
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced electrocatalytic reduction of CO2 to formate via doping Ce in Bi2O3 nanosheets†
Xiao
Li‡
a,
Ningkang
Qian‡
a,
Liang
Ji
a,
Xingqiao
Wu
a,
Junjie
Li
a,
Jingbo
Huang
a,
Yucong
Yan
ac,
Deren
Yang
 a and
Hui
Zhang
*ab
a and
Hui
Zhang
*ab
aState Key Laboratory of Silicon Materials and School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, People's Republic of China. E-mail: msezhanghui@zju.edu.cn
bInstitute of Advanced Semiconductors, Hangzhou Innovation Center, Zhejiang University, Hangzhou, Zhejiang 310027, People's Republic of China
cBTR New Material Group CO., LTD, GuangMing District, Shenzhen 518106, People's Republic of China
First published on 30th March 2022
Abstract
Formate is considered as the most economically viable product of the prevalent electrochemical CO2 reduction (ECR) products. However, most of the catalysts for ECR to formate in aqueous solution often suffer from low activity and limited selectivity. Herein, we report a novel Ce-doped Bi2O3 nanosheet (NS) electrocatalyst by a facile solvothermal method for highly efficient ECR to formate. The 5.04% Ce-doped Bi2O3 NSs exhibited a current density of 37.4 mA cm−2 for the production of formate with a high formate faradaic efficiency (FE) of 95.8% at −1.12 V. The formate FE was stably maintained at about 90% in a wide potential range from −0.82 to −1.22 V. More importantly, density functional theory (DFT) calculations revealed that Ce doping can lead to a significant synergistic effect, which promotes the formation and the adsorption of the OCHO* intermediate for ECR, while significantly inhibiting the hydrogen evolution reaction via depressing the formation of *H, thus helping achieve high current density and FE. This work provides an effective and promising strategy to develop efficient electrocatalysts with heteroatom doping and new insights for boosting ECR into formate.
Introduction
Nowadays, excessive CO2 emission has caused many severe problems related to the resources, environment, and climate, known as the “greenhouse effect”.1 Therefore, besides reducing CO2 production, converting CO2 into fuels or value-added chemicals seems to be necessary, which is also an urgent focus of research.2–5 There are several routes for the chemical conversion of CO2, including thermochemical,6 photochemical,7 biochemical,8 and electrochemical reactions.9–11 Among these conversion approaches, electrochemical reduction of CO2 powered by renewable energies is more attractive because it enables better sustainable development of energy and the environment. Nevertheless, some huge challenges remain in electrochemical CO2 reduction (ECR) in aqueous media, such as the low activity, even when electrocatalysts and high electrode reduction potential are applied; the low selectivity, caused by the competitive hydrogen evolution reaction (HER); and low stability of catalysts.12–14 Therefore, it's highly desired to develop efficient electrocatalysts for ECR.Formate is viewed as the most economically viable product of the prevalent ECR products, and it is promising for energy and industrial related applications.15–19 Recently, various metallic catalysts have proved to be effective to form formate (or HCOOH) through ECR.20–24 Among these electrocatalysts explored to date, Bi-based materials have been widely investigated because of their low toxicity, earth-abundance, and high activity. However, various Bi-based electrocatalysts usually suffer from undesirable activity with a narrow potential window of high selectivity, a low current density for the formation of formate and relatively poor selectivity for formate production.15,20,24–28 Up to now, a lot of effort has been devoted to improving the ECR performance of Bi-based electrocatalysts, such as tuning the size,29 controlling the morphology,25–28,30,31 and heterostructure engineering.32–36 Among these strategies, element doping to regulate the electronic structure of catalysts has been regarded as a powerful way to enhance the activity of ECR. The introduction of heteroatoms (e.g. Sn, S, and B) can modify the electronic structures of the active sites and result in optimal adsorption energy of the reaction intermediates.37–40 Cerium-based and ceria oxide-based catalysts are not ideal ECR electrocatalysts because of their poor intrinsic activity and the existence of the competitive HER. But some reports have proved that the activity of some ECR electrocatalysts can be effectively improved by combining with ceria through rational regulation.41–45 Therefore, it is reasonable to expect that doping Ce in Bi-based oxide may improve the activity towards the ECR.
Herein, we report the facile synthesis of Ce-doped Bi2O3 nanosheets (NSs) via a facile solvothermal method. The obtained electrocatalysts showed a remarkable electrocatalytic activity for the production of formate through ECR in a CO2-saturated 0.5 M KHCO3 electrolyte in a H-cell. 5.04% Ce-doped Bi2O3 NSs show the highest faradaic efficiency (FE) (95.8%) with a current density of 37.4 mA cm−2 for formate at −1.12 V versus the reversible hydrogen electrode (RHE), which is better than those of undoped Bi2O3 NSs (89.1%, 26.6 mA cm−2). This sample also possessed a wider negative potential range (from −0.82 to −1.22 V), in which the FE of formate was about 90%. Furthermore, 5.04% Ce-doped Bi2O3 NSs show a good catalytic stability over 10 h. Indeed, density functional theory (DFT) calculations suggest the mechanism for the enhancement in activity. Finally, we integrate 5.04% Ce-doped Bi2O3 NSs with a dimensionally stable anode (DSA) and achieve battery-driven CO2/H2O splitting to formate/O2 with excellent activity.
Results and discussion
The Ce-doped Bi2O3 NSs and undoped Bi2O3 NSs were prepared in polyol media by a solvothermal method. The details of the synthetic procedure are shown in Experimental section.† X-ray diffraction (XRD) patterns (Fig. S1†) show that these four samples all display a crystalline phase of cubic Bi2O3 (JCPDS no. 27-0052), and four obvious diffraction peaks can be assigned to the (111), (200), (220) and (311) planes. There is no diffraction peak associated with CeO2 or other types of Ce-based oxide to be observed in the XRD patterns, indicating that no Ce-based oxide coexists in Ce-doped Bi2O3 NSs. Moreover, the transmission electron microscopy (TEM) and high angle annular darkfield scanning transmission electron microscopy (HAADF-STEM) images show that the undoped Bi2O3 product exhibited a sheet-like nanostructure (Fig. S2a and b†). The high-resolution TEM (HRTEM) images (Fig. S2c†) of the undoped Bi2O3 NSs show a lattice spacing of about 0.276 nm that corresponds to the {200} facets of cubic Bi2O3. The HAADF-STEM-EDX images in Fig. S2d† demonstrate the uniform distribution of Bi and O elements in the undoped NSs. The sheet-like shape is well preserved after doping 5.04% atomic percentage of Ce in Bi2O3 NSs, as shown in the TEM (Fig. 1a) and HAADF-STEM (Fig. 1b) images. The HRTEM image of 5.04% Ce-doped Bi2O3 NSs in Fig. 1c shows the fringes with a lattice spacing of 0.278 nm that also corresponds to the {200} facets of Bi2O3. The expansion of the lattice fringes may result from the larger atomic radius of Ce compared with that of Bi. Additionally, elemental mapping images of 5.04% Ce-doped Bi2O3 NSs in Fig. 1d demonstrate that the Bi, Ce and O elements are uniformly distributed throughout the NSs without any element aggregation. Furthermore, the morphological, structural and compositional characterization of the 2.28% and 7.96% Ce-doped Bi2O3 NSs was also carried out (Fig. S3 and S4†). It should be noted that 2.28%, 5.04% and 7.96% represent the atomic percentage of Ce, and they were determined by using inductively coupled plasma atomic emission spectrometry (ICP-AES). The valence states of Bi and Ce were analyzed by X-ray photoelectron spectroscopy (XPS), as shown in Fig. S5 and S6.† The only two main peaks in the Bi 4f spectra (Fig. S6a, c, e, and g†) demonstrate a single oxidation state of Bi3+ for the undoped Bi2O3 NSs and three Ce-doped Bi2O3 NSs. The XPS spectra of Ce 3d (Fig. S6b, d, f, and h†) were fitted into 6 peaks corresponding to the Ce 3d5/2 and Ce 3d3/2 states. The V0, U0, V1 and U1 peaks are assigned to Ce4+ species, and the remaining two peaks are associated with Ce3+ species.46 This result demonstrates that the valence states of Ce in Ce-doped Bi2O3 NSs are a mixture of Ce3+ and Ce4+ (the proportions of Ce3+ and Ce4+ are also shown in Fig. S6†). | ||
| Fig. 1 (a) TEM, (b) HAADF-STEM, (c) HRTEM, and (d) elemental mapping images of 5.04% Ce-doped Bi2O3 NSs. | ||
To evaluate the catalytic properties of the Ce-doped Bi2O3 NSs including undoped Bi2O3 NSs toward CO2 electroreduction, the electrocatalytic reactions are carried out in homemade H-type cells, and the linear sweep voltammetry (LSV) measurements are conducted in 0.5 M KHCO3 at normal temperature and pressure. As shown in Fig. S7,† the negative current densities for these electrocatalysts in CO2 are much higher than those in an Ar atmosphere, indicating their excellent intrinsic activities for reduction of CO2 and a more favorable ECR activity over the competitive HER. To identify the reduction products and current densities at various potentials, the controlled potential electrolysis is carried out for the four electrocatalysts (Fig. S8†). The gas-phase products are detected using a gas chromatograph (GC), and the liquid products are quantitatively analyzed by ion chromatography (IC). Formate was measured by ion chromatography according to the standard curve (Fig. S9†). The summarized FEs of all products at various potentials in Fig. S10† demonstrate that formate is the only liquid product and also the predominant one among the whole CO2 reduction products. As shown in Fig. 2a, the total negative current density (jtotal) of Ce-doped Bi2O3 NSs remarkably increases compared with that of undoped Bi2O3 NSs. Among them, 5.04% Ce-doped Bi2O3 NSs show the highest current density, indicating the best performance for ECR. Moreover, 5.04% Ce-doped Bi2O3 NSs exhibit the highest FE for formate (Fig. 2b), achieving a maximum FE value of 95.8% at −1.12 V versus RHE. In addition, the formate FE stably remains at about 90% in a wide negative potential range of 400 mV (from −0.82 to −1.12 V). It should be noted that H2 FEs decrease significantly after doping Ce in Bi2O3 NSs (Fig. S10†), which demonstrates that doping Ce in Bi2O3 NSs is helpful to suppress the competing reaction of the HER. Fig. 2c and d show the formate partial current density (jHCOO−) and formate production rate of these four electrocatalysts at various potentials, respectively. Obviously, all three Ce-doped Bi2O3 NSs exhibit the higher jHCOO− and faster formate production rate at all potentials compared with the undoped one. In particular, 5.04% Ce-doped Bi2O3 NSs achieved a jHCOO− of 43.1 mA cm−2 and a formate production rate of 0.805 mmol cm−2 h−1 at −1.32 V, which were 1.55 times as high as those of undoped Bi2O3 NSs (27.8 mA cm−2 and 0.520 mmol cm−2 h−1). Fig. 2e shows the energy efficiency for formate (ΦHCOO−) of the four electrocatalysts at different applied potentials. After Ce doping, the ΦHCOO− of Ce-doped Bi2O3 NSs increases significantly compared with that of undoped Bi2O3 NSs. The ΦHCOO− of 5.04% Ce-doped Bi2O3 NSs exceeded 50% at potentials ranging from −0.72 to −1.22 V, while that of undoped Bi2O3 NSs is −0.82 to −1.12 V. To the best of our knowledge, the ECR performance of 5.04% Ce-doped Bi2O3 NSs outperforms many other reported electrocatalysts in the H-cell (Table S1†).
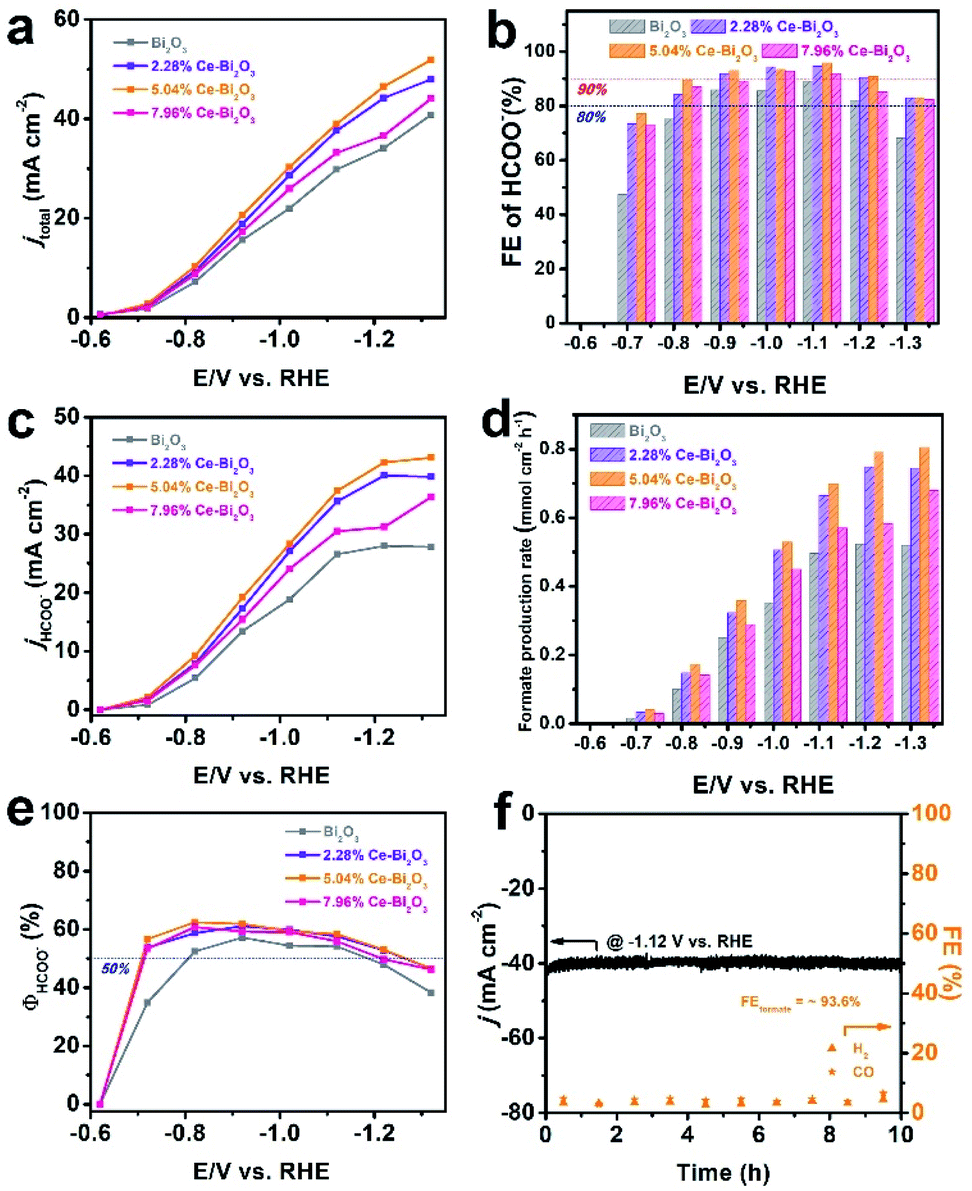 | ||
| Fig. 2 (a) Total current densities, (b) potential-dependent FE for formate, (c) formate partial current densities, (d) formate production rate and (e) energy efficiency for formate for the four samples. (f) Long-term chronoamperometry test of 5.04% Ce-doped Bi2O3 NSs at potentials of −1.12 V, and corresponding FEs for H2 and CO. | ||
The stability of 5.04% Ce-doped Bi2O3 NSs is tested through a long-term chronoamperometry test at −1.12 V. As shown in Fig. 2f, the current density and FE of H2 and CO have no obvious change over 10 h of electrolysis, demonstrating the favorable stability of the electrocatalyst. The initial current density was 42.1 mA cm−2, and the final current density was 40.2 mA cm−2, a drop of less than 5%. The average FE of formate during 10 h reached ∼93.6%, suggesting that the ECR selectivity was well maintained. After electrolysis, the morphology of 5.04% Ce-doped Bi2O3 NSs is largely preserved (Fig. S11a†). In addition, unavoidable reduction of Bi2O3 to metallic Bi happens during electrolysis, as evidenced by the XRD pattern (Fig. S11b†). This phenomenon has been reported in other studies on Bi2O3-based electrocatalysts as well.47–49 Consequently, Ce-doped Bi2O3 NSs have high durability and selectivity towards formate generation in ECR.
To explore the factors for the enhanced electrocatalytic ability of Ce-doped Bi2O3 NSs, some measurements together with DFT calculations were performed. As shown in Fig. 3a, 5.04% Ce-doped Bi2O3 NSs possess better CO2 adsorption capacity compared with the undoped one, which could improve the intermediate CO2*− formation before further reduction. To gain deeper insight into the kinetic mechanism for ECR, the corresponding Tafel slope is fitted (Fig. 3b), which is an indicator of the rate-determining step (RDS). Obviously, the Tafel slopes for both electrocatalysts are close to the theoretical value of 118 mV dec−1, suggesting that the first electron transfer process is the rate-determining step (RDS).50,51 Furthermore, 5.04% Ce-doped Bi2O3 NSs exhibited a lower Tafel slope of 124 mV dec−1 than that of undoped Bi2O3 NSs (133 mV dec−1), implying its better performance for ECR. Moreover, the electrochemical surface area (ECSA) of these catalysts was measured (Fig. S12†), and Ce-doped Bi2O3 NSs exhibited a little larger slope than that of undoped Bi2O3 NSs (Fig. S13†). This result suggests a slightly increased number of electrochemical active sites originating from doping Ce. These samples' activity normalized to the ECSA (the value of Cdl) is further calculated (Fig. S14†), which reveals that Ce-doped Bi2O3 NSs show its intrinsically better electrocatalytic activity for ECR.
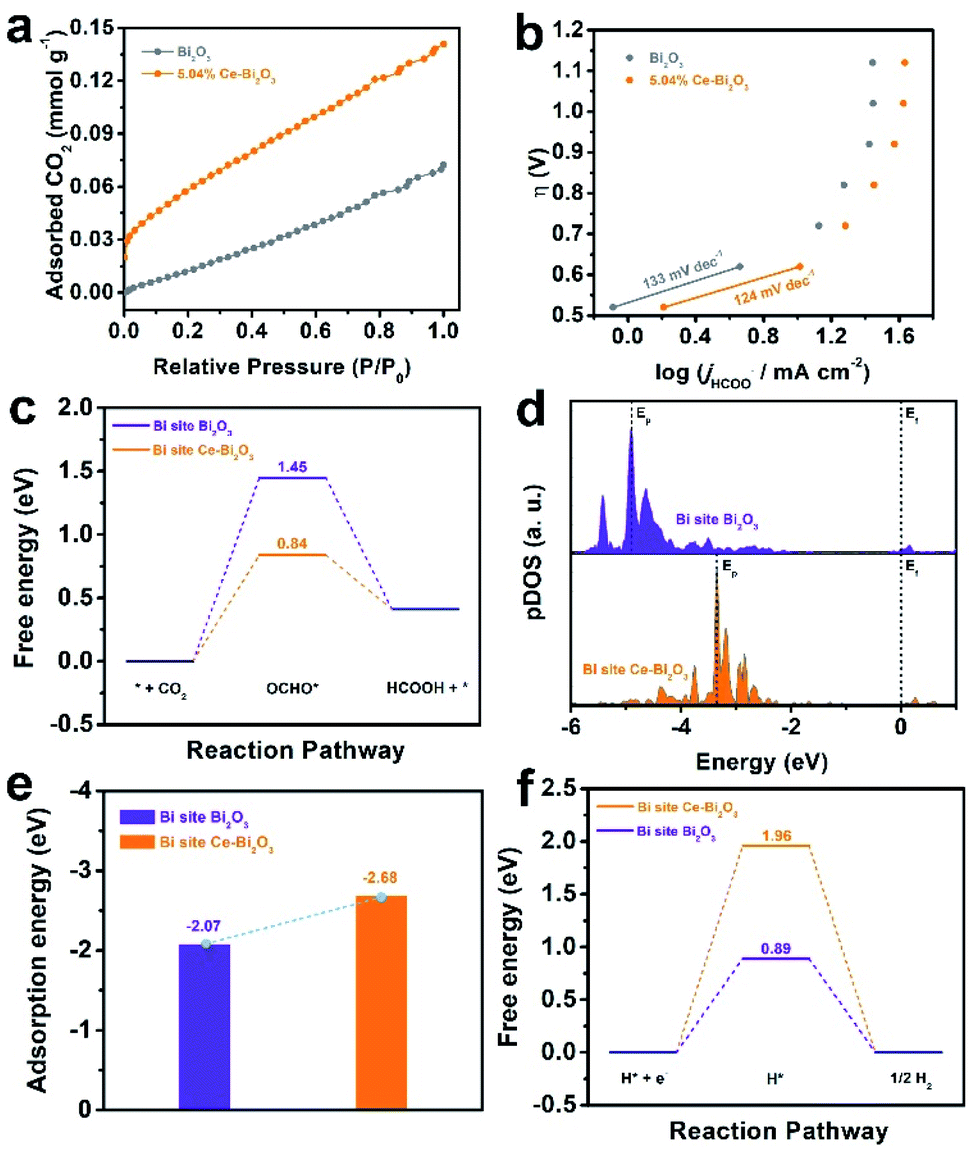 | ||
| Fig. 3 (a) CO2 adsorption isotherm and (b) Tafel plots for undoped Bi2O3 NSs and 5.04% Ce-doped Bi2O3 NSs. (c) Calculated free-energy diagram of the OCHO* intermediate, (d) projected p-orbital DOS of OCHO*, (e) calculated adsorption energy of OCHO*, and (f) calculated free-energy diagram of H* for the Bi site on Bi2O3 and Ce-doped Bi2O3. | ||
DFT calculations are also employed to explore the origin of enhanced ECR activity of doping Ce in Bi2O3 NSs (calculation details are shown in the ESI, and the simulation models of undoped Bi2O3 and Ce-doped Bi2O3 are shown in Fig. S15†). Fig. 3c shows the free energy diagrams for the conversion of CO2 to OCHO* and eventually HCOOH over the Bi-site on the undoped Bi2O3 and Ce-doped Bi2O3. Both undoped Bi2O3 and Ce-doped Bi2O3 show the largest energy barrier for OCHO* formation, demonstrating that the initial proton-coupled electron transfer is the potential limiting step. Encouragingly, after doping Ce, the energy barrier for OCHO* formation decreases from 1.45 eV for undoped Bi2O3 to 0.84 eV for Ce-doped Bi2O3. That is, it is easier to form the OCHO* intermediate on the surface of Ce-doped Bi2O3 compared to undoped Bi2O3. Besides, p-projected density of states (pDOS) analysis is shown in Fig. 3d. After OCHO* adsorption, compared with that of undoped Bi2O3, the highest peak of total DOS (Ep) for Ce-doped Bi2O3 is much closer to the Fermi level (Ef), indicating a higher binding strength of Ce-doped Bi2O3 to OCHO*.52,53 Additionally, the adsorption energy of OCHO* on the electrocatalysts’ surface was further studied (Fig. 3e). The adsorption energy of OCHO* on Ce-doped Bi2O3 was more negative than that on undoped Bi2O3. This result demonstrates that OCHO* adsorption on the surface of Ce-doped Bi2O3 is more energetically favorable than that on undoped Bi2O3, which coincides with the calculation results of pDOS analysis. Meanwhile, the free energies of H*on both electrocatalysts are calculated and shown in Fig. 3f. It is revealed that Ce-doped Bi2O3 possesses a higher energy barrier than the undoped one, indicating that Ce-doped Bi2O3 has lower activities toward H2 production. Taken together, the origin of improved activity in Ce-doped Bi2O3 NSs can be attributed to the significant synergistic effect caused by doping Ce in Bi2O3, that is, the promotion of formation and adsorption of the OCHO* intermediate for ECR and depression of the formation of H* leading to suppression of the HER.
As a step further, we pursued AA-size alkaline battery-driven CO2/H2O splitting to explore practical application. A commercial DSA was employed as the anode for the oxygen evolution reaction (OER). The commercial DSA used here consists of a thin IrO2 layer coated on Ta-coated-Ti foil, and it was chosen as the benchmark material to couple with 5.04% Ce-doped Bi2O3 NS ECR. Its OER polarization curve was first collected using the standard three-electrode setup and is shown in Fig. 4a. Obviously, the anodic current density reaches 10 mA cm−2 at ∼1.7 V in 0.5 M KHCO3 electrolyte, which shows a decent OER activity. Full cells were then constructed by pairing the 5.04% Ce-doped Bi2O3 NS cathode and DSA anode in a two-compartment cell, and the corresponding typical polarization curve is depicted in Fig. 4b. The ECR-OER couple became turned on under an external voltage of ∼2.1 V and was found to achieve a current density of 7.6 mA cm−2 at 3 V. Furthermore, two AA-size alkaline batteries in series were used as the power source (with an open-circuit potential of ∼3.1 V) to drive the full-cell CO2/H2O splitting (Fig. 4c). A source-meter was connected to continuously monitor the current evolution. The current density starts at 9.5 mA cm−2, slightly decreases and stabilizes at 8.5 mA cm−2 till the end of the 5 h evaluation (Fig. 4d). Analysis of the reduction product shows that the average formate FE is 88.9%, and the FEs of H2 and CO are also stable. The current density of the device is relatively low because the potential on the cathode is not negative enough. If the external power source can be designed properly, the practical feasibility for the ECR-OER device will be further improved.
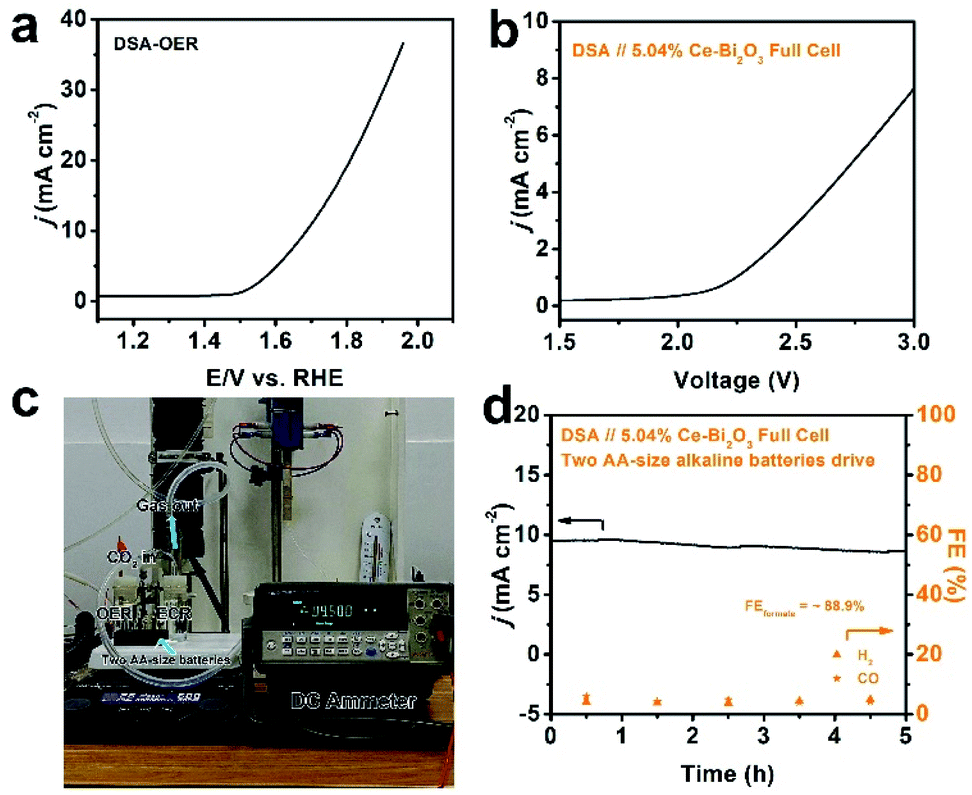 | ||
| Fig. 4 Full-cell electrolysis by coupling 5.04% Ce-doped Bi2O3 NSs ECR with the DSA OER: (a) OER polarization curve of DSA in 0.5 M KHCO3, (b) polarization curve for the ECR-OER full-cell electrolysis, (c) photograph of the setup of two AA-size alkaline batteries driving ECR-OER electrolysis and (d) current evolution and corresponding FEs of H2 and CO for the battery-powered ECR-OER electrolysis. | ||
Conclusions
In summary, we constructed a series of Ce-doped Bi2O3 NS electrocatalysts with different atomic percentages of Ce through a facile solvothermal approach. The 5.04% Ce-doped Bi2O3 NSs possess superior activity and selectivity for CO2 electroreduction to formate in aqueous solution. They exhibited a current density of 37.4 mA cm−2 with a high FE of 95.8% for formate at −1.12 V, and the formate FE was stably maintained at about 90% in a wide potential range from −0.82 V to −1.22 V. Furthermore, some measurements together with DFT calculations were performed to explore the factors for the enhanced electrocatalytic ability. Our work demonstrates a facile doping strategy to fabricate novel Bi-based electrocatalysts, and we expect that this study can be extended to other types of highly efficient electrocatalysts for ECR.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The work on electron microscopy was carried out in the Center for Electron Microscopy of Zhejiang University. This work was supported by the National Science Foundation of China (51871200), National Program for Support of Top-notch Young Professionals, National Key R&D Program of China (2018YFB2200102), and Foundation for Innovative Research Groups of the National Natural Science Foundation of China (61721005).Notes and references
- M. Reichstein, M. Bahn, P. Ciais, D. Frank, M. D. Mahecha, S. I. Seneviratne, J. Zscheischler, C. Beer, N. Buchmann, D. C. Frank, D. Papale, A. Rammig, P. Smith, K. Thonicke, M. van der Velde, S. Vicca, A. Walz and M. Wattenbach, Nature, 2013, 500, 287–295 CrossRef CAS PubMed.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981 RSC.
- L. Zhang, Z. Zhao, T. Wang and J. Gong, Chem. Soc. Rev., 2018, 47, 5423 RSC.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- L. Zhang, Z. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- C. Long, X. Li, J. Guo, Y. Shi, S. Liu and Z. Tang, Small Methods, 2019, 3, 1800369 Search PubMed.
- Y. Quan, J. Zhu and G. Zheng, Small Sci., 2021, 1, 2100043 CrossRef.
- J. Li, Y. Kuang, Y. Meng, X. Tian, W. Hung, X. Zhang, A. Li, M. Xu, W. Zhou, C. Ku, C. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS PubMed.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2019, 10, 1902338 CrossRef.
- S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC.
- W. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Nat. Commun., 2020, 11, 3633 CrossRef CAS PubMed.
- M. G. Mura, L. D. Luca, G. Giacomelli and A. Porcheddu, Adv. Synth. Catal., 2012, 354, 3180–3186 CrossRef CAS.
- P. Ding, H. Zhao, T. Li, Y. Luo, G. Fan, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC.
- Y. Zhou, R. Zhou, X. Zhu, N. Han, B. Song, T. Liu, G. Hu, Y. Li, J. Lu and Y. Li, Adv. Mater., 2020, 32, 2000992 CrossRef CAS PubMed.
- Z. Li, A. Cao, Q. Zheng, Y. Fu, T. Wang, K. T. Arul, J. Chen, B. Yang, N. M. Adli, L. Lei, C. Dong, J. Xiao, G. Wu and Y. Hou, Adv. Mater., 2020, 33, 2005113 CrossRef PubMed.
- Z. Zhang, F. Ahmad, W. Zhao, W. Yan, W. Zhang, H. Huang, C. Ma and J. Zeng, Nano Lett., 2019, 19, 4029–4034 CrossRef CAS PubMed.
- J. Zhu, J. Fan, T. Cheng, M. Cao, Z. Sun, R. Zhou, L. Huang, D. Wang, Y. Li and Y. Wu, Nano Energy, 2020, 75, 104939 CrossRef CAS.
- Y. Qiao, W. Lai, K. Huang, T. Yu, Q. Wang, L. Gao, Z. Yang, Z. Ma, T. Sun, M. Liu, C. Lian and H. Huang, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1088 CrossRef PubMed.
- X. Zhang, X. Sun, S. Guo, A. M. Bond and J. Zhang, Energy Environ. Sci., 2019, 12, 1334–1340 RSC.
- Z. Zhang, M. Chi, G. M. Veith, P. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- H. Xie, T. Zhang, R. Xie, Z. Hou, X. Ji, Y. Pang, S. Chen, M. Titirici, H. Weng and G. Chai, Adv. Mater., 2021, 33, 2008373 CrossRef CAS PubMed.
- J. Fan, X. Zhao, X. Mao, J. Xu, N. Han, H. Yang, B. Pan, Y. Li, L. Wang and Y. Li, Adv. Mater., 2021, 33, 2100910 CrossRef CAS PubMed.
- Y. Duan, K. Liu, Q. Zhang, J. Yan and Q. Jiang, Small Methods, 2020, 4, 1900846 CrossRef CAS.
- Z. Chen, K. Mou, X. Wang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 12790–12794 CrossRef CAS PubMed.
- Q. Lia, X. Zhang, X. Zhou, Q. Li, H. Wang, J. Yi, Y. Liu and J. Zhang, J. CO2 Util., 2020, 37, 106–112 CrossRef.
- S. Liu, X. Lu, J. Xiao, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS PubMed.
- F. Meng, Q. Zhang, K. Liu and X. Zhang, Chem.–Eur. J., 2020, 26, 4013–4018 CrossRef CAS PubMed.
- X. Li, X. Wu, J. Li, J. Huang, L. Ji, Z. Leng, N. Qian, D. Yang and H. Zhang, Nanoscale, 2021, 13, 19610–19616 RSC.
- Y. Zhao, X. Liu, Z. Liu, X. Lin, J. Lan, Y. Zhang, Y. Lu, M. Peng, T. Chan and Y. Tan, Nano Lett., 2021, 21, 6907–6913 CrossRef CAS PubMed.
- S. Liu, M. Gao, R. Feng, L. Gong, H. Zeng and J. Luo, ACS Catal., 2021, 11, 7604–7612 CrossRef CAS.
- X. Chen, H. Chen, W. Zhou, Q. Zhang, Z. Yang, Z. Li, F. Yang, D. Wang, J. Ye and L. Liu, Small, 2021, 17, 2101128 CrossRef CAS PubMed.
- Y. Duan, Y. Zhou, Z. Yu, D. Liu, Z. Wen, J. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2021, 60, 8798–8802 CrossRef CAS PubMed.
- R. Pang, P. Tian, H. Jiang, M. Zhu, X. Su, Y. Wang, X. Yang, Y. Zhu, L. Song and C. Li, Natl. Sci. Rev., 2021, 8, nwaa187 CrossRef CAS PubMed.
- S. B. Varandili, J. Huang, E. Oveisi, G. L. D. Gregorio, M. Mensi, M. Strach, J. Vavra, C. Gadiyar, A. Bhowmik and R. Buonsanti, ACS Catal., 2019, 9, 5035–5046 CrossRef CAS.
- H. Dong, L. Zhang, L. Li, W. Deng, C. Hu, Z. Zhao and J. Gong, Small, 2019, 15, 1900289 CrossRef PubMed.
- S. Ning, Z. Guo, J. Wang, S. Huang, S. Chen and X. Kang, ChemElectroChem, 2021, 8, 2680–2685 CrossRef CAS.
- S. Li, D. Bao, M. Shi, B. Wulan, J. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1700001 CrossRef PubMed.
- D. Wu, G. Huo, W. Chen, X. Fu and J. Luo, Appl. Catal., B, 2020, 271, 118957 CrossRef CAS.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef PubMed.
- C. W. Lee, J. S. Hong, K. Yang, K. Jin, J. H. Lee, H. Y. Ahn, H. Seo, N. E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS.
- D. Zhu, J. Liu and S. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- Y. Duan, F. Meng, K. Liu, S. Yi, S. Li, J. Yan and Q. Jiang, Adv. Mater., 2018, 30, 1706194 CrossRef PubMed.
- Z. Chen, X. Zhang, M. Jiao, K. Mou, X. Zhang and L. Liu, Adv. Energy Mater., 2020, 10, 1903664 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2na00141a |
| ‡ These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2022 |