
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-steroidal anti-inflammatory drugs: recent advances in the use of synthetic COX-2 inhibitors
Mohsen
Ahmadi
*a,
Sander
Bekeschus
a,
Klaus-Dieter
Weltmann
ab,
Thomas
von Woedtke
 abc and
Kristian
Wende
*a
abc and
Kristian
Wende
*a
aLeibniz Institute for Plasma Science and Technology (INP Greifswald), Center for Innovation Competence (ZIK) plasmatis, Felix-Hausdorff-Straße 2, 17489 Greifswald, Germany. E-mail: mohsen.ahmadi@inp-greifswald.de; kristian.wende@inp-greifswald.de
bLeibniz Institute for Plasma Science and Technology (INP Greifswald), Felix-Hausdorff-Straße 2, 17489 Greifswald, Germany
cUniversity Medicine Greifswald, Institute for Hygiene and Environmental Medicine, Walther-Rathenau-Straße 49A, 17489, Germany
First published on 14th February 2022
Abstract
Cyclooxygenase (COX) enzymes comprise COX-1 and COX-2 isoforms and are responsible for prostaglandin production. Prostaglandins have critical roles in the inflammation pathway and must be controlled by administration of selective nonsteroidal anti-inflammatory drugs (NSAIDs). Selective COX-2 inhibitors have been among the most used NSAIDs during the ongoing coronavirus 2019 pandemic because they reduce pain and protect against inflammation-related diseases. In this framework, the mechanism of action of both COX isoforms (particularly COX-2) as inflammation mediators must be reviewed. Moreover, proinflammatory cytokines such as tumor necrosis factor-α and interleukin (IL)-6, IL-1β, and IL-8 must be highlighted due to their major participation in upregulation of the inflammatory reaction. Structural and functional analyses of selective COX-2 inhibitors within the active-site cavity of COXs could enable introduction of lead structures with higher selectivity and potency against inflammation with fewer adverse effects. This review focuses on the biological activity of recently discovered synthetic COX-2, dual COX-2/lipoxygenase, and COX-2/soluble epoxide hydrolase hybrid inhibitors based primarily on the active motifs of related US Food and Drug Administration-approved drugs. These new agents could provide several advantages with regard to anti-inflammatory activity, gastrointestinal protection, and a safer profile compared with those of the NSAIDs celecoxib, valdecoxib, and rofecoxib.
 Mohsen Ahmadi | Mohsen Ahmadi received his Ph.D. (Dr. rer. nat.) in Bioinorganic Chemistry from the University of Greifswald (Germany). His research activities are concerned with developing and mimicking the chemical synthesis of active sites of molybdoenzymes to address the molybdenum cofactor deficiency. He then joined the group of Dr. Kristian Wende at the Leibniz Institute for Plasma Science and Technology (INP), where he worked on drug design and development to treat inflammatory diseases. His main research interests concern drug and prodrug discovery and implementation of artificial enzyme mimetic systems to develop novel therapeutic agents. |
 Sander Bekeschus | Sander Bekeschus is a human biologist trained in immunology with a particular focus on plasma and redox medicine. After short-term scientific missions in New Zealand and the USA, he obtained his Ph.D. from the University of Greifswald (Germany) before starting a third-party funded junior research group at the Leibniz Institute for Plasma Science and Technology (INP) in 2016. Successful results led to the consolidation into a permanent research group at INP in 2021. His main interests are sophisticated cellular and translational research models and enabling the engagement of the immune system using medical plasmas in dermatology, oncology, and redox medicine. |
 Klaus-Dieter Weltmann | Klaus-Dieter Weltmann received his Ph.D. in applied physics working on nonlinear dynamics in low-temperature plasmas from the University of Greifswald (Germany). Since 2003 he has been Chairman of the Board and Scientific Director of the Leibniz Institute for Plasma Science and Technology (INP) and Professor at Greifswald University. His research topics are plasma source development, plasma medicine, modeling, and applications of atmospheric pressure plasmas. |
 Thomas von Woedtke | Thomas von Woedtke studied Pharmacy at the University of Greifswald, Germany. He received the doctoral degree in 1995 in Pharmaceutical Technology, followed by a habilitation degree in 2007. Since 2008 he has been Research Program Manager in Plasma Medicine at the Leibniz Institute for Plasma Science and Technology (INP) Greifswald, Germany, and since 2020 a member of the board of the INP. In addition, he has held a professorship for Plasma Medicine at Greifswald University Medicine since 2011. |
 Kristian Wende | Kristian Wende received his MSc degree (natural compound chemistry) in 1998 and his Ph.D. degree (natural compound analytics and toxicology) in 2003 from the University of Greifswald (Germany). Since 2017 he has been a research group leader at the Leibniz Institute for Plasma Science and Technology (INP). His current research comprises analytical and biophysical methods to determine biomolecule oxidation in the context of redox biology. |
Introduction
Coronavirus disease 2019 (COVID-19) is caused by an infection with severe acute respiratory syndrome coronavirus (SARS-CoV-2). COVID-19 was first reported in Wuhan (China) in December 2019. As of July 2021, 208.9 million people have been infected with SARS-CoV-2, and 4.3 million people have died worldwide. The genome sequence of SARS-CoV-2 shows high similarity with that of SARS-CoV and Middle East respiratory syndrome coronavirus,1,2 promising a way to the fast identification of potential prophylactic and therapeutic intervention drugs that ultimately did not pay off in full. A common and early-identified condition connected to (severe) infection with SARS-CoV-2 is significant inflammation, and various cyclooxygenase (COX) inhibitors have been applied during the COVID-19 pandemic with mixed success.3The spike protein (glycoprotein) of the surface of the enveloped virus binds to the host-cell receptor and mediates virus entry. This is achieved through angiotensin-converting enzyme (ACE)-2 receptors of the host and cleavage of the spike protein by different cellular proteases depending on the viral strain and cell type.4 If SARS-CoV-2 comes into contact with a host cell, it can release the nucleocapsid into the host and produce viral genetic material instead of regular host products. SARS-CoV-2 binds to the COX-2 promoter and leads to its overexpression, and boosts prostaglandin (PG) production as a significant part of an overwhelming inflammatory response that paves the way to a dangerous cytokine storm.3 To maintain control of the inflammation and for pain relief, non-steroidal anti-inflammatory drugs (NSAIDs) were a predominant choice. In addition, the inhibition of cyclooxygenase affects COVID-19 pathogenesis by altering the expression of the initial virus receptor ACE2, reducing the risk of infection and SARS-CoV-2 replication in host cells, and through regulation of the immune response to SARS-CoV-2.3 Robb et al.5 noted that the elevated expression of proinflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-8 during the cytokine storm worsens COVID-19 severity. Therefore, inhibition of the cyclooxygenase activity, especially the COX-2, and the subsequently alleviated expression of pro-inflammatory cytokines by (selective) NSAIDs interferes with the COVID-19 pathogenesis and reduces the severity of the clinical symptoms.6 COX-2 is an inducible enzyme and has been focused upon as a drug target to reduce pain through selective inhibition. The first generation of COX-2 inhibitors, rofecoxib (Vioxx™) and valdecoxib (Bextra™), were withdrawn from the market7 due to severe side-effects, such as cardiovascular disease, risk of stroke, and cardiac arrest.8 US Food and Drug Administration-approved drugs, such as celecoxib (Celebrex™: side-effect boxed warning, but available in the USA), have potent selectivity towards COX-2. Non-selective NSAIDs such as ibuprofen, naproxen, nimesulide, diclofenac, and sulindac cause severe side effects.9 Gastrointestinal bleeding, peptic ulcers, duodenal ulcers, hypertension, dyspepsia, and stroke are widespread side effects that are major challenges in pain management.10 COX-1 is expressed mainly on platelets, in the kidneys, gastric mucosa, and lungs. COX-2 shows a low constitutive expression in the brain, kidney, GI tract, and thymus that is induced further by inflammatory stimuli.11 Elevated expression of COX-2 reduces the pain threshold through the production of PG and in the long run paves the way for inflammation-related diseases.11 A complex regulatory pathway controls inflammation, with the two (main) COX isoforms playing a modulatory and in part controversial role. A reduction of the prostaglandin PGG2 and PGH2 synthesis via COX1/2 inhibition by non-selective NSAIDs and the subsequent reduction of mucosa function paves the way for gastrointestinal tract damage.12 On the other hand, COX-2 plays an essential part in regulating the renal function.13 Hence, in patients carrying a risk of renal ischemia, liver cirrhosis, renal insufficiency, cardiovascular disorders, and congestive heart failure, COX-2 inhibitors show severe side effects, and vigilance is required.9 When prostaglandin synthesis is blocked by COX inhibition, arachidonic acid (AA) is metabolized by the alternative lipoxygenase (LOX) pathway. The generated leukotrienes are associated with asthma and allergic reactions and must be considered for the safety profile of NSAIDs.14 Given the significant pro-inflammatory situation in many patients, selective COX-2 inhibitors have been among the most widely used drugs during the COVID-19 pandemic, indicating the high relevance of safe and efficient drugs and the necessity of further drug development. The current study will primarily focus on recent compounds/drugs that inhibit COX-2 activity from structural and mechanistic viewpoints, provide insight into their structure–activity relationships and outline future research needs for medicinal chemists and biologists.
Inflammation pathways
The cleavage of arachidonic acid (AA) from phospholipids through phospholipase A2 fuels three major inflammation pathways (Fig. 1): cytochrome P450 monooxygenase, lipoxygenase (LOX), and cyclooxygenases (COXs), which is the major inflammation pathway in mammals.14 Cyclooxygenases are bifunctional enzymes converting long-chain (C20⋯C24) polyunsaturated monocarboxylic acid into oxidized cyclic products, mainly prostaglandins (PGs) and thromboxanes (TXs).11 This action occurs via the introduction of two oxygen atoms into the C–H bonds of AA to form a bicyclic peroxide intermediate, PGG2, which is reduced rapidly to PGH2 and then produces the PGs E2, D2, I2, F2, and TXA2 as a response to stimuli (Fig. 1).11 Various signal molecules, such as interleukins, TNF-α, lipopolysaccharide, transforming growth factor-α, interferon-γ, platelet-activating factor, endothelin-1, forskolin, retinoic acid, and AA itself, induce COX-2 expression,15,16 and their respective functions of signal molecules are not fully understood. Leukotrienes (Leu), epoxyeicosatrienoic acids (EETs), and hydroxyeicosatetraenoic acids (HETEs) are also produced from AA metabolism by distinct enzymatic pathways via LOX and soluble epoxide hydrolase (sEH), which have essential roles in inflammation-associated signaling pathways and allergic diseases. For instance, it has been reported that selective COX-2 inhibitors can reduce the level of antithrombotic PGD2 during cardiovascular events.11 Proinflammatory cytokines such as TNF-α and IL-6 are related indirectly to inflammation duration, which induces COX-2 expression in a cascade of production of PGs and IL-6.17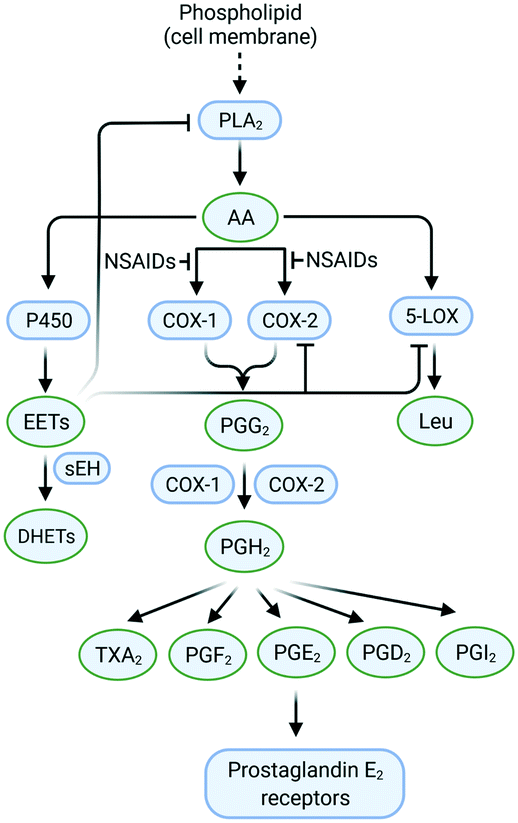 | ||
| Fig. 1 Inflammation pathways and inhibition by targeting of the enzymes COX-2, 5-LOX, and sEH. Created with BioRender.com. | ||
The chemical transformation of arachidonic acid is illustrated in Fig. 2. AA as a natural substrate diffuses into the active-site cavity (ASC) to start the catalytic conversion to PGG2. Tyrosine (Tyr)385 is located in the vicinity of a heme cofactor and is essential for hydrogen (H) atom abstraction according to the redox states of the iron center. The transformation into PGH2 is catalyzed by a peroxygenase. Tyr385 is also necessary for the abstraction of a second H atom. PG production in diverse processes in human tissues leads to various regulations of biological activity that are determined by the type of PG receptors needed for binding.18 PGs have a central role in the regulation of inflammatory processes. PGI2 and PGE2 are involved in vascular permeability, tissue swelling, and secretion of gastric mucus, which are the typical symptoms of inflammation.11
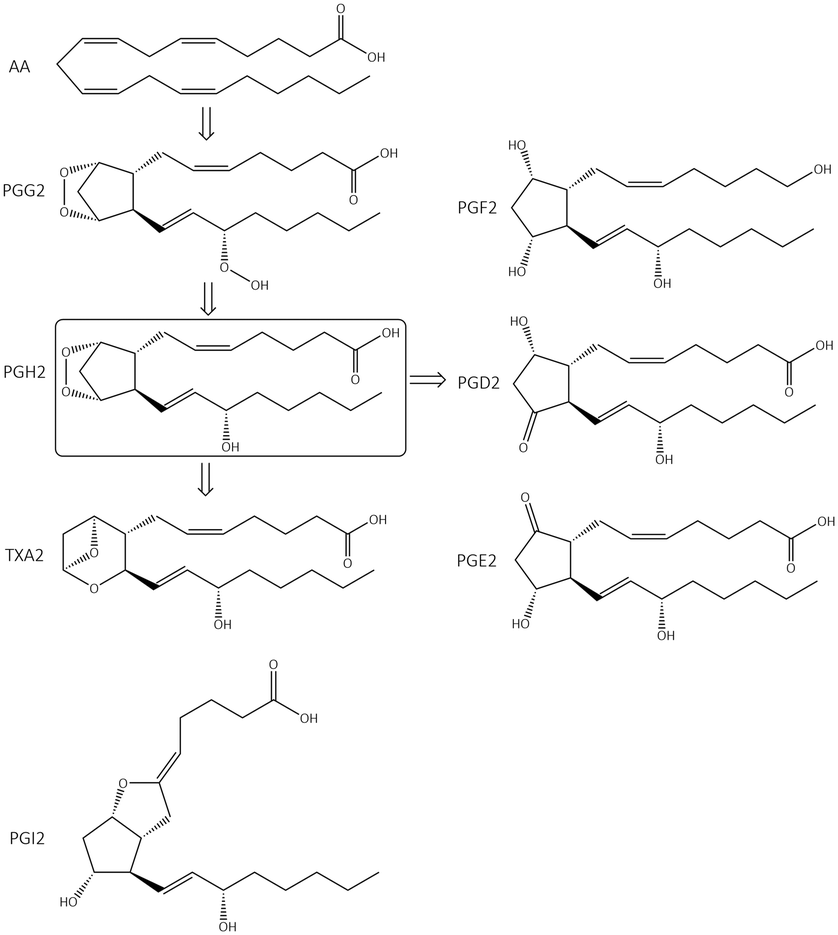 | ||
| Fig. 2 Arachidonic acid (AA) cascade. PG = prostaglandin, TX = thromboxane. | ||
PGD2 regulates bronchoconstriction and impairs innate and adaptive immunity to SARS-CoV-2.19 PGF2 is associated with several reproductive processes.20 TXA2 production by COX-1 is involved in platelet activation/aggregation through their distinct receptors.21 Moreover, PGs have critical roles in diverse regulatory processes, such as body thermoregulation, renal blood flow (renal perfusion), gastrointestinal integrity and the immunomodulatory response.12
Mechanism of action
Oxidoreductase enzymes (EC 1 category) catalyze redox reactions by transferring H atoms and oxygen atoms or electrons from one substance to another.22 COXs are bifunctional oxidoreductase enzymes that catalyze arachidonic acid deoxygenation, and COX-1 and COX-2 are the two (main) isoforms of COX. There is also a splice variant of COX-1 termed COX-3 that was discovered by Simmons and coworkers.23 Both COX1 and COX-2 enzymes are homodimers with 72 kDa subunits comprising three domains: an epidermal growth factor domain, a membrane-binding domain, and an active-site cavity (ASC) for catalysis.24 Both isoforms are internal membrane-bound enzymes with almost identical amino acid sequences in the active site that possess four amphipathic helices with hydrophobic interactions near the ASC. The hydrophobic “pocket” of COX-2 is located at the deep end of the ASC, whereas the ASC entrance is surrounded by the amino acid residues arginine (Arg)120, Tyr355, and glutamic acid (Glu)524, and is known as the “lobby” center (Fig. 3).25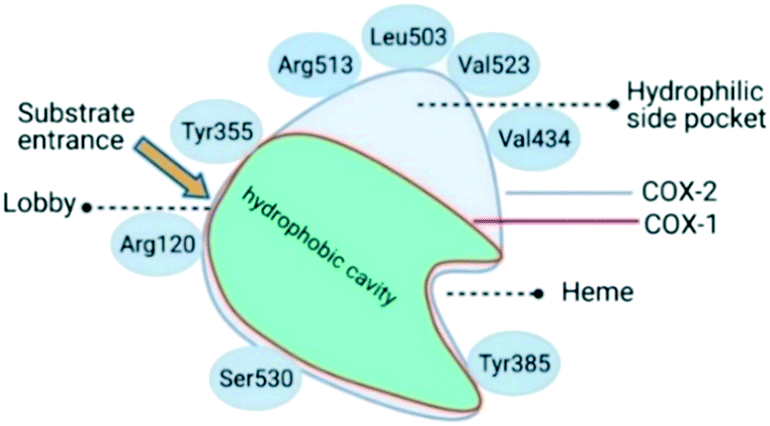 | ||
| Fig. 3 Active-site cavity of COX-1 and COX-2 (schematic). Created with BioRender.com. | ||
The substrate-binding site is located from Arg120 to near Tyr385. COX-2 has a larger binding cavity (∼17%) comprising the amino acids valine (Val)523, Val434, leucine (Leu)503, and Arg513 compared with COX-1, which has the amino acids isoleucine (Ile)523, Ile434, L-phenylalanine (Phe)503, and histidine (His)513 (Fig. 3). The exchange of Val at position 523 with a sterically hindered Ile residue causes an additional sub-pocket so that structurally bulkier inhibitors can fit into the ASC and no longer inhibit COX-1. Arg513 can interact with polar moieties within the ASC of COX-2. Simultaneously, the swing-out of Val434 might increase further access to the active site compared with that of COX-1 with the Ile434 residue. For instance, the interactions of celecoxib within the ASC of COX-1 and COX-2 at the molecular level using the X-ray crystal structure of COX-1 (Protein Databank (PDB) 3KK6, resolution of 2.75 Å)26 and COX-2 (PDB 3LN1, resolution of 2.40 Å)27 are shown in Fig. 4. Celecoxib is involved in different interactions in the ASC of COX isoforms, whereas the pyrazole ring is located in the center of the hydrophobic pocket. The pyrazole ring comprises a CF3 group established with mostly π-stacking interactions with Val116, Val349, Leu359, and alanine (Ala)527 residues in COX-1, whereas the amino acids Arg106, Leu517, Val335, and Ala513 can establish several interactions within the ASC of COX-2. A sulfonamide moiety interacts with glutamine (Gln)192, Leu352, His90, and serine (Ser)516 residues in COX-1, whereas COX-2 interacts with the amino acids Arg499, Leu338, Gln178, and Ser339 through H-bonding. Hence, slight structural differences in the active site modulate the selectivity profile of the compounds. Therefore, analyzing the key amino acids of COX-2 interacting with the inhibitor's chemical structure enables the introduction of potent, selective, and safe COX-2 inhibitors with an improved risk profile.
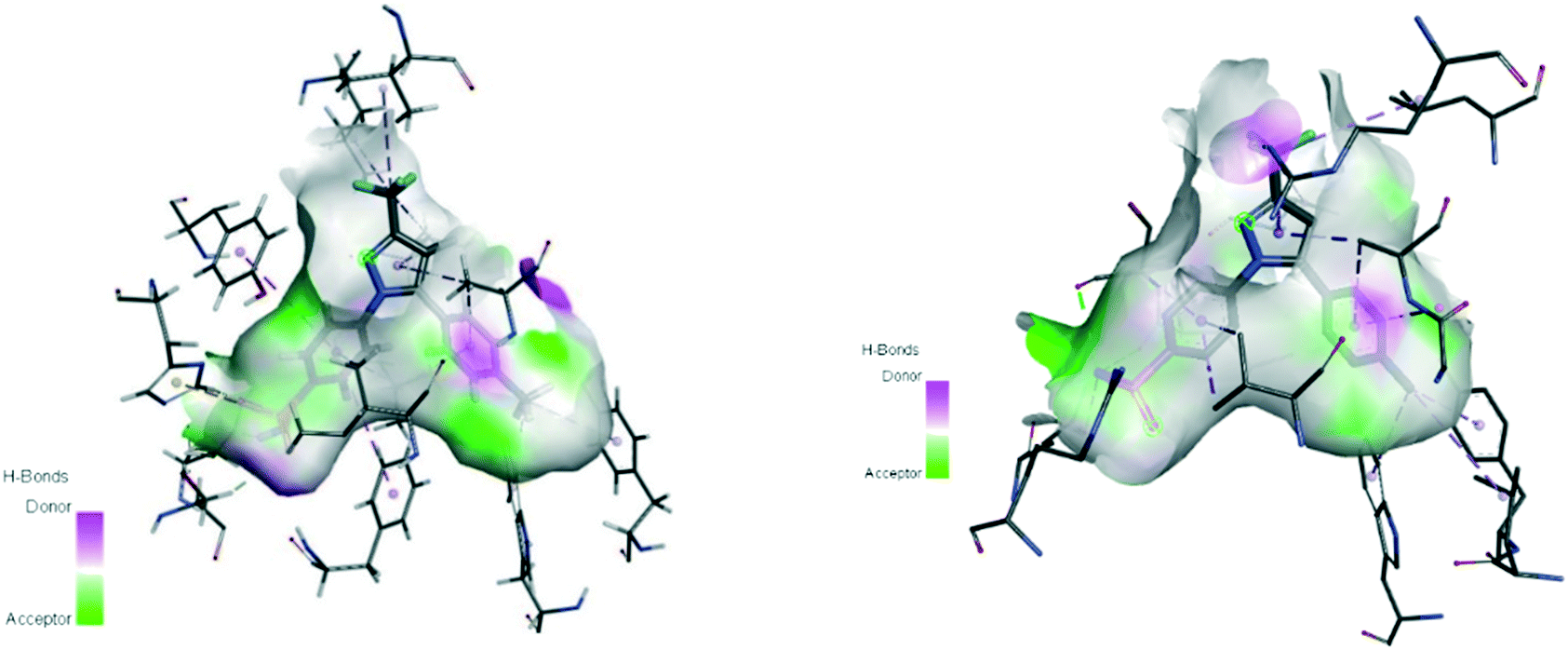 | ||
| Fig. 4 Active site cavity of COX-1 (PDB 3KK6; left) and COX-2 (PDB 3LN1; right) in complex with celecoxib. Figures were generated by Discovery Studio Visualizer v20.1.0.19295. | ||
Classification of COX-2 inhibitors (structural selectivity)
COX-1 is expressed in all tissues, while COX-2 is found in many cell types such as the brain, kidney, and endothelial cells as well as reproductive tissues, inflamed tissues, and tumor cells.28 The relative COX-1/COX-2 selectivity degree by different NSAIDs is assessed in vitro using whole-blood assays and expressed by the ratio of half-maximal inhibitory concentration (IC50) values as depicted in Fig. 5.29 The concentration-dependent relationship of COX-1 and COX-2 will help to assess the relative selectivity and IC50 ratios (selectivity index (SI) = IC50 COX-1/IC50 COX-2).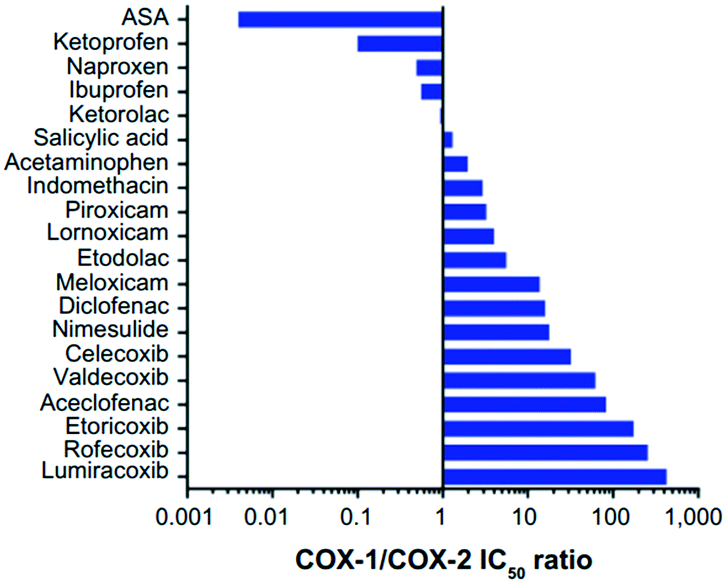 | ||
| Fig. 5 Relative COX-1/COX-2 selectivity of NSAIDs. IC50 >1 = higher selectivity for COX-2, IC50 <1 = higher selectivity for COX-1. ASA = aspirin. Reproduced from ref. 29 with permission from Dove Press Ltd, copyright 2021 (https://creativecommons.org/licenses/by-nc/3.0/).29 | ||
The NSAIDs are divided into three categories based on the kinetics of the interaction with COX-1 and COX-2:30 (i) irreversible inhibitors, e.g., aspirin; (ii) time-dependent and slowly reversible inhibitors, e.g., diclofenac and celecoxib; (iii) freely reversible, e.g., ibuprofen and piroxicam. Acetylsalicylic acid (aspirin) is a COX-1 inhibitor (Fig. 6). Aspirin inhibits COX-1 irreversibly via a salt bridge with Arg120 and interacts with Ser530 by its acetyl group within the ASC.25 Indeed, Ser530 is located close to the COX-1 active site, and the acetyl group blocks the ASC entrance, thereby preventing arachidonic acid access to Tyr385 by its steric hindrance. In contrast, acetylated COX-2 alters the reaction specificity from COX to LOX, resulting in mono-oxygenated 15R-hydroxy-eicosatetraenoic acid (15R-HETE) production.30–32 However, the lowest effective dose of aspirin can permanently inhibit COX-1 and thereby suppress PG and TXA2 production as a promoter of platelet aggregation.33,34
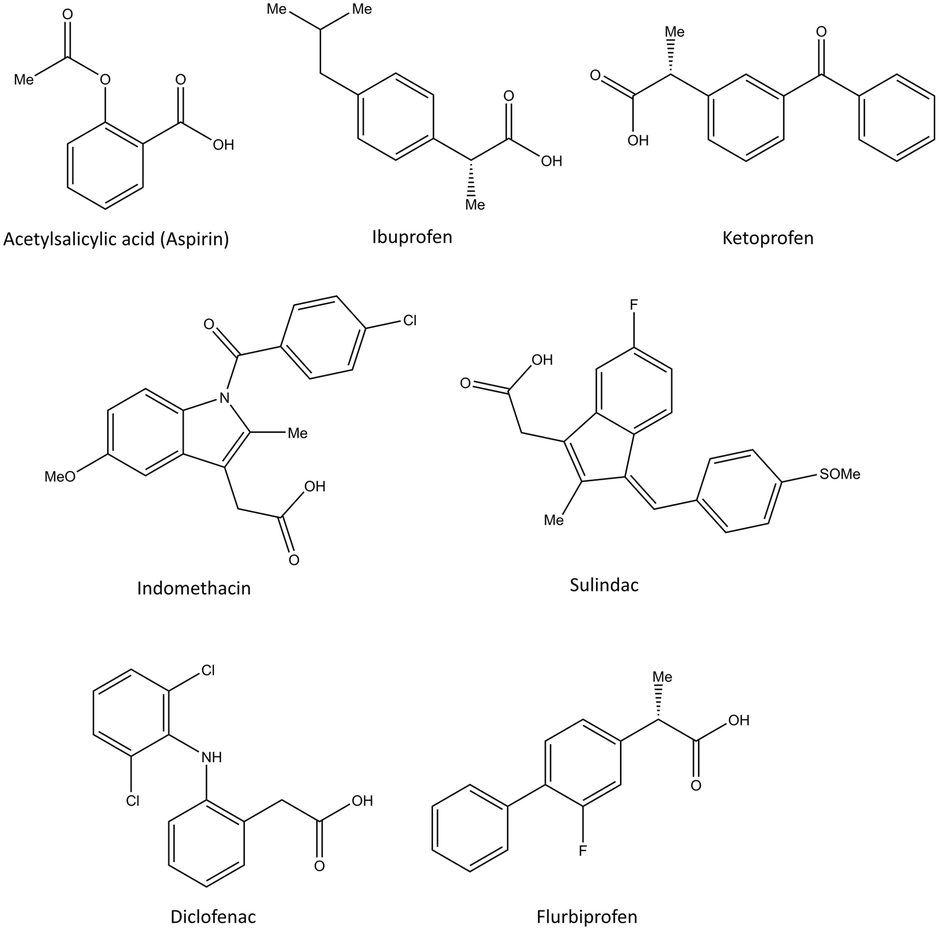 | ||
| Fig. 6 Chemical structure of the slightly selective and reversible COX inhibitors containing carboxylic acid. | ||
Ibuprofen, ketoprofen, flurbiprofen, indomethacin, sulindac, and diclofenac are classical NSAIDs. They are slowly reversible COX inhibitors and are applied widely for anti-inflammation therapy (Fig. 6).25
Ibuprofen can reversibly bind in the ASC of COX-1 and form a salt bridge of its carboxylic acid with Arg120. Indomethacin has been reported to be a time-dependent COX inhibitor that binds to the hydrophobic pocket by the amino acids Ser530, Ala527, Val349, and Leu531. Notably, two chlorine atoms and the carbonyl group of diclofenac are involved in several H-bonds with Ser530 and Tyr385 that slightly enhance selectivity compared with that of other classical NSAIDs.35
Pyrazolidine-3,5-dione (oxicam) derivatives (e.g., piroxicam is an inhibitor of non-carboxylic acid function and contains thiazine and carboxamide moieties) are non-selective COX-2 inhibitors except for meloxicam, which has a slight inhibitory activity on COX-2 (see Fig. 5 and 7).36 The physicochemical properties of oxicams (keto–enol tautomerism) are involved mainly in enzyme inhibition. Hence, several H-bonds have been observed between thiazine and carboxamide moieties with Ser530, Tyr385, Arg120, and Tyr355 residues. Interestingly, oxicam binding within the ASC is accompanied by changes in protein conformation, which is rare because classical NSAIDs undergo rotation of the Leu531 residue and have access to the side binding pocket.37 However, the new generation of COX-2 inhibitors lack a carboxylic-acid group, which leads to enzyme inhibition mainly through H-bond interactions with Ser530 and without salt-bridge interactions with Arg120. Paracetamol is used to treat pain and fever; it does not contain a carboxylic acid substructure and is associated with non-selective COX inhibition (Fig. 7).38,39
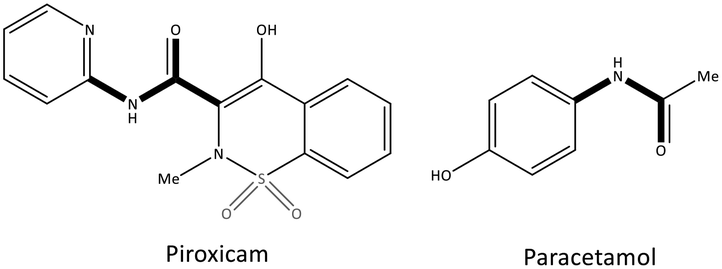 | ||
| Fig. 7 Chemical structure of piroxicam and paracetamol as non-selective COX inhibitors without a carboxylic acid functional group. | ||
The exact mechanism of paracetamol action remains elusive as recently reviewed by Przybyła et al.40 The unusual pharmacological profile of this compound, i.e., the strong antipyretic activity paired with a weak anti-inflammatory activity, suggests a target beyond COX-1/2. Some data indicate that the inhibition of a COX-1 splice variant (COX-3) assumed to be expressed in the human pituitary gland and the hypothalamus may be relevant for the activity of paracetamol. However, the concept of COX-3 had to be rejected.23,41,42 The current view assumes a mixed COX-1/2 inhibition in the central nervous system due to the lipophilicity of the drug. The diaryl heterocyclic coxibs (e.g., celecoxib, valdecoxib, rofecoxib, etoricoxib) have also been developed as selective COX-2 inhibitors (Fig. 8).38,43,44 SC-558 has the same pyrazole-based structure as celecoxib, but bromine is substituted on the phenyl ring instead of a methyl group (Fig. 9). This category of COX inhibitors has substituted diaryls on a central core of five- and six-membered heterocycle scaffolds, including pyrazole (in celecoxib), furanone (in rofecoxib), isoxazole (in valdecoxib), and bipyridine (in etoricoxib). Sulfonamide and methanesulfonyl groups have vital roles in selective COX-2 inhibition. In celecoxib (a diaryl pyrazole-based COX-2 inhibitor), the binding of the –SO2NH2 group in the side pocket with amino acids His90 and Arg513 through sulfonyl oxygen atoms and amino acid Phe518 via carbonyl oxygen atoms can facilitate additional substitution interactions with Val434 and Arg513 residues. The slight structural modifications of COX-2 inhibitors such as celecoxib highlight the importance of the binding affinity to the ASC of COX-2 to maximize potency and selectivity via introduction of steric hindrance between the sulfonamide oxygen and the Ile523 residue in COX-1. Lumiracoxib has a different structure than other coxibs. Lumiracoxib is a diclofenac analog (one of the chlorine groups is replaced with fluorine and phenylacetic acid has a methyl group in the meta position) that creates a salt bridge-based inhibitory structure (Fig. 8).25,45 Diaryl-substituted heterocycles (e.g., thiazole, imidazole, triazole, oxazole, oxadiazole, pyrrole, thiophene) have been widely investigated as selective COX-2 inhibitors.38,46 Thiazole- and thiazolidinone-based derivatives were reviewed by Liaras et al.47 as COX/5-LOX inhibitors. Pyrimidine-based and fused bicyclic-based COX-2 inhibitors have also been reported to have selective COX-2 inhibition.38,48 Acyclic non-diaryl heterocyclic molecules are a new class of COX-2 inhibitors without a central heterocycle core.38,49 Acyclic inhibitors have diverse backbones (e.g., olefin, acetylene, azo, imine, unsaturated ketones), which are substituted with di- and tri-aryl (or -alkyl) functions. Moreover, a wide range of natural compounds, such as xanthines, alkaloids, stilbenes, flavonoids, terpenoids, and quinones, have COX-2 inhibitory activity.50,51
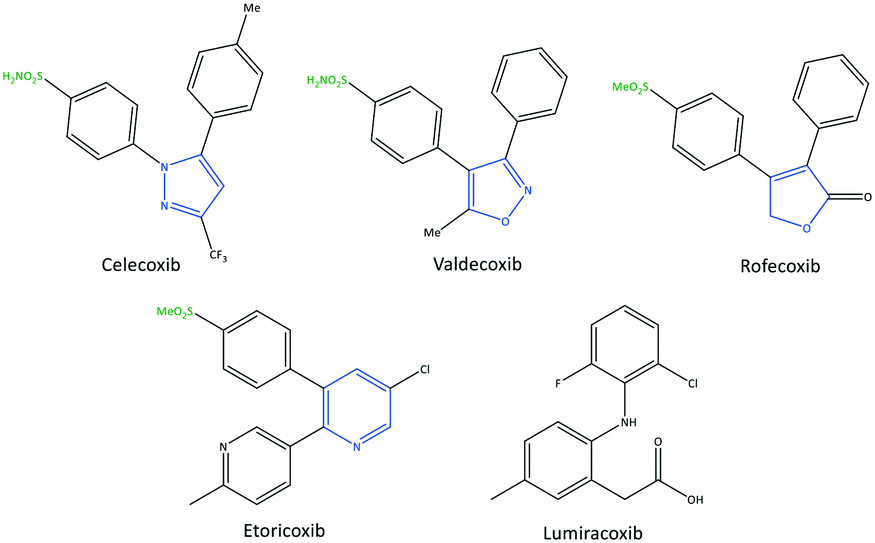 | ||
| Fig. 8 Chemical structure of diarylheterocycle coxibs. | ||
 | ||
| Fig. 9 Chemical structure of SC-558. | ||
Several strategies have been employed to introduce lead structures that selectively inhibit COX-2 with an improved pharmacokinetically safe profile. However, for COX-2 inhibitors at the site of inflamed tissues, one must consider (i) structural differences between the active sites of COX-2 and COX-1 in the drug-discovery process; (ii) structural features of drug candidates using structure–activity relationships; and (iii) oral bioavailability, first-pass metabolism, clearance, and other pharmacokinetic parameters. These considerations could pave the way for painless therapy to reduce the risk of chronic diseases and minimize the adverse effects of drugs.
Recent developments with respect to selective COX-2 inhibitors
Thiazole-based inhibitors
Thiazole is a five-membered heterocyclic ring. It is incorporated into the central core of the structure of compounds that have been used as drugs such as tiazofurin and alpelisib (anticancer), cefiderocol (antibiotic), and cobicistat (for the treatment of human immunodeficiency virus infection).52,53 However, a wide range of synthetic thiazole-based derivatives have been studied for their anti-inflammatory, antitumor, and antioxidant properties.47,52 Recently, Saglik et al.54 reported on thiazolylhydrazine methyl sulfonyl analogs comprising diaryl moieties as selective COX-2 inhibitors with an IC50 of 0.14–0.24 μM (selectivity index (SI) = 10.00–714.28) (Fig. 10). The SI was reported based on the ratio IC50 of COX-1/IC50 of COX-2. It was reported that a compound containing a hydroxy group in the meta position (compound 1: R2 = OH, R1, R3, R4 = H) displayed potent inhibition (IC50 = 0.14 μM) that was comparable with that of celecoxib (IC50 = 0.13 μM). Docking simulations showed that the amino and sulfur oxygen of –SO2NH2 established several H-bond interactions with Leu338, Ser339, Arg499, and Phe504 residues in the ASC similar to that of celecoxib, indicating their role in a selective COX-2 inhibition. The authors reported that the nitrogen atom of the thiazole moiety formed an extra H-bond interaction with the Val335 residue. The thiazole ring was involved in a cation–π interaction with Arg106, whereas the phenyl ring carrying the sulfonamide established π–π interactions with Tyr341. Interestingly, the hydroxyl group in compound 1 established an H-bond with the carbonyl group of methionine (Met)508. As a result, the steric hindrance on phenyl substituents (tri-substitution) did not allow extra interactions in the ASC compared with those in meta-substituted derivatives. In another study, a series of 2-aminothiazole derivatives (2) was reported by Hussein et al.55 to be potent phosphodiesterase type 5 (PDE5), COX-1, and COX-2 inhibitors (Fig. 10). In addition, in vitro anti-inflammatory activity towards COX-2 was presented using colorimetric immunoassays to screen isozyme-specific inhibition.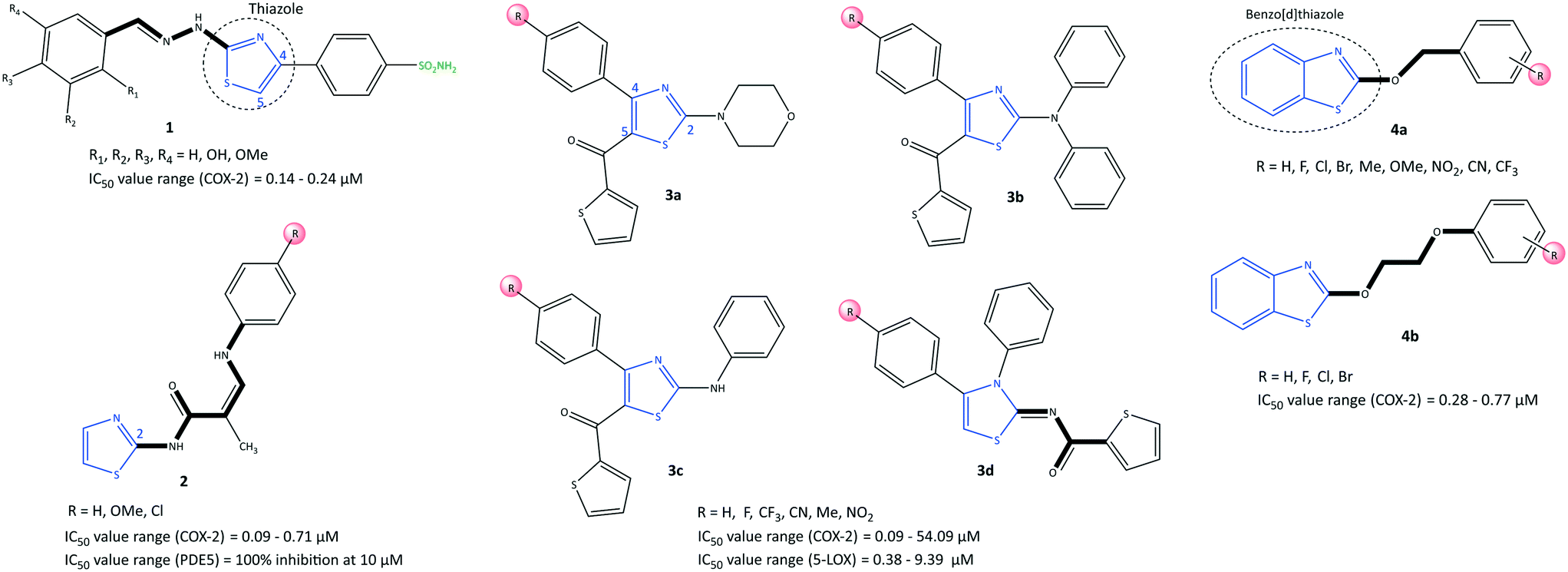 | ||
| Fig. 10 Thiazole-based derivatives 1, 2, 3, and 4. | ||
The authors reported an IC50 of 0.09–0.71 μM (SI = 3.03–16), which is comparable with that of celecoxib (IC50 = 0.83, SI = 8.68). The enamides 2 (R = H, OMe and Cl) exhibited 100% inhibition of PDE5 at 10 μM without a notable decrease in mean arterial blood pressure; 2 were considered to be the first regulators and enhancers of PDE5. Docking simulation showed that the enamides 2 (R = H, OMe) formed H-bond interactions with Tyr341, Leu338, and His75 residues. The authors simulated a “Y” shape of inhibitor within the ASC of COX-2. Notably, the enamide compound 2 (R = Cl) was mostly involved in hydrophobic–hydrophobic interactions.
LOXs are also key enzymes in arachidonic acid metabolism. Thus, dual inhibition of COX-2 and LOXs (mostly 5-LOX and 15-LOX) has become a research “hotspot” for treatment of inflammatory diseases.56,57 Jacob et al.58 reported a series of thiazole derivatives (3) as dual COX-2 and 5-LOX inhibitors with IC50 of 0.09–54.09 μM and 0.38–9.39 μM, respectively (Fig. 10). Anti-inflammatory activity was also reported. The thiazole derivatives reduced the expression of COX-2 and 5-LOX genes and also reduction of PGE2 and LTB4 levels. A diphenyl-amino thiazole compound 3b (R = Me) exhibited the highest inhibitory activity against COX-2 (IC50 = 0.09 μM, SI = 61.66), which is comparable with that of etoricoxib (IC50 = 0.07 μM, SI = 91.28). The substitution environment (electron withdrawing vs. electron donating) and substitution pattern (mostly at the para position) on the phenyl ring influenced the selectivity and inhibitory activity. Monophenyl- and diphenyl-amino thiazoles showed higher COX-2 inhibition than the morpholine derivatives 3a, which was related to more π–π interactions. Docking simulations showed that the carbonyl group was involved in H-bond interactions with His351. Simultaneously, diphenyl-amino moieties oriented within the hydrophobic pocket and stabilized the inhibitor within the ASC of COX-2 through the amino acids His90, threonine (Thr)94, proline (Pro)514, aspartic acid (Asp)515, Pro191, Tyr355, glycine (Gly)354, Gln192, and Ser353. The para phenyl group and a 5-thiophene-2-carbonyl group in derivatives 3d comprising thiazole and thiazolidine motifs fitted within the ASC of COX-2. The authors noted that the 2-substituted secondary amine and 5-thiophenyl-2-carbonyl group in the thiazole ring provided H-bond and hydrophobic interactions, respectively, at the ASC of 5-LOX.
The benzo[d]thiazole analogs (4) were synthesized and evaluated by He et al.59 as selective COX-2 inhibitors (IC50 = 0.28–0.77 μM, SI = 7.2–18.6) with significant in vivo anti-inflammatory activity (percent inhibition = 1.95–94.6%) (Fig. 10). The benzyloxy thiazole analogs (4a) bearing fluorine groups at meta and para positions on the phenyl ring exhibited relatively high COX-2 inhibitory activity. The benzyloxy analogs (4a; R = meta-fluorine) displayed the highest activity among all tested compounds (IC50 = 0.28 μM, SI = 18.6), which is comparable with that of celecoxib (IC50 = 0.27 μM, SI = 19.7). The benzyloxy analogs 4a contained electron-donating groups on the phenyl ring that presented good anti-inflammatory activity. The phenoxyl analogs 4b possessed halogens as electron-withdrawing substituents at the para position of the phenyl ring, which resulted in most of the anti-inflammatory effects. However, anti-inflammatory activity was not reported for the analogs 4a bearing CF3, NO2, and CN groups on the phenyl ring.
Chalcone-based inhibitors
A chalcone scaffold containing an α,β-unsaturated ketone is an important pharmacophore because it demonstrates anti-inflammatory, anticancer, and antioxidant activities.60 Çavuşoğlu et al.61 reported acyclic chalcone-based derivatives (5) possessing a –SO2Me moiety as moderately selective COX-2 inhibitors (IC50 = 0.18–0.34 μM) compared with celecoxib (IC50 = 0.12 μM), ibuprofen (IC50 = 5.33 μM), and nimesulide (IC50 = 1.68 μM) (Fig. 11). Compound 5 with a chloro group (di-substitution in meta–ortho and meta–para positions) and chloro–fluoro groups (in meta positions) exhibited the best inhibitory activity against COX-2. Docking simulations showed almost identical H-bond and π–π interactions similar to those of celecoxib in the ASC of COX-2 through the amino acids Arg499, Phe504, Met508, Ser516, Tyr341, and tryptophan (Trp)373. The –SO2Me group in the para position of the phenyl ring was oriented within the ASC and had several interactions with Arg499 and Phe504 residues. | ||
| Fig. 11 Chalcone-based inhibitors 5 and 6. | ||
In contrast, the phenyl ring involved in π–π interactions supported higher stabilization. Moreover, halogen-bond interactions with the carbonyl group of Met508 and the hydroxyl group of Ser516 represented extra inhibitory activity for chloro-substituted analogs.
Huang et al.62 reported that quinolone chalcone-based analogs (6) had anti-depressant, anti-inflammatory, and COX-2 inhibitory activities (Fig. 11). Compounds 6a, 6b and 6c displayed good in vitro inhibitory activity (IC50 = 0.21–0.29 μM, SI = 27.97–48.11) compared with that of celecoxib (IC50 = 0.19 μM, SI = 93.40). The anti-inflammatory activities of analogs 6a–6c more strongly decreased the duration of immobility related to the anti-depressant activity (85.0%, 92.0%, and 86.8%, respectively). Docking simulation of 6c showed two H-bond interactions with Arg120 and Tyr355 residues through the carbonyl group within the ASC of COX-2. Interestingly, the amino acids Arg513, His90, Ser353, Ser530, Gln192, and Met522 established several π-interactions within the hydrophobic cavity.
Pyrazole-based inhibitors
Pyrazole has been reported to be an active core scaffold that possesses a wide range of biological activities.63 Many NSAIDs (e.g., SC-558, celecoxib) have a pyrazole-ring core that has been shown (via structural diversification) to inhibit COX-2 selectively. Celecoxib (as a pyrazole-based COX-2 inhibitor) was first synthesized by Claisen condensation followed by cyclocondensation reactions to give an overall yield of 50%.64 Recently, Scholtz et al.65 reported improved production of celecoxib (7) using flow synthesis to reach higher purity and yield up to 96% (Fig. 12). The –SO2NH2 moiety in celecoxib has a significant role in providing several H-bond interactions, mainly with the key amino acid Arg513, to anchor the molecule within the ASC of COX-2. Sulfonamide (–SO2NH2) and methylsulfonyl (–SO2Me) are the most well-known motifs in the molecular structure of COX-2 inhibitors, which is due to the high binding interactions inside the hydrophobic pocket. Therefore, typically the sulfone-aryl substitution in the para position attached to pyrazole, isoxazole, and pyridine analogs is chosen to improve selectivity towards COX-2.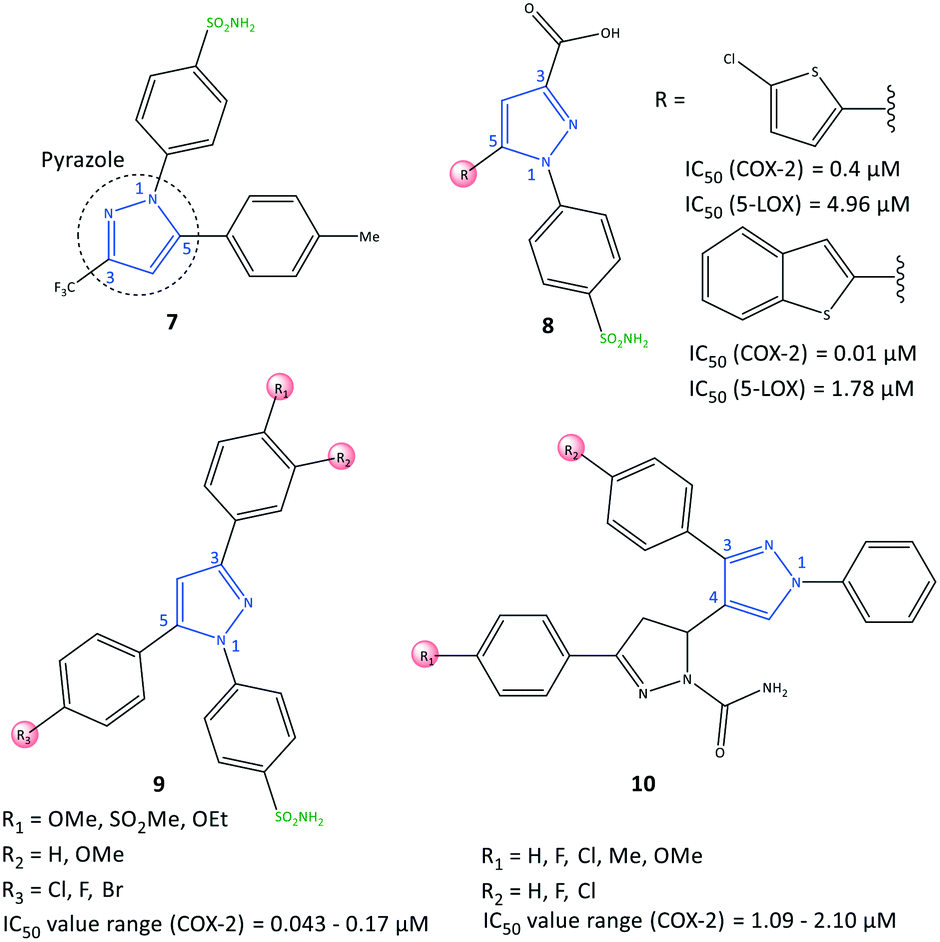 | ||
| Fig. 12 Pyrazole-based inhibitors 7, 8, 9, and 10. | ||
A hybridization strategy by Gedawy et al.66 aimed to combine the binding features of sulindac (an arylalkanoic acid class of NSAIDs with non-selective activity toward COXs [but 5-LOX inhibitory activity]) with celecoxib (selective COX-2 inhibitor) to achieve dual inhibitory activity towards COX-2/5-LOX. The active motifs of licofelone, sulindac sulfide, pyridone, and celecoxib were combined to design pyrazole sulfonamide derivatives (8) that fulfilled the binding requirement for both enzymes (Fig. 12). Derivatives of pyrazole sulfonamide carboxylic acids with an R group comprising 2-chlorothiazole and benzothiophen-2-yl showed the most potent dual anti-inflammatory activities with IC50 = 0.4 μM (SI = 29.73) and 0.01 μM (SI = 344.56) towards COX-2, and 4.96 and 1.78 μM towards 5-LOX. The aryl ring was involved in hydrophobic interactions with Leu338, and a –COOH moiety was directed towards the amino acids Arg106 and Tyr341 with ionic and H-bond interactions, respectively, within the ASC of COX-2. In addition, the sulfonamide group established H-bond interactions with the Ser339 residue.
Abdellatif et al.67 reported a new generation of halogenated triaryl-based pyrazole derivatives (9) based on the structural modification of celecoxib (as a selective COX-2 inhibitor) with IC50 = 0.043–0.17 μM (SI = 50.6–311.6) (Fig. 12). The authors noted that fluorinated pyrazole derivatives exhibited superior anti-inflammatory activity (edema inhibition = 42.1–87.9%) and a better gastric profile than indomethacin. They also studied the effect of the sulfonamide group as a COX-2 pharmacophore, para-halogen substitution as well as alkoxy and methylsulfonyl (–SO2Me) substitutions (in ortho and para positions) on the phenyl ring on COX-2 inhibitory activity. These substitutions led to significant interactions with key amino acids within the ASC of COX-2. Investigation of the effect of variation in electron-withdrawing and electron-donating groups of the phenyl rings on inhibitory activity showed that fluorinated pyrazoles exhibited remarkable potency close to that of celecoxib (IC50 = 0.055 μM) but with a higher SI. Docking simulations of the target triarylpyrazoles showed that the nitrogen atom of the pyrazole ring, –SO2Me, and –SO2NH2 moieties were primarily involved in several H-bond interactions within the ASC.
COX-2 is also released by cancer cells in the tumor microenvironment and leads to inflammation, which has a significant role in promoting cancer progression.68 Anti-inflammatory drugs with a dual-inhibition mechanism could inhibit COX-2/LOXs that catalyze PGs and leukotrienes. Therefore, “hybrid” molecules with active motifs of known COX-2 inhibitors on the market could help to introduce multi-targeting drugs with numerous biological activities.69 Pyrazole-containing drugs such as celecoxib, deracoxib, lonazolac, and tepoxalin are NSAIDs used to treat inflammation and cancer. A series of pyrazole–pyrazoline analogs (10) were reported by Akhtar et al.70 for their in vitro anticancer, anti-inflammatory, and COX inhibitory activities (Fig. 12). The COX-2 inhibition activity of targeted hybrid molecules was evaluated by immunoassays (IC50 = 1.09–2.10 μM, SI = 63.56–80.03). The authors found that compound 10 (R1 = OMe, and R2 = H) had a high spectrum of activity against COX-2 (IC50 = 1.09 μM, SI = 80.03), anti-inflammatory activity based on protein albumin denaturation assays (IC50 = 43.47 μM), and in vitro anticancer activity against MCF7, A549, SiHa, COLO205, HepG2, and HaCaT cell lines. The electron-withdrawing groups in the R2 position had higher inhibitory activity, whereas the electron-donating groups in the R1 position had reduced activity.
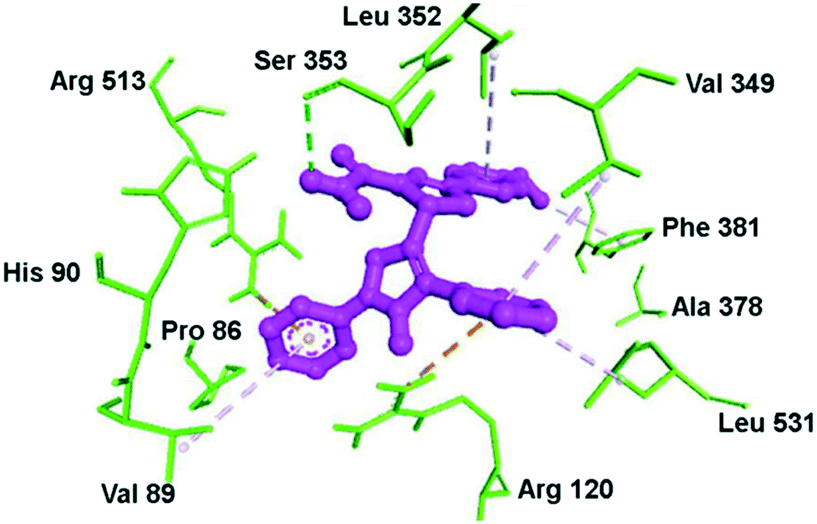
Docking simulation of pyrazole–pyrazoline compound 10 within the ASC of COX-2 is depicted in Fig. 13. The phenyl and methoxy groups provided a platform for π–alkyl interactions with Leu352 and Phe381 residues, respectively. The substituted amide moiety on the pyrazoline ring formed H-bond interactions with the Ser353 residue. Val349 provided a platform for π–alkyl interaction through stabilization of the phenyl ring substited in position 1 of the pyrazole ring. The key amino acids Arg120 and Arg513 were involved in π–cation interactions.
 | ||
| Fig. 13 Docking simulations of the pyrazole–pyrazoline compound 10 (R1 = OMe, R2 = H) within the active-site cavity of COX-2. The broken line in green: H-bond interactions; orange lines: π–cation interactions; purple lines: hydrophobic interactions. Reproduced from ref. 70 with permission from Wiley-VCH GmbH, Weinheim, copyright 2021.70 | ||
The third major metabolite of arachidonic acid is the cytochrome P450 pathway and leads to formation of EET regioisomers.71 sEH can catalyze the conversion of EETs into the corresponding diols and dihydroxyeicosatrienoic acids.72 Recently, Abdelazeem et al.73 reported on urea- and amide-linked diarylpyrazole derivatives (11) with dual inhibitory activity towards COX-2 and sEH (IC50 of COX-2 = 1.24–4.12 μM, SI = 2.85–7.03; IC50 of sEH = 0.40–4.00 nM) (Fig. 14). The authors reported high anti-inflammatory activities against edema of up to 93.79%. sEH inhibitors have been developed to treat inflammatory and cardiovascular diseases through diminished degradation of EETs. A combination of a diaryl-pyrazole moiety with urea and amide linkages composed of active pharmacophores of celecoxib, SC-558, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid, and GSK2256294 into a single hybrid diarylpyrazole scaffold showed a more favorable cardiovascular profile than that of celecoxib with fewer risks of cardiovascular toxicity. The urea-linked derivative 11a showed higher inhibitory activity against COX-2 (SI = 5.27–7.03), whereas the amide-linked derivative 11b was more potent towards sEH. The urea-linked diarylpyrazole compound 11a comprising trifluoromethyl phenyl as an R group was reported to be the most potent dual COX-2/sEH inhibitor (COX-2: IC50 = 1.24 μM, SI = 7.03; sEH: IC50 = 0.40 nM).
 | ||
| Fig. 14 Pyrazole-based inhibitors 11, 12, and 13 comprising amide and urea linkers. | ||
A series of pyrazole derivatives linked to aminophosphonate (12) were reported by Zhang et al.74 to be selective COX-2 inhibitors using enzyme-linked immunosorbent assays (IC50 = 0.22–5.84 μM, SI = 5.84–179.18) (Fig. 14). The amide-linked compound 12 (R1 = naphthalen-1-yl; R2 = F) displayed significant inhibitory activity towards COX-2 (IC50 = 0.22 μM, SI = 179.18; 2-fold higher selectivity compared with that of celecoxib (IC50 = 0.39 μM)) and anti-proliferative activity against MCF-7 cells (IC50 = 4.37 μM). The authors further evaluated these compounds comprising various functional groups (R2–R5), on the ortho and para positions of two phenyl rings, as shown in Fig. 14. Substitution with naphthalene on the pyrazole ring instead of thiophene led to higher inhibitory activity, particularly if the N-1 pyrazole phenyl ring was functionalized at the para position (–F > –SO2NH2 > –Br).
In another study, Zhang et al.75 also reported a series of pyrazole-based derivatives containing aminophosphonate and sulfonamide (13) to be selective COX-2 inhibitors (IC50 = 0.28–6.32 μM, SI = 5.41–172.32) and to have proliferative capability against cancer cell lines (IC50 = 2.34–16.43 μM) (Fig. 14). The authors examined several substituted compounds with various functional groups at the ortho, meta, and para positions of phenyl rings. Substitution of 4-bromophenyl phenyl in the R2 position and the methoxy group (R1) in the meta position elicited the highest inhibitory activity towards COX-2 (IC50 = 0.28 μM, SI = 172.32). The docking simulation predicted several H-bond interactions of the –SO2NH2 moiety with Arg106 and Val102 residues and a phosphonate function with the Tyr371 residue. Notably, Tyr341, Arg499, and Val509 residues were mostly involved in several π interactions.
Benzenesulfonamide derivatives (14) with a bumetanide main scaffold bearing pyrazole and triazole moieties were reported by Ibrahim et al.76 to be selective COX-2 inhibitors with IC50 = 0.17–4.79 μM (SI = 4.84–115.82) (Fig. 15). The 1,2,4-triazole derivatives 14e (R = 4-Br–C6H4; IC50 = 0.17 μM) and 1,2-pyrazole derivatives 14a (R = Ph; IC50 = 0.32 μM) had the highest inhibitory activity and were 12-fold to 23-fold more selective than celecoxib, respectively.
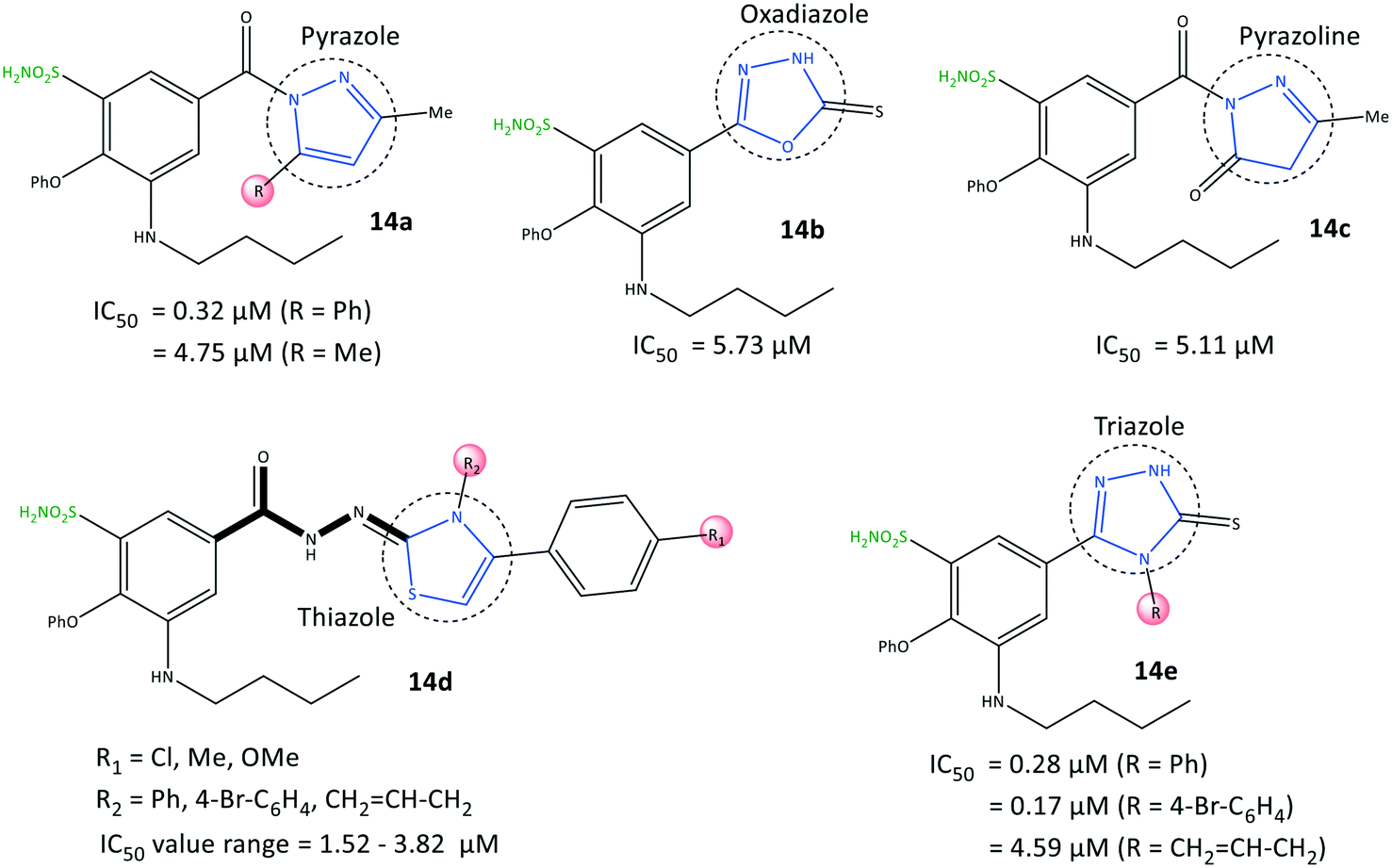 | ||
| Fig. 15 Benzenesulfonamide derivatives 14. | ||
The phenoxy group was bound into the hydrophobic pocket and had π–π interactions with the Trp373 residue and hydrophobic interactions with Leu338 or Tyr371 residues. The –SO2NH2 group was involved in H-bond interactions with Gly512 and Ala513 residues, whereas the carbonyl group did not show any H-bonds due to different aromaticity. Various hydrophobic interactions were observed through aromatic groups attached to heterocycles within the ASC of COX-2 with the amino acids Ala513, Val102, Leu517, Tyr341, and Arg106. The authors also noted that the butylamino group bound into the hydrophobic pocket was accessible only in COX-2 via Val509, and this was the main reason for the higher SI. In vitro and in vivo results exhibited high anti-inflammatory activity of the target inhibitors against peptic ulcers.
Triazole-based inhibitors
Assali et al.77 reported on diaryl-based pyrazole and triazole derivatives (15) synthesized based on Vilsmeier–Haack and click reactions (Fig. 16). In vitro COX-2 inhibition assays revealed significant inhibitory activity (IC50 = 0.002–3.324 μM, SI = 0.008–162.5).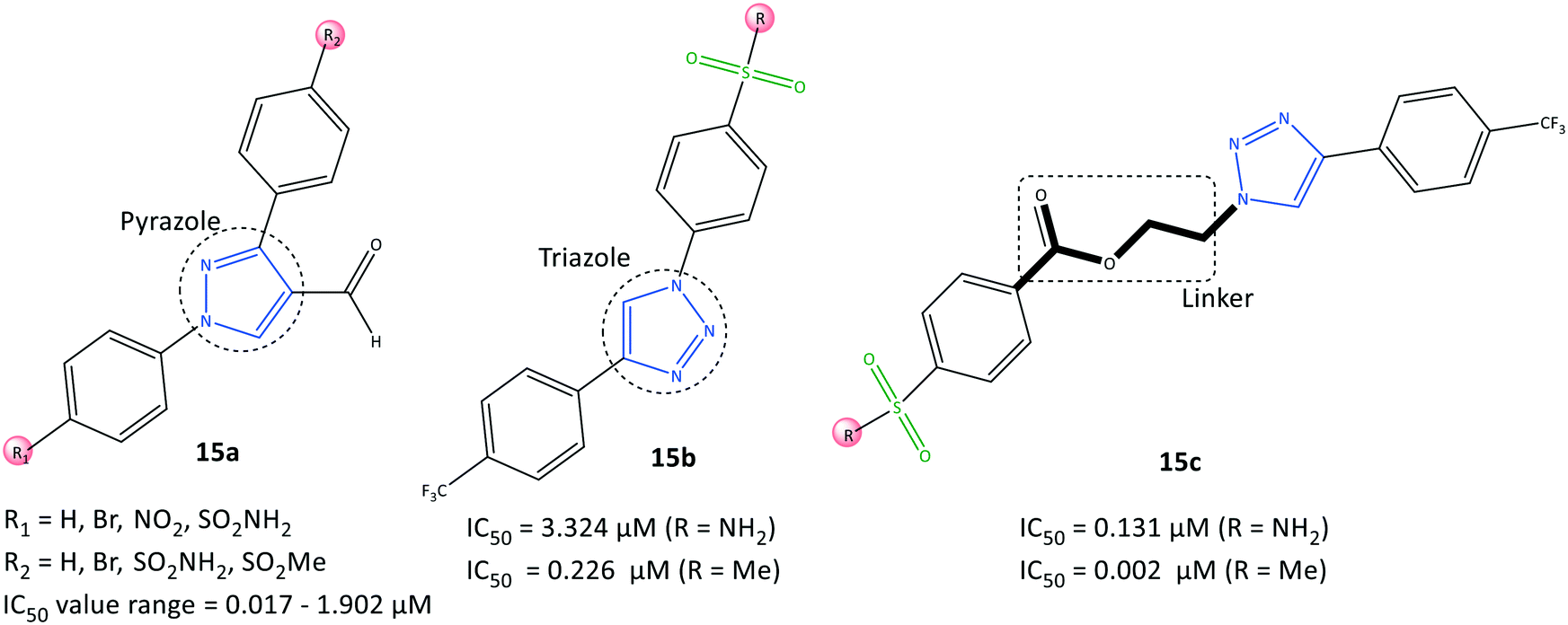 | ||
| Fig. 16 Diaryl-based pyrazole and triazole derivatives 15. | ||
Diarylpyrazole-substituted analogs 15a comprising sulfonamide and sulfone groups on the phenyl rings, and formaldehyde on the pyrazole ring, were reported to be the most selective COX-2 inhibitors (IC50 = 0.098 μM, SI = 54.847). The diaryltriazole substituted analogs 15c with –SO2Me on the phenyl carboxylate linker and CF3 groups on the phenyl ring (IC50= 0.002 μM, SI = 162.5) exhibited the highest inhibitory activity and selectivity for COX-2.
A diaryltriazole compound with a carboxylate linkage fitted better within the hydrophobic pocket due to its flexibility, and showed 2-fold higher inhibitory activity compared with that of celecoxib. The sulfonamide group of diarylpyrazole analogs 15a established H-bond interactions mostly with Ser530, Tyr348, and Tyr385 residues. The sulfonamide group of analogs 15b stabilized the inhibitor/enzyme complex within the ASC of COX-2 through several H-bond interactions with the amino acids Ser530, His90, Arg513, Ile517, and Phe518.
The more robust inhibition profile of 15b was due mainly to more favored interactions within the hydrophobic pocket with Leu96 and Val116 residues and lack of H-bond restrictions with the polar backbone comprising Ile517 and Phe518 residues.
Li et al.78 reported a series of 1,2,4-triazole-3-carboxylate derivatives (16) to be selective COX-2 inhibitors (IC50 = 0.00712–100 μM) and COX-2 inhibitors with anti-inflammatory activity on nitric oxide inhibition (IC50 <7.0 μM), which are comparable with values for celecoxib and indomethacin (Fig. 17). The tendency of COX-2 inhibition with substitution on the phenyl ring was reported to be meta > para > ortho. Electron-withdrawing groups were more favorable than ED groups in the meta position. Compounds bearing a fluoro group in meta and para positions had significant inhibitory activity (IC50 = 7.12 and 17.9 nM, respectively). A 1,2,4-triazole-3-carboxylate compound with a para-fluoro substituent on the N-1 phenyl ring exhibited higher selectivity towards COX-2 (SI = 1080), elicited significant inhibition and exhibited fewer gastrointestinal side-effects. The tendency order of substitution on the aryl group was reported as follows: F > Cl > CF3 > Me > CN > Br.
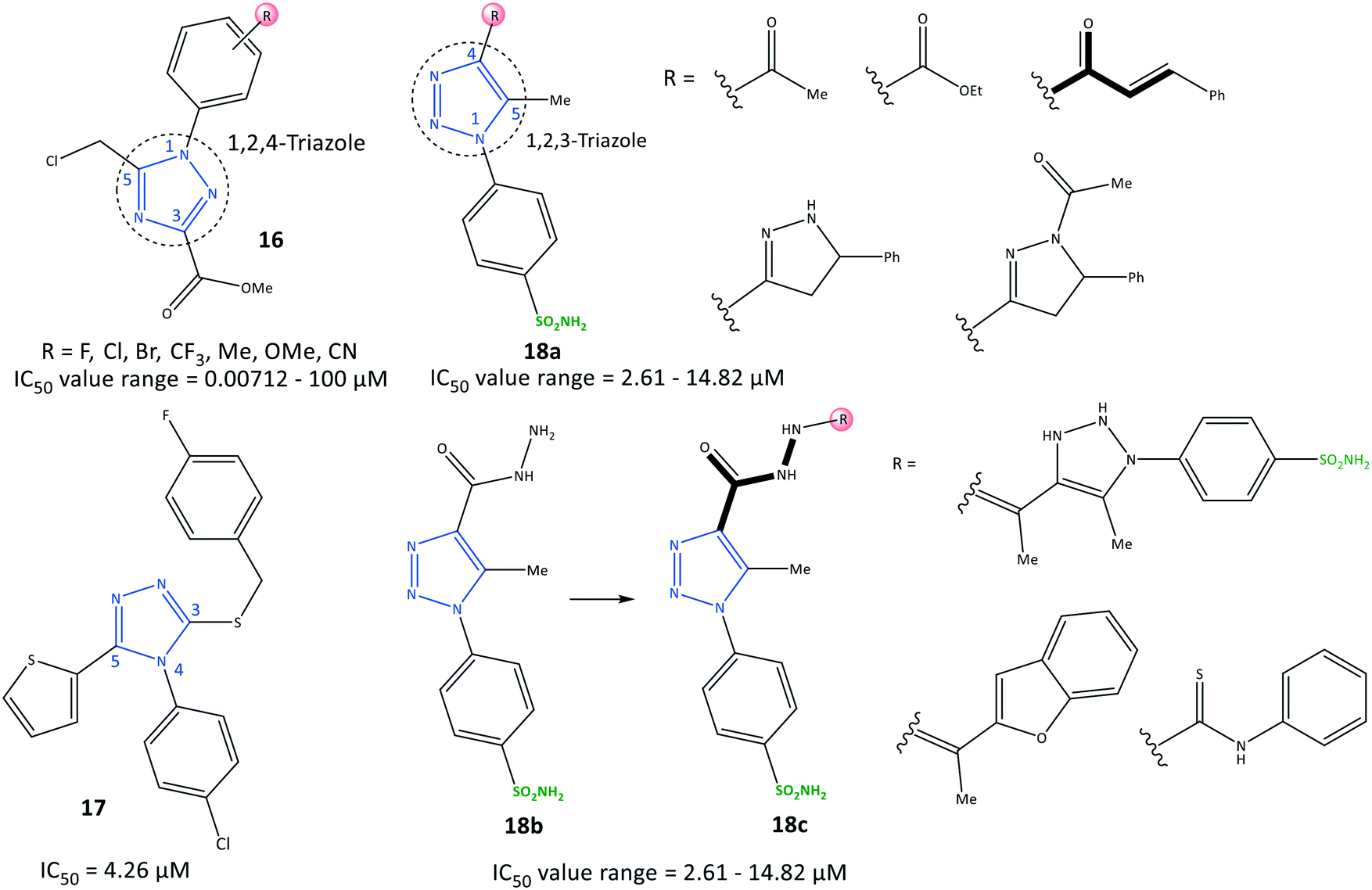 | ||
| Fig. 17 Triazole derivatives 16, 17 and 18. | ||
H-bond interactions with His75, Leu338, and Phe504 residues were predicted by docking simulation. The N-4 position of the 1,2,4-triazole scaffold interacted with the Leu338 residue. The carbonyl group formed two H-bonds with His75 and Phe504 residues. Several van der Waals interactions were identified with Val335, Leu338, Ser339, Try341, Phe504, Val509, Gly512, and Ala513 residues within the ASC. The authors showed that the weak binding energy of Ile523 within the ASC of COX-1 with a para-fluoro substituent on the N-1 phenyl ring was the main reason for higher selectivity towards COX-2. Furthermore, the level of the proinflammatory mediators TNF-α and IL-6 decreased upon administration of para-fluoro-substituted compounds.
Recently, Al-Wahaibi et al.79 reported a selective triazole-based COX-2 inhibitor (17) with IC50 = 4.26 μM (SI = 1.89) as compared with celecoxib (IC50 = 0.07 μM, SI = 308.57) (Fig. 17). Docking simulations displayed two cation–π stacking interactions of the Tyr355 residue with p-Cl–C6H4– and thiophene rings. The crystal structure of 4-(4-chlorophenyl)-3-[(4-fluorobenzyl)sulfanyl]-5-(thiophen-2-yl)-4H-1,2,4-triazole revealed the role of different noncovalent interactions, such as a rare chalcogen H-bond, and unorthodox S⋯C(π) and F⋯C(π) interactions in the self-assembly process to stabilize supramolecular sheets.
In another study, Bekheit et al.80 reported a 1,4,5-trisubstituted triazole-based selective COX-2 inhibitor bearing a sulfonamide moiety (18) with IC50 = 2.61–14.82 μM (SI = 1.95–6.98) (Fig. 17). Compounds 18a (R = 5-phenyl-pyrazoline) and 18b showed the highest inhibitory activities towards COX-2, with IC50 = 2.61 and 2.92 μM, respectively. Docking simulations showed that the phenyl-sulfonamide ring occupied the hydrophilic side pocket of COX-2, whereas the sulfonamide was oriented towards the amino acids Gln178, Arg499, Phe504, Val509, and Leu338. Phe504 and Gln178 residues were involved in H-bond interactions with oxygen and amine groups. Notably, compound 18b established two additional H-bonds with the guanidine group of Arg499 and Ser516 through sulfonamide and carbonyl moieties, respectively.
Arylidene-based inhibitors
Namera et al.81 revealed that arylidene derivatives (19) contained pyrazole and sulfonamide moieties similar to those in celecoxib, and reported 19 to be selective COX-2 inhibitors with IC50 = 0.150–0.308 μM (Fig. 18). Docking simulation predicted several H-bond interactions, mainly with the amino-acid residues His75, Arg499, Ser339, Gln178, and Phe504 within the ASC. The number of H-bonds was vital for stabilizing the inhibitor/enzyme complex but did not guarantee the highest inhibitory activity or selectivity towards COX-2. Most of the H-bond interactions were established through –SO2NH2 and the nitrogen atom of the pyrazole moiety. A wide range of imidazolone triaryl derivatives comprising benzoate or sulfonamide moieties were reported by Metwally et al.82 to have high anti-inflammatory activity as well as activity against dihydrofolate reductase (DHFR) and COX-2.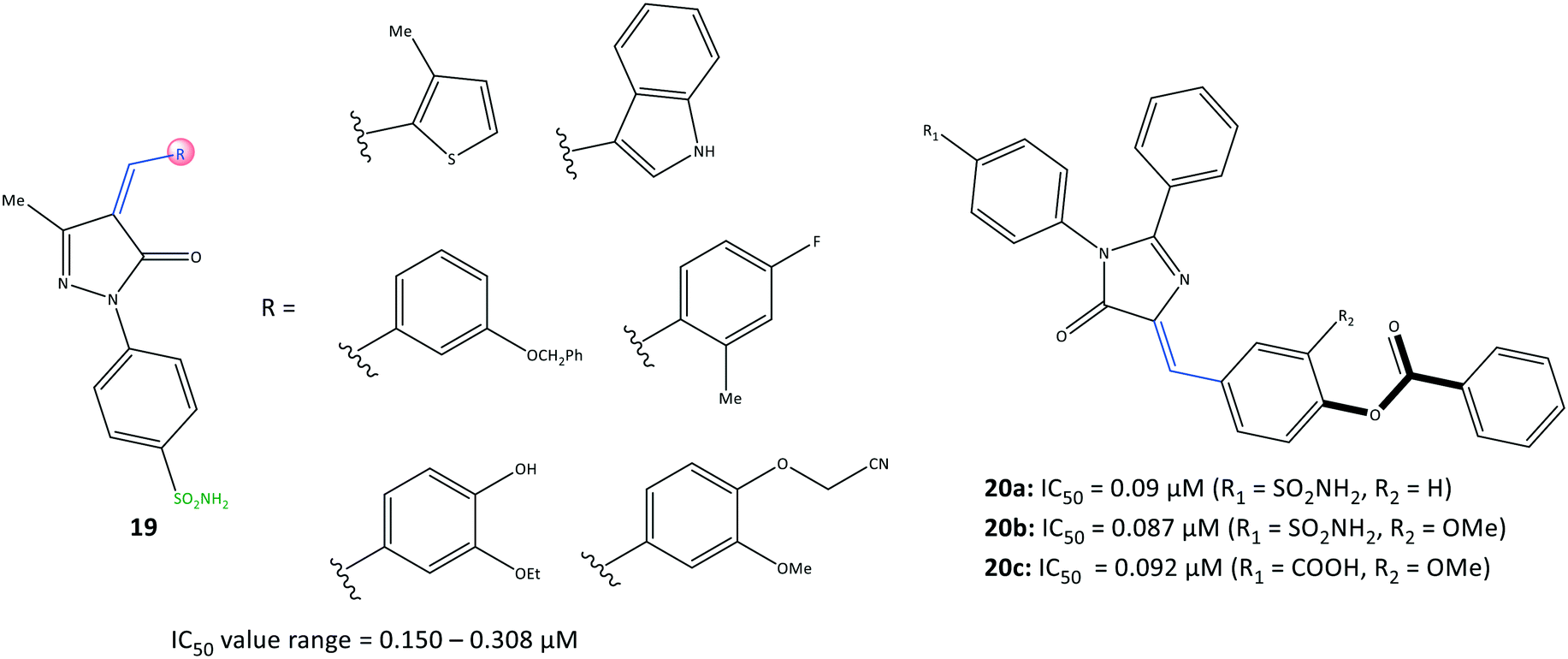 | ||
| Fig. 18 Arylidene-based derivatives 19 and 20. | ||
The authors made several arylidene derivatives,81 and among them, the in vitro inhibitory activity of imidazole-5-one derivatives 20a (R1 = SO2NH2, R2 = H), 20b (R1 = SO2NH2, R2 = OMe), and 20c (R1 = COOH, R2 = OMe) was evaluated using celecoxib as a standard reference (Fig. 19). The COX-2-inhibitory activity of the target inhibitors was reported to be up to 0.087 μM, which is superior to that of celecoxib (IC50 = 0.11 μM). DHFR inhibitors are used to combat malaria and treat cancer as well as fungal and bacterial infections.83 DHFR catalyzes the nicotinamide adenine dinucleotide phosphate-dependent reduction of dihydrofolate to tetrahydrofolate in the folate cycle.83
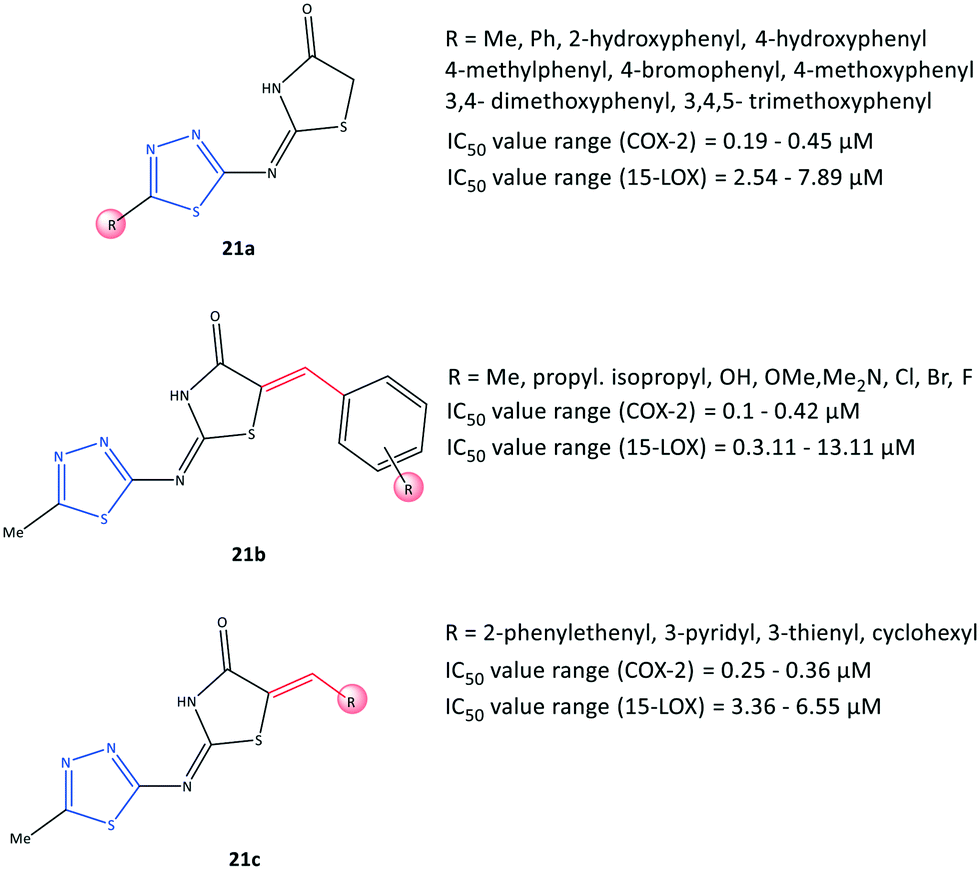 | ||
| Fig. 19 1,3,4-Thiadiazole-thiazolidinone hybrids 21. | ||
Hence, they also evaluated each target compound individually and in combination with sulfadiazine to inhibit DHFRs.81 Good binding affinity towards DHFR (IC50 = 0.28–0.79 μM), compared with that towards sulfadiazine (IC50 = 0.13 μM), was noted. The binding mode of targeted compounds comprising –SO2NH2 and –COOH moieties into the ASC of COX-2 revealed several H-bond interactions. The sulfonamide moiety interacted with the Tyr385 residue, whereas the benzoate moiety containing –COOH was involved in H-bond interactions with Arg513 and His90 residues. The benzoate moiety was oriented near the substrate-binding site of COX-2. Notably, the phenyl rings were involved in π–π interactions with the Arg120 residue.
Thiadiazole and thiazolidinone inhibitors
Omar et al.84 reported a series of 1,3,4-thiadiazole-thiazolidinone hybrids (21) with COX-2 inhibitory activity (IC50 = 0.1–0.45 μM, SI = 8.80–151.10) and 15-LOX inhibitory activity (IC50 = 2.54–13.11 μM), which are comparable with that of celecoxib (IC50 = 0.049 μM, SI = 308.16) and zileuton (IC50 = 15.6 μM), respectively (Fig. 19). The authors reported that the tested compounds were located within the ASC of COX-2 by carrying out various substitutions on the C-5 position of thiadiazole and thiazolidinone moieties. 15-LOX is converted from arachidonic acid into 15-hydroxyeicosatetraenoic acid and other proinflammatory mediators, and this should be considered when designing anti-inflammatory drugs.85 Significant inhibitory activity against COX-2 and 15-LOX was reported in relation to the presence of a number of substitutions on the thiadiazole ring (analogs 21a). Substitution with a phenyl group displayed the highest inhibitory activity towards 15-LOX (IC50 = 2.54 μM), whereas a compound bearing 3,4,5-trimethoxyphenyl exhibited the highest potency towards COX-2 (IC50 = 0.19 μM) among the tested compounds. Within the arylidene series, 21b (R = p-Me) exhibited the highest potency (IC50 = 3.11 μM) towards 15-LOX, whereas a tested compound bearing 3,4,5-trimethoxy and pentafluoro moieties on the phenyl ring displayed the highest COX-2 inhibitory activity (IC50 = 0.1 μM for both) and highest SI (123.40 and 151.10, respectively). The authors also noted that the substitution with the aromatic phenyl ring via electron-withdrawing groups and bulky substituents provided more selectivity and potency towards COX-2, whereas 15-LOX preferred less rigid substitutions with more ED groups. Nevertheless, dual balanced inhibition of COX-2 and 15-LOX must be considered when developing safe anti-inflammatory hybrid agents.Docking simulations showed that all compounds were involved in H-bond interactions within the ASC of COX-2 between the 1,3,4-thiadiazole ring and the Arg513 residue, except the arylidene analogs (21b) bearing Br, Cl, and isopropyl groups in the para position of the phenyl ring, which interacted with the Arg120 residue.
Tetrazole-based inhibitors
Tetrazole is a five-membered heterocyclic ring. It is applied frequently for the design and development of drugs. It has a wide range of biological and pharmacological applications due to its nitrogen-rich, multi-electron conjugated system. The novel tetrazole-based selective COX-2 inhibitors (22) reported by Labib et al.86 were based on the bioisosteric replacement of the –SO2NH2 moiety in celecoxib with a tetrazole ring incorporating the active motifs of pyrazole (22a), chalcone (acyclic α,β-unsaturated ketone moiety: 22b) and isoxazole (22c) (Fig. 20). The authors concluded that replacement of the –SO2NH2 moiety with a less acidic tetrazole moiety in celecoxib and rofecoxib analogs could lead to higher water solubility, in vitro selectivity, and in vivo anti-inflammatory activity.86 The tetrazole-based derivatives exhibited in vitro COX-2 inhibitory activity (IC50 = 0.039–0.31 μM, SI= 24.40–317.95). These analogs displayed a high SI compared with that of celecoxib (IC50 = 0.045 μM, SI = 282.22) and much higher SI compared with that of indomethacin (IC50 = 0.045 μM, SI = 1.25). The substituted aryl ring attached to pyrazole and an α,β-unsaturated ketone with a methoxy group in meta, ortho, and para positions showed high anti-inflammatory activity and SI towards COX-2. Among the most active compounds in this study, compounds 22c and 22a (Ar = trimethoxyphenyl) displayed significant in vivo anti-inflammatory activity (edema inhibition = 29.209–42.643%) and potent inhibitory effect on the production of PGE2 (inhibition = 81.042–82.724%). Notably, the compound 22a displayed a significant decrease in serum concentration of TNF-α ( inhibition = 55.349%) and IL-6 (inhibition = 61.561%). Docking simulation of active compounds displayed several H-bond interactions with His90, Ser530, and Tyr385 residues within the ASC of COX-2. The tetrazole moiety was involved mainly in H-bond interactions with His90, whereas methoxy groups were engaged in H-bond interactions with Ser530 and Tyr385 residues and elicited high anti-inflammatory activity. Notably, H-bond interactions with Arg513 were not reported due to replacing the –SO2NH2 group with a tetrazole moiety.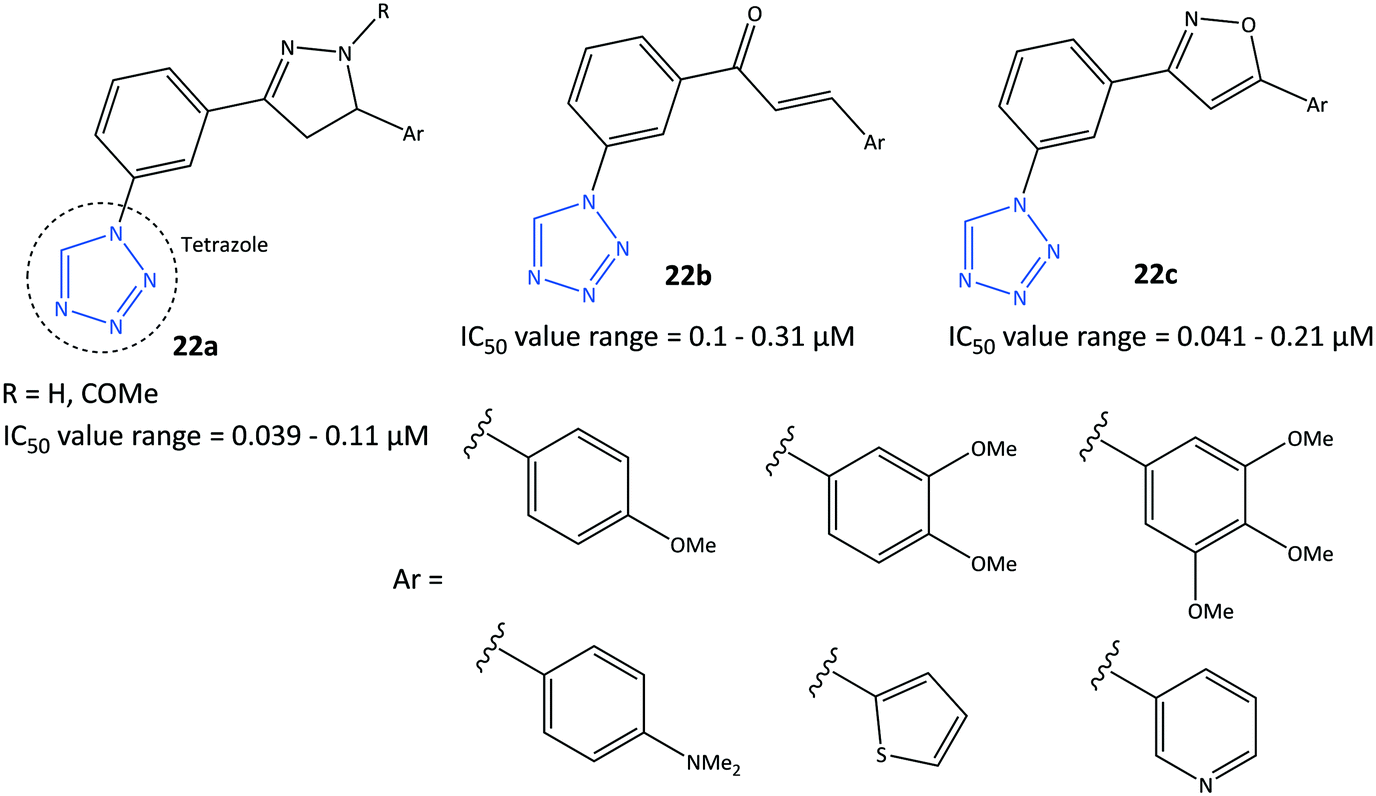 | ||
| Fig. 20 Tetrazole-based derivatives 22. | ||
Oxadiazoles-based inhibitors
Oxadiazole is a five-membered heterocyclic ring. It has three isomers, 1,2,4, 1,2,5-, and 1,3,4-oxadiazole, which have numerous biological activities such as antitumor, anti-viral, antibacterial, and antioxidant activity.87 The anticancer activity of derivatives of 1,3,4-oxadiazoles, as inhibitors of the epidermal growth factor receptor (EGFR) and COX-2, were reported by Sever et al.88 (Fig. 21). Among them, 2-[(5-((1H-indol)methyl)-1,3,4-oxadiazol-2-yl)thio]N-(6-ethoxybenzothiazol-2-yl)acetamide (23) comprising thiazole and benzothiazole scaffolds displayed selective COX-2 inhibitory activity (IC50 = 37.5 μM, SI = 1.96) compared with celecoxib (IC50 = 2.75 μM, SI = 3.23) and indomethacin (IC50 = 0.575 μM, SI = 0.21).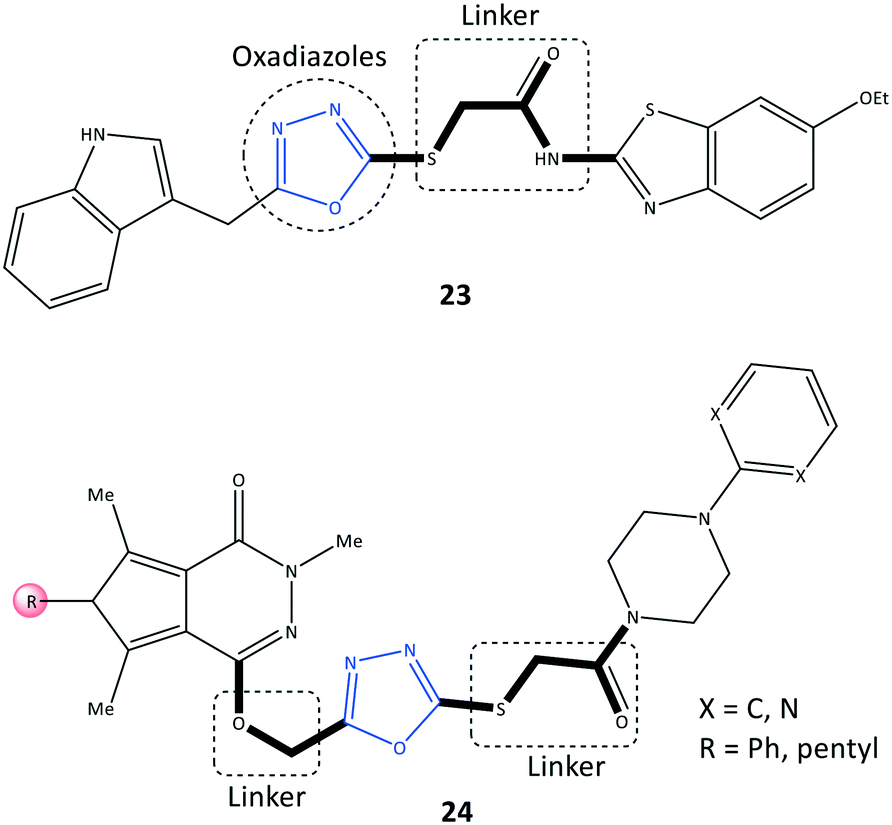 | ||
| Fig. 21 Oxadiazole-based derivatives 23 and 24. | ||
The oxadiazole ring was involved in CH–π interactions with Ser339 and Val509 residues within the ASC of COX-2, whereas oxadiazole could not interact within the ASC of COX-1. These specific binding interactions with COX-2 could explain the SI. The benzothiazole side chain established two H-bonds with His75 and Leu338 residues. Notably, the target molecule also exhibited significant activity against various cancer cell lines (IC50 = 6.43–9.62 μM compared with that using erlotinib (IC50 = 17.86–23.81 μM)). Mechanistic studies revealed that the EGFR was triggered via PGE2 in the inflammation pathway. The oxadiazoles-based derivative 23 demonstrated significant inhibition of EGFR expression (IC50 = 2.80 μM) compared with that using erlotinib (IC50 = 0.04 μM). Erlotinib is used to stop the progression of non-small-cell lung cancer.89
Pyrrolo[3,4-d]pyridazinone derivatives (24) bearing a 4-aryl-1-(1-oxoethyl)piperazine pharmacophore were reported by Szczukowski et al.90 to be selective COX-2 inhibitors. They had promising antioxidant activity and had active motifs comparable with those of pyrrolo[3,4-d]pyridazinone, valdecoxib (isoxazole group), celecoxib (pyrazole group), and 1,3,4-oxadiazole derivatives of diclofenac (Fig. 21). A combination of the aryl piperazine pharmacophore through a flexible 2-oxoethyl linker to the main scaffold enhanced the analgesic and anti-inflammatory activities of 24. An oxadiazole thione moiety with biodiverse biological activities was selected by the authors as a core to introduce the above-mentioned active motifs to increase selectivity and inhibitory activity. The reported IC50 (6.8–15.0 μM) is comparable with that of meloxicam (IC50 = 2.4 μM). Docking simulations and spectroscopic investigations indicated that the H-bond interactions within the ASC of COX-2 with Arg208 and Lys211 were mostly through the carbonyl group and nitrogen atom of the pyridazine moiety, respectively.
Succinimide-based inhibitors
Succinimide is a heterocyclic imide core that is considered a privileged scaffold in drug design and has diverse therapeutic applications such as anti-inflammatory, antiseizure, and antitumor.91N-Substituted pyrrolidine-2,5-dione derivatives (25) were reported by Jan et al.92 to be multi-target anti-inflammatory agents to inhibit COX-2 (IC50 = 0.98–113.44 μM, SI = 0.52–31.5) and 5-LOX (IC50 = 0.81–130.5 μM) (Fig. 22). The anti-inflammatory potential of test compounds comprising cycloalkyls (25a), alkyls (25b), and aryl carbonyls (25c) on the main pyrrolidine-2,5-dione scaffold showed COX-2 inhibitory activity in the micromolar range. A methylene linker coordinated to aryl and heteroaryl carbonyl groups provided flexibility to orient the molecule with the ASC. Hence, the analog 25c (R = Me, X = CH) displayed high inhibitory activity towards COX-2 (IC50 = 0.98 μM, SI = 31.5). The authors also reported that among all tested compounds, the analogs 25c (R = H, X = N) and (R = Me, X = CH) exhibited the highest inhibitory activity towards 5-LOX, attaining IC50 values of 0.81 and 0.86 μM respectively, which are comparable with that of zileuton (IC50 = 0.63 μM).
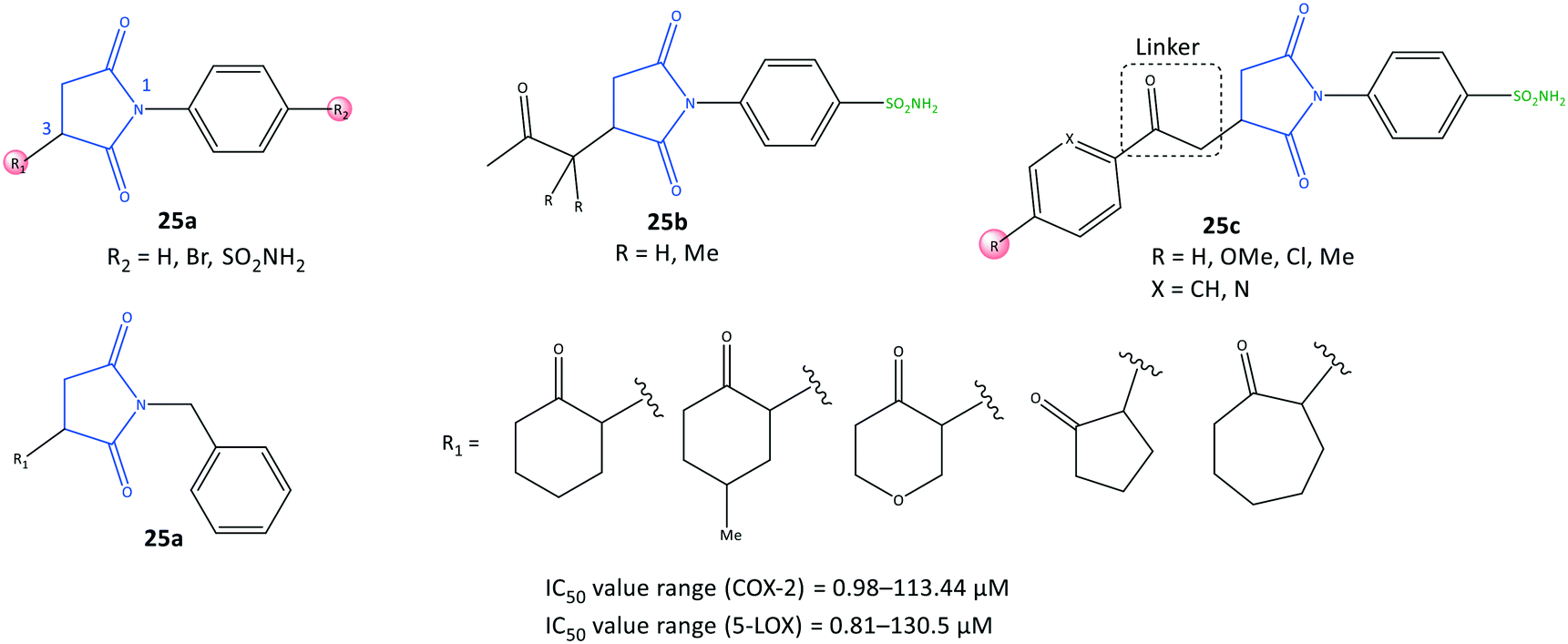 | ||
| Fig. 22 N-Substituted pyrrolidine-2,5-dione derivatives 25. | ||
Docking simulation of analog 25c (R = Me, X = CH) predicted several interactions with the amino acids His90, Leu352, and Ser353 located in the secondary pocket, whereas the aryl carbonyl group was oriented towards the amino acids Tyr385, Leu384, and Arg120 (Fig. 23).92
 | ||
| Fig. 23 Ribbon model of the superimposed binding poses of derivatives 25c (R-isomer) inside the active-site cavity of COX-2 (PDB 1CX2). Secondary pocket residues are shown as red spheres. In comparison, some other essential residues are shown as yellow spheres. Reproduced from ref. 92 with permission from MDPI, copyright 2008 (https://creativecommons.org/licenses/by/4.0/).92 | ||
Indole-based inhibitors
Indole is a biologically important fused-ring scaffold and is used to screen various receptors. Amide indomethacin analogs (26) were reported by Abdellatif et al.93 to have significant selective inhibitory activity towards COX-2 (IC50 = 0.09–0.4 μM, SI = 4.07–6.33), which is comparable with that of celecoxib (IC50 = 0.89 μM, SI = 3.52) (Fig. 24). They also reported moderate anti-inflammatory activity for this series of indole derivatives. The reported amide indomethacin derivatives had four types of amide linkages (thiourea, cyanoacetyl, pyridinone, cyanopyridone) instead of the carboxylic acid group in position 3 of indomethacin. Replacement of the –COOH moiety of indomethacin by an amide linkage led to almost identical inhibitory activity to that of indomethacin (IC50 = 0.087 μM) but with a higher SI towards COX-2. The authors reported that the thiourea analogs 26a (X = H, Me, OMe) linked to the central indole scaffold by an amido linkage exhibited significant inhibitory activity (IC50 = 0.09–011 μM) comparable with that of analog 26d comprising an amido-cyanopyridone linkage (X = Cl) (IC50 = 0.09 μM).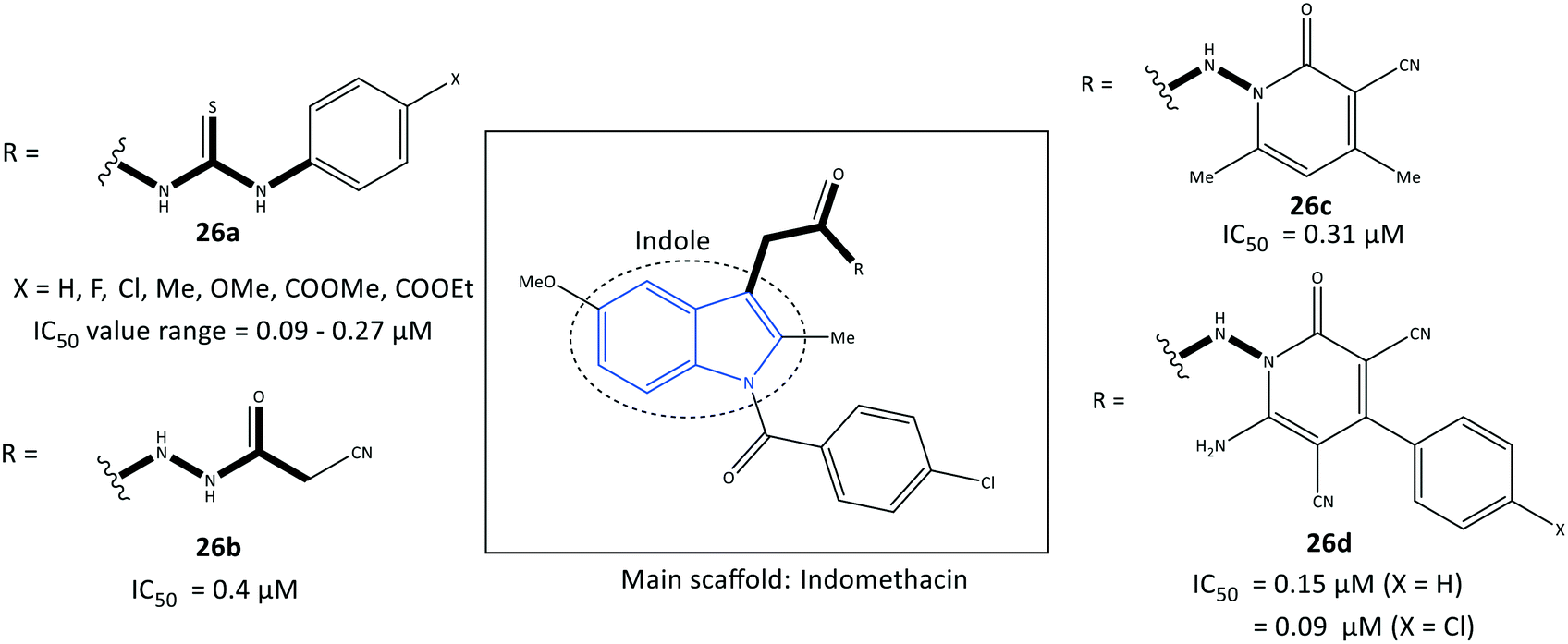 | ||
| Fig. 24 Amide indomethacin derivatives 26. | ||
Notably, the analogs 26a were almost 3-fold more selective than the analogs 26d. Several H-bond interactions of analogs 26a (X = H, Me, OMe), 26b, and 26c predicted by docking simulation with amino acids Arg120, Val116, Ser530, Glu524, and lysine (Lys)83 represented slightly different binding modes compared with those of indomethacin (H-bond interactions: Arg120 and Arg513) and celecoxib (Arg513 and Tyr355).
Fused-ring inhibitors
Bilavendran et al.94 reported a series of pyrazolopyridine derivatives (27) as anti-inflammation medications by measuring the inhibition of expression of pro- and anti-inflammatory cytokines and COX-2 inhibition (Fig. 25). Docking simulations of pyrazolopyridine analogs 27 (R = H, OH, NO2) showed good binding energies and significant affinity towards COX-2 in the nanomolar range. Those results are in accordance with values for in vitro COX-2 inhibition (IC50 = 0.002–0.01 μM; up to 98% inhibition). A compound bearing a hydroxyl group with IC50 = 2.25 nM was comparable with celecoxib (IC50 = 10.25 nM, 90.25% inhibition). This compound was analyzed further by docking simulations and displayed H-bond interactions with the Tyr385 residue and π–π interactions between the phenyl ring and the His207 residue. Moreover, the authors reported that the two compounds bearing hydroxyl and nitro groups could considerably decrease the levels of the proinflammatory cytokines TNF-α, IL-1β, and IL-6, and increase the levels of the anti-inflammatory cytokine IL-10, which were higher than those observed for indomethacin (positive control). Significant in vitro anti-inflammatory activity was also reported for the amide indomethacin derivatives 26.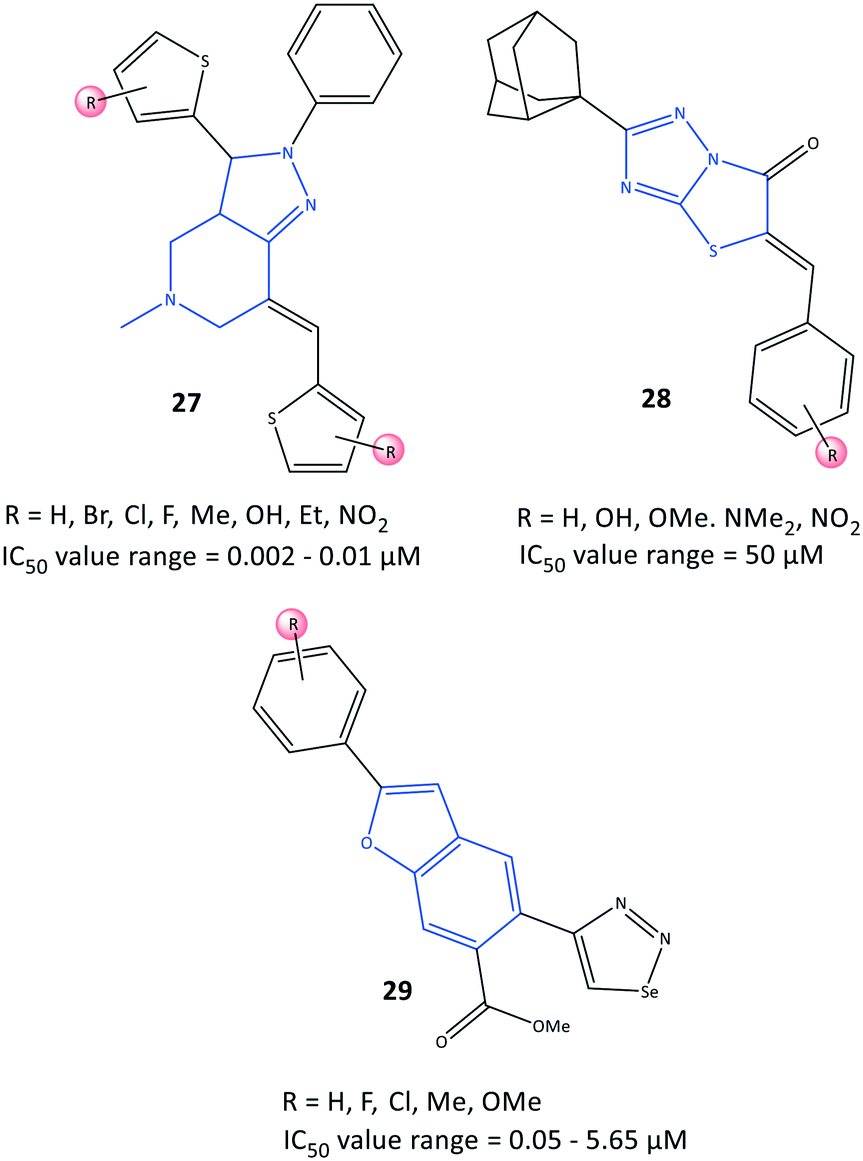 | ||
| Fig. 25 Fused-ring derivatives 27, 28, and 29. | ||
Substituted 5-benzylideno-2-adamantylthiazol[3,2-b][1,2,4]triazol-6(5H)ones (28) were evaluated by Tratrat et al.95 for their anti-inflammatory activity (Fig. 25). In general, all compounds showed moderate-to-low inhibitory activity towards COX-2. Nevertheless, an analog bearing a hydroxyl group in the meta position and an analog with a hydroxyl group in the para position and two methoxy groups in meta positions exhibited 65% inhibitory activity towards COX-2 with IC50 = 50 μM compared with that of naproxen (IC50 = 50 μM).
The authors found that the aryl ring was involved in hydrophobic interactions with Leu338, whereas the carboxylic-acid moiety directed towards Arg106 and Tyr341 had ionic and H-bond interactions, respectively. The sulfonamide established H-bond interactions with the Ser339 residue.
A series of benzofuran-selenadiazole hybrids 29 were reported by Olomola et al.96 to have inhibitory activity towards α-glucosidase (IC50 = 1.03–1.97 μM) and COX-2 (IC50 = 0.05–5.65 μM) as well as antioxidant activity (capacity to scavenge free radicals) due to selenium-containing organic scaffolds (Fig. 25).
Transformation of selenadiazoles on the 2-aryl-benzofuran led to 1,2,3-selanadiazole hybrid compounds. Compound 29a substituted with the more polar lipophilic p-Me–C6H4 moiety on the furan ring resulted in significantly increased inhibitory activity towards COX-2 (IC50 = 0.05 μM) compared with that of celecoxib (IC50 = 2.27 μM). The authors identified several hydrophobic interactions of compounds 29 (R = p-OMe) and 29b (R = H) with the lipophilic amino acids Ala527, Leu352, Val116 Tyr355, Ala527, Val523, and Gly526 via docking simulations. The selenadiazole ring in 29b was involved in several interactions with Trp387, Met522, Gly526, and Leu384 residues, whereas compound 29a established π interactions with the Val523 residue. Interestingly, the selenadiazole ring in 29b was involved in H-bond interactions with Tyr382, whereas the p-Me–C6H4 moiety in 29a interacted with the Ser530 residue.
Due to the efficacy and selectivity of selenium-based organic scaffolds against cancer cells, the authors also screened compounds 29a and 29b for anti-proliferative activity against breast cancer cells (MCF-7), which is related indirectly to COX-2 overexpression.
Pyrazoloquinazoline derivatives (30) reported by Shaaban et al.97 to be dual COX-2 and 5-LOX inhibitors with moderate anti-inflammatory activities are depicted in Fig. 26. Chemical modification and mono-, di- and trisubstitution on the phenyl ring (in meta, ortho, and para positions) coordinated to the pyrazoloquinazoline ring led to diverse pyrazoloquinazoline derivatives with significant inhibitory activity towards COX-2 (IC50 = 0.047–0.488 μM, SI = 0.19–14.2) and moderate inhibitory activity towards 5-LOX (IC50 = 0.6–4.3 μM). Combination of a quinazolinone-based scaffold with a pyrazole moiety comprising a para-methoxy substituted on the phenyl ring inhibited 5-LOX through suppression of an inflammation pathway when compared with zileuton (IC50 = 0.8 μM). A 3,4,5-tri-methoxy-fused-ring compound 30 (substituted in meta and para positions) was the most promising derivative, with IC50 = 0.047 μM towards COX-2, which is almost 7-fold higher selectivity compared with that of celecoxib (SI = 14.2).
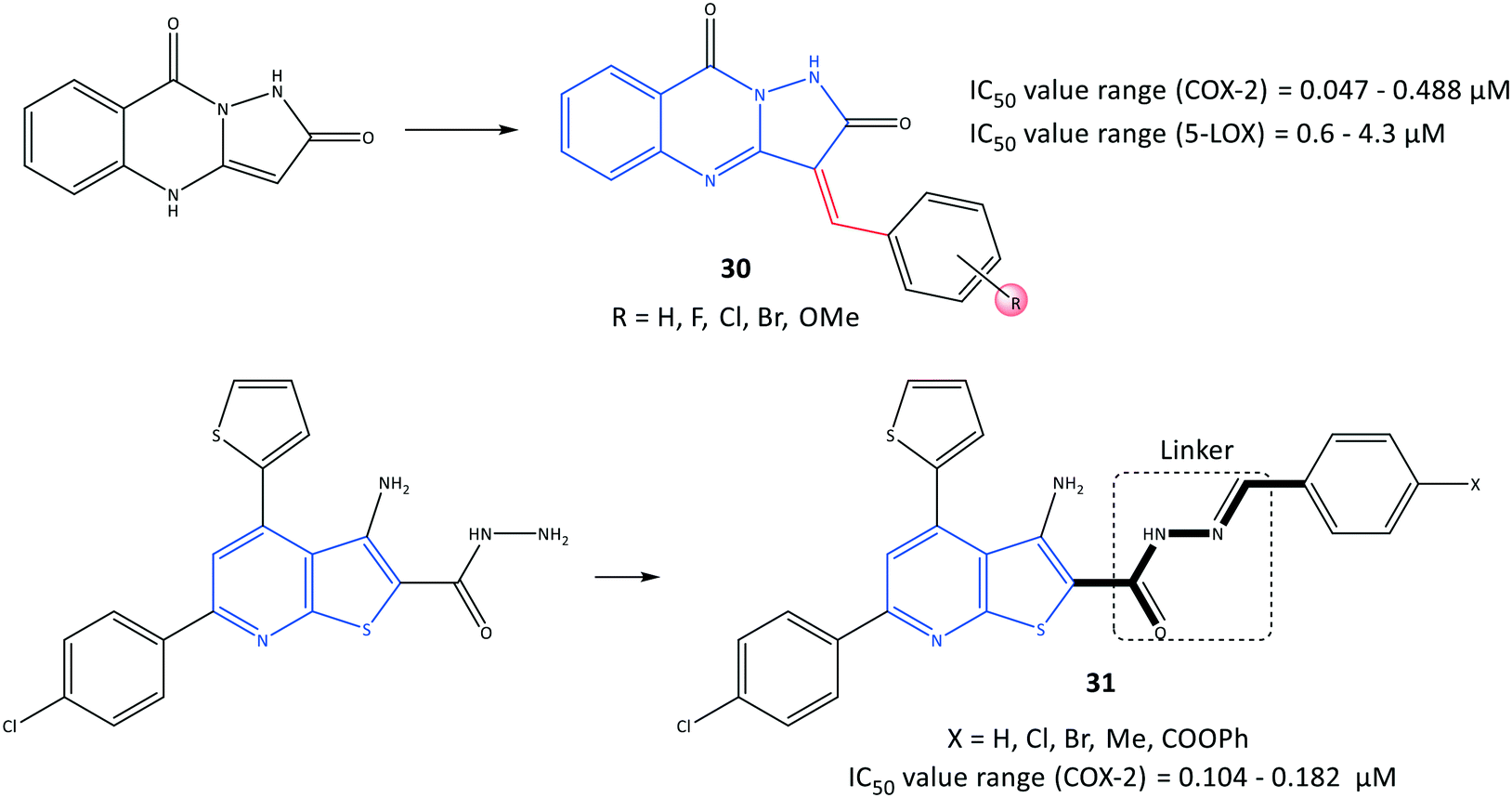 | ||
| Fig. 26 Fused-ring derivatives 30 and 31. | ||
The authors identified that the pyrazole moiety formed hydrophobic interactions with amino acid residues within the ASC of COX-2. The phenyl ring established mainly van der Waals interactions. Notably, the pyrimidine moiety was involved in H-bond interactions to anchor the molecule within the ASC. Alignment of docking poses of pyrazoloquinazoline derivatives 30 (line) with SC-558 (stick) within the ASC of COX-2 is depicted in Fig. 27.97
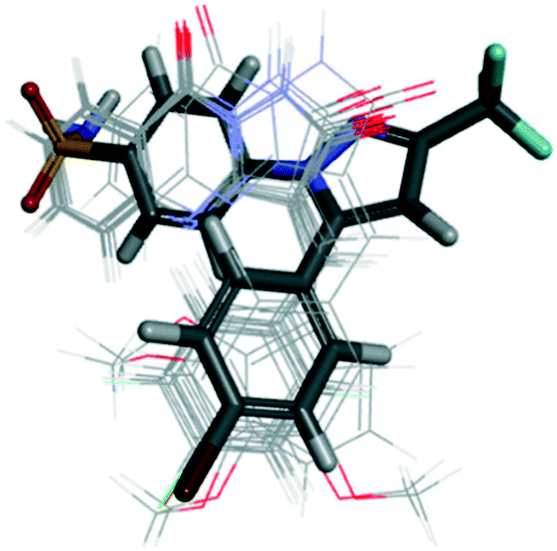 | ||
| Fig. 27 Alignment of docking poses of pyrazoloquinazoline derivatives 30 (line) with SC-558 (stick) inside the active-site cavity of COX-2. Reproduced from ref. 97 with permission from Wiley-VCH GmbH, Weinheim, copyright 2021.97 | ||
A series of fused-ring derivatives incorporating a thienopyridine moiety were reported by Sanad et al.98 to be COX-2 inhibitors with significant antibacterial activity (Fig. 26). The analogs 31 displayed suitable inhibitory activities towards COX-2 with IC50 = 0.104–0.182 μM, which are comparable with that of celecoxib (IC50 = 0.115 μM). Docking simulation of analog 31 comprising a methyl group in the para position of the phenyl ring displayed several H-bond interactions with the amino acids Arg44 and Tyr130 within the ASC of COX-2 through hydrazone-NH, thienopyridine-S, carbonyl-O, and hydrazine-N.
In another study, a series of semi-synthetic sclerotiorin derivatives (32) were reported by Chen et al.99 to be COX-2 inhibitors (inhibition ≤70%) and to have activity against various cancer cell lines (Fig. 28). Among all tested compounds, compound 32a displayed the highest in vitro inhibition (70.6%) compared with that of sclerotiorin (78.9%). However, the esterification products 32c displayed low COX-2 inhibitory activity. The authors also reported that compound 32a exhibited high toxicity against breast cancer cells (IC50 = 6.39 μM), superior to COX-2 inhibitory activity, among the other derivatives. The ester group of compound 32a was involved in H-bond interactions with the Gln92 residue within the ASC of COX-2.
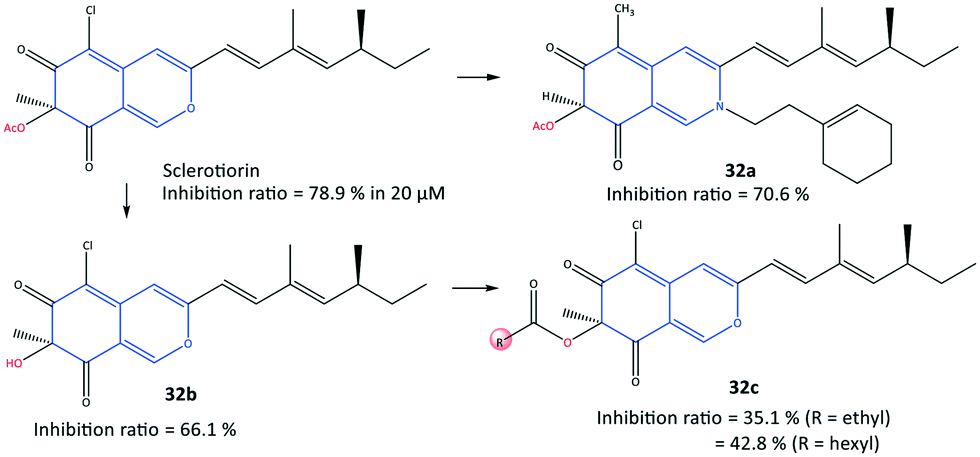 | ||
| Fig. 28 Semi-synthetic sclerotiorin-based derivatives 32. | ||
Uddin et al.100 reported novel harmaline analogs (33) to be selective COX-2 inhibitors (IC50 = 0.2 to >4 μM) (Fig. 29). A para-chlorobenzyl substitution on the pyrrole NH improved in vitro and in vivo metabolic stability. Compound 33b bearing a 6-OMe substituent on the fused phenyl ring as well as a methyl group in the C-1 position of the 4,9-dihydro-3H-pyrido[3,4-]indole ring was reported to have the highest inhibitory activity among the compounds evaluated. The target compound inhibited 2-arachidonoylglycerol oxygenation selectively over arachidonic acid oxygenation by COX-2. The authors reported substrate-selective COX-2 inhibitory activity with IC50 = 0.022 μM compared with that of arachidonic acid (>1 μM). Compounds 33b showed good selectivity towards COX-2, whereas other harmaline derivatives showed no selectivity (IC50 for COX-2 = IC50 for COX-1 > 4 μM). A crystal complex of COX-2 with compound 33b (R = 6-OMe) showed that small structural changes due to steric hindrance of a tricyclic indole and Leu531 residue within the ASC contributed to its substrate-selectivity (Fig. 29).
 | ||
| Fig. 29 (Top left) Harmaline derivatives 33. (Top right) Stereodiagram of the X-ray co-crystal structure of 33b (R = 6-OMe) bound within the active-site cavity of COX-2 with PDB code 6V3R (this figure is reproduced from ref. 100 with permission from the American Chemical Society, copyright 2020). (Bottom left) Polycyclic polyprenylated acylphloroglucinol analog derivatives 34 (ref. 101). (Bottom right) Docking simulation of compound 34a within the active-site cavity of COX-2 (ref. 101). | ||
Xie et al.101 reported unusual polycyclic polyprenylated acylphloroglucinol analogs (34) with a tricyclo-[7.3.1.03,7]tridecane core bearing a 5/7/6 carbon skeleton to be potent COX-2 inhibitors (Fig. 29). Compounds 34a and 34b exhibited COX-2 inhibitory activity with IC50 of 5.27 and 8.32 μM, respectively. They also reported the anti-inflammatory and enzyme inhibitory activities of lipopolysaccharide-induced RAW 264.7 cells upon addition of 34a.
The authors also noted that PGE2 production, along with TNF-α and IL-1β levels, was significantly reduced compared to celecoxib as positive control.101 Docking analyses of the interactions between COX-2 with key amino acid residues showed similar binding modes to those of isoxicam. Several H-bond interactions with the amino acids Arg120, Ser353, and Met522 were predicted. Notably, Tyr355 faced with an olefin bond of 34a influenced the inhibition potency directly (Fig. 29).
Acyclic inhibitors
Acyclic compounds have a central backbone possessing olefin, azo, imine, and α,β-unsaturated carbonyl-based structures. Various substitution patterns on the phenyl ring linked to a central core, as an integral motif of COX-2 inhibitors, are crucial for achieving the highest selectivity and potency. Analyses of the substitution patterns and spatial positions (meta-, ortho-, and para-substituted arrangements) on the phenyl ring are essential to regulate the steric hindrance of drug candidates in the ASC and, consequently, affect COX-2 selectivity. Ahmed et al.102 reported a series of pyridazine derivatives 35 as selective COX-2 inhibitors with IC50 = 0.0438–0.0672 μM (SI = 0.6–11.5). They exhibited 2-fold and 11-fold higher inhibitory activity than that of celecoxib and indomethacin, respectively (Fig. 30). Dihydropyridazinone and pyridazinone cores equipped with various electronic and lipophilic environments influenced the selectivity and potency of COX-2 inhibition. The pyridazine derivatives 35a comprising methoxy (in ortho and para positions) and chlorine (in the para position) and 35b consisting of a methoxy group at the meta position of the phenyl ring displayed the highest inhibitory activities towards COX-2. Compound 35a comprising two methoxy groups in ortho and para positions showed high anti-inflammatory activity compared with that of indomethacin.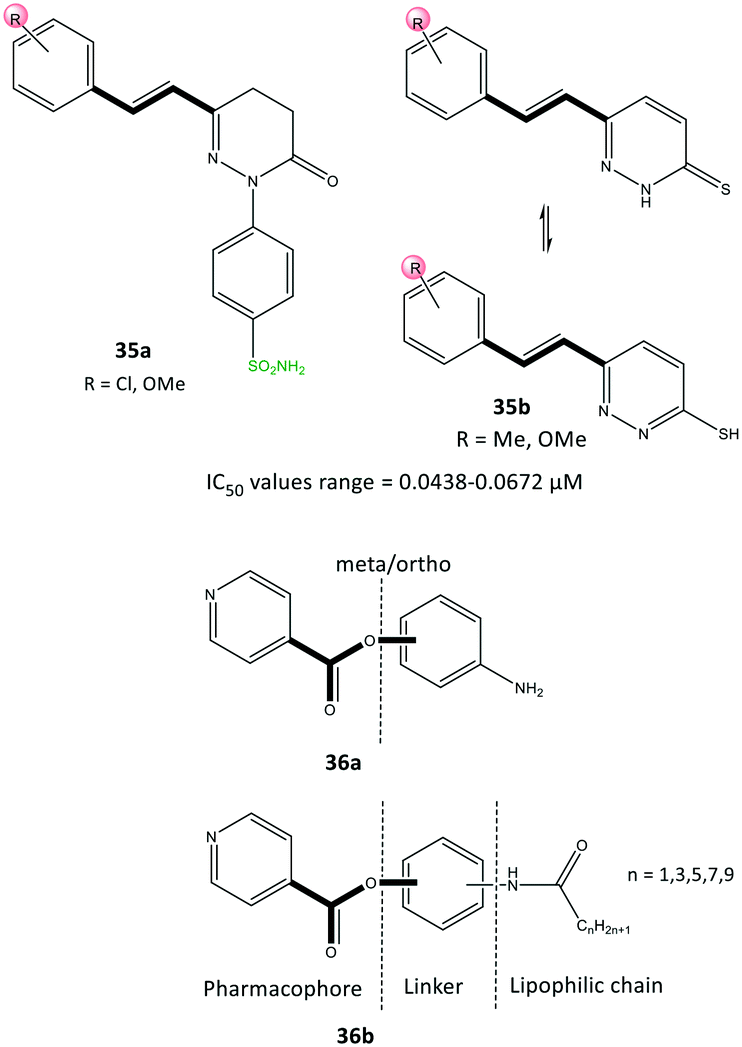 | ||
| Fig. 30 Pyridazine derivatives 35 and isonicotinic acid derivatives 36. | ||
Docking simulations visualized the orientation of the pyridazinone ring in the central region of the ASC, whereas the phenyl sulfonamide ring occupied the secondary pocket. The amino acids His90 and Arg513 established H-bond interactions with the –SO2NH2 moiety close to the ASC entrance, whereas the pyridazinone ring was involved in several interactions with Tyr355 and Arg120 residues.
The authors also identified two methoxy groups involved in H-bond interactions with Tyr385 and Ser530 residues. The docking outcomes indicated favorable interactions within the ASC of COX-2 compared with that of celecoxib.
Yaqoob et al.103 evaluated the in vitro anti-inflammatory and reactive-oxygen-species inhibitory activities of a series of isonicotinic acid analogs (36) (Fig. 30). Among the tested compounds, 36a with meta-aminophenol on the isonicotinate scaffold (main pharmacophore) without a lipophilic chain exhibited high anti-inflammatory activity with IC50 = 1.42 μg mL−1 (percent inhibition = 95.9%), which is 8-fold higher than that of ibuprofen (11.2 μg mL−1). Compound 36b with a lipophilic chain (n = 3) exhibited the highest anti-inflammatory activity (inhibition = 85.4%, IC50 = 3.7 μg mL−1). The authors, therefore, assumed that the anti-inflammatory activity of the isonicotinic acid analogs might be correlated to their inhibitory effect on COX-2. Docking simulations predicted several H-bond interactions of the amide NH and pyridine-N with Arg121 and Tyr356 residues within the ASC. Moreover, the authors reported that the amino acid residues Val350, Leu353, Tyr356, Leu360, Tyr386, Trp388, Val524, and Ala528 in the hydrophobic pocket interacted prominently with target inhibitors.
Modified NSAIDs
Conjugation of NSAIDs to biologically relevant molecules such as peptides is an emerging area for targeted drug delivery.104 Moreira et al.105 reported a series of naproxen–dehydrodipeptide conjugates (37) using a library of dehydrodipeptides. They contained N-terminal canonical amino acids (Phe, Tyr, Trp, Ala, Asp, Lys, Met) N-capped with naproxen and linked to a C-terminal dehydroaminoacid (ΔPhe, ΔAbu) that blunted 5-LOX and COX-2 expression as dual inhibitors (Fig. 31). The analogs 37 afforded IC50 = 48.9–67.4 μM, which represents lower inhibitory activity than that of naproxen as the parent molecule (IC50 = 22 μM).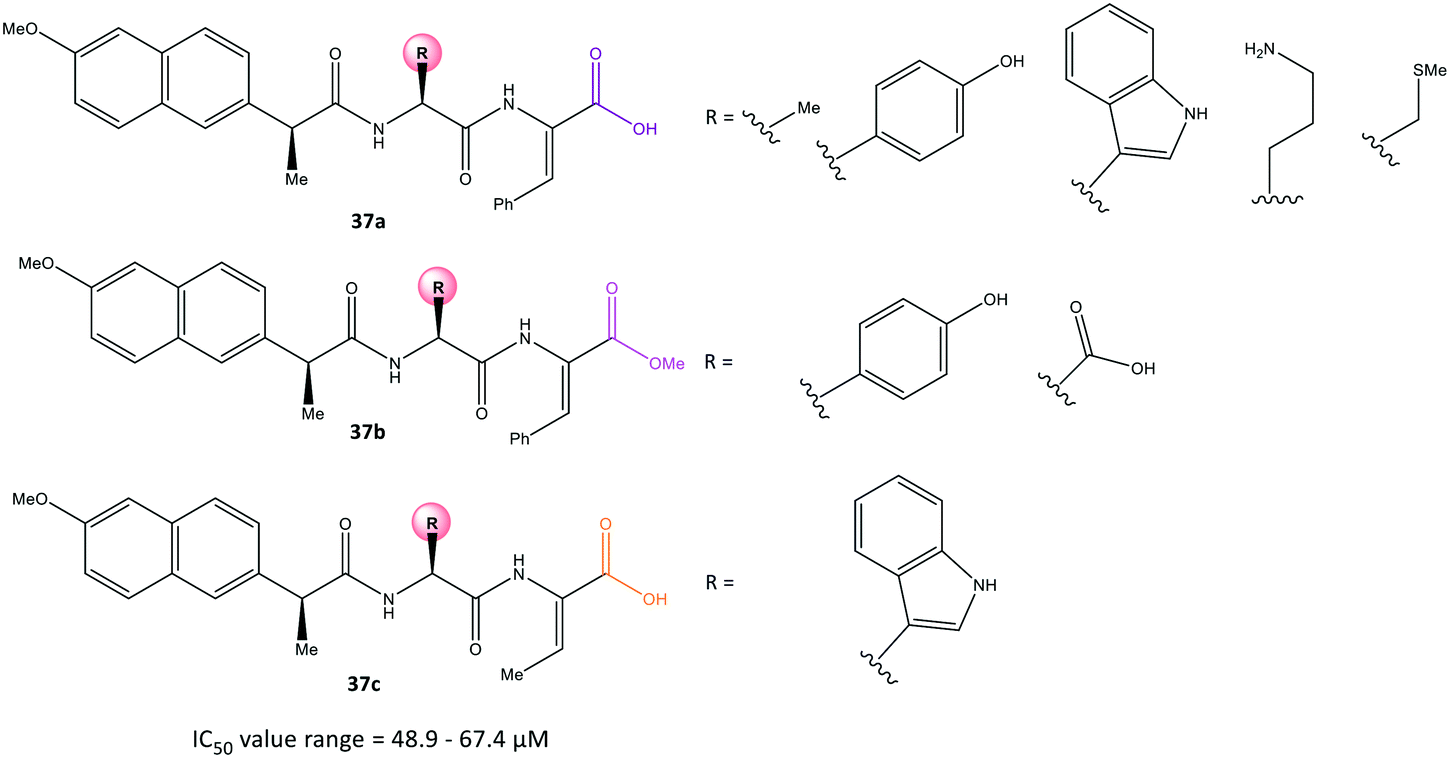 | ||
| Fig. 31 Naproxen–dehydrodipeptide derivatives 37. | ||
Interestingly, compound 37a (R = Me) displayed significant COX-2 inhibition (inhibition = 66%) similar to that of naproxen (inhibition = 61%). All tested compounds inhibited 5-LOX significantly except analog 37a (R = N-terminal charged Lys residue and propylamine). The analog 37a provided different environmental interactions from those of arachidonic acid as a natural substrate due to its high polarity. Docking simulations predicted that the amide-NH would coordinate to the naproxen moiety. The N-terminal amino acid in compound 37a would be involved in H-bond interactions with Arg120 and Tyr355 residues in a similar way as that seen for the –COOH group of naproxen. Notably, the authors reported that the C-terminal dehydrophenylalanine residue occupied the ASC of COX-2 lined by H and Pro residues.
In another study, mefenamic acid was modified structurally by Madduluri et al.106via biologically relevant molecules such as glycopeptides (Fig. 32). The COX-2 inhibitory activity of D-glucose-derived N-glycopeptides comprising mefenamic acid (38) were evaluated by immunoassays and exhibited percent inhibition of 45–85% compared with that of mefenamic acid (83%). The interaction of a tryptophan analog inside the ASC was predicted via docking simulations. Several H-bond interactions were displayed through NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Trp and NH and OH of glucose with the amino acids Arg120 and Tyr355. The indole ring was involved mainly in π–π interactions with the phenyl ring of Tyr355. The authors also reported that mefenamic acid was oriented within the hydrophobic pocket of COX-2.
O of Trp and NH and OH of glucose with the amino acids Arg120 and Tyr355. The indole ring was involved mainly in π–π interactions with the phenyl ring of Tyr355. The authors also reported that mefenamic acid was oriented within the hydrophobic pocket of COX-2.
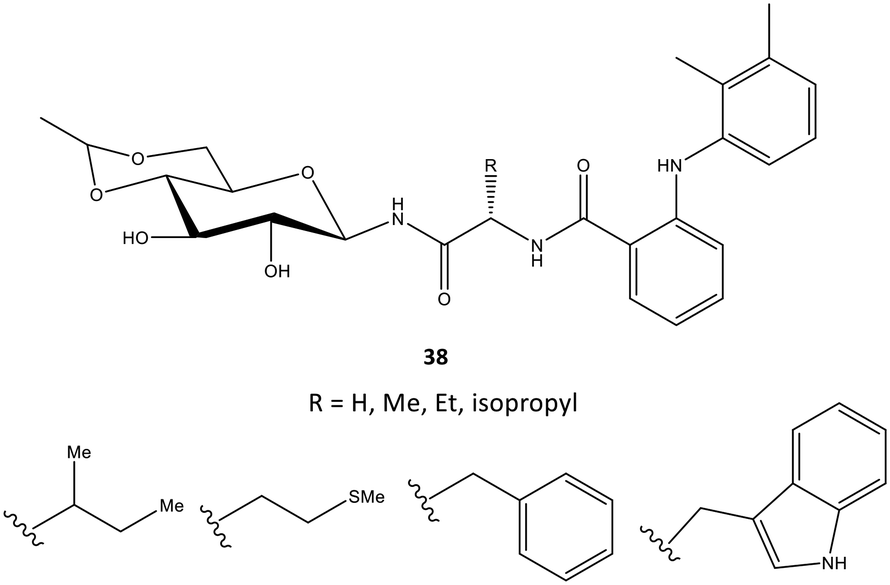 | ||
| Fig. 32 D-Glucose-derived N-glycopeptides containing derivatives of mefenamic acid 38. | ||
Perrone et al.107 reported a series of hybrid inhibitors comprising mofezolac-spacer-mofezolac (39a) and mofezolac-spacer-arachidonic acid (AA) (39b) that formed COX-1 homodimers (Fig. 33). Depending upon the nature of the spacer (conformationally flexible or rigid structures), selectivity and inhibitory activity towards COX-1 and COX-2 differed. They also reported that inhibition of AA induced platelet aggregation, in vitro coagulation, and cytotoxicity studies. The highest COX-1 selectivity was exhibited by the mofezolac-benzidine-mofezolac compound (IC50 = 0.08 μM, SI = 625) compared with that of mofezolac as a COX-1 inhibitor ((IC50 = 0.0079 μM, SI = 6392). The mofezolac-spacer-AA compounds 39b comprising butane (–(CH2)4–) and biphenyl spacers inhibited COX-2 selectively with IC50 = 0.8 μM (SI = 20) and 0.09 μM (SI = 189), respectively, whereas compound 39b with a phenylene spacer inhibited COX-1 selectively (IC50 = 0.05 μM, SI > 1000). The mofezolac-spacer-mofezolac compound 39a with an ethane spacer (–(CH2)2–) inhibited COX-2 selectively (IC50 = 0.12 μM, SI = 12).
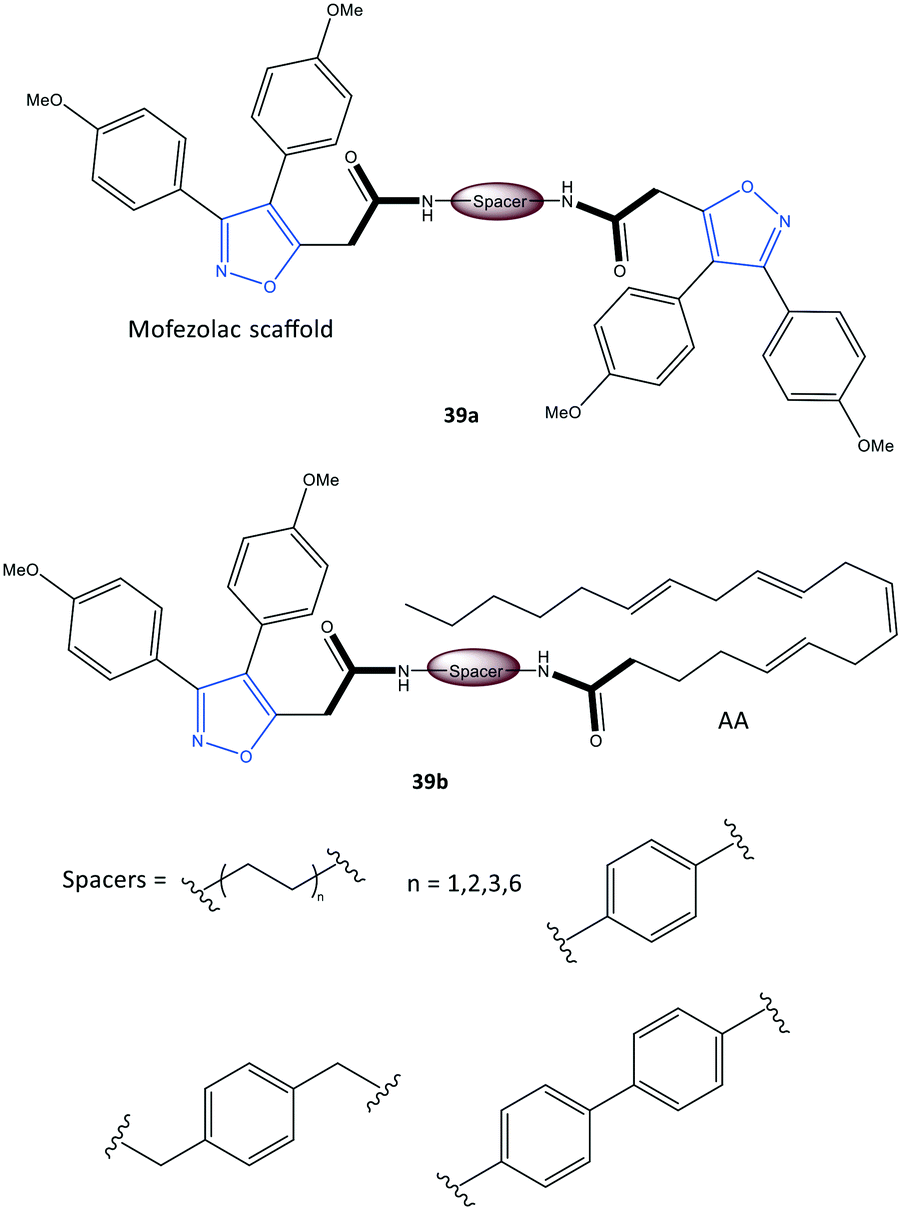 | ||
| Fig. 33 Mofezolac-spacer-mofezolac derivatives 39a and mofezolac-spacer-AA derivatives 39b. AA = arachidonic acid. | ||
The authors simulated the three-dimensional binding mode of derivatives 39a and 39b using molecular docking (Fig. 34). They reported the isoxazole ring to be involved in H-bond interactions with the Ser530 residue. An additional H-bond network was displayed between Arg120/Tyr355 with a carbonyl group and the outer isoxazole ring of derivatives 39a and 39b with a butane spacer at the ASC. Mofezolac was oriented differently in compound 39b with biphenyl spacers and established π–π interactions with Met522 and Tyr385 residues.
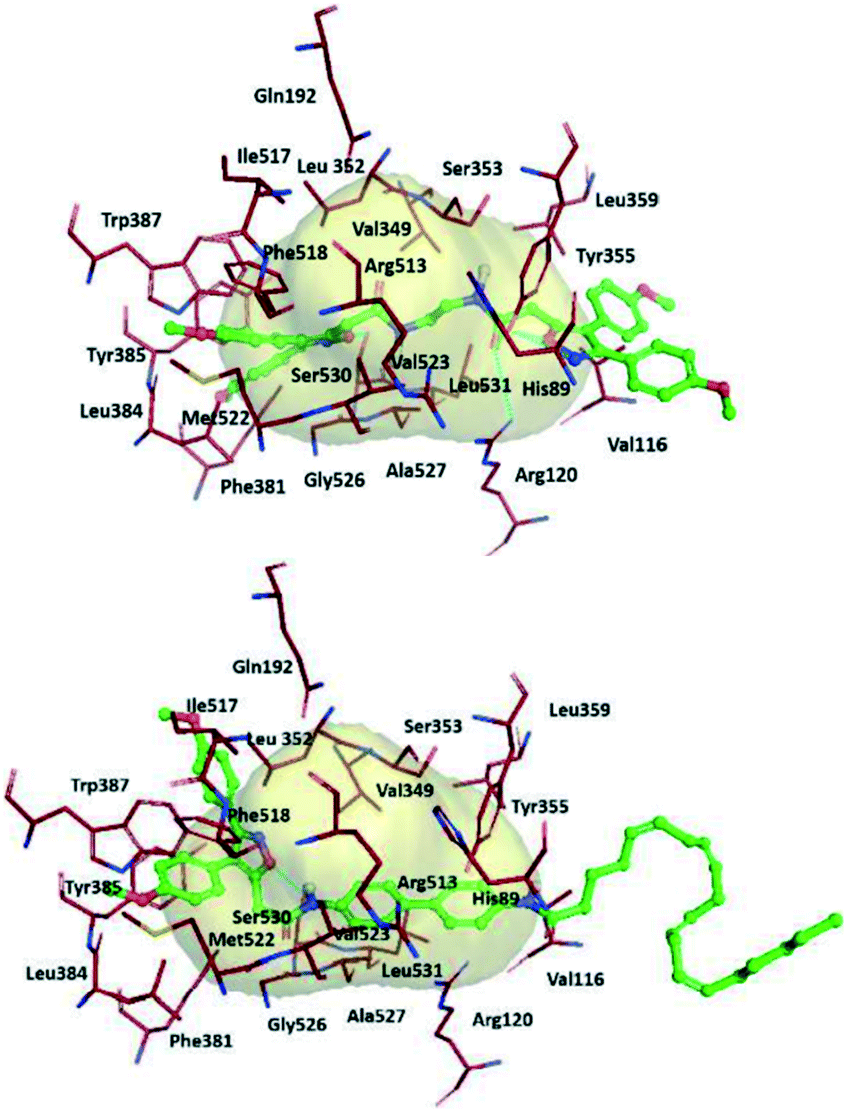 | ||
| Fig. 34 Proposed 3D binding mode inside the active-site cavity of COX-2. (Top) mof-spacer-mof derivative 39a (spacer = ethane), (bottom) mof-spacer-AA derivative 39b (spacer = biphenyl). Reproduced from ref. 107 with permission from Elsevier, copyright 2021.107 | ||
Notably, Ser530 and Arg120 were involved in H-bond interactions with the isoxazole ring, inner amidic NH, and carbonyl group of AA, respectively.
Zhang et al.108 reported a new class of carborane-based analogs 40 to inhibit COX-2 (Fig. 35). Analogs 40 were also tested for efficacious treatment of squamous cell carcinoma of the tongue by boron neutron capture therapy (BNCT). Such tumor cell-targeted radiotherapy was based on increasing the sensitivity of tumor cells to radiation. Nedunchezhian and coworkers reviewed the concept of BNCT for cancers comprehensively.109 However, a combination of a carborane scaffold with active motifs of COX-2 inhibitors (A1: selective COX inhibitors; A2: non-selective COX inhibitors) was introduced by Zhang et al.108 to explore lead structures with dual efficacy. These carborane-based analogs with various R groups showed inhibitory activity towards COX-2 (IC50 = 0.17–18.67 μM, SI = 0.86–108.06). Compound 40a bearing celecoxib as the A1 motif exhibited high COX-2 selectivity (IC50 = 0.17 μM, SI = 108.06) compared with that of celecoxib (IC50 = 0.06 μM, SI = 254.33) and low cytotoxicity (higher selectivity) against CAL27 cells than sodium borocaptate.
 | ||
| Fig. 35 Carborane-based derivatives 40. | ||
Computational modeling and molecular dynamic simulations
Identifying potential selective COX-2 inhibitors and their mechanism of action is vital to exploring novel lead compounds with high selectivity and few side effects to combat inflammation. Computational modeling and molecular dynamic simulations can help to achieve this goal. Docking simulations aid understanding of the essential features and motifs required to inhibit COX-2. Beura et al.110 reported a virtual screening method of potential inhibitors with various molecular structures (pyrazole, indole, pyrimidine, oxazole, pyridine, and pyrrole scaffolds) through ranking their best docking scores.The pharmaceutical bioactivity of validated molecules was evaluated using Lipinski's rule of five, and then the most active inhibitors were subjected to docking simulations. The authors predicted that structure 41a would show high selectivity towards COX-2 with a docking score of −8.16 kcal mol−1 (H-bond interactions with Thr212 and Asn382 residues and π–π interactions with the His207 residue) in comparison with structure 41b (Fig. 36).
 | ||
| Fig. 36 Potential COX-2 inhibitors 41, 42 and 43. | ||
Araújo et al.111 reported a molecular-modeling approach of potential COX-2 inhibitors through a quantitative structure–activity relationship using the ZINC database. The authors predicted the inhibitory activity (IC50) of the test compounds obtained with virtual screening through analyses of physiochemical, biological, and toxicological properties. Thus, virtual screening of rofecoxib analogs (including various molecular structures and functionalization based on pharmacophores and quantitative structure–activity relationships) was undertaken. The authors identified three proposed structures (42a–c) that showed high selectivity towards COX-2 (predicted –log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50 = 7.94–9.52) with a docking score of −9.5 kcal mol−1 (Fig. 36). Similar H-bond interactions were reported with the amino acids Arg513, His90, Ser530, and Phe518 within the ASC of COX-2-proposed structures compared with those of rofecoxib. Structure 42b was reported to be the most promising given all the applied predictions.
IC50 = 7.94–9.52) with a docking score of −9.5 kcal mol−1 (Fig. 36). Similar H-bond interactions were reported with the amino acids Arg513, His90, Ser530, and Phe518 within the ASC of COX-2-proposed structures compared with those of rofecoxib. Structure 42b was reported to be the most promising given all the applied predictions.
Leão et al.112 also reported a computational strategy to predict new rofecoxib-based COX-2 inhibitors. A ligand-based virtual screening study was applied by considering the pharmacokinetics, toxicological, and molecular-dynamic features. As a result, they predicted that structures 43a–c could be considered as putative COX-2 inhibitors (Fig. 36).
Perspective overview
An enzyme inhibitor is distinguished by its potency and specificity (selectivity) to minimize side effects and toxicity. Structural diversification of Food and Drug Administration-approved drugs, such as core scaffolds, substitution on subunit platforms, and molecular flexibility through applying linkers and spacers, are vital parameters to orient and locate target inhibitors within the ASC of the enzyme to improve pharmacokinetics and efficacy.Our review provides a fresh look at recent heterocyclic and acyclic derivatives included in drug discovery of potent and selective COX-2 inhibitors and hybrid drugs that in addition to COX-2 inhibit further enzymes (e.g., LOXs, sEH, 2, DHFR, α-glucosidase, EGFR, PDE5). We also highlight the role of synthetic COX-2 inhibitors as potential NSAIDs in pathways involving upregulated expression of pro-inflammatory cytokines (PGE2, IL-1β, IL-6, IL-10, TNF-α) and leukotrienes. Finally, we aimed to review the use of very recent synthetic COX-2 inhibitors introduced during the COVID-19 pandemic by highlighting structure–activity relationships and the mechanism of action within the ASC. The advanced and rational drug design is vital for their efficiency and selectivity that are useful for medicinal chemists and biologists.
The molecular design of synthetic COX-2 inhibitors modulates the hydrophobic and hydrophilic interactions with key amino acid residues such as Arg120, Arg513, Leu503, Val523, Val434, Tyr385, Ser530, Tyr255, and Ala513 within the ASC, determining the potency (IC50) and selectivity (SI) of COX-2 inhibition. Reported studies on the structure–activity relationships of various derivatives and hybrid structures have shown that aryl sulfonyl groups (–SO2NH2 and –SO2Me) at the para position were involved in H-bond interactions with Leu338, Ser339, Arg499, Phe504, Arg513, and Arg106 residues as well as π–π interactions mostly with Tyr341 and Val509 residues. The amino acids Arg513, Gln192, Ser353, Ser530, Arg499, His90, Tyr341, and Val102 are involved primarily in hydrophobic interactions. Electron-withdrawing substituents such as halogens and CF3 at the para position of the phenyl ring coordinate directly with central heterocyclic cores, provide the best orientation within the ASC, and enhance potency. Moreover, ortho–para and meta–para are the most favorable substitution patterns on the phenyl ring as an integral motif and could influence selectivity by occupying the secondary pocket of COX-2.
Conversely, flexible aliphatic linkers are more favored than aromatic linkers and impact directly on selectivity due to their adaptability in anchoring themselves within the ASC and secondary side pocket of COX-2.
Modification of known COX-2 inhibitors with linkers or spacers can enhance the SI significantly. Thiazole-based inhibitors exhibit potent inhibitory activity, whereas triaryl pyrazole-based inhibitors show stronger potency with a notable SI. Triazole derivatives exhibit slightly superior inhibitory activity and selectivity compared with that of other derivatives. Interestingly, aryledene scaffolds coordinated to a heterocyclic core exhibit high potency. Fused-ring heterocyclic derivatives and hybrid molecules have relatively high inhibitory activity compared with that of others and provide dual inhibition.
Furthermore, studies on structure–activity relationships could aid the prediction of structures capable of higher potency, selectivity, and a better benefit–risk balance. However, the potent inhibition of COX-2 by approved or withdrawn NSAID is connected to cardiovascular side effects.30 COX-2 inhibitors, e.g., valdecoxib and rofecoxib, were associated with a higher risk of stroke, while etoricoxib can increase blood pressure.29 These side effects seem to be due to the inhibition of PG production by the COX-2 expressed by the vascular endothelium.33 It is essential to mention that PGI2 counteracts the action of TXA2 and maintains a balance on vascular homeostasis.30,33 Therefore, improving our knowledge on the binding mechanism and structure relationships in the ASC are doubtless a vital predisposition for reducing the risk of side effects. It is necessary to mention that pharmacokinetic half-life, frequency, dose and duration of use and degree of inhibition of COX-2 (selectivity index) by NSAIDs have a direct effect on their side effects.30 As an exit strategy, hybrid molecules with multi-target enzyme inhibitory activity were assumed to overcome these obstacles and challenges. Dual inhibitors, e.g., hybrid COX-2/LOX and COX-2/sEH can block multiple pathways. We hope that the strategies described here might pave the way for structure-based drug discovery to develop effective, selective, safe, and pharmacokinetically suitable COX-2 inhibitors.
Notes and abbreviations
| ASC | Active-site cavity |
| COVID-19 | Coronavirus disease 2019 |
| COX | Cyclooxygenase |
| NSAID | Nonsteroidal anti-inflammatory drug |
| LOX | Lipoxygenase |
| sEH | Epoxide hydrolase |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| ACE | Angiotensin-converting enzyme |
| PG | Prostaglandin |
| TX | Thromboxane |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| AA | Arachidonic acid |
| EETs | Epoxyeicosatrienoic acids |
| EGFR | Epidermal growth factor receptor |
| PDE5 | Phosphodiesterase type 5 |
| BNCT | Boron neutron capture therapy |
| SI | Selectivity index |
| IC50 | Half-maximal inhibitory concentration |
| DHFR | Dihydrofolate reductase |
| μM | Micromolar |
| nM | Nanomolar |
| Tyr | Tyrosine |
| Val | Valine |
| Leu | Leucine |
| Ile | Isoleucine |
| Phe | L-Phenylalanine |
| His | Histidine |
| Arg | Arginine |
| Glu | Glutamine |
| Ser | Serine |
| Thr | Threonine |
| Pro | Proline |
| Asp | Aspartic acid |
| Gly | Glycine |
| Trp | Tryptophan |
| Lys | Lysine |
| Met | Methionine |
| Ala | Alanine |
| Gln | Glutamine |
Author contributions
M. A. reviewed the articles, analyzed data, and drafted the manuscript. M. A. and K. W. designed the review procedure. K. W. and S. B. revised the manuscript and provided general oversight. K.-D. W. and T. v. W. undertook overall management of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The processing charges of this review article were funded by the German Federal Ministry of Educations and Research (grant number: 03Z22DN12 to K. W.).References
- Z. Zhu, X. Lian, X. Su, W. Wu, G. A. Marraro and Y. Zeng, Respir. Res., 2020, 21, 224 CrossRef CAS PubMed.
- Z. Abdelrahman, M. Li and X. Wang, Front. Immunol., 2020, 11, 552909 CrossRef CAS PubMed.
- J. S. Chen, M. M. Alfajaro, R. D. Chow, J. Wei, R. B. Filler, S. C. Eisenbarth and C. B. Wilen, J. Virol., 2021, 95, e00014–e00021 CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Kruger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Muller, C. Drosten and S. Pohlmann, Cell, 2020, 181, 271–280 e278 CrossRef CAS PubMed.
- C. T. Robb, M. Goepp, A. G. Rossi and C. Yao, Br. J. Pharmacol., 2020, 177, 4899–4920 CrossRef CAS PubMed.
- S. Baghaki, C. E. Yalcin, H. S. Baghaki, S. Y. Aydin, B. Daghan and E. Yavuz, Int. J. Infect. Dis., 2020, 101, 29–32 CrossRef CAS PubMed.
- B. Sibbald, CMAJ, 2004, 171, 1027–1028 Search PubMed.
- S. X. Sun, K. Y. Lee, C. T. Bertram and J. L. Goldstein, Curr. Med. Res. Opin., 2007, 23, 1859–1866 CrossRef CAS PubMed.
- I. L. Meek, M. A. Van de Laar and H. E. Vonkeman, Pharmaceuticals, 2010, 3, 2146–2162 CrossRef CAS PubMed.
- C. Mattia and F. Coluzzi, Minerva Anestesiol., 2005, 71, 461–470 CAS.
- E. Ricciotti and G. A. FitzGerald, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 986–1000 CrossRef CAS PubMed.
- C. Gunaydin and S. S. Bilge, Eurasian J. Med., 2018, 50, 116–121 CAS.
- R. C. Harris and M. D. Breyer, Am. J. Physiol., 2001, 281, F1–F11 CrossRef CAS PubMed.
- B. Wang, L. Wu, J. Chen, L. Dong, C. Chen, Z. Wen, J. Hu, I. Fleming and D. W. Wang, Signal Transduction Targeted Ther., 2021, 6, 94 CrossRef CAS PubMed.
- J. M. Zhang and J. An, Int. Anesthesiol. Clin., 2007, 45, 27–37 CrossRef PubMed.
- P. Wojdasiewicz, L. A. Poniatowski and D. Szukiewicz, Mediators Inflammation, 2014, 2014, 561459 Search PubMed.
- S. Kany, J. T. Vollrath and B. Relja, Int. J. Mol. Sci., 2019, 20, 6008 CrossRef CAS PubMed.
- E. Ricciotti and G. A. FitzGerald, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 986–1000 CrossRef CAS PubMed.
- A. Gupta and K. Chander Chiang, Med. Hypotheses, 2020, 143, 110122 CrossRef CAS PubMed.
- M. G. Papich, in Saunders Handbook of Veterinary Drugs, ed. M. G. Papich and W. B. Saunders, St. Louis, 2016, pp. 689–690, DOI:10.1016/b978-0-323-24485-5.00491-5.
- E. M. Smyth, Clin. Lipidol., 2010, 5, 209–219 CrossRef CAS PubMed.
- C. Schulzke, Eur. J. Inorg. Chem., 2011, 2011, 1189–1199 CrossRef.
- N. V. Chandrasekharan, H. Dai, K. L. T. Roos, N. K. Evanson, J. Tomsik, T. S. Elton and D. L. Simmons, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13926–13931 CrossRef CAS PubMed.
- R. G. Kurumbail, J. R. Kiefer and L. J. Marnett, Curr. Opin. Struct. Biol., 2001, 11, 752–760 CrossRef CAS PubMed.
- A. L. Blobaum and L. J. Marnett, J. Med. Chem., 2007, 50, 1425–1441 CrossRef CAS PubMed.
- G. Rimon, R. S. Sidhu, D. A. Lauver, J. Y. Lee, N. P. Sharma, C. Yuan, R. A. Frieler, R. C. Trievel, B. R. Lucchesi and W. L. Smith, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 28–33 CrossRef CAS PubMed.
- J. L. Wang, D. Limburg, M. J. Graneto, J. Springer, J. R. Hamper, S. Liao, J. L. Pawlitz, R. G. Kurumbail, T. Maziasz, J. J. Talley, J. R. Kiefer and J. Carter, Bioorg. Med. Chem. Lett., 2010, 20, 7159–7163 CrossRef CAS PubMed.
- N. Zidar, K. Odar, D. Glavac, M. Jerse, T. Zupanc and D. Stajer, J. Cell. Mol. Med., 2009, 13, 3753–3763 CrossRef PubMed.
- K. Brune and P. Patrignani, J. Pain Res., 2015, 8, 105–118 CrossRef PubMed.
- S. Tacconelli, A. Bruno, R. Grande, P. Ballerini and P. Patrignani, Expert Opin. Drug Saf., 2017, 16, 791–807 CrossRef CAS PubMed.
- M. J. Lucido, B. J. Orlando, A. J. Vecchio and M. G. Malkowski, Biochemistry, 2016, 55, 1226–1238 CrossRef CAS PubMed.
- J. A. Giménez-Bastida, W. E. Boeglin, O. Boutaud, M. G. Malkowski and C. Schneider, FASEB J., 2019, 33, 1033–1041 CrossRef PubMed.
- B. Wang, L. Wu, J. Chen, L. Dong, C. Chen, Z. Wen, J. Hu, I. Fleming and D. W. Wang, Signal Transduction Targeted Ther., 2021, 6, 94 CrossRef CAS PubMed.
- I. D'Agostino, S. Tacconelli, A. Bruno, A. Contursi, L. Mucci, X. Hu, Y. Xie, R. Chakraborty, K. Jain, A. Sacco, M. Zucchelli, R. Landolfi, M. Dovizio, L. Falcone, P. Ballerini, J. Hwa and P. Patrignani, Pharmacol. Res., 2021, 170, 105744 CrossRef PubMed.
- A. M. Rayar, N. Lagarde, C. Ferroud, J. F. Zagury, M. Montes and M. Sylla-Iyarreta Veitia, Curr. Top. Med. Chem., 2017, 17, 2935–2956 CrossRef CAS PubMed.
- S. Xu, C. A. Rouzer and L. J. Marnett, IUBMB Life, 2014, 66, 803–811 CrossRef CAS PubMed.
- S. Xu, D. J. Hermanson, S. Banerjee, K. Ghebreselasie, G. M. Clayton, R. M. Garavito and L. J. Marnett, J. Biol. Chem., 2014, 289, 6799–6808 CrossRef CAS PubMed.
- A. Zarghi and S. Arfaei, Iran. J. Pharm. Res., 2011, 10, 655–683 CAS.
- B. J. Anderson, Paediatr. Anaesth., 2008, 18, 915–921 CrossRef PubMed.
- G. W. Przybyła, K. A. Szychowski and J. Gmiński, Clin. Exp. Pharmacol. Physiol., 2021, 48, 3–19 CrossRef PubMed.
- P. C. A. Kam and A. So, Curr. Anaesth. Crit. Care, 2009, 20, 50–53 CrossRef.
- B. Kis, J. A. Snipes and D. W. Busija, J. Pharmacol. Exp. Ther., 2005, 315, 1–7 CrossRef CAS PubMed.
- G. A. FitzGerald and C. Patrono, N. Engl. J. Med., 2001, 345, 433–442 CrossRef CAS PubMed.
- P. Sooriakumaran, Postgrad. Med. J., 2006, 82, 242–245 CrossRef CAS PubMed.
- A. L. Blobaum and L. J. Marnett, J. Biol. Chem., 2007, 282, 16379–16390 CrossRef CAS PubMed.
- S. M. I. Mahboubi Rabbani and A. Zarghi, Expert Opin. Ther. Pat., 2019, 29, 407–427 CrossRef CAS PubMed.
- K. Liaras, M. Fesatidou and A. Geronikaki, Molecules, 2018, 23, 685 CrossRef PubMed.
- H. U. Rashid, M. A. U. Martines, A. P. Duarte, J. Jorge, S. Rasool, R. Muhammad, N. Ahmad and M. N. Umar, RSC Adv., 2021, 11, 6060–6098 RSC.
- A. K. Chakraborti, S. K. Garg, R. Kumar, H. F. Motiwala and P. S. Jadhavar, Curr. Med. Chem., 2010, 17, 1563–1593 CrossRef CAS PubMed.
- P. Chandel, R. K. Rawal and R. Kaur, Future Med. Chem., 2018, 10, 2471–2492 CrossRef CAS PubMed.
- G. G. Ambati and S. M. Jachak, Curr. Med. Chem., 2021, 28, 1877–1905 CrossRef CAS PubMed.
- T. C. Mahesh, P. Shivani, M. Palmi and S. B. Pathik, Curr. Top. Med. Chem., 2016, 16, 2841–2862 CrossRef PubMed.
- A. Petrou, M. Fesatidou and A. Geronikaki, Molecules, 2021, 26, 3166 CrossRef CAS PubMed.
- B. N. Saglik, D. Osmaniye, S. Levent, U. A. Cevik, B. K. Cavusoglu, Y. Ozkay and Z. A. Kaplancikli, Eur. J. Med. Chem., 2021, 209, 112918 CrossRef CAS PubMed.
- A. H. M. Hussein, A. A. Khames, A. B. A. El-Adasy, A. A. Atalla, M. Abdel-Rady, M. I. A. Hassan, M. T. M. Nemr and Y. Elshaier, RSC Adv., 2020, 10, 29723–29736 RSC.
- G. A. Czapski, K. Czubowicz, J. B. Strosznajder and R. P. Strosznajder, Neurochem. Res., 2016, 41, 243–257 CrossRef CAS PubMed.
- C. Charlier and C. Michaux, Eur. J. Med. Chem., 2003, 38, 645–659 CrossRef CAS PubMed.
- P. J. Jacob and S. L. Manju, Bioorg. Chem., 2020, 100, 103882 CrossRef PubMed.
- L. Y. He, S. S. Zhang, D. X. Peng, L. P. Guan and S. H. Wang, Bioorg. Med. Chem. Lett., 2020, 30, 127376 CrossRef CAS PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed.
- B. Kaya Çavuşoğlu, B. N. Saglik, U. Acar Cevik, D. Osmaniye, S. Levent, Y. Ozkay and Z. A. Kaplancikli, Arch. Pharm., 2021, 354, e2000273 CrossRef PubMed.
- Z. H. Huang, L. Q. Yin, L. P. Guan, Z. H. Li and C. Tan, Bioorg. Med. Chem. Lett., 2020, 30, 127173 CrossRef CAS PubMed.
- M. J. Naim, O. Alam, F. Nawaz, M. J. Alam and P. Alam, J. Pharm. BioAllied Sci., 2016, 8, 2–17 CrossRef CAS PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- C. Scholtz and D. L. Riley, React. Chem. Eng., 2021, 6, 138–146 RSC.
- E. M. Gedawy, A. E. Kassab and A. M. El Kerdawy, Eur. J. Med. Chem., 2020, 189, 112066 CrossRef CAS PubMed.
- K. R. A. Abdellatif, E. K. A. Abdelall, M. B. Labib, W. A. A. Fadaly and T. H. Zidan, Bioorg. Chem., 2020, 105, 104418 CrossRef CAS PubMed.
- N. Hashemi Goradel, M. Najafi, E. Salehi, B. Farhood and K. Mortezaee, J. Cell. Physiol., 2019, 234, 5683–5699 CrossRef CAS PubMed.
- J. J. P, S. L. Manju, K. R. Ethiraj and G. Elias, Eur. J. Pharm. Sci., 2018, 121, 356–381 CrossRef PubMed.
- W. Akhtar, A. Marella, M. M. Alam, M. F. Khan, M. Akhtar, T. Anwer, F. Khan, M. Naematullah, F. Azam, M. A. Rizvi and M. Shaquiquzzaman, Arch. Pharm., 2021, 354, e2000116 CrossRef PubMed.
- A. A. Spector and H. Y. Kim, Biochim. Biophys. Acta, 2015, 1851, 356–365 CrossRef CAS PubMed.
- C. Pereira-Leite, C. Nunes, S. K. Jamal, I. M. Cuccovia and S. Reis, Med. Res. Rev., 2017, 37, 802–859 CrossRef PubMed.
- A. H. Abdelazeem, A. G. Safi El-Din, M. M. Abdel-Fattah, N. H. Amin, S. M. El-Moghazy and M. T. El-Saadi, Eur. J. Med. Chem., 2020, 205, 112662 CrossRef CAS PubMed.
- B. Zhang, X. T. Hu, K. M. Zhou, Y. S. Yang and H. L. Zhu, Bioorg. Chem., 2020, 102, 104096 CrossRef CAS PubMed.
- B. Zhang, X. T. Hu, J. Gu, Y. S. Yang, Y. T. Duan and H. L. Zhu, Bioorg. Chem., 2020, 105, 104390 CrossRef CAS PubMed.
- T. S. Ibrahim, I. M. Salem, S. M. Mostafa, O. I. El-Sabbagh, M. K. M. ElKhamisi, L. Hegazy and B. Elgendy, Bioorg. Chem., 2020, 100, 103878 CrossRef CAS PubMed.
- M. Assali, M. Abualhasan, H. Sawaftah, M. Hawash and A. Mousa, J. Chem., 2020, 2020, 6393428 Search PubMed.
- S. M. Li, S. E. Tsai, C. Y. Chiang, C. Y. Chung, T. J. Chuang, C. C. Tseng, W. P. Jiang, G. J. Huang, C. Y. Lin, Y. C. Yang, M. T. Fuh and F. F. Wong, Bioorg. Chem., 2020, 104, 104333 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, B. Rahul, A. A. B. Mohamed, M. S. M. Abdelbaky, S. Garcia-Granda, A. A. El-Emam, M. J. Percino and S. Thamotharan, ACS Omega, 2021, 6, 6996–7007 CrossRef CAS PubMed.
- M. S. Bekheit, H. A. Mohamed, B. F. Abdel-Wahab and M. A. Fouad, Med. Chem. Res., 2021, 30, 1125–1138 CrossRef CAS.
- D. L. Namera, S. S. Thakkar, P. Thakor, U. Bhoya and A. Shah, J. Biomol. Struct. Dyn., 2020, 39, 7150–7159 CrossRef PubMed.
- N. H. Metwally and M. S. Mohamed, Bioorg. Chem., 2020, 99, 103438 CrossRef CAS PubMed.
- M. V. Raimondi, O. Randazzo, M. La Franca, G. Barone, E. Vignoni, D. Rossi and S. Collina, Molecules, 2019, 24, 1140 CrossRef PubMed.
- Y. M. Omar, S. G. Abdel-Moty and H. H. M. Abdu-Allah, Bioorg. Chem., 2020, 97, 103657 CrossRef CAS PubMed.
- S. Leone, A. Ottani and A. Bertolini, Curr. Top. Med. Chem., 2007, 7, 265–275 CrossRef CAS PubMed.
- M. B. Labib, A. M. Fayez, E. El-Nahass, M. Awadallah and P. A. Halim, Bioorg. Chem., 2020, 104, 104308 CrossRef CAS PubMed.
- A. Siwach and P. K. Verma, BMC Chem., 2020, 14, 70 CrossRef CAS PubMed.
- B. Sever, M. D. Altintop, A. Ozdemir, G. Akalin Ciftci, D. E. Ellakwa, H. Tateishi, M. O. Radwan, M. A. A. Ibrahim, M. Otsuka, M. Fujita, H. I. Ciftci and T. F. S. Ali, Molecules, 2020, 25, 5190 CrossRef CAS PubMed.
- C. Gridelli, P. Maione, M. A. Bareschino, C. Schettino, P. C. Sacco, R. Ambrosio, V. Barbato, M. Falanga and A. Rossi, Anticancer Res., 2010, 30, 1301–1310 CAS.
- L. Szczukowski, E. Krzyzak, A. Zborowska, P. Zajac, K. Potyrak, K. Peregrym, B. Wiatrak, A. Marciniak and P. Swiatek, Int. J. Mol. Sci., 2020, 21, 9623 CrossRef CAS PubMed.
- Z. Zhao, J. Yue, X. Ji, M. Nian, K. Kang, H. Qiao and X. Zheng, Bioorg. Chem., 2021, 108, 104557 CrossRef CAS PubMed.
- M. S. Jan, S. Ahmad, F. Hussain, A. Ahmad, F. Mahmood, U. Rashid, O. U. Abid, F. Ullah, M. Ayaz and A. Sadiq, Eur. J. Med. Chem., 2020, 186, 111863 CrossRef CAS PubMed.
- K. R. A. Abdellatif, E. K. A. Abdelall, H. A. H. Elshemy, E. El-Nahass, M. M. Abdel-Fattah and Y. Y. M. Abdelgawad, Arch. Pharm., 2021, 354, e2000328 CrossRef PubMed.
- J. D. Bilavendran, A. Manikandan, P. Thangarasu and K. Sivakumar, Bioorg. Chem., 2020, 94, 103484 CrossRef PubMed.
- C. Tratrat, M. Haroun, A. Paparisva, C. Kamoutsis, A. Petrou, A. Gavalas, P. Eleftheriou, A. Geronikaki, K. N. Venugopala, H. Kochkar and A. B. Nair, Molecules, 2021, 26, 659 CrossRef CAS PubMed.
- T. O. Olomola, M. J. Mphahlele and S. Gildenhuys, Bioorg. Chem., 2020, 100, 103945 CrossRef CAS PubMed.
- M. A. Shaaban, A. M. Kamal, S. I. Faggal, N. A. Farag, N. M. Aborehab, A. E. Elsahar and K. O. Mohamed, Arch. Pharm., 2020, 353, e2000027 CrossRef PubMed.
- S. M. H. Sanad and A. E. M. Mekky, J. Iran. Chem. Soc., 2020, 17, 3299–3315 CrossRef CAS.
- T. Chen, Y. Huang, J. X. Hong, X. K. Wei, F. Zeng, J. L. Li, G. T. Ye, J. Yuan and Y. H. Long, Mar. Drugs, 2021, 19, 12 CrossRef CAS PubMed.
- M. J. Uddin, S. Xu, B. C. Crews, A. M. Aleem, K. Ghebreselasie, S. Banerjee and L. J. Marnett, ACS Med. Chem. Lett., 2020, 11, 1881–1885 CrossRef CAS PubMed.
- S. Xie, C. Qi, Y. Duan, Q. Xu, Y. Liu, Y. Huang, X. Yin, W. Sun, Y. Zhou and Y. Zhang, Org. Chem. Front., 2020, 7, 1349–1357 RSC.
- E. M. Ahmed, M. S. A. Hassan, A. A. El-Malah and A. E. Kassab, Bioorg. Chem., 2020, 95, 103497 CrossRef CAS PubMed.
- S. Yaqoob, N. Nasim, R. Khanam, Y. Wang, A. Jabeen, U. Qureshi, Z. Ul-Haq, H. R. El-Seedi, Z. H. Jiang and F. A. Khan, Molecules, 2021, 26, 1272 CrossRef CAS PubMed.
- A. Carvalho, J. Gallo, D. M. Pereira, P. Valentão, P. B. Andrade, L. Hilliou, P. M. T. Ferreira, M. Bañobre-López and J. A. Martins, Nanomaterials, 2019, 9, 541 CrossRef CAS PubMed.
- R. Moreira, P. J. Jervis, A. Carvalho, P. M. T. Ferreira, J. A. Martins, P. Valentao, P. B. Andrade and D. M. Pereira, Pharmaceutics, 2020, 12, 122 CrossRef CAS PubMed.
- V. K. Madduluri and A. K. Sah, ChemistrySelect, 2020, 5, 2197–2200 CrossRef CAS.
- M. G. Perrone, M. Miciaccia, P. Vitale, S. Ferorelli, C. Araujo, G. S. de Almeida, T. F. Souza Domingos, L. da Silva, M. de Padula, L. M. Cabral, P. C. Sathler, C. Bonaccorso, C. G. Fortuna and A. Scilimati, Eur. J. Med. Chem., 2021, 209, 112919 CrossRef CAS PubMed.
- T. Zhang, S. Du, Y. Wang, Y. Guo, Y. Yi, B. Liu, Y. Liu, X. Chen, Q. Zhao, D. He, Z. Wang, H. Zhang and Q. Ma, ChemistrySelect, 2020, 5, 14652–14660 CrossRef CAS.
- K. Nedunchezhian, N. Aswath, M. Thiruppathy and S. Thirugnanamurthy, J. Clin. Diagn. Res., 2016, 10, ZE01–ZE04 Search PubMed.
- S. Beura and P. Chetti, J. Mol. Struct., 2020, 1216, 128271 CrossRef CAS.
- P. H. F. Araújo, R. S. Ramos, J. N. da Cruz, S. G. Silva, E. F. B. Ferreira, L. R. de Lima, W. J. C. Macedo, J. M. Espejo-Roman, J. M. Campos and C. B. R. Santos, Molecules, 2020, 25, 4183 CrossRef PubMed.
- R. P. Leão, J. V. Cruz, G. V. da Costa, J. N. Cruz, E. F. B. Ferreira, R. C. Silva, L. R. de Lima, R. S. Borges, G. B. Dos Santos and C. B. R. Santos, Pharmaceuticals, 2020, 13, 209 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |