
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe doped NiS nanosheet arrays grown on carbon fiber paper for a highly efficient electrocatalytic oxygen evolution reaction†
Wenrui
Li‡
,
Haofei
Zhao‡
,
Hao
Li
and
Rongming
Wang
 *
*
Beijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory for Magneto-Photoelectrical Composite and Interface Science, School of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China. E-mail: rmwang@ustb.edu.cn
First published on 18th January 2022
Abstract
Developing efficient and low-cost non-noble metal catalysts for the oxygen evolution reaction (OER) is important for hydrogen production through water electrolysis. Herein, Fe doped NiS nanosheets directly grown on conductive carbon fiber paper (Fe–NiS@CFP) were fabricated through a two-step hydrothermal process. The microstructure, interface and electronic states of the prepared sample were modulated by Fe doping, exhibiting small internal and interface charge-transfer resistance. Benefiting from these factors, Fe–NiS@CFP shows superior electrocatalytic performance with an overpotential of 275 mV at 100 mA cm−2 and maintains the activity for at least 50 h as a working electrode for the OER. This work may provide insights into the design and fabrication of non-noble metal sulfide electrocatalysts.
Introduction
The increasing energy demand and imminent threat of global warming have prompted researchers to explore sustainable green energies.1 Hydrogen, as a high density (∼282 kJ mol−1) and zero-carbon energy source, is an ideal candidate.2,3 Electrocatalytic water splitting can provide a feasible method for hydrogen production, and it can be divided into the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode.4,5 The OER involving a four-electron transfer is a very slow kinetic process, thus efficient electrocatalysts are essential to improve the overall water-splitting efficiency.6,7 At present, the commercial OER catalysts are noble-metal-based materials (RuO2, IrO2, etc.), but the high cost and scarcity limit their wide application.8 Therefore, developing efficient and non-noble metal oxygen evolution catalysts is highly desirable.9Transition metal compounds have been widely considered for their earth abundance and similar electronic structure to noble metals. Among the potential candidates, nickel sulfides (NiS, NiS2, Ni3S4, Ni3S2, etc.) have been studied extensively due to their considerable electrocatalytic performances.10–14 However, these materials suffer from low conductivity; one possible strategy is combining them with conductive substrates, but the binder would introduce additional resistance in the prepared electrode through a postcoating process.12,15 Direct growth of these nanomaterials on conducting substrates such as carbon paper, nickel foam, and so forth can increase the internal conductivity, thus improving the catalytic performance.16,17 Zhu et al. reported that NiS nanosheets supported by nickel foam exhibit enhanced mass transport and charge mobility, and need an overpotential of 320 mV (20 mA cm−2) when employed as an OER electrode in an alkaline medium.12 Guo et al. showed that NiS2 on CFP without a binder requires a small overpotential of 246 mV (10 mA cm−2) for the OER.18 Meanwhile, heteroatom doping is an effective strategy to tune the catalytic performance of electrocatalysts through microstructure and electronic state modulation.19,20 Han et al. found that Fe doping could boost the OER activity of Ni3S2 by triggering a synergic effect combining size decrease, electronic state modification, and strain engineering.21 Liu et al. studied the electronic state reconfiguration of V-doped pyrite NiS2, which exhibited superior electroactivity for overall water splitting.22 Thus, doped samples directly grown on conductive substrates may exhibit superior electroactivity and should be ideal for the study of heteroatom doping and interface modulation.
Herein, 3D interconnected Fe-doped NiS nanosheets in situ grown on carbon fiber paper (Fe–NiS@CFP) were fabricated through a two-step hydrothermal process. Benefitting from the low resistance that resulted from direct growth on a conductive substrate and the modulation of the microstructure and electronic states by Fe doping, Fe–NiS@CFP exhibits excellent OER performance in 1 M KOH. The overpotential of Fe–NiS@CFP is only 275 mV at the current density of 100 mA cm−2, and the current density is still stable after a 50 hour chronopotentiometry test at ∼100 mA cm−2.
Experimental
Synthesis of the samples
The sample was synthesized by a two-step hydrothermal process. A piece of carbon fiber paper (CFP) (1 cm × 1.2 cm) was pre-washed ultrasonically with acetone, ethanol, and deionized water respectively for 10 min and dried for 8 h. Firstly, 1 mmol iron(II) acetate (Fe(CH3COO)2), 2 mmol nickel acetate (Ni(CH3COO)2·4H2O), 10 mmol urea and 4 mmol ammonium fluoride (NH4F) were dissolved in 40 mL deionized water under stirring for 30 min to form a green solution. The green solution was transferred into a 50 mL Teflon-lined stainless-steel autoclave with cleaned CFP and heated at 120 °C for 6 h in an oven. After cooling to room temperature, the CFP was taken out, washed with deionized water and ethanol several times, and subsequently dried at 50 °C for 8 h. Then the NiFe-layered double hydroxide (LDH) product was formed on CFP, and the sample is denoted as NiFe LDH@CFP.Secondly, the as-obtained NiFe LDH@CFP was put into 40 mL deionized water containing 1 M Na2S·9H2O and heated at 150 °C for 8 h in an oven. The product was rinsed repeatedly with deionized water and ethanol several times after naturally cooling to room temperature, and then dried at 50 °C for 8 h. Fe doped NiS was synthesized on CFP and is denoted as Fe–NiS@CFP. The mass loading of Fe–NiS@CFP was measured as 1.3 mg cm−2.
NiS@CFP was synthesized without an Fe source by a similar process for comparison.
Microstructure characterization
The prepared samples were investigated with scanning electron microscopy (SEM), Brunauer–Emmett–Teller (BET) surface area analysis, X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and inductively coupled plasma optical emission spectrometry (ICP-OES). SEM was performed on a Zeiss SUPRA55 SEM. The XRD patterns were acquired with a Rigaku Smartlab(3) diffractometer with Cu Kα radiation (λ = 0.15406 nm). FTIR spectra were collected on a PerkinElmer spectrum GX. The TEM characterizations were made on a JEOL JEM-2200FS and an FEI ETEM Titan G80-300. XPS measurements were made on a PHI5000 Versaprobe III X-ray photoelectron spectrometer using Al Kα as the excitation source (hν = 1486.6 eV). ICP-OES measurements were performed on an American Agilent 5110. BET surface area data were obtained using a pore size analyzer (Tristar II 3020).Electrochemical measurements
The electrochemical experiments were conducted with a CHI760E electrochemical workstation (CH Instruments, China) using a three-electrode system in 1 M KOH solution. The prepared samples served as the working electrode, while Hg/HgO and platinum were used as the reference electrode and the counter electrode, respectively. All potentials were calibrated to the reversible hydrogen electrode (RHE) using the following equation:| ERHE = EHg/HgO + 0.098 V + 0.0592 × pH |
Linear sweep voltammetry (LSV) measurements were conducted with a scan rate of 5 mV s−1 to minimize the capacitive current. All the measured currents were 100% iR-corrected. Tafel plots were extracted from LSV curves and fitted to the Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj + a, where η is the overpotential, j is the current density, and b is the Tafel slope). Electrochemical impedance spectroscopy (EIS) measurements were conducted in the range of 100 kHz to 0.0005 Hz with an AC voltage amplitude of 5 mV. A simplified Randles equivalent circuit was used to fit the impedance data to obtain the concrete charge-transfer resistances. The turnover frequency (TOF) measurements were calculated from the following equation: TOF = j × A/(4 × F × n), where A is the geometric area of the samples, j is the current density at a constant overpotential, F is the Faraday constant and n is the number of moles of the active materials. For n calculation, we quantify the surface concentration of the active sites by electrochemistry according to the previous report.23 The electrochemically active surface areas (ECSAs) of the samples were estimated from their double layer capacitance in the non-faradaic region using scan rates from 40 to 200 mV s−1.
logj + a, where η is the overpotential, j is the current density, and b is the Tafel slope). Electrochemical impedance spectroscopy (EIS) measurements were conducted in the range of 100 kHz to 0.0005 Hz with an AC voltage amplitude of 5 mV. A simplified Randles equivalent circuit was used to fit the impedance data to obtain the concrete charge-transfer resistances. The turnover frequency (TOF) measurements were calculated from the following equation: TOF = j × A/(4 × F × n), where A is the geometric area of the samples, j is the current density at a constant overpotential, F is the Faraday constant and n is the number of moles of the active materials. For n calculation, we quantify the surface concentration of the active sites by electrochemistry according to the previous report.23 The electrochemically active surface areas (ECSAs) of the samples were estimated from their double layer capacitance in the non-faradaic region using scan rates from 40 to 200 mV s−1.
Results
The 3D interconnected Fe–NiS@CFP nanosheet arrays were obtained through a facile two-step hydrothermal procedure, as illustrated in Scheme 1. Firstly, the NiFe LDH nanosheets were prepared by a typical hydrothermal method using metal acetate, urea, and NH4F. The presence of urea promotes the formation of the LDH structure by releasing OH− and CO32− ions during the synthesis process. Moreover, NH4F acts as the morphology-controlling agent to build the nanosheet structure by retarding the nucleation reaction.24 Subsequently, hydrothermal treatment in 1 M Na2S solution led to the formation of the sulfide.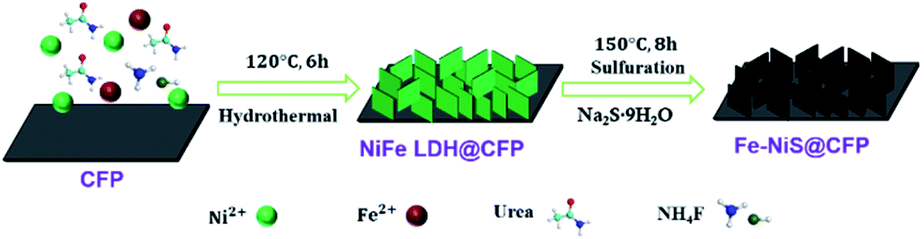 | ||
| Scheme 1 Schematic illustration for the stepwise preparation of Fe–NiS@CFP. | ||
The morphologies of the precursors and sulfides were observed by scanning electron microscopy (SEM). SEM image shows that the CFP as the substrate for the growth of the electrocatalyst has a smooth surface (Fig. S1†). After the first hydrothermal process, the fiber surfaces are evenly covered by 3D interconnected nanosheet arrays (Fig. 1a). After Fe doping and sulfurization, the samples retain the interconnected nanosheet morphology (Fig. 1b–d), and the surface of the nanosheets becomes rougher as shown in high magnification images (insets). The 3D interconnected nanosheet structures with excellent structural stability and high surface area could increase the contact area with the electrolyte and facilitate the release of products during the catalytic process.25 The nanosheets become smaller after Fe doping and sulfurization. By measuring the sizes of the nanosheets, the average length decreased from ∼2.2 μm for NiS@CFP to ∼0.7 μm for Fe–NiS@CFP. The average thickness of the nanosheets of Fe–NiS@CFP (∼20 nm) is also lower than that of NiS@CFP (∼60 nm). The BET measurement results reveal that the specific surface area of Fe–NiS@CFP is 2.8 m2 g−1, larger than that of NiFe LDH@CFP (2.3 m2 g−1), NiS@CFP (1.9 m2 g−1) and Ni LDH@CFP (1.3 m2 g−1). The corresponding N2 adsorption–desorption isotherm distribution results are shown in Fig. S2.†
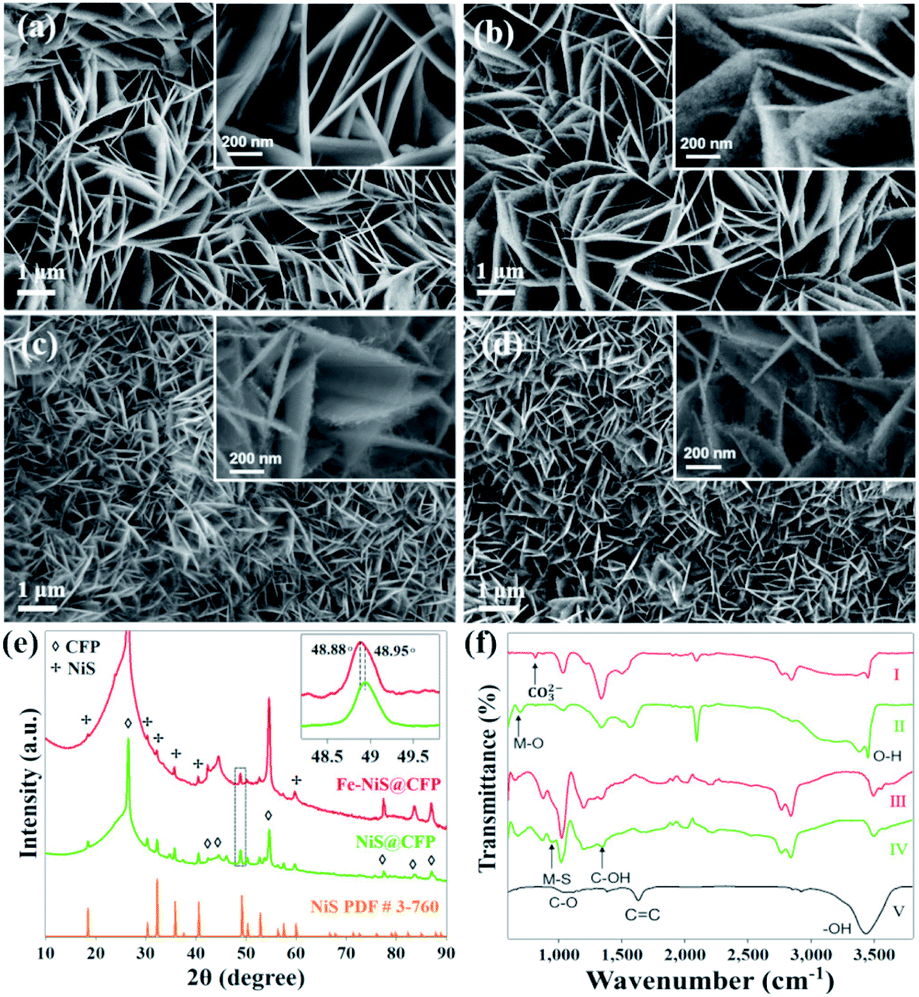 | ||
| Fig. 1 SEM images of (a) Ni LDH@CFP, (b) NiS@CFP, (c) NiFe LDH@CFP, and (d) Fe–NiS@CFP; insets: high-magnification SEM images representing the typical morphology of the nanosheets. (e) XRD spectra of Fe–NiS@CFP and NiS@CFP; inset: magnified (131) peak from the dashed square showing a blue shift of the peak after Fe doping. (f) FTIR spectra of NiFe LDH@CFP (I), Ni LDH@CFP (II), Fe–NiS@CFP (III), NiS@CFP (IV) and CFP (V). | ||
The crystal structure of the samples was examined by X-ray diffraction (XRD). The acquired patterns of the precursors are shown in Fig. S3,† and the diffraction peaks agree well with that of Ni(Fe) LDH (JCPDS no. 40-125). Besides the peaks of the CFP substrate, the diffraction peaks of the prepared sulfides at 18.5°, 30.4°, 32.2°, 35.8°, 40.5°, 48.9°, 52.7° and 57.6° correspond to the (110), (101), (300), (021), (211), (131), (401) and (330) planes of rhombohedral NiS (JCPDS no. 3-760), respectively, as shown in Fig. 1e. It should be mentioned that there are some minor peaks at 34.7°,45.9° and 53.7° in the pattern of NiS@CFP, which could be indexed to the (101), (102) and (110) planes of the hexagonal NiS (JCPDS no. 75-613). The formation of multi-phase NiS was also observed previously.26 Moreover, the peak position of Fe–NiS@CFP (48.88°) shifts to a lower angle compared with NiS@CFP (48.95°), which could be clearly distinguished in the magnified (131) peak as shown in the inset. This could be ascribed to the larger radius of Fe2+ ion.27 The ICP-OES results suggest that the Fe/(Ni + Fe) ratio in Fe–NiS@CFP is about 16.9%. So, the Fe doped NiS has been successfully prepared by a simple two-step hydrothermal method.
The bonds of CFP and the prepared samples were analysed by FTIR, the acquired spectra are shown in Fig. 1f. The FTIR spectrum of CFP shows the characteristic IR vibrations at 1058 cm−1, 1386 cm−1, 1631 cm−1 and 3545 cm−1 corresponding to C–O, C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and –OH functional groups, respectively.28 The abundant –OH groups could provide sites for the LDH precursors to nucleate and grow. The FTIR spectra for Ni LDH@CFP and NiFe LDH@CFP show a peak at 662 cm−1, which is ascribed to metal–oxygen (M–O) stretching and bending vibrations.29 The peaks at 3545 cm−1 and 3613 cm−1 are the O–H stretching band, arising from interlayer water molecules.29,30 Additionally, the peak at 834 cm−1 results from the bending mode of the interlayer carbonate group.31 The coexistence of water molecules and CO32− confirms the formation of LDHs. For sulfides, the band at 960 cm−1 can be attributed to the symmetrical and asymmetrical stretch of the metal–sulfide (M–S) band.32
C and –OH functional groups, respectively.28 The abundant –OH groups could provide sites for the LDH precursors to nucleate and grow. The FTIR spectra for Ni LDH@CFP and NiFe LDH@CFP show a peak at 662 cm−1, which is ascribed to metal–oxygen (M–O) stretching and bending vibrations.29 The peaks at 3545 cm−1 and 3613 cm−1 are the O–H stretching band, arising from interlayer water molecules.29,30 Additionally, the peak at 834 cm−1 results from the bending mode of the interlayer carbonate group.31 The coexistence of water molecules and CO32− confirms the formation of LDHs. For sulfides, the band at 960 cm−1 can be attributed to the symmetrical and asymmetrical stretch of the metal–sulfide (M–S) band.32
We further investigated the prepared samples with transmission electron microscopy (TEM). Fig. 2a and d are representative bright-field TEM images of NiS@CFP and Fe–NiS@CFP, respectively. There are many nanoparticles on the nanosheets, and the corresponding selected area electron diffraction (SAED) patterns (Fig. 2a and d insets) could be well indexed with NiS (JCPDS no. 3-760), consistent with the XRD results. The diffraction rings in the pattern of Fe–NiS@CFP are more obvious, suggesting that the orientations of the nanoparticles in the Fe-doped sample are more irregular. The high-resolution TEM (HRTEM) images (Fig. 2b and e) reveal that the sulfides are composed of many crystallites of several nanometers. The interplanar spacings are 3.0, 2.2, 1.8 and 1.6 Å, which can be ascribed to the (101), (211), (131) and (330) planes of NiS, respectively. Moreover, holes with sizes of several nanometers and defects are also observed on the surface in the HRTEM images, and they can provide more active sites and regulate the electronic states, thus improving the electrocatalytic performance.7,10,33–36 The high-angle annular dark-field scanning TEM (HAADF-STEM) and elemental mapping results suggest that Ni, S and Fe elements are homogeneously distributed throughout the nanosheets in the sulfides (Fig. 2c and f), which further confirms the formation of Fe doped NiS.
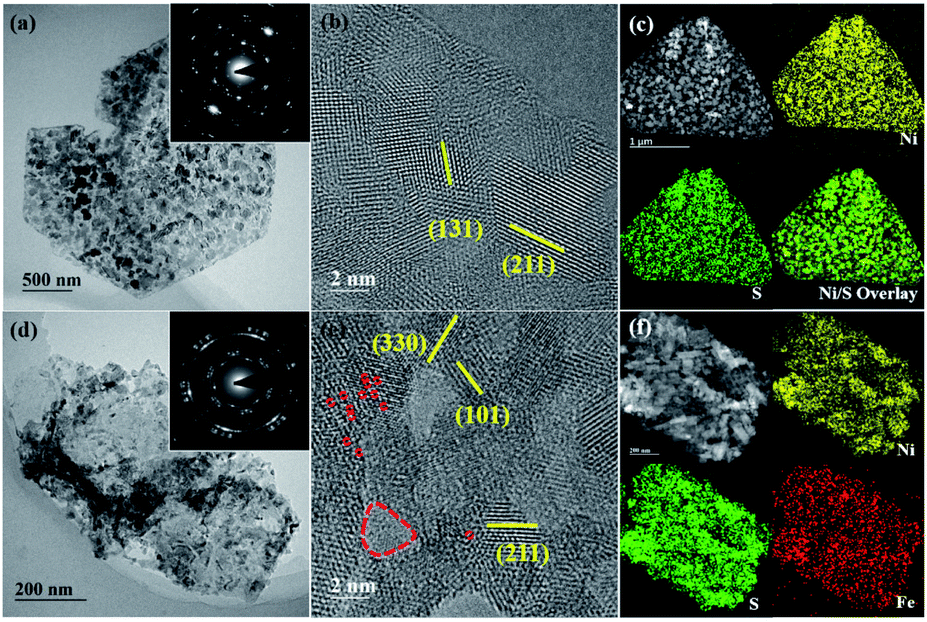 | ||
| Fig. 2 Representative TEM images of (a–c) NiS@CFP and (d–f) Fe–NiS@CFP. (a and d) Bright-field images, insets are the corresponding SAED images; (b and e) high-resolution TEM images revealing the existence of holes and defects, which are marked with red dashed circles; (c and f) HAADF-STEM images and the corresponding elemental map of Ni, S and Fe. | ||
The composition and electronic states of NiS@CFP and Fe–NiS@CFP were investigated by XPS. The survey spectrum of Fe–NiS@CFP (Fig. 3a) verifies the presence of Ni, Fe, and S. The carbon and oxygen elements may result from the adsorbed surface organic matter. In the high resolution spectra of Ni 2p (Fig. 3b), there are two peaks located at 853.7 and 855.8 eV ascribed to Ni2+ and Ni3+, respectively.2,17,37 Besides, the presence of Ni3+ may be due to the oxidation of the surface.33,38–40 Compared with NiS@CFP (853.36 eV), the peaks for Ni 2p in Fe–NiS@CFP are shifted to the higher binding energies (853.54 eV), which may be related to electronic interactions between Ni, Fe and S in Fe–NiS@CFP.41,42 As shown in Fig. 3c, the S 2p spectra could be deconvoluted into four peaks: 161.7, 162.9, 162.4 and 168.8 eV, corresponding to S 2p3/2, S 2p1/2 of S2−, the bond between metal and sulfur (M–S)43–45 and sulfate species resulting from surface oxidation of sulfide in air,37,46 respectively. It is noteworthy that the peak position of S2− in Fe–NiS@CFP is lower than that of the undoped sample, indicating the increase of electron density near the S atom,47,48 conforming to the result of Ni 2p spectra. The binding energy range of the Fe 2p signal overlaps with the Ni LMM signal.48 However, the spectrum of Fe–NiS@CFP exhibits an obvious peak at 706.7 eV (Fig. 3d), which could be ascribed to the Fe–S bond,42,49–51 suggesting the presence of Fe element. The above results confirm that the Fe-doped sample has been successfully synthesized.
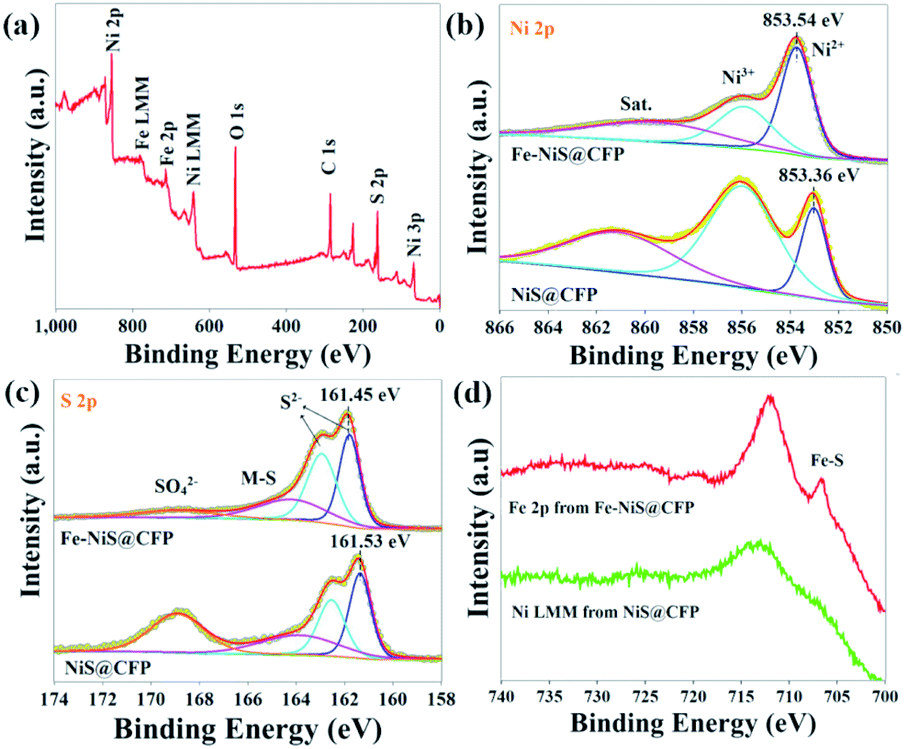 | ||
| Fig. 3 (a) Survey spectrum of Fe–NiS@CFP. High-resolution (b) Ni 2p and (c) S 2p spectra with deconvolution of peaks of NiS@CFP and Fe–NiS@CFP. (d) Spectra acquired at binding energies between 700 eV and 740 eV. | ||
The as-prepared samples can be directly used as a working electrode. In order to evaluate the electrocatalytic performances of the prepared samples, LSV was carried out at a scanning rate of 5 mV s−1 in an O2-saturated 1 M KOH electrolyte using a standard three-electrode system. Blank CFP was tested under the same conditions and exhibited negligible electrocatalytic activity. The iR-compensated LSV curves are shown in Fig. 4a. Due to the presence of an obvious Ni oxidation peak, it is difficult to determine the overpotential when the current density is 10 mA cm−2. Hence, we used the overpotential at 100 mA cm−2 (η100) as an indicator of electrochemical performance. The η100 of Ni LDH@CFP is 524 mV, while the overpotential is reduced to 305 mV after Fe doping. This may be attributed to the modulation of the local electron density of Ni sites by Fe.30,52,53 After sulfurization, the η100 of Fe–NiS@CFP is further reduced to 275 mV, which even outperformed the state-of-the-art catalysts (355 mV). The mass loading of RuO2@CFP is equivalent to that of Fe–NiS@CFP. The activity of RuO2@CFP was similar to that presented in previous papers.8,44 Furthermore, the electrocatalytic activity of Fe–NiS@CFP is close and even superior to those of the previously reported supported transition metal sulfide electrocatalysts in alkaline media, such as Ni3S2@NiV-LDH/NF (320 mV), FeCo2S4/CC (317 mV), Zn–Ni3S2/NF (300 mV), Cr3–Co6S8/NF (313 mV), Ni2.3%–CoS2/CC (370 mV), NiCO2S4/CC (340 mV) and (Ni,Fe)S2/(Ni,Fe)3S4 (320 mV).23,25,44,54–57 Besides, Fe–NiS@CFP exhibits the smallest Tafel slope of 57.2 mV dec−1 (Fig. 4b), implying faster OER reaction kinetics. And a Tafel slope of about 60 mV dec−1 suggests that the fourth step in the OER reaction path is the rate-determining step.58,59
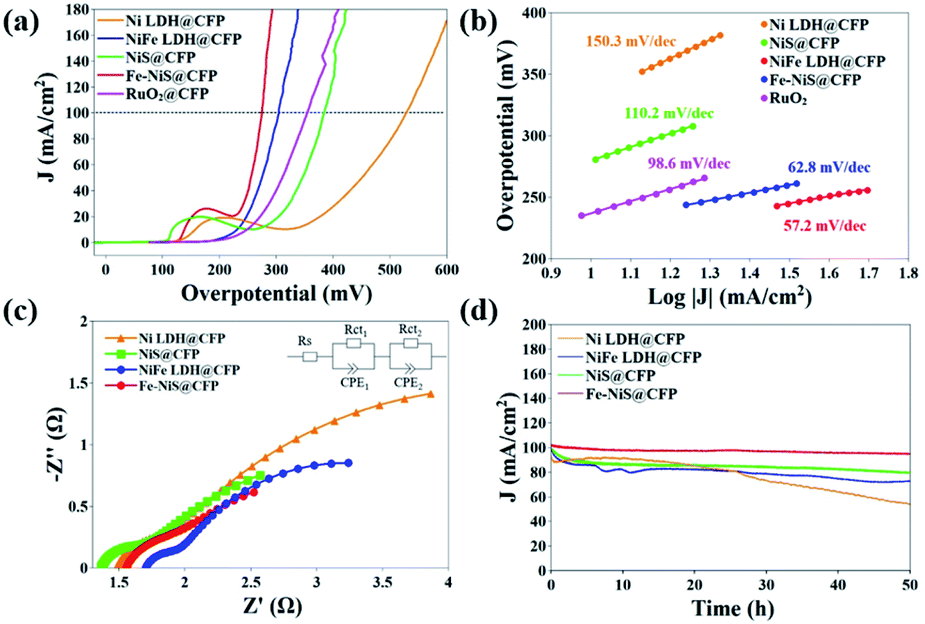 | ||
| Fig. 4 Electrocatalytic performances of the prepared samples: (a) LSV curves, (b) Tafel plots, (c) Nyquist plots and (d) stability tests. For comparison, RuO2 dispersed on CFP (RuO2@CFP) was tested under the same conditions and the results are shown in (a) and (b). The EIS data were fitted with a simplified Randles equivalent circuit as shown in the inset of (c). | ||
Fig. 4c shows the Nyquist plots of the samples fitted with a Randles equivalent circuit model (inset). The semicircles in the high and low frequency regions are ascribed to the internal charge-transfer resistances (Rct1) of electrodes and the interface resistances between the electrode and the electrolyte (Rct2), respectively.60 The fitted parameters are shown in Table S1.† The Rct1 values are as low as ∼0.3 Ω indicating a fast charge-transfer in the electrode, which should result from the direct growth of the samples on the substrates. The total charge-transfer resistances of Ni LDH@CFP, NiFe LDH@CFP, NiS@CFP and Fe–NiS@CFP are 4.54 Ω, 3.75 Ω, 2.81 Ω and 2.52 Ω, respectively.
The electrochemical active surface area (ECSA) of these catalysts was evaluated through Cdl by measuring cyclic voltammograms at various scan rates in a nonfaradaic region (Fig. S4†).61–63 The Cdl value of Fe–NiS@CFP is 1.48 mF cm−2, larger than that of NiFe LDH@CFP (0.73 mF cm−2) and NiS@CFP (0.58 mF cm−2), but smaller than that of Ni LDH@CFP (2.24 mF cm−2), which demonstrates that the superior electrocatalytic activity of Fe–NiS@CFP should be ascribed to its intrinsic activity. Furthermore, the TOF defined as the number of moles of O2 produced per second at each active site, is used to estimate the intrinsic properties of the samples.64 The TOF value of Fe–NiS@CFP is 1.25 s−1 at an overpotential of 400 mV, larger than that of NiFe LDH@CFP (0.47 s−1), NiS@CFP (0.30 s−1) and Ni LDH@CFP (0.21 s−1). It is clear that Fe doping and sulfurization facilitate the charge transfer, thus contributing to the improvement of the intrinsic OER activity for Fe–NiS@CFP.
The stability of the electrocatalysts was evaluated with chronoamperometry at the overpotential of η100. As presented in Fig. 4d, Fe–NiS@CFP demonstrates outstanding stability with less than 6% decay of the current density after a 50 h test, while the decay of NiS@CFP, NiFe LDH@CFP and Ni LDH@CFP is 20%, 30% and 50%, respectively. The η100 of Fe–NiS@CFP derived from the LSV curve only increased 12 mV after the test, as shown in Fig. S5.†
After the long-term stability test (∼100 mA cm−2, 50 h), Fe–NiS@CFP retains the 3D interconnected nanosheet morphology with a relatively rougher surface of the nanosheets (Fig. 5a). The chemical composition and microstructure may undergo severe changes during the reaction as revealed by in situ investigations.65,66 In the EDX spectrum (Fig. S6†), the peak of S almost disappeared, and the peak of O significantly increased, which suggests that the sulfide transforms into the (oxy)hydroxide during the electrocatalytic process. The Ni 2p3/2 region of Fe–NiS@CFP XPS spectrum could be deconvoluted into three peaks at 857.9 eV, 856.1 eV and 862.3 eV (Fig. 5b), corresponding to NiOOH, Ni(OH)2 and the satellite peak,6,38,62 respectively. The peaks at 531.4 eV and 533.5 eV in the O 1s spectrum belong to –OH and adsorbed H2O or –OOH, respectively (Fig. 5c).67,68 Meanwhile, the diffuse rings in the SAED pattern could be indexed with γ-NiOOH (JCPDS no. 6-75) (Fig. S7†). According to previous studies, oxyhydroxides should be the real OER active species,55,69,70 but they are unstable and may transform into Ni(OH)2 after the voltage is withdrawn: NiOOH + e− + H2O → Ni(OH)2 + OH−.71,72 Moreover, the S 2p signal is significantly weak and the peaks of S2− and M–S bond are obviously reduced after the OER test (Fig. 5d), consistent with the EDX result. The residual S may also contribute to the enhanced OER activity.73
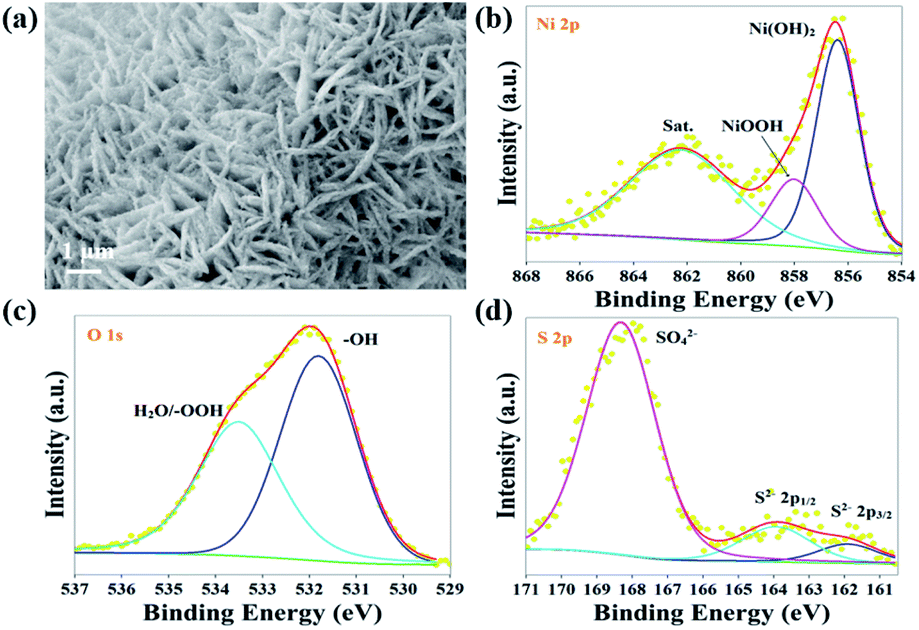 | ||
| Fig. 5 Fe–NiS@CFP catalyst after the chronopotentiometry test at ∼100 mA cm−2 for 50 h: (a) SEM image, XPS spectra of (b) Ni 2p, (c) O 1s and (d) S 2p. | ||
The superior performance of Fe–NiS@CFP could be attributed to the following factors: (1) the 3D interconnected nanosheets directly grow on conductive substrates, resulting in small internal and electrode–electrolyte interface resistances, thus facilitating the charge transfer during the electrochemical reaction; (2) Fe doping and sulfurization result in finer grains, introduction of defects and holes, and the modulation of electronic states, thus contributing to the superior intrinsic electroactivity; (3) the (oxy)hydroxide, obtained by in situ electrochemical formation, is a highly efficient species for the electrocatalytic oxygen evolution reaction.
Conclusions
In summary, Fe doped NiS nanosheets directly grown on carbon fiber paper have been synthesized by a simple two-step hydrothermal method. The prepared Fe–NiS@CFP exhibits outstanding electrocatalytic activity toward the OER with a low overpotential of 275 mV to achieve a current density of 100 mA cm−2, a small Tafel slope of 57.2 mV dec−1, and high stability (less than 6% decay at ∼100 mA cm−2 for 50 h) in 1.0 M KOH. This study may contribute to the development of transition-metal sulfides as electrocatalysts for water splitting.Author contributions
Li W. and Zhao H. wrote the paper with support from Wang R.; all authors contributed to the general discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Beijing Natural Science Foundation (Grant No. 2212034), National Natural Science Foundation of China (No. 51971025 and 12034002) and the Fundamental Research Funds for the Central Universities (FRF-IDRY-19-028).Notes and references
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 4661–4672 CrossRef CAS.
- X. Zhang, H. Xu, X. Li, Y. Li, T. Yang and Y. Liang, ACS Catal., 2015, 6, 580–588 CrossRef.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- K. Zhang, X. Min, T. Zhang, M. Si, J. Jiang, L. Chai and Y. Shi, ACS Appl. Mater. Interfaces, 2020, 12, 54553–54562 CrossRef CAS PubMed.
- L. Du, Y. Sun and B. You, Mater. Rep. Energy, 2021, 1, 100004 Search PubMed.
- Z. Wu, X. Wang, J. Huang and F. Gao, J. Mater. Chem. A, 2018, 6, 167–178 RSC.
- S. Y. Lim, S. Park, S. W. Im, H. Ha, H. Seo and K. T. Nam, ACS Catal., 2019, 10, 235–244 CrossRef.
- J. Lin, P. Wang, H. Wang, C. Li, X. Si, J. Qi, J. Cao, Z. Zhong, W. Fei and J. Feng, Adv. Sci., 2019, 6, 1900246 CrossRef PubMed.
- D. Cao and D. Cheng, Chem. Commun., 2019, 55, 8154–8157 RSC.
- J.-T. Ren and Z.-Y. Yuan, ACS Sustainable Chem. Eng., 2017, 5, 7203–7210 CrossRef CAS.
- Y. Zhu, H. Yang, K. Lan, K. Iqbal, Y. Liu, P. Ma, Z. Zhao, S. Luo, Y. Luo and J. Ma, Nanoscale, 2019, 11, 2355–2365 RSC.
- Y. Lv, S. Duan, Y. Zhu, H. Guo and R. Wang, Chem. Eng. J., 2020, 382, 122794 CrossRef CAS.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, e1806326 CrossRef PubMed.
- S. Shit, S. Chhetri, W. Jang, N. C. Murmu, H. Koo, P. Samanta and T. Kuila, ACS Appl. Mater. Interfaces, 2018, 10, 27712–27722 CrossRef CAS PubMed.
- X. Chen, D. Chen, X. Guo, R. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 18774–18781 CrossRef CAS PubMed.
- Y. Guo, D. Guo, F. Ye, K. Wang and Z. Shi, Int. J. Hydrogen Energy, 2017, 42, 17038–17048 CrossRef CAS.
- Y. Li, J. Yin, L. An, M. Lu, K. Sun, Y. Q. Zhao, D. Gao, F. Cheng and P. Xi, Small, 2018, 14, e1801070 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, 1–12 CrossRef PubMed.
- C. Han, W. Li, C. Shu, H. Guo, H. Liu, S. Dou and J. Wang, ACS Appl. Energy Mater., 2019, 2, 5363–5372 CrossRef CAS.
- H. Liu, Q. He, H. Jiang, Y. Lin, Y. Zhang, M. Habib, S. Chen and L. Song, ACS Nano, 2017, 11, 11574–11583 CrossRef CAS PubMed.
- Q. Liu, L. Xie, Z. Liu, G. Du, A. M. Asiri and X. Sun, Chem. Commun., 2017, 53, 12446–12449 RSC.
- D. Lim, C. Lim, M. Hwang, M. Kim, S. E. Shim and S.-H. Baeck, J. Power Sources, 2021, 490, 229552 CrossRef CAS.
- Q. Liu, J. Huang, Y. Zhao, L. Cao, K. Li, N. Zhang, D. Yang, L. Feng and L. Feng, Nanoscale, 2019, 11, 8855–8863 RSC.
- G. B. Shombe, M. D. Khan, C. Zequine, C. Zhao, R. K. Gupta and N. Revaprasadu, Sci. Rep., 2020, 10, 3260 CrossRef CAS PubMed.
- Z. Zhang, T. Zhang and J. Y. Lee, Ind. Eng. Chem. Res., 2019, 58, 13053–13063 CrossRef CAS.
- G. Sahoo, N. Sarkar, D. Sahu and S. K. Swain, RSC Adv., 2017, 7, 2137–2150 RSC.
- G. E. Nyongombe, Int. J. Electrochem. Sci., 2020, 4494–4502, DOI:10.20964/2020.05.33.
- R. Li, Z. Hu, X. Shao, P. Cheng, S. Li, W. Yu, W. Lin and D. Yuan, Sci. Rep., 2016, 6, 18737 CrossRef CAS PubMed.
- J. Liu, J. Song, H. Xiao, L. Zhang, Y. Qin, D. Liu, W. Hou and N. Du, Powder Technol., 2014, 253, 41–45 CrossRef CAS.
- E. Darezereshki, A. B. Vakylabad, A. Hassanzadeh, T. Niedoba, A. Surowiak and B. Koohestani, Minerals, 2021, 11, 419 CrossRef CAS.
- Y. Zhang, H. Guo, X. Li, J. Du, W. Ren and R. Song, Chem. Eng. J., 2021, 404, 126483 CrossRef CAS.
- C. Lei, W. Zhou, Q. Feng, Y. Lei, Y. Zhang, Y. Chen and J. Qin, Nano-Micro Lett., 2019, 11, 45 CrossRef CAS PubMed.
- Q. Wang, X. Xue, Y. Lei, Y. Wang, Y. Feng, X. Xiong, D. Wang and Y. Li, Small, 2020, 16, e2001571 CrossRef PubMed.
- C. Wang, C. Li, J. Liu and C. Guo, Mater. Rep. Energy, 2021, 1, 100006 Search PubMed.
- C. Liu, D. Jia, Q. Hao, X. Zheng, Y. Li, C. Tang, H. Liu, J. Zhang and X. Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 27667–27676 CrossRef CAS PubMed.
- X. Du, H. Su and X. Zhang, J. Catal., 2020, 383, 103–116 CrossRef CAS.
- M. Yao, B. Sun, L. He, N. Wang, W. Hu and S. Komarneni, ACS Sustainable Chem. Eng., 2019, 7, 5430–5439 CrossRef CAS.
- F. Jing, Q. Lv, J. Xiao, Q. Wang and S. Wang, J. Mater. Chem. A, 2018, 6, 14207–14214 RSC.
- M. Zhou, Q. Weng, X. Zhang, X. Wang, Y. Xue, X. Zeng, Y. Bando and D. Golberg, J. Mater. Chem. A, 2017, 5, 4335–4342 RSC.
- P. Xu, J. Li, J. Luo, L. Wei, D. Zhang, D. Zhou, W. Xu and D. Yuan, Sci. Rep., 2018, 8, 9425 CrossRef PubMed.
- S. Li, W. Huang, Y. Yang, J. Ulstrup, L. Ci, J. Zhang, J. Lou and P. Si, J. Mater. Chem. A, 2018, 6, 20480–20490 RSC.
- X. Hu, R. Wang, P. Sun, Z. Xiang and X. Wang, ACS Sustainable Chem. Eng., 2019, 7, 19426–19433 CrossRef CAS.
- Y. Huang, Y. Zhao, J. Bao, J. Lian, M. Cheng and H. Li, J. Alloys Compd., 2019, 772, 337–347 CrossRef CAS.
- K. Fan, Z. Jin, H. Yang, D. Liu, H. Hu and Y. Bi, Sci. Rep., 2017, 7, 7710 CrossRef PubMed.
- D. Lim, E. Oh, C. Lim, S. E. Shim and S.-H. Baeck, Electrochim. Acta, 2020, 361, 137080 CrossRef CAS.
- G. Zhang, Y.-S. Feng, W.-T. Lu, D. He, C.-Y. Wang, Y.-K. Li, X.-Y. Wang and F.-F. Cao, ACS Catal., 2018, 8, 5431–5441 CrossRef CAS.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- N. Yang, C. Tang, K. Wang, G. Du, A. M. Asiri and X. Sun, Nano Res., 2016, 9, 3346–3354 CrossRef CAS.
- J. Yu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 15838–15844 RSC.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- L. Lei, D. Huang, C. Zhou, S. Chen, X. Yan, Z. Li and W. Wang, Coord. Chem. Rev., 2020, 408, 213177 CrossRef CAS.
- X. Du, H. Su and X. Zhang, J. Alloys Compd., 2020, 824, 153965 CrossRef CAS.
- W. Fang, D. Liu, Q. Lu, X. Sun and A. M. Asiri, Electrochem. Commun., 2016, 63, 60–64 CrossRef CAS.
- D. Liu, Q. Lu, Y. Luo, X. Sun and A. M. Asiri, Nanoscale, 2015, 7, 15122–15126 RSC.
- P. Hao, Y. Xin, J. Tian, L. Li, J. Xie, F. Lei, L. Tong, H. Liu and B. Tang, Sci. China: Chem., 2020, 63, 1030–1039 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed.
- Y. Lu, H. Zhang, E. H. Ang, Z. Nie, H. Liu, Y. Du, C. Han, J. Zhu and W. Huang, Mater. Today Phys., 2021, 16, 100303 CrossRef CAS.
- Y. Lu, H. Zhang, Y. Du, C. Han, Z. Nie, Z. Sun, K. Rui, J. Zhu and W. Huang, ACS Appl. Energy Mater., 2020, 3, 6240–6248 CrossRef CAS.
- B. L. Li, C.-B. Gong, W. Shen, J.-D. Peng, H. L. Zou, H. Q. Luo and N. B. Li, J. Mater. Chem. A, 2021, 9, 11056–11063 RSC.
- J. Hu, Y. Ou, Y. Li, D. Gao, Y. Zhang and P. Xiao, ACS Sustainable Chem. Eng., 2018, 6, 11724–11733 CrossRef CAS.
- R. Wang, Nat. Catal., 2020, 3, 333–334 CrossRef CAS.
- Y. Zhu, H. Zhao, Y. He and R. Wang, J. Phys. D: Appl. Phys., 2021, 54, 443002 CrossRef CAS.
- J. Lv, X. Yang, H.-Y. Zang, Y.-H. Wang and Y.-G. Li, Mater. Chem. Front., 2018, 2, 2045–2053 RSC.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- W. Zhu, Z. Yue, W. Zhang, N. Hu, Z. Luo, M. Ren, Z. Xu, Z. Wei, Y. Suo and J. Wang, J. Mater. Chem. A, 2018, 6, 4346–4353 RSC.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- K. Zhang, X. Xia, S. Deng, Y. Zhong, D. Xie, G. Pan, J. Wu, Q. Liu, X. Wang and J. Tu, Nano-Micro Lett., 2019, 11, 21 CrossRef CAS PubMed.
- D. Di Girolamo, M. Piccinni, F. Matteocci, A. G. Marrani, R. Zanoni and D. Dini, Electrochim. Acta, 2019, 319, 175–184 CrossRef CAS.
- T. Wang, G. Nam, Y. Jin, X. Wang, P. Ren, M. G. Kim, J. Liang, X. Wen, H. Jang, J. Han, Y. Huang, Q. Li and J. Cho, Adv. Mater., 2018, 30, e1800757 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2na00004k |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2022 |