
Exploiting valuable supramolecular materials from waste plastics†
Xuehui
Liu
,
Xu
Zhao
,
Wenli
An
,
Rongcheng
Du
,
Gang
Wu
,
Shimei
Xu
 *,
Fan
Zhang
and
Yu-Zhong
Wang
*
*,
Fan
Zhang
and
Yu-Zhong
Wang
*
The Collaborative Innovation Center for Eco-Friendly and Fire-Safety Polymeric Materials (MoE), National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), College of Architecture and Environment, State Key Laboratory of Polymer Materials Engineering, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: yzwang@scu.edu.cn; xushimei@scu.edu.cn
First published on 30th September 2022
Abstract
A new family of supramolecular materials is exploited from waste thermosets via a one-step retrosynthetic approach, which exhibits distinguished adhesion properties in dry/wet environments, good corrosion resistance and dynamic reversibility. This work opens up a wide design space for supramolecular materials with excellent performances and proposes a new strategy for efficient utilization of hybrid degraded products.
New conceptsExploiting novel and high-performance materials from post-consumer wastes is an innovative and far-reaching work whereas it has been manifested to be extremely challenging. Herein, we demonstrate upcycling of waste thermosets into supramolecular materials based on low-molecular-weight molecules via one-step oxidation, thus realizing the long-term circulation of commercial polymers from petrochemical resources, which is rarely reported. Driven by supramolecular interactions, degraded products even with a complex composition are directly and efficiently reutilized without sophisticated separation/purification. It is so different from traditional recovery strategies, which focus on reconstructing new products using covalent bonds after breaking waste polymers, on most occasions, with low utilization. Our method realizes for the first time the reconstruction of small-molecule degraded products using non-covalent bonds on the basis of full use of the wastes. What is more, this waste-based material exhibits distinguished adhesion properties in dry/wet environments. It can act as a sealing and oil–water separation material based on spontaneous formation of a solidified layer in air or at the oil/water interface. This work is of great significance not only in waste recovery where degraded products are fully used with a high value being added but also in designing unprecedented supramolecular materials where distinguished and unique performance is obtained. |
Introduction
Since 1950, plastics have been in widespread use, and 75% of 8.3 billion metric tons of plastics produced worldwide have become wastes, but only 8.8% of them have been recycled, causing a serious threat to the ecological environment and large consumption of non-renewable resources.1–3 At present, much effort is devoted to recycling plastic wastes into value-added products, but this field seems to be still in its infancy.4 Physical recovery of plastic wastes is simple but has serious limitations including inferior properties and limited categories of regenerative products.5 Chemical recovery can be used to recycle monomers6,7 or added-value chemicals8,9/polymers10–13 from plastic wastes and is regarded as a promising method. Recently, most of the efforts have been focused on recycling of disposable thermoplastics (especially polyolefin),14–16 but less attention has been paid to recycle waste thermosets. Compared to thermoplastics, thermosets are harder to recycle because of their stable cross-linking network. In order to overcome these problems, one method is to design a new type of recyclable and malleable thermoset by introducing dynamic or cleavable bonds,17,18 and the other is to recycle thermosets that are commercially available and widely used in various fields. The former is an effective way, but there is still a long way to go to meet actual application requirements of thermosets. Thus, it becomes more important for the recovery of commercial thermosets before its service time comes to an end.Epoxy resin (EP), as a typical thermoset, has been used widely in different industrial applications, including adhesives and paintings, electronics, wind blades, due to its excellent performance. Previously, the recovery strategies mainly focused on breaking down thermosets to recycle reinforcing fibers but neglected further utilization of the degraded products.19–21 Recent developments reported that the degraded products can be used as additive components, but the performances of final materials might be weakened in high addition of degraded products.22–24 Furthermore, dense cross-linked networks and various C–C, C–O and C–N bonds of EP make it extremely difficult to recover chemicals or polymers with high purity and quality. Several EP recovery examples to recycle raw chemicals such as bisphenol A have been reported but with limited productivity.25,26 Consequently, diverse structures, complicated separation/purification processes and incomplete reutilization of the degraded products lead to a huge waste of energy and resources and even cause severe secondary pollution. In particular, for commercial plastics which contain various additives, such as plasticizers, flame retardants and so on, it is more difficult to make full use of the degraded products. To address these problems and challenges, some new strategies are highly desired.
Molecular building blocks of modern polymeric materials are connected by covalent bonds or noncovalent interactions. The polymers cross-linked by covalent bonds generally require structural units with strict screening and an appropriate design. It often leads to multi-step processing and high cost. By contrast, supramolecular materials are generated by assembling molecules into larger structures via noncovalent interactions, while introducing unique and fascinating properties, such as dynamic reversibility, adaptivity and stimulus responsiveness to the materials.27,28 Now, it is an extremely promising and emerging area in materials chemistry.29 The functional groups of the molecular building blocks are the key to realize this goal. This may provide a new opportunity for high-value and efficient utilization of degraded products with complex compositions. Yet, the approaches to construct supramolecular systems are still based on fresh structural units, and the utilization of renewable resources is very challenging.
Herein, we demonstrate the conversion of waste amine-cured EP into a high-performance supramolecular material, which not only uses a new and universal upcycling method by introducing supramolecular interactions into the reconstruction of degraded products but also introduces a viable path to construct supramolecular materials. The new strategy converts waste materials directly into new high-performance materials, avoiding complex and energy-consuming separation. To our best knowledge, this is the first report on supramolecule reconstruction instead of traditional chemical reconstruction, which makes full use of degraded products. Our approach used hydrogen peroxide alone as a reaction reagent to decompose waste EP without the assistance of additional catalysts or organic solvents. The degraded amine-cured EP (DEP) was easily separated from water solution by filtering due to its insolubility. During the degradation process, OH radicals attack the C–N cross-linking points to break the three-dimensional networks in EP, while the disconnected epoxy units are oxidized into oligomers with ketones or carboxyl groups. This end-functionalized DEP with intermolecular hydrogen bonds can be directly used as a novel low-molecular-weight monomer (LMWM)-based supramolecular adhesive. It exhibits excellent and reversible adhesion properties for various substrates and good corrosion resistance to water, acid, and salt and alkane organic solvents. Even DEP also shows strong adhesion properties under aqueous conditions with a maximum lap-shear strength of 5.8 MPa, which is higher than those of most of the underwater adhesives previously reported. The success of this work provides new insights into upcycling of plastic wastes and opens up a new path for designing supramolecular materials.
Results and discussion
13C-NMR spectra of amine-cured EP and DEP (Fig. 1a) show that the C–N bond is partially broken because of its relatively low bond energy, while the benzene ring and C–O–C bond remain intact. Meanwhile a new peak corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is observed in the spectrum of DEP. Similar results can be observed in the Fourier transform infrared (FTIR) spectra (Fig. S1, ESI†). In the 1H-NMR spectrum of DEP, a characteristic peak of H belonging to carboxylic acid appears in the range of 9–13 ppm (Fig. S2, ESI†). Besides, the C 1s spectrum of DEP shows an additional peak attributed to CO (288.4 eV), and the O 1s spectrum exhibits a new peak corresponding to O–CO (531.8 eV), further confirming the existence of carboxyl groups (Fig. 1b). Meanwhile, owing to the cleavage of some C–N bonds in the amine-cured EP backbone, peaks attributed to pyrrolic-N and oxidized-N appear in the N 1s spectrum of DEP. As a result, an increase in the O/C ratio is observed in DEP (Fig. S3 and Table S1, ESI†). All of these results support strongly the breakup of the cross-linked structure of EP by H2O2 alone.
O is observed in the spectrum of DEP. Similar results can be observed in the Fourier transform infrared (FTIR) spectra (Fig. S1, ESI†). In the 1H-NMR spectrum of DEP, a characteristic peak of H belonging to carboxylic acid appears in the range of 9–13 ppm (Fig. S2, ESI†). Besides, the C 1s spectrum of DEP shows an additional peak attributed to CO (288.4 eV), and the O 1s spectrum exhibits a new peak corresponding to O–CO (531.8 eV), further confirming the existence of carboxyl groups (Fig. 1b). Meanwhile, owing to the cleavage of some C–N bonds in the amine-cured EP backbone, peaks attributed to pyrrolic-N and oxidized-N appear in the N 1s spectrum of DEP. As a result, an increase in the O/C ratio is observed in DEP (Fig. S3 and Table S1, ESI†). All of these results support strongly the breakup of the cross-linked structure of EP by H2O2 alone.
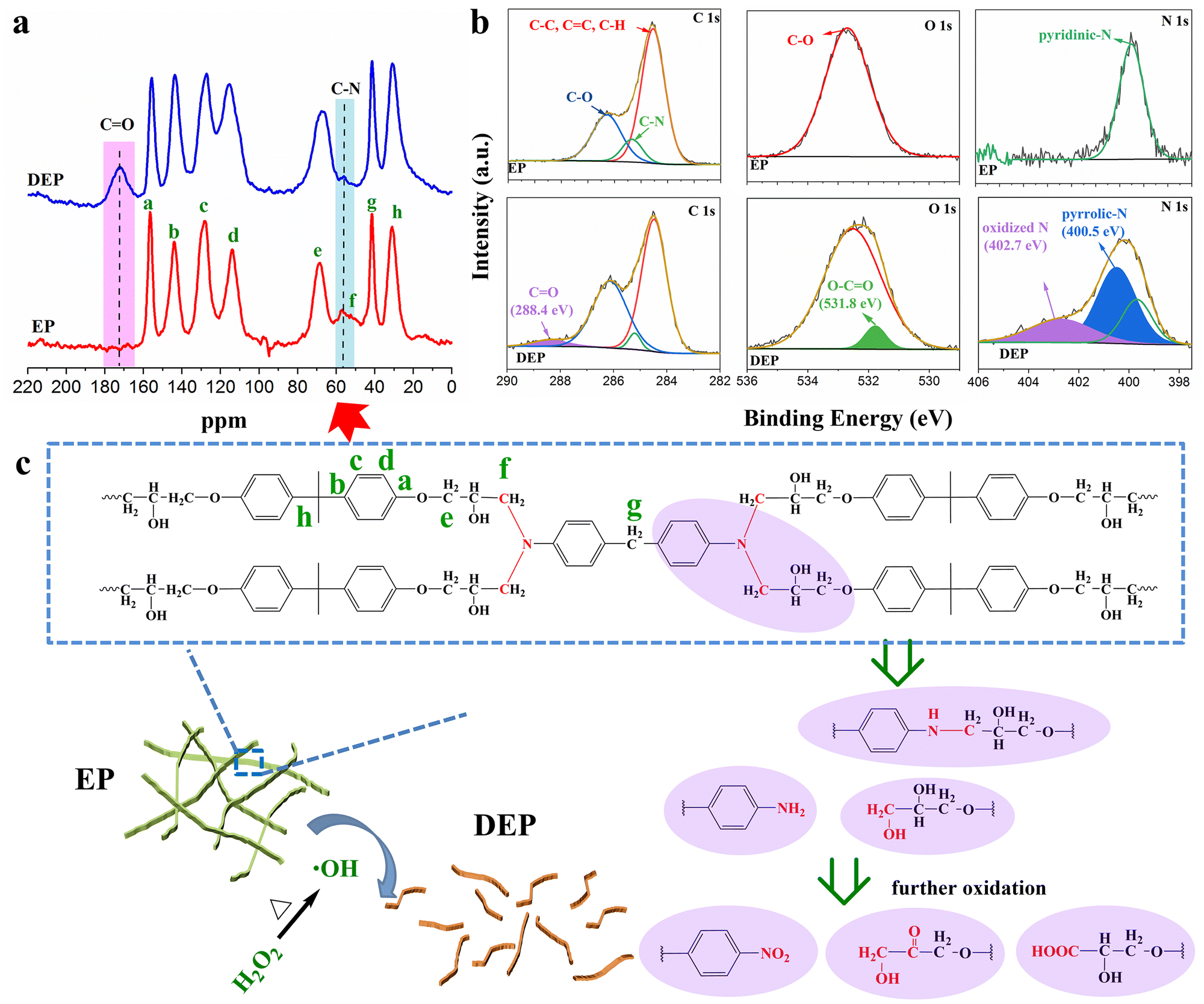 | ||
| Fig. 1 Chemical and structural characterization of DEP. (a) 13C-NMR spectra of amine-cured EP and DEP. (b) High-resolution C 1s, O 1s, and N 1s spectra of EP and DEP. (c) Possible decomposition mechanism of EP in H2O2. | ||
The results of gel permeation chromatography (GPC) show that the weight-average molecular weight of DEP is 709 with a polydispersity index of 1.69 (Fig. S4, ESI†), indicating that DEP can be a monomer or a multimer. The possible molecular structures of DEP were investigated by electrospray ionization mass spectrometry, and it shows that the compounds in DEP were similar to the monomers, dimers and trimers of diglycidyl ether of bisphenol A (DGEBA). This finding further confirms that EP is degraded through the cleavage of C–N cross-linked points. Different from the molecular ions of DGEBA, oligomers rich in hydroxyl, carboxyl, amino and nitro functional groups are clearly detected in DEP (Fig. S5 and S6, ESI†). From the molecular fragments, hydroxyl groups tend to oxidize, and ketone/aldehyde and carboxyl groups are produced. Amino groups may be oxidized to nitro groups. Additionally, pyrolysis-GC/MS was used to determine the structure of the final decomposed products. The signal of the phenolic derivative from DEP is observed after pyrolysis at 700 °C, proving that the benzene rings are retained during the degradation process (Fig. S7, ESI†).
According to the above results, the possible decomposition mechanism of the amine-cured EP in H2O2 is proposed (Fig. 1c). First, hydroxyl radicals from H2O2 mainly attacked C–N bonds to form compounds containing hydroxyl and amino groups, without destroying benzene rings and other resin skeleton structures. Owing to strong oxidation capacity of H2O2, the secondary and primary hydroxyl groups could be further oxidized to form ketones or carboxyl groups, respectively. Besides, amino groups can also be oxidized to nitro groups. Therefore, the cross-linked network in amine-cured EP was transformed into linear short chains, which can be dissolved in DMF (Fig. S8h, ESI†). But it was interesting to find that DEP particles stick together to form solid blocks after the reaction due to abundant hydrogen bonds formed in DEP. To verify the above mechanism, comparative reactions were designed by adding tert-butanol or ethanol as a radical scavenger. The obtained degraded products were still solid powders and insoluble in DMF (Fig. S8g and i, ESI†) and exhibited markedly different chemical structures (Fig. S9, ESI†). These results can be explained as that the hydroxyl radicals from H2O2 played a leading role in the degradation of the amine-cured EP.
The amine-cured EP exhibits high initial thermal decomposition temperature and glass transition temperature (Tg). After degradation, the DEP still possesses excellent thermal stability since it starts to decompose as the temperature exceeds 210 °C and shows a Tg of 85 °C (Fig. S10 and S11, ESI†). Temperature-dependent FTIR spectroscopy was used to explore the assembly mechanism of DEP (Fig. 2a and Fig. S12a, ESI†). The bands in the region of 3500–3200 cm−1, which are assigned to the stretching vibration of hydroxy and amino groups, gradually shift to high wavenumbers upon heating, accompanied by their intensity reduction. It suggests the generation of “free” groups. At the same time, the N–H bending vibration (1607 cm−1) and C–OH stretching vibration (1041 cm−1) gradually shift to lower wavenumbers. Moreover, with increasing temperature, the peak intensity of the 1748 cm−1 (“free” CO groups) peak gradually increases, revealing the rupturing of hydrogen bonds related to carboxyl groups.30,31 Meanwhile, C–O–C and C–N also show complex changes upon heating from 25 to 130 °C. The spectral intensity of the 1244 cm−1 (C–O–C) and 1108 cm−1 (fatty C–N) peaks gradually decreases, while the peak at 1294 cm−1 (aromatic C–N) exhibits a red shift during the heating process. These results confirm that various types of hydrogen bonds exist in DEP.
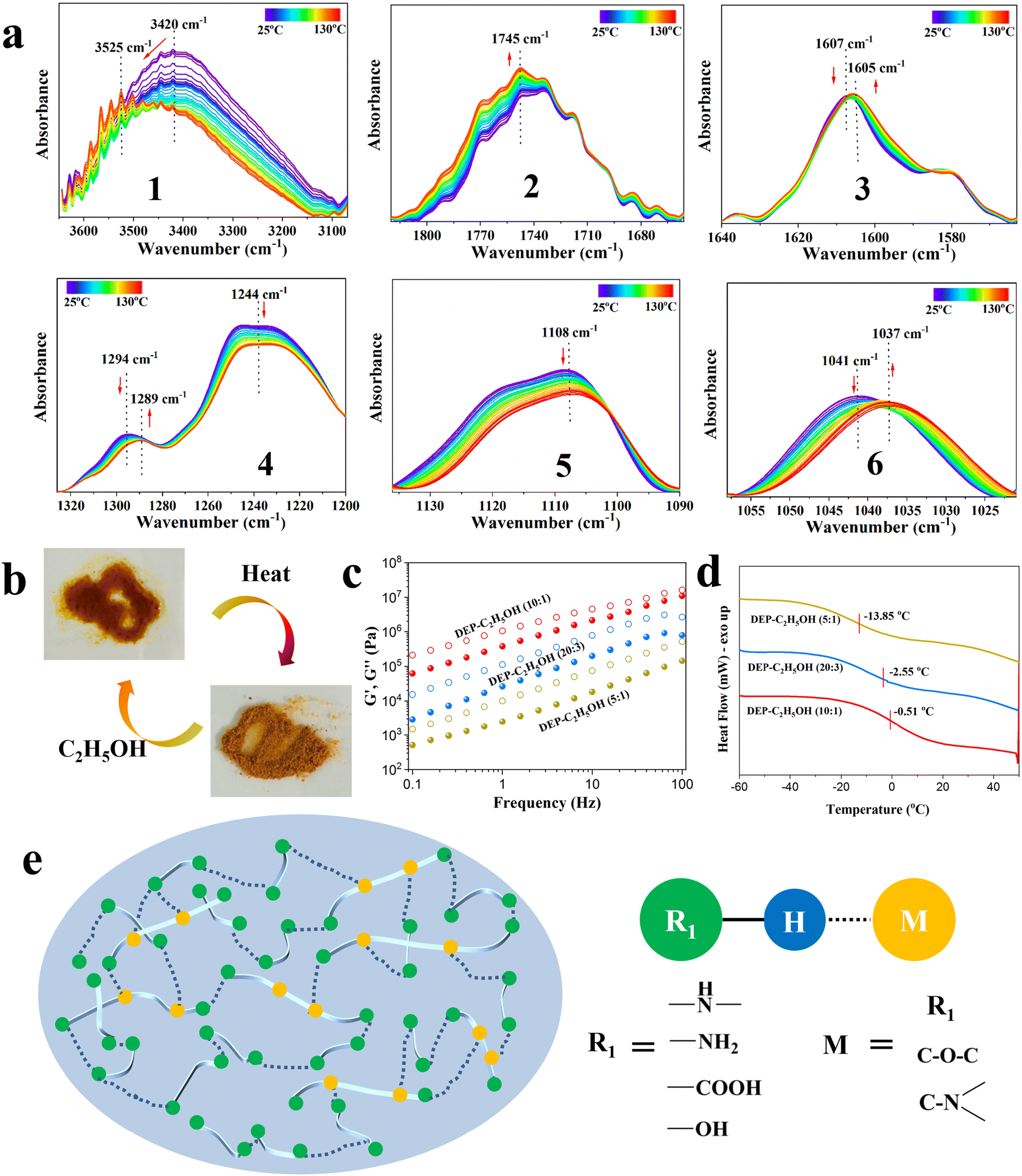 | ||
| Fig. 2 Supramolecular interaction of DEP. (a) Temperature-dependent FTIR spectra of DEP upon heating from 25 to 130 °C: 3650–3070 cm−1 (1), 1820–1656 cm−1 (2), 1640–1563 cm−1 (3), 1326–1200 cm−1 (4), 1136–1090 cm−1 (5), and 1058–1021 cm−1 (6). (b) Photographs of DEP before and after C2H5OH treatment. DEP transformed from powders to a viscous liquid with the addition of an appropriate amount of C2H5OH. (c) Frequency sweep of the storage (solid dots, G′) and loss (hollow dots, G′′) modulus of DEP-C2H5OH. (d) Tg of DEP with different ethanol contents. (e) Schematic illustration of supramolecular multiple hydrogen-bond networks in DEP. | ||
In addition, we also observed that powdery DEP became a viscous liquid after adding hot ethanol or immersing in hot water (Fig. 2b and Fig. S13, ESI†). No carbon skeleton changes in DEP are found after treating with C2H5OH (DEP-C2H5OH) and H2O (DEP-H2O) (Fig. S14 and S15, ESI†). However, even after sufficient drying, an obvious increase in the O/C ratio is observed in DEP-H2O and DEP-C2H5OH (Table S2, ESI†). This result is due to the formation of new hydrogen bonds between DEP and water/ethanol molecules as the hydrogen bonds between DEP break. The interaction between ethanol and DEP is also confirmed from the rheological behaviors of C2H5OH-DEP. All samples are a viscous liquid with G′′ > G′ over the frequency sweep and temperature sweep (Fig. 2c and Fig. S16, ESI†). Upon increasing the content of ethanol, DEP exhibits a much lower G′, G′′, complex viscosity and Tg, proving the role of ethanol in breaking the hydrogen bonds in DEP (Fig. 2c, d and Fig. S17, ESI†). These results further verify that multiple hydrogen bonds exist in DEP, and the possible supramolecular network is shown in Fig. 2e (Fig. S12b, ESI†).
Impressively, DEP melted in the range of 145 to 210 °C and solidified as the temperature decreased. It behaves like the melting transitions of commercially available hot-melt adhesives such as the ethylene vinyl acetate copolymer (EVA) and amorphous poly alpha olefin (APAO) (Fig. S18, ESI†). Thus, DEP can be used as a candidate for hot-melt adhesives. Its adhesive strength reaches 2.31 MPa, which is about four times that of EVA (0.64 MPa) and three times that of APAO (0.72 MPa) (Fig. 3a). Encouraged by these results, the adhesive properties of DEP with the assistance of ethanol were further investigated to lower its operating temperature. As expected, DEP after adding ethanol exhibits an obvious stringing phenomenon when stretched between the two glass slices (Fig. 3b). The adhesive properties of DEP with different ethanol contents were investigated to imitate the adhesion process of DEP-C2H5OH with the extension of the drying time. As the ethanol evaporates, the adhesive strength of DEP gradually increases due to the reformation of intermolecular hydrogen bonds in DEP (Fig. 3b and Fig. S19, ESI†).
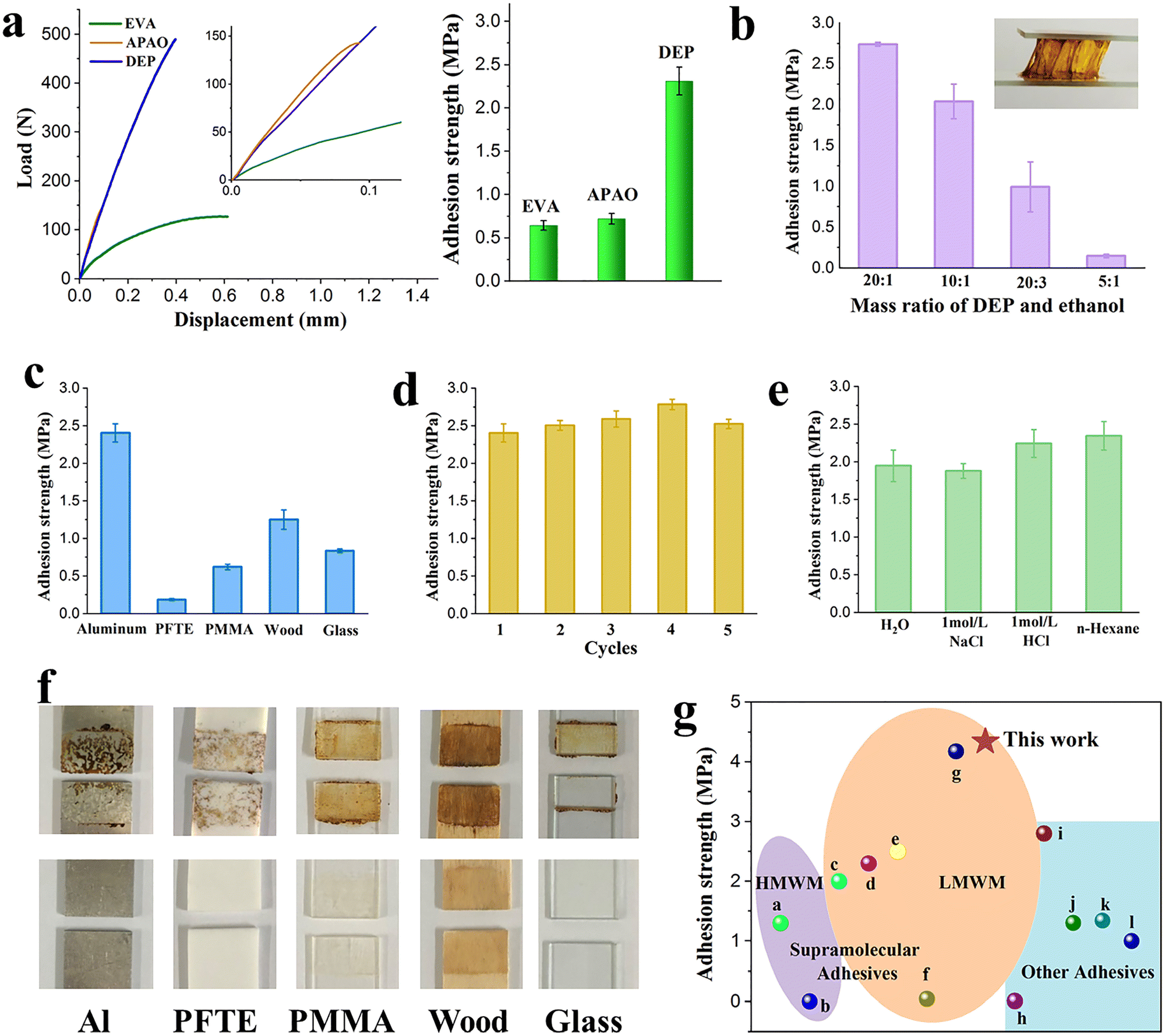 | ||
| Fig. 3 Adhesion properties of DEP. (a) Lap-shear strength curves and adhesion strength of EVA, APAO and DEP on Al slides (layer size: 2 × 1 cm2). Inset: Lap-shear strength curves of EVA, APAO and DEP at a displacement of 0 to 0.1 mm. (b) Adhesion strength of DEP with different ethanol contents (layer size: 2 × 1 cm2). Inset: DEP shows an obvious sticky behavior after ethanol is added. (c) Adhesion strength of DEP on different substrate surfaces (layer size: 2 × 1 cm2). (d) Reversible adhesion of DEP on Al slides. (e) Adhesion strength of DEP after being immersed in various solutions for 48 h. (f) Photographs of DEP after failure on different substrates and no DEP remains on the substrate surfaces after ethanol cleaning. (g) Comparison of adhesion strength among different adhesives. | ||
Benefiting from versatile functional groups, DEP shows excellent adhesion to various substrates, including hydrophilic glass and hydrophobic PTFE (Fig. 3c and Fig. S20 and S21, ESI†). Considering the remarkable and excellent adhesive strength to wood (1.25 MPa), DEP is expected to be a substitute for phenolic resin adhesive to reduce the use of formaldehyde. Interestingly, the soft PDMS after being cut can also be completely bonded and further bent over (Fig. S22, ESI†). The adhesive strength of DEP to aluminum slides remains at 2.5 MPa even after five cycles (Fig. 3d and Fig. S23, ESI†), which is equivalent to the initial value, exhibiting excellent regenerative nature. Furthermore, DEP shows excellent corrosion resistance to different aqueous or organic solutions. Notably, the adhesion strength of DEP remains above 2 MPa, and no small molecules are released from DEP even after immersing in different solvents for 48 h, proving the excellent stability of DEP (Fig. 3e and Fig. S24–S28, ESI†).
Different from most existing adhesives, DEP is easily detached from the substrate surface by implanting ethanol. In this way, the separated substrate could be restored to its original state and reused again (Fig. 3f). Aluminum slides are taken as an example to demonstrate the adhesion in various DEP layer sizes (Fig. S29, ESI†), and the adhesive strength for a DEP layer size of 1 cm2 is up to 4.34 MPa, beyond the property space of the previously reported LMWM-based supramolecular adhesives, and even higher than that of the previously reported high-molecular-weight molecule (HMWM)-based adhesives (Fig. 3g, details are in Table S3, ESI†).32–43 As the DEP layer size increased, greater force capacity can be observed. Furthermore, upon increasing the layer size to 3 cm2, the load remained but the displacement increased instead according to the lap-shear strength curve. This was because the bonded aluminum slides slipped between the clamps before they were pulled off due to the strong adhesion of DEP.
Developing adhesives that can adhere to wet surfaces or even be used underwater is extremely challenging. In this work, DEP exhibits excellent underwater adhesion. The introduction of a small amount of ethanol permits DEP-C2H5OH kneading casually, which can be shaped into balls, cubes, stars and other shapes (Fig. 4a). It shows a plasticizing effect and thus facilitates accurate delivery of the adhesive to the underwater target location. The target object was picked up from the water as the temperature first increased to 80 °C and then decreased to 20 °C (Fig. 4c and Videos S2, ESI†), while no adhesive behavior was observed at 20 °C (Fig. 4b and Videos S1, ESI†), indicating that DEP can be used as an underwater adhesive via temperature control.
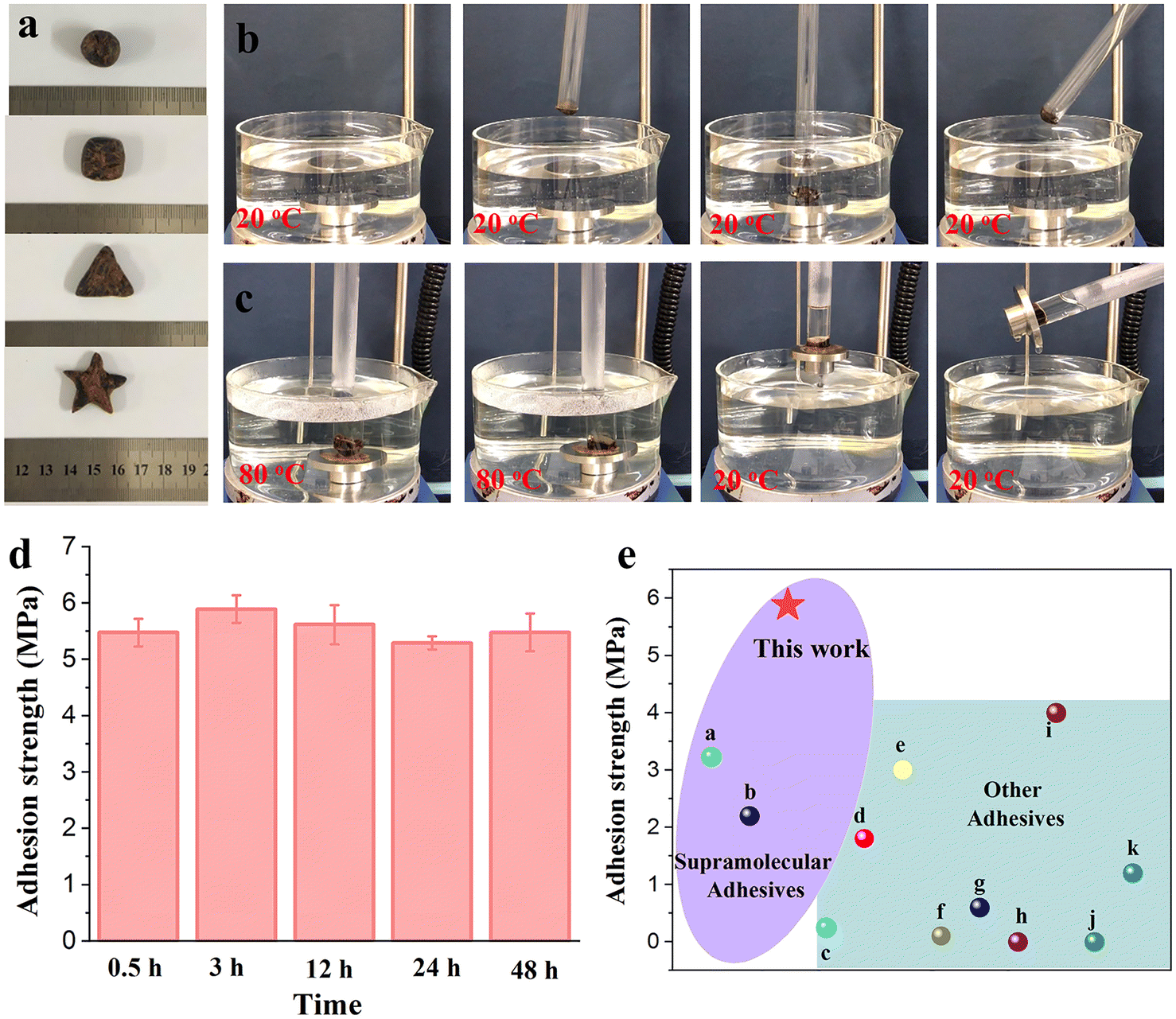 | ||
Fig. 4 Underwater adhesion performances of DEP. (a) DEP-C2H5OH (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) is similar to plasticine and can be kneaded into different shapes at room temperature. (b) and (c) Underwater adhesion performance of DEP at (b) 20 °C and (c) 80 °C first and then at 20 °C. The metal block can be lifted from the water when the water is cooled from 80 to 20 °C. (d) Changes in underwater force capacity and adhesion strength of DEP-C2H5OH (20:1) versus immersion time (layer size: 2 × 0.5 cm2). (e) Comparison of adhesion strength among different underwater adhesives. :1) is similar to plasticine and can be kneaded into different shapes at room temperature. (b) and (c) Underwater adhesion performance of DEP at (b) 20 °C and (c) 80 °C first and then at 20 °C. The metal block can be lifted from the water when the water is cooled from 80 to 20 °C. (d) Changes in underwater force capacity and adhesion strength of DEP-C2H5OH (20:1) versus immersion time (layer size: 2 × 0.5 cm2). (e) Comparison of adhesion strength among different underwater adhesives. | ||
The underwater adhesive performances of DEP are dependent on the content of ethanol. Without the addition of ethanol, DEP cannot transform into a completely fluid state in 80 °C water and tends to show poor adhesion performance due to insufficient contact with the substrates. With an increase in the ethanol content, the adhesion strength was inclined to be lower due to the weakened hydrogen-bond network of DEP (Fig. S30, ESI†). Additionally, the underwater adhesion performances of DEP with different layer sizes were investiagted. The adhesion strength is up to 5.88 MPa at a layer size of 1 cm2. As the layer size increases to 3 cm2, the bonded Al slides remain stable without breaking under this test condition, which is associated with the excellent underwater adhesion of DEP (Fig. S31, ESI†). Moreover, the adhesion strength of DEP does not decay (5.5 MPa) even if it is placed underwater for 48 h, suggesting the desirable stability of DEP for long-term use (Fig. 4d and Fig. S32, ESI†). Compared with previously reported underwater adhesives, the adhesion strength of DEP is higher, and it can be used as a promising candidate for an ideal wet adhesive (Fig. 4e, details are in Table S4, ESI†).38,44–53
Taking advantage of the desired water resistance, adhesion to a variety of substrates and underwater adhesion performance, DEP can be also developed as sealing and separating materials (Fig. 5a). Upon heating, the powdered DEP sticks together due to the supramolecular interaction between the components. After cooling, a strong sealing layer was formed on the water surface while the water was encapsulated in the test tube. Even after standing upside down, the water does not flow out (Fig. 5b and Video S3, ESI†). Such an excellent performance makes it possible for DEP to be applied in preventing liquid leakage and other fields. Furthermore, when DEP was added to an oil–water mixture, such as an n-hexane–water mixture, it floated at the interface of oil and water due to the lipophilicity and hydrophobicity of DEP. Similarly a DEP sealing layer was formed through heating/cooling cycles so the oil–water mixtures were separated successfully (Fig. 5c and Video S4, ESI†). To our best knowledge, this is the first report on such adhesiveness–separation bifunctional materials, which avoids the disadvantages of traditional filtration or adsorption separation methods, such as complicated equipment, low flux and easy contamination.
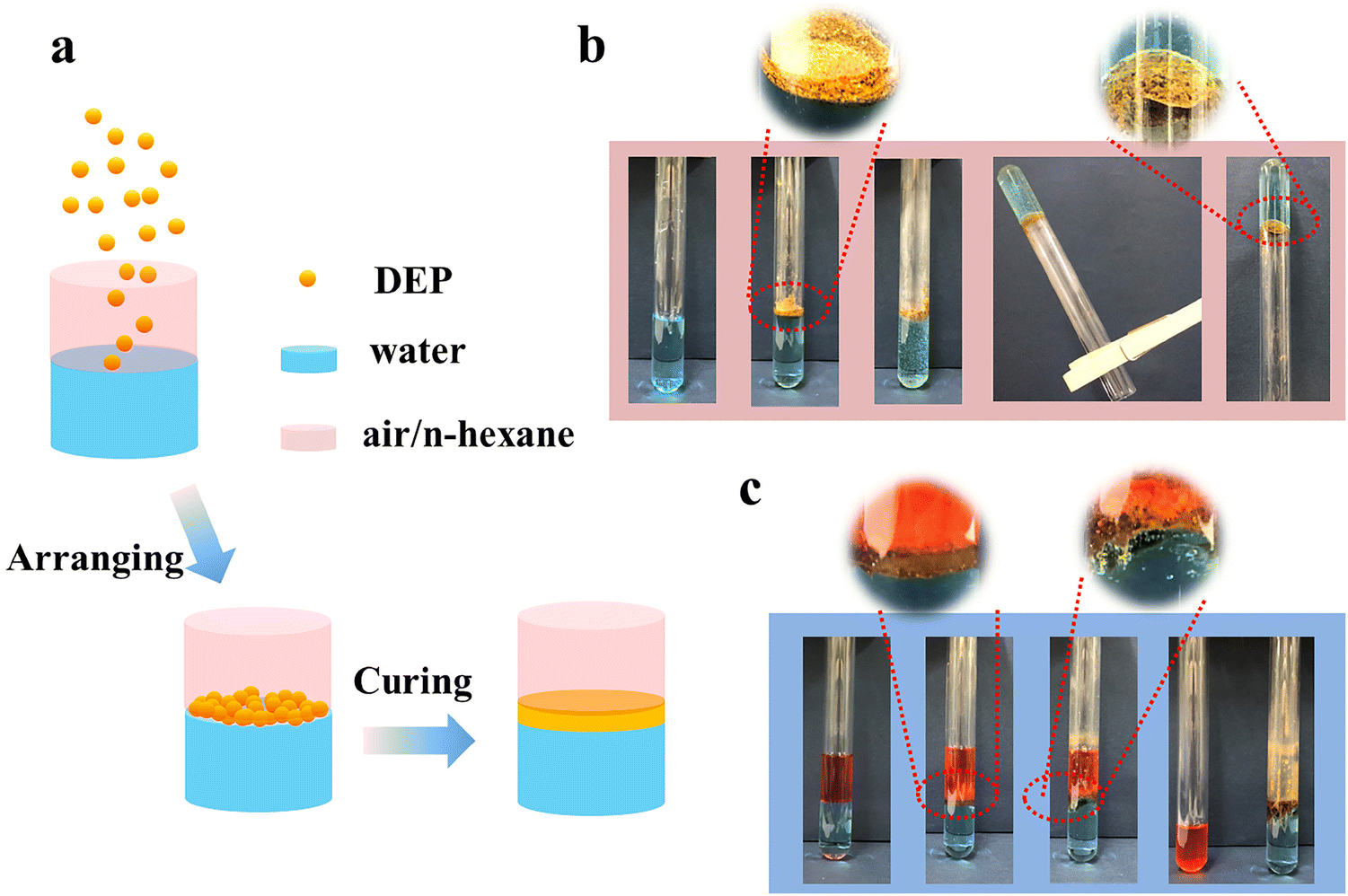 | ||
| Fig. 5 Other applications. (a) Schematic representation of DEP at the two-phase (air/water, oil/water) interface. (b) Photographs of DEP used as a sealing layer. After the heating/cooling cycle, the water existed in the inverted test tube without falling. (c) Photographs depict that DEP can be used in the separation of oil–water mixtures. | ||
Conclusions
In summary, we presented a new idea for one-step upcycling of waste thermosets to high-performance materials by mild degradation and supramolecular reconstruction. The waste amine-cured EP was completely degraded into oligomers containing carboxyl, amino and hydroxyl groups in aqueous solution under mild conditions, and all the collected degraded products were fully utilized as supramolecular materials. Taking advantage of the multiple hydrogen bonds, DEP exhibits distinguished adhesion properties to various substrates, excellent reusability and easy removability from surfaces. Moreover, DEP shows strong resistance to water and various organic solvents. More impressively, DEP is also used as an underwater adhesive with an enviable underwater adhesion strength of 5.88 MPa and distinguished long-term stability. Furthermore, DEP shows bright prospects in sealing and oil–water separation applications. But before considering a broader rollout of such a material, its safety issues and health impacts still have to be investigated. To our best knowledge, this is the first example of high-value utilization of the whole low-molecular-weight degraded products without separation or other supporting components. We believe that our findings not only open a significant path toward waste disposal but also provide a new design concept for preparing supramolecular materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 51721091), the State Key Laboratory of Polymer Materials Engineering Open Fund project (sklpme2020-1-02) and the Fundamental Research Funds for the Central Universities.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 25–29 CrossRef PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 69, 66–69 CrossRef PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manag., 2017, 69, 24–58 CrossRef CAS.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed.
- N. A. Rorrer, S. Nicholson, A. Carpenter, M. J. Biddy, N. J. Grundl and G. T. Beckham, Joule, 2019, 3, 1006–1027 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS.
- X.-H. Yue, F.-S. Zhang, C.-C. Zhang and P. Qian, J. Hazard. Mater., 2022, 432, 128746 CrossRef CAS PubMed.
- X. Liu, F. Tian, X. Zhao, R. Du, S. Xu and Y. Z. Wang, Appl. Surf. Sci., 2020, 529, 147151 CrossRef CAS.
- W. An, X. L. Wang, Y. Yang, H. Xu, S. Xu and Y. Z. Wang, Green Chem., 2019, 21, 3006–3012 RSC.
- X. L. Wang, W. L. An, Y. Yang, Z. Y. Hu, S. Xu, W. Liao and Y. Z. Wang, Chem. Eng. J., 2019, 361, 21–30 CrossRef CAS.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y. H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Nat. Catal., 2020, 3, 893–901 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Cell Reports Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- O. Zabihi, M. Ahmadi, C. Liu, R. Mahmoodi, Q. Li and M. Naebe, Composites, Part B, 2020, 184, 107750 CrossRef CAS.
- C. Pei, P. yu Chen, S. C. Kong, J. Wu, J. H. Zhu and F. Xing, Sep. Purif. Technol., 2022, 278, 119591 CrossRef CAS.
- D. H. Kim, A. Yu and M. Goh, J. Ind. Eng. Chem., 2021, 96, 76–81 CrossRef CAS.
- D. H. Kim, M. Lee and M. Goh, ACS Sustainable Chem. Eng., 2020, 8, 2433–2440 CrossRef CAS.
- M. Das, R. Chacko and S. Varughese, ACS Sustainable Chem. Eng., 2018, 6, 1564–1571 CrossRef CAS.
- Y. Ma, C. A. Navarro, T. J. Williams and S. R. Nutt, Polym. Degrad. Stab., 2020, 175, 109125 CrossRef CAS.
- J. Li, P. L. Xu, Y. K. Zhu, J. P. Ding, L. X. Xue and Y. Z. Wang, Green Chem., 2012, 14, 3260–3263 RSC.
- C. A. Navarro, Y. Ma, K. H. Michael, H. M. Breunig, S. R. Nutt and T. J. Williams, Green Chem., 2021, 23, 6356–6360 RSC.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- J. Cao, C. Zhou, G. Su, X. Zhang, T. Zhou, Z. Zhou and Y. Yang, Adv. Mater., 2019, 31, 1900042 CrossRef PubMed.
- X. Liu, G. Su, Q. Guo, C. Lu, T. Zhou, C. Zhou and X. Zhang, Adv. Funct. Mater., 2018, 28, 1–10 Search PubMed.
- A. C. Ferahian, D. K. Hohl, C. Weder and L. Montero de Espinosa, Macromol. Mater. Eng., 2019, 304, 1–10 CrossRef.
- Y. Wang, L. Li, Y. Ma, Y. Tang, Y. Zhao, Z. Li, W. Pu, B. Huang, X. Wen, X. Cao, J. Chen, W. Chen, Y. Zhou and J. Zhang, ACS Nano, 2020, 14, 8202–8219 CrossRef CAS PubMed.
- Z. Wu, C. Ji, X. Zhao, Y. Han, K. Müllen, K. Pan and M. Yin, J. Am. Chem. Soc., 2019, 141, 7385–7390 CrossRef CAS PubMed.
- J. Zhu, X. Lu, W. Zhang and X. Liu, Macromol. Rapid Commun., 2020, 41, 1–6 Search PubMed.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579–2591 CrossRef CAS PubMed.
- A. del Prado, D. K. Hohl, S. Balog, L. M. de Espinosa and C. Weder, ACS Appl. Polym. Mater., 2019, 1, 1399–1409 CrossRef CAS.
- Q. Zhang, C. Y. Shi, D. H. Qu, Y. T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2020, 142, 5371–5379 CrossRef PubMed.
- J. Yang, S. Chen, J. Luo, C. Persson, H. Cölfen, K. Welch and M. Strømme, ACS Appl. Mater. Interfaces, 2020, 12, 7403–7410 CrossRef CAS.
- X. Li, Y. Deng, J. Lai, G. Zhao and S. Dong, J. Am. Chem. Soc., 2020, 142, 5371–5379 CrossRef CAS PubMed.
- M. D. Davidson, E. Ban, A. C. M. Schoonen, M. H. Lee, M. D’Este, V. B. Shenoy and J. A. Burdick, Adv. Mater., 2019, 18, 1315–1320 Search PubMed.
- J. K. Román and J. J. Wilker, J. Am. Chem. Soc., 2019, 141, 1359–1365 CrossRef PubMed.
- S. Roh, A. H. Williams, R. S. Bang, S. D. Stoyanov and O. D. Velev, Nat. Mater., 2019, 18, 1315–1320 CrossRef CAS PubMed.
- J. Liu and O. A. Scherman, Adv. Funct. Mater., 2018, 28, 1–6 Search PubMed.
- C. Cui, C. Fan, Y. Wu, M. Xiao, T. Wu, D. Zhang, X. Chen, B. Liu, Z. Xu, B. Qu and W. Liu, Adv. Mater., 2019, 31, 1–9 Search PubMed.
- B. Jin, G. Zhang, J. Lian, Q. Zhang, X. Zhan and F. Chen, J. Mater. Chem. A, 2019, 7, 12266–12275 RSC.
- F. Pan, S. Ye, R. Wang, W. She, J. Liu, Z. Sun and W. Zhang, Mater. Horiz., 2020, 7, 2063–2070 RSC.
- A. Narayanan, J. R. Menefee, Q. Liu, A. Dhinojwala and A. Joy, ACS Nano, 2020, 14, 8359–8367 CrossRef CAS PubMed.
- Z. Wang, L. Guo, H. Xiao, H. Cong and S. Wang, Mater. Horiz., 2020, 7, 282–288 RSC.
- X. Liu, Q. Zhang, L. Duan and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 6644–6651 CrossRef CAS PubMed.
- J. Yu, B. Cheng and H. Ejima, J. Mater. Chem. B, 2020, 8, 6798–6801 RSC.
- X. Li, Z. Du, Z. Song, B. Li, L. Wu, Q. Liu, H. Zhang and W. Li, Adv. Funct. Mater., 2018, 28, 1–8 Search PubMed.
- Y. Mu, X. Wu, D. Pei, Z. Wu, C. Zhang, D. Zhou and X. Wan, ACS Biomater. Sci. Eng., 2017, 3, 3133–3140 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh00781a |
| This journal is © The Royal Society of Chemistry 2022 |