
19F chemical library and 19F-NMR for a weakly bound complex structure†
Shoko
Shinya‡
a,
Ritsuko
Katahira‡
a,
Kyoko
Furuita‡
a,
Toshihiko
Sugiki
a,
Young-Ho
Lee
 abcd,
Yoshikazu
Hattori
a,
Kohei
Takeshita
a,
Atsushi
Nakagawa
a,
Aoi
Kokago
e,
Ken-ichi
Akagi
f,
Muneki
Oouchi
g,
Fumiaki
Hayashi
g,
Takanori
Kigawa
h,
Midori
Takimoto-Kamimura
*i,
Toshimichi
Fujiwara
a and
Chojiro
Kojima
*ae
abcd,
Yoshikazu
Hattori
a,
Kohei
Takeshita
a,
Atsushi
Nakagawa
a,
Aoi
Kokago
e,
Ken-ichi
Akagi
f,
Muneki
Oouchi
g,
Fumiaki
Hayashi
g,
Takanori
Kigawa
h,
Midori
Takimoto-Kamimura
*i,
Toshimichi
Fujiwara
a and
Chojiro
Kojima
*ae
aInstitute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565-0871, Japan
bResearch Center for Bioconvergence Analysis, Korea Basic Science Institute, Chungbuk 28119, South Korea
cBio-Analytical Science, University of Science and Technology, Daejeon 34113, South Korea
dGraduate School of Analytical Science and Technology, Chungnam National University, Daejeon 34134, South Korea
eGraduate School of Engineering Science, Yokohama National University, Tokiwadai 79-5, Hodogaya-ku, Yokohama 2408501, Japan. E-mail: kojima-chojiro-xk@ynu.ac.jp
fNational Institute of Biomedical Innovation, Health and Nutrition, 7-6-8 Saito Asagi, Ibaraki-city, Osaka 567-0085, Japan
gRIKEN Spring-8 Center, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan
hRIKEN Center for Biosystems Dynamics Research, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan
iQuantum-Structural Life Science Laboratories, CBI Research Institute, 3-11-1 Shibaura, Minato-ku, Tokyo 108-0023, Japan. E-mail: kamimura@cbi-society.org
First published on 22nd July 2022
Abstract
Fragment-based drug discovery (FBDD), which involves small compounds <300 Da, has been recognized as one of the most powerful tools for drug discovery. In FBDD, the affinity of hit compounds tends to be low, and the analysis of protein–compound interactions becomes difficult. In an effort to overcome such difficulty, we developed a 19F-NMR screening method optimizing a 19F chemical library focusing on highly soluble monomeric molecules. Our method was successfully applied to four proteins, including protein kinases and a membrane protein. For FKBP12, hit compounds were carefully validated by protein thermal shift analysis, 1H–15N HSQC NMR spectroscopy, and isothermal titration calorimetry to determine dissociation constants and model complex structures. It should be noted that the 1H and 19F saturation transfer difference experiments were crucial to obtaining highly precise model structures. The combination of 19F-NMR analysis and the optimized 19F chemical library enables the modeling of the complex structure made up of a weak binder and its target protein.
Introduction
Fragment-based drug discovery (FBDD) has emerged in the past two decades as a powerful tool.1,2 FBDD uses small compounds (molecular weight < 300 Da) that bind target proteins specifically. These compounds are optimized by increasing their size and/or linking them together to improve the binding affinity. In FBDD, the detection of a weak interaction is crucial because small compounds possess weak affinity (>micromolar) in many cases. NMR spectroscopy is useful for analyzing high-concentration solutions and detecting weak interactions and is thus frequently used in FBDD for screening and hit validation.3,419F possesses a natural abundance of 100% and high sensitivity for NMR observation, corresponding to 83% of the 1H sensitivity. The narrow linewidth and large chemical shift dispersion in 19F-NMR enable the use of solution cocktails of more than 20 compounds without the overlapping of chemical shifts. 19F-NMR screening is performed using fluorine-containing compounds to detect hit compounds through the disappearance of 19F-NMR signals and requires a special chemical library composed of soluble fluorine-containing compounds.5
Structural information of the protein–drug complex is essential to optimize a small compound.6 X-ray crystallography is most widely used to obtain the protein–drug complex structure; however, it does not work well for weakly bound complexes. Therefore, obtaining the structural information of the weakly binding complex is still a significant bottleneck in FBDD. NMR analysis provides structural information about the protein–drug complex. For example, a drug-binding site is easily identified by heteronuclear single quantum coherence (HSQC) titration,7,81H saturation transfer difference (STD),8,9 and 19F STD10 experiments, and a complex structure is generated using a docking program, such as HADDOCK.11
In this report, we constructed a chemical library composed of small, fluorine-containing, highly water-soluble, and monomeric compounds. This library was successfully applied to the 19F-NMR screening of four proteins composed of one soluble protein, two protein kinases, and one membrane protein. Hit compounds obtained by 19F-NMR screening were validated by protein thermal shift (PTS) analysis, 1H–15N HSQC NMR spectroscopy, and isothermal titration calorimetry (ITC), and the values of the dissociation constants (Kd) were determined. The complex structure of one of the small weak binders was modeled based on 1H and 19F saturation transfer difference (STD) experiments and chemical shift perturbation HSQC experiments.
Experimental
Sample preparation for proteins
Human FKBP12 with a glutathione S-transferase (GST) tag encoded in the pKT7 vector (a gift from Prof. Toshiyuki Kohno, MITILS and Kitasato University) was overexpressed in Escherichia coli Rosetta™ (DE3) (Novagen, Madison, WI, USA) and grown in LB medium. For uniform 15N labelling, Escherichia coli cells were grown in M9 minimal medium with 0.5 g L−1 15NH4Cl. Protein expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside when the optical density at 600 nm reached 0.6. After induction, cells were incubated for 5 h at 37 °C. Cells were harvested by centrifugation, suspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 0.5 mM ethylenediaminetetraacetic acid, and 1 mM dithiothreitol (DTT)), and sonicated. After the cell debris was removed by centrifugation (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 30 min), the supernatant was applied to an affinity chromatography column comprising 5 ml of Glutathione-Sepharose 4B (GE Healthcare, Waukesha, WI, USA). The resin was washed with lysis buffer and the protein was eluted with lysis buffer containing 30 mM reduced glutathione. Following cleavage of the GST tag overnight incubation at 4 °C with HRV 3C protease, the solution was applied to a HiLoad 26/600 Superdex 75 pg (GE Healthcare) size-exclusion chromatography column equilibrated with NMR buffer (50 mM sodium phosphate, 50 mM NaCl, and pH 6.8) to remove GST and other impurities. Finally, the solution was concentrated using an Amicon Ultra-15 filter unit with a molecular weight cutoff of 3 kDa (EMD Millipore, Billerica, MA, USA).
000 rpm at 4 °C for 30 min), the supernatant was applied to an affinity chromatography column comprising 5 ml of Glutathione-Sepharose 4B (GE Healthcare, Waukesha, WI, USA). The resin was washed with lysis buffer and the protein was eluted with lysis buffer containing 30 mM reduced glutathione. Following cleavage of the GST tag overnight incubation at 4 °C with HRV 3C protease, the solution was applied to a HiLoad 26/600 Superdex 75 pg (GE Healthcare) size-exclusion chromatography column equilibrated with NMR buffer (50 mM sodium phosphate, 50 mM NaCl, and pH 6.8) to remove GST and other impurities. Finally, the solution was concentrated using an Amicon Ultra-15 filter unit with a molecular weight cutoff of 3 kDa (EMD Millipore, Billerica, MA, USA).
GSK3 was purchased from Upstate Biotechnology (Lake Placid, New York, USA). JAK3 was purchased from Carna Biosciences Inc (Kobe, Japan). VSOP was prepared as reported.12
19F fragment screening
Fluorine {proton decoupled} NMR spectra were recorded on a Bruker AVANCE III HD 400 spectrometer equipped with a 5 mm BBFO probe at 298 K. Fragment screening was performed by an R2-filter (140 ms filter period) (Table 1) and normal 1D experiments.13 The data were collected with a spectral width of 110 ppm. The acquisition and repetition times were 0.79 and 4.79 s for the R2-filter experiment, respectively. The binding of the fragments was evaluated by taking the ratio of the signal intensities of the R2-filter experiments (Ic/If), where Ic and If denote the signal intensities in the presence and absence of protein, respectively. Ic and If were normalized by the signal intensity of 40 μM sodium trifluoroacetate (Sigma-Aldrich Co., Ltd) added to the sample. The final concentrations of DMSO-d6 and D2O were 1% and 10%, respectively.| Purpose | Experiment | Brief description about the experiment |
|---|---|---|
| Screening | R 2-filtered 19F-NMR | A 19F-observed 1D NMR experiment with the R2 filter, which attenuates the signal of macromolecules. When a compound is mixed with a protein, the signal of compounds that bind to the protein is attenuated but that of free compounds is not. Therefore, the signal of compounds that bind to proteins is easily identified |
| 19F 2D DOSY | A 19F-observed 2D NMR experiment which measures translational diffusion coefficients (D). The change in D of a compound with or without a protein is used to evaluate whether the compound binds to the protein or not | |
| Hit validation and obtaining binding information | PTS | Protein thermal shift assay: this assay examines the change in protein denaturation temperature in the presence or absence of a compound. When the compound binds to the protein, the protein is thermally stabilized due to complex formation. The degree of the increase in protein denaturation temperature is directly related to the strength of the binding |
| 1H–15N HSQC | A 1H-observed 2D NMR experiment which provides the correlation between 1H and directly attached 15N nucleus. In the case of proteins, backbone amide group of each residue gives a peak in the 1H–15N HSQC spectrum. By examining the change in 2D 1H–15N HSQC spectra of the protein with and without compounds, the compound binding to the protein, the compound binding site on the protein, and the Kd value of the protein–compound complex are determined | |
| ITC | Isothermal titration calorimetry measures the enthalpy change (ΔH) induced by the interaction between a protein and a compound. In experiment the compound is titrated to the protein, or vice versa. Other thermodynamic parameters (e.g., TΔS and ΔG), the binding constant, and the stoichiometry are determined by this titration | |
| 1H STD | A 1H-observed 1D NMR experiment, in which, the spectrum is measured after irradiating specific 1H atoms (usually methyl groups of the protein) with a sample of compounds mixed with a small amount of a protein. The signals of 1H close to the irradiated 1H are detected as signal intensity change. Information on the compound binding to the protein and the binding site are obtained | |
| 19F STD | A 1H-observed 1D NMR experiment, in which the spectrum is measured after irradiating specific 19F atoms (usually fluorine group of the compound) with a sample of compounds mixed with a small amount of a protein. The signals of 1H close to the irradiated 19F are detected. Information on the compound binding to the protein and the binding site are obtained | |
| TrNOE | A 1H-observed 2D NMR experiment, in which a 2D NOESY spectrum is measured with a sample of compounds mixed with a small amount of protein. When the compound binds to the protein, the compound shows negative NOE signals. Free compound gives positive NOE signals, so the compound binding to the protein is easily identified. The negative NOE signals provide distance information on the compound when bound to the protein | |
| Water-LOGSY | A 1H-observed 1D NMR experiment, in which only the water signal is excited and the 1D NOESY spectrum is measured with a sample of compounds mixed with a small amount of protein. The sign of the compound's signal changes due to negative NOE when the compound binds to the protein. So information on the compound binding to the protein is obtained |
For FKBP12, 40 μM mixtures were diluted in buffer (50 mM sodium phosphate, 50 mM NaCl, pH 6.8) with or without 11 μM protein. Competition binding experiments were performed by the addition of 10 μM FK506 (Sigma-Aldrich Co., Ltd) to the solution of the hit fragment and FKBP12.
For JAK3, 40 μM mixtures were diluted in buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10% glycerol, 5 mM DTT) with or without 5.6 μM protein. Competition binding experiments were performed by the addition of 20 μM tofacitinib (Funakoshi) to the solution of the hit fragment and JAK3. For GSK3, 40 μM mixtures were diluted in buffer (20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 5% glycerol, 2 mM DTT) with or without 4 μM protein. Competition binding experiments were performed by the addition of 8 μM SB216763 (Funakoshi) to the solution of the hit fragment and GSK3. For VSOP, 40 μM mixtures were diluted in buffer (20 mM HEPES-Na, 200 mM NaCl, 0.2% 5-cyclohexyl-1-pentyl-β-D-maltoside) with or without 5 μM protein.
19F DOSY
NMR samples for the 19F 2D diffusion-ordered spectroscopy (DOSY) experiments16 (Table 1) consisted of 10 or 11 fluorine library compounds (36 or 40 μM) and sodium trifluoroacetate (40 μM) with or without 15N-labeled FKBP12 (11 μM) dissolved in 89.2% H2O/9.8% D2O/1% DMSO-d6 containing 50 mM sodium phosphate (pH 6.8) and 50 mM sodium chloride. The 19F DOSY NMR experiments were performed on an AVANCE III HD 600 MHz (proton frequency) spectrometer equipped with a 5 mm QCI 1H–13C/15N/19F CryoProbe and a SampleJet (Bruker). The measurements were performed using the pulse sequence described by Vulpetti et al., which uses a broadband adiabatic refocusing pulse to cover a large bandwidth.14 The spectra were measured at 25 °C. The carrier frequency was −95 ppm, and the spectral width was 90 ppm. The strength of the pulsed field gradient was linearly increased from 2.65 G cm−1 to 50.35 G cm−1 in 32 steps. Sixty-four scans were recorded for each increment. The delay for the length of the gradients was set to 1.5 ms, and the delay for diffusion was set to 80 ms. The data were processed by TOPSPIN 3.2 (Bruker), and the diffusion coefficients were calculated using the Dynamics Center (Bruker).Thermal shift assay (Table 1)
All reaction mixtures were prepared in each well of a MicroAmp Fast 96-well reaction plate (ABI) on ice. The total liquid volume of the reaction mixtures was adjusted to 20 μL as follows. For the no protein control (NPC) sample, 5 μL of protein thermal shift buffer (ABI), 2.5 μL of 1000×-diluted protein thermal shift™ dye (ABI), and 12.5 μL of solvent, which was used for dissolving FKBP12 in the other samples, were mixed. For the test samples, 7 μL of 240 μM FKBP12, 0.4 μL of the 4 mM analyte compound cocktail containing 10 structurally diverse various small molecule fragments, and 5.1 μL of solvent were added instead of the 12.5 μL of solvent that was used in the NPC sample to adjust the final concentration of FKBP12 and analyte compounds to 84 μM and 80 μM, respectively. As a positive control sample, in the same manner, FKBP12 and FK506 were mixed at final concentrations of 84 μM and 150 μM, respectively. Following sufficient mixing of the reaction by gentle pipetting, the reaction plate was completely sealed with MicroAmp optical adhesive film (ABI), and the reaction mixtures were gently spun down. Protein melt reactions were started after 2 h of incubation at room temperature. The running of the reactions and the detection of fluorescence emission were performed by using StepOnePlus Real-time PCR equipment and StepOne software V 2.2.2 (ABI). The sample temperature was gradually increased from 25 to 98 °C at a constant rate of 1 °C min−1. The fluorescence emission of the dye in each reaction was detected in accordance with the elevating sample temperature in a real-time manner using a 490 nm excitation filter (SYBR Green filter) and a 575 nm emission filter (ROX filter). All samples were measured in quintuplet, and the standard error of the mean of the thermal denaturing temperature (Tm) values was calculated. The fluorescence intensity of the dye in each reaction was plotted against the sample temperature, and the midpoint of Tm for FKBP12 was estimated by curve fitting to the folded-unfolded transition moiety of the raw melting curves with a simple Boltzmann equation by using Protein Thermal Shift software™ (ABI),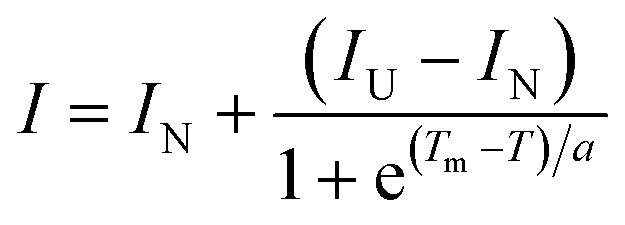 | (1) |
Titration experiment of 15N HSQC NMR spectroscopy
NMR samples for the chemical shift perturbation experiments consisting of 15N-labeled FKBP12 (11 μM) with or without the mixture solution (40 μM) or hit compound (40 μM) were prepared. The 15N HSQC experiments (Table 1) were performed on an AVANCE III HD 600 MHz spectrometer equipped with a 5 mm QCI-P CryoProbe (Bruker). The spectra were measured at 25 °C.To determine the dissociation constant, Kd, the compounds were added to a solution of 15N-labeled FKBP12 (11 μM), and a series of 15N HSQC spectra were recorded at various compound concentrations in the 0–400 μM range. Δδobs values relative to the maximum shift (Δδmax) were plotted against the free compound concentration, and Kd was estimated by nonlinear curve-fitting based on eqn (2),15
| Δδobs = Δδmax{[P]t + [L]t + Kd − [([P]t + [L]t + Kd)2 − 4[P]t[L]t]1/2}/2[P]t | (2) |
ITC
Isothermal titration calorimetry (ITC) (Table 1) was conducted to examine the binding of FKBP12 to the ligands in 50 mM phosphate buffer (pH 6.8) containing 50 mM NaCl and 1% DMSO by using a VP-ITC instrument (Malvern, UK) at 25 °C. Considering the low solubility of the ligands, reverse titrations of 2.1 mM of FKBP12 in a syringe to #4–1, #8–1, or #8–2 in the cell were conducted, as was our previous study.16 The concentrations of ligands and FKBP12 were 25–50 μM and 1.5–2.1 mM, respectively. The FKBP12 titration comprised 22 or 38 injections with a spacing time of 300 s and a stirring speed of 307 rpm. The injection volume was 2 or 7 μL for each sample. Binding isotherms were analyzed with the theoretical curve of eqn (3),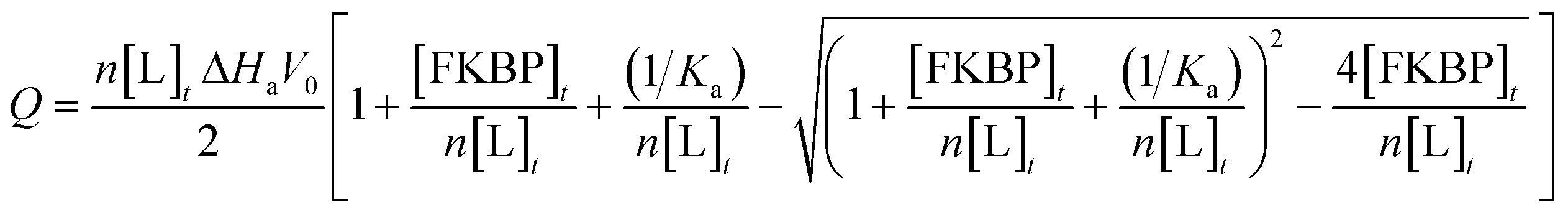 | (3) |
By using the two values of ΔHa, the association constant (Ka) obtained by the nonlinear fitting to eqn (3) and the following thermodynamic relationships (eqn (4) and (5)), the change in Gibbs free energy (ΔGa) and entropy (ΔSa) for the intermolecular association was calculated,
| ΔGa = −RTlnKa | (4) |
| ΔGa = ΔHa − TΔSa | (5) |
TrNOE and water-LOGSY
NMR samples for the transferred nuclear Overhauser effect (TrNOE) experiments18 (Table 1) consisted of 1066 μM #8–1, 1600 μM #4–1 and 44 μM FKBP12 dissolved in buffer (50 mM sodium phosphate, 50 mM NaCl, pH 6.8). TrNOE NMR experiments were performed on an AVANCE II US2 800 MHz spectrometer equipped with a 5 mm TXI CryoProbe (Bruker) at 288 K. TrNOE were recorded as 2D NOESY spectra with 16 scans. The mixing time (τm) was set to 500 ms, and the relaxation delay was set to 1.5 s. NMR samples for water-LOGSY experiments consisted of 530 μM #8–1, 800 μM #4–1 and 44 μM FKBP12 dissolved in buffer (50 mM sodium phosphate, 50 mM NaCl, pH 6.8). Water-LOGSY experiments (Table 1) were performed on an AVANCE III HD 600 MHz spectrometer equipped with a 5 mm QCI-P CryoProbe (Bruker) at 10 °C. The spectra were recorded with 1024 scans.Docking simulation
Model structures of the complex of FKBP12 and #8–1 were generated using HADDOCK.11,19 The protein structure of FKBP12 was prepared by using PDB-deposited structures (2PPN, crystal structure of apo FKBP12; 1J4I, crystal structure of the FKBP12–000308 complex; 1FKJ, crystal structure of the FKBP12–FK506 complex; 2DG3, crystal structure of the FKBP12–rapamycin complex; 1FKT, NMR structure of apo FKBP12; and 1F40, NMR structure of the FKBP12–GPI-1046 complex). These 6 structures of FKBP12 were used as the starting structures for HADDOCK calculations. An initial structure of #8–1 was prepared by ChemDraw Professional 16.0 (PerkinElmer Informatics, Waltham, MA, USA). Initially, 1000 structures were determined by rigid body docking. Then, simulated annealing (SA) was conducted with the 400 lowest-energy structures using the default force field parameters. Calculated model structures were clustered on the basis of the similarity of the poses and ranked on the basis of total score.1D 1H STD NMR spectroscopy (1H STD)
1H STD experiments (Table 1) were performed on an AVANCE III HD 600 MHz spectrometer equipped with a 5 mm QCI-P CryoProbe (Bruker) at 10 °C, as described in a previous publication.20 The spectra were recorded with 2048 scans and selective saturation of the protein resonances at 0.054 ppm for the γ-methyl group of Val55 or −0.259 ppm for the δ-methyl group of Ile91, and 30 ppm for the reference spectra using a pulse train of 40 Gaussian shaped pulses of each 50 ms in length. A 30 ms spin-lock pulse was applied after excitation to reduce the intensity of the protein resonances.19F saturated 1H NMR spectroscopy (19F STD)
19F STD experiments (Table 1) were performed on an AVANCE III HD 600 MHz equipped with QCI 1H–13C/15N/19F CryoProbe (Bruker) at 10 °C. The spectra were recorded as described in a previous publication with the exception that selective saturation was applied to 19F nuclei and 1H nuclei detected. The spectra were recorded with 8192 scans and selective saturation of compound resonances at −67245.33 Hz and −1840.76 Hz for reference spectra using a train of 80 rectangular pulses with a length of 50 ms.Results & discussion
Design of the 19F chemical library
A total of 253 fluorine-containing compounds showing high logS (log solubility) values were selected from a commercially available chemical library (Kishida chemicals 19F FBDD KP χ vol-2) (Kishida Chemical Co., Ltd., Tokyo, Japan). These compounds were selected from the ChEMBL21 and the original Kishida building-block based on molecular weight (<300 Da), solubility (estimated LogS > 3.0), and similarity to substructures of compounds with known biological activity.22 Compounds that induce split NMR peaks, such as alicyclic compounds possessing stereoisomers, were excluded. Each compound was prepared in a concentrated (40 or 80 mM) stock solution using dimethyl sulfoxide (DMSO)-d6. To check the solubility in aqueous solution, each stock solution at a concentration of 40 μM was dissolved in aqueous buffer (50 mM sodium phosphate, 50 mM NaCl, pH 6.8) and subjected to 19F-NMR measurements with 1H decoupling. Thirty-one out of the 253 compounds were excluded from the library due to significantly low solubility, low signal intensity (<30%) suggesting aggregation, or the possession of minor NMR signals. Two types of solution cocktails (10- and 20-mix) were prepared possessing 10–11 and 19–21 compounds for each cocktail, respectively. For the 10- or 20-mix cocktails, an equal aliquot of 10–11 or 19–21 compounds each was mixed and named cocktail #1–#11 or #12–#22, where 40 or 80 mM stock solutions for 111 or 220 compounds was used, respectively. The combination of the compounds was designed to avoid the signal overlaps, the signal intensities, and the chemical shift changes caused by the mixing.
The solubility of the prepared 19F chemical library was evaluated, as highly soluble compounds are required for advanced NMR analysis, such as the TrNOE18 and INPHARMA.23 Based on the ratios of the signal heights of each 40 μM compound to sodium trifluoroacetate in the #12–#22 cocktails, 36 out of 220 compounds were determined to be less soluble. The same evaluation was performed using 1 mM compound solution, and 179 compounds were confirmed to be highly soluble.
19F-NMR screening using the 19F chemical library
R 2-filtered 19F-NMR13 was employed for screening using the 19F chemical library. 19F-NMR spectra of each cocktail were measured with or without target proteins. The compounds giving weak signal intensity without protein were excluded from the screening because of self-aggregation and/or association with other molecules. Hit compounds were evaluated by a decrease in the 19F-NMR signal intensity.To test this screening system, three soluble proteins (12 kDa FK506-binding protein, FKBP12; Janus kinase 3, JAK3; and glycogen synthase kinase-3, GSK-3) and one membrane protein (voltage-sensor only protein, VSOP) were examined (Table 2). In the presence of FKBP12, three compounds, #4–1, #8–1, and #8–2 present in cocktails #4, #8, and #8, respectively, showed reduced signal intensities, as shown in Fig. S1.† In an effort to estimate the binding site of these compounds, 30 μM FK506 was used as a competitive binder of FKBP12. The signal intensities of these compounds were recovered by the addition of FK506 (Fig. 1); thus, the binding site of these compounds was judged to be, at least in part, the FK506-binding site of FKBP12. For JAK3 and GSK-3, 9 and 16 compounds showed reduced signal intensities, respectively, and their signal intensities were recovered by the addition of tofacitinib, an inhibitor of JAK3, and SB216763, an inhibitor of GSK-3, respectively (Table 2). For VSOP, the signals of 14 compounds were disappeared (Table 2). Because VSOP requires a detergent (0.2% 5-cyclohexyl-1-pentyl-D-maltoside) in buffer, the spectra of 19F compounds were measured in buffer containing the detergent without VSOP. Compounds whose peak intensities were reduced in the R2-filtered spectra in the absence of VSOP were excluded from the screening because of off-target binding with the detergent. The signal intensities of the compounds in the R2-filtered spectra drastically decreased for VSOP comparing to soluble proteins. This suggests that the screening for membrane proteins may require different experimental conditions, such as lower protein concentration and shorter R2-filter time than those for soluble proteins. 4 out of 14 hit compounds specifically bound to VSOP, 8 out of 14 bound to one or a few out of 11 examined soluble proteins in our lab, and 2 out of 14 were non-specific. These data suggest our screening procedure is applicable to membrane protein.
| Hits | Hit rate | I c/If | |
|---|---|---|---|
| FKBP12 | 3 | 1.4% | <0.6 |
| GSK3 | 16 | 7.2% | <0.6 |
| JAK3 | 9 | 4.1% | <0.6 |
| VSOP | 14 | 6.3% | =0 |
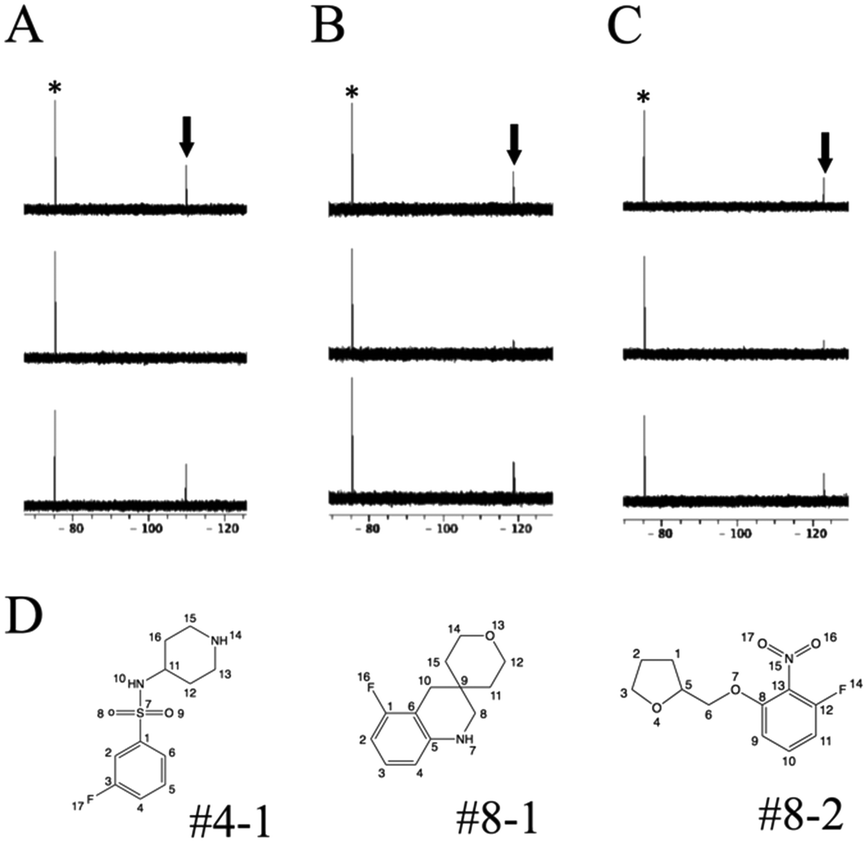 | ||
| Fig. 1 R 2-filtered 19F-NMR spectra recorded with proton decoupling for fragment #4–1 (A), #-8–1 (B), and #8–2 (C) in the fragment alone (bottom), in the presence of FKBP12 (middle), and in the presence of FKBP12 and FK506 (top). The asterisks indicate the signals from sodium trifluoroacetate. The arrows indicate the signals from the compounds. The chemical structures of #4–1, #8–1 and #8–2 with numbering (D). | ||
19F 2D DOSY was further used for screening using the 19F chemical library-based screening to compare with the R2-filtered 19F-NMR results. The DOSY spectrum of each cocktail was measured in the presence or absence of FKBP12, and their translational diffusion coefficients (D) were compared. Since the D value depends on the size of the molecule, the comparison between the D values can tell us about the binding between the compound and the target protein. In our experiment, the observed diffusion coefficients of each cocktail did not change after the addition of FKBP12 (Fig. S2†), indicating that screening based on the 19F DOSY experiment did not give a hit compound. This result is consistent with a previous report indicating that the sensitivity of 19F 2D DOSY screening is lower than R2-filter 19F-NMR.14
Hit validation
For FKBP12, three chemical compounds, #4–1, #8–1, and #8–2, were identified as hit compounds from the 19F-NMR screening (see above). Each hit compound was mixed with FKBP12 and subjected to a protein thermal shift (PTS) assay. The melting temperature of FKBP12 increased by approximately +3 °C in the presence of the three compounds (Fig. S3,†Table 3). The increase in the thermal denaturing temperature (Tm) values validates the binding of these compounds to FKBP12.| Compounds | T m (°C) | ΔTmc (°C) |
|---|---|---|
| a T m values evaluated by protein thermal shift (PTS) assay with the standard error of the mean for the Tm values calculated by quintuple experiments. b T m value of FKBP12 in the absence of any compounds. c Difference of the average Tm values of FKBP12 in the presence and absence of the analyte compounds (ΔTm = Tmb − Tm). d Positive control. | ||
| None | 65.57 ± 0.04b | |
| #4–1 | 68.35 ± 0.09 | +2.78 ± 0.13 |
| #8–1 | 68.55 ± 0.07 | +2.98 ± 0.11 |
| #8–2 | 68.27 ± 0.08 | +2.70 ± 0.22 |
| FK506d | 78.81 ± 0.06 | +13.20 ± 0.10 |
For further validation, each hit compound was titrated into a 15N-labeled FKBP12 solution and monitored by 1H–15N HSQC spectral analysis. The chemical shifts of the backbone resonances (15N and amide protons) were assigned based on a previous report.24 In the presence of #4–1, perturbed resonances (>0.02 ppm) were observed for R42, I56, and I90 (Fig. 2A and S4A†). For #8–1, perturbed resonances (>0.03 ppm) were observed for S38, S39, D41, R42, G51, I56, W59, E61, V63, and F99 (Fig. S4B†), and significantly large perturbations (>0.06 ppm) were observed for R42, G51, and I56 (Fig. 2B). For #8–2, no perturbed resonance was observed (Fig. 2C). To identify the binding sites of compounds #4–1 and #8–1 on FKBP12, perturbed amino acid residues were mapped on the FKBP12 structure (PDB: 2PPN) (Fig. 2D and E). Most of the perturbed residues (Fig. 2D and E) were located near the FK506 binding site (Fig. 2F). These results are consistent with the 19F-NMR screening experiments showing that compounds #4–1 and #8–1 competed with FK506.
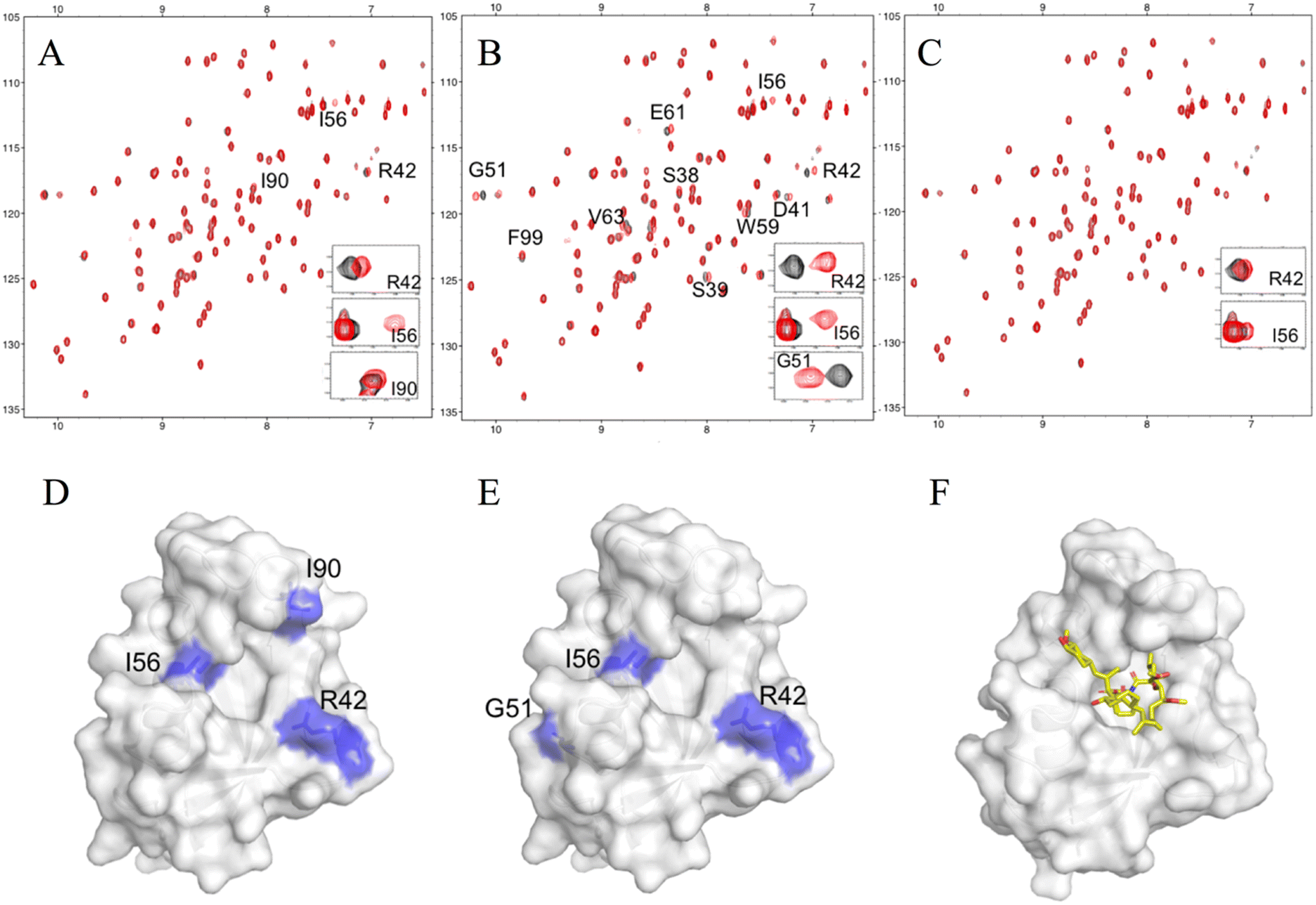 | ||
| Fig. 2 NMR titrations between FKBP12 and the hit compounds. Overlays of the 15N HSQC spectra of 11 μM FKBP12 in the absence (black) or presence (red) of 40 μM #4–1 (A), #8–1 (B), and #8–2 (C). Mapping of the chemical shift perturbations on the surface representations of FKBP12 (PDB: 2PPN) by #4–1 binding (D) and #8–1 binding (E). The complex structure of FKBP12 with FK506 (as shown as yellow sticks) (F). | ||
To obtain more clues for the binding of the three ligands to FKBP12, we performed ITC measurements (Fig. S5†). A series of titrations of FKBP12 to #4–1 were accompanied by large exothermic heats (Fig. S5A†), which were larger than the heats of dilution (Fig. S5D†), indicating complex formation between FKBP12 and #4–1. Similarly, the thermograms of the FKBP12 titration to #8–1 showed exothermic reactions (Fig. S5B†), which also suggested the binding of #8–1 to FKBP12. The titration of FKBP12 to #8–2 showed a unique thermogram with exothermic heats. The thermogram appeared to consist of several reactions, indicating multiple binding reactions between FKBP12 and #8–2 (Fig. S5C†). Proteins that accommodate multiple binding sites for metal ions have shown similar thermograms.25 Although detailed analyses to obtain various thermodynamic parameters were difficult, the exothermic reactions clearly suggested the binding of #8–2 to FKBP12.
K d value of hit compounds
Using 1H–15N HSQC titration experiments, the binding affinity values of the hit compounds to FKBP12 were determined by plotting the chemical shift change of each residue for #8–1 and #4–1 against the compound concentration. Each titration curve was fitted by eqn (2), and the dissociation constant (Kd) was obtained (Fig. S6,†Table 4). The determined Kd values were 564 ± 37 μM for #8–1 and 200 ± 12 μM for #4–1.| Compound | K d (μM) | ΔGaa (kcal mol−1) | ΔHaa (kcal mol−1) | TΔSaa (kcal mol−1) |
|---|---|---|---|---|
| a ΔGa, ΔHa, and TΔSa values were determined by ITC for the FKBP-compound association. b n.d. indicates values that were not obtained due to the weak intermolecular interaction. | ||||
| #4–1 | 690 ± 39 (ITC) | −4.3 ± 0.0 | −8.2 ± 0.5 | −3.9 ± 0.6 |
| 564 ± 37 (NMR) | ||||
| #8–1 | 300 ± 19 (ITC) | −4.8 ± 0.1 | −8.4 ± 1.2 | −3.6 ± 1.3 |
| 200 ± 12 (NMR) | ||||
| #8–2 | n.d.b | n.d.b | n.d.b | n.d.b |
Using ITC experiments, the Kd values of #8–1 and #4–1 for FKBP12 binding were determined to be 300 ± 19 μM (corresponding to a ΔGa of −4.8 ± 0.1 kcal mol−1) and 690 ± 39 μM (corresponding to a ΔGa of −4.3 ± 0.0 kcal mol−1), respectively, which were in good agreement with the values obtained by the 1H–15N HSQC analysis. That is, compound #8–1 binds more strongly than compound #4–1. It is noted that recent 19F-NMR progress enables Kd determination in protein–drug interaction system.26
All of the thermodynamic parameters obtained are summarized in Table 4. Interestingly, although hydrophobic interactions between the hydrophobic pockets of FKBP12 and the hydrophobic ligands were expected to be a driving force through an increase in entropy due to dehydration (i.e., positive TΔSa), both binding systems showed negative ΔHa and TΔSa values (Table 4). Consequently, the binding reactions were purely driven by enthalpy (i.e., negative ΔHa). This purely enthalpy-driven complex formation was also observed for FK506 (ref. 15). This may result from an increase in noncovalent intramolecular interactions, which in turn can reinforce the thermal stability of FKBP12 (Table 4) through a loss of conformational entropy. Purely enthalpy-driven complex formation and an increase in the thermal stability were also observed for the binding of hydrophobic ligands to the hydrophobic cavity of L-PGDS.27
Binding ability evaluated by TrNOE and water-LOGSY experiments
As shown above, the binding affinity of the hit compounds was relatively low. TrNOE and water-LOGSY experiments are suitable for such weak binders because TrNOE and water-LOGSY experiments require fast exchange conditions between free and bound states of the ligand.28 For the TrNOE experiment, a 1H–1H nuclear Overhauser effect spectroscopy (NOESY) spectrum was measured for FKBP12 with two hit compounds, #8–1 and #4–1 (Fig. 3A). Compound #8–1 showed a negative NOE, which is the same sign as the diagonal peaks, indicating that compound #8–1 binds to FKBP12. Compound #4–1 showed a positive NOE, which is the opposite sign to the diagonal peaks, indicating that compound #4–1 does not bind or very weakly binds to FKBP12.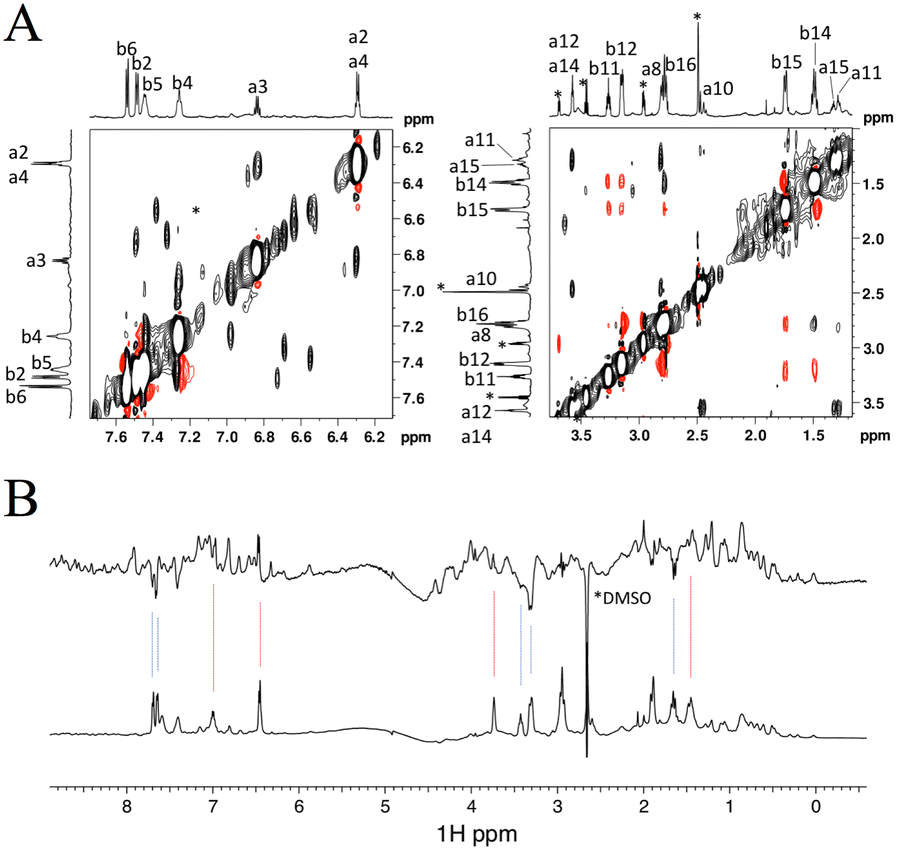 | ||
| Fig. 3 Aromatic (left) and aliphatic (right) region of the 1H–1H NOESY spectrum measured for 44 μM FKBP12 with 1600 μM #4–1 and 1066 μM #8–1. The numbers from a1 to a15 and from b1 to b16 correspond to the 1H atom numbers of #8–1 and #4–1 shown in Fig. 1D, respectively. Positive and negative peaks are plotted in black and red, respectively. Asterisks indicate impurities (A). Water LOGSY spectrum (top) measured for 44 μM FKBP12 with 800 μM #4–1 and 530 μM #8–1. The bottom spectrum is the reference 1D. Blue and red lines indicate the signals of #4–1 and #8–1, respectively. Negative peaks are from free compounds (B). | ||
In water-LOGSY experiments, the proton signal of bulk water is saturated, and the saturated magnetization is transferred to the free ligand if the ligand binds to the protein.
In Fig. 3B, a water-LOGSY spectrum is shown where hit compounds #4–1 (800 μM) and #8–1 (530 μM), were mixed with 44 μM FKBP12. Positive signals were observed for #8–1, indicating that #8–1 binds to FKBP12. Negative signals were observed for #4–1, indicating that compound #4–1 does not bind or very weakly binds to FKBP12. These TrNOE and water-LOGSY data were consistent with the 1H–15N HSQC titration experiments and ITC experiments in the sense that compound #8–1 binds stronger than compound #4–1.
Model structure of the FKBP12-compound complex based on 1H and 19F STD experiments
Although all experiments revealed the binding of compound #8–1 to FKBP12, the complex of FKBP12 with #8–1 was not crystallizable. In an effort to model the complex structure between FKBP12 and #8–1, HADDOCK 2.2.11 The active residues for the HADDOCK calculation were selected to be S38, S39, D41, R42, G51, I56, W59, E61, V63, and F99, which are the residues that showed significant chemical perturbation (>0.03 ppm in the 1H–15N HSQC titration experiment). As the final complex structure depended on the starting structure, 6 structures (2PPN, crystal structure of apo FKBP12; 1J4I, crystal structure of the FKBP12–000308 complex; 1FKJ, crystal structure of the FKBP12–FK506 complex; 2DG3, crystal structure of the FKBP12–rapamycin complex; 1FKT, NMR structure of apo FKBP12; and 1F40, NMR structure of the FKBP12–GPI-1046 complex) were used as starting structures. For 2PPN, 1J4I, 1FKJ, 2DG3, 1FKT, and 1F40, 8, 9, 7, 11, 9, and 7 clusters of complex structures were obtained, respectively (Table S1†).To select the appropriate complex model, 1H STD NMR spectra were obtained at 283 K for 30 μM FKBP12 with 1.0 mM compound #8–1 (Fig. 4A). The Val55 γ and Ile91 δ methyl groups of FKBP12 were selectively irradiated by Gaussian-shaped pulses at 0.054 and −0.259 ppm, respectively. When 0.054 and −0.259 ppm were irradiated, strong STD signals were observed for atoms 12, 14 and 2, 4 of compound #8–1, respectively. Assuming that the atom pairs showing strong 1H{1H} STD signals are within 5 Å and the distances between the attached carbons are less than 7 Å, the 5 clusters of the modeled structures were selected (Fig. 5A, S7† and Table 5).
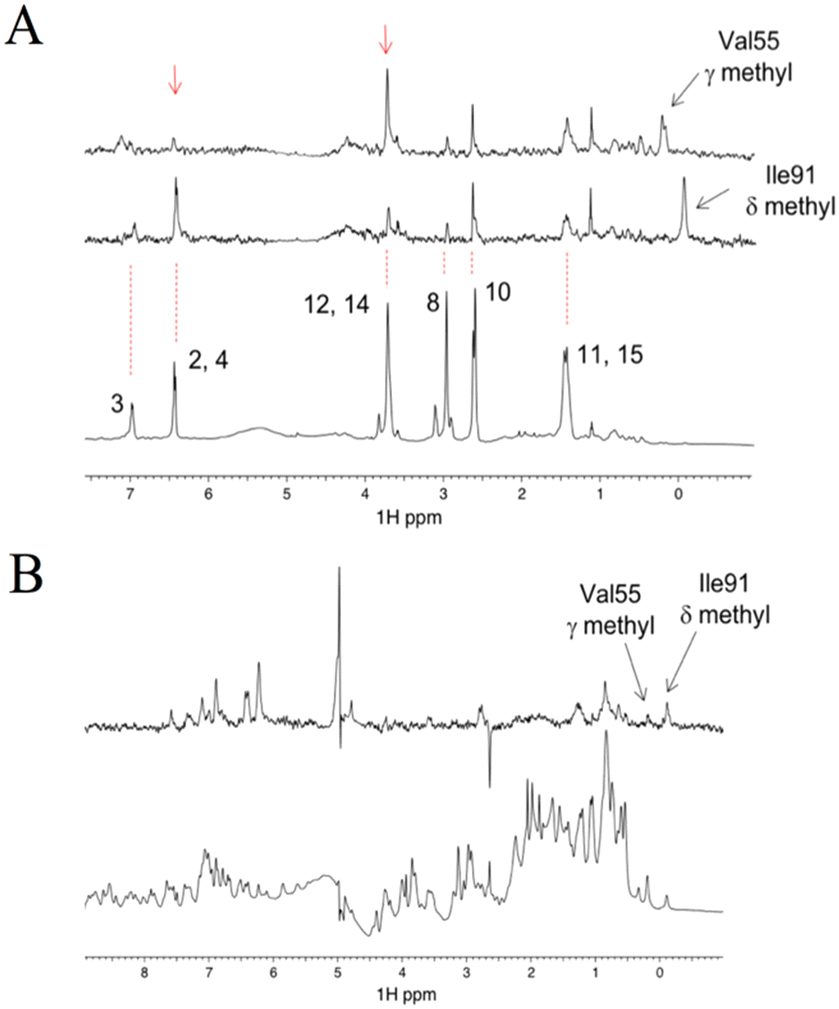 | ||
| Fig. 4 1H selective STD spectra of 30 μM FKBP12 and 1000 μM #8–1 measured with irradiation at the Val55 γ methyl (top) and Ile91 δ methyl (middle). The bottom spectrum is the reference 1D. Red arrows indicate signals from atoms 2/4 and 12/14 of #8–1 (A). 19F STD spectrum of 250 μM FKBP12 and 250 μM #8–1 measured with irradiation at the 19F signal of #8–1 (top). The bottom spectrum is the reference 1D (B). | ||
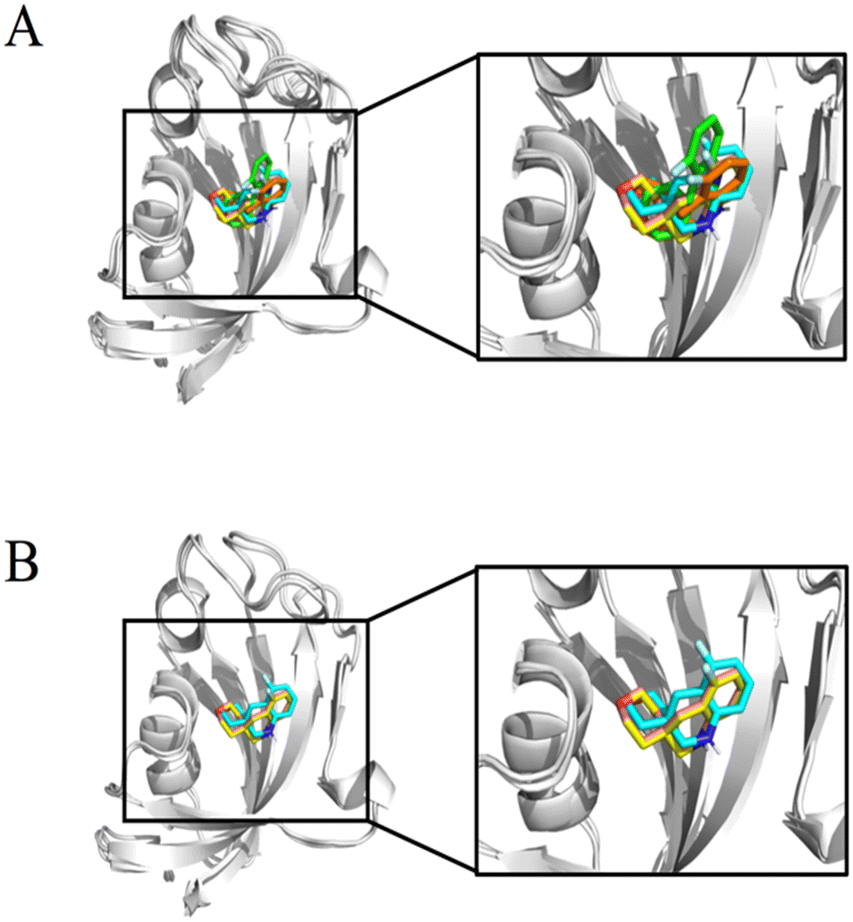 | ||
| Fig. 5 The overlay of HADDOCK-derived structural models of FKBP12 and #8–1 complex structures. The displayed structures were selected based on 1H STD signals (Table 4) starting from the structures of PDB 2PPN (pink), 1J4I (yellow), 1FKJ (orange), 2DG3 (green), and 1F40 (cyan) (A). The structures selected based on the 19F STD signals in addition to 1H STD signals (Table 4) when the HADDOCK calculation started from the structure of PDB 2PPN (pink), 1J4I (yellow), and 1F40 (cyan) (B). | ||
| Amino acid residue & group | Atom no. of compound | 2PPN cluster 4a | 1J4I cluster 4a | 1FKJ cluster 2 | 2DG3 cluster 7 | 1F40 cluster 6a |
|---|---|---|---|---|---|---|
| a Carbon–carbon distances of <7 Å are selected from Table S3.† b Carbon–fluorine distances of <6 Å are shown by bold letter. | ||||||
| Val55 Cγ1 | C12 | 3.746 | 3.746 | 3.225 | 3.530 | 4.179 |
| Val55 Cγ1 | C14 | 4.933 | 4.933 | 5.374 | 4.307 | 5.706 |
| Ile91 Cδ1 | C2 | 4.685 | 4.685 | 5.747 | 5.180 | 4.254 |
| Ile91 Cδ1 | C4 | 6.484 | 6.484 | 6.048 | 3.934 | 6.617 |
| Ile91 Cδ1 | F16 | 4.574 | 4.574 | 7.048 | 7.351 | 3.984 |
For further selection, a 19F STD NMR spectrum was obtained at 283 K for 0.25 mM FKBP12 with 0.25 mM compound #8–1 (Fig. 4B). The 19F STD spectra do not have the artefacts originated from off-target saturation and spin diffusion, often seen in the 1H STD spectra, and do provide more reliable distance information than the 1H STD spectra.10 This is because the number of fluorine atom of the compound was one or two and 1H–19F saturation transfer between the compound and the protein strongly depended on the relative position between the fluorine atom of the compound and the hydrogen atoms of the protein.
In this experiment, the 19F nucleus in compound #8–1 was selectively irradiated by a rectangular pulse to transfer the saturation to the bound FKBP12. A strong STD signal was observed for the Ile91 δ methyl group. Assuming that the atom pairs showing strong 1H{19F} STD signals are within 5 Å and the distances between the attached carbons and fluorine (Ile91 δ methyl group and 19F nucleus) are less than 6 Å, 3 (Fig. 5B and S8†) out of the 5 (Fig. 5A) clusters of the modeled structures were selected, as shown by bold letters in Table 5. The convergence of the compound positions in the modeled structures, including the orientation, was significantly improved by 19F STD. Additionally, selective irradiation for the 19F STD experiment is much easier than for the 1H STD experiment.
Yu et al. measured 1H{19F} STD HMQC spectra of the complex between a protein and 19F-containing compound, which were measured over 3.5 days using 1 mM 13C-labeled protein.10 When we applied this 2D 1H{19F} STD HMQC to our sample, many artificial peaks were observed presumably due to low S/N ratio. Therefore, to detect the 1H{19F} STD signal, we employed a 1D experiment instead of 2D. The 1D 1H{19F} STD experiment allowed us to obtain the signal in 3 hours using 0.25 mM protein sample. We observed many signals in the 1D 1H{19F} STD spectrum but did not use these signals for obtaining distance information because their assignments were ambiguous due to chemical shift overlapping except for Val55 γ and Ile91 δ. A measurement of the buildup curve may provide more precise distance information although we did not try it for our sample.
NMR is suitable for cocktail screening
The use of compound cocktails is necessary for efficient 19F-NMR screening. In this report, R2-filtered 19F-NMR was successfully applied to cocktail screening, and the model structure of the drug–protein complex was successfully obtained. However, cocktail screening is a controversial procedure under certain circumstances. For example, if the compounds interact with each other and/or more than two compounds within each cocktail bind to one target protein, the screening result becomes complex and can be misread. In our 19F chemical library, the self-associating compounds were carefully removed by NMR-based examination of the solubility and concentration-dependent change. The compounds that showed an interaction with different compounds were easily identified by comparing the NMR spectra of free and cocktail states, and such inter-compound interactions were carefully excluded by redesigning the compound cocktail combination. In our final cocktails, no such interactions were observed by NMR. Therefore, the problem of cocktail screening that comes from inter-compound interactions was unlikely in our 19F chemical library and cocktail.Verification of false positives and false negatives
If more than two compounds bind a target protein, artificial effects may occur as pseudo-positives and pseudo-negatives due to synergistic and/or competitive effects of the protein–compound interaction. In our 19F-NMR screening, pseudo-positives were carefully excluded at the hit validation step, but not pseudo-negatives. Typical pseudo-negatives are the disappearance of the apparent perturbation upon binding, that is, one compound gives a positive perturbation and the other compound gives a negative perturbation, or one large perturbation hides the other small perturbation. To discuss the artificial effects, we performed cocktail screening employing two different methods, a protein-based NMR approach and a PTS assay using the same 19F chemical library.For the protein-based NMR approach, the 1H–15N HSQC spectra of 15N-labeled FKBP12 were measured in the presence and absence of the cocktails (Fig. 6 and S11†). Chemical shift perturbations were observed for cocktails of #4 and #8 containing the 19F-NMR screening hits of #4–1, #8–1, and #8–2 (Fig. 6). Compared to Fig. 2, the perturbations induced by the cocktails were almost the same as those induced by each hit compound. Therefore, the pseudo-negatives from the cocktail screening coming from multicompound binding with FKBP12 are unlikely for cocktails #4 and #8. two different methods, a protein-based NMR approach and a PTS assay using the same 19F chemical library.
 | ||
| Fig. 6 NMR titrations between FKBP12 and the cocktails containing hit compounds. Overlay of 15N HSQC spectra of FKBP12 in the absence (black) or presence (red) of Mix4 (A) and Mix8 (B). Insets indicate expanded views of Arg42, Ile56, and Ile90 for Mix4 and Arg42, Ile56, and Gly51 for Mix8. | ||
By using the PTS assay, the thermal stability of FKBP12 was measured in the presence and absence of each cocktail. The Tm values of FKBP12 increased in excess of 1 °C for cocktails #2, #4, #7, #8, and #10 (Fig. S10,† Table S2), although hit compounds were not included in cocktails #2, #7, and #10. PTS screening has been reported to possess lower hit ratios than water-LOGSY screening29 due to the pseudo-positives of nonspecific binders, which improve the thermal stability of the target proteins, such as salts, sugars, osmolytes, and kosmotropes. Thus, our PTS screening conceivably possesses false positives. In fact, the 1H–15N HSQC spectra of FKBP12 for cocktails #2, #7, and #10 showed much smaller perturbations than for #4 and #8.
Conclusions
A 19F chemical library composed of small fluorine-containing compounds was constructed as 11 cocktails containing 40 μM each 10–11 or 19–21 compounds using 40 or 80 mM stock solutions of 111 or 220 compounds, respectively. Among them, 179 compounds were judged to be highly soluble as monomeric molecules in a 1 mM aqueous solution of each compound. This library has the distinguishing feature that it is composed of compounds that have been experimentally confirmed to be highly soluble (>1 mM for >80% compounds).The R2-filter method was successfully applied to the 19F-NMR screening of FKBP12, JAK3, GSK-3, and VSOP using a 19F chemical library. For FKBP12, three hit compounds were obtained and validated by PTS assay, 1H–15N HSQC, and ITC. The dissociation constants of the two hit compounds were determined within the range of 200–690 μM. The binding affinity of two of the hit compounds was further evaluated by TrNOE and water-LOGSY experiments, and one of the two compounds weakly but significantly bound to FKBP12. The use of compound cocktails for screening was evaluated and discussed, as it is necessary for efficient 19F-NMR screening.
Using the bound compound, the FKBP12-compound complex structure was modeled based on 1H STD, 19F STD and HSQC experiments. About the 19F STD experiment, the original 2D 19F STD method10 was not popular maybe due to its low sensitivity and requiring 13C-enriched protein sample. Here we succeeded in increasing the sensitivity of 19F STD by performing it as a 1D experiment and obtaining distance information as well as the original 2D method. The distance information derived from 1D 19F STD was necessary to refine the modeled structure. It is noted that 1D 19F STD experiments we performed have the advantage of not requiring the 13C-labeling.
Based on the results of this study, we propose a scheme for FBDD using 19F-NMR (Fig. 7). The scheme consists of the identification of hit compounds by 19F-NMR screening, validation of hit compounds and collection of distance information by 1H–15N HSQC, 1H{1H} STD and 1H{19F} STD, and construction of a model structure of protein–compound complex.
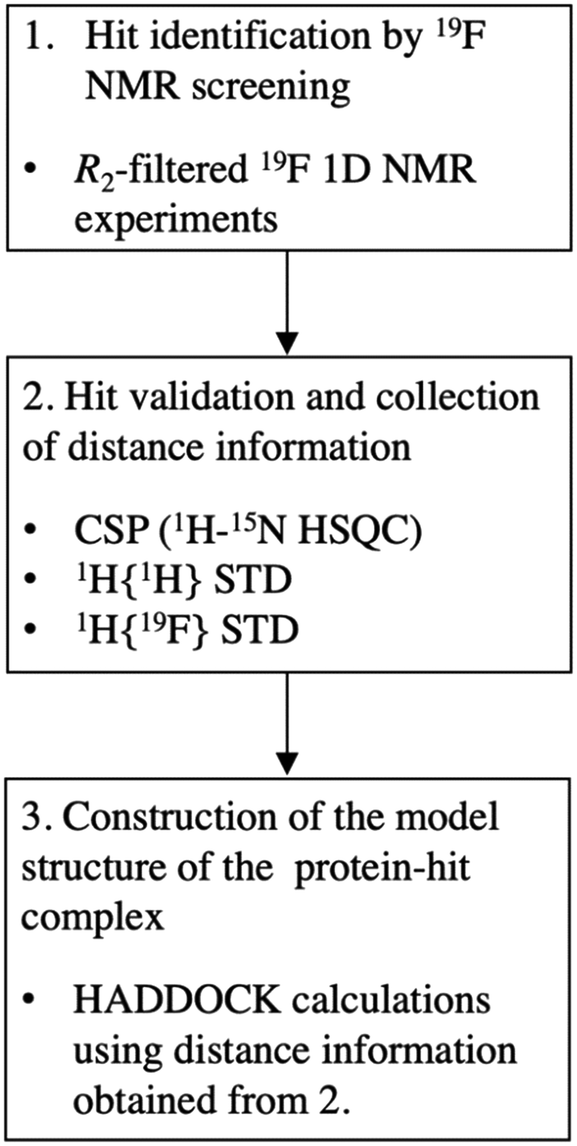 | ||
| Fig. 7 The scheme for FBDD using 19F-NMR. The recommended experiments in each step are indicated. CSP, chemical shift perturbation experiment. | ||
In this report, the contribution of 19F-NMR in drug discovery and the importance of a multi-disciplinary approach to obtain reliable results are clearly explained. The combination of 19F-NMR spectroscopy and the optimized 19F chemical library enables the modeling of the precise complex structure of a weakly binding small compound (Kd ∼ 200–300 μM) to a target protein and therefore removes the bottleneck of FBDD.
Author contributions
Conceptualization: MTK, CK. Data curation: SS, RK, KF, TS, YHL, AK, FH, TK, CK. Formal analysis: SS, RK, KF, TS, YHL, AK, KA, MO, FH, CK. Funding acquisition: TK, TF, CK. Investigation: SS, RK, KF, TS, YHL, AK, KA, MO, FH. Methodology: SS, RK, KF, TS, AK, MO, FH, MTK, TF, CK. Project administration: CK. Resource: YH, KT, AN. Supervision: TF, CK. Validation: SS. Visualization: SS, RK, KF, TS, YHL, CK. Writing – original draft: SS, RK, KF, TS, YHL, CK. Writing – review & editing: SS, RK, KF, TS, YHL, YH, KT, AN, AK, KA, MO, FH, TK, MTK, TF, CK.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank M. Yoneyama for sample preparation. This study was supported in part by MEXT/JSPS KAKENHI, Grant-in-Aid for Scientific Research on Innovative Areas (17H05836, 19H04856, and 22H05536 to C.K.), Grant-in-Aid for Scientific Research (B) (number 20H03191 to C.K.), Platform for Drug Discovery, Informatics, and Structural Life Science, and Project for Advanced Research Infrastructure Sharing (Creation of Research Platforms) (JPMXS04101) by MEXT, Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research, BINDS; number JP21am0101072) by AMED, the Collaborative Research Program of the Institute for Protein Research, Osaka University (CR-20-02), the National Research Foundation of Korea (NRF) grant funded by the Korean government [NRF-2019R1A2C1004954 and NRF-2022R1A2C1011793 (Y.-H.L.)], and KBSI fund [C220000, C230130, and C280320] (Y.-H.L.).References
- C. W. Murray and D. C. Rees, The rise of fragment-based drug discovery, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- Q. Li and C. Kang, Perspectives on Fragment-based Drug Discovery: A Strategy Applicable to Diverse Targets, Curr. Top. Med. Chem., 2021, 21, 1099–1112 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Twenty years on: The impact of fragments on drug discovery, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- B. Diethelm-Varela, ChemMedChem, 2021, 16, 725–742 CrossRef CAS PubMed.
- C. Dalvit, Ligand- and substrate-based 19F NMR screening: Principles and applications to drug discovery, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 243–271 CrossRef CAS.
- P. J. Hajduk and J. Greer, A decade of fragment-based drug design: strategic advances and lessons learned, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed.
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Discovering High-Affinity Ligands for Proteins: SAR by NMR, Science, 1996, 274, 1531–1534 CrossRef CAS PubMed.
- T. Sugiki, K. Furuita, T. Fujiwara and C. Kojima, Current NMR Techniques for Structure-Based Drug Discovery, Molecules, 2018, 23, 148 CrossRef PubMed.
- J. L. Wagstaff, S. L. Taylor and M. J. Howard, Recent developments and applications of saturation transfer difference nuclear magnetic resonance (STD NMR) spectroscopy, Mol. BioSyst., 2013, 9, 571–577 RSC.
- L. Yu, P. J. Hajduk, J. Mack and E. T. Olejniczak, Structural studies of Bcl-xL/ligand complexes using 19F NMR, J. Biomol. NMR, 2006, 34, 221–227 CrossRef CAS PubMed.
- G. C. P. Van Zundert, J. P. G. L. M. Rodrigues, M. Trellet, C. Schmitz, P. L. Kastritis, E. Karaca, A. S. J. Melquiond, M. Van Dijk, S. J. De Vries and A. M. J. J. Bonvin, The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS PubMed.
- K. Takeshita, S. Sakata, E. Yamashita, Y. Fujiwara, A. Kawanabe, T. Kurokawa, Y. Okochi, M. Matsuda, H. Narita, Y. Okamura and A. Nakagawa, X-ray crystal structure of voltage-gated proton channel, Nat. Struct. Mol. Biol., 2014, 21, 352–357 CrossRef CAS PubMed.
- S. Meiboom and D. Gill, Modified Spin-Echo Method for Measuring Nuclear Relaxation Times, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- A. Vulpetti and C. Dalvit, Design and generation of highly diverse fluorinated fragment libraries and their efficient screening with improved (19) F NMR methodology, ChemMedChem, 2013, 8, 2057–2069 CrossRef CAS PubMed.
- L. Fielding, NMR methods for the determination of protein–ligand dissociation constants, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 219–242 CrossRef CAS.
- M. A. Wear, A. Patterson and M. D. Walkinshaw, A kinetically trapped intermediate of FK506 binding protein forms in vitro: chaperone machinery dominates protein folding in vivo, Protein Expression Purif., 2007, 51, 80–95 CrossRef CAS PubMed.
- S. Keller, C. Vargas, H. Zhao, G. Piszczek, C. A. Brautigam and P. Schuck, High-precision isothermal titration calorimetry with automated peak-shape analysis, Anal. Chem., 2012, 84, 5066–5073 CrossRef CAS PubMed.
- F. Ni, Recent developments in transferred NOE methods, Prog. Nucl. Magn. Reson. Spectrosc., 1994, 26, 517–606 CrossRef CAS.
- S. J. de Vries, M. van Dijk and A. M. J. J. Bonvin, The HADDOCK web server for data-driven biomolecular docking, Nat. Protoc., 2010, 5, 883–897 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Characterization of ligand binding by saturation transfer difference NMR spectroscopy, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS PubMed.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Krüger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, The ChEMBL bioactivity database: an update, Nucleic Acids Res., 2014, 42, D1083–D1090 CrossRef CAS PubMed.
- The Kishida Chemical Online, http://www.kishida.co.jp/files/a6754a063d94f4880ee23bd3fa6f9470_1.pdf.
- V. Sánchez-Pedregal, The INPHARMA Method: Protein-Mediated Interligand NOEs for Pharmacophore Mapping, Angew. Chem., 2005, 44, 4172–4175 CrossRef PubMed.
- M. K. Rosen, S. W. Michnick, M. Karplus and S. L. Schreiber, Proton and nitrogen sequential assignments and secondary structure determination of the human FK506 and rapamycin binding protein, Biochemistry, 1991, 30, 4774–4789 CrossRef CAS PubMed.
- Y. Chao and D. Fu, Thermodynamic studies of the mechanism of metal binding to the Escherichia coli zinc transporter YiiP, J. Biol. Chem., 2004, 279, 17173–17180 CrossRef CAS PubMed.
- C. Dalvit, A. Parent, F. Vallée, M. Mathieu and A. Rak, Fast NMR Methods for Measuring in the Direct and/or Competition Mode the Dissociation Constants of Chemical Fragments Interacting with a Receptor, ChemMedChem, 2019, 14, 1115–1127 CrossRef CAS PubMed.
- S. Kume, Y.-H. Lee, M. Nakatsuji, Y. Teraoka, K. Yamaguchi, Y. Goto and T. Inui, Fine-tuned broad binding capability of human lipocalin-type prostaglandin D synthase for various small lipophilic ligands, FEBS Lett., 2014, 588, 962–969 CrossRef CAS PubMed.
- W. Becker, K. C. Bhattiprolu, N. Gubensäk and K. Zangger, Investigating Protein–Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy, ChemPhysChem, 2018, 19, 895–906 CrossRef CAS PubMed.
- N. Basse, J. L. Kaar, G. Settanni, A. C. Joerger, T. J. Rutherford and A. R. Fersht, Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant, Chem. Biol., 2010, 17, 46–56 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S3 and Fig. S1–S11. See DOI: https://doi.org/10.1039/d2md00170e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |