
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinctive optical transitions of tunable multicolor carbon dots†
Hyeong Seop
Shim
 ,
Jun Myung
Kim
,
Seonghyun
Jeong
,
Youngwon
Ju
,
Sung Jae
Won
,
Jeongyun
Choi
,
Sangwon
Nam
,
Aniruddha
Molla‡
,
Joohoon
Kim
* and
Jae Kyu
Song
*
,
Jun Myung
Kim
,
Seonghyun
Jeong
,
Youngwon
Ju
,
Sung Jae
Won
,
Jeongyun
Choi
,
Sangwon
Nam
,
Aniruddha
Molla‡
,
Joohoon
Kim
* and
Jae Kyu
Song
*
Department of Chemistry, Kyung Hee University, Seoul 02447, Republic of Korea. E-mail: jkim94@khu.ac.kr; jaeksong@khu.ac.kr
First published on 28th December 2021
Abstract
Three types of carbon dots (CDs) are synthesized from isomers of phenylenediamine to develop multicolor nanomaterials with low toxicity, high stability, and high quantum yield. The distinctive electronic structures of CDs lead to the characteristic optical transitions, such as three colors of blue, green, and red, which are primarily attributed to the difference in configurations, despite the similar basic structures of conjugated systems. The excitation-independent emission and the single exponential decay of CDs indicate the single chromophore-like nature in each type of CD. In addition, the two-photon luminescence of CDs exhibits a comparable shape and time profile to the typical photoluminescence with high photostability. Although the surface-related defect states are observed by intragap excitation, the contribution of defect states is barely observed in the emission profile upon band gap excitation. Consequently, the controllability of optical transitions in CDs enhances the potential of tunable multicolor nanomaterials for various applications as alternatives to quantum dots containing toxic elements.
1. Introduction
Tunable multicolor nanomaterials have attracted much attention, because they hold high potential for various applications, such as bioimaging, light-emitting diodes, and optoelectronic devices.1–3 In the past decades, abundant multicolor materials have been developed, such as quantum dots and molecular nanomaterials, although practical applications are often limited by high toxicities, low stabilities, and low quantum yields (QYs).1–5 Accordingly, carbon dots (CDs) are emerging as alternatives due to low toxicities, high stabilities, and high QYs with unique optical properties.6–15 In general, CDs are represented by the quasi-zero-dimensional nanomaterials with small-sized, sp2-hybridized carbon frameworks, whose surfaces contain hydrophilic functional groups to ensure the solubility in water.10–15Numerous approaches have been reported for fabrications of CDs by varying precursor materials and synthesis procedures,6–15 while most CDs share common features of small sizes and bright emissions. Besides, several successful works have realized tunable photoluminescence (PL) from blue to red, which is independent of excitation energy, although the corresponding mechanism still remains under debate.16–25 The tunable emission is often attributed to the involvement of surface states,16–20 whereas the quantum confinement effects are also proposed for CDs with small sizes.21–25 Furthermore, both confinement effects and surface states, in addition to their strong interactions, are suggested to be responsible for tunable emission.17,18 Recently, the electronic structures estimated by the first-principle calculations disclose that the band gap energies of CDs are influenced by configurations of skeletal frameworks, which is verified by the experimental spectra.26,27 Therefore, the electronic structures of CDs, which afford the tunable and excitation-independent feature of emission, should be understood to control the multicolor emission.
CDs have been prepared by various bottom-up methods, which include hydrothermal synthesis and microwave-assisted reaction.10–15 However, the optical features of CDs synthesized by the bottom-up approach are often inconsistent, while some behaviors are even contradictory.16–25 In particular, the excitation-energy dependent shape of PL suggests the existence of multiple chromophores,7,21 which is supported by the broad spectral shapes of as-prepared CDs.19,25 Moreover, PL QYs of CDs are frequently lower than those of well-known quantum dots,1–5 but the enhancement of QY is not easily achieved in CDs. Certainly, many studies have proposed the strategies to increase QY,28,29 although the underlying mechanism has not been fully consented. Hence, in addition to the development of facile methods to prepare stable and tunable emission of CDs with controlled sizes and shapes, the systematic approach is imperative to investigate the characteristics of optical transitions in CDs, which would provide innovative ways to enhance QY of CDs.
The biocompatible feature with low toxicity is essential for bioimaging applications. Consequently, CDs are suitable for bioimaging, because the surface of CDs coated with hydrophilic groups leads to the high solubility as well as the low toxicity toward biosystems.10–15 The near-IR excitation is also conducive to bioimaging, because near-IR excitation provides minimal photobleaching, low autofluorescence, and deep tissue penetration. Furthermore, two-photon absorption of near-IR gives rise to the visible emission to enhance the imaging efficiency, while the color coding with multicolor materials is beneficial to the imaging of complex biosystems.25,30 Accordingly, the multicolor emission of CDs by two-photon absorption of near-IR would assist the imaging of biosystems, when the optical transition of each color is controllable with high QYs and stabilities.
In this study, we fabricate three kinds of CDs using isomers of phenylenediamine (PD) to develop multicolor nanomaterials. The optical transitions are distinctive in the three types of CDs, which are mainly attributed to the configurational difference, because CDs show the comparable sizes, element ratios, and functional groups. The excitation-independent emission and the single exponential decay of PL suggest the characteristics of single chromophore in each type of CDs. Two-photon luminescence by near-IR excitation presents the similar shape and time profile to typical PL of CDs with high photostability against intense excitation. Although the excitation-dependent PL is observed by intragap excitation, the low intensity indicates only a minor contribution of defect states in the emission profile. The detailed study on optical transitions unravels the unique features of absorption, excitation, and emission, demonstrating the potentials for multicolor nanomaterials as alternatives to conventional quantum dots.
2. Experimental section
Three types of CDs were synthesized using microwave-assisted reactions of PDs and subsequent purifications through silica gel column chromatography.31 Briefly, 0.25 g of PD isomers, such as ortho-phenylenediamine (OPD), meta-phenylenediamine (MPD), and para-phenylenediamine (PPD), was dissolved in 10 mL of deionized water to prepare the individual solutions of PDs. Each solution was subjected to the microwave irradiation cycle, which included the heating to 220 °C, maintaining for 10 min, and cooling to 55 °C, in a microwave synthesis reactor (Monowave 300, Anton Paar GmbH). The resulting solutions, such as the aqueous solutions of ortho-type carbon dots (OCD), meta-type carbon dots (MCD), and para-type carbon dots (PCD), were purified by silica gel column chromatography (230–400 mesh) using mixtures of ethyl acetate and hexane as eluents. Additional purification of CDs was also performed by centrifugal ultrafiltration (Amicon ultra-4 centrifugal filter, MWCO 3 K) as a control experiment. After drying purified solutions, CDs were dispersed in ethanol (0.25 mg mL−1) for subsequent characterizations. Transmission electron microscopy (TEM) was performed using Tecnai G2 F30ST (FEI Co) in the Center for Advanced Functional Ceramics (Gachon University, Korea). X-ray photoelectron spectroscopy (XPS) and Fourier-transform infrared (FTIR) spectra were acquired using K-Alpha X-ray photoelectron spectrometer (Thermo Electron) and Alpha II (Bruker Co), respectively. Raman spectra were obtained using a home-made spectrometer at the excitation wavelength of 532 nm. UV-visible absorption spectra were attained using Agilent 8453 (Agilent Tech). Photoluminescence excitation (PLE) and PL spectra were obtained using FluoroMate FS-2 (Scinco Co). QYs were also estimated using FluoroMate FS-2. Time-resolved PL spectra were measured using a home-made spectrometer using femtosecond pulses from a cavity-dumped oscillator (Mira/PulseSwitch, Coherent) and time-correlated single photon counter (PicoHarp 300, PicoQuant).32,333. Results and discussion
Three kinds of CDs (MCD, OCD, and PCD) were synthesized from three isomers of PDs (MPD, OPD, and PPD), respectively. TEM images of CDs revealed the well-dispersed but rather large-sized CDs with the average diameters of 17.6 ± 4.9 nm, 19.6 ± 3.6 nm, and 16.4 ± 4.1 nm for MCD, OCD, and PCD, respectively (Fig. S1†).19,29 The large sizes were frequently observed in CDs synthesized from PDs.23,27 The chemical compositions estimated by XPS showed that CDs were mainly composed of carbon, nitrogen, and oxygen (Fig. S2†). The element ratio ([C]/[N]/[O]) was comparable, such as 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:4 of MCD, 89:7:4 of OCD, and 92:4:4 of PCD, where the dominant constituent of carbon suggested the basic structures of CDs.24,26 Furthermore, carbon was predominantly in sp2-hybridized (C–C/C
:5:4 of MCD, 89:7:4 of OCD, and 92:4:4 of PCD, where the dominant constituent of carbon suggested the basic structures of CDs.24,26 Furthermore, carbon was predominantly in sp2-hybridized (C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) states in the high-resolution spectra (Fig. S2†), indicating the conjugated carbon systems of CDs. Nitrogen was mainly in pyridinic states and oxygen was mostly in carboxyl and hydroxyl groups (CO and C–O). The chemical bonds revealed by FTIR spectroscopy indicated the similar chemical structures and functional groups (Fig. S3†), including the peaks at 2900 cm−1 (C–H), 1730 cm−1 (COOH), 1630 cm−1 (CN), and 1250 cm−1 (C–O).18,19,26 Notably, the characteristic peaks of CDs were barely found in PDs (Fig. S3†), implying the intermolecular reactions of PDs, such as the decomposition, intermolecular cyclization, and condensation processes, to form CDs by the heating and microwave treatment. Raman spectra presented D-band at 1350 cm−1 and G-band at 1580 cm−1 (Fig. S4†), which were attributed to the disordered and graphitic structures of conjugated carbon systems, respectively. The intensity ratio of two bands (ID/IG) was comparable in the range of 0.95–1.11,18,19 indicating the similar basic structures of conjugated systems in CDs.
C) states in the high-resolution spectra (Fig. S2†), indicating the conjugated carbon systems of CDs. Nitrogen was mainly in pyridinic states and oxygen was mostly in carboxyl and hydroxyl groups (CO and C–O). The chemical bonds revealed by FTIR spectroscopy indicated the similar chemical structures and functional groups (Fig. S3†), including the peaks at 2900 cm−1 (C–H), 1730 cm−1 (COOH), 1630 cm−1 (CN), and 1250 cm−1 (C–O).18,19,26 Notably, the characteristic peaks of CDs were barely found in PDs (Fig. S3†), implying the intermolecular reactions of PDs, such as the decomposition, intermolecular cyclization, and condensation processes, to form CDs by the heating and microwave treatment. Raman spectra presented D-band at 1350 cm−1 and G-band at 1580 cm−1 (Fig. S4†), which were attributed to the disordered and graphitic structures of conjugated carbon systems, respectively. The intensity ratio of two bands (ID/IG) was comparable in the range of 0.95–1.11,18,19 indicating the similar basic structures of conjugated systems in CDs.
UV-visible spectra (Fig. 1) showed the absorption bands of CDs in deep-UV region (<300 nm), which were also found in PDs (Fig. S5†) and thus ascribed to π–π* transitions.21,25 Moreover, the distinctive absorption bands were observed in the long-wavelength region (>300 nm), such as 345 nm of MCD, 430 nm of OCD, and 510 nm of PCD, which were not observed in PDs. The different peak positions suggested that the electronic structures were influenced by the isomer types of PDs employed as precursors,23,27 despite the analogous sizes and basic chemical structures. PLE spectra obtained at the corresponding emission maxima exhibited the analogous positions of excitation bands to absorption bands (Fig. 1), supporting the characteristic electronic structures. PL spectra also showed the distinctive bands (Fig. 1), such as the bands at 405 nm of MCD, 530 nm of OCD, and 600 nm of PCD, which were different from the corresponding PDs (Fig. S5†). Indeed, PL of CDs was closely correlated to the absorption band with the similar Stokes shift, suggesting that PL was the band gap emission.23–27 Consequently, MCD, OCD, and PCD were represented as the blue-emitting CDs (BCD), green-emitting CDs (GCD), and red-emitting CDs (RCD), respectively. The estimated QYs of BCD, GCD, and RCD were 12%, 35%, and 8%, respectively, at the peak wavelengths of PLE, which were comparable to the previous results.23,27 In addition, the excitation-independent emission shapes suggested the nature of single chromophore with the characteristic band structure in each type of CDs (Fig. 2). The time-resolved PL profiles evidenced the feature of single chromophore, because the single exponential decays were observed with lifetimes of 2.2, 3.7, and 9.2 ns in BCD, GCD, and RCD (Fig. 1d), respectively.26,27 Notably, upon UV excitation (355 nm), CDs showed the primary colors of band gap emission, such as blue, green, and red (insets of Fig. 1), with the suppressed reabsorption due to the large Stokes shift.20,24
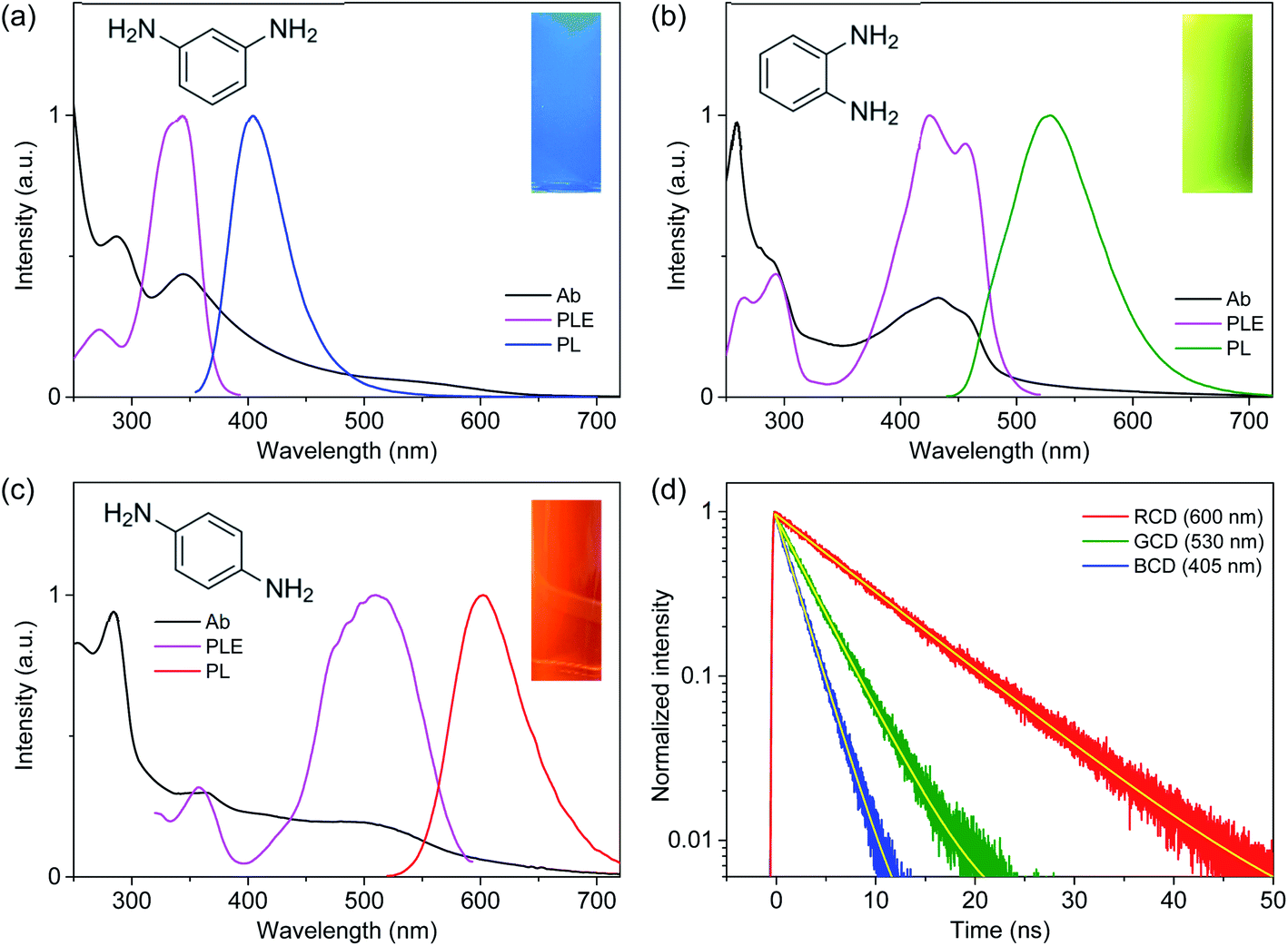 | ||
| Fig. 1 Absorption (Ab), photoluminescence excitation (PLE), and photoluminescence (PL) spectra of (a) MCD (BCD), (b) OCD (GCD), and (c) PCD (RCD). The intensities are adjusted for comparison. The left insets show the molecular structures of meta-, ortho-, and para-phenylenediamine precursors. The right insets present the photographs of PL from CDs in solution. (d) Time-resolved PL profiles of CDs are fitted by the single-exponential model (yellow lines). | ||
 | ||
| Fig. 2 PL spectra of (a) BCD, (b) GCD, and (c) RCD at various excitation wavelengths show the excitation-independent emission shapes. | ||
The optical transition energies of CDs have been affected by the synthesis conditions and precursors, which were generally ascribed to the quantum confinement effect and the degree of surface oxidation.16–25 In other words, the electronic structures of chromophores were determined by the sizes and chemical compositions of CDs,21–25 which were further influenced by the surface environments such as hydroxyl and carboxyl groups.16–20 Certainly, three colors of blue, green, and red were previously reported in CDs synthesized from MPD, OPD, and PPD, respectively,23,27 where the tunability of PL was attributed to the quantum confinement effects and element ratios. However, the optical transition energies in the present study were not closely correlated to the average sizes of CDs (Fig. S1†), despite the nearly identical absorption and emission wavelengths in the three types of CDs to the previous study.23 Besides, the average sizes were larger than the reported values, indicating that the tunable PL might not be ascribed to the quantum confinement effects. The optical transition energies were not also associated to the element ratio of oxygen (Fig. S2†), because the ratio was nearly identical (∼4%). Moreover, the ratio of nitrogen was not linearly dependent on optical transition energies, because the ratio increased and then decreased in the narrow range (5% to 7% and to 4%) with the decreasing transition energy.
Since impurities and unreacted species in CDs might affect the observed optical features, an additional purification procedure was employed, although the silica gel column chromatography has been widely used to separate and reduce the impurities.19,23,26 After the separation through the column chromatography, CDs were further purified by centrifugal ultrafiltration. Nevertheless, the difference was hardly observed in the optical shapes of absorption, excitation, and emission spectra (Fig. S6†), indicating that the effect of impurities was not significant in the present study. The functional groups at surface were also similar in CDs (Fig. S3†), suggesting another aspect to influence the electronic structures of CDs.
Recently, the optical transition energies were estimated for three molecular configurations synthesizable from isomers of PDs by the first-principle calculations using the density functional theory (DFT).26 The difference in configuration led to the change of transition energies, which was supported by experimental spectra. To investigate the molecular geometries of chromophores in blue-, green-, and red-emitting CDs correlated to isomers of PDs, the electronic band structures were calculated for various forms of PDs using Gaussian 09 program suite.34 Stable geometries of the ground and excited states were calculated using DFT at the level of wB97XD/aug-cc-pVDZ and the energies were estimated using the zero-point vibrational energy corrections through frequency calculations. Among various forms of PDs (Fig. S7†), the energy gaps between the ground and excited states in the trimeric forms of PDs were in the comparable feature to the measured optical transition energies (Fig. S8†), such as the appropriate red-shift from BCD to GCD and to RCD, supporting the configurational effects correlated to isomers of PDs.
However, the calculated energies were slightly different from the experimental results (Fig. 3),26 implying additional contribution on the electronic band structures. Hence, the functional groups observed in FTIR spectra were included in the first-principle calculations. Although the functional groups influenced the energy gaps in the trimeric forms of PDs (Fig. S9†), the effect of functional groups was not predominant. Therefore, the distinctive electronic structures in the three types of CDs were mainly attributable to the configurational effects, while the development of electronic structures was also dependent on the sizes, chemical compositions, and functional groups of CDs. Consequently, the well-defined electronic band structures, which were influenced by the molecular configurations and stabilized by the interactions with surface conditions,16–20 were responsible for the excitation-independent optical features with the single exponential decay. Certainly, the disordered structures were not negligible in Raman spectra (Fig. S4†), compared to the graphitic structures of conjugated systems, implying that the disordered structures correlated to the difference in molecular configurations could affect the electronic structures and the trimeric forms of PDs would be the skeletal frameworks of CDs.27 Although the interactions with surface conditions would be also indispensable for the evolution of electronic structures, the similar types and densities of functional groups were not primarily responsible for the difference in electronic structures of CDs.
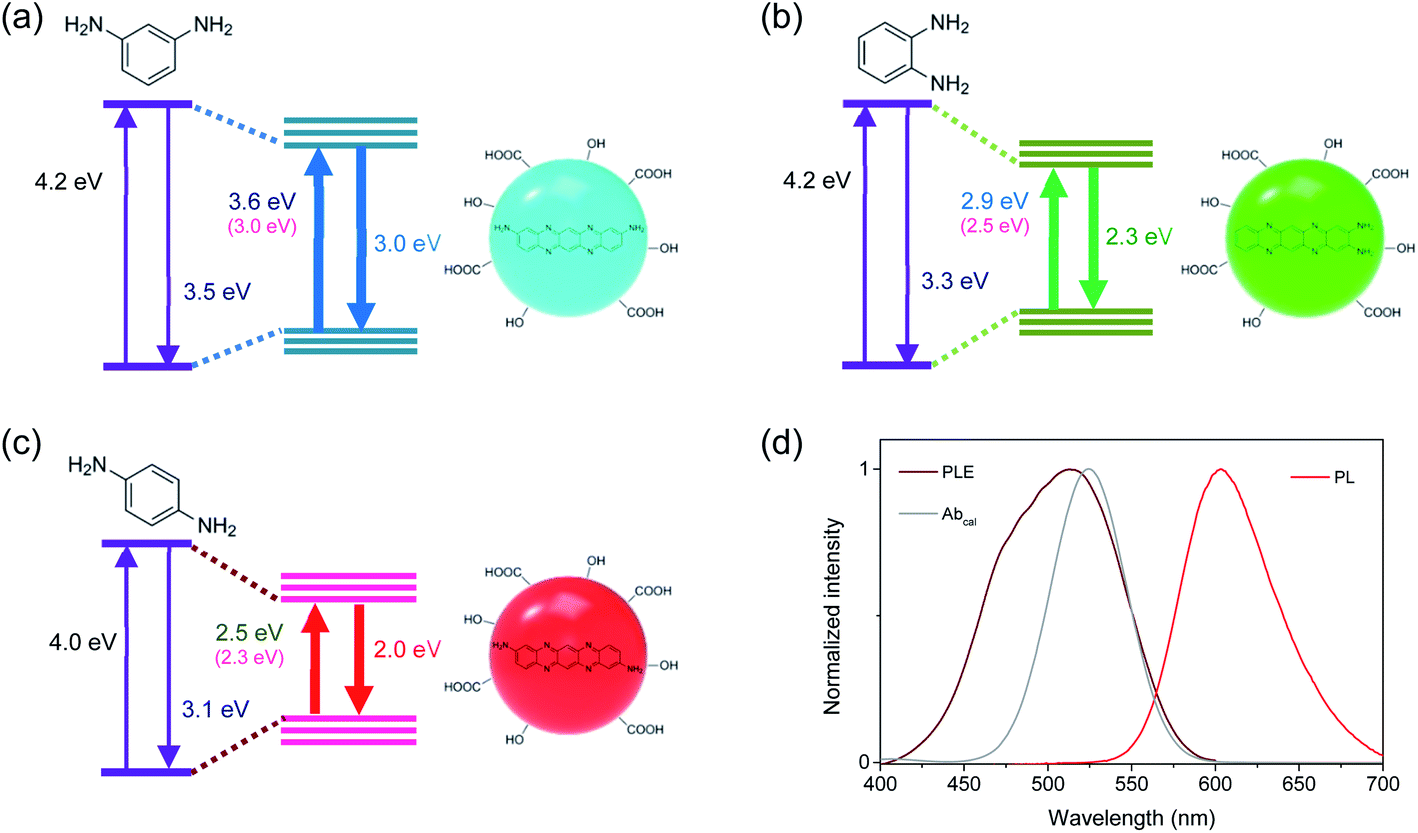 | ||
| Fig. 3 Schematic band structures of (a) BCD, (b) GCD, and (c) RCD. Energy gaps between the ground and excited states of CDs are reduced by intermolecular reactions of PDs, which are influenced by configurations and stabilized by surface groups. The energy values denote the experimentally obtained values at the maximum positions of absorption and PL spectra. The energy values in parentheses denote the calculated values for trimeric forms of PDs. (d) The calculated absorption spectrum (Abcal) of RCD is compared to the experimental PLE and PL spectra. | ||
Since near-IR excitation is widely employed to probe biosystems,35–37 the optical properties of CDs were investigated by near-IR excitation to evaluate the possibility of multicolor applications.25,30 Upon excitation of 710 nm pulses, CDs exhibited the comparable emission to the typical PL (Fig. 4a), despite a lower excitation energy (1.75 eV) than the band gap energy. The emission intensities were in the quadratic dependence on excitation intensities (Fig. S10†), indicating two-photon luminescence (TPL). In addition to the similar shapes, the time profiles of TPL were equivalent to those of PL (Fig. 4b), which confirmed that TPL was the band gap emission. Since the stability against photobleaching was essential for bioimaging,22,23 the photostability of CDs was examined with the intense near-IR excitation. Even when subjected to pulsed excitation of 710 nm at a high repetition rate (1.0 × 106 Hz) and a high excitation intensity (50 μJ cm−2 pulse−1), TPL spectra of CDs remained nearly unchanged for long-time exposure (Fig. S11†), while the intensities of TPL were slightly reduced (<3%) until 3.6 × 109 excitation (1 hour) under ambient conditions. Likewise, the intensities of the typical PL did not decrease noticeably for the long-time exposure of UV (355 nm) even at the high excitation power of 1.0 W cm−2 (Fig. S11†). Such stability was comparable to the previous results,25,30 demonstrating the high photostability suitable for multicolor applications.
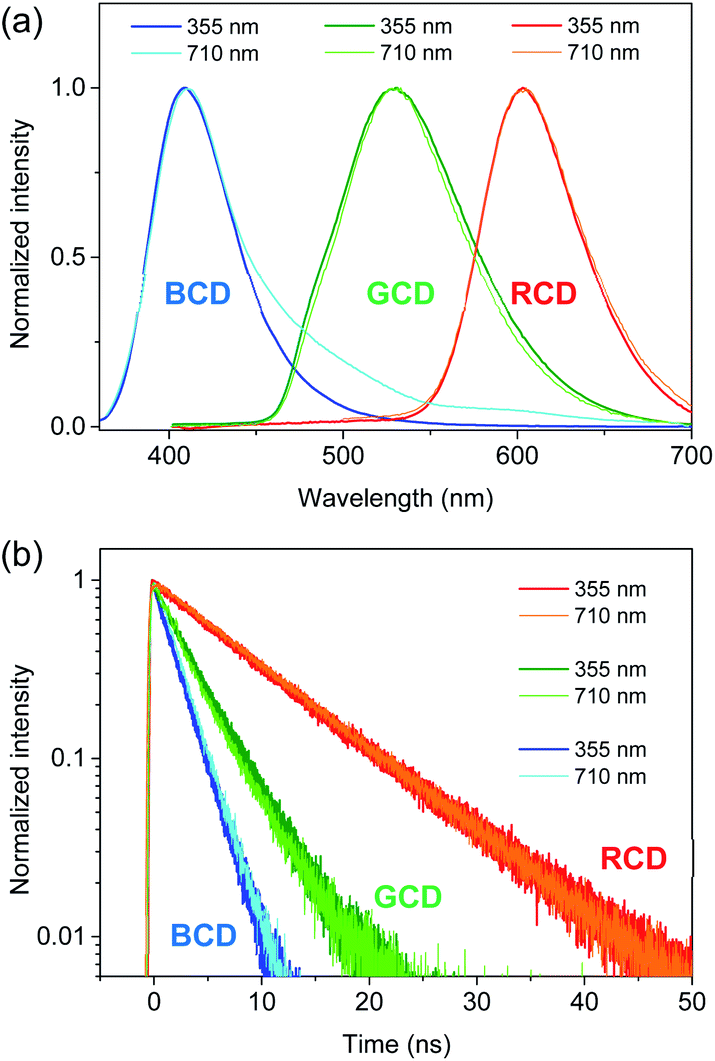 | ||
| Fig. 4 (a) PL spectra of CDs obtained by excitation of 355 and 710 nm are normalized for comparison. (b) Time-resolved PL profiles of CDs obtained by excitation of 355 and 710 nm are normalized. | ||
However, TPL of BCD displayed a tail in the long wavelength region (>430 nm), which was different from the typical PL (Fig. 4a), although the band maximum was not changed by the excitation wavelength (λex = 355 and 710 nm). To understand the tail region of emission, PL spectra were further obtained with increasing excitation wavelength. Evidently, the shape of PL was excitation-independent (Fig. 2) above the band gap energy of BCD (λex ≤ 360 nm). On the other hand, with increasing excitation wavelength to the tail region of absorption (Fig. 1a), PL became red-shifted (inset of Fig. 5a). Accordingly, the excitation-dependent property of PL was observed by the excitation of long wavelengths (λex > 360 nm),7,20 whereas the intensity of PL was considerably reduced (Fig. 5a). For instance, upon excitation of 380 nm, the band maximum was observed at 435 nm, which was red-shifted from the band maximum of 405 nm (λex ≤ 360 nm). Indeed, the emission peak of BCD was also observed at 435 nm upon excitation of 365 nm in the previous study.23 The time profile of 435 nm (λex = 380 nm) showed the bi-exponential decay with the time constants of 0.8 and 4.5 ns (Fig. 5b), which was different from the single exponential decay of 405 nm with lifetime of 2.2 ns (λex = 355 nm). The band maximum was further shifted to ∼500 nm (inset of Fig. 5a) by excitation of a longer wavelength in near-IR region (800 nm), and the time profile of 500 nm (λex = 800 nm) was different from that of 405 nm (λex = 710 nm).
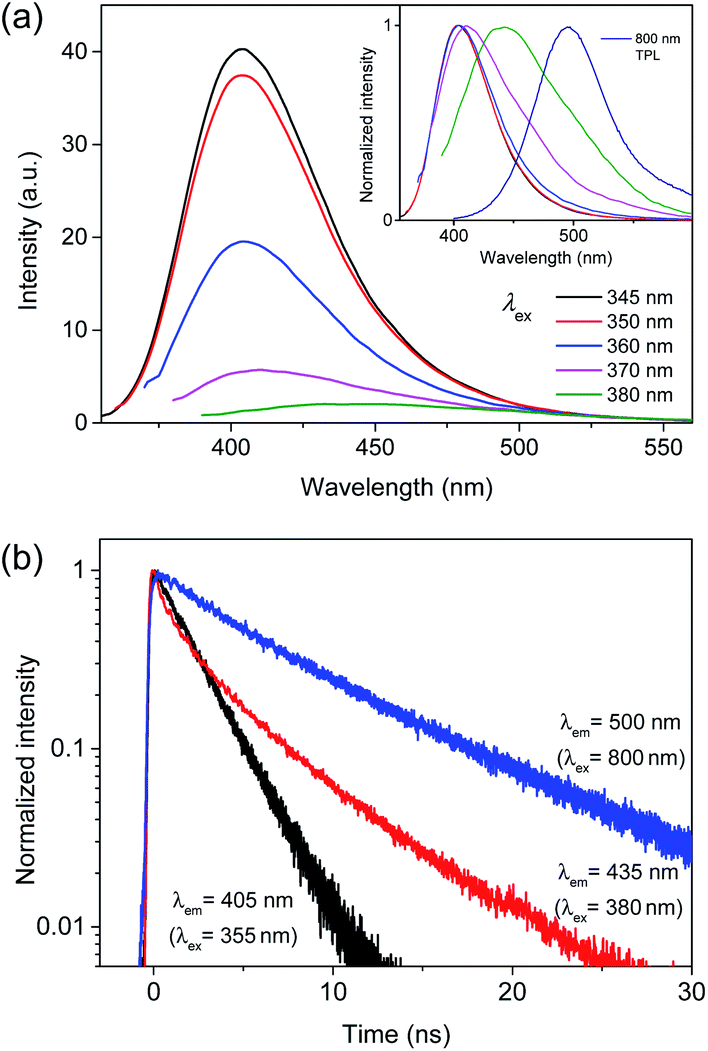 | ||
| Fig. 5 (a) PL spectra of BCD with increasing excitation wavelength to the tail region of absorption. The inset shows the normalized PL spectra. (b) Time-resolved PL profiles of BCD at 405, 435, and 500 nm obtained by excitation of 355, 380, and 800 nm, respectively. | ||
The excitation-dependent PL spectra indicated the existence of multiple chromophores in the intragap region of BCD, which were supported by the multi-component time profiles upon the intragap excitation.16,17 The multiple chromophores in CDs have been attributed to several types of trapped states, such as self-trapped exciton states and surface-related defect states, whose energy levels were influenced by local environments.38–41 The contribution of trapped states in optical transitions was evidenced by the comparison of absorption to PLE spectra of BCD (Fig. S12†). The absorption profile showed the tail in the long wavelength region (>360 nm), which was much reduced in the PLE profile. Accordingly, the absorbance in the long wavelength region was ascribed to the involvement of trapped states,42,43 which were responsible for the excitation-dependent PL spectra by the intragap excitation. Besides, the reduced intensity suggested the low emission efficiency of trapped states (<5%),20 because the absorbance in the long wavelength region indicated the nontrivial density of trapped states. In addition, the emission of trapped states was not easily discernible from the emission of band gap states by excitation of band gap region (Fig. 2). This implied that the energy transfer from band gap states to trapped states did not occur effectively, despite lower energy levels, suggesting the localized nature of trapped states.16,29
The low emission efficiency and ineffective energy transfer indicated that the trapped states would be the surface-related defect states, rather than the self-trapped exciton states.39–41 Since the energy levels of surface states were strongly influenced by local environments,16–20 the appearance of multiple chromophores was correlated to various local conditions at the surface of CDs. Therefore, a few surface-related defect states were involved in the intragap optical transitions of BCD, although the contribution of defect states was much lower than that of band gap states in the emission profile. Notably, the ratio of two bands (ID/IG) in Raman spectra of BCD was slightly higher than the ratios of GCD and RCD (Fig. S4†), despite the similar basic structures of conjugated systems. The enhanced intensity of D-band supported the contribution of defect states in BCD, because D-band was correlated to the defect states as well as the disordered structures.18
On the other hand, TPL (λex = 710 nm) exhibited the nearly identical shape to the typical PL in GCD and RCD (Fig. 4a). Furthermore, TPL obtained by another wavelength of near-IR (λex = 800 nm) showed the comparable profile (Fig. S13†). In addition, the shapes of PL were barely changed even with increasing excitation wavelength to the tail region of absorption (Fig. S13†), suggesting excitation-independent features of GCD and RCD. Moreover, the absorption profiles of GCD and RCD did not significantly deviate from the PLE profiles near the onset (Fig. S12†), supporting the low contribution of the defect states in the optical transitions.
Despite the nontrivial density of defect states, the low emission efficiency of defect states, to which the energy transfer from band gap states was not active, was responsible for the excitation-independent spectra with only a minor contribution of defect states, when the band gap region was excited in BCD (Fig. 6). However, the optical transition of defect states was discernible by the intragap excitation, where the band gap emission was absent (Fig. 5a). Besides, the coexistence of a few defect states led to the multiple components in the time profiles (Fig. 5b), suggesting that the interaction between the defect states was not strong with the localized features (Fig. 6).39–41 Nevertheless, the shape in the long wavelength region of TPL was different from that of PL in BCD (Fig. 4a), although two-photon energy of 710 nm was identical to single-photon energy of 355 nm. The appearance of the tail in the long wavelength was ascribed to the oscillator strength of two-photon process, which was not equal to that of single-photon process.30,44 The optical transition of defect states, which was relatively enhanced for two-photon absorption, could be detectable to some extent even by the band gap excitation (Fig. 6). Still, the oscillator strength for two-photon process of 710 nm of band gap states was much higher than that of defect states, leading to a higher emission intensity of band gap states than that of defect states in the TPL spectrum of BCD (Fig. 4a).
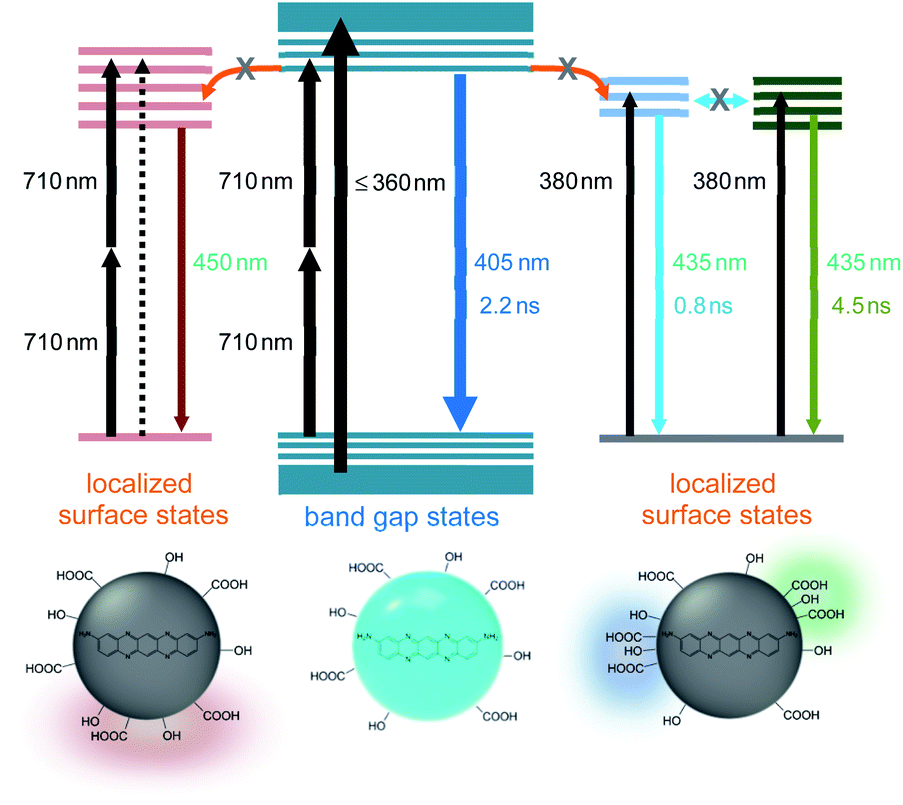 | ||
| Fig. 6 Schematic band structures of BCD (middle). A few localized surface states are accessible by direct absorption, leading to the multiple components in the time profiles of PL (right). The optical transition to surface states is enhanced for two-photon absorption of 710 nm (left). | ||
4. Summary
Three types of CDs were synthesized from isomers of PDs to develop multicolor nanomaterials with unique optical transitions. CDs showed the comparable sizes, element ratios, and functional groups, indicating the similar basic structures of conjugated systems. However, the distinctive optical transitions suggested the different electronic structures in the three types of CDs, which were supported by excitation-independent emission shapes and the single exponential decays. The calculation results indicated that characteristic electronic structures were primarily attributable to the configurational effects, while the constitution of band structures was also influenced by sizes, chemical compositions, and functional groups of CDs. TPL obtained by near-IR excitation exhibited the comparable shape and time profile to typical PL with high photostability. The excitation-dependent property was observed in BCD by the intragap excitation, although the emission intensity was significantly reduced. The low emission efficiency and ineffective energy transfer suggested the contribution of surface-related defect states, whose energy levels were influenced by local environments. Overall, the detailed studies of optical transitions in CDs enhance the controllability of tunable nanomaterials for various applications, such as multicolor bioimaging and optoelectronic devices, with alleviated concern of toxicity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2021R1A2C1003767, 2021R1A4A5032876, and 2020R1A2C2013790).References
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- Y. Lee and Y. Lo, Adv. Funct. Mater., 2009, 19, 604–609 CrossRef.
- Y. Rong, C. Wu, J. Yu, X. Zhang, F. Ye, M. Zeigler, M. E. Gallina, I. Wu, Y. Zhang, Y. Chan, W. Sun, K. Uvdal and D. T. Chiu, ACS Nano, 2013, 7, 376–384 CrossRef CAS PubMed.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS.
- A. Patra, C. G. Chandaluri and T. P. Radhakrishnan, Nanoscale, 2012, 4, 343–359 RSC.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- Y. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS PubMed.
- S. Qu, D. Zhou, D. Li, W. Ji, P. Jing, D. Han, L. Liu, H. Zeng and D. Shen, Adv. Mater., 2016, 28, 3516–3521 CrossRef CAS PubMed.
- Z. Yang, M. Wang, A. M. Yong, S. Y. Wong, X. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- P. Hsu and H. Chang, Chem. Commun., 2012, 48, 3984–3986 RSC.
- K. Holá, M. Sudolská, S. Kalytchuk, D. Nachtigallová, A. L. Rogach, M. Otyepka and R. Zbořil, ACS Nano, 2017, 11, 12402–12410 CrossRef PubMed.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118–5120 RSC.
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed.
- S. Sun, L. Zhang, K. Jiang, A. Wu and H. Lin, Chem. Mater., 2016, 28, 8659–8668 CrossRef CAS.
- C. Chien, S. Li, W. Lai, Y. Yeh, H. Chen, I. Chen, L. Chen, K. Chen, T. Nemoto, S. Isoda, M. Chen, T. Fujita, G. Eda, H. Yamaguchi, M. Chhowalla and C. Chen, Angew. Chem., Int. Ed., 2012, 51, 6662–6666 CrossRef CAS PubMed.
- L. Wang, S. Zhu, H. Wang, S. Qu, Y. Zhang, J. Zhang, Q. Chen, H. Xu, W. Han, B. Yang and H. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- L. Bao, C. Liu, Z. Zhang and D. Pang, Adv. Mater., 2015, 27, 1663–1667 CrossRef CAS PubMed.
- H. Ding, S. Yu, J. Wei and H. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS PubMed.
- Y. Chen, H. Lian, Y. Wei, X. He, Y. Chen, B. Wang, Q. Zeng and J. Lin, Nanoscale, 2018, 10, 6734–6743 RSC.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. Tsang, X. Yang and S. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- B. Zhu, S. Sun, Y. Wang, S. Deng, G. Qian, M. Wang and A. Hu, J. Mater. Chem. C, 2013, 1, 580–586 RSC.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS PubMed.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436 CrossRef PubMed.
- W. Kuo, X. Shen, C. Chang, H. Kao, S. Lin, J. Wang and P. Wu, ACS Nano, 2020, 14, 11502–11509 CrossRef CAS PubMed.
- S. Song, K. Liu, J. Wei, Q. Lou, Y. Shang and C. Shan, Nano Lett., 2019, 19, 5553–5561 CrossRef CAS PubMed.
- D. Rodríguez-Padrón, A. D. Jodlowski, G. de Miguel, A. Puente-Santiago, A. M. Balu and R. Luque, Green Chem., 2018, 20, 225–229 RSC.
- M. Wu, J. Zhan, B. Geng, P. He, K. Wu, L. Wang, G. Xu, Z. Li, L. Yin and D. Pan, Nanoscale, 2017, 9, 13195–13202 RSC.
- T. Lee, S. Won, Y. Park and W. Kwon, ACS Appl. Nano Mater., 2021, 4, 2462–2469 CrossRef CAS.
- W. Kuo, Y. Shao, K. Huang, T. Chou and C. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 14438–14446 CrossRef CAS PubMed.
- A. Molla, H. Lee, Y. Ju, J. Choi and J. Kim, Dyes Pigm., 2022, 197, 109883 CrossRef CAS.
- S. Jeong, M. Ko, S. Jeong, S. Y. Shin, S. M. Park, Y. R. Do and J. K. Song, J. Phys. Chem. C, 2020, 124, 14400–14408 CrossRef CAS.
- H. W. Choi, U. Lee, S. Jeong, Y. Kim and J. K. Song, J. Phys. Chem. C, 2020, 124, 25521–25528 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. Li, W. Yao, J. Liu, Q. Tian, L. Liu, J. Ding, Q. Xue, Q. Lu and W. Wu, J. Mater. Chem. C, 2017, 5, 6512–6520 RSC.
- W. Yao, Q. Tian and W. Wu, Adv. Opt. Mater., 2019, 7, 1801171 CrossRef.
- Y. Wu and W. Wu, Adv. Opt. Mater., 2021, 9, 2100281 CrossRef CAS.
- S. Kim, B. Yoo, Y. Choi, B. Kim and O. Kwon, Phys. Chem. Chem. Phys., 2018, 20, 11673–11681 RSC.
- L. Xiao, Y. Wang, Y. Huang, T. Wong and H. Sun, Nanoscale, 2017, 9, 12637–12646 RSC.
- H. S. Shim, M. Ko, S. Jeong, S. Y. Shin, S. M. Park, Y. R. Do and J. K. Song, J. Phys. Chem. C, 2021, 125, 9965–9972 CrossRef CAS.
- S. Ghosh, A. M. Chizhik, N. Karedla, M. O. Dekaliuk, I. Gregor, H. Schuhmann, M. Seibt, K. Bodensiek, I. A. T. Schaap, O. Schulz, A. P. Demchenko, J. ö. Enderlein and A. I. Chizhik, Nano Lett., 2014, 14, 5656–5661 CrossRef CAS PubMed.
- A. Podborska, B. Gaweł, Ł. Pietrzak, I. B. Szymańska, J. K. Jeszka, W. Łasocha and K. Szaciłowski, J. Phys. Chem. C, 2009, 113, 6774–6784 CrossRef CAS.
- S. J. Ikhmayies and R. N. Ahmad-Bitar, J. Mater. Res. Technol., 2013, 2, 221–227 CrossRef CAS.
- G. Nagamine, H. B. Nunciaroni, H. McDaniel, A. L. Efros, C. H. de Brito Cruz and L. A. Padilha, Nano Lett., 2018, 18, 6353–6359 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S13. See DOI: 10.1039/d1na00811k |
| ‡ Present Address: Department of Chemistry and Chemical Engineering, Education and Research Center for Smart Energy and Materials, Inha University, Incheon 22212, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2022 |