
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploration of piperidine 3D fragment chemical space: synthesis and 3D shape analysis of fragments derived from 20 regio- and diastereoisomers of methyl substituted pipecolinates†
S. Paul
Jones
a,
James D.
Firth
 a,
Mary C.
Wheldon
a,
Masakazu
Atobe
ab,
Roderick E.
Hubbard
ac,
David C.
Blakemore
d,
Claudia
De Fusco
e,
Simon C. C.
Lucas
f,
Stephen D.
Roughley
c,
Lewis R.
Vidler
g,
Maria Ann
Whatton
h,
Alison J.-A.
Woolford
i,
Gail L.
Wrigley
j and
Peter
O'Brien
*a
a,
Mary C.
Wheldon
a,
Masakazu
Atobe
ab,
Roderick E.
Hubbard
ac,
David C.
Blakemore
d,
Claudia
De Fusco
e,
Simon C. C.
Lucas
f,
Stephen D.
Roughley
c,
Lewis R.
Vidler
g,
Maria Ann
Whatton
h,
Alison J.-A.
Woolford
i,
Gail L.
Wrigley
j and
Peter
O'Brien
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: peter.obrien@york.ac.uk
bAsahi Kasei Pharma Corporation, 632-1 Mifuku, Izunokuni, Shizuoka 410-2321, Japan
cVernalis (R&D) Ltd., Granta Park, Abington, Cambridge, CB21 6GB, UK
dMedicine Design, Pfizer Inc., 445 Eastern Point Road, Groton, CT 06340, USA
eBayer AG, Research and Development, Pharmaceuticals, Synthetic Modalities, 13353, Berlin, Germany
fHit Discovery, Discovery Sciences, R&D, AstraZeneca, Cambridge, CB4 0WG, UK
gAmphista Therapeutics, The Cori Building, Granta Park, Great Abington, Cambridge CB21 6GQ, UK
hEvotec (UK) Ltd, Dorothy Crowfoot Hodgkin Campus, 114 Innovation Drive, Milton Park, Abingdon, Oxon, OX14 4RZ, UK
iAstex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA, UK
jMedicinal Chemistry, Oncology R&D, AstraZeneca, Cambridge, UK
First published on 11th October 2022
Abstract
Fragment-based drug discovery is now widely adopted for lead generation in the pharmaceutical industry. However, fragment screening collections are often predominantly populated with flat, 2D molecules. Herein, we report the synthesis of piperidine-based 3D fragment building blocks – 20 regio- and diastereoisomers of methyl substituted pipecolinates using simple and general synthetic methods. cis-Piperidines, accessed through a pyridine hydrogenation were transformed into their trans-diastereoisomers using conformational control and unified reaction conditions. Additionally, diastereoselective lithiation/trapping was utilised to access trans-piperidines. Analysis of a virtual library of fragments derived from the 20 cis- and trans-disubstituted piperidines showed that it consisted of 3D molecules with suitable molecular properties to be used in fragment-based drug discovery programs.
Introduction
Fragment based drug discovery (FBDD) is now one of the main approaches for drug discovery in both pharmaceutical and academic laboratories.1 Over the last few years, six marketed drugs are listed as having their origins in FBDD methodology.2–7 Fragment sized chemical space can be broadly categorized by the ‘rule-of-three’: MW (molecular weight) < 300 Da; Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P < 3; number of hydrogen bond acceptors (HBA) and donors (HBD) ≤ 3.8 Recently, Astex have adopted a narrower definition of fragment space with MW 140–230 Da (approximately 10–16 heavy atoms) and ClogP 0–2.9 The relatively low MW of fragments means that there is a more efficient sampling of chemical space and so fragment libraries are much smaller in size than typical high-throughput screening libraries.10 We11 and others12 have become increasingly interested in the exploration of 3D fragment chemical space. To some degree, the developing interest in 3D fragments can be traced back to Lovering et al.'s ‘Escape from Flatland’ paper,13 even though this work was not specifically concerned with fragment space and utilised Fsp3 (fraction of sp3 hybridised carbon atoms) as a metric (which we have recently shown is not a particularly good descriptor for 3D shape11c).
P < 3; number of hydrogen bond acceptors (HBA) and donors (HBD) ≤ 3.8 Recently, Astex have adopted a narrower definition of fragment space with MW 140–230 Da (approximately 10–16 heavy atoms) and ClogP 0–2.9 The relatively low MW of fragments means that there is a more efficient sampling of chemical space and so fragment libraries are much smaller in size than typical high-throughput screening libraries.10 We11 and others12 have become increasingly interested in the exploration of 3D fragment chemical space. To some degree, the developing interest in 3D fragments can be traced back to Lovering et al.'s ‘Escape from Flatland’ paper,13 even though this work was not specifically concerned with fragment space and utilised Fsp3 (fraction of sp3 hybridised carbon atoms) as a metric (which we have recently shown is not a particularly good descriptor for 3D shape11c).
To illustrate the structural and shape diversity offered by ‘saturation’ of a scaffold, without significantly increasing its overall MW, Lovering et al. highlighted the isomeric composition of dimethyl pyridine 1versus dimethyl piperidine 2 (Fig. 1A).13 For dimethyl pyridine 1, there are six isomers which are of 2D shape but do at least provide different regioisomeric vectors for further functionalisation. In contrast, for dimethyl piperidine 2 (including N-methyl compounds), Lovering indicated that there were 34 isomers. As it turns out, this was incorrect as some of the depicted isomers were in fact the same compounds – there are 30 isomers which reduces to 25 isomers if we consider only NH piperidine compounds (Fig. 1A). Whichever number of isomers is considered, ‘saturation’ clearly provides a significant increase in shape diversity and complexity with only a small increase in MW (of 6). The 25 isomers of dimethyl NH piperidines 2 (see ESI†) comprise five achiral compounds (two of which are meso) and ten enantiomeric pairs of chiral compounds (cis- and trans-configurations in four cases).
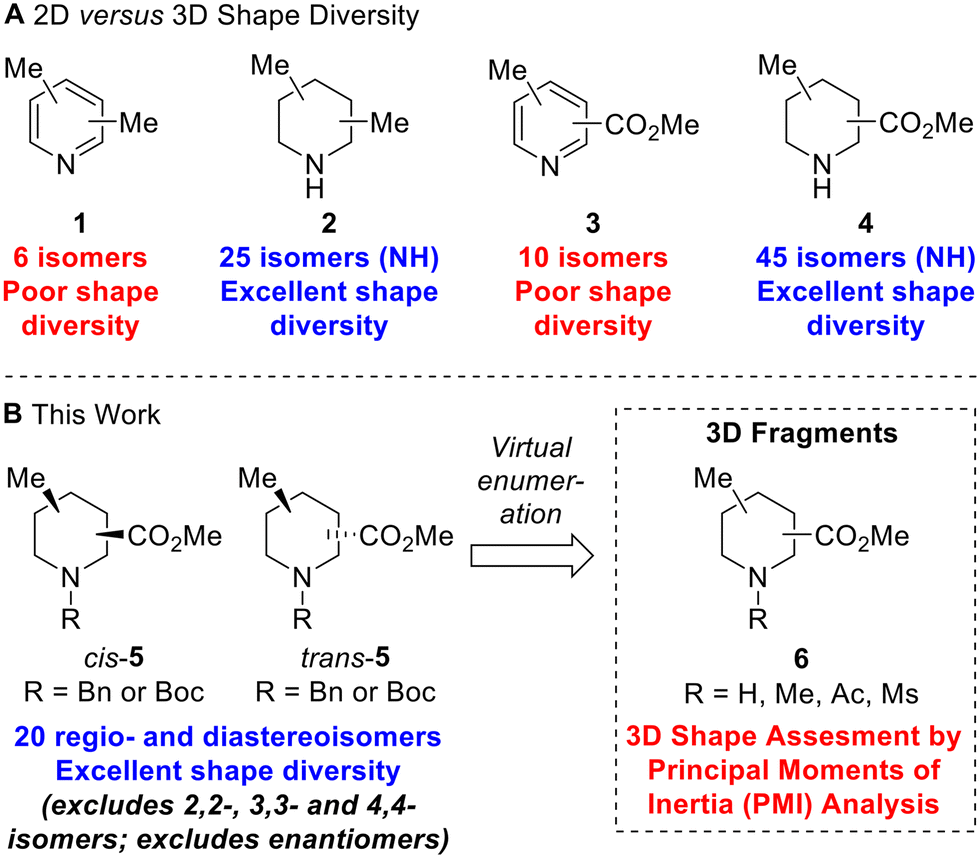 | ||
| Fig. 1 2D versus 3D shape diversity in disubstituted pyridines and piperidines and virtual 3D fragments presented in this work. | ||
Given our interest in 3D fragments, we extended Lovering's analysis of pyridines and NH piperidines by furnishing the scaffolds with two different substituents: a methyl group and a methyl ester. In this case, the increase in 3D shape diversity was even more pronounced going from a 2D to a 3D scaffold: there are ten 2D isomers for methyl picolinates 3 and 45 3D isomers for methyl pipecolinates 4 (Fig. 1A). Of the 45 isomers for methyl pipecolinates 4, five are 2,2-, 3,3- or 4,4-disubstituted piperidines and there are two enantiomeric series of 20 cis- and trans-disubstituted piperidines. We considered that these 20 racemic cis- and trans-disubstituted methyl pipecolinates 4 would be interesting building blocks for use in medicinal chemistry and could form the key scaffold of novel 3D fragments (with N-functionalisation providing an additional point of diversity). Indeed, such piperidine motifs have found utility in drug development programs, including the development of PDK1 inhibitors14 and orexin receptor antagonists.15 However, for such 3D shape diversity to be realised in practice, viable synthetic routes to all possible isomers would be needed since a number of these substitution patterns had not been reported previously.
In this paper, we report three distinct pieces of synthetic methodology that has allowed access to each of the 20 regio- and diastereoisomers of methyl substituted pipecolinates cis- and trans-5 where the N-substituent is protected with either a benzyl or Boc group (Fig. 1B). It was not necessary to include the 2,2-, 3,3- or 4,4-disubstituted piperidines in this study since we had previously reported the synthesis of each of the N-Boc methyl pipecolinates (via enolate methylation).11c Our synthetic approach towards the 20 regio- and diastereoisomers of methyl substituted pipecolinates cis- and trans-5 utilises pyridine hydrogenation and base-mediated epimerisation (via enolate formation) as well as Boc-directed α-lithiation-trapping (vide infra). This is the first report of the systematic synthesis of each isomer of methyl substituted pipecolinates cis- and trans-5 and means that fragments based on such piperidines can now be considered in drug discovery programmes. To highlight the 3D shape diversity provided by these 20 piperidines, each was enumerated with NH, NMe, N-acetamide and N-mesyl groups to generate a virtual library of 80 synthetically accessible 3D fragments 6, and their 3D shape was assessed using a principal moment of inertia (PMI) plot.16 Herein, we present our results.
Results and discussion
Our strategy for the synthesis of all 20 regio- and diastereoisomers of methyl substituted pipecolinates cis- and trans-5 (R = benzyl or Boc) is set out in Scheme 1. It was envisaged that hydrogenation of disubstituted pyridines 3 and subsequent N-protection would deliver methyl substituted pipecolinates cis-5 diastereoselectively.17 Then, subsequent base-mediated epimerisation of piperidines cis-5 (via enolate formation and re-protonation) should deliver diastereoisomeric piperidines trans-5. However, the epimerisation stage is not as straightforward as might appear and we anticipated that judicial choice of the N-protecting group would allow us to carry out the necessary epimerisations. For example, with 2,3-disubstitued N-benzyl piperidine cis-5a, one substituent will be axial and one equatorial. Thus, epimerisation of cis-5a under thermodynamic conditions should generate trans-5a driven by relieving the unfavourable 1,3-diaxial interaction (Scheme 1). Using a N-benzyl group, this strategy should be successful for the preparation of 2,3-trans, 2,5-trans and 3,4-trans isomers (together with 3,5-cis).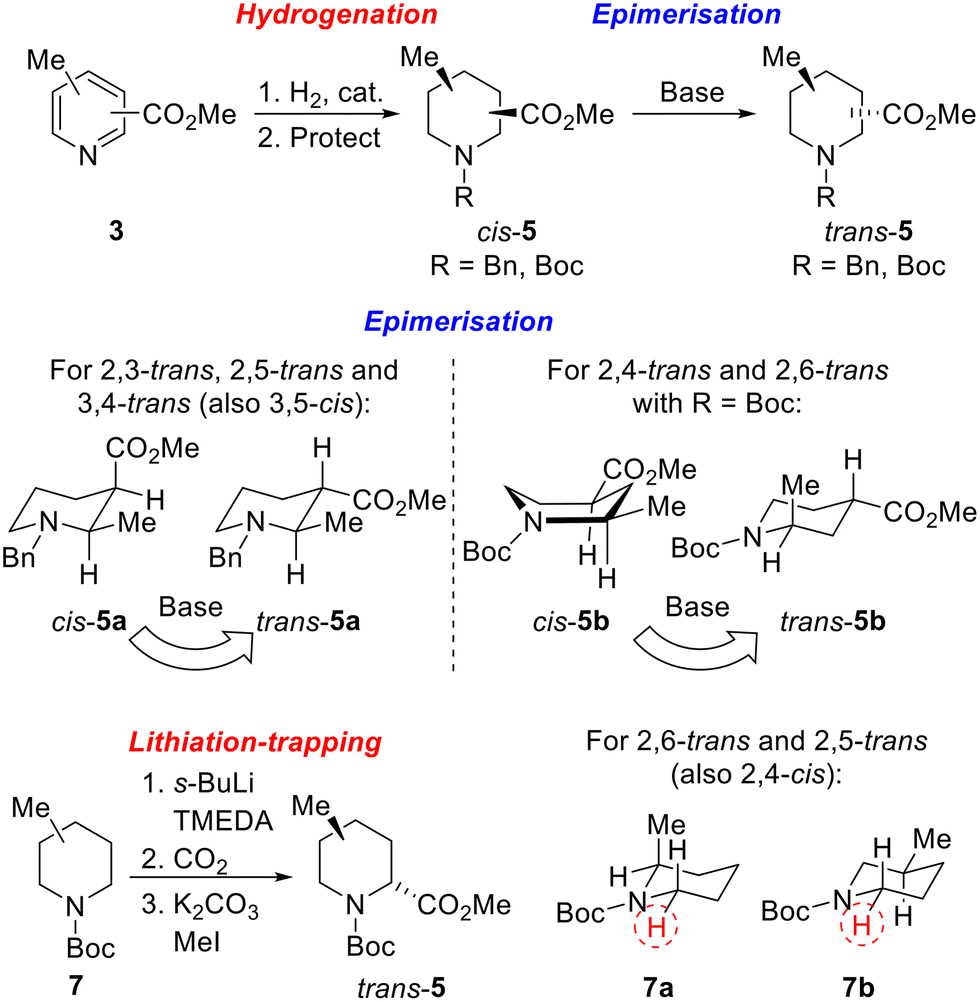 | ||
| Scheme 1 Synthetic strategy towards 20 regio- and diastereoisomers of methyl substituted pipecolinates: hydrogenation and epimerisation. | ||
In contrast, when R = Boc in methyl pipecolinates, there are different effects which control the lowest energy conformations and this would impact on the epimerisation event. For example, with 2,4-disubstitued N-Boc piperidine cis-5b, the lowest energy conformation will likely be a twist-boat conformation with both substituents in pseudo-equatorial positions.18 In contrast, the epimeric 2,4-disubstituted N-Boc piperidine trans-5b will adopt a lower energy conformation, with an equatorial 4-substituent and the 2-substituent adopting an axial orientation to avoid unfavourable A1,3-type strain between the Boc group and the 2-substituent.18a Thus, base-mediated epimerisation under thermodynamic conditions should convert cis-5b into trans-5b (Scheme 1). With a N-Boc substituent, this approach should be successful for the formation of 2,4-trans and 2,6-trans isomers. Clearly, either benzyl or Boc groups would be suitable for thermodynamically driven epimerisation of piperidines without 2-substituents (i.e. to 3,4-trans isomers).
Finally, an alternative route to selected trans-isomers would be the use of Beak's α-lithiation-trapping methodology,18a,19 with which we have much experience.11a,20 For this, it was envisaged starting with N-Boc methyl piperidines 7 and carrying out α-lithiation (using s-BuLi/TMEDA) and subsequent trapping with carbon dioxide (followed by methylation) to give methyl pipecolinates trans-5 (Scheme 1). For N-Boc 2-methyl piperidine 7a, equatorial lithiation from the lowest energy conformation with an axial 2-methyl group (to avoid unfavourable A1,3-type strain) should give the 2,6-trans isomer.19a In contrast, for N-Boc 3-methyl piperidine 7b, the lowest energy conformation will have an equatorial methyl group (to avoid 1,3-diaxial interactions) and equatorial lithiation should then deliver the 2,5-trans isomer.18a A similar rationale would also allow access to the 2,4-cis-isomer if desired.
Although the transition metal catalysed hydrogenation of several regioisomers of disubstituted pyridines 3 is known,21 no general methods exist. These reductions are often carried out at high pressure and/or high temperature and use high catalyst loadings or mixed metal catalyst systems. Furthermore, in many cases, specialist equipment such as Parr reactors, microwaves21b or flow equipment21c is required. We wanted to identify a general and simple hydrogenation process in which all 10 possible isomers of 3 could be reduced without the need for especially high catalyst loadings or specialist equipment.
The use of PtO2 as a hydrogenation catalyst was initially explored. To our delight, use of a comparatively low loading of 10 mol% PtO2 and a balloon of hydrogen successfully reduced acetic acid solutions of all but one pyridine 3 in under 16 h; complete reduction of 3h required the use of 30 mol% PtO2. Following protection with either a benzyl or Boc group, piperidines cis-5 were isolated in 50–90% yields as single diastereomers (Scheme 2). In all but one case, protected piperidines cis-5 were the major products, being formed in 65:35 to >95:5 dr (the drs reported are the ratio of diastereoisomers obtained from the 1H NMR spectrum of the crude reaction product). Of particular note, 2,4-substituted systems cis-5b and cis-5g were isolated in 90% and 85% yield respectively, with each being formed in >95:5 dr.
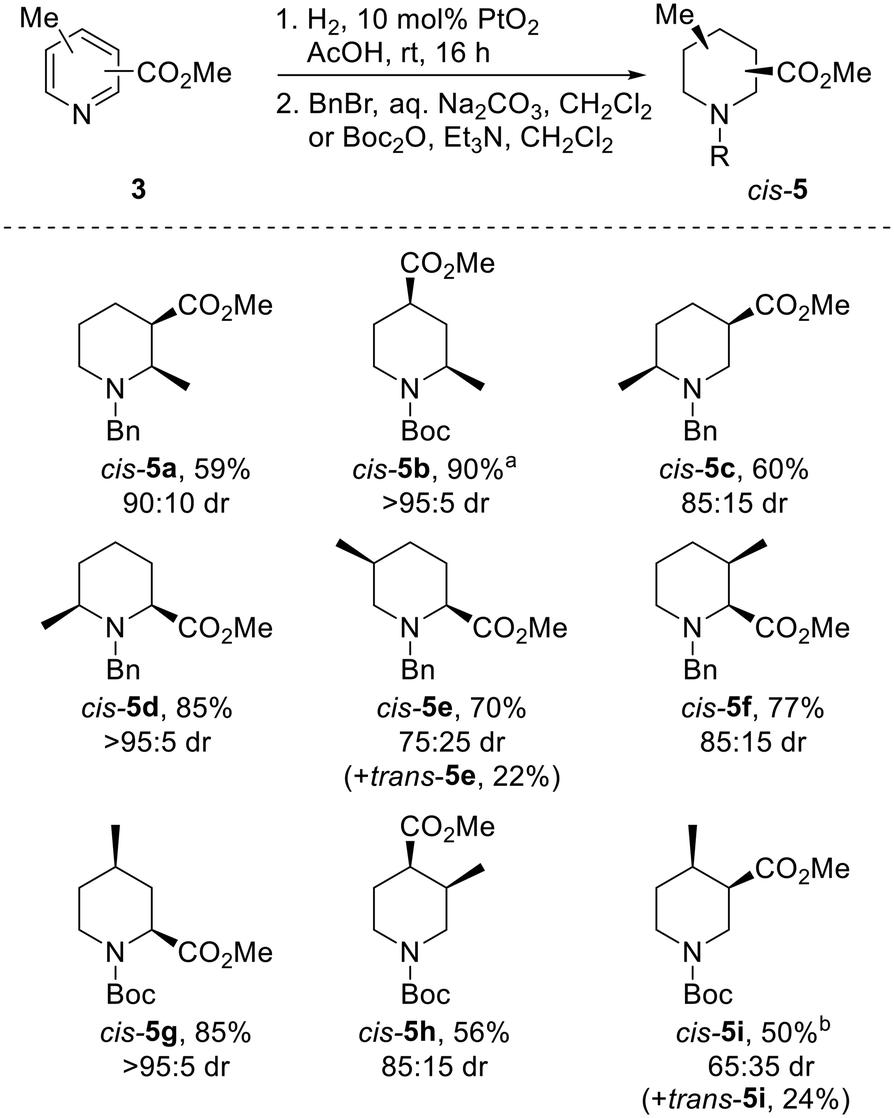 | ||
| Scheme 2 Reduction of pyridines 3 to piperidines cis-5. All products are racemic and the drs reported are the ratio of diastereoisomers obtained from the 1H NMR spectrum of the crude reaction product. a Methyl 2-chloro-6-methylpyridine-4-carboxylate was used; b 30 mol% PtO2 used. | ||
Surprisingly, hydrogenation of 3,5-substituted pyridine 3j using 10% Pd/C led to preferential formation of the trans-isomer in 70:30 dr, with trans-5j and cis-5j being isolated in 51% and 17% yields after N-benzylation (Scheme 3). Use of 10% PtO2 resulted in 60:40 dr with trans-5j as the major product. Interestingly, Kappe has shown that the diastereoselectivity of the hydrogenation of 3j under flow conditions is dependent upon the catalyst employed, reaction temperature and pressure.21c Given that cis-isomer cis-5j is likely to be the thermodynamically favoured product from an epimerisation experiment (vide infra), preferential formation of N-benzyl piperidine trans-5jvia hydrogenation was fortuitous.
 | ||
| Scheme 3 Reduction of pyridine 3j to piperidines cis-5. Both products are racemic and the dr reported is the ratio of diastereoisomers obtained from the 1H NMR spectrum of the crude reaction product. | ||
The relative stereochemistry of cis-5h was confirmed by single-crystal X-ray diffraction of the N-tosyl derivative.11c The relative stereochemistry of all other cis-piperidines cis-5 was confirmed by comparison with known compounds or by analysis of J values in the 1H NMR spectra (see ESI† for full details).
Next, the epimerisation of the ester groups of cis-piperidines cis-5 to the epimeric piperidines trans-5 (and of trans-5j to cis-5j) were explored. Isolated examples of the epimerisation of piperidines 5 have been reported,21d,i,22 but we were keen to identify general conditions that allowed access to all 10 possible epimers. Treatment of eight diastereomerically pure piperidines cis-5 with potassium tert-butoxide in THF at −78 °C for 2 h21i resulted in epimerisation to give trans-piperidines trans-5, formed in 50:50–95:5 dr, isolated in 40–90% yields (Scheme 4). Of note, epimerisation of cis-5g to trans-5g required the use of LDA as a base due to a significant amount of trans-esterification when using potassium tert-butoxide. In this case, the observed 80:20 dr may be due to kinetic selectivity in the enolate re-protonation event. When 3,5-disubituted piperidine trans-5j was subjected to the standard conditions, cis-5j was formed in 85:15 dr and isolated in 62% yield.
 | ||
| Scheme 4 Epimerisation of di-substituted piperidines 5. All products are racemic and the drs reported, unless stated otherwise, are the ratio of diastereoisomers obtained from the 1H NMR spectrum of the crude reaction product. a LDA was used; b isolated as a 90:10 mixture of trans- and cis-isomers. | ||
Based on the analysis shown in Scheme 1, all of the epimerisations except that for piperidine trans-5e proceeded towards the expected thermodynamic product. Surprisingly, for trans-5e, a 50:50 mixture of trans- and cis-5e was generated, from which a 40% yield of trans-5e was obtained.
Unfortunately, we were unable to install the requisite Boc group on to the cis-2,6-disubstituted system (cf. cis-5d) due to steric constraints. Therefore, the trans-2,6-system was not accessible using the epimerisation strategy and diastereoselective lithiation/trapping methodology was thus utilised to synthesise the single outstanding diastereomer trans-5d. Using a modification of Beak's conditions for the α-lithiation of substituted piperidines,18a,19N-Boc 2-methyl piperidine 7a was treated with s-BuLi and TMEDA in Et2O at −40 °C for 90 min. Subsequent trapping with CO2 gave 2,6-disubstitiuted piperidine trans-8d as a single regio- and diastereoisomer in 82% yield. Methylation of trans-8d gave piperidine trans-5d in 97% yield (Scheme 5).
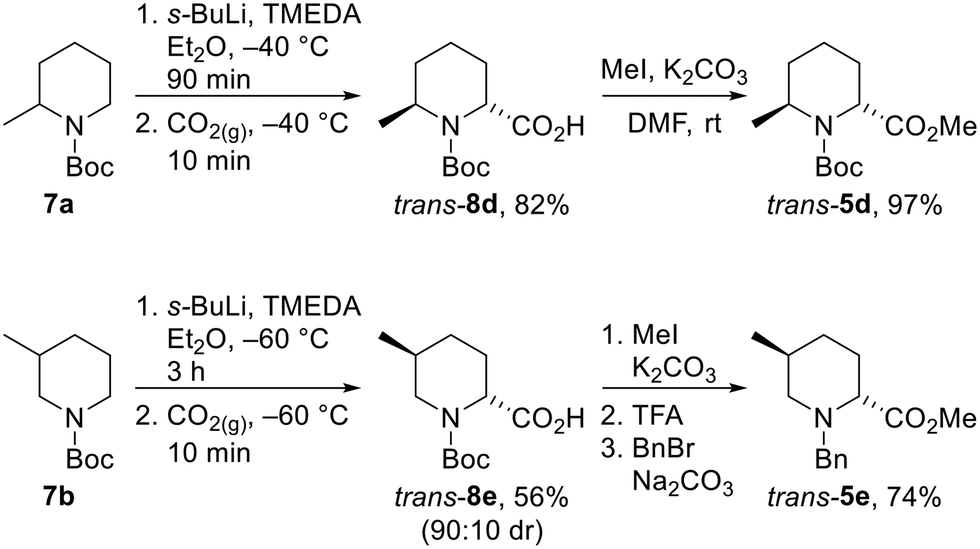 | ||
| Scheme 5 Synthesis of trans-5d and trans-5evia lithiation/trapping. All products are racemic and the dr of trans-8e reported is the ratio of diastereoisomers obtained from the 1H NMR spectrum of the crude reaction product and after chromatography. | ||
Due to the poor diastereoselectivity (50:50 dr), and hence yield (40%) in the epimerisation of cis-5e to trans-5e (see Scheme 4), the lithiation/trapping methodology was explored as an alternative method for the synthesis of trans-5e.22 Treatment of N-Boc 3-methyl piperidine 7b with s-BuLi and TMEDA in Et2O at −60 °C for 3 h and subsequent trapping with CO2 gave an inseparable 90:10 mixture of trans-8e and cis-8e in 56% yield. Methylation followed by Boc group removal and N-benzylation resulted in trans-5e being isolated in 74% yield (41% overall from 6b) (Scheme 5).
Thus, by using three different pieces of synthetic chemistry, all 20 regio- and diastereoisomers of methyl substituted picolinates 5 were prepared. Next, the suitability of the 20 piperidines 5 for the synthesis of 3D fragments for use in FBDD projects was assessed. First, we virtually removed the N-Boc or benzyl groups from 5 to give secondary amines 6a. Then, the amines 6a were virtually capped with one of three small groups (Me, Ac or Ms) to generate a virtual library of 80 fragments 6a–d (Fig. 2). The Me, Ac, and Ms substituents were selected as they are the simplest representatives of reductive amination, amide formation and sulfonamide formation reactions at the piperidine nitrogen atom, which are very commonly used in medicinal chemistry,23 and also provide mutually distinct exit vectors for substituents. Furthermore, Me, Ac, and Ms substituents featured in our previous 3D fragments11c and thus provide a suitable comparison. In addition, methodology for attaching these substituents has been established in this previous work. Importantly, all fragments fit within Astex's updated guidelines for the physicochemical properties of fragments (HAC: 10–16 and logP: 0–2)9 (see ESI†). The 3D shape of the fragments was analysed using normalised principal moments of inertia (PMI) for the molecular mechanics-generated lowest energy conformation, as introduced by Sauer and Schwarz.16 The three vertices of these plots correspond to rod, disc and spherical shapes. Many current fragment libraries consist predominantly of flat (hetero)aromatic compounds that populate the rod-disc axis.12b In comparison, our virtual fragments occupy a wide area of chemical space away from the rod-disc axis (Fig. 2, red dots), with 79 of the 80 fragments deemed to be 3D (by Firth's definition: NPR1 + NPR2 ≥ 1.07)11d,24 and would make suitable 3D fragments for a FBDD project. For comparison, the ten 2D isomers for methyl picolinates 3 lie on the rod-disc axis (Fig. 2, blue dots). Finally, the suitability of the piperidine fragment scaffolds as building blocks for medicinal chemistry was validated using the open-access tool LLAMA25 (see ESI† for full details).
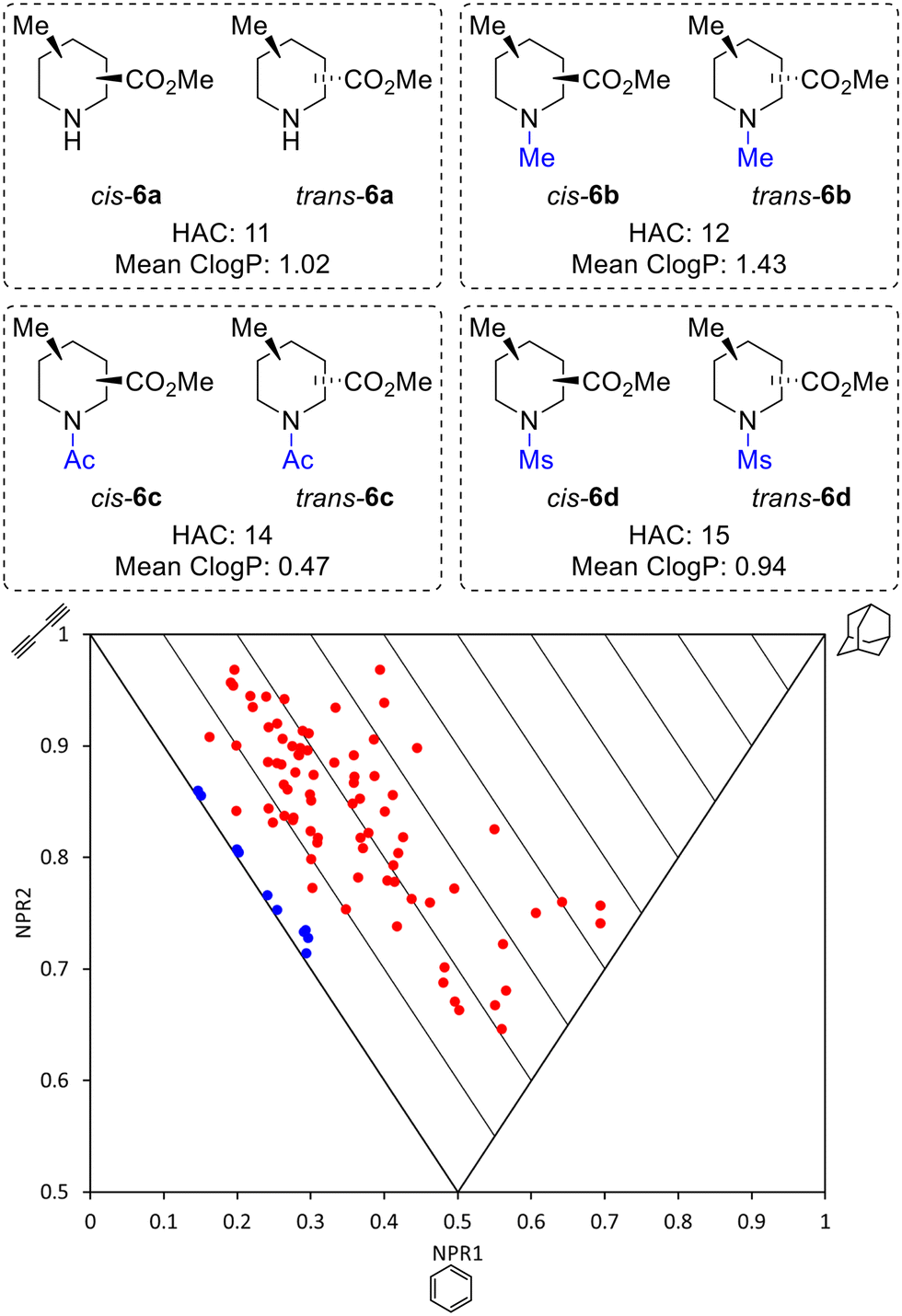 | ||
| Fig. 2 Molecular properties and PMI plot of the virtual fragment library: cis- and trans-6a–6d (red dots); pyridines 3 (blue dots) NPR is the normalised principal moment of inertia (see ESI†). | ||
Conclusions
In summary, we have developed simple and practical methods for the synthesis of all 20 regio- and diastereoisomers of cis- and trans-methyl substituted pipecolinates 5. Architecturally simple and easily accessible pyridines 3 were hydrogenated under mild and practical conditions to generate more structurally complex piperidines cis-5 in moderate to excellent yields. Subsequent epimerisation, facilitated by conformational control, using general conditions gave all but one piperidines trans-5; the remaining isomer (trans-5d) was attained using diastereoselective lithiation/trapping methodology in excellent yield. Crucially, we have shown that a virtual library of fragments 6 derived from piperidines 5 are three-dimensional, cover a wide area of 3D fragment space and have excellent molecular properties for fragment-based drug discovery campaigns.Author contributions
All authors contributed to the conceptualization and writing (review & editing). SPJ, JDF, MCW, MA and POB carried out the investigation, methodology, data curation and formal analysis. POB and REH provided supervision and funding acquisition. POB and JDF carried out the writing (original draft).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to AstraZeneca, Astex Pharmaceuticals, Lilly, Pfizer and Vernalis for supporting this venture. When this research was carried out, CDF was employed by AstraZeneca and LRV and MAW were employed by Lilly. MA is currently employed by Modulus Discovery. This project was funded by the EPSRC (MCW), BBSRC (BB/N008332/1) (JDF), University of York (SPJ), Asahi Kasei (MA) and The Royal Society (Industry Fellowship, INF\R1\191028) (POB). We thank Biovia for supplying Pipeline Pilot software.Notes and references
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Nat. Rev. Drug Discovery, 2012, 11, 873–886 CrossRef CAS PubMed.
- A. J. Souers, J. D. Leverson, E. R. Boghaert, S. L. Ackler, N. D. Catron, J. Chen, B. D. Dayton, H. Ding, S. H. Enschede, W. J. Fairbrother, D. C. S. Huang, S. G. Hymowitz, S. Jin, S. L. Khaw, P. J. Kovar, L. T. Lam, J. Lee, H. L. Maecker, K. C. Marsh, K. D. Mason, M. J. Mitten, P. M. Nimmer, A. Oleksijew, C. H. Park, C.-M. Park, D. C. Phillips, A. W. Roberts, D. Sampath, J. F. Seymour, M. L. Smith, G. M. Sullivan, S. K. Tahir, C. Tse, M. D. Wendt, Y. Xiao, J. C. Xue, H. Zhang, R. A. Humerickhouse, S. H. Rosenberg and S. W. Elmore, Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed.
- (a) T. P. S. Perera, E. Jovcheva, L. Mevellec, J. Vialard, D. De Lange, T. Verhulst, C. Paulussen, K. Van De Ven, P. King, E. Freyne, D. C. Rees, M. Squires, G. Saxty, M. Page, C. W. Murray, R. Gilissen, G. Ward, N. T. Thompson, D. R. Newell, N. Cheng, L. Xie, J. Yang, S. J. Platero, J. D. Karkera, C. Moy, P. Angibaud, S. Laquerre and M. V. Lorenzi, Mol. Cancer Ther., 2017, 16, 1010–1020 CrossRef CAS PubMed; (b) C. W. Murray, D. R. Newell and P. Angibaud, Med. Chem. Commun., 2019, 10, 1509–1511 RSC.
- (a) C. Zhang, P. N. Ibrahim, J. Zhang, E. A. Burton, G. Habets, Y. Zhang, B. Powell, B. L. West, B. Matusow, G. Tsang, R. Shellooe, H. Carias, H. Nguyen, A. Marimuthu, K. Y. J. Zhang, A. Oh, R. Bremer, C. R. Hurt, D. R. Artis, G. Wu, M. Nespi, W. Spevak, P. Lin, K. Nolop, P. Hirth, G. H. Tesch and G. Bollag, Proc. Natl. Acad. Sci., 2013, 110, 5689–5694 CrossRef CAS PubMed; (b) W. D. Tap, Z. A. Wainberg, S. P. Anthony, P. N. Ibrahim, C. Zhang, J. H. Healey, B. Chmielowski, A. P. Staddon, A. L. Cohn, G. I. Shapiro, V. L. Keedy, A. S. Singh, I. Puzanov, E. L. Kwak, A. J. Wagner, D. D. Von Hoff, G. J. Weiss, R. K. Ramanathan, J. Zhang, G. Habets, Y. Zhang, E. A. Burton, G. Visor, L. Sanftner, P. Severson, H. Nguyen, M. J. Kim, A. Marimuthu, G. Tsang, R. Shellooe, C. Gee, B. L. West, P. Hirth, K. Nolop, M. van de Rijn, H. H. Hsu, C. Peterfy, P. S. Lin, S. Tong-Starksen and G. Bollag, N. Engl. J. Med., 2015, 373, 428–437 CrossRef CAS PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H.-L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed.
- J. Schoepfer, W. Jahnke, G. Berellini, S. Buonamici, S. Cotesta, S. W. Cowan-Jacob, S. Dodd, P. Drueckes, D. Fabbro, T. Gabriel, J.-M. Groell, R. M. Grotzfeld, A. Q. Hassan, C. Henry, V. Iyer, D. Jones, F. Lombardo, A. Loo, P. W. Manley, X. Pellé, G. Rummel, B. Salem, M. Warmuth, A. A. Wylie, T. Zoller, A. L. Marzinzik and P. Furet, J. Med. Chem., 2018, 61, 8120–8135 CrossRef CAS PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 55, 488–492 CrossRef CAS PubMed.
- G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray and S. D. Pickett, J. Med. Chem., 2016, 59, 8189–8206 CrossRef PubMed.
- (a) M. Lüthy, M. C. Wheldon, C. Haji-Cheteh, M. Atobe, P. S. Bond, P. O'Brien, R. E. Hubbard and I. J. S. Fairlamb, Bioorg. Med. Chem., 2015, 23, 2680–2694 CrossRef PubMed; (b) J. D. Firth and P. O'Brien, Lead- and fragment-oriented synthesis, in Chemical and Biological Synthesis: Enabling Approaches for Understanding Biology, ed. N. J. Westwood and A. Nelson, The Royal Society of Chemistry, Cambridge, 2018, pp. 74–113 Search PubMed; (c) P. O'Brien, T. D. Downes, S. P. Jones, H. F. Klein, M. C. Wheldon, M. Atobe, P. S. Bond, J. D. Firth, N. S. Chan, L. Waddelove, R. E. Hubbard, D. C. Blakemore, C. De Fusco, S. D. Roughley, L. R. Vidler, M. A. Whatton, A. J.-A. Woolford and G. L. Wrigley, Chem. – Eur. J., 2020, 26, 8969–8975 CrossRef PubMed; (d) D. J. Hamilton, T. Dekker, H. F. Klein, G. V. Janssen, M. Wijtmans, P. O'Brien and I. J. P. de Esch, Drug Discovery Today: Technol., 2020, 38, 77–90 CrossRef PubMed; (e) A. Douangamath, D. Fearon, P. Gehrtz, T. Krojer, P. Lukacik, C. D. Owen, E. Resnick, C. Strain-Damerell, A. Aimon, P. Ábrányi-Balogh, J. Brandão-Neto, A. Carbery, G. Davison, A. Dias, T. D. Downes, L. Dunnett, M. Fairhead, J. D. Firth, S. P. Jones, A. Keeley, G. M. Keserü, H. F. Klein, M. P. Martin, M. E. M. Noble, P. O'Brien, A. Powell, R. N. Reddi, R. Skyner, M. Snee, M. J. Waring, C. Wild, N. London, F. von Delft and M. A. Walsh, Nat. Commun., 2020, 11, 5047 CrossRef CAS PubMed; (f) M. Schuller, G. J. Correy, S. Gahbauer, D. Fearon, T. Wu, R. E. Díaz, I. D. Young, L. C. Martins, D. H. Smith, U. Schulze-Gahmen, T. W. Owens, I. Deshpande, G. E. Merz, A. C. Thwin, J. T. Biel, J. K. Peters, M. Moritz, N. Herrera, H. T. Kratochvil, QCRG Structural Biology Consortium, A. Aimon, J. M. Bennett, J. Brandão-Neto, A. E. Cohen, A. Dias, A. Douangamath, L. Dunnett, O. Fedorov, M. P. Ferla, M. R. Fuchs, T. J. Gorrie-Stone, J. M. Holton, M. G. Johnson, T. Krojer, G. Meigs, A. J. Powell, J. G. M. Rack, V. L. Rangel, S. Russi, R. E. Skyner, C. A. Smith, A. S. Soares, J. L. Wierman, K. Zhu, P. O'Brien, N. Jura, A. Ashworth, J. J. Irwin, M. C. Thompson, J. E. Gestwicki, F. von Delft, B. K. Shoichet, J. S. Fraser and I. Ahel, Sci. Adv., 2022, 7, eabf8711 CrossRef PubMed; (g) H. F. Klein, D. J. Hamilton, I. J. P. de Esch, M. Wijtmans and P. O'Brien, Drug Discovery Today, 2022, 27, 2484–2496 CrossRef CAS PubMed.
- (a) A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci., 2011, 108, 6799–6804 CrossRef CAS PubMed; (b) A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert, S. Hoelder, A. Jordan, S. V. Ley, A. Merritt, D. Miller, M. E. Swarbrick and P. G. Wyatt, Drug Discovery Today, 2013, 18, 1221–1227 CrossRef PubMed; (c) K. F. Morgan, I. A. Hollingsworth and J. A. Bull, Chem. Commun., 2014, 50, 5203–5205 RSC; (d) N. C. Tran, H. Dhondt, M. Flipo, B. Deprez and N. Willand, Tetrahedron Lett., 2015, 56, 4119–4123 CrossRef CAS; (e) H. Prevet, M. Flipo, P. Roussel, B. Deprez and N. Willand, Tetrahedron Lett., 2016, 57, 2888–2894 CrossRef CAS; (f) S. L. Kidd, T. J. Osberger, N. Mateu, H. F. Sore and D. R. Spring, Front. Chem., 2018, 6, 460 CrossRef PubMed; (g) D. G. Twigg, N. Kondo, S. L. Mitchell, W. R. J. D. Galloway, H. F. Sore, A. Madin and D. R. Spring, Angew. Chem., Int. Ed., 2016, 55, 12479–12483 CrossRef CAS PubMed; (h) H. Prescher, G. Koch, T. Schuhmann, P. Ertl, A. Bussenault, M. Glick, I. Dix, F. Petersen and D. E. Lizos, Bioorg. Med. Chem., 2017, 25, 921–925 CrossRef CAS PubMed; (i) P. Garner, P. B. Cox, U. Rathnayake, N. Holloran and P. Erdman, ACS Med. Chem. Lett., 2019, 10, 811–815 CrossRef CAS PubMed; (j) R. Zhang, P. J. McIntyre, P. M. Collins, D. J. Foley, C. Arter, F. von Delft, R. Bayliss, S. Warriner and A. Nelson, Chem. – Eur. J., 2019, 25, 6831–6839 CrossRef CAS PubMed; (k) A. Sveiczer, A. J. P. North, N. Mateu, S. L. Kidd, H. F. Sore and D. R. Spring, Org. Lett., 2019, 21, 4600–4604 CrossRef CAS PubMed; (l) U. Grädler, D. Schwarz, M. Blaesse, B. Leuthner, T. L. Johnson, F. Bernard, X. Jiang, A. Marx, M. Gilardone, H. Lemoine, D. Roche and C. Jorand-Lebrun, Bioorg. Med. Chem. Lett., 2019, 29, 126717 CrossRef PubMed; (m) N. S. Troelsen, E. Shanina, D. Gonzalez-Romero, D. Danková, I. S. A. Jensen, K. J. Śniady, F. Nami, H. Zhang, C. Rademacher, A. Cuenda, C. H. Gotfredsen and M. H. Clausen, Angew. Chem., 2020, 59, 2204–2210 CrossRef CAS PubMed; (n) A. R. Hanby, N. S. Troelsen, T. J. Osberger, S. L. Kidd, K. T. Mortensen and D. R. Spring, Chem. Commun., 2020, 56, 2280–2283 RSC; (o) B. Cox, V. Zdorichenko, P. B. Cox, K. I. Booker-Milburn, R. Paumier, L. D. Elliott, M. Robertson-Ralph and G. Bloomfield, ACS Med. Chem. Lett., 2020, 11, 1185–1190 CrossRef CAS PubMed; (p) A. K. Pandey, S. E. Kirberger, J. A. Johnson, J. R. Kimbrough, D. K. D. Partridge and W. C. K. Pomerantz, Org. Lett., 2020, 22, 3946–3950 CrossRef CAS PubMed; (q) I. P. Silvestri and P. J. J. Colbon, ACS Med. Chem. Lett., 2021, 12, 1220–1229 CrossRef CAS PubMed; (r) M. J. Caplin and D. J. Foley, Chem. Sci., 2021, 12, 4646–4660 RSC.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- J. R. Medina, C. J. Becker, C. W. Blackledge, C. Duquenne, Y. Feng, S. W. Grant, D. Heerding, W. H. Li, W. H. Miller, S. P. Romeril, D. Scherzer, A. Shu, M. A. Bobko, A. R. Chadderton, M. Dumble, C. M. Gardiner, S. Gilbert, Q. Liu, S. K. Rabindran, V. Sudakin, H. Xiang, P. G. Brady, N. Campobasso, P. Ward and J. M. Axten, J. Med. Chem., 2011, 54, 1871–1895 CrossRef CAS PubMed.
- (a) M. Girardin, S. G. Ouellet, D. Gauvreau, J. C. Moore, G. Hughes, P. N. Devine, P. D. O'Shea and L.-C. Campeau, Org. Process Res. Dev., 2013, 17, 61–68 CrossRef CAS; (b) R. Jiang, X. Song, P. Bali, A. Smith, C. R. Bayona, L. Lin, M. D. Cameron, P. H. McDonald, P. J. Kenny and T. M. Kamenecka, Bioorg. Med. Chem. Lett., 2012, 22, 3890–3894 CrossRef CAS PubMed.
- W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comput. Sci., 2003, 43, 987–1003 CrossRef CAS PubMed.
- M. G. P. Buffat, Tetrahedron, 2004, 60, 1701–1729 CrossRef CAS.
- (a) P. Beak and W. K. Lee, J. Org. Chem., 1993, 58, 1109–1117 CrossRef CAS; (b) S. Seel, T. Thaler, K. Takatsu, C. Zhang, H. Zipse, B. F. Straub, P. Mayer and P. Knochel, J. Am. Chem. Soc., 2011, 133, 4774–4777 CrossRef CAS PubMed; (c) A. Choi, A. J. H. M. Meijer, I. P. Silvestri and I. Coldham, J. Org. Chem., 2022, 87, 8819–8823 CrossRef CAS PubMed.
- (a) P. Beak and W. K. Lee, J. Org. Chem., 1990, 55, 2578–2580 CrossRef CAS; (b) D. J. Gallagher and P. Beak, J. Org. Chem., 1995, 60, 7092–7093 CrossRef CAS.
- (a) G. Carbone, P. O'Brien and G. Hilmersson, J. Am. Chem. Soc., 2010, 132, 15445–15450 CrossRef CAS PubMed; (b) D. Stead, G. Carbone, P. O'Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260–7261 CrossRef CAS PubMed; (c) G. Barker, J. L. McGrath, A. Klapars, D. Stead, G. Zhou, K. R. Campos, P. O'Brien and P. O'Brien, J. Org. Chem., 2011, 76, 5936–5953 CrossRef CAS PubMed; (d) N. S. Sheikh, D. Leonori, G. Barker, J. D. Firth, K. R. Campos, A. J. H. M. Meijer, P. O'Brien and I. Coldham, J. Am. Chem. Soc., 2012, 134, 5300–5308 CrossRef CAS PubMed; (e) P. J. Rayner, P. O'Brien and R. A. J. Horan, J. Am. Chem. Soc., 2013, 135, 8071–8077 CrossRef CAS PubMed; (f) D. Stead, P. O'Brien and A. Sanderson, Synlett, 2015, 26, 2381–2384 CrossRef CAS; (g) J. D. Firth, P. O'Brien, L. Ferris, J. D. Firth, P. O'Brien and L. Ferris, J. Am. Chem. Soc., 2016, 138, 651–659 CrossRef CAS PubMed; (h) J. D. Firth, P. O'Brien and L. Ferris, J. Org. Chem., 2017, 82, 7023–7031 CrossRef CAS PubMed; (i) J. D. Firth, G. Gelardi, P. J. Rayner, D. Stead and P. O'Brien, Heterocycles, 2018, 97, 1288–1303 CrossRef CAS; (j) A. Kwong, J. D. Firth, T. J. Farmer and P. O'Brien, Tetrahedron, 2021, 81, 131899 CrossRef CAS.
- (a) C. Subramanyam, S. Chattarjee and J. P. Mallamo, Tetrahedron Lett., 1996, 37, 459–462 CrossRef CAS; (b) L. Piras, E. Genesio, C. Ghiron and M. Taddei, Synlett, 2008, 1125–1128 CAS; (c) M. Irfan, E. Petricci, T. N. Glasnov, M. Taddei and C. O. Kappe, Eur. J. Org. Chem., 2009, 1327–1334 CrossRef CAS; (d) R. J. Snow, R. Baker, R. H. Herbert, I. J. Hunt, K. J. Merchant and J. Saunders, J. Chem. Soc., Perkin Trans. 1, 1991, 409–420 RSC; (e) D. Barawkar, T. Bende, R. Zahler, A. Bandyopadhyay, R. S. Sarangthem, J. Doshi, Y. Waman, R. Jadhav and U. P. Singh, WO2012127506A1, 2012, Advinus Therapeutics Limited; (f) S. Allerheiligen, A. Buchmüller, K. Engel, C. Gerdes, K. M. Gericke, M. Gerisch, S. Heitmeier, A. Hillisch, T. Kinzel, P. Lienau, B. Riedl, S. Röhrig, M. V. Schmidt, J. Strassburger and A. Tersteegen, WO2014195230A1, 2014, Bayer Pharma Aktiengesellschaft; (g) P. A. Barsanti, C. Hu, X. Jin, S. C. Ng, K. B. Pfister, M. Sendzik and J. Sutton, WO2012101064A1, 2012, Novartis Ag; (h) J. R. Henry, WO2016077161A1, 2016, Eli Lilly And Company; (i) J. R. Hauske, WO2008024371A2, 2008, Prexa Pharmaceuticals, Inc.; (j) E. Lorthiois, I. Marek and J. F. Normant, J. Org. Chem., 1998, 63, 566–574 CrossRef CAS PubMed.
- Z. Zhang, G. M. Giampa, C. Draghici, Q. Huang and M. Brewer, Org. Lett., 2013, 15, 2100–2103 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- N. C. Firth, N. Brown and J. Blagg, J. Chem. Inf. Model., 2012, 52, 2516–2525 CrossRef CAS PubMed.
- (a) I. Colomer, C. J. Empson, P. Craven, Z. Owen, R. G. Doveston, I. Churcher, S. P. Marsden and A. Nelson, Chem. Commun., 2016, 52, 7209–7212 RSC; (b) https://llama.leeds.ac.uk/, (accessed June 2022).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data and properties analysis. See DOI: https://doi.org/10.1039/d2md00239f |
| This journal is © The Royal Society of Chemistry 2022 |