn ions generated cation vacancies at the tetrahedral sites in Zn0.6Co0.4Al2O4, which improved the OER catalytic activity.
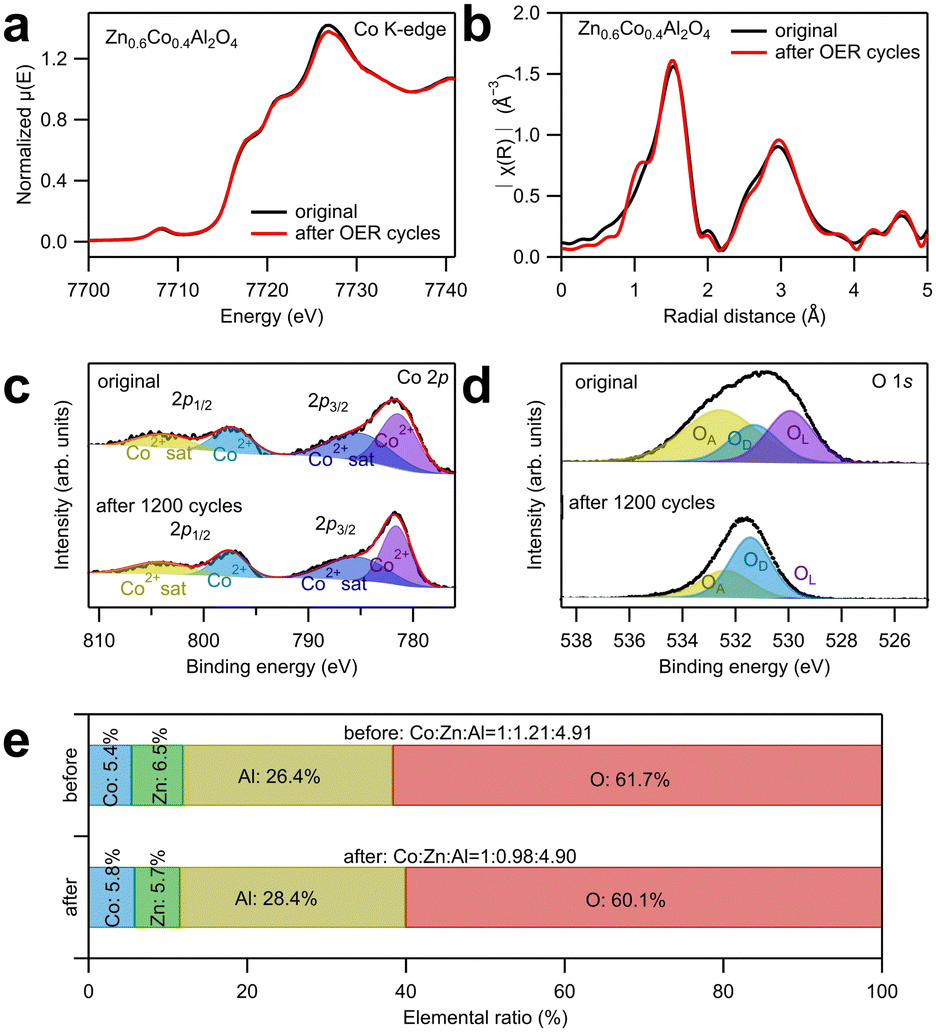 | ||
| Fig. 4 (a) X-Ray absorption near edge structure (XANES) and (b) extended X-ray absorption fine structure (EXAFS) spectra around Co K-edge, and XPS spectra in (c) Co 2p region and (d) O 1s region for Zn0.6Co0.4Al2O4 before and after the electrochemical tests for 1200 cycles. (e) Atomic concentration ratios of elements in Zn0.6Co0.4Al2O4 before and after the 1200 CV cycles. | ||
Herein, the effects of cation vacancies at the tetrahedral sites in Co spinel oxides for OER catalysis are discussed further. The Tafel plots for Zn0.6Co0.4Al2O4 and CoAl2O4 are displayed with that for a reference catalyst Co3O4 in Fig. 5a. A comparison of the logarithmic values of current densities in the x-axis revealed that Co3O4 shows a larger current density than CoAl2O4 and Zn0.6Co0.4Al2O4 at the same potential. This indicates that the Co ions at the octahedral sites play a more active role than those at the tetrahedral sites in OER catalysis. However, this does not imply that modification at the tetrahedral sites is ineffective in improving the OER activity.
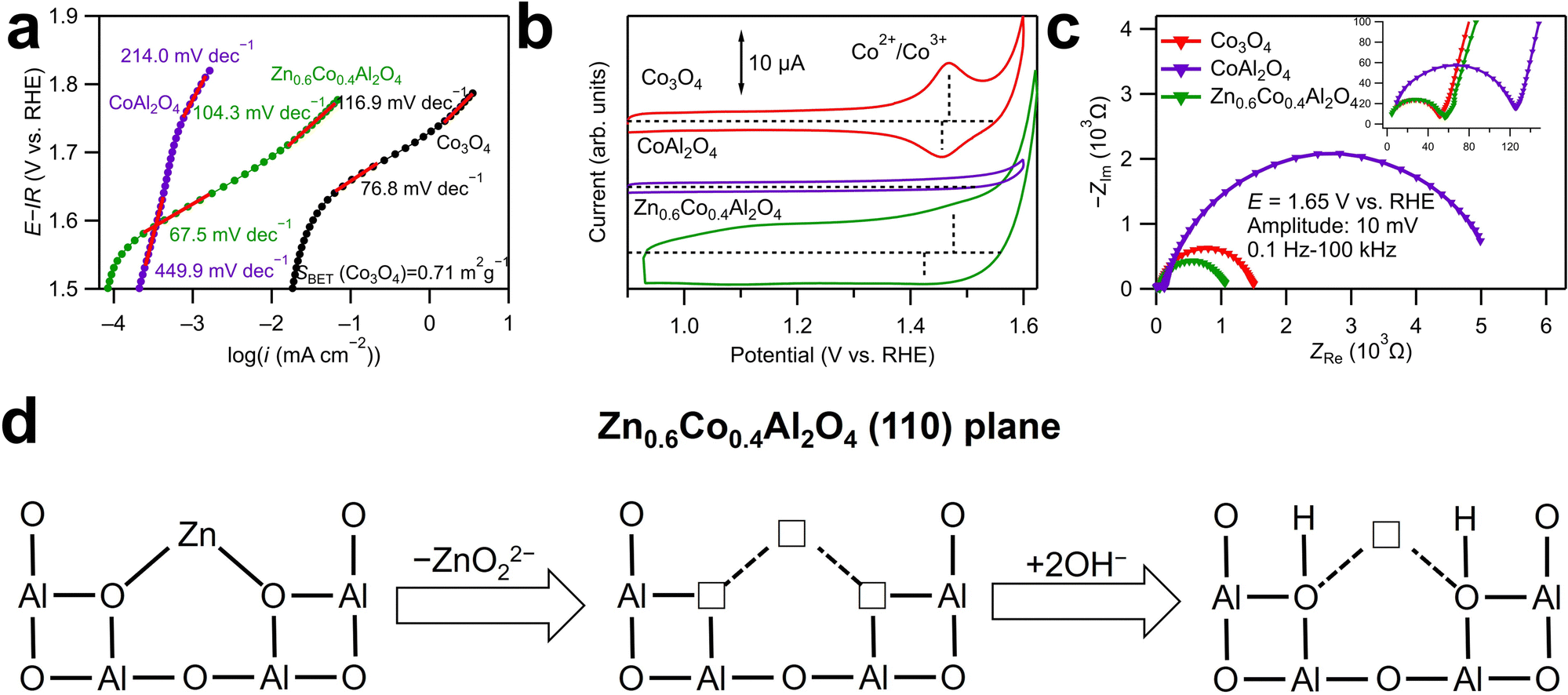 | ||
| Fig. 5 (a) Tafel plots for Zn0.6Co0.4Al2O4, CoAl2O4 and Co3O4. The currents for Co3O4 were standardized by the determined BET surface area. (b) Unaveraged cyclic voltammograms for Co3O4, CoAl2O4 and Zn0.6Co0.4Al2O4 in 0.1 M KOH aqueous solution with a scan rate of 10 mV s−1. (c) Nyquist plots for Co3O4, Zn0.6Co0.4Al2O4 and CoAl2O4 measured using EIS at 1.65 V vs. RHE. (d) Illustration of charge compensation mechanism for Zn dissolution by OH− adsorption. | ||
During the anodic sweeps, Co2+ ions at the tetrahedral sites in Co spinel oxides are oxidized into Co3+ ions at approximately 1.4–1.5 V. In the cyclic voltammograms presented in Fig. 5b, only Co3O4 shows a clear Co redox peak (Co2+ to Co3+) at approximately 1.5 V, whereas the Co oxidation peak for CoAl2O4 cannot be identified and that for Zn0.6Co0.4Al2O4 is indistinct. The difference in the redox peaks was because of the Al3+ ions at the octahedral sites, which stabilized the Co2+ ions at the tetrahedral sites. Consequently, CoAl2O4 exhibited an extremely large Tafel slope (449.9 mV dec−1) at 1.55–1.65 V vs. RHE, whereas those for Zn0.6Co0.4Al2O4 (67.5 mV dec−1) and Co3O4 (76.8 mV dec−1) were smaller. The smallest Tafel slope for Zn0.6Co0.4Al2O4 implies that even Co2+ ions cannot be oxidized as easily as that in Co3O4, Moreover, Zn dissolution in Zn0.6Co0.4Al2O4 facilitates OER kinetics by generating cation vacancies. The smallest semicircle in low-frequency range in Fig. 5c indicates the fastest adsorption/desorption rate of OER intermediates. In addition, the largest double layer capacitance of Zn0.6Co0.4Al2O4 can be confirmed in Fig. S10 (ESI†) compared to those of Co3O4 and CoAl2O4. In addition, Fig. 5a shows that when the potential increased over 1.7 V, the Tafel slope of CoAl2O4 decreased to 214 mV dec−1, which may be because Co2+ to Co3+ oxidation can occur at the high potential, and the increased Co3+ ions enhanced OER kinetics. In contrast, Tafel slopes both increased with the increase in potential for Co3O4 (116.9 mV dec−1) and Zn0.6Co0.4Al2O4 (104.3 mV dec−1), possibly resulting from a different rate-determining step on the surface of Co4+ ions, which are generated from Co3+ oxidation.
Fig. 5d illustrates the charge compensation mechanism for the cation vacancies due to Zn dissolution at the tetrahedral sites in Zn0.6Co0.4Al2O4. Generally, when cation vacancies are generated, the charges may be compensated through two possible routes: increases in the valence states of other cations or desorption of negatively charged ions. For the spinel oxides examined in this study, the tetrahedral cations bonded with four surrounding oxygen ions and four octahedral Al3+ ions. Charge compensation by Al3+ ions is difficult as Al3+ has no variable valence states. In contrast, Zn ions dissolve into the solution together with adjacent O2− ions, leading to very unstable oxygen vacancies. In the strong alkaline electrolyte, there are plenty of OH− ions and they will fill in the oxygen vacancies immediately. The existence of excessive adsorbed OH− and oxygen vacancies is also supported by XPS analysis of O 1s, as shown in Fig. 4d. During the anodic sweeps, excessive adsorption of OH− facilitates Co oxidation and subsequent OER catalysis, which is also evident from the ambiguous Co oxidation peak of Zn0.6Co0.4Al2O4 in Fig. 5b.16 In this case, generation of cation vacancies leads to excessive OH− adsorption, further resulting in enhanced OER activity. Consequently, Zn0.6Co0.4Al2O4 with less Co ions shows higher catalytic activity than CoAl2O4 and comparable OER kinetics (Tafel slopes) with Co3O4.
Experimental
Synthesis of ZnxCo1−xAl2O4
Synthesis of the spinel oxides follows a typical sol–gel method.16 Herein, we consider the example of synthesizing CoAl2O4. All the reagents in the method were purchased from Nacalai Tesque Inc. (Kyoto, Japan) without further purification. First, 0.01 mol Co(NO3)2·6H2O and 0.02 mol Al(NO3)3·9H2O were weighed and dissolved in 20 mL deionized water. Subsequently, 0.1 mol anhydrous citric acid and 2 mL 60 wt% concentrated nitric acid were added to the solution, followed by 20 mL ethylene glycol. The solution was stirred at 160 °C for 12 h. After the solution transformed into a gel, the mixture was transferred to a muffle furnace and heated at 800 °C for 6 h. The obtained foam-like material was ground thoroughly to obtain the final product. A part of or the entirety of Co(NO3)2·6H2O was replaced by Zn(NO3)2·6H2O with different stoichiometric ratios for synthesizing other spinel oxides. Co3O4 (Practical Grade) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and used as the reference catalyst without any purification.Electrochemical characterization
A rotating glassy carbon disk electrode (4 mm in diameter) was used as the working electrode for electrochemical tests. A Hg/HgO (0.1 M KOH) electrode and a Pt wire were used as the reference and counter electrodes, respectively. Prior to measurement, the catalysts were used to prepare catalyst inks, which were coated onto the disk electrode to modify its surface. Moreover, 0.1 M KOH aqueous solution and proton-type Nafion solution (45% water and 5% mass in a mixture of lower aliphatic alcohols, purchased from Sigma Aldrich) were mixed at a volume ratio of 1:2 to obtain a K+-exchanged Nafion solution. Subsequently, 10 mg of acetylene black, 0.3 mL of the K+-exchanged Nafion solution, and 50 mg of a catalyst were mixed in a 10 mL volumetric flask, followed by filling the flask to 10 mL with tetrahydrofuran (Nacalai Tesque Inc.).
Electrochemical measurements were conducted using a rotating (1600 rpm) ring-disk electrode system (RRDE-3a, BAS Inc., Tokyo, Japan). Before the experiment, O2 was bubbled to prepare an O2-saturated 0.1 M KOH aqueous solution. IR correction was conducted for the linear sweep voltammograms using the resistance determined by the EIS analysis shown in Table S5 in ESI.†29 All the potentials measured versus the Hg/HgO, that is, the electrode E (vs. Hg/HgO) were converted to the potential values versus the RHE, that is, E (vs. RHE) using the following equation:
| E(vs. RHE) = E(vs. Hg/HgO) + 0.926 V |
Material characterization
The morphologies of the samples were observed by SEM with an integrated energy-dispersive X-ray spectroscopy analyzer (JSM-6010 LA, JEOL Ltd., Tokyo, Japan). The structures of the products were analyzed by SXRD at the beamline BL02B2, SPring-8 (Hyogo, Japan). Rietveld refinement of the SXRD patterns was conducted using the RIETAN-FP program (© MateriApps).30,31 XAS spectra of the as-synthesized catalyst powders were acquired in the transmission mode at the beamline BL14B2, SPring-8 with a Si(111) double-crystal monochromator. The obtained spectra were calibrated by the XAS spectra of a Co foil. The XANES and EXAFS spectral data were processed using Athena and Artemis, respectively, with an IFEFFIT software package.32 The surface area of the samples was evaluated by BET analysis of the N2 adsorption/desorption isotherms obtained by a surface area and pore size distribution analyzer (BELSORP-max II, MicrotracBEL Corp., Osaka, Japan). The XAS spectrum of Zn0.6Co0.4Al2O4 after 1200 cycles of CV test was measured in the fluorescence mode, where the sample was loaded onto a carbon paper (SIGRACET® Gas Diffusion Media, Type: GDL10BC, SGL carbon, Germany), and the modified carbon paper was directly used as the working electrode in the CV test. The XPS analysis was conducted using a PHI Quantera (ULVAC-PHI Inc., Kanagawa, Japan) for Zn0.6Co0.4Al2O4 before and after 1200 cycles of the CV test using the following parameters (photoemission angle: 45°, X-ray energy of Al Kα: 1486.6 eV, pass energy: 55 eV, energy step: 0.05 eV). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was conducted to detect the cation concentration in the electrolyte for Zn0.6Co0.4Al2O4 before and after 1200 cycles of the CV test using an ICP-AES equipment (SPS3500, Hitachi, Tokyo, Japan).Conclusions
In this study, a series of Zn- and Al-substituted Co spinel oxides, ZnxCo1−xAl2O4 (x = 0, 0.2, 0.4, 0.6, 0.8, and 1), were synthesized and their catalytic activities on OER were evaluated in 0.1 M KOH aqueous solution. According to the results from Rietveld refinement of SXRD patterns and XAS spectra, Zn and Co ions primarily occupy the tetrahedral sites with Al ions at the octahedral sites. Zn0.6Co0.4Al2O4 shows the highest OER catalytic activity among all the samples. During the 1200 OER cycles, numerous Zn ions in Zn0.6Co0.4Al2O4 dissolved, whereas the Co and Al ions were preserved, and the spinel oxide structure was maintained. Although the intrinsic activity of Zn0.6Co0.4Al2O4 is inferior to that of Co3O4, the kinetic rate of Zn0.6Co0.4Al2O4 was comparable to that of Co3O4. This was attributed to an increased adsorption of OH− ions from cation vacancies at the tetrahedral sites generated by Zn dissolution. Thus, the present study clarified the mechanism by which cation vacancies enhance OER activity, which can contribute to future catalyst design.Author contributions
W. Liu: conceptualization, formal analysis, investigation, visualization, writing – original draft; M. Kamiko: formal analysis, writing – review & editing; I. Yamada: funding acquisition, resources, writing – review & editing; S. Yagi: funding acquisition, resources, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Toru H. Okabe and Dr. Takanari Ouchi for their support in ICP-AES measurements. This research was financially supported by a Grant-in-Aid for the University of Tokyo Excellent Young Researcher and a Grant-in-Aid for Scientific Research (20H02825, 22H04497) commissioned by the Japan Society for the Promotion of Science and JST SPRING (JPMJSP2108). Synchrotron X-ray diffraction data were obtained at the BL02B2 beamline (Proposal No. 2020A0583) and BL14B2 beamline (Proposal No. 2021B1902) at SPring-8 with support from the Japan Synchrotron Radiation Research Institute.Notes and references
- T. Schuler, T. Kimura, T. J. Schmidt and F. N. Buchi, Energy Environ. Sci., 2020, 13, 2153–2166 RSC.
- J. M. Hu, J. Q. Zhang and C. N. Cao, Int. J. Hydrogen Energy, 2004, 29, 791–797 CrossRef CAS.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1636–1641 CrossRef CAS PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- S. Hirai, S. Yagi, A. Seno, M. Fujioka, T. Ohno and T. Matsuda, RSC Adv., 2016, 6, 2019–2023 RSC.
- I. Yamada, A. Takamatsu, K. Asai, T. Shirakawa, H. Ohzuku, A. Seno, T. Uchimura, H. Fujii, S. Kawaguchi, K. Wada, H. Ikeno and S. Yagi, J. Phys. Chem. C, 2018, 122, 27885–27892 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- E. Cossar, F. Murphy and E. A. Baranova, J. Chem. Technol. Biotechnol., 2022, 97(7), 1611–1624 CrossRef CAS.
- Y. Li, F. Li, X. Meng, S. Li, J. Zeng and Y. Chen, ACS Catal., 2018, 8(3), 1913–1920 CrossRef CAS.
- Z. Xiao, Y. Huang, C. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142(28), 12087–12095 CrossRef CAS PubMed.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24(18), 3567–3573 CrossRef CAS.
- H. Zeng, M. Oubla, X. Zhong, N. Alonso-Vante, F. Du, Y. Xie, Y. Huang and J. Ma, Appl. Catal., 2021, 281, 119535 CrossRef CAS.
- Y. Li, L. Zhang, J. Peng, W. Zhang and K. Peng, J. Power Sources, 2019, 433, 226704 CrossRef CAS.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29(23), 1606800 CrossRef PubMed.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef PubMed.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- W. Liu, M. Kamiko, I. Yamada and S. Yagi, RSC Adv., 2022, 12, 8731–8736 RSC.
- W. Liu, J. Han, I. Yamada and S. Yagi, J. Catal., 2021, 394, 50–57 CrossRef CAS.
- X. Wang, T. Ouyang, L. Wang, J. Zhong and Z. Liu, Angew. Chem., 2020, 132, 6554–6561 ( Angew. Chem., Int. Ed. , 2020 , 59 , 6492–6499 ) CrossRef.
- J. Popovic, E. Tkalcec, B. Grzeta, S. Kurajica and B. Rakvin, Am. Mineral., 2009, 94(5–6), 771–776 CrossRef CAS.
- F. Bosi, U. Halenius, V. Dlppolito and G. B. Andreozzi, Am. Mineral., 2012, 97(11–12), 1834–1840 CrossRef CAS.
- C. Meng, Y. Gao, X. Chen, Y. Li, M. Lin and Y. Zhou, ACS Sustainable Chem. Eng., 2019, 7(21), 18055–18060 CrossRef CAS.
- D. Antipin and M. Risch, arXiv preprint, 2021, 2112, 04747 Search PubMed.
- F. R. Costa, D. V. Franco and L. M. Da Silva, Electrochim. Acta, 2013, 90, 332–343 CrossRef CAS.
- C. McCrory, S. Jung, J. Peters and T. Jaramillo, J. Am. Chem. Soc., 2013, 135(45), 16977–16987 CrossRef CAS PubMed.
- X. Duan, M. Pan, F. Yu and D. Yuan, J. Alloys Compd., 2011, 509, 1079–1083 CrossRef CAS.
- X. Peng, J. Cheng, J. Yuan, N. Jin, J. Kang, Y. Hou and Q. Zhang, Adv. Appl. Ceram., 2018, 117(5), 303–311 CrossRef CAS.
- M. Zhu, J. Yang, X. Duan, D. Zhang, S. Wang, B. Yuan and M. Fu, Chem. Eng. Sci., 2020, 397, 125339 CrossRef CAS.
- I. Yamada, H. Fujii, A. Takamatsu, H. Ikeno, K. Wada, H. Tsukasaki, S. Kawaguchi, S. Mori and S. Yagi, Adv. Mater., 2017, 29(4), 1603004 CrossRef PubMed.
- F. Izumi and T. Ikeda, Mater. Sci. Forum, 2000, 321–324, 198–205 CAS.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15–20 CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00729k |
| This journal is © The Royal Society of Chemistry 2022 |