
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post-polymerisation approaches for the rapid modification of conjugated polymer properties
Martina
Rimmele
 a,
Florian
Glöcklhofer
*a and
Martin
Heeney
*ab
a,
Florian
Glöcklhofer
*a and
Martin
Heeney
*ab
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, London, W12 0BZ, UK. E-mail: f.glocklhofer@imperial.ac.uk
bKAUST Solar Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: martin.heeney@kaust.edu.sa
First published on 11th August 2022
Abstract
Post-polymerisation functionalisation provides a facile and efficient way for the introduction of functional groups on the backbone of conjugated polymers. Using post-polymerisation functionalisation approaches, the polymer chain length is usually not affected, meaning that the resulting polymers only differ in their attached functional groups or side chains, which makes them particularly interesting for investigating the influence of the different groups on the polymer properties. For such functionalisations, highly efficient and selective reactions are needed to avoid the formation of complex mixtures or permanent defects in the polymer backbone. A variety of suitable synthetic approaches and reactions that fulfil these criteria have been identified and reported. In this review, a thorough overview is given of the post-polymerisation functionalisations reported to date, with the methods grouped based on the type of reaction used: cycloaddition, oxidation/reduction, nucleophilic aromatic substitution, or halogenation and subsequent cross-coupling reaction. Instead of modifications on the aliphatic side chains of the conjugated polymers, we focus on modifications directly on the conjugated backbones, as these have the most pronounced effect on the optical and electronic properties. Some of the discussed materials have been used in applications, ranging from solar cells to bioelectronics. By providing an overview of this versatile and expanding field for the first time, we showcase post-polymerisation functionalisation as an exciting pathway for the creation of new conjugated materials for a range of applications.
Introduction
The remarkable optical and electronic properties of conjugated polymers (CPs), combined with their good processability and mechanical flexibility, has prompted much interest in their use for a variety of applications, ranging from energy storage to optoelectronic devices.1,2 Among the most important applications are organic photovoltaics and photodetectors (OPVs and OPDs), organic light-emitting diodes (OLEDs), sensors, and organic field-effect transistors (OFETs).3–6 The utilisation of CPs can enable new device properties, such as mechanical flexibility and optical transparency, as well as facilitate large-scale fabrication via potentially lower-cost printing based approaches.7–10For all CP applications, a number of molecular design features need to be considered. Importantly, CPs usually consist of the following parts (Fig. 1a–c): (1) the π-conjugated backbone that determines most of the optical and electronic properties, (2) aliphatic side chains that influence the solid-state packing and render the polymers soluble, crucial for processing the polymers from solution, and – optionally – (3) additional functional groups for fine-tuning of the optical and electronic properties, either electron-withdrawing groups (EWGs) or electron-donating groups (EDGs) with stronger electron-donating properties than the aliphatic side chains.11–13 The side chains and the additional functional groups are often combined into the same substituents attached to the π-conjugated backbone (Fig. 1c). Each of the three parts needs to be carefully selected, as they determine the properties of the polymers, most importantly the frontier molecular orbital (HOMO and LUMO) energy levels and, consequently, the band gap. The band gap is important as it determines the wavelength of light emitted in OLED applications but also the fraction of the solar spectrum that is absorbed in OPV/OPD applications. However, in addition to band gap requirements, the frontier molecular orbital energy levels of conjugated polymers also need to be carefully controlled in order to provide suitable matching with the work function of the electrodes or to facilitate desired charge-separation or -transfer processes between component materials.14
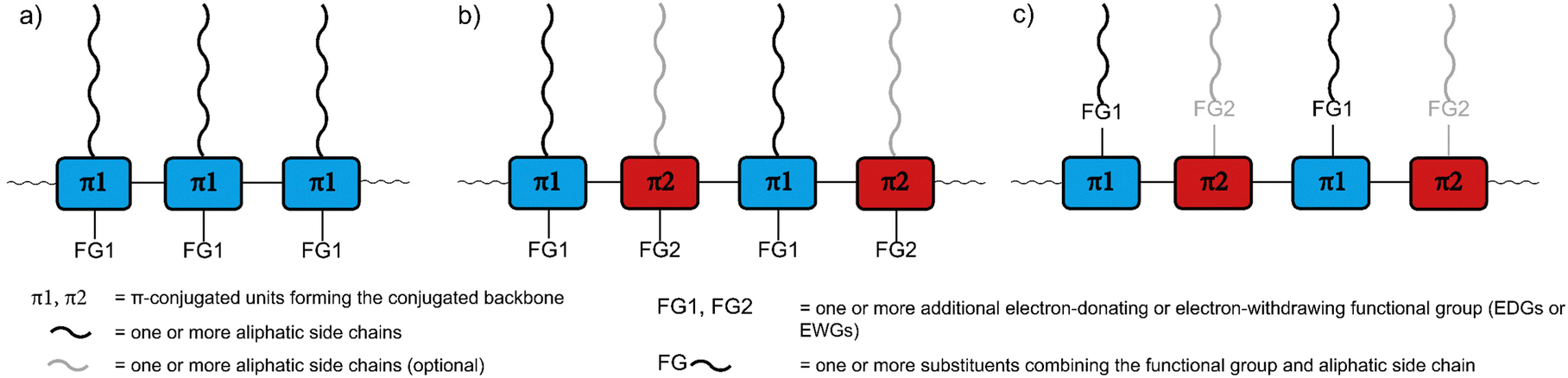 | ||
| Fig. 1 Schematic representation of conjugated polymers with (a) a single type of π-conjugated unit (π1) forming the conjugated backbone as well as aliphatic side chains and (optional) additional functional groups (FG1) as the other two parts of the conjugated polymer, (b) alternating donor and acceptor units (π1 and π2) forming the conjugated backbone as well as aliphatic side chains and (optional) additional functional groups, which are not necessarily present in both units, and (c) aliphatic side chains and additional functional groups combined into the same substituents. | ||
Besides the HOMO and LUMO energy levels and band gap, other properties such as good film-forming ability and high charge carrier mobility need to be considered in the polymer design.15,16 By thoughtfully selecting the three parts of CPs and designing the monomers accordingly, all these properties can be tuned and controlled. For a narrow band gap, the HOMO energy level needs to be raised or the LUMO energy level lowered (or both). Functional groups can be attached to the monomers to achieve this effect, making use of mesomeric and inductive effects to modify the electronic character: The attachment of EDGs can increase the HOMO and LUMO energy level of the polymer, while the attachment of EWGs can decrease the energy levels. In a different approach, co-polymerisation of electron-rich donor (D) and electron-deficient acceptor (A) monomers can be used to obtain alternating D–A polymers (Fig. 1b and c).15,17,18 In such D–A polymers, the bandgap can be significantly narrowed as a result of the hybridisation of the energy levels of the donor and acceptor units. D–A interactions are often proposed to lead to increased double bond character between the D and A units and therefore to increased backbone planarity, but we note that in a recently reported crystal structure of a D–A dimer such effects could not be observed.19 Regarding the polymer design, it is noteworthy that the electron-rich or -deficient character of the D and A units can be an inherent property of their conjugated system, but EDGs or EWGs can be attached to further strengthen this character and shift the energy levels.20–24
Another important aspect to consider in the polymer design is the self-assembly of the polymers, as the backbone orientation and packing structure can have a significant impact on the charge carrier mobility and, hence, on the device performance.25,26 The choice of side chains, which are needed to render the polymers soluble, has a particularly strong impact here. The side chains lead to spatial separation of the backbones, diminishing the molecular orbital overlap and, therefore, limiting the hopping of charges between polymer chains. While bulky side chains can additionally lead to twisting of the polymer backbone, preventing the formation of ordered domains, linear side chains can enable interdigitation and, consequently, closer packing and reduced spatial separation of the backbones, leading to better charge carrier mobility.27 Moreover, the formation of ordered domains can also be enhanced by regioregularity, which generally has a positive effect on the charge carrier mobilities.25,28,29 On the other hand, increased spatial separation of the backbones can be favourable in OLED devices, where close packing can lead to excimer formation, one of the processes that can result in quenching of the excited state in the solid state.
Traditionally, both the side chains and the additional functional groups (for tuning the self-assembly and the optical and electronic properties, respectively) are introduced in the monomers. These monomers are then (co-)polymerised using a broad variety of techniques that enable the formation of carbon–carbon single bonds. However, the synthesis of these functionalised monomers is often laborious, which inhibits facile changes to the attached groups. If researchers wish to attach a different side chain or functional group to the polymer, they usually need to start again early in the synthesis sequence, namely with the synthesis of the monomer(s).21,30,31 In addition, the range of functional groups that can be introduced via the available polymerisation techniques is limited; some functional groups cause side reactions, lead to a loss of control over the molecular weight, or completely inhibit the polymerisation reaction.
Post-polymerisation functionalisation can provide a solution to this problem. The approach dates back to Hermann Staudinger, the 1953 Nobel laureate in chemistry, who reported what he called “polymer analogous reactions”, describing them as reactions that lead to polymer derivatives with similar molecular weight as the starting polymers.32–35 The idea is that polymers that contain certain functional groups or attachment points on the repeat units are available for functionalisation in a consecutive step. Such functionalisation after the polymerisation step, nowadays known as post-polymerisation functionalisation or modification, offers several advantages, including facilitated introduction of a range of different functional groups, which enables the generation of large sets of polymers with less synthetic effort. Importantly, the polymer chain length is usually not affected by these functionalisations, making it a very powerful approach for creating libraries of comparable functionalised polymers.
It should be noted that reactions suitable for post-polymerisation functionalisation need to be very efficient and high yielding, with side reactions not being tolerated, as side reactions that occur on the polymer lead to defects that cannot be corrected by removal of the side product. Furthermore, high reactivity is crucial, as the accessibility of the polymer backbones is lowered by the sterically demanding nature of the polymers, and low reactivity can lead to complex mixtures of polymers with varying degrees of functionalisation.32 Today, very efficient transformations are available that can be used for post-polymerisation functionalisation, but so far, most of the effort has focussed on post-polymerisation functionalisation of non-conjugated polymers or on the side chains of conjugated polymers.32,36–46 Unfortunately, the optical and electronic properties of conjugated polymers are usually not significantly influenced by reactions on the side chains, which limits the usefulness of these modifications of conjugated polymers for certain applications.47,48 However, there are now a number of reactions that meet the above requirements and take place directly on the conjugated polymer backbone or in very close proximity. For this review, we focus exclusively on these functionalisations, as they can alter the electronic and optical properties significantly. We divided the review in sections based on the type of reaction used for these post-polymerisation functionalisations – cycloaddition, oxidation/reduction, nucleophilic aromatic substitution, as well as halogenation and subsequent cross-coupling reaction – and focus our discussion on how these modifications affect the energy levels and optical properties of the polymers.
We further focus on the modification of the chemical composition of the backbone, thereby excluding for example the (reversible) oxidation/reduction of the backbone in doping reactions. In addition, a large body of work has been reported towards the preparation of conjugated ladder polymers, in which adjacent monomers within the conjugated backbone are reacted intramolecularly to provide a rigid link.49 These are a fascinating and highly promising class of materials,50–53 but we did not consider these reactions here. There are a number of relevant reviews on the preparation of ladder-type conjugated polymers for the interested reader.54–57
Cycloaddition
Cycloaddition reactions are frequently used for the functionalisation of polymers and materials.58,59 The cycloaddition reactions used for functionalising polymers are usually classified as ‘click reactions’. Click reactions are per definition “modular and orthogonal as well as proceeding under simple and mild reaction conditions while affording high yields of a single product with facile purification”,60 which makes click chemistry a powerful approach in polymer chemistry and the development of functional materials.38,58,59,61–63 The orthogonality of the reactions and the high yields make them particularly useful for post-polymerisation functionalisation. Fig. 2 provides a list of cycloaddition reactions used for post-polymerisation functionalisation of conjugated polymers discussed in this review. | ||
| Fig. 2 Schematic representation and list of cycloaddition reactions used for post-polymerisation functionalisation of conjugated polymers. | ||
Using a click reaction, Adronov and co-workers demonstrated that the polyimine P1 with 5,6-didehydro-11,12-dihydrodibenzo[a,e]cyclooctene units can be functionalised via strain-promoted azide–alkyne cycloaddition (SPAAC) (Fig. 3, top).64 SPAAC has been widely used for the functionalisation or preparation of polymers as well as the formation of supramolecular structures.65 Compared to copper-catalysed AAC reactions (CuAAC), SPAAC has lower activation energies and hence requires lower temperatures and proceeds without copper catalysts.66 The mild reaction conditions and the non-necessity of a metal catalyst are beneficial for biological systems (for bioorthogonal reactions), enabling in vivo applications.67 The authors reported that the functionalisation of polymer P1 proceeds quantitatively in just 30 minutes at room temperature using toluene as the solvent and a 1.5-fold excess of azide. The polymer was functionalised with a variety of aryl azides, containing EDGs and EWGs on the aromatic ring (Fig. 3, bottom).
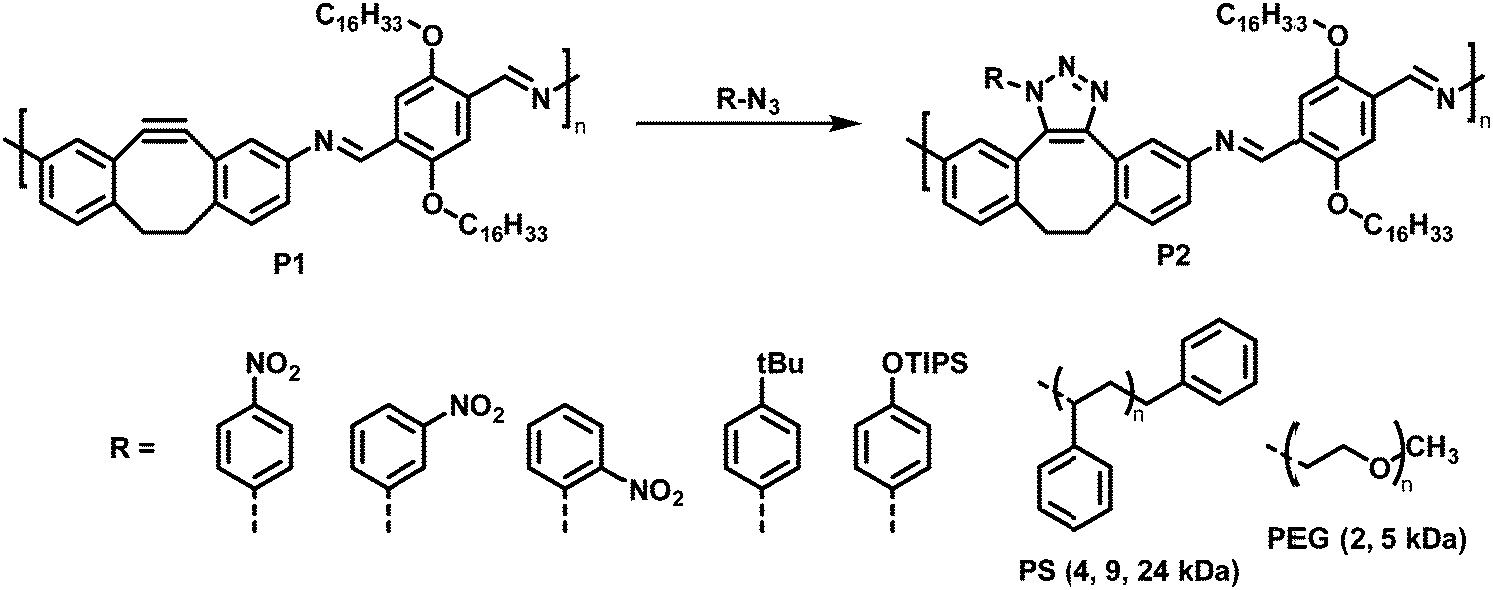 | ||
| Fig. 3 Post-polymerisation functionalisation of polyimine P1 with 5,6-didehydro-11,12-dihydrodibenzo[a,e]cyclooctene units using azides. Various aryl azides as well as azide-terminated polymers were attached to the polymer backbone via SPAAC.64 | ||
Studying the photophysical properties of the functionalised polymers in solution (THF), P2 showed that the post-polymerisation functionalisation led to small changes in the UV-vis absorption spectrum; the nature of the attached group in P2 has no influence on the UV-vis absorption spectrum. However, in contrast to the alkyne-containing starting polymer P1, the functionalised derivatives did not exhibit any fluorescence. In addition, the authors demonstrated the grafting of azide-terminated polystyrenes with molecular weights of up to 24 kDa, as well as azide-terminated polyethylene glycol monomethyl ether (N3-PEG-OMe) of 2 kDa and 5 kDa onto the polyimine under mild reaction conditions (12 h and 60 °C). The coupling of one polymer chain end to the backbone of another polymer can be considered the gold standard in terms of testing the efficacy of any post-polymerisation chemistry, because of the paucity of the polymer end-groups and the sterically challenging environment. Here grafting of the polymers occurred quantitatively in all cases, as indicated by 1H NMR and Raman spectroscopy, highlighting the power of the approach. In the case of grafting with N3-PEG-OMe, significant changes in the solubility were observed. In contrast to the starting polymer P1, which only exhibited solubility in apolar solvents, these grafted polymers showed good solubility in MeOH (2 kDa N3-PEG-OMe) and even water (5 kDa N3-PEG-OMe).
In a follow-up work, the authors demonstrated the photoclick functionalisation of a 9,10-phenanthrenquinone-containing conjugated polymer (P3) with electron-rich vinyl ethers (Fig. 4a).68 LED illumination of a solution of the polymer and butyl vinyl ether (BVE) resulted in conversion to the cycloaddition adducts P4 in just 5–10 min. Extensive IR spectroscopy experiments verified the quantitative conversion. The authors also studied the influence of the functionalisation on the photophysical properties. They found that the fluorescence intensity of the photoclick product P4 in solution (toluene) was remarkably increased in comparison to the starting material P3. The reaction was also amenable to photopatterning via the use of a P3/silicon elastomer blend which was exposed to BVE (Fig. 4b). Only the parts of the samples exposed to BVE and visible light showed emission when examined by confocal fluorescence microscopy, in accordance with the properties of the photoclicked polymer P4 (Fig. 4c). The versatility of the method was further demonstrated by coupling with an end-functionalised polymer (vinyl ether-terminated polyethylene glycol monomethyl ether (PEG-VE)) to afford a water-soluble graft co-polymer.
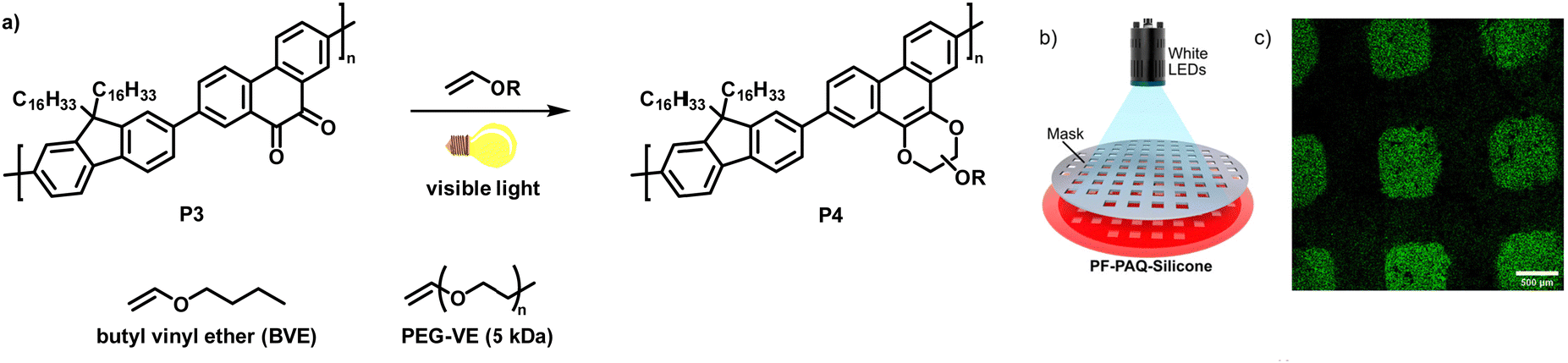 | ||
| Fig. 4 (a) Functionalisation of a 9,10-phenanthrenquinone-containing polymer with vinyl ethers.68 (b) Photopatterning of a sample of P3 embedded in a silicon elastomer and exposed to butyl vinyl ether (BVE). (c) Fluorescence microscopy image showing that only the parts of the sample exposed to BVE and light were functionalised. Adapted from ref. 68 with permission, Copyright 2020, American Chemical Society. | ||
Thermal [2+2] cycloaddition-retroelectrocyclisation (CA–RE) reaction is another approach to post-polymerisation functionalisation via click chemistry.69 In this reaction, electron-rich alkynes in the polymer backbone are reacted with electron-deficient alkenes to form what are usually non-planar donor–acceptor chromophores. In the first step of the reaction, a four-membered ring is formed as an intermediate, which subsequently rearranges by ring-opening leading to the formation of two double bonds (Fig. 5), resulting in the formation of a cross-conjugated donor–acceptor polymer. Strong electron donors need to be present in the polymer backbone in order to achieve sufficient electron density on the alkyne to enable the reaction. Several examples of polymers obtained by this post-polymerisation functionalisation method are shown in Fig. 6 and discussed below. Table 1 provides an overview of their properties.
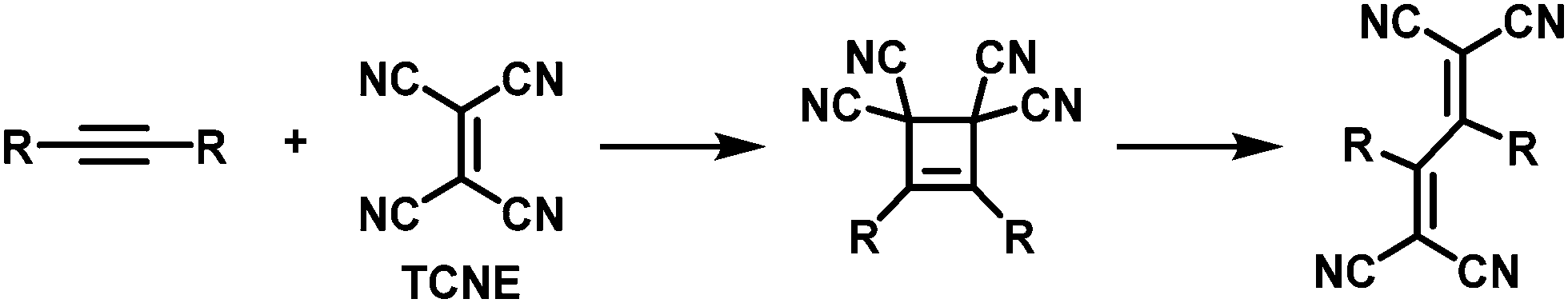 | ||
| Fig. 5 Mechanism of the thermal [2+2] cycloaddition–retroelectrocyclisation (CA–RE) reaction, using tetracyanoethylene (TCNE) as the electron-deficient alkene in this example. | ||
 | ||
| Fig. 6 Polymers obtained by post-polymerisation functionalisation via CA–RE reaction.70–75 | ||
| Polymer | Degree of functionalisation x | E opt (eV) | E ox1,onset (V vs. Fc/Fc+) | E red1,onset (V vs. Fc/Fc+) | Δ|Eox1,onset−Ered1,onset| (eV) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|---|---|---|
a From UV-vis absorption spectroscopy in CHCl3 solution.
b From UV-vis absorption spectroscopy in o-dichlorobenzene solution.
c From UV-vis absorption spectroscopy in CH2Cl2 solution.
d From cyclic voltammetry (CV) in 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M nBu4NClO4 in CH2Cl2.
e From CV in 0.1 M nBu4NClO4 in CH3CN using polymer thin films cast on a glassy carbon electrode.
f From CV in 0.1 M nBu4NPF6 in CH2Cl2. M nBu4NClO4 in CH2Cl2.
e From CV in 0.1 M nBu4NClO4 in CH3CN using polymer thin films cast on a glassy carbon electrode.
f From CV in 0.1 M nBu4NPF6 in CH2Cl2.
|
|||||||
| P5 | 0.75 | 1.46a | 0.30d | −0.86d | 1.16 | ||
| P6 | 0.30 | 1.18b | 0.76e | −0.52e | 1.28 | −5.56 | −4.28 |
| P7 | 0.19 | 1.02b | 0.92e | −0.01e | 0.93 | −5.72 | −4.79 |
| P8 | 0.56 | 1.00c | 0.42d | −0.61d | 1.03 | ||
| P9 | 0.72 | 1.46c | 0.42d | −0.82d | 1.24 | −5.22 | −3.98 |
| P10 | Quant. | 1.40c | 0.48f | −0.62f | 1.10 | −5.42 | −4.32 |
| P11 | Quant. | 1.83b | 0.63e | −0.82e | 1.45 | ||
| P12 | n.a. | 1.39c | 1.13f | −0.27f | 1.40 | −5.72 | −4.32 |
As a first example, Shigehara and co-workers demonstrated the post-polymerisation functionalisation of a ferrocene-containing poly(aryleneethynylene), featuring alkoxy substituents at the arylene units that contribute to the electron-rich character of the polymer.70 Functionalisation of the polymer by reaction with tetracyanoethylene (TCNE) in 1,2-dichlorobenzene at 120 °C yielded polymer P5 (with a degree of functionalisation of 75%). The functionalised polymer exhibited charge-transfer bands in the visible region, a characteristic of donor–acceptor chromophores.
Thiophene was reported to be another sufficiently strong backbone donor unit to enable the alkynes to undergo reactions with TCNE for the formation of polymer P6.71 As an alternative to TCNE, the authors also functionalised the polymer with 7,7,8,8-tetracyanoquinodimethane (TCNQ), giving polymer P7. As thiophene is a weaker electron donor than ferrocene, the alkynes are less reactive and the reaction was thus performed under microwave irradiation (120 °C, 4 h) to achieve satisfying conversion. The reactions were similarly effective in both cases, with degrees of functionalisation of 30% with TCNE and 19% with TCNQ. The formation of donor–acceptor chromophores in the polymers was confirmed by the appearance of a new broad charge-transfer band (>500 nm) in the visible region of the UV-vis absorption spectrum in solution (measured in o-dichlorobenzene) and by the quenching of the fluorescence (measured in CH2Cl2). Further measurements showed that the electrochemical gap was narrowed, with a lower lying LUMO and an elevated HOMO level.
Michinobu and co-workers studied the functionalisation of a poly(o-phenylenebutadiynylene) with TCNE to obtain polymer P8.72 They observed the expected shift of the absorption onset to longer wavelengths (in o-dichlorobenzene solution) as well as the occurrence of a charge transfer peak in the vis/NIR-region (596 nm). Titration experiments suggested a maximum of 56% functionalisation of the backbone with TCNE. In contrast to the synthesis of P8, the authors reported the appearance of side-reactions when testing the functionalisation of a structurally related poly(o-phenyleneethynylene). Similar functionalisation has been reported on a carbazole-containing poly(arylenebutadiynylene), resulting in a narrowed optical gap and the characteristic charge transfer band in the visible region (∼515 nm) for polymer P9 in solution (CH2Cl2).73 A degree of functionalisation of 72% was reported for this polymer.
Poly(arylene ethynylene)s containing both electron-donating dialkylamino groups and electron-withdrawing ester groups are also amenable to functionalisation (P10), with quantitative functionalisation.74 A broad charge transfer band was observed at ∼578 nm in solution (CH2Cl2), which is characteristic for the intramolecular interactions of the dialkylamino and 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) groups. These intramolecular charge transfer interactions also explain the small electrochemical gap of 1.10 eV (determined via cyclic voltammetry vs. Fc/Fc+).
The scope of polymers that can be functionalised via the CA–RE reaction was further broadened by Michinobu and co-workers, who functionalised a platinum-containing polyyne in a reaction with TCNE, resulting in polymer P11.75 Only one alkyne moiety per repeat unit underwent reaction with TCNE. This indicates that, upon reaction of the first alkyne, the electron density on the other alkyne is lowered, preventing further reaction. The method can also be utilised to functionalise electron rich alkynes attached to the conjugated polymer backbone. For example, alkyne containing poly(vinylene sulfide) was reacted with TCNE, resulting in the formation of donor–acceptor chromophores (P12) with strong intramolecular charge-transfer interactions.76
Overall, the CA–RE approach clearly results in significant changes to the optical and electronic properties of the polymers. However, the requirement for very electron-rich starting alkynes and the limited degree of functionalisation in some cases are drawbacks. The loss of backbone planarity may also be undesirable in some cases.
Benzannulation or cyclopentannulation has been shown to be another attractive tool for the synthesis of donor–acceptor polymers by post-polymerisation functionalisation, in particular for the synthesis of polyarylenes. Typically, the preparation of donor–acceptor polyarylenes relies on co-polymerisation of arylene monomers, but this often results in low molecular weight polymers due to steric or electronic influence.77 The preparation of polyarylenes via a post-polymerisation modification approach can be beneficial, as high molecular weight precursor polymers, synthesised via well-studied polymerisation techniques, can be used for the conversion into high molecular weight, low-bandgap copolymers.78
Making use of a copper-catalysed benzannulation reaction,79 Dichtel and colleagues synthesised sterically demanding polyarylenes from a poly(phenyleneethynylene) precursor (Fig. 7).80 In this post-polymerisation functionalisation, the polymer was reacted with o-(phenylethynyl)benzaldehyde (2 equiv.) with the aid of CF3COOH (3 equiv.) and Cu(OTf)2 (0.05 equiv.). The formation of the benzannulated product was confirmed by a combination of FT-IR and an isotopic labelling NMR study. Samples with 13C-enriched alkyne moieties were prepared, and the authors observed a clean shift of the alkyne signals after the benzannulation reaction with no remaining alkyne signals in the 13C NMR spectrum, indicating complete conversion. They observed an increase in solubility of the benzannulated product P14 compared to the precursor P13, attributed to steric hindrance in the product. The limited conformational freedom prevents the polymer from adopting a planar conformation. As a result, the behaviour of P14 in solution differs from that of P13, as was observed by size exclusion chromatography. The authors suggest that P14 adopts a more compact structure in solution compared to the non-annulated precursor P13 and is therefore retained longer on the column, despite its higher molecular weight. Molecular dynamics simulations are in accordance with these experimental findings. In UV-vis absorption measurements, a blue shift of the absorption of P14 (λmax = approx. 280 nm) of about 140 nm was observed compared to the precursor P13 (λmax = approx. 420 nm), which can also be explained by the nonplanarity of the backbone.
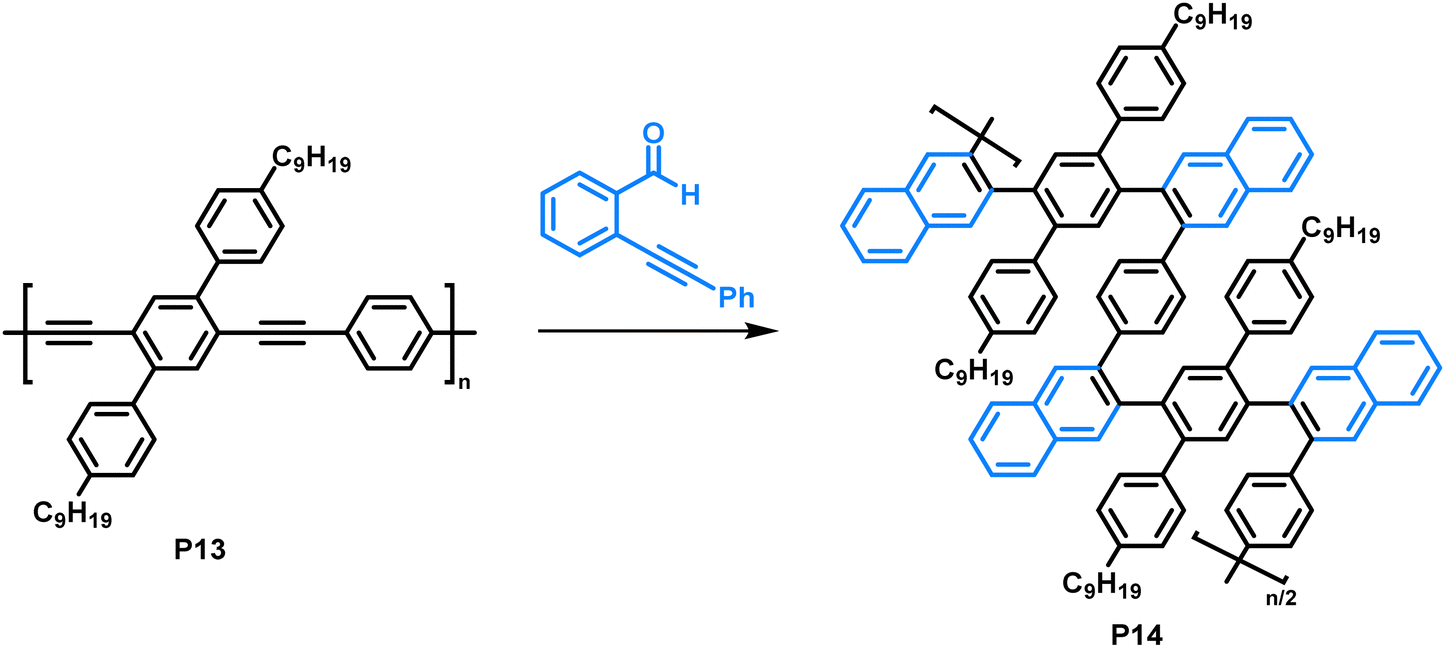 | ||
| Fig. 7 Copper-catalysed benzannulation of poly(phenyleneethynylene) (P13) with o-(phenylethynyl)benzaldehyde.80 | ||
The scope of the annulation approach has been expanded by Plunkett and colleagues. They earlier reported the palladium-catalysed cyclopentannulation of molecular precursors in order to obtain 1,2,6,7-tetraarylcyclopenta[hi]aceanthrylenes.81 Adapting this reaction for the functionalisation of acetylene-containing conjugated polymers (Fig. 8), an excess of aryl bromide was used in order to achieve a high degree of functionalisation, giving polymers P15–17.78 Analysis of polymers enriched with 13C (analogues of P15) by 13C NMR spectroscopy showed that unreacted alkynes were still present after functionalisation, but ”near complete conversion” was reported. The incomplete functionalisation was attributed to the reduced access to the polymer backbone, owing to a more folded structure upon functionalisation. The occurrence of side reactions was ruled out as a possible explanation, as the alkyne functionalities would be consumed likewise. GPC data showed similar trends as reported by Dichtel et al.; upon annulation the conformation of the polymer changes significantly to a more compact structure, resulting in longer retention times than observed for the precursor polymer. Formation of the cyclopenta[hi]aceanthrylene unit bathochromically shifted the thin film absorption onset from approx. 600 nm in the precursor to above 800 nm in the functionalised polymer. The donor strength of the respective aromatic moiety (increasing from fluorene to carbazole to dialkoxyphenylene) did not affect the optical and electronic properties of the polymers significantly (Table 2), with all three polymers, P15, P16 and P17, having very similar optical bandgaps of ∼1.5 eV.
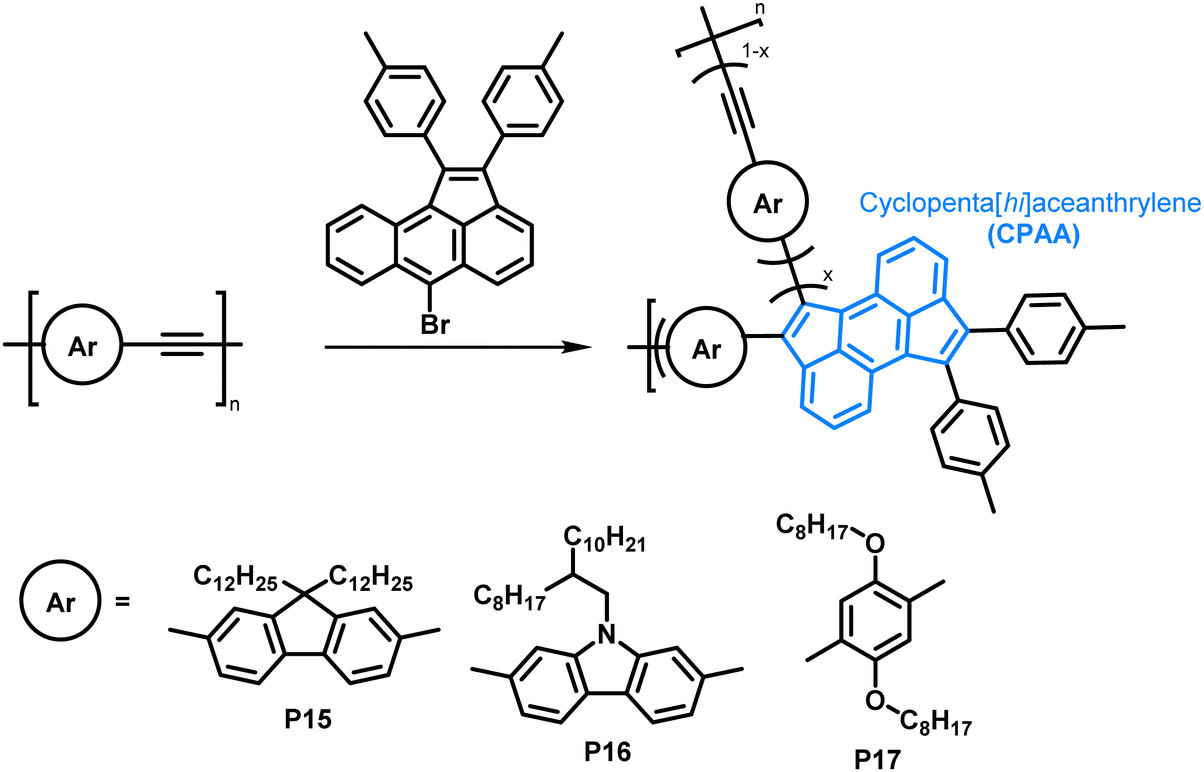 | ||
| Fig. 8 Cyclopentannulation of poly(arylene ethynylene)s as a post-polymerisation functionalisation approach. Polymers containing donor groups of increasing donor strength (from fluorene to carbazole to dialkoxyphenylene).78 | ||
| Polymer | λ onset (nm) | E g (opt.) (eV) | E ox1,onset (V vs. Fc/Fc+) | E red1,onset (V vs. Fc/Fc+) | Δ|Eox1,onset−Ered1,onset| (eV) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|---|---|---|
| P15 | 817 | 1.52 | 0.70 | −1.37 | 2.07 | −5.50 | −3.43 |
| P16 | 821 | 1.51 | 0.64 | −1.39 | 2.03 | −5.44 | −3.41 |
| P17 | 824 | 1.50 | 0.61 | −1.42 | 2.03 | −5.41 | −3.38 |
An interesting cycloaddition approach reported by Hayashi and colleagues involved the reaction of diene containing cross-conjugated precursor polymers with a dienophile via Diels–Alder reaction (Fig. 9).821H NMR spectra showed full conversion of the diene in the polymer backbone (by the disappearance of the signals of the diene protons). Similarly, full conversion was also confirmed for the subsequent rearomatisation step. The obtained all-(Z)-alkene- and ortho-arylene-containing polymers exhibited remarkable Stokes shifts (∼150 nm) in solution (THF), attributed to the π–π interactions between the arylene moieties.
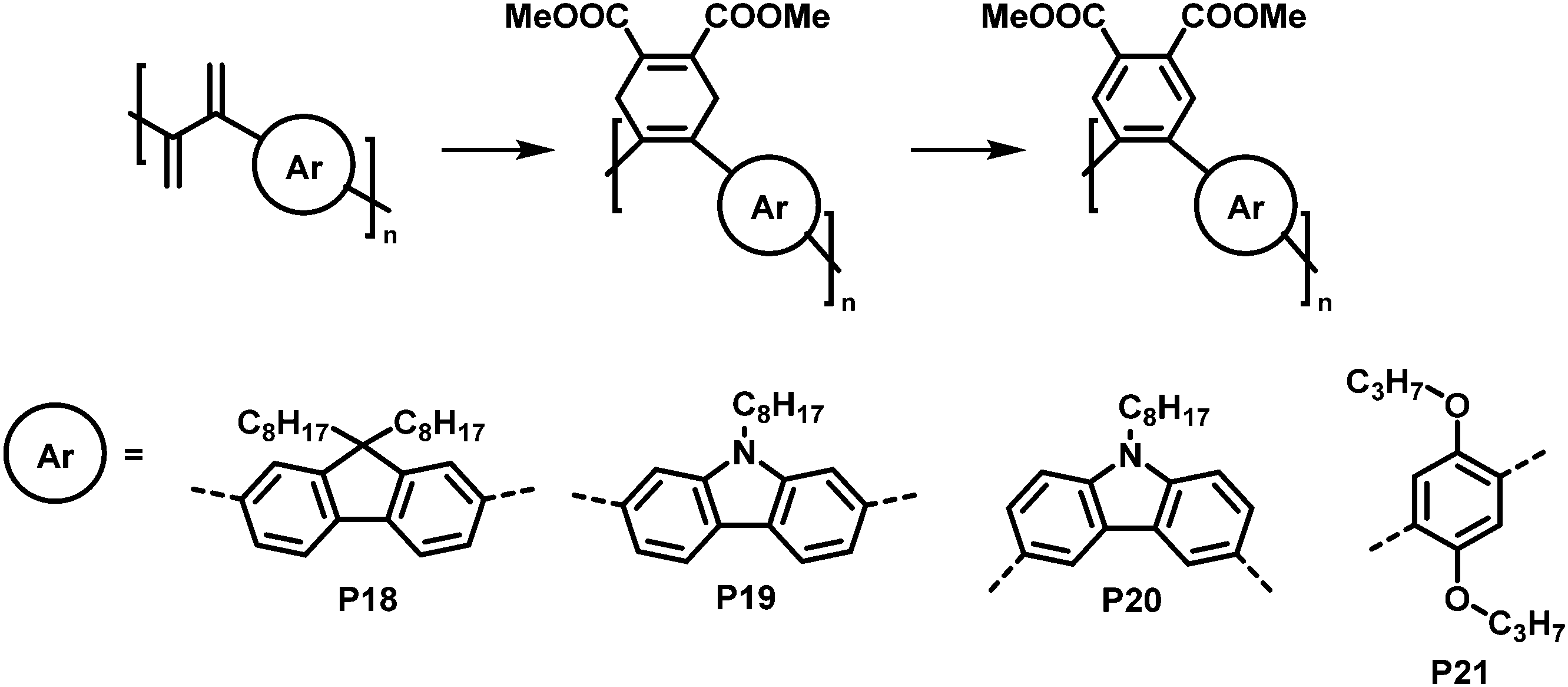 | ||
| Fig. 9 Post-polymerisation functionalisation of a diene-containing polymer via Diels–Alder reaction. Four different aromatic units were incorporated in the main chain of the conjugated polymer. After Diels–Alder reaction, the polymers were rearomatized in an additional step.82 | ||
Similarly, tetrazine containing polymer P22 is amenable to inverse electron demand Diels–Alder (IEDDA) reactions with a variety of cyclooctenes (Fig. 10).83 The reaction takes place rapidly at room temperature in solution, using only a small excess of the respective electron-rich dienophile. Using seven different trans-cyclooctene derivatives (see R in Fig. 10), the authors exemplified how conveniently different groups can be quantitatively introduced via this approach. It needs to be noted that the cycloaddition products P23 are always a mixture of two regioisomers. In a following step, the 1,4-dihydropyridazine rings were oxidised using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to rearomatize and hence restore conjugation in polymer P24. The emission intensity in solution (THF) was substantially increased upon functionalisation with trans-cyclooctenes.
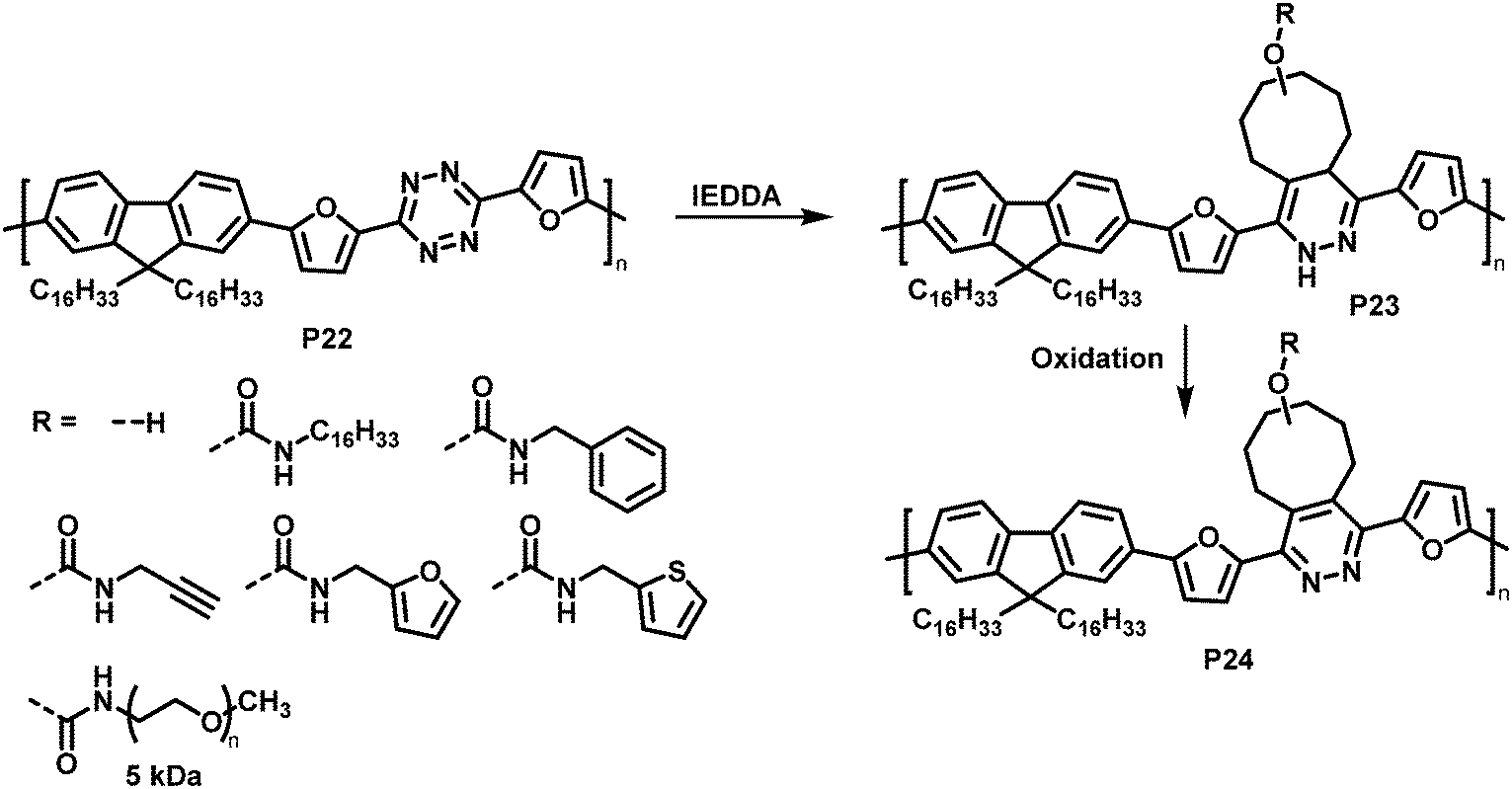 | ||
| Fig. 10 Post-polymerisation functionalisation of a tetrazine-containing polymer via inverse electron demand Diels–Alder (IEDDA) reaction and subsequent oxidation to rearomatise the unit. | ||
Oxidation and reduction
Fig. 11 provides a list of oxidation and reduction reactions used for post-polymerisation functionalisation of conjugated polymers discussed in this review. The first reaction – the oxidation of sulfur atoms in the backbone or in functional groups attached to the backbone to the corresponding sulfoxides or sulfones – has been shown to be a particularly fast and selective approach for post-polymerisation functionalisation of conjugated polymers.84–88 The approach is especially useful to obtain polymers with significantly altered properties from a starting polymer in just one step, as the electron-donating sulfur is transformed into electron-withdrawing groups by the oxidation. Fig. 12 shows sulfone-substituted polymers P25–31 obtained by this approach and Table 3 provides an overview of the reported optical and electronic properties. By making use of this post-polymerisation oxidation, n-type conjugated polymers can be obtained that may not be accessible by polymerisation of the oxidised monomer.88 Using this approach, a continuous junction design for organic electronic devices relying on post-processing oxidation of electron-donor polymer thin films has been proposed, as discussed in more detail below.85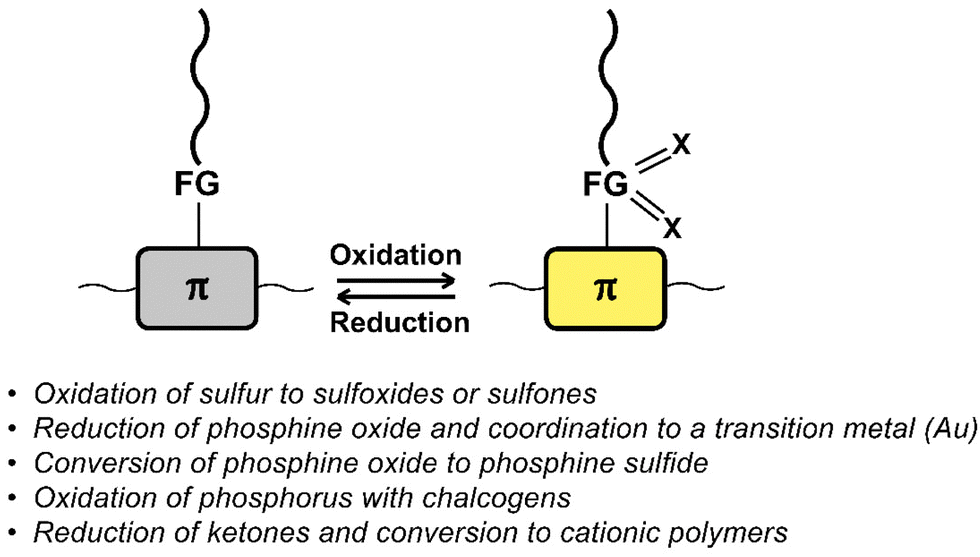 | ||
| Fig. 11 Schematic representation and list of oxidation and reduction reactions used for post-polymerisation functionalisation of conjugated polymers. | ||
 | ||
| Fig. 12 Sulfone-substituted polymers P25–31 prepared via post-polymerisation oxidation of sulfur atoms in polythiophenes and alkylthio-substituted polymers.84–88 | ||
In 2014, Campos and colleagues reported the use of Rozen's reagent (HOF·CH3CN) for the oxidation of thiophene in order to obtain thiophene-1,1-dioxide (TDO)-containing polymers P25.84 The reaction occurred rapidly at room temperature, and 60% of the polymeric thiophene was converted to TDO by reaction with 4 equiv. of Rozen's reagent. The addition of more than 4 equiv. of the reagent did not increase the extent of oxidation, but rather enhanced the occurrence of side reactions. The absorption onset of the polymer (measured in CHCl3 solution) was shifted significantly to longer wavelengths by the oxidation, and ultraviolet photoelectron spectroscopy (UPS) measurements of thin films found that both the HOMO and LUMO energy levels were lowered, with a larger influence on the latter. The electron accepting character of the oxidised polymer was further confirmed by its ability to effectively quench the fluorescence of poly(3-hexylthiophene) (P3HT). The authors also demonstrated that the backbone oxidation could be performed in the presence of potentially reactive end groups, such as hydroxy groups, opening up the possibility of further end-group modification.
In the same year, Fröhlich, Glöcklhofer and co-workers introduced dimethyldioxirane (DMDO) as a suitable oxidising agent for post-polymerisation oxidation.86 DMDO is a versatile and mild reagent that gives acetone as the only by-product, facilitating purification after the modification. Excellent efficiency and selectivity further support the use of this reagent in post-polymerisation functionalisation reactions. Initially, they used DMDO for the oxidation of polymers prepared by microwave-assisted CuAAC.86 A series of monomers containing selenium, tellurium and sulfur were prepared and polymerised, and the sulfur-containing polymer was fully converted to the sulfone-containing substituted polymer P26. Measuring the polymers in CH2Cl2 solution, no change in the absorption wavelengths was observed in this case, but the emission was red shifted by ∼80 nm, from blue to yellow.
| Polymer | E g (opt.) (eV) | E ox1,onset (V vs. Fc/Fc+) | E red1,onset (V vs. Fc/Fc+) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|---|
| a R = 75% H, 25% 2-ethylhexyl. b Determined via ultraviolet photoelectron spectra (UPS). c Estimated based on Eox1,onset and Eg (opt.) d R = 2-ethylhexyl. e Optical gap (Eopt), from UV-vis absorption spectroscopy in CHCl3 solution. | |||||
| P25 | 1.76e | — | — | −5.79b | −4.03b |
| P26 | 3.2 | 0.79 | −2.4c | −5.59 | −2.4 |
| P27 | 3.2 | 1.17 | −2.0c | −5.97 | −2.8 |
| P31 | 2.47 | — | −1.14 | −6.13 (opt.) | −3.66 |
OPV devices usually rely on the separation of tightly bound electron–hole pairs, so called excitons, at the interface of a donor and an acceptor material.14 In order to increase the interface area and, thus, the efficiency of the charge separation, most OPV devices make use of the bulk heterojunction (BHJ) concept, where the two materials are mixed to form a bicontinuous network. A new concept was proposed by Fröhlich and colleagues in 2015, which is based on post-processing oxidation of alkylthio-substituted donor polymers for the in situ preparation of acceptor polymers.85 Treating donor polymer thin films with DMDO solution was suggested to result in an oxidation gradient within the films, which would gradually change the electronic character by the conversion of electron-donating alkylthio groups (+M effect) into electron-withdrawing sulfoxide and sulfone groups (−M effect). One of the advantages of such a continuous junction would be that only one material needs to be prepared by polymerisation, which significantly lowers the synthetic effort. The authors prepared polymers P26 and P27 in order to test the conversion and to compare the properties to those of the starting polymers.85 Cyclic voltammetry measurements confirmed that the oxidised polymer exhibited lower lying HOMO and LUMO energy levels. However, the energy offset of the LUMO levels was too small to ensure efficient exciton dissociation with these polymers, and the low degree of polymerisation combined with the presence of triazole units in the backbone limited charge carrier mobility. In some more recent follow-up work, the use of DMDO to alter the photophysical properties of alkylthio-substituted conjugated polymers was demonstrated in donor–acceptor type materials (P28–30) as well as alkylthio-substituted poly(p-phenylene vinylene)s P31.87,88
In a different approach, Rupar and colleagues reported the post-polymerisation modification of phosphafluorene oxide-containing copolymers (Fig. 13).89 The initially prepared donor–acceptor type copolymer P32 could undergo three different modifications at the phosphorus centre: (i) reduction of the P-centre with HSiCl3, although the resulting polymer rapid re-oxidized in presence of air, (ii) quantitative reduction and coordination to a transition metal such as gold (AuCl), enabling air-stability of the resulting polymer P33, and (iii) quantitative conversion of the phosphine oxide to phosphine sulfide with Lawesson's reagent, giving polymer P34. Gold complexation resulted in a significant change in optical and electronic properties, compared to the parent P32, whereas changing the phosphine oxide to the phosphine sulfide had a more subtle effect (Table 4).
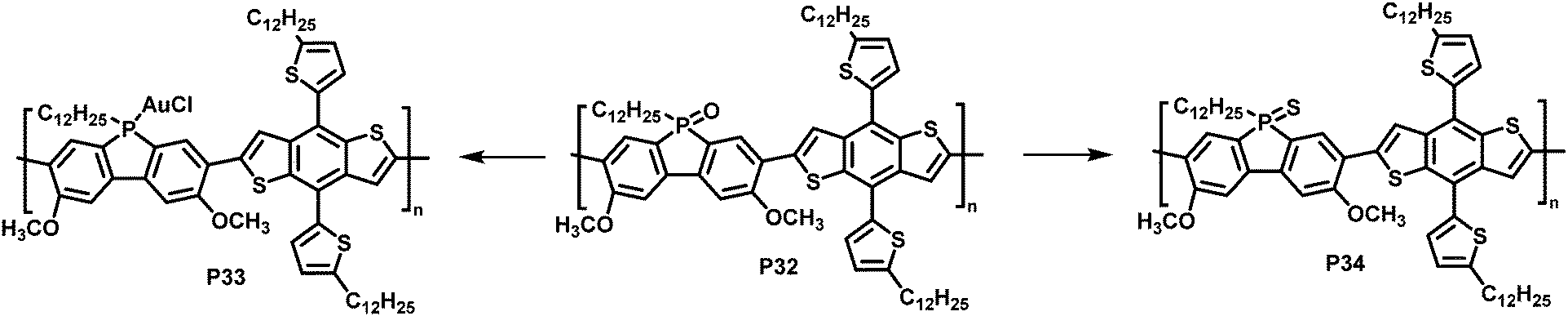 | ||
| Fig. 13 Modification of phosphafluorene oxide-containing polymer P32.89 | ||
| Polymer | λ max,ab (nm) | λ max,em (nm) | λ onset (nm) | LUMOb (eV) | HOMOb (eV) | E g,CV (eV) |
|---|---|---|---|---|---|---|
| a From UV-vis absorption spectroscopy in CH2Cl2 solution. b From CV measurements in 0.1 M nBu4NClO4 in CH3CN measuring polymer thin films cast onto the working electrode. | ||||||
| P32 | 465 | 521 | 538 | −2.99 | −5.56 | 2.57 |
| P33 | 530 | 550 | 555 | −3.43 | −5.23 | 1.80 |
| P34 | 466 | 520 | 534 | −3.03 | −5.46 | 2.43 |
In a related, albeit inverted approach, Réau et al. prepared phosphole-containing polymer P35 (Fig. 14),90 which was oxidised with chalcogens such as sulfur and selenium, giving polymers P36 and P37. 31P MAS NMR and SEM-EDX confirmed the almost quantitative oxidation of the phosphorus-centres with sulfur. Reaction with sulfur or selenium was found to influence the optical and electronic properties of the conjugated polymers, enabling potential sensing applications.
 | ||
| Fig. 14 Modification of phosphole-containing polymers by oxidation with chalcogens such as sulfur and selenium.90 | ||
In another interesting approach, Chiechi and co-workers reduced the cross-conjugated polymer P38, initially yielding a non-conjugated polymer with ether groups. Subsequent treatment with acid formed a charged, fully conjugated polymer P39 (Fig. 15), which could be switched reversibly.91 Very large changes in the absorption onset of the polymer (in CH2Cl2 solution) were observed upon modification. The absorption onset shifted from 3.26 eV (380 nm) for cross-conjugated P38 to 1.55 eV (800 nm) for P39. Subsequent work on related materials demonstrated how they could be fabricated into proof-of-concept bilayer solar cells from aqueous formic acid.92
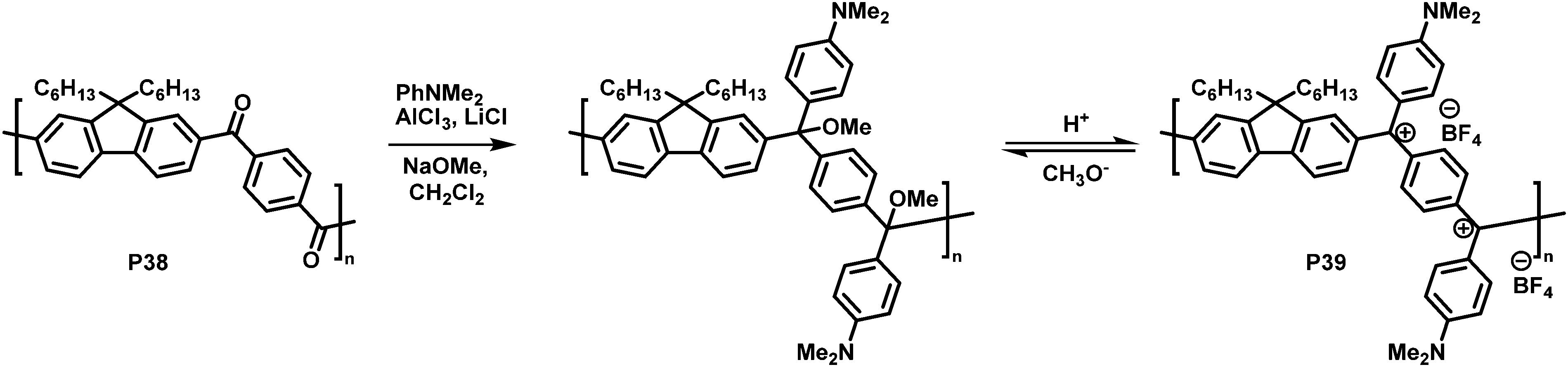 | ||
| Fig. 15 Conversion of a cross-conjugated polymer into a fully conjugated cationic polymer.91 | ||
Nucleophilic aromatic substitution (SNAr)
The interchange of functional groups on the polymer backbone is another approach to introduce varying functionality (Fig. 16), with recent work demonstrating the usefulness of nucleophilic aromatic substitution (SNAr) reactions in post-polymerisation functionalisation.93 Electron deficient aromatics containing halide leaving groups are particularly amenable to SNAr reactions, with fluorinated leaving groups being particularly active, due to their strong electron withdrawing effect. For example, fluorinated 2,1,3-benzothiadiazole has been shown to be active to SNAr reactions under mild conditions.94 Incorporation of such monomers into a conjugated backbone allows the displacement to occur post polymerisation. Thus, reaction of fully conjugated poly(9,9-dioctylfluorene-alt-5-fluoro-2,1,3-benzothiadiazole) (F8FBT) P40 with a series of thiols and alcohols in the presence of base affords polymers P41 with different X–R groups (Fig. 17). Due to the change from an electron withdrawing fluorine substituent to a donating alkoxy or thioether group, substitution was shown to have significant effects on the optical gap. The reaction conditions even enabled introduction of sidechains containing orthogonal functional groups, which can be used for further functionalisation. For example, azide-containing substituents were readily introduced via SNAr, and semiconducting polymer nanoparticles were prepared of the resulting polymers. Surface azides were then shown to undergo CuAAC or SPAAC, reactions widely used in materials chemistry and bioconjugation.45,59 The authors showed that the degree of substitution can simply be varied by the amount of nucleophile present, as indicated by 1H NMR and UV-vis absorption spectroscopy. Remarkably, this reaction also enables the preparation of multi-functionalised polymers in a one-pot procedure, for instance polymers containing not only azide groups but also carboxylic acid and silane groups. | ||
| Fig. 16 Schematic representation of nucleophilic aromatic substitution used for post-polymerisation functionalisation of conjugated polymers. All reactions in this section relied on the substitution of fluorides, but various different nucleophiles were used, such as thiols, alcohols and carbazole. | ||
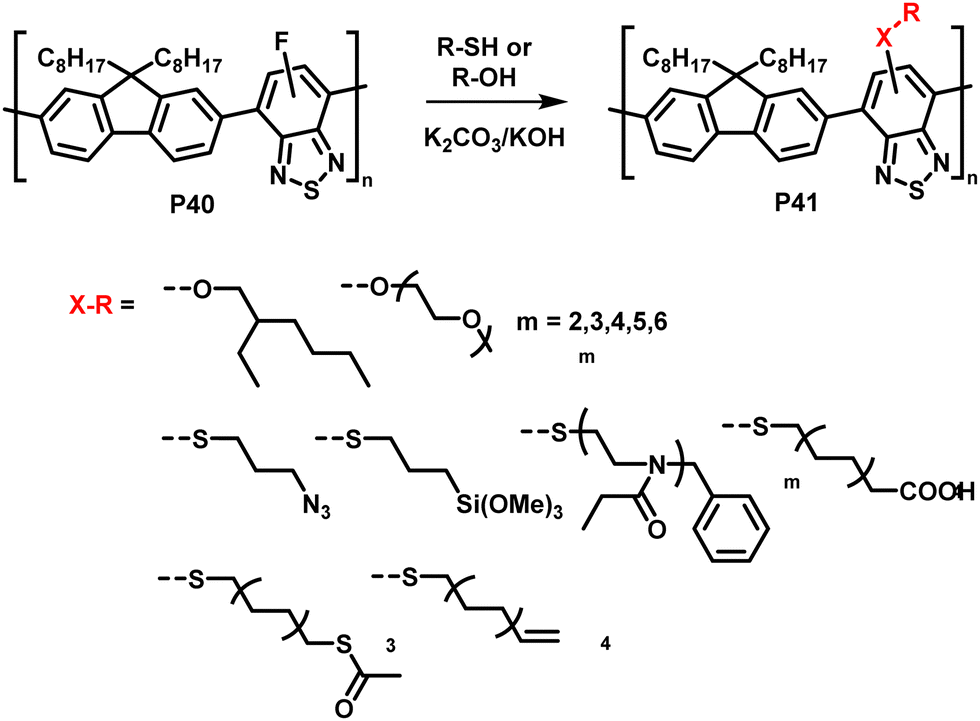 | ||
| Fig. 17 Nucleophilic aromatic substitution of fluorinated, fully conjugated F8FBT.93,95 | ||
In addition, the authors have shown the substitution of two fluorides on 5,6-difluoro-2,1,3-benzothiadiazole units (P42 and P43 in Fig. 18), using an excess of thiol. Furthermore, the reaction has also been proven to be feasible on other fluorinated acceptor units, such as alkylated 5-fluoro-1,2,3-benzotriazole (P44 in Fig. 18), which is less electron accepting in nature, and fluorinated thieno[3,4-b]thiophene (P45 in Fig. 18). In both cases, complete substitution was reported.
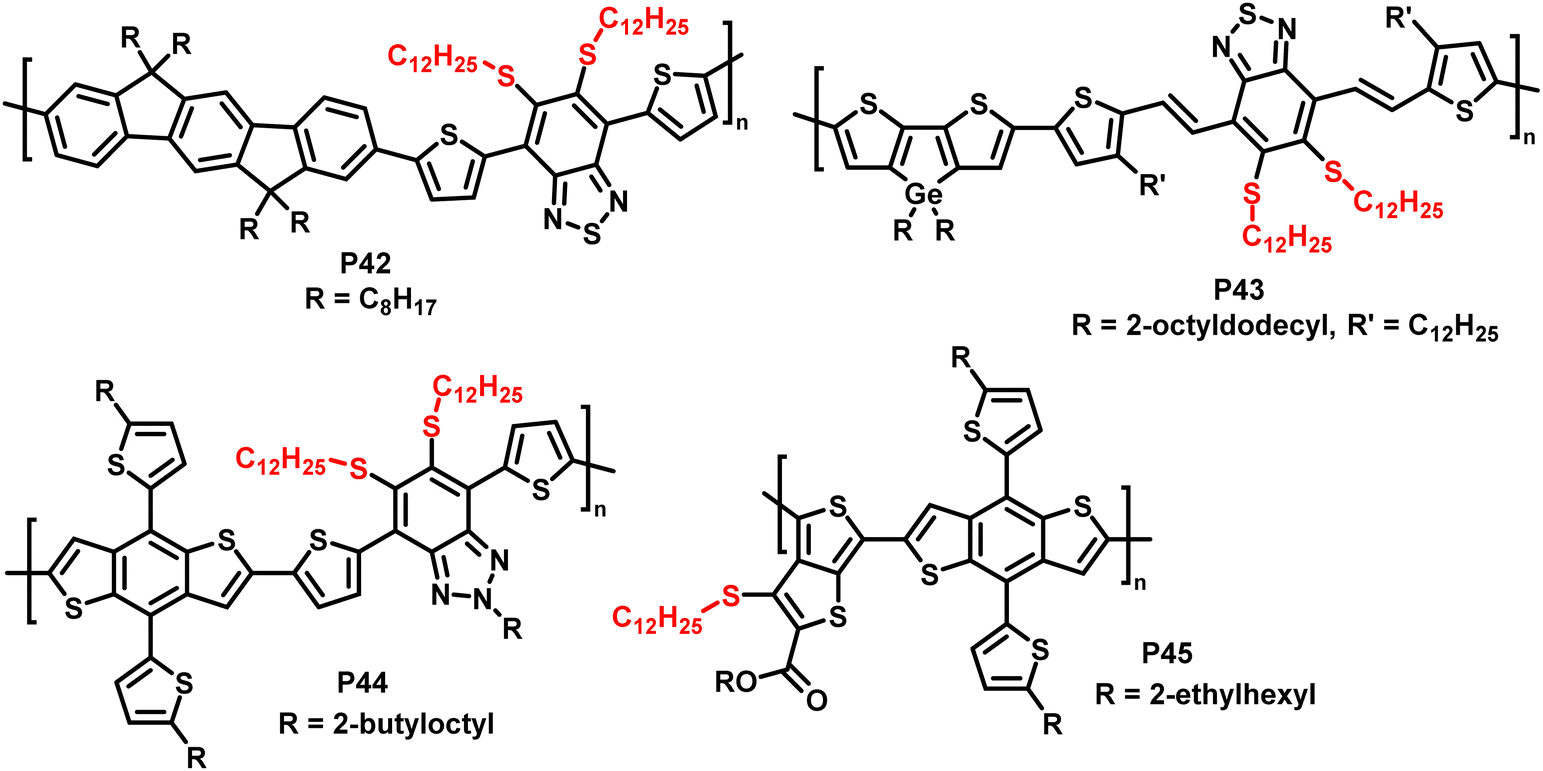 | ||
| Fig. 18 Polymers obtained by nucleophilic aromatic substitution, showing that the substitution approach is amenable to polymers with a variety of acceptor units such as fluorinated benzotriazole and thieno[3,4-b]thiophene.93 | ||
In a later work, the functionalisation of fluorinated F8FBT with a series of ethylene glycol oligomers as well as polyethylene glycol (PEG) by SNAr was demonstrated.95 Typically, conjugated polymers are hydrophobic and therefore insoluble in highly polar solvents. However, for some applications it is desirable that the materials feature hydrophilic functionalities, which renders them soluble in polar, greener solvents such as water and alcohols,96 and explains the authors’ interest in introducing hydrophilic (poly)ethylene glycol side chains. Moreover, the increased wettability of polymers substituted with such side chains enables efficient contact with cell tissue.97 The hydrophilicity of the polymers was gradually increased with increasing length of the ethylene glycol sidechains, as indicated by contact angle measurements and solubility tests. The photophysical properties are, however, not influenced by the increasing length of ethylene glycol chains.95
Inagi and colleagues broadened the scope of polymer backbones that can undergo SNAr reactions.98,99 They prepared a tetrafluorophenylene-fluorene copolymer P46 and subsequently functionalised it with a variety of S-, O- and N-nucleophiles to yield polymers P47–49 (Fig. 19), facilitating control of the photophysical properties. Similarly, intramolecular reactions of amine functionalised co-polymers have been reported.100
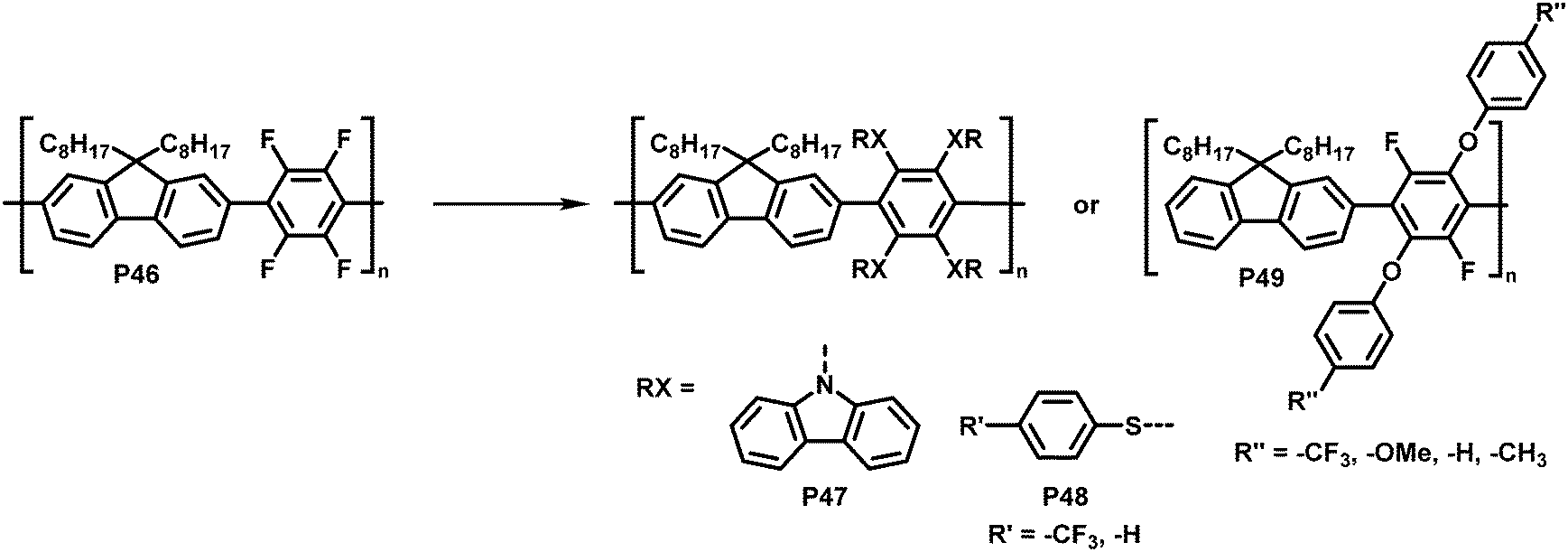 | ||
| Fig. 19 Functionalisation of tetrafluorophenylene-containing polymers via SNAr with S-, O- and N-nucleophiles.98 | ||
To demonstrate what a helpful tool for quick modification of polymer characteristics post-polymerisation functionalisation can be, Chen et al. used nucleophilic aromatic substitution to improve the performance of a conjugated polymer in photocatalytic H2-evolution.101 Various substituents of different hydrophilicity, ranging from methoxy groups to glycol chains, were introduced into hyperbranched polymer P50 (Fig. 20). The authors showed that by increasing the affinity to water, the hydrogen evolution rate (HER) increases. This was demonstrated by comparing contact angles and the HER. Compared to the starting polymer containing difluorinated BT units (P50), the HER was increased almost 2.5-fold, from 126 to 311 μmol h−1 for P51g with a glycol chain.
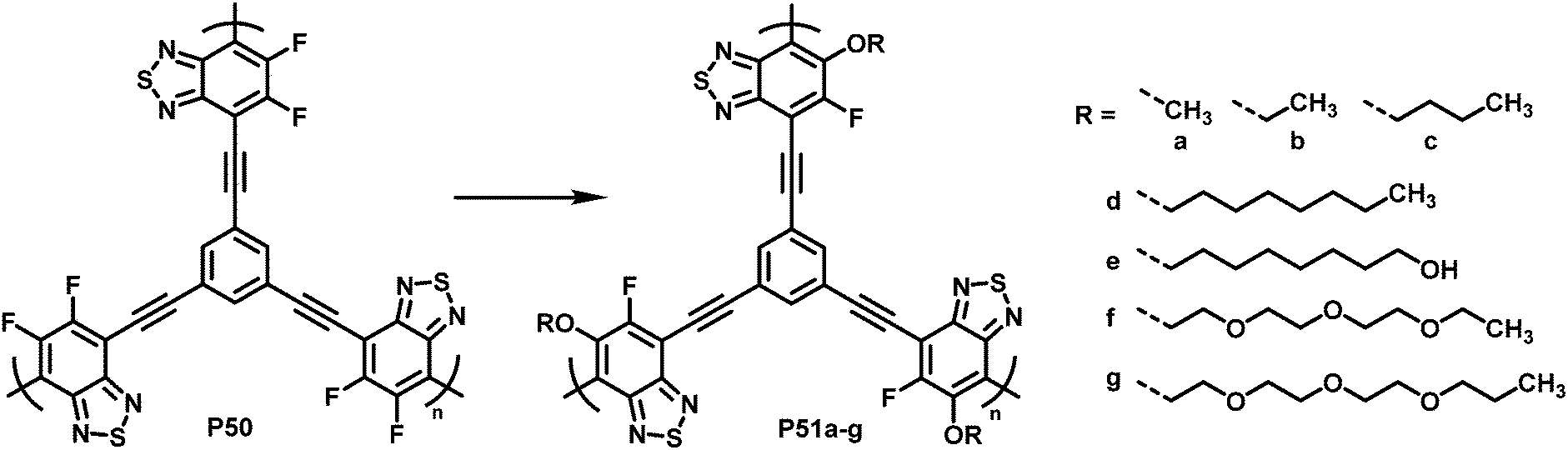 | ||
| Fig. 20 BT-based polymers for photocatalytic hydrogen evolution after functionalisation with various sidechains R.101 | ||
Halogenation & other electrophilic aromatic substitution (SEAr) and Pd-catalysed cross-coupling
The halogenation of conjugated backbones has been demonstrated on a wide variety of polymers via both electrophilic and nucleophilic approaches. Halogenation has the potential to alter the optical and electronic properties of the polymer, but it also allows for many subsequent functionalisation reactions. Fig. 21 provides a list of halogenation/electrophilic substitution and cross-coupling reactions used for post-polymerisation functionalisation of conjugated polymers that are discussed in this review.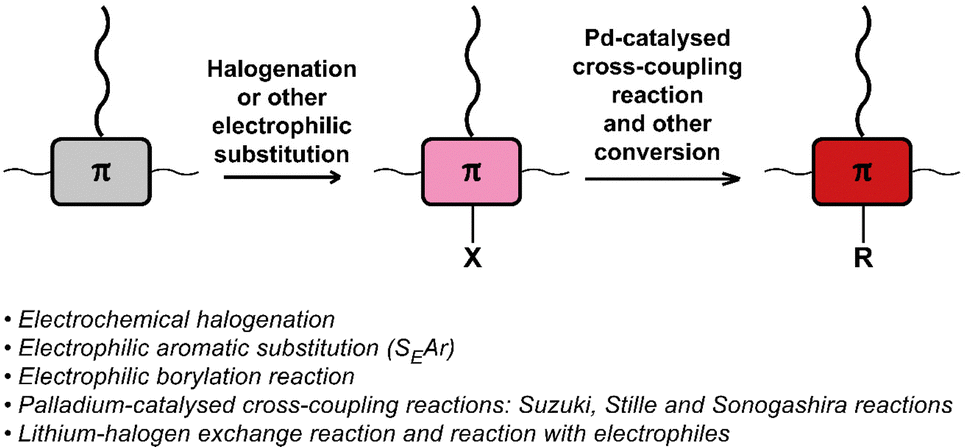 | ||
| Fig. 21 Schematic representation and list of (i) halogenation and (other) electrophilic substitution reactions used for post-polymerisation functionalisation and (ii) Pd-catalysed cross-coupling reactions and other conversion of halogenated conjugated polymers. | ||
One of the earliest reports utilised the inherent electroactive nature of the conjugated backbone, showing that electrochemical oxidation of various conjugated polymers in the presence of halides can result in backbone halogenation.102 Interestingly, the reaction was performed on the solid film, itself prepared via electrochemical polymerisation. For example, film oxidation in a 0.1 M Et4NCl/CH3CN solution resulted in 100% chlorination of poly(3-methylthiophene) and polythiophene after repeated oxidations. Other halides were also explored for the substitution on poly(3-methylthiophene); 50% bromination was achieved with Bu4NBr under the same conditions. The authors also tested iodination but found that the reactivity of the iodide nucleophile is not sufficient, in agreement with the nucleophilicity of halides in aprotic solvents (Cl− > Br− > I−). Moreover, alkoxylation with 3-bromopropanol was shown to be feasible with approximately 25% substitution on poly(3-methylthiophene). The same approach was also used for the chlorination of poly(3-(4-fluorophenyl)thiophene) on the conjugated backbone.103 Co-polymers of thiophene and fluorene are also amenable to electrochemical bromination (Br introduced per thiophene unit: 1.1) and chlorination (Cl introduced per thiophene unit: 1.7).104 Again, attempts to iodinate the polymer were unsuccessful, further confirming the insufficient nucleophilicity of iodide. Lastly, the presence of a Lewis acid has been shown to facilitate such reactions, via shifting of the oxidation onset potential of the conjugated polymer.105 This was shown to be effective for anodic chlorination of P3HT, poly(p-phenylene) and poly(9,9-dioctylfluorene) using an AlCl3/acetonitrile system, with high current efficiency and high degrees of chlorination (79% for P3HT at optimum conditions).
Backbone functionalisation under electrophilic aromatic substitution (SEAr) conditions has also been successful, as reported for P3HT by Holdcroft and co-workers in early 2001.106 Treatment with N-bromosuccinimide (NBS), N-chlorosuccinimide (NCS), or fuming nitric acid (HNO3) resulted in 100% substitution in the 4-position with bromo (P52), chloro (P53) and nitro groups (P54) produced in high yield (Fig. 22). Functionalisation resulted in a widening of the optical gap in all cases, attributed to the large size of the substituents causing backbone twisting. In follow-up work, they applied this electrophilic substitution approach to poly(2,5-dihexyloxy-1,4-phenylenevinylene) (DHO-PPV).107 In this case, the reaction occurred not only on the phenylene units but also on the vinylene bridges. 1H NMR spectroscopy indicated that 30% of the reaction with NBS occurs at the bridges, the remaining 70% at the phenylene units. For chlorination, similar results were obtained: 22% of the reaction with NCS occurs at the vinylene bridges, 78% at phenylene units. The reaction at the vinylenes leads to a rupture of the conjugation and therefore decreases the conjugation length. It was demonstrated that by modifying the ratio of reagent (NBS or NCS) to vinylene units it is possible to control the absorption and emission maximum wavelengths as well as the fluorescence efficiency.
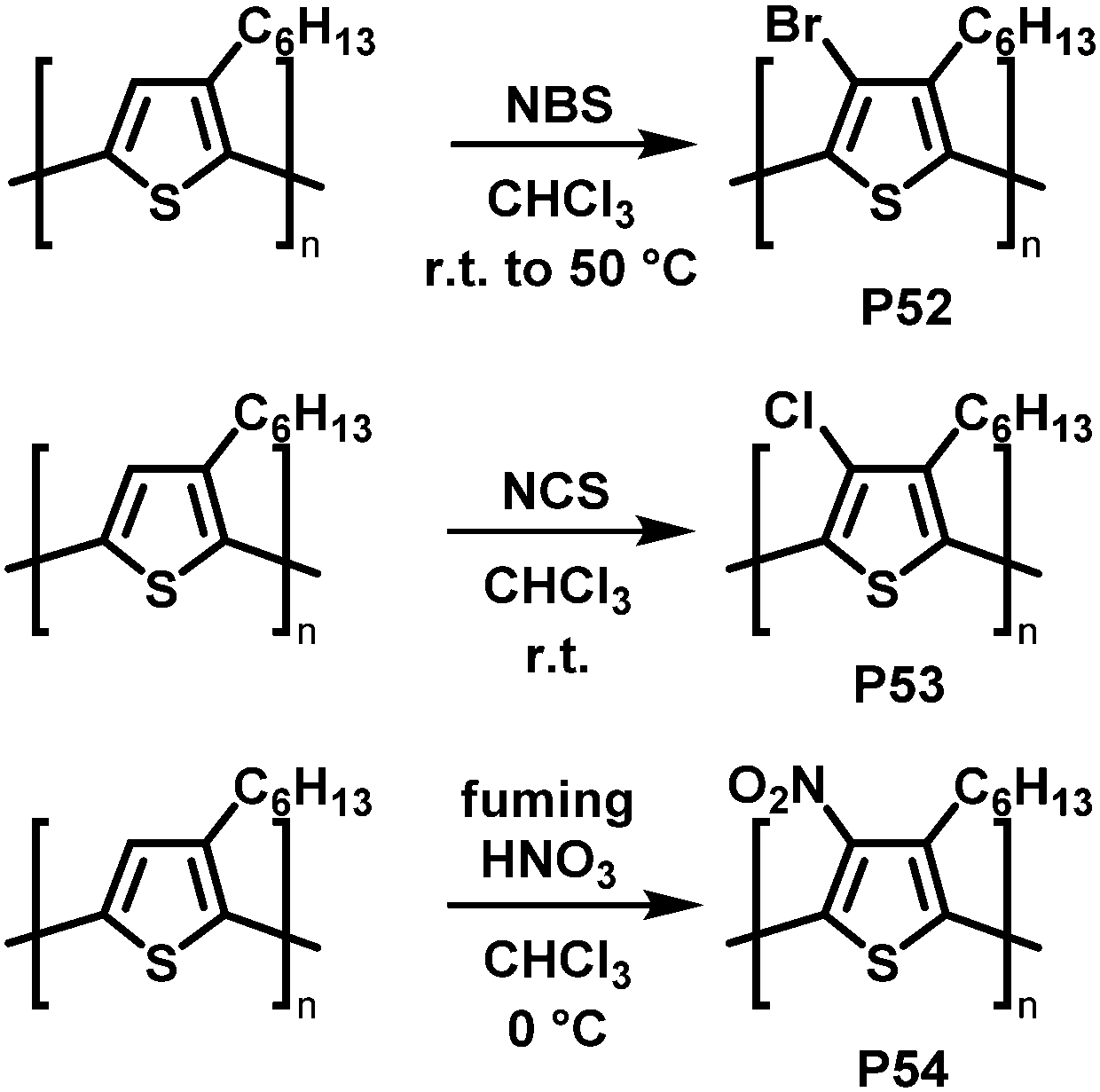 | ||
| Fig. 22 Selected examples of P3HT functionalisation.106 | ||
An unusual electrophilic substitution of the polymer backbone has been reported by Ingleson and co-workers who utilised an electrophilic borylation reaction of F8BT (P55) with BCl3, followed by reaction with diaryl zinc to prepare near-infrared emitting polymer P56 (Fig. 23).108 The electron-accepting boryl group adjacent to the 2,1,3-benzothiadiazole unit results in the formation of a dative B–N bond. The resulting planarization of the conjugated backbone in combination with the electron withdrawing effect of the introduced functional group resulted in a significant red shift of the emission of the functionalised polymers (in solution and thin film). The percentage of borylation was readily controlled by varying the equivalents of BCl3 added in the synthesis (x = 0.1 to 1 in P56). The solid-state emission maximum of the fully borylated polymer was redshifted by about 200 nm to a wavelength of 757 nm and a photoluminescence quantum yield of 11% was determined. The electrochemical gap becomes considerably smaller by the C–H borylation, from ∼2.60 eV to 1.94 eV (determined by CV in 0.1 M nBu4NPF6 in CH2Cl2). Those characteristics combined with excellent stability to protic species suggest that the polymers are applicable for in vivo biological imaging. The authors prepared encapsulated nanoparticles and confirmed excellent photophysical properties and stability as well as good biocompatibility.109
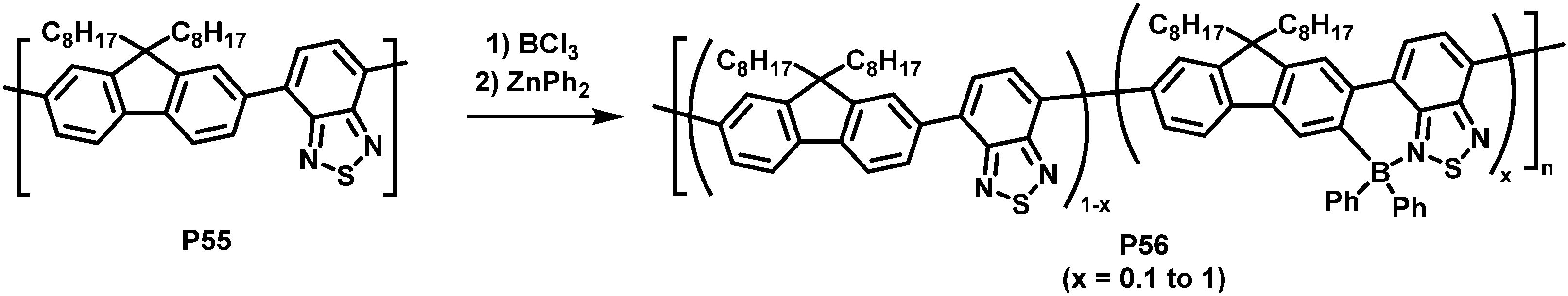 | ||
| Fig. 23 Introduction of heteroatoms as a strategy to alter the optical and electronic properties of conjugated polymers.108 | ||
An attractive feature of backbone bromination is the ability to subsequently engage in palladium-catalysed cross-coupling reactions, such as Suzuki, Stille and Heck reactions. Holdcroft and colleagues demonstrated this with their brominated P3HT, resulting in a number of functionalised derivatives of P3HT (P57) in excellent yields.110 The introduction of aromatic groups (Y in Fig. 24) resulted in a remarkably increased fluorescence efficiency in the solid state (9–22%) compared to unfunctionalised P3HT (1.6%) and a red shift in the absorption and emission maxima.111,112 Moreover, with the use of a Suzuki coupling reaction, perylene diimide (PDI) containing P3HT derivative P58 was realised.113 This polymer was used to assist in templated crystal growth of free PDI, demonstrating a useful approach for obtaining supramolecular structures.
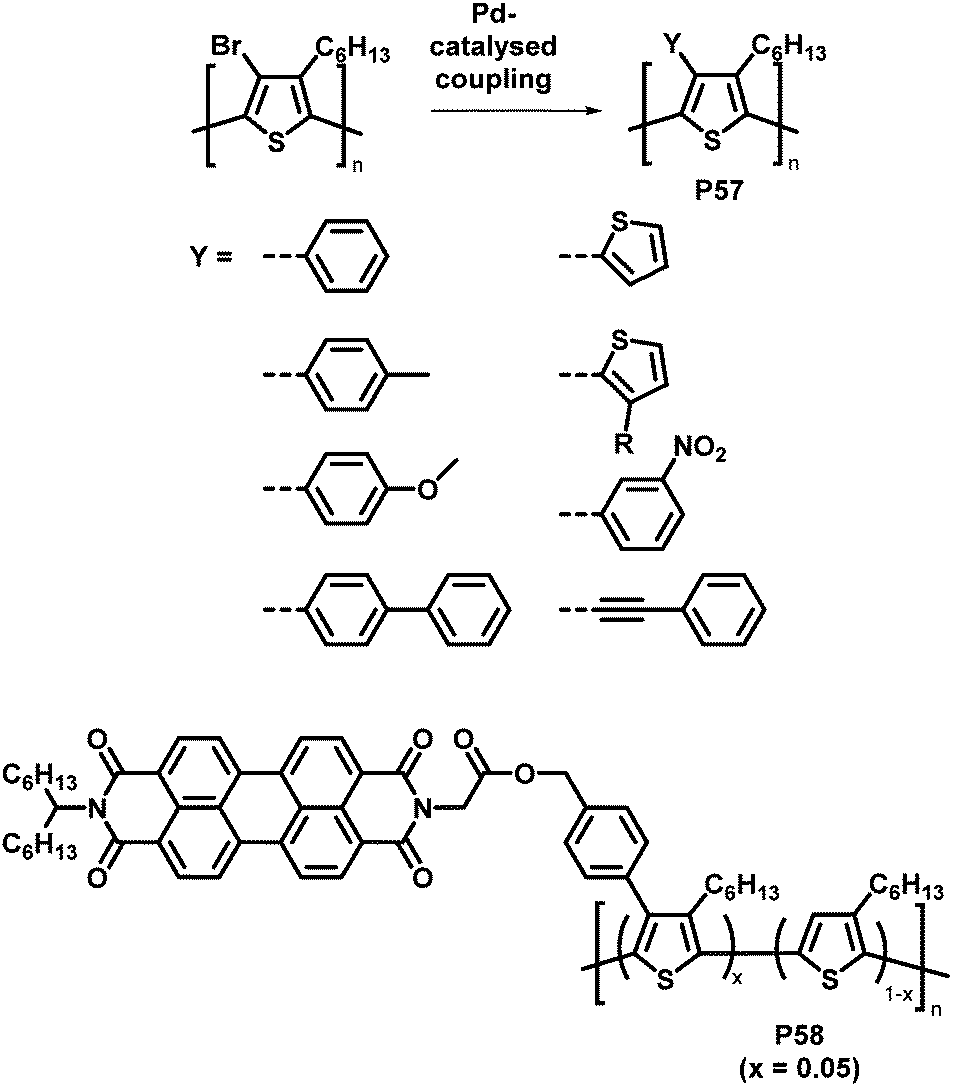 | ||
| Fig. 24 Products of Pd-catalysed coupling reactions of halogenated P3HT with different aromatic groups Y (top)112 and P3HT-bearing perylene diimide (PDI) units introduced by Suzuki cross-coupling (bottom).113 | ||
The combination of consecutive post-polymerisation reactions demonstrates the power of the approach. For example, bromination and subsequent Stille reaction of P3HT affords polymer P59 (Fig. 25).114 This was then used in a [2+2] CA–RE reaction with TCNE, a reaction discussed in detail in the section on cycloaddition above, resulting in an electron-accepting TCBD unit (P60). Interestingly electrochemical reduction of P60 created stable poly(anionic radical)s. Evidence for the stability at room temperature of the poly(anionic radical)s was presented in the form of electron spin resonance (ESR) measurements.
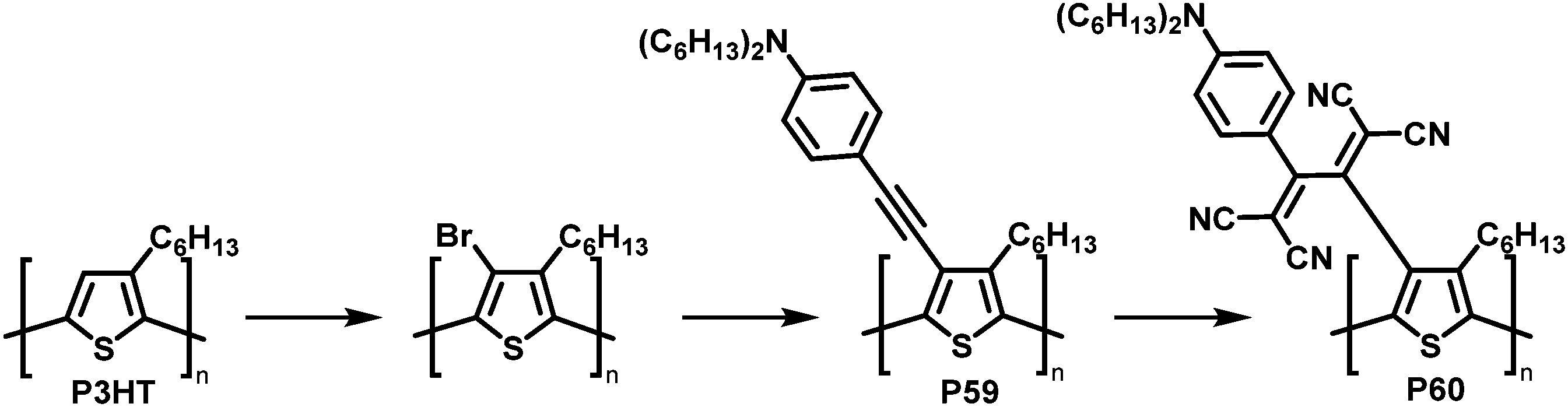 | ||
| Fig. 25 Post-polymerisation functionalisation of P3HT: halogenation followed by Stille coupling for the synthesis of P59 and subsequent reaction with TCNE giving P60.114 | ||
The utility of brominated P3HT was further highlighted by Swager and co-workers, who used it for a lithium–halogen exchange reaction.115 A nearly quantitative exchange was observed and the resulting lithiated P3HT was reacted with a variety of functional electrophiles. Thus, a broad range of functionalised P3HT was obtained, including polymers with functional groups such as ketones, alcohols (primary and secondary) and azides, as well as silanes and fluoride moieties. The functional groups enabled the authors to perform further reactions, such as a click reaction of the azides with alkynes.
Much of the work in conjugated backbone functionalisation has been driven by the challenge of creating bicontinuous networks of electron-rich donor materials (most commonly a conjugated polymer such as P3HT) and electron-deficient acceptor materials (such as fullerenes) for BHJ solar cells.14,116,117 Often, due to poor miscibility, the interfacial area between donor and acceptor is small, which can cause poor charge separation and transport.118,119 Yang and co-workers addressed this issue by functionalising P3HT with side-chains bearing groups that can improve the contact between the donor and acceptor material and decrease macrophase separation of the two materials.120–122 They prepared a soluble P3HT derivative bearing fullerenes (P61 in Fig. 26) via a post-polymerisation functionalisation route.121 P3HT was brominated with NBS with a degree of functionalisation between 12 and 20% in the first step, followed by a Suzuki coupling reaction with 4-formylphenylboronic acid and a subsequent Prato reaction with C60, yielding C60-Ph-P3HT (P61) (100% of Br converted in the coupling reaction). The fluorescence in solution is almost fully quenched (compared to P3HT or P3HT:C60 blend), indicating that electron transfer is readily occurring. However, X-ray powder diffraction (XRD) studies suggested the lamellar packing of P3HT was slightly perturbed by the presence of the tethered fullerene.
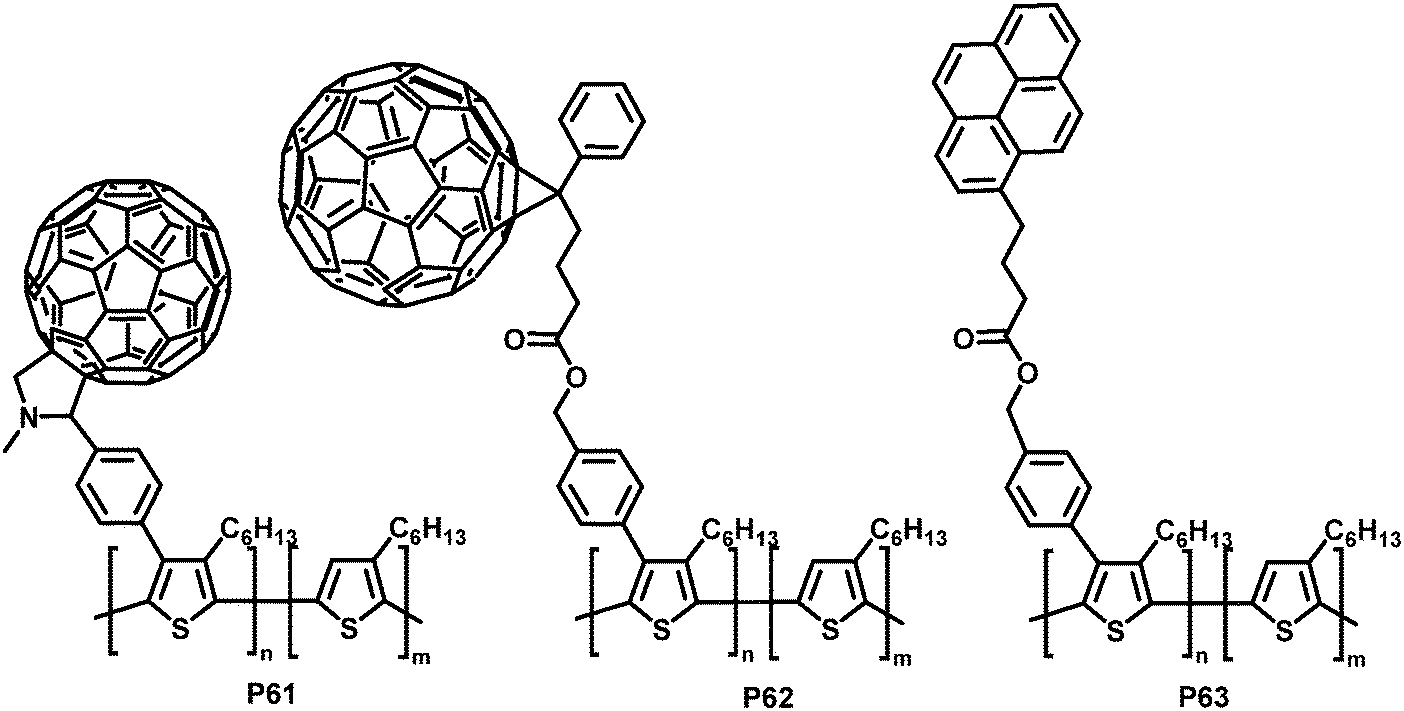 | ||
| Fig. 26 P3HT functionalised with carbon moieties that are able to interact with the fullerene acceptor materials or nanotubes and can therefore improve device performance.120–122 | ||
Similarly, PCBM ([6,6]-phenyl-C61-butyric acid methyl ester) was introduced to P3HT using the same post-polymerisation functionalisation strategy and was shown to have a similar effect on the fluorescence.120 Here it was found that the bicontinuous interpenetrating network morphology of a P3HT:PCBM blend was improved by adding PCBM-Ph-P3HT (P62 in Fig. 26) to the blend, as indicated by AFM morphology studies. Fabrication of photovoltaic devices indicated an increase in PCE by 12% at the optimum ratio (1 wt% PCBM-Ph-P3HT), albeit with relatively low PCE in all cases (Table 5).
SWCNTs (single-walled carbon nanotubes) are another interesting electron acceptor material that has been investigated as an alternative to fullerene-based acceptors.123 Yang et al. prepared pyrene-functionalised P3HT derivative P63 (Fig. 26, 15% substitution) by post-polymerization modification and observed increased interactions with SWCNTs as a result of the introduction of the pyrene units.122
With the same intention of improving the chemical compatibility between donor and acceptor material, Hadziioannou and colleagues investigated a grafting-onto strategy to obtain brush-type copolymers by post-polymerisation functionalisation of P3HT.124 P3HT was brominated and propionyl bromide groups were introduced as polymerisation initiators via Suzuki coupling and further modification. A quinoline containing vinyl monomer (SDPQ) was then polymerised onto the polymer under atom transfer radical polymerisation (ATRP) conditions. The authors found that upon addition of the resulting copolymer P64 (Fig. 27) to a blend of P3HT:PSDPQ, the surface roughness and phase separation were reduced significantly, therefore improving the overall morphology.
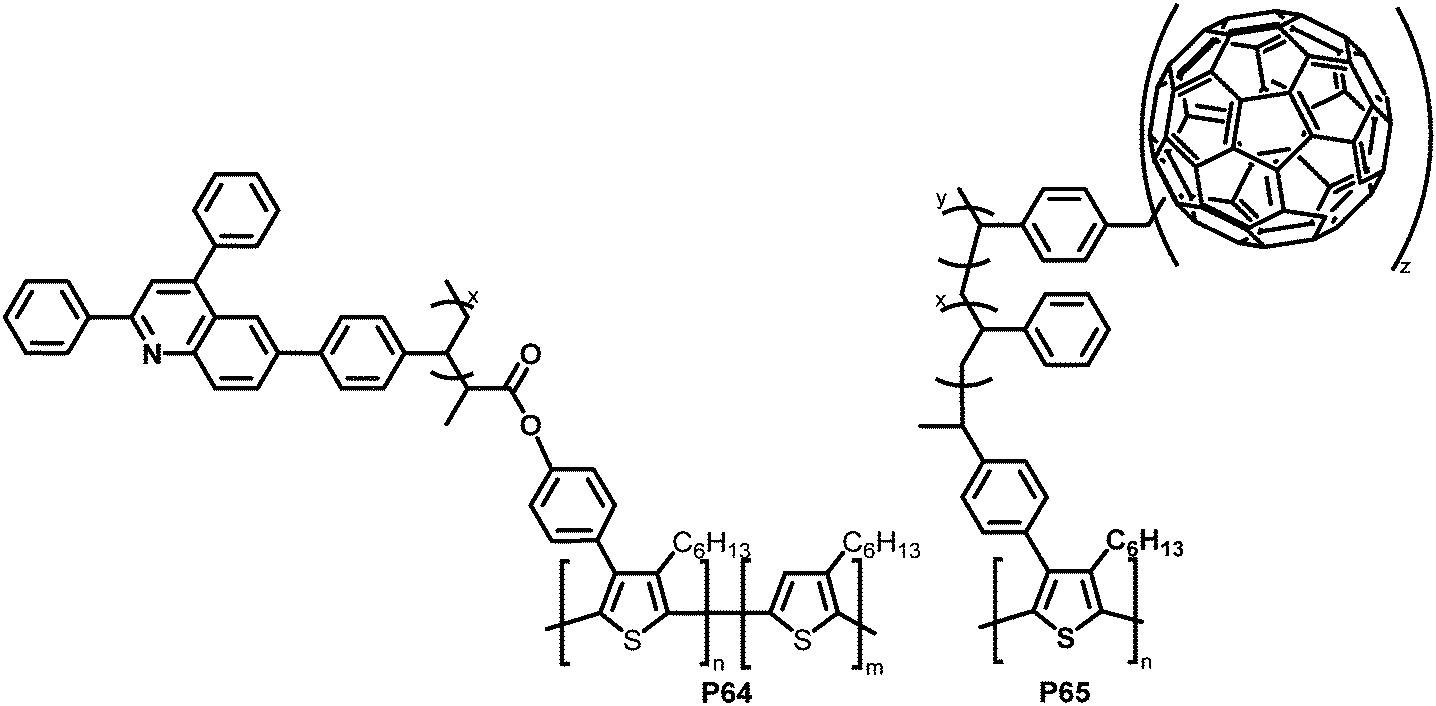 | ||
| Fig. 27 Brush-type graft-copolymers, obtained via Suzuki coupling followed by radical polymerisation techniques.124–126 | ||
Using a related post-polymerisation functionalisation approach, the grafting of fullerene containing sidechains onto P3HT was presented as another strategy to prevent the undesired large-scale phase separation and the ensuing reduction of the donor–acceptor interfacial area over time.125–127 P3HT was brominated, followed by Pd-catalysed cross-coupling with a nitroxide-bearing moiety that is capable of initiating a radical polymerisation (nitroxide-mediated radical polymerisation (NMRP)) with styrene (ST) and 4-chloromethylstyrene (CMS). The polymerised CMS could then be further functionalised with fullerenes via atom transfer radical addition (ATRA) to give P65 (Fig. 27).125 The photoluminescence in solution was completely quenched upon introduction of the fullerene, which indicates the donor–acceptor characteristics of the graft polymer. In a subsequent work, it was shown that it is also feasible to substitute the chlorides in the polymerised CMS units by azides and carry out a cycloaddition reaction of the azides and fullerenes/PCBM.126 The grafting density was carefully controlled between 1 and 30%. Investigations of the film microstructure demonstrated that the graft copolymers form a network with very small feature sizes (∼5 nm). It was shown that the domain sizes were decreased in comparison to blends of P3HT and PCBM. Due to the covalent link between the fullerene and the graft polymer, no diffusion and aggregation of fullerene molecules was observed. Even after extensive annealing no changes in domain sizes occurred. However, BHJ-PSC devices fabricated using the copolymer exhibited poor photovoltaic activities, which was attributed to less crystalline P3HT domains and the lower content (wt%) of active materials (P3HT and PCBM) in the active layer compared to conventional blends. In the graft-polymer, the active materials are diluted due to the presence of 36–60 wt% inactive side chains (poly(CMS-stat-ST)).
In the examples discussed thus far, backbone halogenation post-polymerisation has been utilised. However, it is also possible to introduce the halogen handle in the monomer and utilise it in a post-polymerisation reaction. For example, Bunz and colleagues reported the post-polymerisation functionalisation of a hyperbranched conjugated polymer containing iodine groups that are available for Sonogashira coupling reactions with alkynes.128 This allowed them to introduce a multitude of functional groups onto the backbone. The emission spectrum depended on the introduced functional groups, which enabled the authors to obtain polymers with emission maximum wavelengths ranging from 510 nm to 602 nm in solution and a strong bathochromic shift in the solid state.
Functionalisation of linear conjugated polymers containing iodo groups has also been reported,129 with polymers P66 and P67 (Fig. 28, X = I) containing m-phenylene units. The presence of the m-phenylene impedes the conjugation in the polymer chain, and the iodo handle offers the possibility for Pd-catalysed coupling reactions to further functionalise the polymer. Three different units were introduced by coupling reactions, yielding polymers with different characteristics depending on the units (Fig. 28, X = A, P, Fe). P66-A, a crosslinked polymer that was expected to exhibit pockets around the terphenyl unit was tested for the detection of volatile organic compounds such as 2,6-dinitrotoluene (DNT). It should be noted that the polymer was most likely not fully crosslinked, so unreacted alkyne groups can be assumed to be present. Thin films of P66-A showed a fast response to DNT vapour, with its fluorescence quenched by half in just 30 s. The authors also tested the polymers in regard to their photoluminescence (PL) response to metal ions, as potential environmental sensors. In particular, the influence of different metal ions on the PL of polymers P66-P and P67-P, both bearing pyridyl groups, was investigated. Depending on the frontier orbital match of the metal ion-binding sidechain and the polymer backbone, the PL in solution was either quenched or intensified.
 | ||
| Fig. 28 Polymers prepared via post-polymerisation functionalisation using Sonogashira coupling reactions.129 | ||
Post-polymerisation functionalisation was also possible using brominated poly(thienylene vinylene)s (PTVs) and poly(selenylene vinylene)s (PSVs) for Stille and Sonogashira cross-coupling reactions (Fig. 29 and 30).130,131 Both, PTVs and PSVs, were prepared by acyclic diene metathesis (ADMET) polymerisation of brominated precursors. A variety of alkyne-containing groups were introduced quantitatively to PTVs by the cross-coupling reactions and further modification, allowing tuning of the optical gap and energy levels.130 A reduction in the gap of up to 0.3 eV was found in comparison to the parent PTV. Whilst OPV devices exhibited relatively modest performance (highest PCE ∼1% for P72) due to the amorphous nature of the polymers, the energy levels of the polymers could be tuned to the reduction potential of hydrogen, allowing photocatalytic hydrogen evolution. Blends of functionalised PTVs (P68–72) with graphitic carbon nitride (g-C3N4), exhibited better photocatalytic activity than g-C3N4 alone, but the pristine PTV exhibited the best performance. This was attributed to the energy misalignment between the LUMO level of the PTVs and the conduction band edge of g-C3N4, together with the reduced crystallinity in the functionalised polymers.
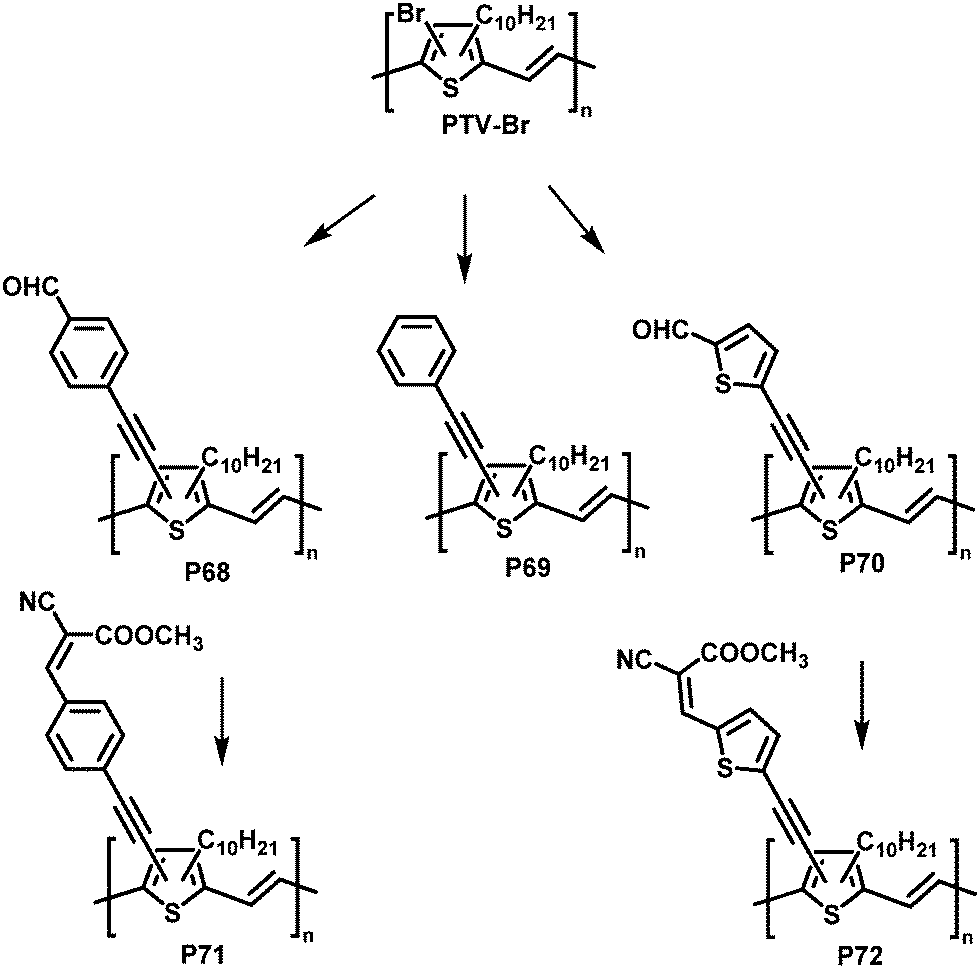 | ||
| Fig. 29 Post-polymerisation functionalisation of poly(thienylene vinylene)s (PTVs) with alkyne containing groups.130 | ||
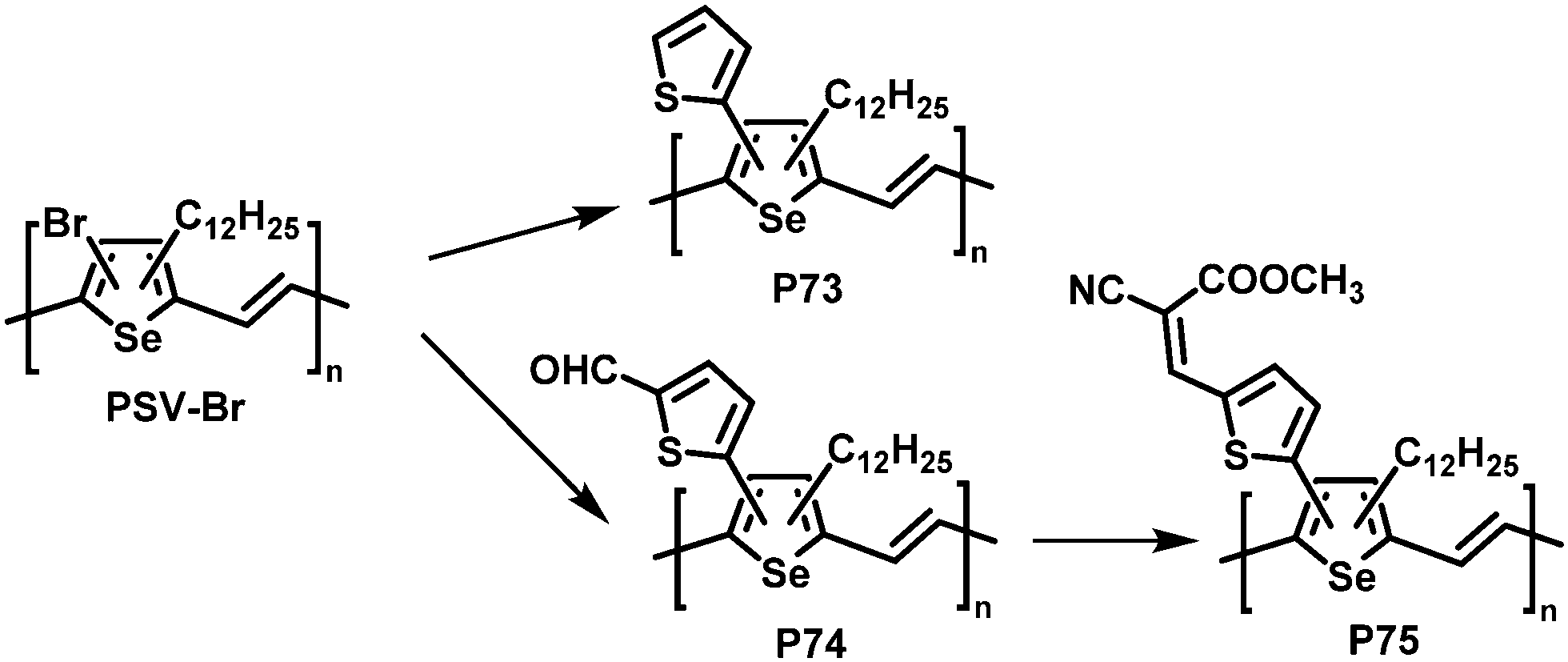 | ||
| Fig. 30 Poly(selenylene vinylene)s (PSVs) bearing thienyl moieties obtained by post-polymerisation functionalisation.131 | ||
Similar results were reported for post-polymerisation-functionalised PSVs (Fig. 30).131 Thienyl derivatives were introduced to the backbone with a degree of functionalisation close to 100%, resulting in lower lying energy levels of polymers P73–75 due to inductive and conjugation effects of the substituents. However, due to the amorphous nature of P73–75 the performance in OPV devices was again low, with a maximum PCE of 0.74% for P75.
Summary and conclusions
In this review, we have presented and discussed the different types of reactions and approaches for post-polymerisation functionalisation of conjugated polymer backbones. It is clear that the introduction of functional groups on the backbone by post-polymerisation functionalisation methods can have pronounced effects on the properties of conjugated polymers, which can be used to change, adapt, and optimise the properties for specific applications. Backbone functionalisation can also facilitate the preparation of both donor and acceptor materials for the use in optoelectronic devices or enable the preparation of graft co-polymers, which can improve the morphology and, hence, the performance of the devices. A broad range of applications and strategies on how to improve the device performance and morphology using post-polymerisation functionalisation approaches have been presented. However, trends in the nature of the changes of the properties upon functionalisation are not always clearcut, due to differences in the nature of each conjugated polymer and the competing effect of steric and electronic influences. For example, sulfur oxidation results in a consistent red shift in the optical gap when applied to the heteroatom of thiophene containing polymers, as the resulting thiophene-1,1-dioxide is more electron accepting and steric interactions with adjacent heterocycles are not significantly changed. However, when applied to thioether groups attached directly to the polymer backbone, as in the case for S-PPV (P31), the optical bandgap is widened, likely due to the steric effects of the larger sulfone group influencing backbone planarization and solid-state packing effects. Nevertheless, trends can be observed with a consistent series of conjugated polymers. For example, increasing displacement of fluoride from F8FBT (P40) with thioether groups results in a consistent decrease in intensity of the long-wavelength absorption band together with the growth of a new optical transition in the blue region of the spectra. This can be precisely controlled by the degree of reaction.Cycloaddition as well as halogenation and subsequent Pd-catalysed cross-coupling are the most frequently used types of reactions for post-polymerisation functionalisation of conjugated polymers directly on the backbone. Such reactions often proceed rapidly with high selectivity, making them ideal for the functionalisation of polymers, where defects are obtained if side reactions occur. However, it is noted that in some of these cases a full degree of functionalisation could not be achieved. Although there are many applications where this is not problematic, good control over the degree of functionalisation is desirable from a reproducibility standpoint. Oxidation (or reduction) as well as nucleophilic aromatic substitutions are two types of reactions that allow for such good control of the degree of functionalisation, prompting the recent interest in these reactions.
Most of the presented reactions used 1–2 equivalents of reagent per reactive site, but for some reactions a larger excess of reagents was used to maximise the degree of functionalisation within the reaction time. This is particularly true for the presented cyclopentannulation (5 equiv.), Diels–Alder (5 equiv.) and SNAr (up to 10 equiv.) reactions. It should be noted, however, that many of the reagents used for these reactions are low-cost, such as the alcohols and thiols used for the SNAr reactions, which limits the impact on the cost of the reactions. Furthermore, post-polymerisation functionalisation can facilitate the direct introduction of reactive groups which would not survive the polymerisation conditions, removing the requirement for additional protection/deprotection steps or functional groups interconversions. For example, traditional approaches to the introduction of azide containing sidechains require the polymerisation of alkyl bromide containing polymers, followed by a subsequent interconversion to the azide.132 In contrast, post-polymerisation SNAr reactions can directly introduce azide containing sidechains. Similarly, SNAr has been shown to directly introduce highly moisture sensitive cross-linkable sidechains, such as trimethoxysilanes.93 These are extensively used as surface modifiers and cross-linkers in non-conjugated polymers but have barely been investigated in the world of conjugated polymers due to their lack of compatibility with typical polymerisation conditions.
There are many possible directions for future research in the field, but we believe it should focus where post-polymerisation functionalisation can particularly show its strengths, namely the introduction of functional groups or moieties that cannot easily be introduced via other strategies as well as the creation of sets of structurally related conjugated polymers – or a combination of both. For example, many approaches to tune the performance of conjugated polymers rely on the copolymerisation of appropriate monomeric materials. However, the establishment of structure–property relationships in such materials is often complicated by the need to synthesise batches of varying comonomer ratios. It is therefore difficult to exclude the influence of variations in the molecular weight, end-groups and dispersities from changes in comonomer ratio. Performing post-functionalisation chemistry on single batches of consistent weight and dispersity may help to overcome these issues. Furthermore, the development of post-polymerisation chemistries which can be utilised to selectively functionalise the surface of a polymer film or nanoparticle is an exciting direction. Such approaches could potentially tune the energy gradient within such systems, potentially facilitating exciton migration and harvesting, or allow the attachment of receptor moieties for sensor applications. These functionalisations may rely on reactions presented in this review, focussing on the development of new polymers that can be obtained with these reactions, but future research should also aim to identify new reactions suitable for post-polymerisation functionalisation. The many opportunities that still need to be explored and the wide range of potential applications – ranging from organic electronics to biological applications – make post-polymerisation functionalisation of conjugated polymer backbones a truly exciting field for further research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the EPSRC (grant EP/V048686/1), the Austrian Science Fund (FWF) (project number J 4463), and the Royal Society and Wolfson Foundation for financial support.References
- Z. Qiu, B. A. G. Hammer and K. Müllen, Prog. Polym. Sci., 2020, 100, 101179 CrossRef CAS.
- P. Xue, P. Cheng, R. P. S. Han and X. Zhan, Mater. Horiz., 2022, 9, 194–219 RSC.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef PubMed.
- B. Van der Zee, Y. Li, G.-J. A. H. Wetzelaer and P. W. M. Blom, Adv. Mater., 2022, 34, 2108887 CrossRef CAS.
- S. Holliday, Y. Li and C. K. Luscombe, Prog. Polym. Sci., 2017, 70, 34–51 CrossRef CAS.
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- G. Dufil, I. Bernacka-Wojcik, A. Armada-Moreira and E. Stavrinidou, Chem. Rev., 2022, 122, 4847–4883 CrossRef CAS PubMed.
- V. V. Brus, J. Lee, B. R. Luginbuhl, S.-J. Ko, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2019, 31, 1900904 CrossRef.
- H.-J. Jhuo, P.-N. Yeh, S.-H. Liao, Y.-L. Li, Y.-S. Cheng and S.-A. Chen, J. Chin. Chem. Soc., 2014, 61, 115–126 CrossRef CAS.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS.
- X. Guo and A. Facchetti, Nat. Mater., 2020, 19, 922–928 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- J. Yang, F. Kang, X. Wang and Q. Zhang, Mater. Horiz., 2022, 9, 121–146 RSC.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743–768 CrossRef CAS.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS.
- T. E. Anderson, E. W. Culver, I. Badía-Domínguez, W. D. Wilcox, C. E. Buysse, M. C. Ruiz Delgado and S. C. Rasmussen, Phys. Chem. Chem. Phys., 2021, 23, 26534–26546 RSC.
- H. J. Son, F. He, B. Carsten and L. Yu, J. Mater. Chem., 2011, 21, 18934–18945 RSC.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 CrossRef CAS.
- H. G. Kim, M. Kim, J. A. Clement, J. Lee, J. Shin, H. Hwang, D. H. Sin and K. Cho, Chem. Mater., 2015, 27, 6858–6868 CrossRef CAS.
- A. Creamer, A. Casey, A. V. Marsh, M. Shahid, M. Gao and M. Heeney, Macromolecules, 2017, 50, 2736–2746 CrossRef CAS.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- R. J. Kline and M. D. McGehee, J. Macromol. Sci., Part C, 2006, 46, 27–45 CrossRef CAS.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, E. H. Jung and W. H. Jo, Org. Electron., 2016, 31, 149–170 CrossRef CAS.
- J. C. Gustafsson-Carlberg, O. Inganäs, M. R. Andersson, C. Booth, A. Azens and C. G. Granqvist, Electrochim. Acta, 1995, 40, 2233–2235 CrossRef CAS.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- H. Staudinger and J. Fritschi, Helv. Chim. Acta, 1922, 5, 785–806 CrossRef CAS.
- H. Staudinger, E. Geiger and E. Huber, Ber. Dtsch. Chem. Ges. A and B, 1929, 62, 263–267 CrossRef.
- H. Staudinger, Nobel Prize Lecture, https://nobelprize.org/nobel_prizes/chemistry/laureates/1953/staudinger-lecture.pdf, (accessed 13/01/2022).
- B. A. Van Horn, R. K. Iha and K. L. Wooley, Macromolecules, 2008, 41, 1618–1626 CrossRef CAS.
- I. Taniguchi, A. M. Mayes, E. W. L. Chan and L. G. Griffith, Macromolecules, 2005, 38, 216–219 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS.
- B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 15020–15021 CrossRef CAS.
- J. Liu, E. N. Kadnikova, Y. Liu, M. D. McGehee and J. M. J. Fréchet, J. Am. Chem. Soc., 2004, 126, 9486–9487 CrossRef CAS.
- D. Quémener, M. L. Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155–173 CrossRef.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- X. Chen and T. Michinobu, Macromol. Chem. Phys., 2022, 223, 2100370 CrossRef CAS.
- J. Duchateau, L. Lutsen, W. Guedens, T. J. Cleij and D. Vanderzande, Polym. Chem., 2010, 1, 1313–1322 RSC.
- C. L. Chochos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326–1414 CrossRef CAS.
- U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 RSC.
- U. Scherf and K. Müllen, Makromol. Chem., Rapid Commun., 1991, 12, 489–497 CrossRef CAS.
- A. C. Grimsdale and K. Müllen, Macromol. Rapid Commun., 2007, 28, 1676–1702 CrossRef CAS.
- B. VanVeller, D. Robinson and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 1182–1186 CrossRef CAS PubMed.
- J. Lee, A. J. Kalin, C. Wang, J. T. Early, M. Al-Hashimi and L. Fang, Polym. Chem., 2018, 9, 1603–1609 RSC.
- Y. C. Teo, H. W. H. Lai and Y. Xia, Chem. – Eur. J., 2017, 23, 14101–14112 CrossRef CAS.
- Z. Cai, M. A. Awais, N. Zhang and L. Yu, Chem, 2018, 4, 2538–2570 CAS.
- S. Che and L. Fang, Chem, 2020, 6, 2558–2590 CAS.
- J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- A. Marrocchi, A. Facchetti, D. Lanari, S. Santoro and L. Vaccaro, Chem. Sci., 2016, 7, 6298–6308 RSC.
- V. Kardelis, R. C. Chadwick and A. Adronov, Angew. Chem., Int. Ed., 2016, 55, 945–949 CrossRef CAS PubMed.
- K. Li, D. Fong, E. Meichsner and A. Adronov, Chem. – Eur. J., 2021, 27, 5057–5073 CrossRef CAS.
- R. C. Chadwick, V. Kardelis, S. Liogier and A. Adronov, Macromolecules, 2013, 46, 9593–9598 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS.
- D. Fong, A. Lang, K. Li and A. Adronov, Macromolecules, 2020, 53, 1760–1766 CrossRef CAS.
- T. Michinobu and F. Diederich, Angew. Chem., Int. Ed., 2018, 57, 3552–3577 CrossRef CAS.
- T. Michinobu, H. Kumazawa, K. Noguchi and K. Shigehara, Macromolecules, 2009, 42, 5903–5905 CrossRef CAS.
- Y. Yuan, T. Michinobu, M. Ashizawa and T. Mori, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1013–1020 CrossRef CAS.
- H. Fujita, K. Tsuboi and T. Michinobu, Macromol. Chem. Phys., 2011, 212, 1758–1766 CrossRef CAS.
- T. Michinobu and H. Fujita, Materials, 2010, 3, 4773–4783 CrossRef CAS PubMed.
- W. Huang, React. Funct. Polym., 2014, 83, 42–48 CrossRef CAS.
- Y. Yuan and T. Michinobu, Macromol. Chem. Phys., 2012, 213, 2114–2119 CrossRef CAS.
- D. Wang, Q. Guo, H. Gao, Z. Yang, H. Cao, W. He and H. Wang, RSC Adv., 2016, 6, 59327–59332 RSC.
- S. Xu, E. H. Kim, A. Wei and E.-I. Negishi, Sci. Technol. Adv. Mater., 2014, 15, 044201 CrossRef CAS.
- X. Zhu, S. R. Bheemireddy, S. V. Sambasivarao, P. W. Rose, R. Torres Guzman, A. G. Waltner, K. H. DuBay and K. N. Plunkett, Macromolecules, 2016, 49, 127–133 CrossRef CAS.
- N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921–10925 CrossRef CAS PubMed.
- H. Arslan, J. D. Saathoff, D. N. Bunck, P. Clancy and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 12051–12054 CrossRef CAS PubMed.
- S. R. Bheemireddy, P. C. Ubaldo, A. D. Finke, L. Wang and K. N. Plunkett, J. Mater. Chem. C, 2016, 4, 3963–3969 RSC.
- T. Koizumi, T. Kameda, H. Saito, A. Sato and S. Hayashi, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 827–832 CrossRef CAS.
- V. Kardelis, M. M. Denk and A. Adronov, Angew. Chem., Int. Ed., 2021, 60, 2980–2986 CrossRef CAS PubMed.
- S. Wei, J. Xia, E. J. Dell, Y. Jiang, R. Song, H. Lee, P. Rodenbough, A. L. Briseno and L. M. Campos, Angew. Chem., Int. Ed., 2014, 53, 1832–1836 CrossRef CAS.
- F. Glöcklhofer, D. Lumpi, M. Kohlstädt, O. Yurchenko, U. Würfel and J. Fröhlich, React. Funct. Polym., 2015, 86, 16–26 CrossRef.
- F. Glöcklhofer, D. Lumpi, B. Stöger and J. Fröhlich, New J. Chem., 2014, 38, 2229–2232 RSC.
- E. Reichsöllner, A. Creamer, S. Cong, A. Casey, S. Eder, M. Heeney and F. Glöcklhofer, Front. Chem., 2019, 7, 123 CrossRef.
- M. Rimmele, K. Ableidinger, A. V. Marsh, N. J. Cheetham, M. J. Taublaender, A. Buchner, J. Prinz, J. Fröhlich, M. M. Unterlass, M. Heeney and F. Glöcklhofer, Polym. Chem., 2019, 10, 738–750 RSC.
- H. Cao, N. Bauer, S. Bi, D. Li, W. You and P. A. Rupar, Eur. Polym. J., 2018, 104, 157–163 CrossRef CAS.
- M. Sebastian, M. Hissler, C. Fave, J. Rault-Berthelot, C. Odin and R. Réau, Angew. Chem., Int. Ed., 2006, 45, 6152–6155 CrossRef CAS PubMed.
- T. P. Voortman, H. D. de Gier, R. W. A. Havenith and R. C. Chiechi, J. Mater. Chem. C, 2014, 2, 3407–3415 RSC.
- G. Ye, N. Y. Doumon, S. Rousseva, Y. Liu, M. Abdu-Aguye, M. A. Loi, J. C. Hummelen, L. J. A. Koster and R. C. Chiechi, ACS Appl. Energy Mater., 2019, 2, 2197–2204 CrossRef CAS.
- A. Creamer, C. S. Wood, P. D. Howes, A. Casey, S. Cong, A. V. Marsh, R. Godin, J. Panidi, T. D. Anthopoulos, C. H. Burgess, T. Wu, Z. Fei, I. Hamilton, M. A. McLachlan, M. M. Stevens and M. Heeney, Nat. Commun., 2018, 9, 3237 CrossRef PubMed.
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 CrossRef CAS.
- S. Cong, A. Creamer, Z. Fei, S. A. J. Hillman, C. Rapley, J. Nelson and M. Heeney, Macromol. Biosci., 2020, 20, 2000087 CrossRef CAS PubMed.
- Q. Cui and G. C. Bazan, Acc. Chem. Res., 2018, 51, 202–211 CrossRef CAS PubMed.
- K. Feron, R. Lim, C. Sherwood, A. Keynes, A. Brichta and P. C. Dastoor, Int. J. Mol. Sci., 2018, 19, 2382 CrossRef PubMed.
- N. Shida, K. Ninomiya, N. Takigawa, K. Imato, Y. Ooyama, I. Tomita and S. Inagi, Macromolecules, 2021, 54, 725–735 CrossRef CAS.
- K. Ninomiya, N. Shida, T. Nishikawa, T. Ishihara, H. Nishiyama, I. Tomita and S. Inagi, ACS Macro Lett., 2020, 9, 284–289 CrossRef CAS PubMed.
- H. Wang, H. Zhao, S. Chen, L. Bai, Z. Su and Y. Wu, Org. Lett., 2021, 23, 2217–2221 CrossRef CAS.
- B. Chen, N. Yang, P. Wang, Y. Xiang and H. Chen, Appl. Surf. Sci., 2020, 499, 143865 CrossRef CAS.
- Z. Qi, N. G. Rees and P. G. Pickup, Chem. Mater., 1996, 8, 701–707 CrossRef CAS.
- P. Soudan, P. Lucas, L. Breau and D. Bélanger, Langmuir, 2000, 16, 4362–4366 CrossRef CAS.
- S. Inagi, S. Hayashi, K. Hosaka and T. Fuchigami, Macromolecules, 2009, 42, 3881–3883 CrossRef CAS.
- T. Kurioka, N. Shida, I. Tomita and S. Inagi, Macromolecules, 2021, 54, 1539–1547 CrossRef CAS.
- Y. Li, G. Vamvounis and S. Holdcroft, Macromolecules, 2001, 34, 141–143 CrossRef CAS.
- Y. Li, G. Vamvounis and S. Holdcroft, Chem. Mater., 2002, 14, 1424–1429 CrossRef CAS.
- D. L. Crossley, L. Urbano, R. Neumann, S. Bourke, J. Jones, L. A. Dailey, M. Green, M. J. Humphries, S. M. King, M. L. Turner and M. J. Ingleson, ACS Appl. Mater. Interfaces, 2017, 9, 28243–28249 CrossRef CAS.
- P. R. Neumann, D. L. Crossley, M. Turner, M. Ingleson, M. Green, J. Rao and L. A. Dailey, ACS Appl. Mater. Interfaces, 2019, 11, 46525–46535 CrossRef CAS PubMed.
- Y. Li, G. Vamvounis, J. Yu and S. Holdcroft, Macromolecules, 2001, 34, 3130–3132 CrossRef CAS.
- S. Holdcroft, Y. Li, G. Vamvounis, H. Aziz and Z. D. Popovic, Chromogenic Phenomena in Polymers, American Chemical Society, 2004, ch. 17, vol. 888, pp. 220–232 Search PubMed.
- Y. Li, G. Vamvounis and S. Holdcroft, Macromolecules, 2002, 35, 6900–6906 CrossRef CAS.
- Z. Zhou, J. L. Brusso and S. Holdcroft, Chem. Mater., 2010, 22, 2287–2296 CrossRef CAS.
- Y. Yuan, W. Choi, H. Nishide and T. Michinobu, Chem. Sci., 2013, 4, 345–350 RSC.
- B. Koo, E. M. Sletten and T. M. Swager, Macromolecules, 2015, 48, 229–235 CrossRef CAS PubMed.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- C. H. Woo, B. C. Thompson, B. J. Kim, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 16324–16329 CrossRef CAS PubMed.
- M. Chen, M. Li, H. Wang, S. Qu, X. Zhao, L. Xie and S. Yang, Polym. Chem., 2013, 4, 550–557 RSC.
- M. Li, P. Xu, J. Yang and S. Yang, J. Mater. Chem., 2010, 20, 3953–3960 RSC.
- M. Li, P. Xu, J. Yang, H. Ying, K. Haubner, L. Dunsch and S. Yang, J. Phys. Chem. C, 2011, 115, 4584–4593 CrossRef CAS.
- J. Geng and T. Zeng, J. Am. Chem. Soc., 2006, 128, 16827–16833 CrossRef CAS PubMed.
- S. P. Economopoulos, C. L. Chochos, V. G. Gregoriou, J. K. Kallitsis, S. Barrau and G. Hadziioannou, Macromolecules, 2007, 40, 921–927 CrossRef CAS.
- X. Chen, B. Gholamkhass, X. Han, G. Vamvounis and S. Holdcroft, Macromol. Rapid Commun., 2007, 28, 1792–1797 CrossRef CAS.
- B. Gholamkhass, T. J. Peckham and S. Holdcroft, Polym. Chem., 2010, 1, 708–719 RSC.
- Z. Zhou, X. Chen and S. Holdcroft, J. Am. Chem. Soc., 2008, 130, 11711–11718 CrossRef CAS PubMed.
- C. Kub, J. Tolosa, A. J. Zucchero, P. L. McGrier, C. Subramani, A. Khorasani, V. M. Rotello and U. H. F. Bunz, Macromolecules, 2010, 43, 2124–2129 CrossRef CAS.
- A. A. Buelt, C. A. Conrad, W. D. Mackay, M. F. Shehata, V. D. Smith and R. C. Smith, Polym. Int., 2015, 64, 730–739 CrossRef CAS.
- Z. Zhang, X. Zong, Z. Sun and Y. Qin, Polymer, 2019, 166, 115–122 CrossRef CAS.
- Z. Zhang and Y. Qin, Polym. Chem., 2019, 10, 1018–1025 RSC.
- D. Fong, J. Yeung, S. A. McNelles and A. Adronov, Macromolecules, 2018, 51, 755–762 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |