
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving sensing of formaldehyde using ZnO nanostructures with surface-adsorbed oxygen†
Sherif Abdulkader
Tawfik‡
 b,
Hang
Tran‡
b and
Michelle J. S.
Spencer
*ab
b,
Hang
Tran‡
b and
Michelle J. S.
Spencer
*ab
aARC Centre of Excellence in Low-Energy Electronics Technologies, School of Science, RMIT University, GPO Box 2476, Melbourne, Victoria 3001, Australia
bSchool of Science, RMIT University, GPO Box 2476, Melbourne, Victoria 3001, Australia. E-mail: michelle.spencer@rmit.edu.au
First published on 3rd December 2021
Abstract
Detection of pollutant gases, such as formaldehyde (HCHO), in our homes and surrounding environment is of high importance for our health and safety. The effect of surface defects and specifically pre-adsorbed oxygen on the gas sensing reaction of HCHO with ZnO nanostructures is largely unknown. Using density functional theory, nonequilibrium Green's function method and ab initio molecular dynamics (AIMD) simulations, we show that the presence of surface oxygen has two key roles in the sensitivity of ZnO towards HCHO: (1) it leads to the presence of charge trap states, which vanish upon the adsorption of HCHO, and (2) it facilitates the dissociative chemisorption of HCHO on the surface. Our ground state and AIMD calculations show that multiple reaction products are produced, which eventually lead to cleaning the surface from the adsorbed species, and hence enhancing the recyclability of the surface. We not only confirm the reaction proposed by experiment, but show that the presence of surface oxygen facilitates other surface reactions as well. Our work provides insights into the gas–surface reaction mechanism of ZnO-nanostructure based gas sensors, not provided before by experiment.
1. Introduction
Zinc oxide (ZnO) is one of the most widely used metal oxides in sensing devices to detect gaseous pollutants such as formaldehyde (HCHO), which is readily present around us. ZnO has traditionally been used as a thin film in sensing devices, which operates by measuring a change in conductivity due to the interaction between the gas and the surface of the sensor material. This change in conductivity can subsequently be used as an indication of the presence or absence of the gaseous pollutant. The development of ZnO and its nanostructures as gas sensors has become of great interest due to its cheap synthesis methods, response time, high structural and electrochemical stability, high mobility of conduction electrons, and non-toxicity.1–3 In particular, ZnO is able to form highly complex shapes such as nanonails, nanobelts, nanoribbons, nanorods, nanowires, nanotubes and flower-shaped nanostructures.4–8 The electronic properties, and hence the gas sensing properties, of ZnO are known to be strongly influenced by the surface morphology and the overall structure of the nanostructure, which means that ZnO offers a platform for experimentalists to fabricate highly tuneable sensing surfaces by varying the nanostructure growth parameters and the surface postprocessing.One of the most common, yet less examined, surface modifications of ZnO is the presence of oxygen species, including O, 2O and O2 on the ZnO surface. These oxygen species act as electron acceptors by capturing electrons from the conduction band of the ZnO.9,10 However, the impact of the surface oxygen on the chemistry of the ZnO surface as a sensor remains largely unclear.
It is known that the formation of the different pre-adsorbed, or ionosorbed, O species is dependent on temperature.9
| O2(ads) + e− → O−2(ads), (<373 K) | (1) |
| O2− + e− → 2O−(ads), (373−573 K) | (2) |
| O− + e− → O2−(ads), (>573 K) | (3) |
During gas sensing, the trapped electrons are returned to the conduction band upon adsorption of the gas of interest and/or by desorption of the adsorbed O, which subsequently decreases the sensor resistance. The following reaction has been proposed for the adsorption of HCHO at an adsorbed O site between 373 and 573 K:9–11
| HCHO + 2O−(ads) → CO2 + H2O + 2e− | (4) |
The release of electrons back to the conduction band causes a decrease in the sensor resistivity due to a decrease in thickness of the depletion layer, allowing the gas to be detected. In order to gain an insight into the reaction mechanisms, and how they depend on the surface morphology of ZnO, quantum mechanical simulations of the HCHO reaction with the adsorbed O species is required.
In this work, we combine density functional theory (DFT) with nonequilibrium Green's function (NEGF) to examine the effects of surface-adsorbed oxygen on the sensing of HCHO on ZnO. We focus on two ZnO morphologies: ZnO nanowires (NWs) and facetted-nanotubes (FNTs). The stable surface orientations, binding energies, vibrational frequencies, charge transfer and electronic structure of the HCHO-surface system are calculated. Ab initio molecular dynamics (AIMD) simulations are used to study the surface reaction at elevated temperatures on the nanostructures and to determine the dissociation species formed during the reaction as suggested by experiment. The findings provide crucial details that will help to better understand the gas sensing mechanism and experimental development of these materials for sensors.
2. Computational methodology
We perform DFT calculations using three different software packages: the Vienna Ab initio Simulation Package (VASP),12–14 the QUANTUM ESPRESSO code15 and the SIESTA code.16 In the VASP calculations, the plane wave pseudopotential approach was adopted with a cut-off energy of 400 eV. A generalised gradient approximation (GGA) with the Perdew, Burke and Ernzerhof (PBE)17 functional was used, and the PAW pseudopotentials as supplied by VASP were implemented. van der Waals forces were calculated using Grimme's D3 method.18 The VASP method is used to be consistent with our previous DFT work on gas adsorption on ZnO surfaces and nanostructures and so comparisons can be made for different gases. A 1 × 1 × 8 mesh was used to perform k-point sampling under the Monkhorst–Pack scheme19 for single-unit cell systems (as described in the next section), while a 1 × 1 × 2 mesh was used to perform k-point sampling for the 3-unit cells systems. In the QUANTUM ESPRESSO calculations, the behavior of the core electrons is approximated using the projector-augmented wave pseudopotentials.2 The valence electronic wave functions are expanded in a plane-wave basis set with a kinetic energy cut-off of 40 Ry, and the k-point sampling, the exchange correlation and the vdW correction are the same as that used in the VASP calculation. The SIESTA calculation is also performed within the PBE functional.20 While both VASP and QUANTUM ESPRESSO are plane-wave basis methods, the basis set in SIESTA comprises numerical atomic orbitals, and approximates the atomic potential in terms of Troullier–Martins21 norm-conserving pseudopotentials. The auxiliary basis uses a real-space mesh with a kinetic energy cut-off of 500 Ry, and the basis functions are radially confined using an energy shift of 0.005 Ry (see ref. 16 for details). In the relaxation calculations of VASP and SIESTA, we allow full atomic relaxation until the forces on the atoms are less than 0.01 eV A−1, while in QUANTUM ESPRESSO, the force tolerance is 5 × 10−3 Ry au−1.Calculations of the ZnO facetted-nanotube and nanowire structures were carried out using the supercell model with periodic boundary conditions (PBCs). The desired nanostructure morphologies were cleaved from a bulk wurtzite ZnO structure with lattice constants of a = 3.268 Å and c = 5.233 Å, and an internal parameter of u = 0.3826, as determined previously.22,23 The pre-adsorbed O (AO) system was created by adsorbing an O atom on the surface of the nanostructures; several sites were modelled and the most stable one was used for adsorption of HCHO.22Fig. 1(a and c) displays the stoichiometric facetted-nanotube and nanowire structures reported previously,22 respectively, and Fig. 1(b and d) displays the O pre-adsorbed ZnO-AO structures, FNTAO and NWAO, respectively.
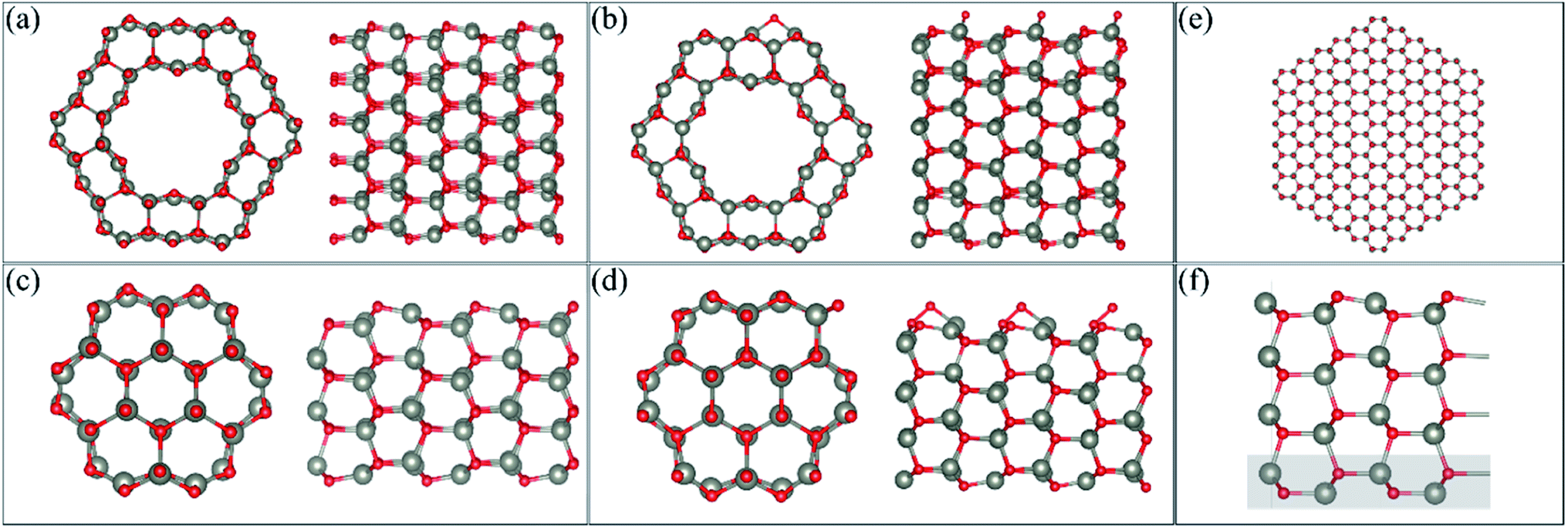 | ||
Fig. 1 Cross-section and side view of the (a) pristine ZnO facetted-nanotube (FNT), (b) ZnO facetted-nanotube with pre-adsorbed O per unit cell (FNTAO), (c) pristine ZnO nanowire (NW), (d) ZnO nanowire with pre-adsorbed O per unit cell (NWAO), and (e) the pristine ZnO nanowire model with a radius of ∼3.7 nm. (f) The slab model of the ZnO (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surface. The atoms shaded with a grey rectangle are frozen in the calculation. 0) surface. The atoms shaded with a grey rectangle are frozen in the calculation. | ||
The single unit-cell of the stoichiometric facetted-nanotube contains 48 Zn and 48 O atoms, and has a diameter of ∼16.63 Å (measured as d(Os–Os) or 16.03 Å from d(Zns–Zns)). The stoichiometric nanowire contains 24 Zn and 24 O atoms, and has a diameter of 9.82 Å (measured as d(Os–Os) or 9.31 Å from d(Zns–Zns)).
For the slab model, we construct the ZnO (100) surface based on a 4-layer thick slab that is 3 × 2 supercells wide, as shown in Fig. 1(f). The structure is then optimized using a 2 × 2 × 1 k-point sampling, where the positions of the atoms in the bottom layer (highlighted in a grey rectangle) are frozen while the rest of the atoms are free to move.
It is important to note that the modelled nanostructures are in fact smaller than the experimentally grown structures. The ZnO nanowire, for example, is modelled with a diameter of 9.88 Å (0.99 nm) but has been experimentally grown to as small as 6.5 nm.4 This decrease in size is necessary to reduce the computational cost of the DFT calculations, while still providing nanostructures with different morphologies as well as the structural details seen experimentally. In order to examine the stability of our FNT and NW structures, we compared the cohesive energy of the structures relative to that of the ZnO nanowire presented in Fig. 1(e), which has a diameter of ∼3.7 nm using the SIESTA code (432 atoms). The reason we used SIESTA for this particular calculation is that it is more computationally scalable than the other two codes, and therefore more efficient for systems with a large number of atoms. We note here that VASP, instead of SIESTA, was used for the remaining calculations because of its superior accuracy in calculating the gas adsorption energies, and because the D3 method is implemented and well-tested in VASP.
As the use of PBCs was employed, vacuum spacers of at least 12 Å for the facetted-nanotube and 15 Å for the nanowire were inserted in the x- and y-directions to ensure the atoms did not interact with each other in adjacent cells. Cells were replicated in the z-direction to emulate the length of the nanostructure. For the ZnO(100) slab, a vacuum of 15 Å was added in the z-direction.
Using VASP, the HCHO molecule was optimised in a 15 × 15 × 15 Å sized cell, with optimised O–C and C–H bond lengths calculated of 1.22 Å and 1.12 Å, respectively. The optimised bond angles were 122° (∠OCH) and 116° (∠HCH). The HCOOH molecule was also optimised using similar settings because it was one of the reaction products. The calculated O–C bond lengths are 1.36 Å and 1.21 Å, while the C–H bond length is 1.11 Å, the O–H bond length is 0.98 Å, which all agree well with the experimental values (within 2%).24
Multiple initial orientations of the gas were modelled on each nanostructure by adsorbing one HCHO molecule on the facetted-nanotube or nanowire at an initial distance of ∼2.5–2.7 Å away from the surface.
Vibrational frequency calculations were performed by diagonalising a finite difference construction of the Hessian matrix with displacements of 0.015 Å, allowing only the adsorbate molecules to relax while atoms in the ZnO nanostructure were fixed. All real vibrational frequencies obtained from these calculations enabled confirmation that the adsorbate was indeed at a minimum energy position on the potential energy surface, and hence a stable structure.
The binding energy (BE) of the HCHO adsorbed on the surface of the nanostructure was calculated using the following formula,
| BE = [Eads/ZnO-AO − (Eads + EZnO-AO)] | (5) |
Bader partial charges on individual atoms were calculated using the method described by Henkelman et al.25 These partial charges were used to determine the charge transfer (Δq), and thus whether the adsorbate behaves as a charge donor or acceptor. FFT grid accuracies of (1×), (2×) and (3×) were tested to determine the accuracy required to achieve converged results. The partial charges were found to be converged to ∼0.01e using the more dense FFT grid (3×). The charge transfer was then calculated for each system using the following formula,
| Δq = Σqads | (6) |
In order to obtain the pathway for the HCHO reaction taking place on the ZnO-AO FNT surface, a constrained optimization calculation was performed using the Quantum Espresso code.15 The constrained optimization approach, in which a bond representing the reaction coordinate is held fixed during the relaxation calculation, while the rest of the atoms are free to move, can yield an estimation for the energy barrier which is reasonably accurate,26 compared to the more rigorous methods such as the nudged elastic band method.27
The transport calculations are performed using the nonequilibrium Green's function (NEGF) method as implemented in the TRANSIESTA code.28 A double-ζ basis set size is used for all of the atoms in the system. The current I through the scattering region, as a function of the bias voltage Vbias across the device can be estimated using the Landauer–Buttiker formula,29
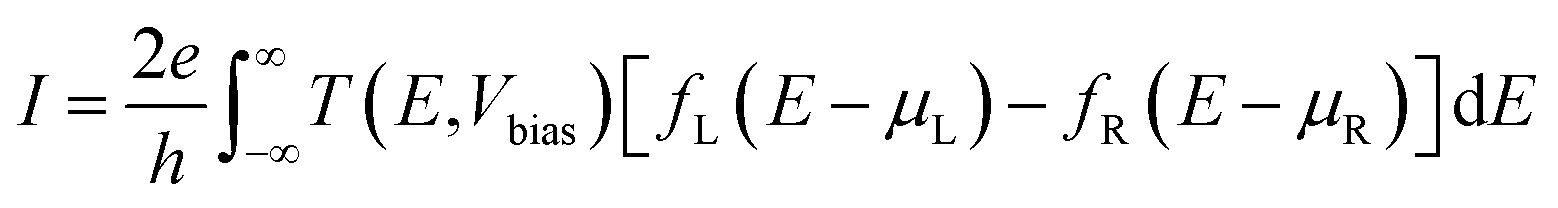 | (7) |
| T(E,Vbias) = Tr[t†t] = Tr[ΓLGΓRG†] | (8) |
AIMD calculations were performed using the VASP code. The Verlet algorithm was employed to integrate the equations of motion, with the temperature being controlled by the Nosé thermostat.30 Simulations were carried out for HCHO reacting with the surface of the facetted-nanotube and nanowire in a nonequilibrium orientation (with HCHO initially positioned ∼2.5–2.7 Å from the surface, i.e. not adsorbed), as well as for the most stable optimised structures at 300, 520 and/or 700 K. These three temperatures were chosen to investigate the surface reaction at approximately room temperature and at the optimal operating temperatures noted for detection of HCHO with ZnO nanosensors in experimental works (see ref. 31 and references therein). A time step of 0.5 fs was used and all atoms were allowed to relax.
3 Results and discussion
3.1 ZnO nanostructures with surface-adsorbed oxygen (ZnO-AO)
The cohesive energy of the stoichiometric ZnO FNT and NW structures and the larger NW structure (displayed in Fig. 1(a, c and e) respectively), per atom were calculated to be −2.75 eV per atom, −2.74 eV per atom, and −2.96 eV per atom, respectively. These values are all very close to the value reported in the literature for bulk ZnO, which is 3.250 Å.32 Therefore, all structures examined in the present work are thermodynamically stable and could potentially be synthesised.The preferred oxygen binding site on the facetted nanotube and nanowire is shown in Fig. 1(b and d), respectively. The adsorption energy of O on the facetted nanotube (FNTAO) and nanowire (NWAO) are −2.02 eV and −2.25 eV, respectively. For FNTAO, the O atom covalently binds to two Zn atoms, forming a bridge defect site with two Zn–O bond lengths of 1.91 Å and 1.98 Å. For NWAO, the O atom binds differently, forming a bridge configuration between a Zn and an O atom along the c-axis of the stoichiometric crystal. The O–O and Zn–O bond lengths are 1.50 Å and 1.89 Å, respectively. The adsorption energy values and bond lengths indicate the O atom is chemisorbed to the surface forming covalent bonds.
3.2 HCHO/ZnO-AO facetted-nanotube
Six unique structures were found for HCHO adsorbed on the facetted-nanotube surface containing pre-adsorbed O (FNTAO) (see Fig. 2 and Table 1). HCHO spontaneously adsorbs dissociatively onto the surface in structures FNTAO-1, FNTAO-2, FNTAO-3, FNTAO-4 and FNTAO-5, to produce adsorbed CHO and H species, according to the following equations, where O* represents the pre-adsorbed O atom and Os a surface O atom: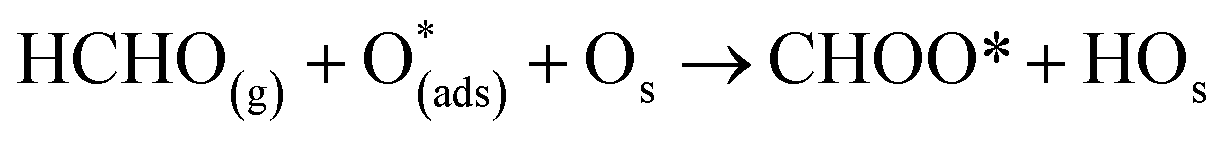 | (9a) |
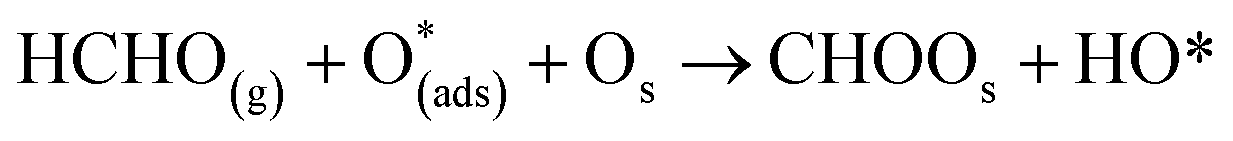 | (9b) |
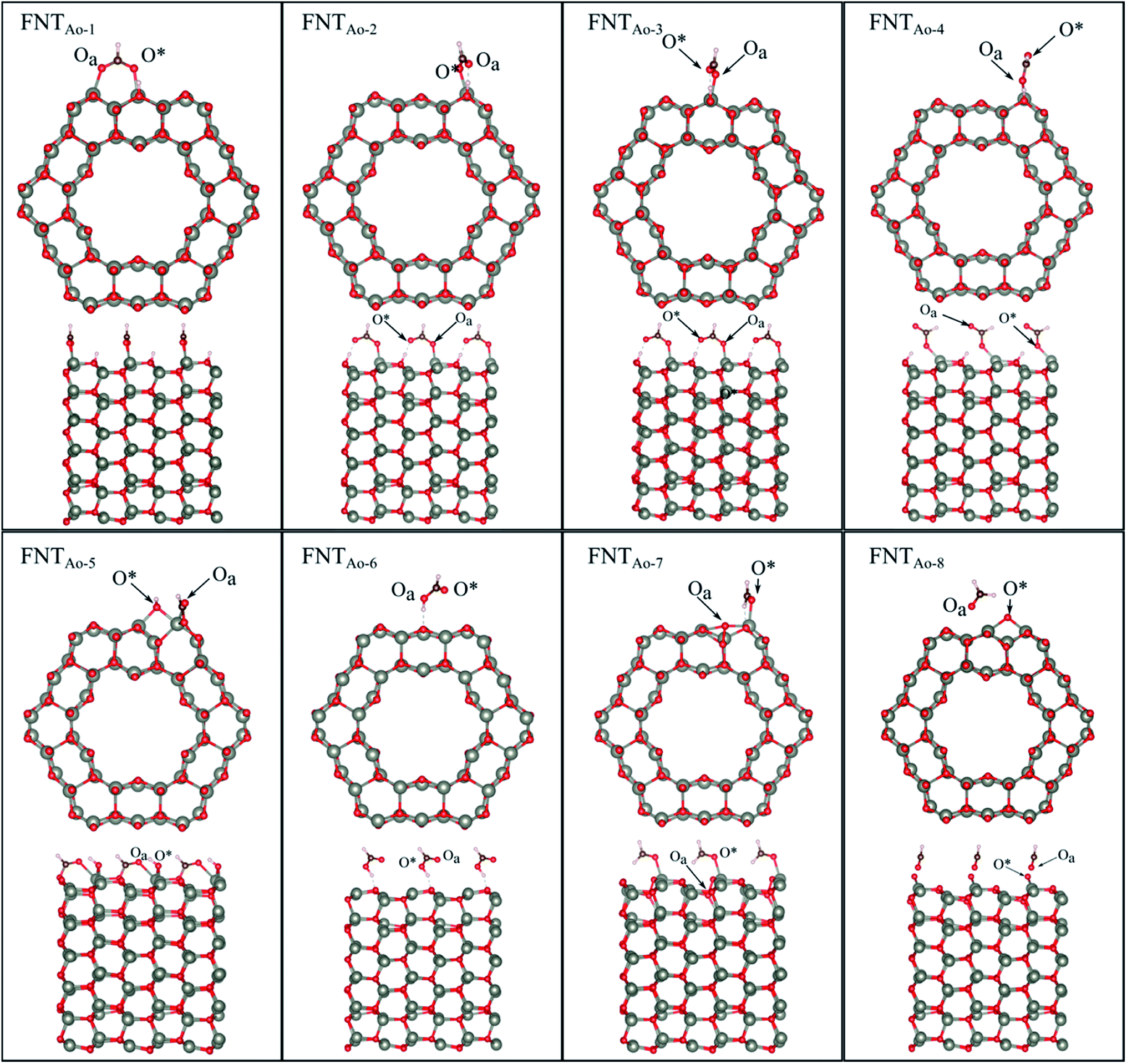 | ||
| Fig. 2 Optimised structures of HCHO adsorbed on the ZnO facetted-nanotube (FNT) with a pre-adsorbed O (FNTAO), showing the cross-section (top) and side (bottom right) views of the nanostructure. | ||
| Structure | BE (eV) | d(Oa–Zns) (Å) | d(O*–Zns) (Å) | d(C–Oa) (Å) | ∠COaZn (°) | Δq (e) |
|---|---|---|---|---|---|---|
a Parameters: binding energy (BE); distance between the O of HCHO and Zn (d(Oa–Zn)); distance between pre-adsorbed O and Zns (d(O*–Zn)); distance between Ca and Oa of HCHO (d(Oa–C)); bond angle between OC and Zn (∠COaZn); antisymmetric C–H stretch (v(C–H)); symmetric C–H stretch (v(C–H)); C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch (v(C–H)); charge transfer (Δq); band gap (Eg). *This adsorption energy was calculated for a system where the HCOOH is adsorbed on the pristine ZnO FNT surface. O stretch (v(C–H)); charge transfer (Δq); band gap (Eg). *This adsorption energy was calculated for a system where the HCOOH is adsorbed on the pristine ZnO FNT surface.
|
||||||
| HCHO | — | — | — | 1.21 | — | — |
| HCOOH | — | — | — | 1.21 | — | — |
| FNTO | — | — | 1.91, 1.98 | — | — | — |
| FN AO-1 | −6.16 | 2.51 | 2.12 | 1.27 | 95 | 1.10 |
| FNTAO-2 | −5.85 | — | 1.94 | 1.25 | 125 | 1.15 |
| FNTAO-3 | −5.84 | — | 1.95 | 1.25 | — | 1.14 |
| FNTAO-4 | −5.42 | — | 1.96 | 1.22 | — | 1.13 |
| FNTAO-5 | −4.96 | 2.32 | 2.02 | 1.26 | 119 | 1.09 |
| FNTAO-6 | −4.85 (−0.62*) | 2.9 | — | 1.34 | — | 1.12 (−0.06*) |
| FNTAO-7 | −1.15 | 2.15 | 2.02 | 1.23 | — | 0.04 |
| FNTAO-8 | −0.51 | 2.42 | 1.98 | 1.22 | 112 | 0.04 |
In structures FNTAO-1, FNTAO-2, FNTAO-3, FNTAO-4, the H atom is bonded to Os (eqn (9a)), whereas in FNTAO-5 it is bonded to O* (eqn (9b)). For structures FNTAO-2 and FNTAO-3 the surface species are stabilised by the formation of a H-bond between the adsorbed H atom and the surface O or O* atom.
A similar dissociation reaction was also shown by Xu et al.33 for adsorption of H2O on the stoichiometric ZnO nanowire surface (where adsorbed OH and H species were formed). While large structural changes of the adsorbate molecule are clearly observed for this reaction, the C–Oa bond length and O–C–H bond angle are similar to those in the gas phase HCHO molecule. The O*–Zn bond lengths only show a small increase of up to ∼0.11 Å compared to the unadsorbed nanostructure, showing little structural change. The difference between structures FNTAO1-4 are due to the relative orientation of the dissociated species on the surface. For structure FNTAO-5, HCHO forms two bonds to the surface of the facetted-nanotube, namely an Oa–Zn and a C–Os bond (where Os is a surface O atom which is different from the pre-adsorbed O atom). This is similar to the doubly bonded structure found on the stoichiometric facetted-nanotube34 and by Li et al.35 for HCHO adsorbed on a TiO2(101) surface doped with platinum (Pt), where the HCHO adsorbed in a bridge-type site on the surface but did not bond with the Pt. The calculated bond lengths and angles of this structure are similar to the values calculated for the doubly coordinated structures found in our stoichiometric study.34 This is as expected due to their similar structural deformation required to form the ‘bridge’-type site bonding: (Zn–Oa)–(C–Os). The bond lengths of Oa–Zn and C–Os were calculated to be 2.32 Å and 1.26 Å respectively. Hence, the gas will still be detected if it collides with the surface in different orientations.
For structure FNTAO-6, a rearrangement occurs during the surface reaction, such that the HCHO removes the pre-adsorbed O atom to form a formic acid (HCOOH) molecule that is adsorbed associatively to the surface through hydrogen bonding between one of the H atoms and a surface O atom. This illustrates the ability of the reducing gas to essentially ‘clean’ the surface of the facetted-nanotube and restore the stoichiometric surface, which can be expressed as:
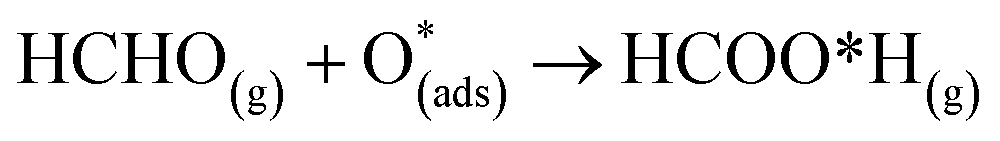 | (10) |
For structure FNTAO-7, the HCHO dissociates into a CH2 and an O species; the CH2 binds to the pre-adsorbed O atom and the O to two surface Zn atoms according to the following reaction:
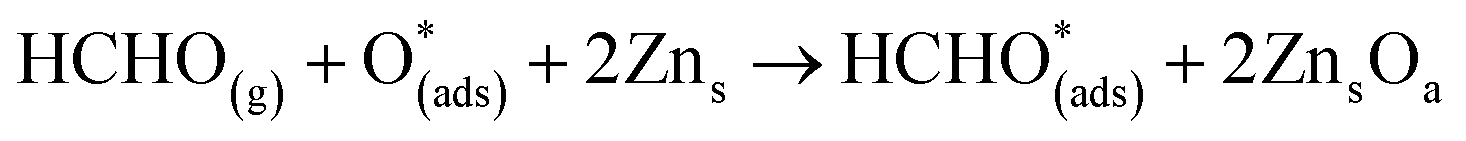 | (11) |
For FNTAO-8, the HCHO molecule weakly physisorbs associatively to the surface forming a H–O* bond of 2.22 Å and a Zn–Oa bond of 2.42 Å as follows:
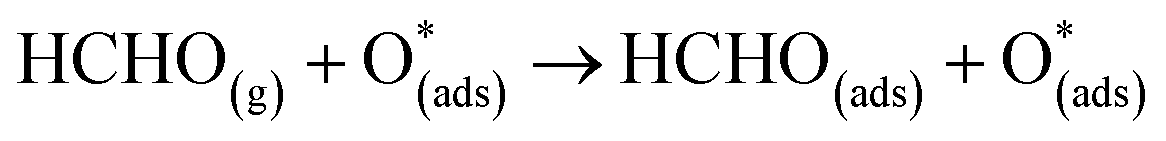
For structures FNTAO-2, FNTAO-3 and FNTAO-7, the surface species are stabilised by the formation of a H-bond between the adsorbed H atom and the surface O or O* atom.
Vibrational frequency values for the HCHO/FNTAO structures are presented in Table 2. The C–H stretch occurs at ∼2900 cm−1 for all structures which corresponds to the typical aldehyde C–H stretch (2830–2695 cm−1), which can be explained by the adsorbed CHO group. The small difference is due to the aldehyde group being adsorbed on the nanostructure surface which shifts the C–H stretch to a higher value. These shifts mean that it should be possible to detect the presence of dissociated HCHO on the surface experimentally using Fourier transform infrared spectroscopy (FTIR), high resolution electron energy loss spectroscopy (HREELS) or reflection absorption infra-red spectroscopy (RAIRS), for example.
| Structure | BE (eV) | d(Oa–Zns) (Å) | d(O*–Zns) (Å) | d(O*–Os) (Å) | d(Ca–Oa) (Å) | ∠CaOaZns (°) | Δq (e) |
|---|---|---|---|---|---|---|---|
|
a Parameters: binding energy (BE); distance between the O atom of HCHO and a surface Zn atom (d(Oa–Zns)); distance between the pre-adsorbed O atom and surface Zn atom (d(O*–Zns)); distance between the pre-adsorbed O atom and a surface O atom (d(O*–Os)); distance between the C and O atoms of HCHO (d(Oa–Ca)); bond angle between the O, Ca and Zns atoms (∠CaOaZns); antisymmetric C–H stretch (v(C–H)); symmetric C–H stretch (v(C–H)); CO stretch (v(C–H)); charge transfer (Δq); band gap (Eg). *This adsorption energy was calculated for a system where the HCOOH is adsorbed to the pristine ZnO NW surface.
|
|||||||
| HCHO | — | — | — | — | 1.22 | — | — |
| HCOOH | — | — | — | — | 1.21 | — | — |
| ZnO NWO | — | — | 1.89 | 1.50 | — | — | — |
| NWAO-1 | −5.57 (−1.56*) | 1.95 | — | — | 1.28 | 125 | 1.14 |
| NWAO-2 | −4.71 | 2.03 | 2.08 | — | 1.26 | 127 | 1.15 |
| NWAO-3 | −1.58 | 1.91 | — | 1.56 | 1.36 | 123 | −0.04 |
| NWAO-4 | −1.49 | 2.16 | — | 1.51 | 1.39 | — | −0.05 |
| NWAO-5 | −1.40 | 1.92 | 1.91 | 1.51 | 1.34 | 125 | −0.17 |
| NWAO-6 | −1.36 | 1.92 | 2.04 | 1.5 | 1.32 | 111 | −0.14 |
| NWAO-7 | −0.94 | 1.97 | 2.18 | 1.51 | 1.35 | 99 | −0.1 |
| NWAO-8 | −0.43 | — | — | 1.51 | 1.22 | — | −0.04 |
The strong BE values calculated for structures FNTAO-1, FNTAO-2, FNTAO-3, FNTAO-4, FNTAO-5 and FNTAO-6 (−6.16 eV, −5.85 eV, −5.84 eV, −5.42 eV, −4.96 eV, and −4.85 eV, respectively), suggests that if HCHCO reacted on the surface to produce adsorbed CHO and OH, these species would likely to remain adsorbed on the surface, blocking new adsorption sites for other HCHO molecules to react, and thus be detected. The reason for the high BE values here is the presence of the pre-adsorbed O atom, which plays an essential part in the dissociative chemisorption. This was in contrast to the pristine FNT surface, which did not cause dissociation.36
The binding energies for FNTAO-7 and FNTAO-8 are −1.15 eV and −0.51 eV, respectively, indicating that HCHO weakly chemisorbs to the surface, and is therefore more likely to desorb and restore the surface for detection of further gas molecules.
For the FNTAO-1, FNTAO-2, FNTAO-3, FNTAO-4, FNTAO-5 and FNTAO-6 structures, HCHO acts as a charge donor (see Table 2) because of the dissociation of H and its subsequent bonding to the surface. Further, the presence of adsorbed oxygen causes the band gap to widen in FNTAO-1 (Fig. 3) after HCHO adsorption to 1.51 eV, compared to 1.43 eV (for the stoichiometric FNT) and 1.44 eV for the FNT with adsorbed oxygen (FNTAO). The band structure exhibits unoccupied spin-polarized localized defect states within the band gap region that are induced by the adsorption of the O atom, at energies of approx. +0.2 eV and +0.5 eV. After adsorption of HCHO, these states are quenched by the adsorbate, as a result of electrons being transferred from HCHO to fill these unoccupied states, resulting in what would be measured as a decrease in resistance experimentally.
 | ||
| Fig. 3 The spin-polarized band structure of (a) ZnO FNTAO and (b) ZnO FNTAO-1, and the atom-resolved partial density of states (PDOS) of (c) ZnO FNTAO and (d) ZnO FNTAO-1. | ||
We note that while the BE of the FNTAO-6 configuration is large (−4.85 eV), this configuration is equivalent to physisorption of a formic acid molecule, HCOOH, on the pristine FNT surface, were the BE is calculated to be −0.62 eV. This means that the HCOOH will only physisorb to the surface of the pristine FNT and will not readily form a covalent interaction. This result also implies that the HCHO gas might “extract” the pre-adsorbed O atom from the surface of the nanostructure, forming the HCOOH/FNT system, therefore allowing another HCHO molecule to be adsorbed and detected. We examine the feasibility of this occurring by applying a constrained optimization calculation. Here, we constrain the distance between the Ca and the O* atoms highlighted as the black arrow in the inset of Fig. 4. The figure displays the reaction energy barrier as the Ca–O* bond distance is varied, from the initial state (FNTAO-8) to the final state configuration (FNTAO-6), including the transition states. The initial state, FNTAO-8, is 4.34 eV higher in energy than the final state, FNTAO-6. Two steps are identified during this reaction: first, the FNTAO-8 structure undergoes a structural change in which one of the C–H bonds is broken, forming an adsorbed CHO and H species on the Zn–O* site (as per the reaction in eqn (3)). Secondly, the system either converts to structure FNTAO-1, which will release an energy of 2.3 eV, or it will require 2.88 eV to form structure (FNTAO-6) where the CHOOH molecule is formed. This second step has a barrier of 2.88 eV, indicating that removal of the pre-adsorbed O atom is high.
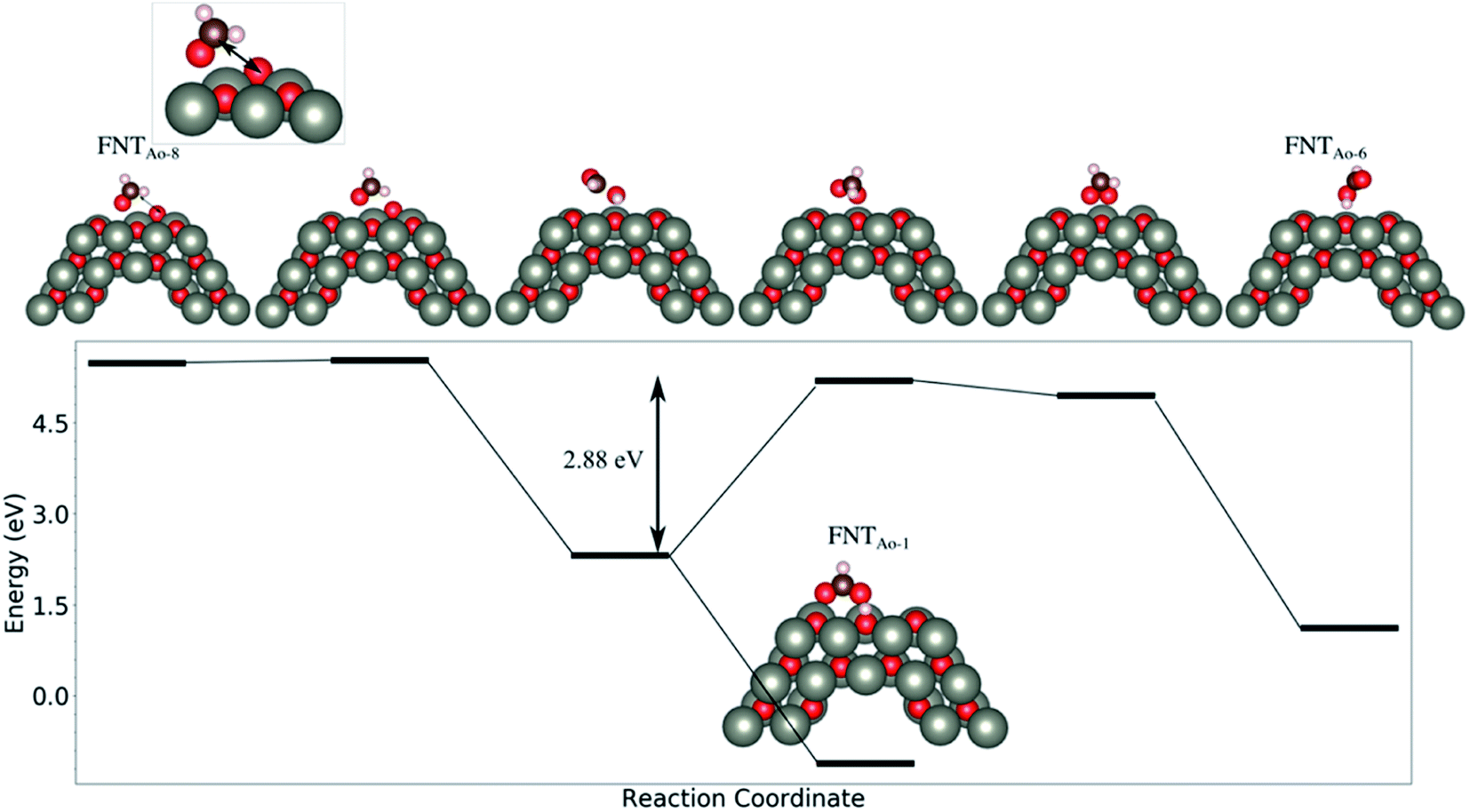 | ||
| Fig. 4 The evolution of the total energy as a function of the reaction coordinate, from the initial state to the final state. The reaction coordinate is proportional to the Ca–O* distance, highlighted with an arrow. | ||
3.3 HCHO/ZnO-AO nanowire
HCHO was found to adsorb in 6 different orientations on the nanowire containing a pre-adsorbed O atom (NWAO) (see Fig. 5 and Table 2). The most stable orientation, NWAO-1, is similar to structure FNTAO-2 where the HCHO dissociates, forming CHO which adsorbs to the pre-adsorbed O atom via the C atom, and H which adsorbs to an adjacent surface O atom. The structure is stabilised by the formation of a H-bond between the adsorbed H atom and the O atom of the adsorbed CHO species. This reaction can be described by eqn (9a).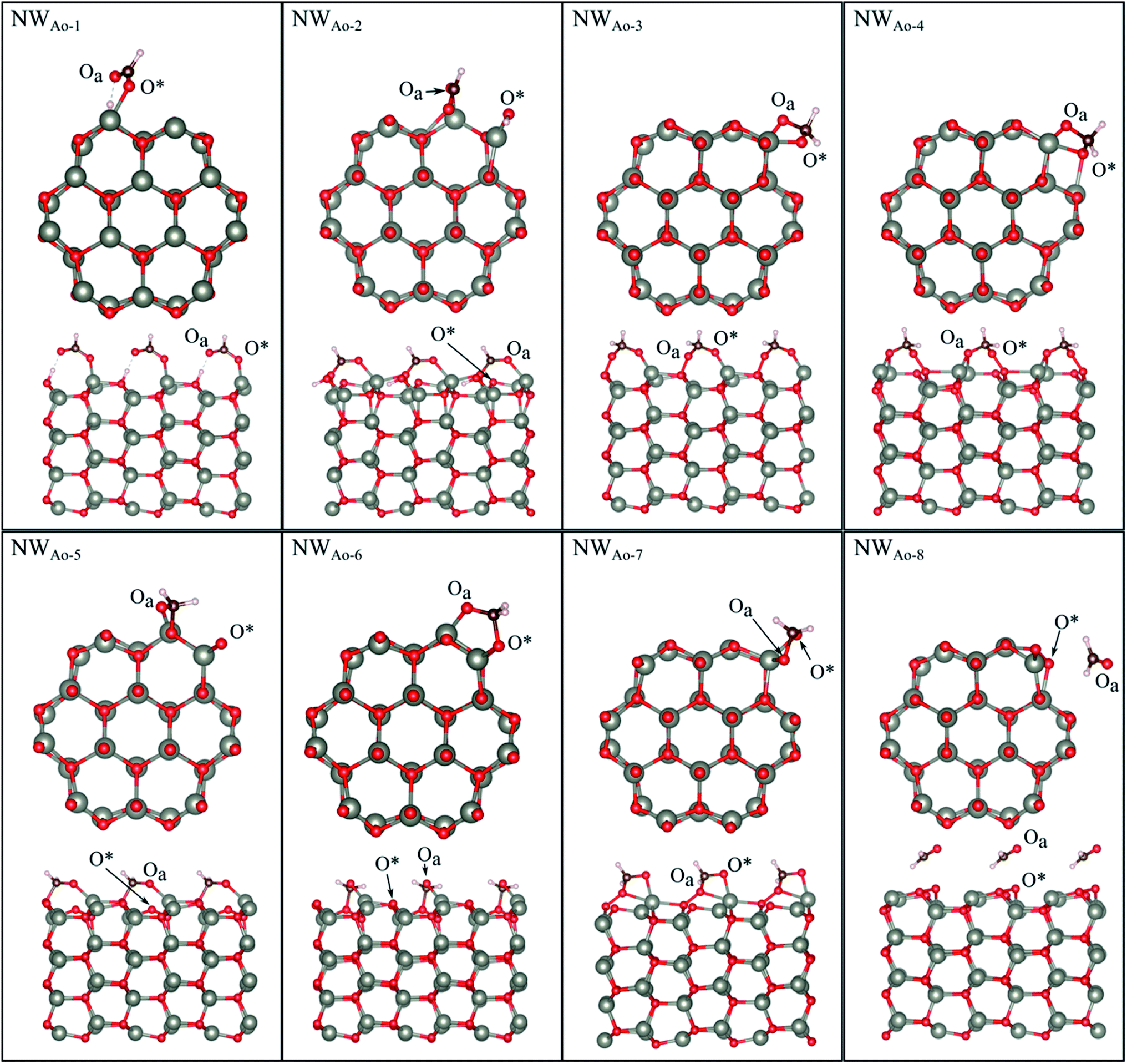 | ||
| Fig. 5 Optimised structures of HCHO adsorbed on the ZnO nanowire with a pre-adsorbed O (NWAO), showing the cross-section (top) and side (bottom right) views of the structures. | ||
For NWAO-2, HCHO also chemisorbs dissociatively like NWAO-1, however, the H atom adsorbs to the pre-adsorbed O atom and the CHO binds to the pre-adsorbed O atom and a surface Zn atom. This orientation is similar to FNTAO-5 and hence has a similar BE of −4.71 eV.
For structures NWAO-3, NWAO-4, NWAO-5, NWAO-6, and NWAO-7, HCHO adsorbs associatively, forming two covalent bonds to the surface. For all structures, one of these bonds is formed between the Oa atom and a surface Zn atom, and the other between the C and the O* atom. The second bond forms in all structures except for NWAO-5. The surface reaction of these structures proceeds viaeqn (5).
The BE values of these configurations are −1.58 eV, −1.49 eV, −1.36 eV and −0.94 eV, which all indicate strong binding. The difference in BE values is due to the orientation of the dissociated species on the NW and indicates that HCHO is stable in multiple sites on the nanostructure surface, making it more readily detected.
Structure NWAO-8 is the weakest of all the structures because HCHO is only physisorbed on the surface, similar to structure FNTAO-8, where HCHO also physisorbed on the surface.
To check whether CHOOH will adsorb to the pristine NW, as is the case for the FNT (structure FNTAO-6 in Fig. 2), a geometry optimization calculation was performed for several initial configurations of CHOOH/NW. Interestingly, unlike on the FNT, it prefers to bind covalently to the NW surface, forming a Zn–Oa bond (NWAO-1 in Fig. 5). As a result, the BE of this structure is −1.56 eV, which is greater than that for CHOOH/FNT indicating it could be a stable structure and formed during the gas sensing reaction.
For NWAO-1 and NWAO-2 the calculated charge transfer is positive (Table 2), while it is negative for the other structures, indicating that HCHO acts as a charge donor in the dissociatively adsorbed configurations (NWAO-1, NWAO-2), but as a charge acceptor when doubly coordinated to the surface (NWAO-3, NWAO-4, NWAO-5, and NWAO-6). As we show that multiple species may be present on the surface at once, the overall charge transfer is likely to still be positive, in agreement with experiment, however, our work shows that the surface chemistry is more complex than is suggested by the experimental evidence.
The band structure and the atom-resolved partial density of states (PDOS) of the most stable structure (NWAO-1), together with the NWAO structure are displayed in Fig. 6. The presence of the surface O atom causes a small widening of the band gap compared to the stoichiometric NW (1.49 compared to 1.47 eV, respectively). Subsequent adsorption of HCHO causes a slight widening of the band gap by 0.02 eV. The band structure (Fig. 6(a)) exhibits occupied localized defect states within the band gap that are induced by the adsorption of the O atom, as seen at energies ∼−0.4 eV and ∼−0.7 eV. These states are spin-unpolarized, which is different from defect states in the FNTAO structure (Fig. 3(a)), where the adsorption of O on the pristine surface induces spin-polarized unoccupied localized defects. These defect states can give rise to charge traps which are likely to result in an increase in resistance of ZnO to photo-generated electric current (photocurrent). The disappearance of these charge trap states suggests a mechanism for the sensing of the HCHO species on O pre-adsorbed FNT and NW surfaces whereby adsorption of HCHO leads to an increase in the photocurrent due to the vanishing charge trap states in the ZnO structure.
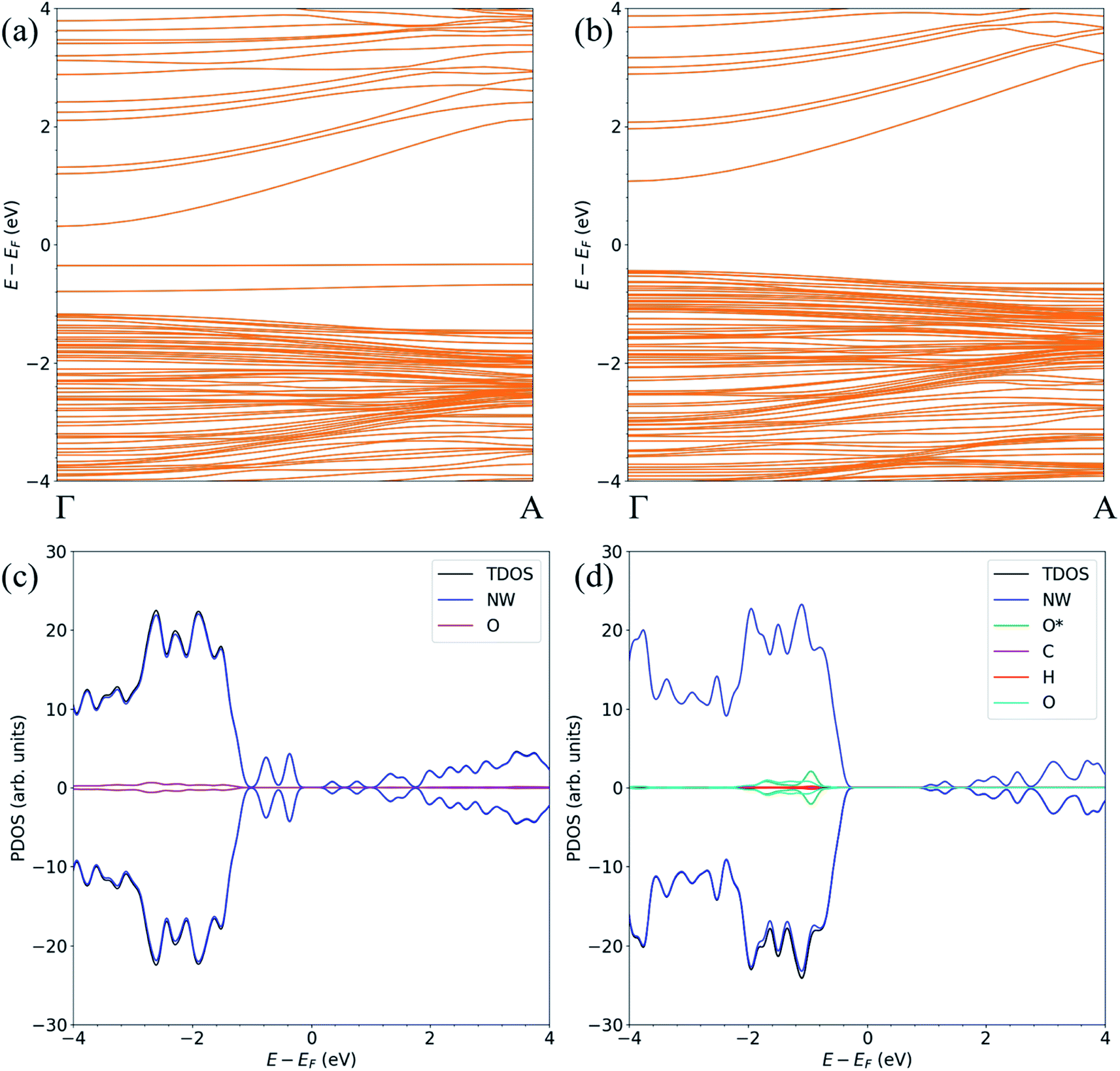 | ||
| Fig. 6 The spin-polarized band structure (upper) and atom-resolved partial density of states (PDOS) (lower) of the ZnO (a, c) NWAO and (b, d) NWAO-1 structures. | ||
3.4 Electronic transport characteristics
To determine the behaviour of the NWAO structure as a resistive sensor, the electronic transport of these systems was calculated using structures NWAO and NWAO-1 (see Fig. 7). These systems are constructed by creating 7 replicas of the NW unit cell, where the middle (fourth) unit cell is either the NWAO or the NWAO-1 structure, as displayed in Fig. 7(a) and (b), respectively. These two structures were re-optimized using SIESTA and using the NW c lattice parameter determined using the same code (5.505 Å).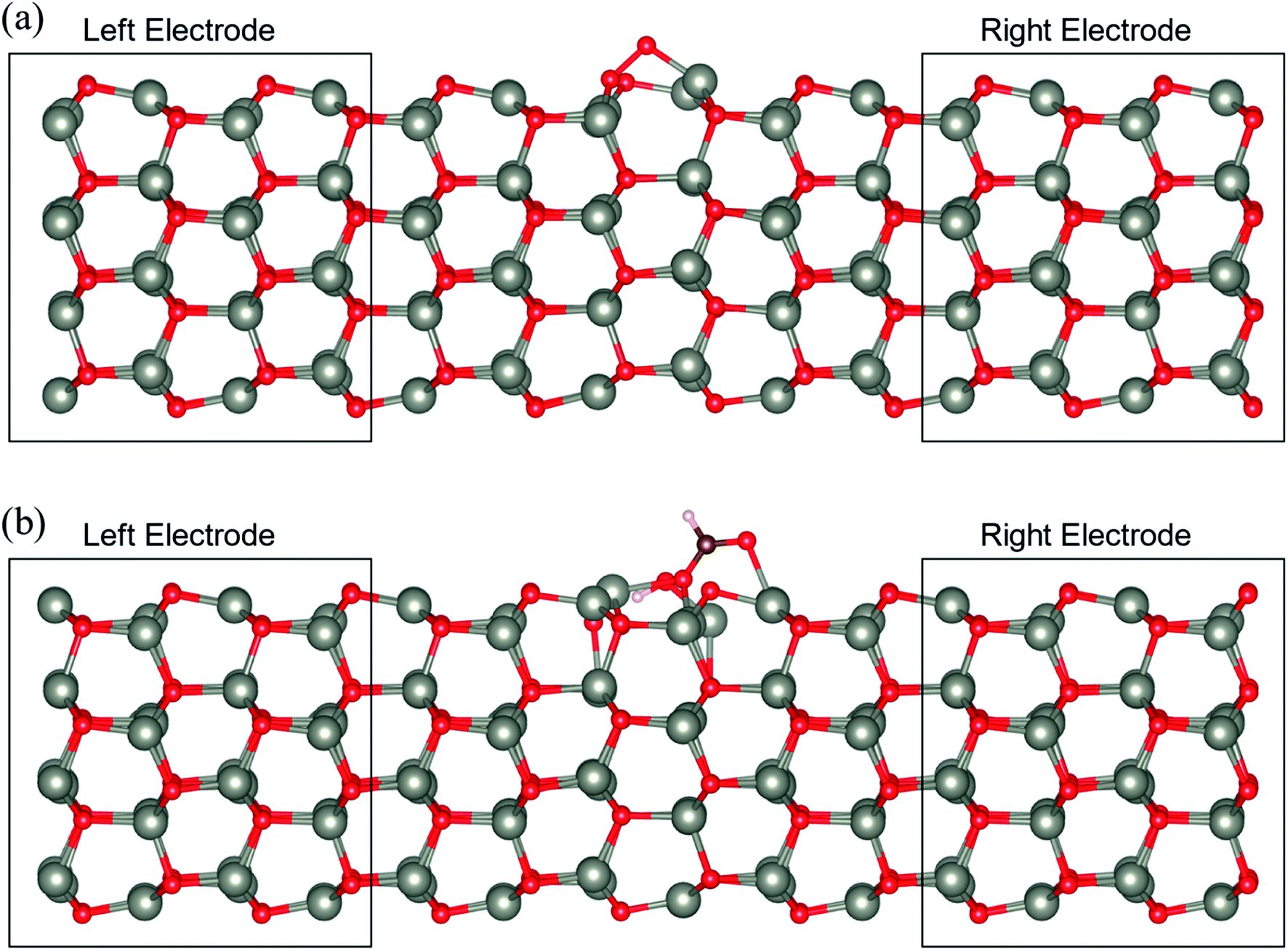 | ||
| Fig. 7 The atomic structure of the ZnO (a) NWAO and (b) NWAO-1 systems. These transport systems are composed of 7 replicas of the NW unit cell, in which the centre is the NWAO and NWAO-1 structures, respectively. | ||
The calculated current−voltage characteristics (IVC) of the complex is shown in Fig. 8. The current is negligible for bias voltages in the range −0.5 V to 0.5 V due to the presence of a band gap in both structures. The magnitude of the current starts increasing beyond this voltage range for both structures. The current in the NWAO system is higher than in the NWAO-1 system, due to the slight widening of the band gap of NWAO-1 upon the adsorption of HCHO. The asymmetry in the electric current at ∼−0.8 V is due to the asymmetry of the NWAO-1 structure (cf.Fig. 7). Even though the magnitude of the calculated electric current after HCHO adsorption is very small (less than 1 nA), it indicates that the system will significantly respond to the adsorption of HCHO and is consistent with experiments.
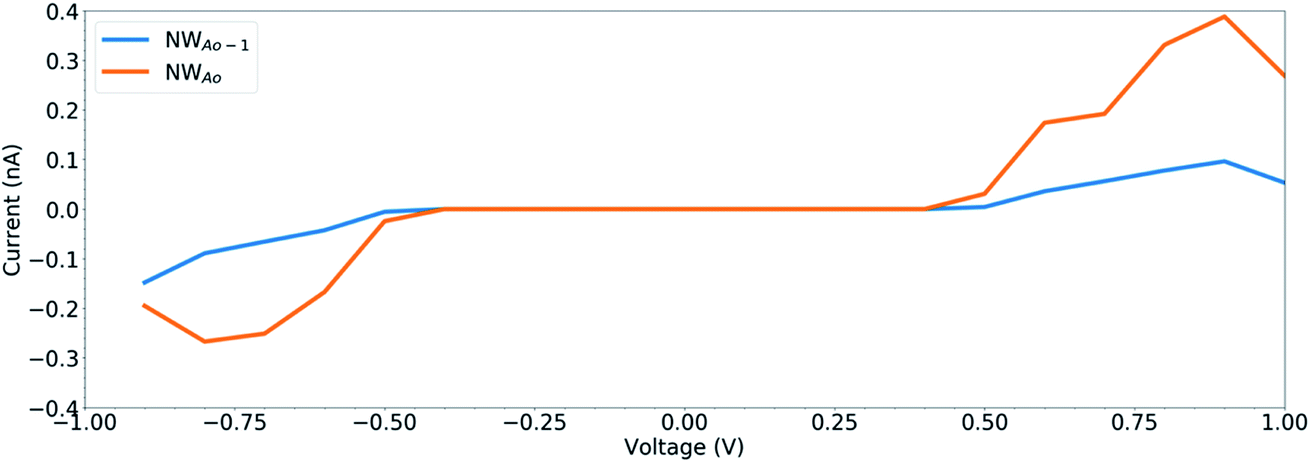 | ||
| Fig. 8 The current–voltage characteristics (IVC) of the structures displayed in Fig. 7. | ||
3.5 Effect of temperature
The calculated binding energies for the HCHO molecule on the various ZnO surface configurations show that the surfaces considered in this work have a high sensing response to HCHO, but are possibly not repeatable due to the strong chemisorption, an undesirable attribute in a sensor that reduces senor recyclability. However, here we show using AIMD simulations the important role of temperature in the recovery of the sensor surface at within very small recovery times (<1 picosecond).AIMD simulations were performed starting with HCO adsorbed as in structures FNTAO-3 and NWAO-2. The evolution of the total energies of the systems are displayed in Fig. S1† showing the thermal equilibration of the simulations. During the AIMD simulation, the adsorbed species in structure FNTAO-3 further dissociates on the surface at the three simulation temperatures examined (300, 520 and 700 K), as illustrated in Fig. 9–11, while we found that the adsorbed species on NWAO-3 undergo significant structural changes at 700 K, as illustrated in Fig. 12.
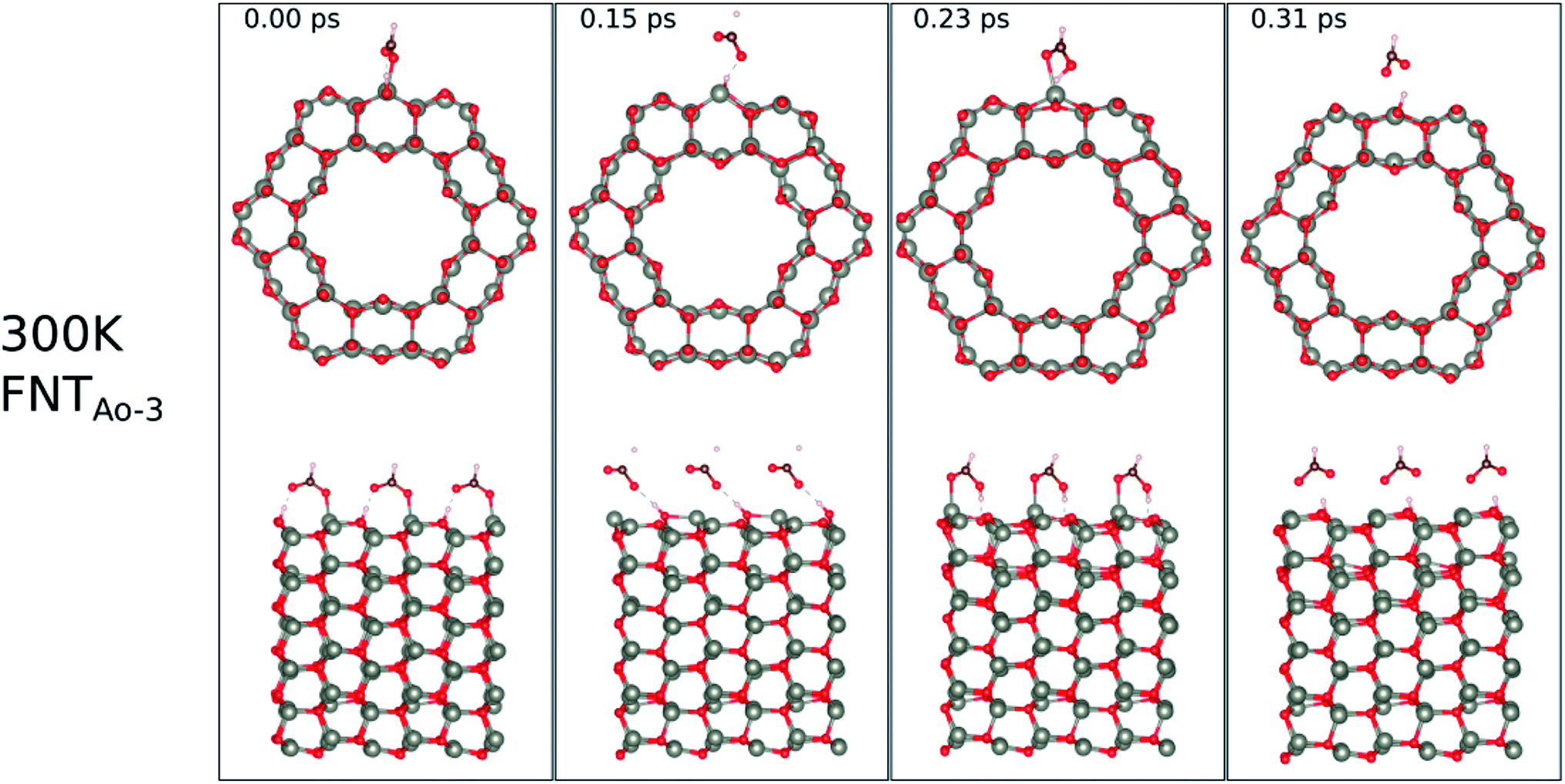 | ||
| Fig. 9 Evolution of the chemical reactions taking place on the surface of FNTAO-3 as function of time, by applying ab initio molecular dynamics at 300 K. | ||
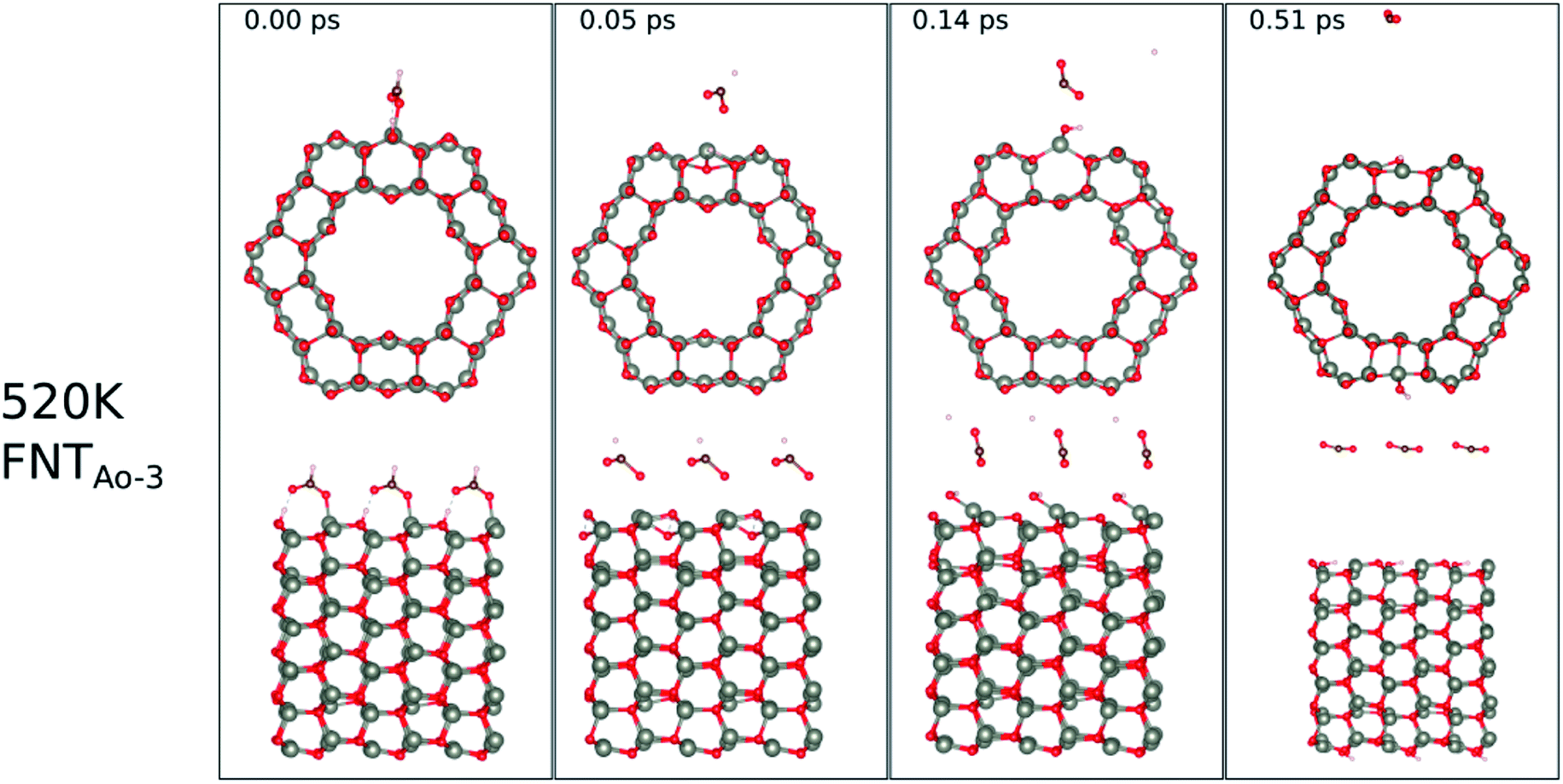 | ||
| Fig. 10 Evolution of the chemical reactions taking place on the surface of FNTAO-3 as function of time, by applying ab initio molecular dynamics at 520 K. | ||
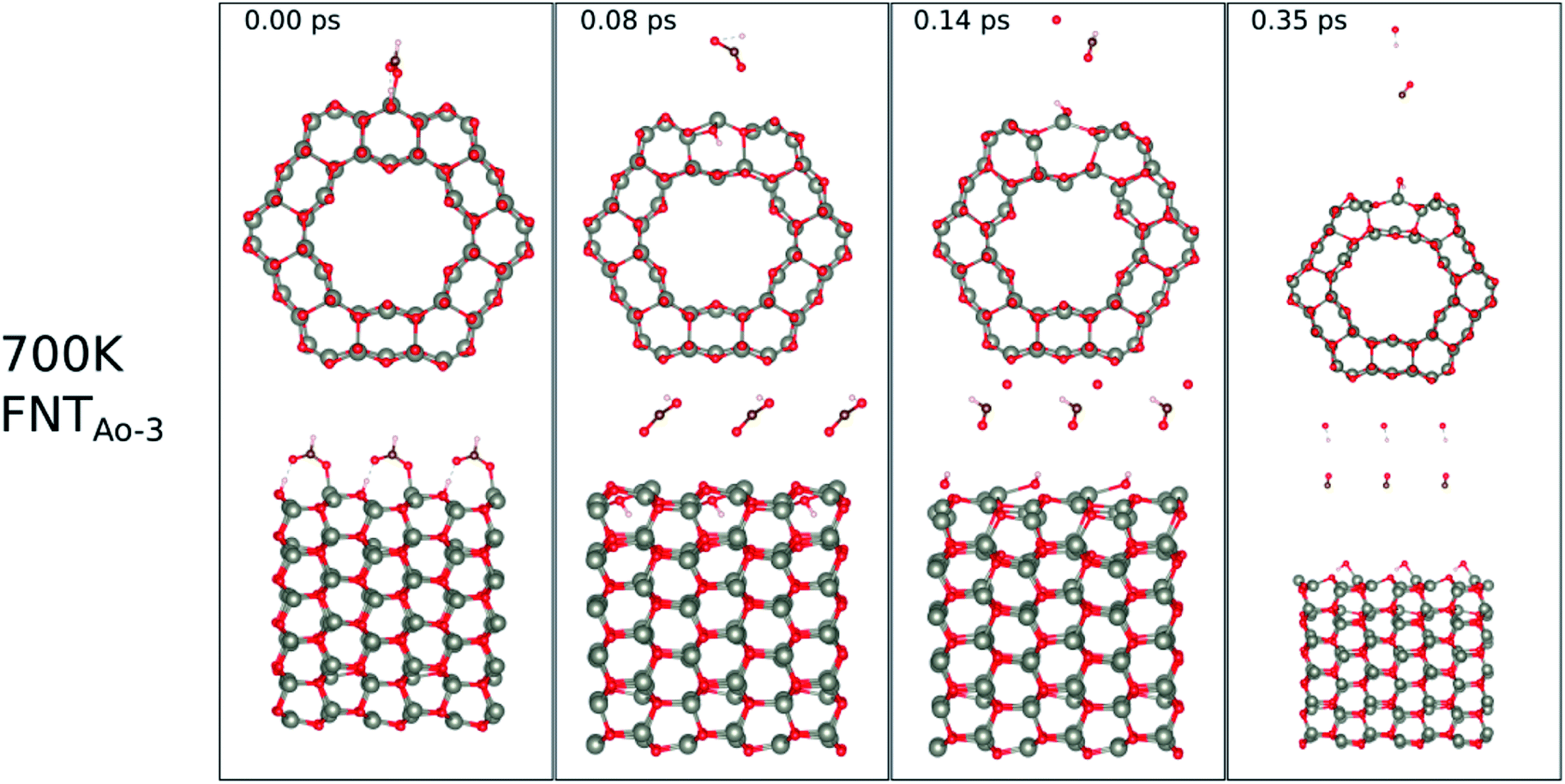 | ||
| Fig. 11 Evolution of the chemical reactions taking place on the surface of FNTAO-3 as function of time, by applying ab initio molecular dynamics at 520 K. | ||
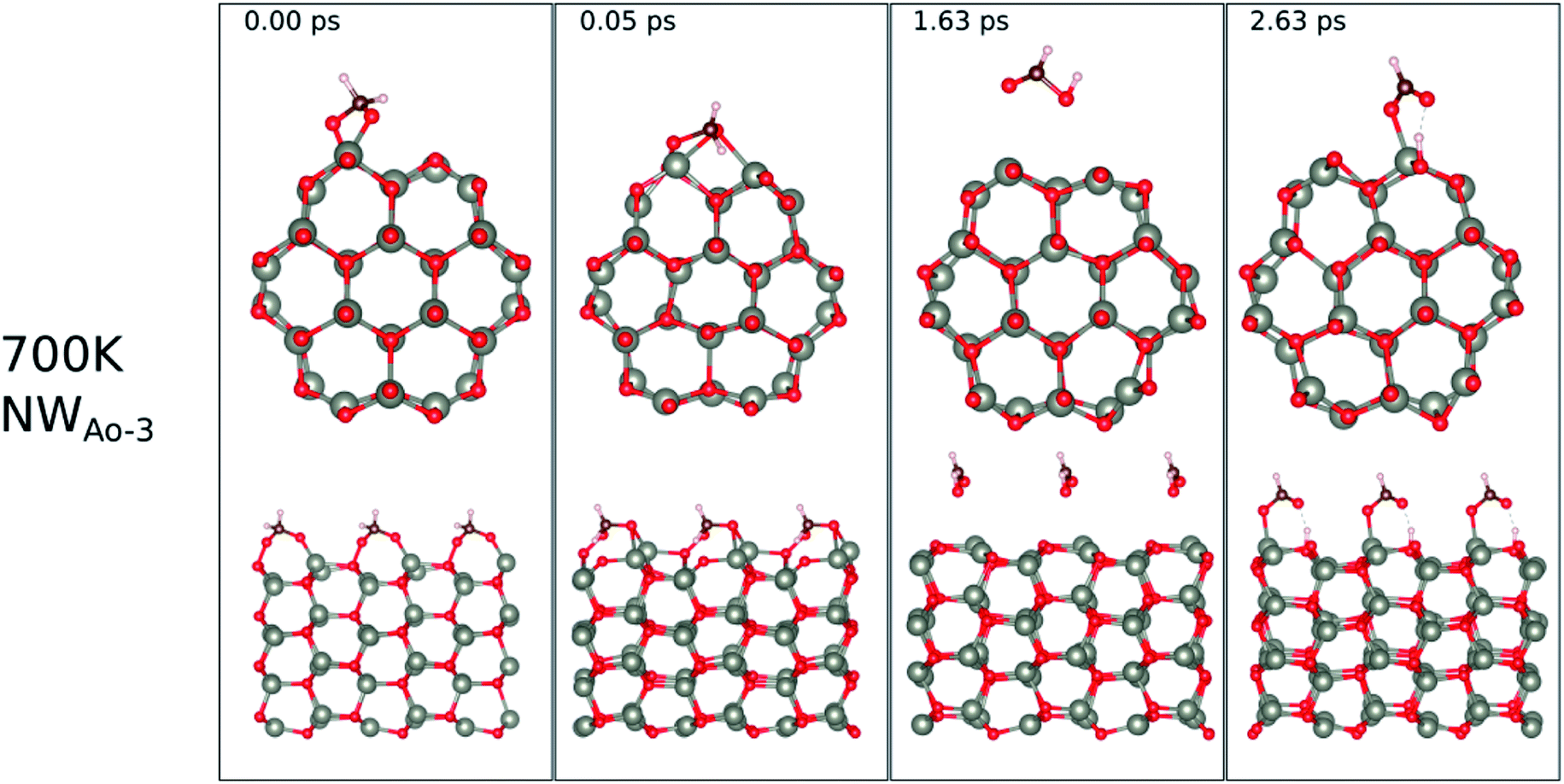 | ||
| Fig. 12 Evolution of the chemical reactions taking place on the surface of NWAO-3 as function of time, by applying ab initio molecular dynamics at 700 K. | ||
For FNTAO-3 at 300 K, the adsorbed HCO only undergoes a small structural change while remaining intact throughout the simulation (as shown in Fig. 9 and in the nearly-steady total energy of the system, shown in Fig. S1(a) in the ESI†). After 0.15 ps and to the end of the simulation, the O* atom comes away from the surface and combines with the CHO to form a formate ion (HCOO−) that stays weakly attached to the surface. Initially, the O*–Zn bond lengthens after 0.15 ps, then becomes slightly shorter at 0.23 ps, then lengthens again after 0.31 ps. This fluctuation in the bonding is akin to the presence of reversible reaction between the formate ion and the ZnO surface as follows:
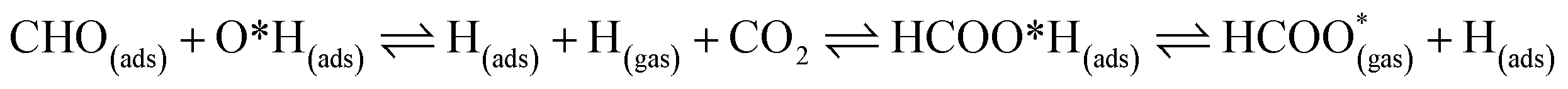 | (12) |
At the higher simulation temperature of 520 K, the surface species combine with the pre-adsorbed O atom and desorb as a formic acid molecule to form carbon dioxide gas and a free H atom that desorbs from the surface after 0.05 ps, as shown in Fig. 10. After a further 0.51 ps, the free H atom adsorbs on the neighbouring image of the FNT structure, leaving a free CO2 molecule, as per the following equation,
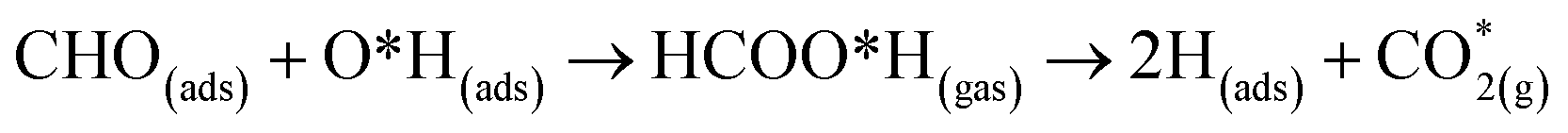 | (13) |
Eqn (13) shows that rapid chemical changes occur in response to a rise in temperature, leaving the ZnO FNTAO-3 in its stoichiometric form (FNT) after a simulation time of 0.51 ps. This indicates that the application of 520 K temperature can deoxidize or “clean” the ZnO FNTAO-3 surface through the reaction with formaldehyde, thus enhancing the recyclability of the ZnO FNT for gas sensing applications.
At 700 K, the adsorbed HCO group dissociates after 0.08 ps as shown in Fig. 11, forming CO and OH after a further 0.06 ps that both desorb from the surface, according to the following equation,
| CHO(ads) + O*H(ads) → HCOO*H(gas) → H(ads) + OH(g) + CO(g) | (14) |
The effect of applying 700 K is similar to that of applying 520 K, in terms of removing the pre-adsorbed O atom from the surface and restoring the stoichiometric surface. However, the structure at 700 K exhibits an importance difference from that at 520 K: the entire nanotube reacts to the rise in temperature by slightly increasing in diameter. Such a change has a significant impact on the catalytic role of the surface.
The surface reactions occurring on the NWAO-3 structure under a temperature of 700 K is displayed in Fig. 12. Here, the adsorbed molecule insignificant structural changes until for 1.63 ps, when the Zn–O* bond breaks, leading to the desorption of formic acid H2CO2 from the surface. Then after 2.63 ps, the formic acid molecule re-adsorbs to the surface, and an H atom dissociates from the formic acid molecule and adsorbs to a surface O atom. The system's energy in Fig. S1(f)† shows that the surface equilibrates. The equation that describes this reaction is as follows:
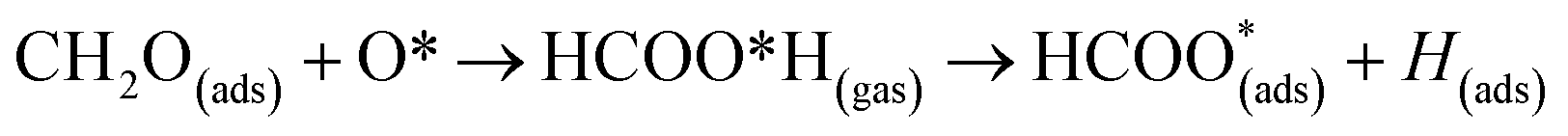 | (15) |
Thus, the adsorption of formaldehyde on the NWAO system results in an adsorbed structure that is stable under high temperature, indicating that it will not be feasible to remove the adsorbed gas from the system thermally, as is the case of the FNTAO system. Interestingly, formic acid was recently reported to result from the interaction between HCHO and SnO2/carbonized melamine foam composites.37
The above simulations show the sub-picosecond recovery times of the FNTAO sensor surface when temperature is applied. Although the application of temperature also removes the pre-adsorbed O species in this surface, it is worth noting that, as shown in eqn (1)–(3), the pre-adsorbed oxygen sensor site can be recovered by applying temperature to the surface.
In order to understand the reaction represented by eqn (4),9–11 we adsorbed two surface O atoms and examined the surface reaction with HCHO (Fig. 13) following a gradual rise in temperature up to 1000 K (300 K, 700 K then 1000 K). The calculations show that the following reaction takes place:
| HCHO + 2O(ads) → O(ads) + CO + H2O | (16) |
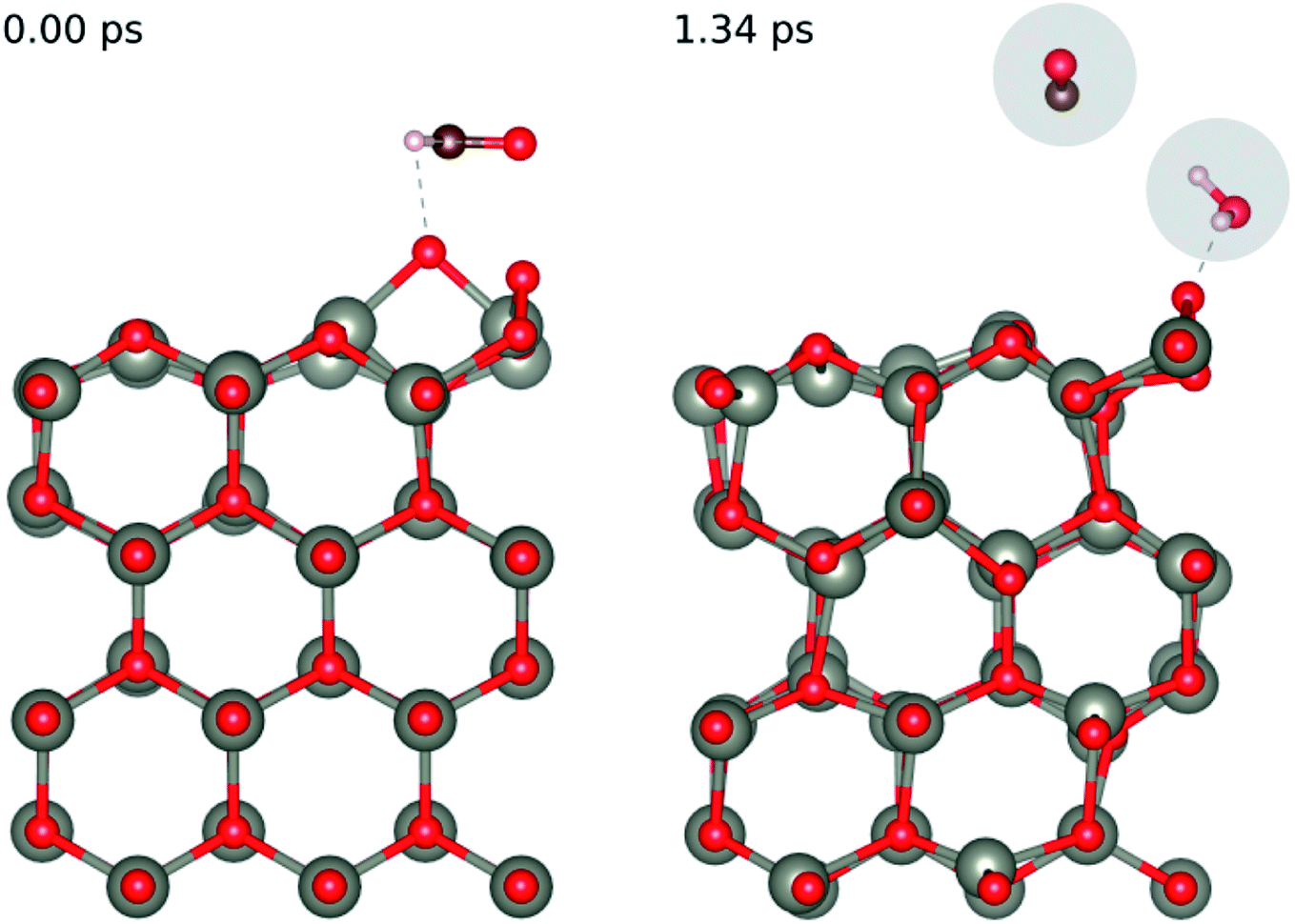 | ||
| Fig. 13 Evolution of the chemical reaction taking place on the ZnO(100) surface as function of time, by applying ab initio molecular dynamics. | ||
Hence, we confirm the formation of H2O during the reaction seen in experiment. While we did not directly see the formation of CO2 during the AIMD simulation we did see the release of CO from the surface, we calculated the enthalpy of formation of the experimentally observed reaction (eqn (4)) and the one we saw in our AIMD simulation (eqn (17)) according to the formula:
| ΔE = ECO2 + EH2O + EZnO − ECH2O − E2O/ZnO | (17) |
| ΔE = ECO + EH2O + EO/ZnO − ECH2O − E2O/ZnO | (18) |
The values we obtained for ΔE are −6.90 eV for eqn (4) (the experimentally observed reaction) and −2.72 eV for eqn (15), indicating that both reactions are feasible and could occur in the sensing reaction.
Overall, we summarise in Fig. 14 the possible reactions that can occur as a result of pre-adsorbed surface oxygen on the ZnO nanostructure. The following gases that are released from the reactions presented in Fig. 14, as well as the reaction in eqn (17), are CO2 (product of reaction in eqn (13)), H2O and CO (products of reaction in eqn (17)) and HCOOH (product of reaction in eqn (4)). These gases can be detected, and therefore enable the verification of the proposed reaction mechanisms.
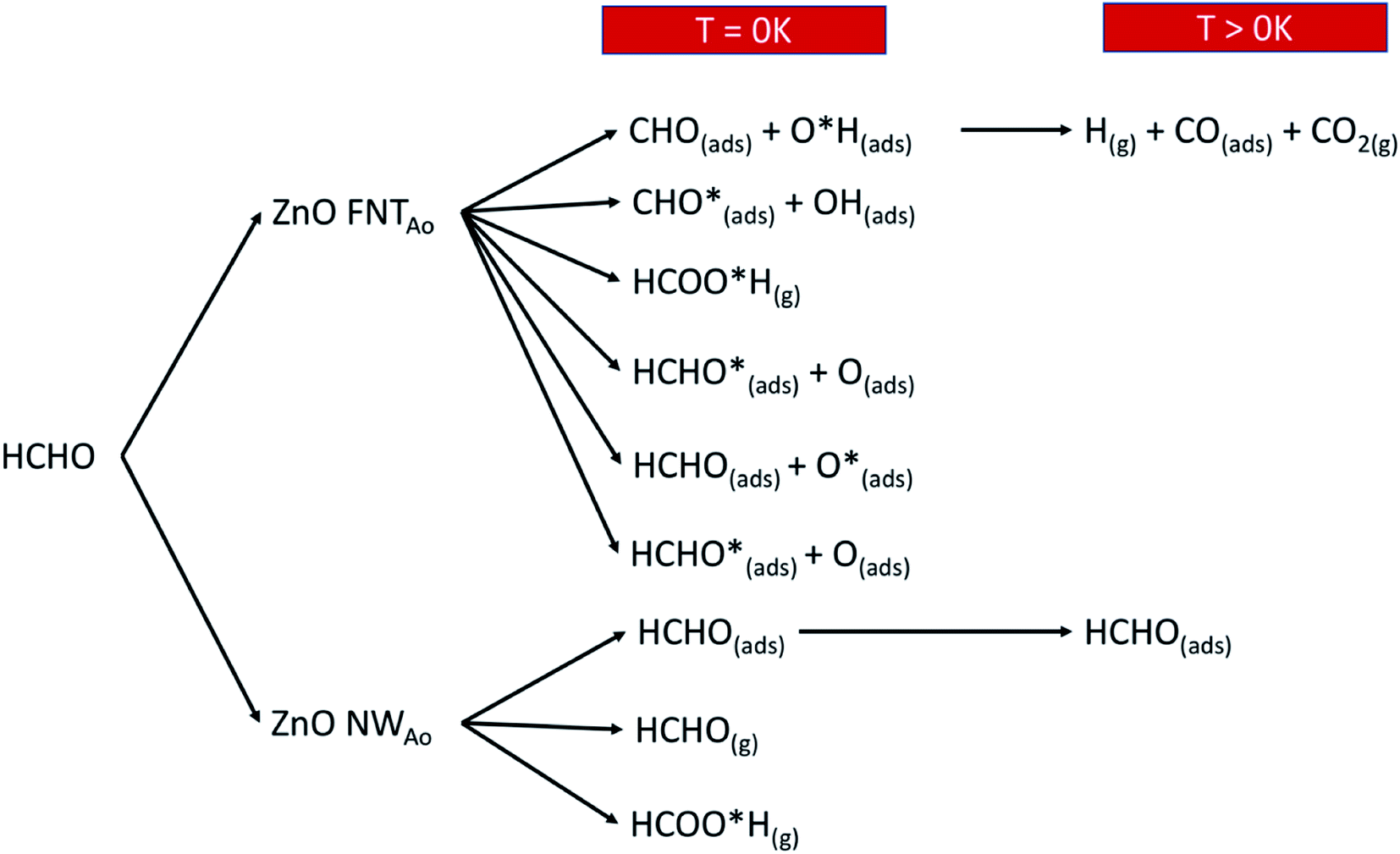 | ||
| Fig. 14 A schematic diagram that shows the various reactions products when HCHO reacts with the ZnO surface with a pre-adsorbed O atom. | ||
4. Conclusion
We show that the presence of pre-adsorbed oxygen on ZnO facetted nanotubes and nanowires plays a significant role in the gas-sensing mechanism of HCHO. The presence of surface oxygen allowed for associative and dissociative adsorption. Both singly and doubly coordinated adsorption geometries of the associatively adsorbed species were determined and found to be thermodynamically and dynamically stable. The main product from dissociation is adsorbed CHO. Compared to the stoichiometric surface, the pre-adsorbed oxygen facilitates dissociation of the HCO molecule leading to a myriad of complex reaction products on the surface. Such products include CHO, H, CO and CO2. The facetted nanotube surface is also found to restore its stoichiometric structure within sub-picosecond recovery times. This means that the ZnO surface is recyclable, which is an important feature in gas sensors. The surface reactions, in the presence of the pre-adsorbed oxygen, are shown to enable the ZnO nanowire to function as a resistive sensor for HCHO. The pre-adsorbed oxygen atom might also enable the ZnO structures to detect the presence of HCHO by variation in the photocurrent. The removal of the pre-adsorbed O atom from the surface, through the formation of formic acid (HCOOH), was also found to occur on the facetted nanotube. This reaction is significant as it explains how the surface may be regenerated for detection of other gas molecules.The facetted nanotube structure has more diverse surface reactions than the nanowire structure due to the occurrence of morphological changes in the facetted nanotube structure during and after adsorption. These changes facilitate complex catalytic reactions of HCHO on the surface.
Overall, this work has shown that the presence of pre-adsorbed oxygen is significant for enhancing the sensitivity of ZnO nanostructures to the presence of HCHO. Importantly, the surface reaction is not simple and cannot be classified using just one reaction and in fact can lead to at least 8 different surface reactions, producing multiple reaction products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computational facilities are gratefully acknowledged from the Australian Government through the National Computational Infrastructure (NCI) Facility and the Pawsey Supercomputing Centre, under the National Computational Merit Allocation Scheme and the Pawsey Energy and Resources Merit Allocation Scheme.References
- D. Farmanzadeh and L. Tabari, Electric field effects on the adsorption of formaldehyde molecule on the ZnO nanotube surface: A theoretical investigation, Comput. Theor. Chem., 2013, 1016, 1–7 CrossRef CAS.
- J. Xu, Y. Zhang, Y. Chen, Q. Xiang, Q. Pan and L. Shi, Uniform ZnO nanorods can be used to improve the response of ZnO gas sensor, Mater. Sci. Eng. B, 2008, 150(1), 55–60 CrossRef CAS.
- G. Korotcenkov, The role of morphology and crystallographic structure of metal oxides in response of conductometric-type gas sensors, Mater. Sci. Eng., R, 2008, 61(1–6), 1–39 CrossRef.
- M. J. S. Spencer, Gas sensing applications of 1D-nanostructured zinc oxide: Insights from density functional theory calculations, Prog. Mater. Sci., 2012, 57(3), 437–486 CrossRef CAS.
- Z. L. Wang, Zinc oxide nanostructures: growth, properties and applications, J. Phys.: Condens. Matter, 2004, 16(25), R829–R858 CrossRef CAS.
- Q. Yu, C. Yu, J. Wang, F. Guo, S. Gao, S. Jiao, H. Li, X. Zhang, X. Wang, H. Gao, H. Yang and L. Zhao, Gas sensing properties of self-assembled ZnO nanotube bundles, RSC Adv., 2013, 3(37), 16619 RSC.
- J. Y. Lao, J. Y. Huang, D. Z. Wang and Z. F. Ren, ZnO Nanobridges and Nanonails, Nano Lett., 2003, 3(2), 235–238 CrossRef CAS.
- S. Nundy, T.-y. Eom, J.-g. Kang, J. Suh, M. Cho, J.-S. Park and H.-J. Lee, Flower-shaped ZnO nanomaterials for low-temperature operations in NOX gas sensors, Ceram. Int., 2020, 46(5), 5706–5714 CrossRef CAS.
- X. Chu, T. Chen, W. Zhang, B. Zheng and H. Shui, Investigation on formaldehyde gas sensor with ZnO thick film prepared through microwave heating method, Sens. Actuators, B, 2009, 142(1), 49–54 CrossRef CAS.
- N. Han, H. Liu, X. Wu, D. Li, L. Chai and Y. Chen, Pure and Sn-, Ga- and Mn-doped ZnO gas sensors working at different temperatures for formaldehyde, humidity, NH3, toluene and CO, Appl. Phys. A: Mater. Sci. Process., 2011, 104(2), 627–633 CrossRef CAS.
- P. Lv, Z. A. Tang, J. Yu, F. T. Zhang, G. F. Wei, Z. X. Huang and Y. Hu, Study on a micro-gas sensor with SnO2–NiO sensitive film for indoor formaldehyde detection, Sens. Actuators, B, 2008, 132(1), 74–80 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(17), 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- P. B. Giannozzi, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al., QUANTUM ESPRESSO: A Modular and Open-source Software Project for Quantum Simulations of Materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, The SIESTA method for ab initio order-N materials simulation, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(17), 13115–13118 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- N. Troullier and J. Martins, A straightforward method for generating soft transferable pseudopotentials, Solid State Commun., 1990, 74, 613–616 CrossRef.
- K. J. Iversen and M. J. S. Spencer, Effect of ZnO Nanostructure Morphology on the Sensing of H2S Gas, J. Phys. Chem. C, 2013, 117(49), 26106–26118 CrossRef CAS.
- T. Morishita, S. P. Russo, I. K. Snook, M. J. S. Spencer, K. Nishio and M. Mikami, First-principles study of structural and electronic properties of ultrathin silicon nanosheets, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(4), 045419 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, 96th edn, 2006 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef.
- R. M. Watwe, R. D. Cortright, J. K. Nørskov and J. A. Dumesic, Theoretical Studies of Stability and Reactivity of C2 Hydrocarbon Species on Pt Clusters, Pt(111), and Pt(211), J. Phys. Chem. B, 2000, 104, 2299–2310 CrossRef CAS.
- J. M. M. Greeley, A First-Principles Study of Methanol Decomposition on Pt(111), J. Am. Chem. Soc., 2002, 124, 7193–7201 CrossRef CAS PubMed.
- M. Brandbyge, J.-L. Mozos, P. Ordejon, J. Taylor and K. Stokbro, Density-functional method for nonequilibrium electron transportMads Brandbyge, José-Luis Mozos, Pablo Ordejón, Jeremy Taylor, and Kurt Stokbro, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef.
- S. Datta, Electronic Transport in Mesoscopic Systems, Cambridge University Press, Cambridge, U.K., 1995 Search PubMed.
- S. Nosé, A Unified Formulation of the Constant Temperature Molecular-Dynamics Methods, J. Chem. Phys., 1984, 81(1), 511–519 CrossRef.
- L. Zhang, J. Zhao, J. Zheng, L. Li and Z. Zhu, Shuttle-like ZnO nano/microrods: Facile synthesis, optical characterization and high formaldehyde sensing properties, Appl. Surf. Sci., 2011, 258(2), 711–718 CrossRef CAS.
- D. Mora-Fonz, J. Buckeridge, A. J. Logsdail, D. O. Scanlon, A. A. Sokol, S. Woodley and C. R. A. Catlow, Morphological Features and Band Bending at Nonpolar Surfaces of ZnO, J. Phys. Chem. C, 2015, 119(21), 11598–11611 CrossRef CAS.
- H. Xu, W. Fan, A. L. Rosa, R. Q. Zhang and T. Frauenheim, Hydrogen and oxygen adsorption on ZnO nanowires: A first-principles study, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79(7) DOI:10.1103/physrevb.79.073402.
- H. T. T. Tran and M. J. S. Spencer, Zinc oxide for gas sensing of formaldehyde: Density functional theory modelling of the effect of nanostructure morphology and gas concentration on the chemisorption reaction, Mater. Chem. Phys., 2017, 193, 274–284 CrossRef CAS.
- S. Li, X. Lu, W. Guo, H. Zhu, M. Li, L. Zhao, Y. Li and H. Shan, Formaldehyde oxidation on the Pt/TiO2(101) surface: A DFT investigation, J. Organomet. Chem., 2012, 704, 38–48 CrossRef CAS.
- H. T. T. Tran and M. J. S. Spencer, Zinc oxide for gas sensing of formaldehyde: Density functional theory modelling of the effect of nanostructure morphology and gas concentration on the chemisorption reaction, Mater. Chem. Phys., 2017, 193, 274–284 CrossRef CAS.
- Y. Li, N. Luo, W. Zhang, Q. Hu, X. Wang, Y. Chen, Z. Cheng and J. Xu, Rational design and in situ growth of SnO2/CMF composites: insightful understanding of the formaldehyde gas sensing mechanism and enhanced gas sensing properties, J. Mater. Chem. C, 2020, 8(36), 12418–12426 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00804h |
| ‡ Current address: Institute for Frontier Materials, Deakin University, Geelong, Victoria 3216, Australia & ARC Centre of Excellence in Exciton Science, School of Science, RMIT University, Melbourne, VIC 3001 Australia. |
| This journal is © The Royal Society of Chemistry 2022 |