
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of atmospheric conditions in the nonradiative recombination in individual CH3NH3PbI3 perovskite crystals†
Ruiyun
Chen
 *ab,
Wenling
Guan
ab,
Wenjin
Zhou
ab,
Zixin
Wang
ab,
Guofeng
Zhang
ab,
Chengbing
Qin
ab,
Jianyong
Hu
ab,
Liantuan
Xiao
*ab and
Suotang
Jia
ab
*ab,
Wenling
Guan
ab,
Wenjin
Zhou
ab,
Zixin
Wang
ab,
Guofeng
Zhang
ab,
Chengbing
Qin
ab,
Jianyong
Hu
ab,
Liantuan
Xiao
*ab and
Suotang
Jia
ab
aState Key Laboratory of Quantum Optics and Quantum Optics Devices, Institute of Laser Spectroscopy, Shanxi University, Taiyuan, Shanxi 030006, China. E-mail: chenry@sxu.edu.cn; xlt@sxu.edu.cn
bCollaborative Innovation Center of Extreme Optics, Shanxi University, Taiyuan, Shanxi 030006, China
First published on 10th October 2022
Abstract
Organic–inorganic metal halide perovskites have been emerging as potential candidates for lightweight photovoltaic applications in space. However, fundamental physics concerning the effect of atmosphere on the radiative and nonradiative recombination in perovskites remains far from well understood. Here, we investigate the creation and annihilation of nonradiative recombination centers in individual CH3NH3PbI3 perovskite crystals by controlling the atmospheric conditions. We find that the photoluminescence (PL) of individual perovskite crystals can be quenched upon exposure from air to vacuum, while the subsequent PL enhancement in air shows a pressure dependence. Further analysis attributes the PL decline in vacuum to the activation of nonradiative trap sites, which is likely due to the lattice distortion caused by the variation of local strain on perovskites. With a gradual increase of the air pressure, the light-assisted chemisorption of oxygen on perovskite will passivate these nonradiative trap sites while simultaneously restoring the lattice imperfection, leading to PL enhancement. The present findings suggest that placing the perovskite in an environment with moderate oxygen content can protect the material from photophysical losses that can be pronounced under inert conditions.
1. Introduction
Organic–inorganic hybrid perovskites (OHPs) have attracted extensive attention for photovoltaic and optoelectronic applications by virtue of their versatile optoelectronic properties, for instance, high photoluminescence (PL) quantum yields, long carrier diffusion length, and remarkable carrier mobility. Moreover, the good solution processability, low fabrication costs, and possibility of fabricating on large-scale flexible substrates open up promising applications, such as lightweight photovoltaic devices performing under space conditions.1 Although previous studies have shown the possibility of operating perovskite solar cells in space extreme environments,2,3 very important challenges remain in maintaining the long-term operational stability of the devices, which is critical for practical applications. It is widely accepted that the surrounding atmosphere plays a significant role in the optoelectronic properties of perovskite materials and, thus, the performance loss of the devices. To systematically compare the results regarding the stability of perovskite-based solar cells, an experimental procedure based on the International Summit on Organic Photovoltaic Stability (ISOS) was proposed,4 which suggested using inert atmospheres or vacuum as a standard operation condition. However, the effects of these inert conditions on the properties of perovskites have not yet been fully understood.Recently, a number of studies have revealed that atmospheric conditions have considerable impact on the photophysical properties of OHPs and device performance. The most commonly reported observation was the drastic, and sometimes transient, enhancement of the PL quantum yields (PLQYs) of perovskite materials under light exposure in air or oxygen,5–8 which was assigned to the annihilation of the defect (trap) states in perovskites.9 Considering that higher PLQYs imply a reduction of nonradiative pathways and, thus, long-lived and long-transported charge carriers in perovskites, this observation indicates that a controlled exposure of perovskite to an oxygen-containing environment could lead to improved device performance. However, almost contradictory results have been also reported, which argued that the presence of oxygen could induce the degradation of perovskite materials.10,11 This degradation is often accompanied by the increase of defect (trap) states which will perturb charge transportation and induce nonradiative recombination, resulting in optical instability of OHPs. A similar contradiction appeared in the study of perovskite materials under inert conditions. It has been reported that vacuum exposure can induce degradation of the perovskite materials.12,13 The degradation mechanism often involves the irreversible decomposition of perovskites which leads to the formation of lead salt.13–15 In contrast, a reversible PL response has also been found both in OHP crystals16 and in all inorganic perovskite crystals17 during vacuum–air exchange, which was attributed to the annihilation of nonradiative recombination states on the surface of the single crystals due to the physisorption of oxygen. However, despite such impressive efforts, the fundamental mechanisms behind the atmosphere-induced photophysics of perovskites remain largely unexplored. Therefore, investigating the interaction between OHPs and different atmospheric conditions, especially during the exchange of ambient and inert conditions, is crucial for fundamental research and technological applications of perovskite.
An investigation on individual perovskite crystals of several nanometers to a sub-micrometer scale would provide detailed information beyond the ensemble averaging picture obtained from bulk and film, making them an ideal platform to study the intrinsic properties of the perovskite materials.18 There is no doubt that different macroscopic operation conditions, particularly a different surrounding ambience, can greatly influence the microscopic behavior of the materials. PL from perovskites at the single-particle level often possesses large fluctuations under the influence of the variation of atmospheric conditions. Therefore, analysis of the evolution of PL properties can be a powerful tool to characterize the effect of atmospheric conditions on radiative decay or degradation in perovskites19–21 and to give insights into their microscopic origins22 that are otherwise convoluted in the ensemble averaging.
In this work, we investigate the atmospheric condition-dependent PL properties of individual CH3NH3PbI3 (in brief, MAPbI3) crystals by using single-particle PL spectroscopy. It is found that the PL intensity of individual MAPbI3 crystals diminishes when changing the surroundings from ambient air to vacuum and does not recover when exposed back to air immediately. However, when the air pressure is gradually raised from vacuum to one atmosphere, the PL can be significantly enhanced, eventually reaching a level of about 3 times higher than the initial PL intensity. Based on the comprehensive optical and X-ray diffraction (XRD) characterization, we attribute the deceased PL under vacuum condition to the increased density of nonradiative recombination centers which is likely associated with the lattice distortions due to variation of the local strain. We propose that the chemisorption of oxygen to perovskite would passivate the nonradiative recombination centers with the participation of light, resulting in PL enhancement. The results indicate that a moderate pressure of oxygen in combination with light exposure assists in improving the performance of OHP materials.
2. Results and discussion
2.1. Morphology and optical properties of MAPbI3 crystals
Individual MAPbI3 crystals, with their morphology depicted in Fig. 1, were synthesized following the procedure reported previously (also see Experimental methods).23 The scanning electron microscopy (SEM) image shows particles with a near-circular morphology and gives the corresponding size histogram with an average size of around 247 nm (Fig. 1b). The height of the crystals was measured using atomic force microscopy (AFM), which gives an average value of 14.2 nm (Fig. S1†). The XRD patterns were clearly resolved for 2θ = 10–60° with peaks at 14.1°(110), 28.4°(220), and 43.2°(330), confirming the tetragonal crystalline phase of the crystals with good purity and crystallinity (Fig. 1c). Fig. 1d shows the PL spectrum of the MAPbI3 crystal in ambient air excited by a pulsed laser (485 nm, repetition rate 2.5 MHz, average power 1.5 μW). All measured MAPbI3 crystals show identical spectra properties with an emission peak at 760 nm.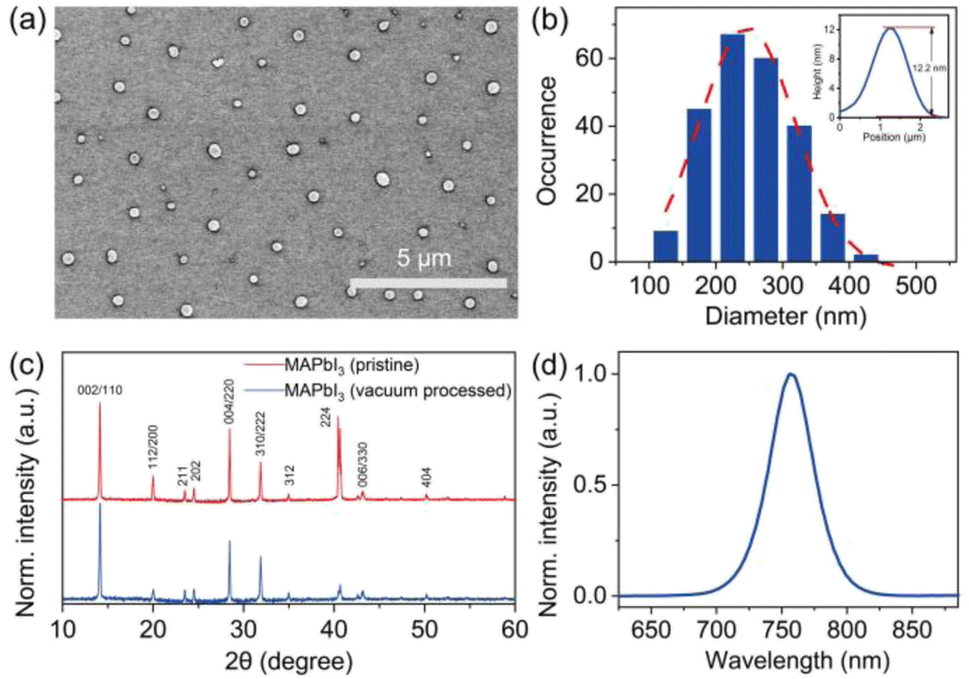 | ||
| Fig. 1 (a) Scanning electron micrograph of the MAPbI3 perovskite crystals. (b) Size histogram extracted from the analysis of over 200 MAPbI3 perovskite crystals, showing near-circular crystals with an average diameter of 247 ± 72 nm. The inset shows the height of an individual MAPbI3 crystal characterized by AFM. (c) X-ray diffraction pattern of bulk MAPbI3 perovskite crystals. (d) Typical PL spectrum of an individual MAPbI3 perovskite crystal. | ||
2.2. Atmospheric condition-dependent PL of individual MAPbI3 crystals
The atmospheric condition-dependent photophysical properties of individual MAPbI3 crystals were measured by using the experimental setup illustrated in Fig. S2.† We first analyzed the evolution of the PL properties of the crystals as the environmental conditions exchanged between ambient air and low vacuum. To do so, a freshly prepared sample with individual MAPbI3 crystals synthesized under ambient conditions was transferred into a chamber. The air pressure in the chamber was controlled to simulate the low vacuum and varied atmospheric conditions (Fig. 2a). First, the PL properties were acquired in ambient air. The chamber was then evacuated to a low-pressure level (0.2 torr) to simulate the vacuum condition and was subsequently refilled with air to ambient pressure.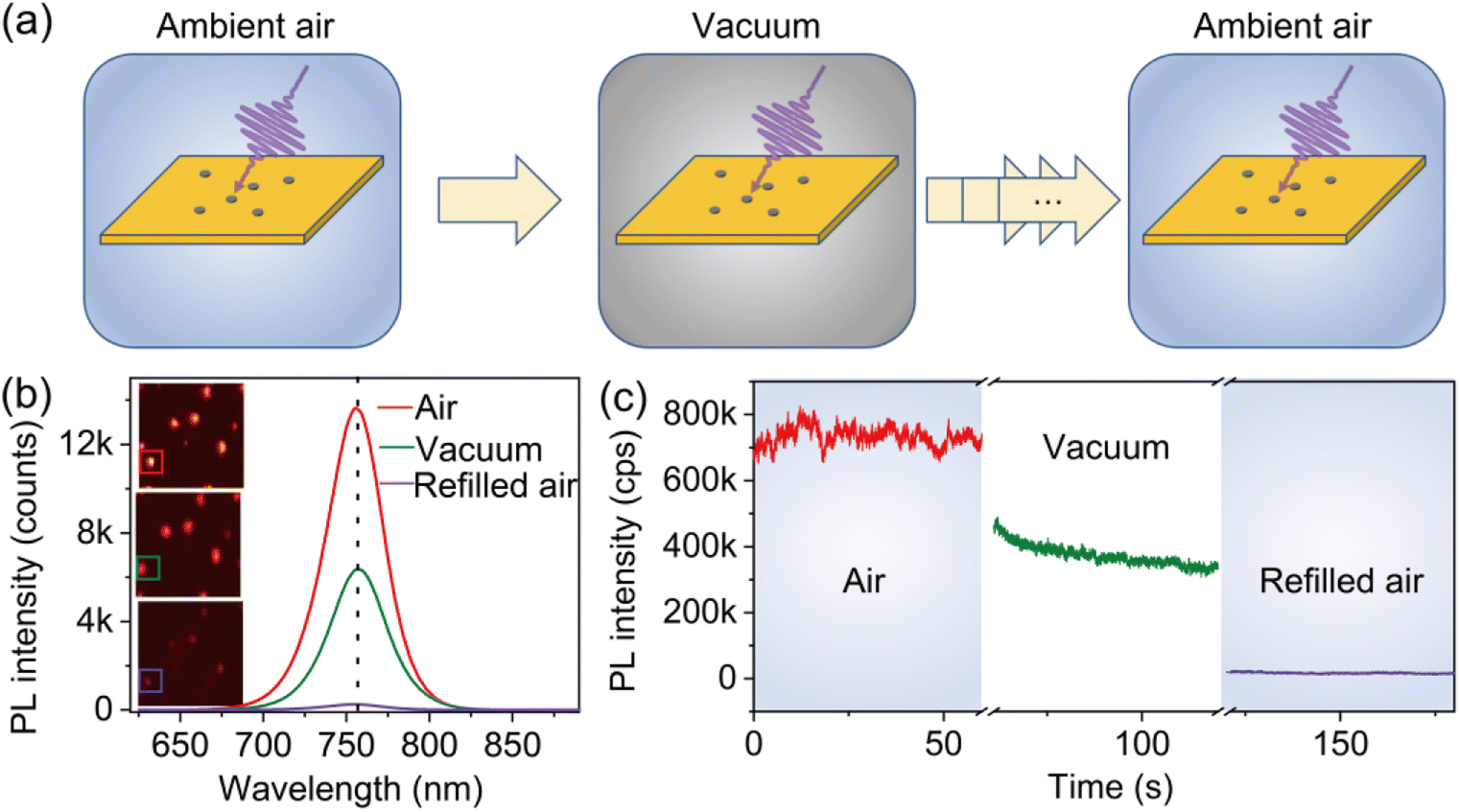 | ||
| Fig. 2 (a) Schematic diagram of the measurement flow of the experiment: the perovskite sample prepared in ambient air is subsequently transferred into a chamber in which the sample is exposed sequentially to ambient air, vacuum, and then back to ambient air. (b) PL spectra of an individual MAPbI3 crystal upon exposure to ambient air, vacuum, and refilled air. The inset shows the corresponding PL imaging of the crystal in different conditions. (c) Evolution of the PL intensity of the individual crystal on exposure to ambient air, vacuum, and refilled air. | ||
Fig. 2b shows the evolution of the PL spectra of a typical MAPbI3 crystal attained at three distinct phases, namely, under ambient air (740 torr), vacuum (0.2 torr), and refilled ambient air (740 torr). The initial PL state of the perovskite crystal showed a relatively high PL intensity (red solid line). When the chamber was evacuated to the low-vacuum condition, the PL intensity decreased clearly (green solid line). Surprisingly, we found a further decrease in PL with the instantaneous refilling of air to ambient pressure (purple solid line). It can be found that the peak position of the PL spectra remains unchanged during the air–vacuum–air exchange, implying that the PL emission under varied atmospheric conditions originates from the same transition mechanism. Fig. 2c illustrates the evolution of the corresponding PL trajectories as the change of the atmospheric condition. The PL intensity is found to decrease by 50% when the chamber is evacuated to 0.2 torr. With the refilling of air, the PL intensity further decreases by 98% of its initial intensity. We also noticed that the decrease of the PL intensity occurs without the participation of light. Specifically, the sample was kept in darkness when the chamber was pumped down to the vacuum condition or refilled with air. When the laser is turned on, the diminished PL intensity is immediately achieved and stabilized at a low level. Furthermore, we found that PL cannot recover when keeping the perovskite in darkness for a long time under ambient conditions (Fig. S3†).
The PL decrease observed under vacuum demonstrates the negative effect of the inert conditions on the photophysical properties of perovskite materials. However, whether this negative effect is reversible or irreversible needs to be further studied, which is crucial to explore the underlying mechanism. Generally, the irreversible effect of the vacuum condition has been found to be accompanied by the decomposition of MAPbI3 and the generation of new products, such as PbI2, which will be further degraded into Pb0 and I2−.15,24 To clarify this issue, we performed an XRD measurement to compare the crystallographic information of the pristine perovskite crystals and that treated with evacuation. By comparing the XRD pattern, neither a shift of the peak position nor the creation of new peaks is found, as depicted in Fig. 1c. This essentially rules out the clear decomposition of the perovskite under the vacuum condition. Such a conclusion can be further supported by the unchanged PL spectra of the crystals during the process (Fig. 2b). Therefore, considering the fact that no obvious sign of sample degradation was found in the perovskite upon vacuum exposure, the irreversibility of PL when exposed to ambient air seems beyond comprehension, which invokes a distinguished mechanism from the permanent degradation of the materials.
In order to provide detailed insight into the underlying mechanisms, we monitored the changes of the photophysics of individual perovskite crystals by gradually increasing the air pressure in the chamber. Fig. 3a shows a typical evolution of the PL intensity of an individual crystal exposed to varied air pressures from vacuum to around 740 torr. The PL intensity of the crystal was measured for 1 min at an interval of 2 min with an increase of the air pressure every 50 torr. In contrast to the PL obtained by immediately returning to ambient air pressure (Fig. 2c), the PLQY of the crystals in this process shows an increase with the step-by-step increase of the air pressure (Fig. 3a). The PL eventually reached a value even higher than the initial PL intensity measured in ambient air before evacuation. We statisticize the results obtained from an ensemble of individual crystals (Fig. 3c). The change of the averaged PL intensity shows a similar trend to that of the individual ones, which implies a common underlying mechanism responsible for the PL fluctuation. We simultaneously measured the PL spectra of the perovskite crystal. Fig. 3b shows the PL spectra of the perovskite crystal corresponding to various air pressure, where it is obvious that the emission peak is located at 760 nm and does not change through the treatment. We also investigated the PL decay dynamics in the perovskite crystals under various air pressure. At an excitation power density of 1.21 kW cm−2 (485 nm pulsed laser with a 2.5 MHz repetition rate), the PL decay time of the crystal shows biexponential behavior. Exposure of the crystal from air to vacuum shows an increase in the averaged PL lifetime, while it decreases gradually with the refilling of air (inset in Fig. 3d). These experimental observations demonstrate a reversible PL dynamic upon exposure to gradually increasing air pressure surrounding the perovskite crystals with the assistance of light illumination, in stark contrast to the unrecovered PL in Fig. 2 when the crystals were exposed back immediately to air with ambient pressure. As a control experiment, we studied the PL response of individual perovskite crystals by increasing the pressure stepwise in dark and measuring the PL when the atmospheric pressure is reached. We found that the PL of the crystals did not recover to its original level (Fig. S4†), which means that the increase of air pressure alone cannot activate the PL of the crystal. We also studied the PL response of the perovskite crystals which were exposed to ambient pressure immediately after evacuation and measured the PL several times with a 1 min acquisition time at an interval of 2 min. However, under the sequential illumination of light at ambient air pressure, the PL of the crystals did not show a significant increase (Fig. 4). Instead, we found a slight blue shift in the PL spectra. This indicates that light illumination alone was also inadequate to induce the PL activation in individual MAPbI3 perovskite crystals that were treated with evacuation.
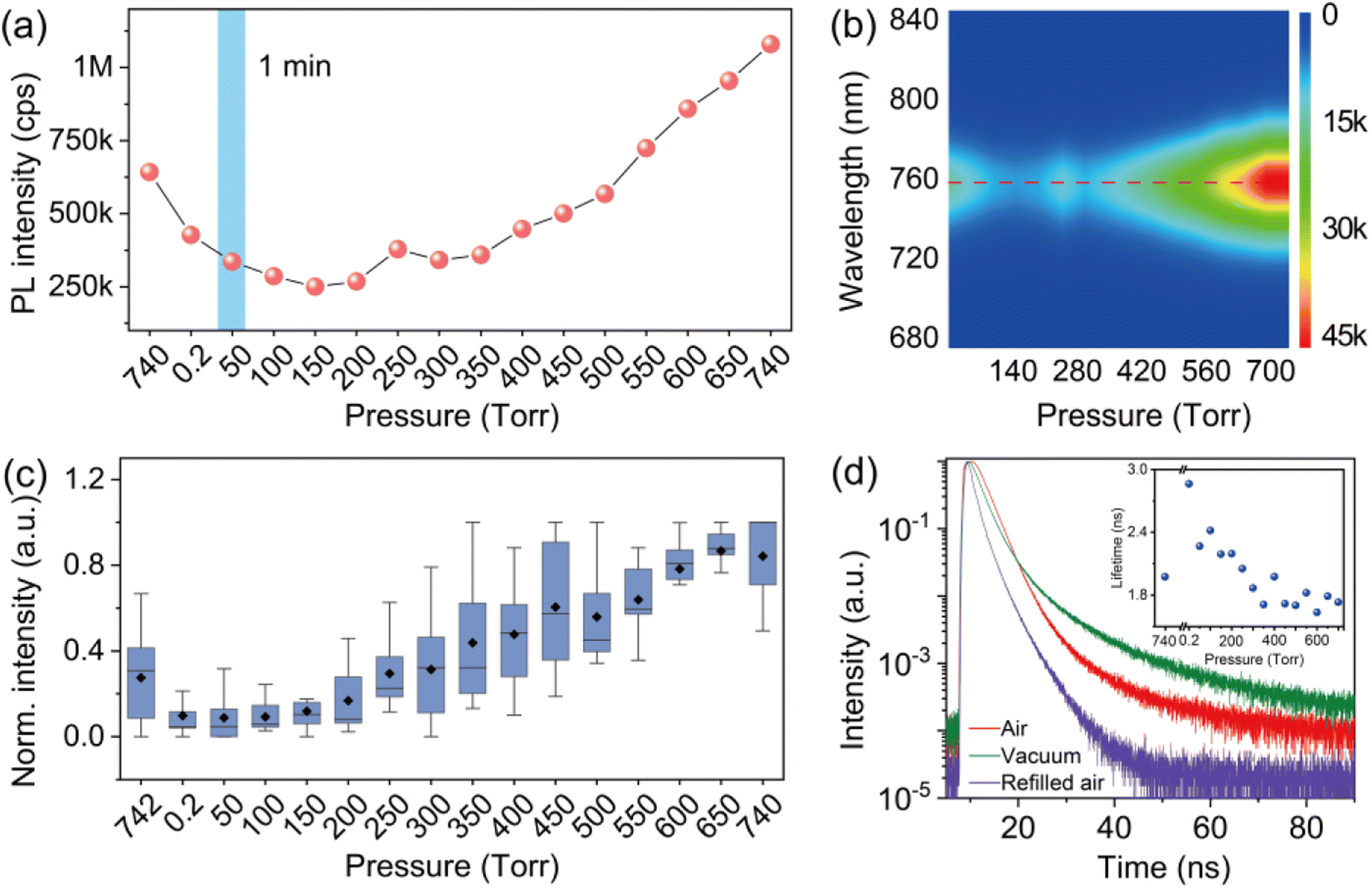 | ||
| Fig. 3 (a) Evolution of the PL intensity of an individual MAPbI3 crystal upon air–vacuum–air exchange. The refilling of air is controlled with an interval of 50 torr for each step and the PL is monitored for 1 min at each pressure. (b) Evolution of the PL spectrum of the crystal. (c) Statistical result of the normalized PL response of an ensemble of individual crystals as a function of air pressure. The data of each crystal is normalized to its maximum PL intensity, respectively. The median and mean value are shown by the line dividing the boxes and the solid black rhombus symbols, respectively. (d) PL decay dynamics of an individual MAPbI3 crystal under ambient air (red), vacuum (green), and refilled air at ambient pressure (purple). The inset shows the averaged lifetime of the crystal as a function of air pressure. | ||
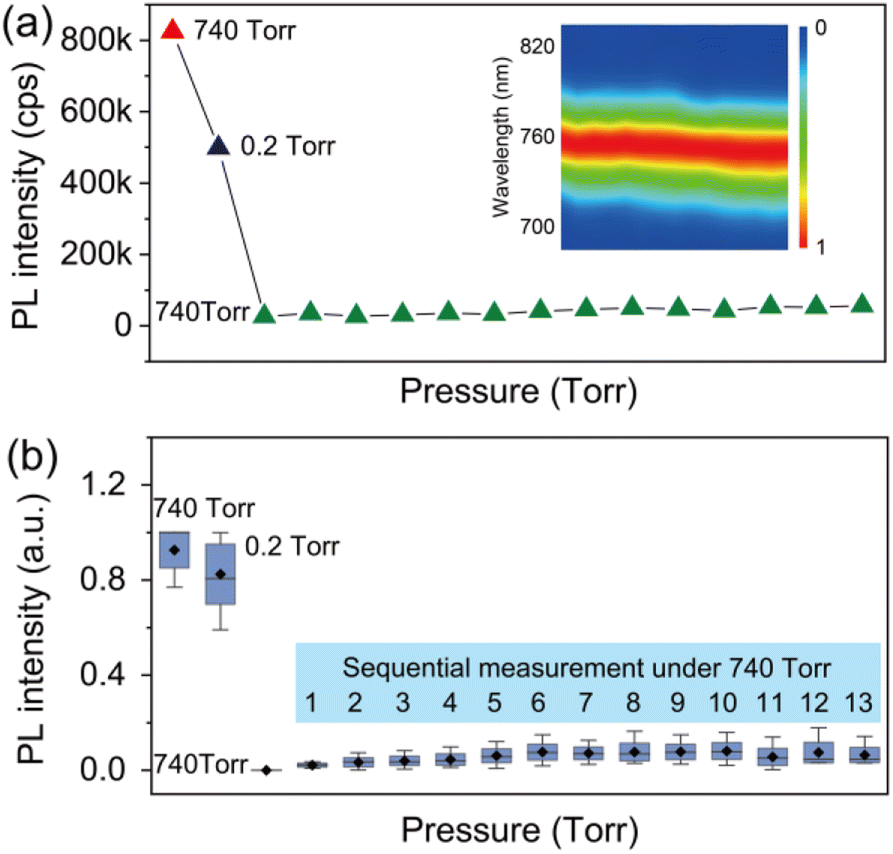 | ||
| Fig. 4 (a) Evolution of the PL from an individual MAPbI3 crystal exposed to ambient air, vacuum, and back to ambient air immediately. The green triangle demonstrates the PL of the crystal measured sequentially with an acquisition time of 1 min for each point. The inset shows the normalized PL spectra of the crystal. (b) Statistic of the PL intensity fluctuation for an ensemble of individual crystals. The numbers from 1 to 13 represent sequential measurements under the same pressure (740 torr). | ||
Given that the interaction of individual perovskite crystals with step increased air facilitates the PL enhancement, it is natural to explore which gas molecule plays a significant role and how. Thus, we performed a control experiment to study the influence of the main components in air. Fig. 5a shows the evolution of the PL trajectory of an ensemble of individual crystals when changing the atmospheric condition from air to pure nitrogen (N2) and back to air. It can be seen that the PL can be quenched upon N2 exposure. However, different from the PL response upon air–vacuum–air exchange, the PL of the crystals shows a reversible and very fast recovery upon exchange from N2 to air. Considering that the exchange of the atmospheric condition from air to pure N2 has a net result of removing O2, the infiltration of N2 is believed to activate the nonradiative recombination centers which were passivated by O2. This can also be confirmed by analyzing the evolution of the PL of the crystals upon exchange of pure O2 and pure N2 (Fig. 5b), indicating that O2 dominates the PL enhancement effect observed in our experiment.
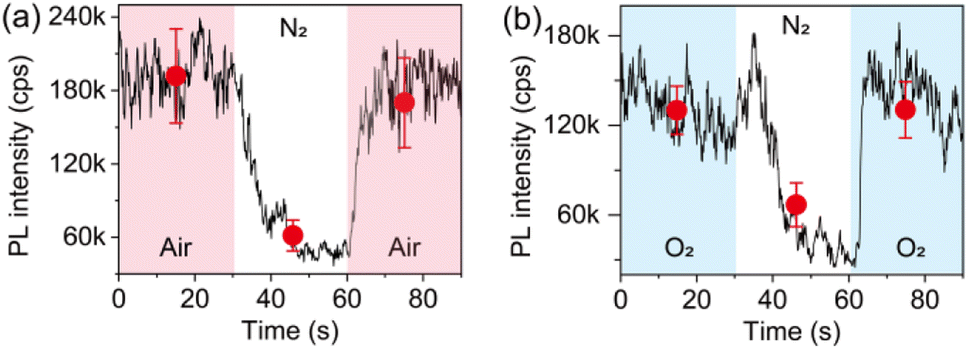 | ||
| Fig. 5 PL response of an ensemble of individual MAPbI3 crystals upon exposure to (a) air–N2–air exchange, and (b) O2–N2–O2 exchange. The solid lines show the PL fluctuation averaged over 20 crystals. The solid circles show the statistical results of the PL intensity averaged over each 30 s for the crystals. | ||
2.3. Mechanistic insights into atmosphere-dependent radiative and nonradiative recombination
Before giving a reasonable explanation of the atmosphere-dependent photophysics, it is critical to discuss the origin of the PL in MAPbI3 crystals. A widely accepted mechanism is the band-to-band recombination of free electrons and holes or the strongly bound exciton recombination.25,26 To clarify this issue, we performed a power-dependent PL measurement of individual MAPbI3 crystals. The corresponding PL intensity shows a power-law dependence on the excitation power (Fig. S5†), indicating an excitonic recombination nature in MAPbI3.27 Defects formed due to structural non-periodicity or crystalline imperfections in perovskite would provide localized electronic states at the energy levels different from the band level, at which the photo-generated electrons or holes will get trapped.22 The trapped charge carriers can be lost though nonradiative recombination, which, thus, decreases the PLQY of the perovskite. As such, we propose that the variation of the nonradiative recombination centres in perovskite due to the influence of the surrounding environment may account for the fluctuation of the PL properties.On the contrary, the annihilation of the nonradiative recombination centers requires the cooperative interaction of both the light illumination and the air condition. This assumption can be confirmed by the light-assisted recovery of the PL shown in Fig. 3. When the chamber is refilled with air to the ambient pressure in darkness and then the laser is switched on, the PL cannot be recovered (Fig. S3 and S4†). It can be seen that the activation of the PL requires the illumination of the sample, as shown in Fig. 3a. This observation highlights the important role of light in the passivation effect of the atmosphere on perovskite materials.5,7,28 However, the overexposure of perovskite crystals to light is detrimental to the photophysics of the perovskite crystals. Fig. S6† shows the results obtained using the same procedure as that in Fig. 3 but with a prolonged illumination of 5 min during acquisition. The PL of the crystals shows an initial increase and a further decrease with the refilling of air step-by-step. This means that prolonged exposure to light and air would induce excess nonradiative recombination centers which further quench the PL of perovskite crystals.
Furthermore, it should be noted that the significant PL recovery can only be found with the gradual increase of the air pressure (Fig. 3), in stark contrast with that measured on the crystals which were exposed to ambient air immediately and illuminated with the same time frame (1 min for each point) (Fig. 4). The dependence of PL recovery on the air pressure indicates that the passivation of nonradiative recombination centers in the crystals studied in this experiment cannot be simply attributed to the physisorption of the gas molecule on the crystal surface that has been supposed to explain the instantaneous recovery of the PL of perovskite materials in air.16,17 Instead, the cooperation effect of light and air pressure indicates a more likely role of the structure or a photochemical process. Previous reports have documented the correlation of pressure-induced structural evolutions with changes in the optical properties of perovskites, both under high hydrostatic pressure29,30 and under low atmospheric pressure,31,32 inspiring the efforts to improve the performance of perovskites through structural engineering.33–35 One of the central goals of these efforts is to release the residual local strain that inherently exists in the solution-processed halide perovskite films. It has been proposed that the local lattice strain in perovskite provides a driving force for defect formation which is directly associated with the creation of nonradiative recombination.36 Therefore, tailoring the local lattice strain would be a useful strategy to improve the PL properties and stability of perovskites.37 Given the hybrid nature and soft lattice of OHPs, their structural changes are susceptible to external driving forces, such as pressure and light. We can, thus, propose that the pressure-dependent PL response observed in our work can be somehow associated with the variation of the local strain in perovskite crystals in which the lattice distortion plays an important role in the activation of nonradiative recombination centers.
Based on these observations, we propose a possible reaction scheme for the creation and annihilation process of trap states in perovskite under the influence of atmospheric conditions (Fig. 6). Here, we attribute the significant PL quenching in vacuum to the activation of nonradiative defects in individual MAPbI3 crystals due to the removal of air in the surroundings, which induces the formation of both deep trap states in the mid-gap and shallow traps near the band edge. We propose that the former dominates the nonradiative decay rate (knr) of the perovskite while the latter mainly influences the radiative decay rate (kr). For a simple scheme, the PLQY (Φ) and PL lifetime (τ) of the perovskite crystals can be related to the radiative and nonradiative rate by the well-known phenomenological expressions: Φ = kr/(kr + knr) = 1/(1 + knr/kr) and τ = 1/(kr + knr). With the activation of trap sites, the increase in the nonradiative decay rate will certainly lead to a decreased PLQY, which is in agreement with a previous report.38 However, we hypothesize that the atmospheric condition also plays a significant role in inducing the fluctuation of the radiative rate due to the involvement of shallow traps. Such an influence is reminiscent of the “delayed” PL kinetics induced by trapping and detrapping of carriers between the shallow traps and the emissive state,39–41 resulting in long-lived PL decay components and a decreased radiative rate with the increase of trap density. Support for this speculation comes from the variation of decay kinetics of individual MAPbI3 crystals during the air–vacuum–air exchange. As shown in Fig. 3d, the extended PL lifetime in vacuum can be a direct manifestation of the reduction of the radiative decay rate kr in vacuum compared to that in air. Also considering the fluctuation of the nonradiative decay rate knr, this can only happen if the decrease in kr is larger than the increase in knr in the crystal. This implies relatively lower formation energies for shallow traps than for deep traps, which is consistent with the arguments in the literature.22,42 With a gradual increase of the air pressure, the O2 exposure under light illumination will passivate the nonradiative trap sites, leading to enhanced Φ and decreased τ, respectively, due to the accelerated kr and suppressed knr. The above observations are in accordance with the point of view which argues that a longer PL lifetime (such as the case in vacuum) does not necessarily mean an improved performance of the perovskite materials; the PLQY should also be well considered.42
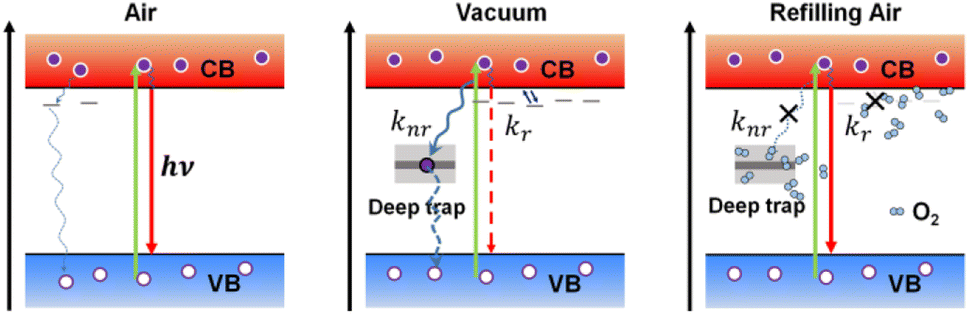 | ||
| Fig. 6 Schematic depiction of proposed radiative and nonradiative dynamics in individual MAPbI3 crystals under air, vacuum, and refilling air. Straight and curly lines present radiative and nonradiative recombination, respectively. In vacuum, both deep traps and shallow traps are activated, resulting in an increased nonradiative recombination rate (knr) owing to deep traps, and a decreased radiative rate (kr) due to trapping and detrapping between an emissive state and shallow traps near the band edge. Nonradiative recombination centers can be passivated with the refilling of air in combination with light illumination, altering both the radiative and nonradiative rate of the crystal. | ||
The unambiguous clarification of the nature of these nonradiative trap sites that can be activated and deactivated by atmospheric conditions remains a daunting task, however, potential candidates are valid based on our observations and the literature. Typically, the shallow traps will involve a relatively small energy difference away from the band edge, corresponding to low formation energies.43 These shallow traps have been widely reported to be halide-ion-related vacancies or interstitials, such as VI and Ii.22,42,44 Due to their low formation energies, the activation and passivation of these shallow traps can be very sensitive to external stimuli such as light and electrical bias,31,45 where photo-induced or bias-induced ion migration has been reported to play an important role. The deep traps responsible for the nonradiative recombination in perovskite showed a complex nature, such as Frenkel defects (complexes consisting of a vacancy and an interstitial, e.g., VI+/Ii−)28 or donor–acceptor pairs.38,46 Furthermore, it is interesting to note that photo-induced hot carriers can be easily captured by shallow traps.47 However, the trapped carriers have a high probability of detrapping, which is not believed to influence the nonradiative recombination dynamics. In contrast, cold carriers mainly interact with deep traps.47 Unfortunately, it is impossible for us to investigate the role of the traps in influencing the radiative and nonradiative recombination of hot and cold carriers in this work, because there would be an ultrafast cooling process for hot carriers,48 which is beyond the scope of our experiment.
The passivation of these trap sites has been repeatedly proven in the literature to be associated with light-induced ion migration, especially in the presence of O2, which accounts for the PL enhancement in perovskites.5,31,49 It has been found that light irradiation can promote the passivation of Frenkel defects in polycrystalline MAPbI3, thus restoring a more perfect crystalline environment.28 Furthermore, it has been proposed that upon light illumination, MAPbI3 can undergo a relaxation of local lattice strain due to lattice expansion, leading to improved PL properties.50 However, in our case, the light-induced PL activation observed showed a strong dependence on the air pressure level. This means that it is only the cooperation of light and pressure that results in the lattice restoration and defect annihilation. The passivation effect of light and oxygen also highlights the role of the photochemical reaction in the trap curing process. This is in agreement with the literature regarding the photoinduced PL enhancement of perovskite, which argues that chemisorption of O2 on perovskite leads to the formation of passivating superoxide species.51–54 Despite this, the creation of the trap sites in vacuum requires a more complex explanation. We speculate that the exchange of the atmospheric condition from air to vacuum induces a variation of the local strain on the crystal lattice. The resulting local distortion leads to a more defective lattice and, thus, a diminished PL intensity. To do so, the local lattice distortion should have the possibility of generating ion migration channels inside perovskites, which is consistent with the argument of Yuan et al.55 The gradual refilling of air could facilitate the light-induced annihilation of the trap sites formed in vacuum with a concomitant slow lattice restoration. But if the crystals are exposed immediately to ambient air pressure or the illumination is maintained for a longer time (Fig. 4 and S6†), the passivation effect may have to compete with the photodegradation of the perovskite, which inhibits the PL activation.
3. Conclusion
In summary, we performed PL measurements on individual MAPbI3 crystals in controlled atmospheric conditions to unveil the role of the environment in radiative and nonradiative recombination dynamics in perovskites. We showed that PL can be quenched upon exposure of individual perovskite crystals to vacuum due to the activation of nonradiative recombination centers, while the cooperation of light and air in moderate pressure facilitates the passivation of these trap sites, resulting in PL recovery. On the basis of in situ PL spectra and PL lifetime characterization of the crystals under exchanged atmospheric conditions and previous well-established facts, we propose that the creation of nonradiative recombination centers likely originated from vacuum-induced slight lattice distortion in the perovskite crystals. The air pressure-dependent PL enhancement supports the hypothesis that the photoinduced formation of superoxide species is capable of passivating nonradiative recombination centers accompanied by lattice restoration. Therefore, it seems crucial to ensure that MAPbI3 perovskite, when incorporated into photovoltaic devices, should be exposed under an appropriate atmospheric condition to improve its performance. Understanding the effect of atmospheric conditions on the photophysical properties of individual perovskite crystals not only helps to reveal the fundamental mechanism regarding the long-term stability of perovskite materials, but also provides suggestions for the preparation of perovskite-based devices.4. Experimental methods
4.1. Preparation of individual perovskite crystals
The perovskite solution was prepared by first mixing the powders of methylammonium iodide (159 mg, Sigma-Aldrich, catalogue no. 793493-5G) and lead iodide (461 mg, Sigma-Aldrich, catalogue no. 211168-50G). Then, the mixed powders were added into 1.25 mL gamma butyrolactone (GBL, Sigma-Aldrich, catalogue no. B0767). The obtained mixture was dissolved and stirred at 60 °C for 2 h at 500 rpm. To prepare individual perovskite crystals, the precursor solution was diluted to 0.05%, 20 μL was drop cast onto a glass coverslip, and then annealed at 80 °C for 20 min under ambient conditions.4.2. Optical characterization
A home-build laser scanning confocal microscope was used to take microscope images, PL spectra, and PL lifetimes of perovskite crystals. A schematic view of the experimental setup can be found in Fig. S2.† Briefly, the sample was sealed in a vacuum cell of a cryostat (Montana Instrument), in which the vacuum degree can be controlled by a vacuum pump. A 485 nm diode laser with pulse repetition rate of 2.5 MHz was used to excite the sample through an objective lens (Attocube, NA = 0.82) mounted in the vacuum cell. The PL of the sample was detected point by point using a single-photon detector and analyzed by a single photon counting module (TCSPC, PicoQuant, HydraHarp 400). To achieve the PL spectra of the perovskite crystals, the PL was separated to a spectrograph (Andor, SR-303i-A) and detected by an electron-multiplying charge-coupled devices (EMCCD, Andor, DU971P-BV).4.3. Morphology and size characterization
SEM images were taken using a microscope (Hitachi, SU8010) with energy-dispersive X-ray spectroscopy (EDS). The size and morphology of the individual perovskite crystals were characterized with SEM at an accelerating voltage of 3 kV. AFM (Nanosurf, C3000) was used to measure the height of the individual crystals.4.4. X-ray diffraction measurements
XRD measurements were performed using an X-ray diffractometer (Rigaku, MiniFlex600) (Cu Kα radiation) with a high-speed 1D array detector D/tex Ultra2. The sample was scanned with a step size of 0.001° and a scan speed of 5.0° min−1.Data availability
The data generated and analyzed in this study are available from the corresponding author on reasonable request.Author contributions
R. C. and L. X. designed and supervised the experiments. R. C. and W. G. carried out the optical experiments. W. Z. and Z. W. prepared and characterized the sample. G. Z. and C. Q. did the investigation. S. J. was responsible for gas management. R. C., W. G., and L. X. wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is sponsored by the National Key Research and Development Program of China (Grant No. 2017YFA0304203), the Natural Nature Science Foundation of China (Grant No. 62127817, 62075122, 62075120, 61875109, 91950109, 62011530133), the research projects supported by Shanxi Scholarship Council of China (Grant No. HGKY2019002) and Department of Science and Technology of Shanxi Province (Grant No. 202104041101021), PCSIRT (Grant No. IRT_17R70), 1331KSC, and 111 Project (Grant No. D18001).References
- Y. Tu, J. Wu, G. Xu, X. Yang, R. Cai, Q. Gong, R. Zhu and W. Huang, Adv. Mater., 2021, 33, 2006545 CrossRef CAS PubMed.
- I. Cardinaletti, T. Vangerven, S. Nagels, R. Cornelissen, D. Schreurs, J. Hruby, J. Vodnik, D. Devisscher, J. Kesters and J. D'Haen, Sol. Energy Mater. Sol. Cells, 2018, 182, 121–127 CrossRef CAS.
- L. K. Reb, M. Böhmer, B. Predeschly, S. Grott, C. L. Weindl, G. I. Ivandekic, R. Guo, C. Dreißigacker, R. Gernhäuser and A. Meyer, Joule, 2020, 4, 1880–1892 CrossRef CAS.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan and D. Di Girolamo, Nat. Energy, 2020, 5, 35–49 CrossRef.
- Y. Tian, M. Peter, E. Unger, M. Abdellah, K. Zheng, T. Pullerits, A. Yartsev, V. Sundström and I. G. Scheblykin, Phys. Chem. Chem. Phys., 2015, 17, 24978–24987 RSC.
- J. F. Galisteo-López, M. Anaya, M. Calvo and H. Míguez, J. Phys. Chem. Lett., 2015, 6, 2200–2205 CrossRef.
- D. W. DeQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulović, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 11683 CrossRef CAS.
- Y. Tian, A. Merdasa, E. Unger, M. Abdellah, K. Zheng, S. McKibbin, A. Mikkelsen, T. Pullerits, A. Yartsev and V. Sundstrom, J. Phys. Chem. Lett., 2015, 6, 4171–4177 CrossRef CAS.
- S. G. Motti, M. Gandini, A. J. Barker, J. M. Ball, A. R. S. Kandada and A. Petrozza, ACS Energy Lett., 2016, 1, 726–730 CrossRef CAS.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., 2015, 127, 8326–8330 CrossRef.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef.
- R. Guo, D. Han, W. Chen, L. Dai, K. Ji, Q. Xiong, S. Li, L. K. Reb, M. A. Scheel and S. Pratap, Nat. Energy, 2021, 6, 977–986 CrossRef CAS.
- Y. J. Hofstetter, I. García-Benito, F. Paulus, S. Orlandi, G. Grancini and Y. Vaynzof, Front. Chem., 2020, 8, 66 CrossRef CAS PubMed.
- I. Deretzis, A. Alberti, G. Pellegrino, E. Smecca, F. Giannazzo, N. Sakai, T. Miyasaka and A. La Magna, Appl. Phys. Lett., 2015, 106, 131904 CrossRef.
- R. K. Gunasekaran, D. Chinnadurai, A. R. Selvaraj, R. Rajendiran, K. Senthil and K. Prabakar, ChemPhysChem, 2018, 19, 1507–1513 CrossRef CAS PubMed.
- H.-H. Fang, S. Adjokatse, H. Wei, J. Yang, G. R. Blake, J. Huang, J. Even and M. A. Loi, Sci. Adv., 2016, 2, e1600534 CrossRef.
- Y. Wang, Y. Ren, S. Zhang, J. Wu, J. Song, X. Li, J. Xu, C. H. Sow, H. Zeng and H. Sun, Commun. Phys., 2018, 1, 96 CrossRef CAS.
- I. G. Scheblykin, Adv. Energy Mater., 2020, 10, 2001724 CrossRef CAS.
- J. R. Vicente, A. R. Miandashti, K. W. E. Sy Piecco, J. R. Pyle, M. E. Kordesch and J. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 18034–18043 CrossRef CAS PubMed.
- A. Mukherjee, M. Roy, N. Pathoor, M. Aslam and A. Chowdhury, J. Phys. Chem. C, 2021, 125, 17133–17143 CrossRef CAS.
- R. Chen, B. Xia, W. Zhou, G. Zhang, C. Qin, J. Hu, I. G. Scheblykin and L. Xiao, Adv. Photonics Res., 2022, 3, 2100271 CrossRef.
- H. Jin, E. Debroye, M. Keshavarz, I. G. Scheblykin, M. B. Roeffaers, J. Hofkens and J. A. Steele, Mater. Horiz., 2020, 7, 397–410 RSC.
- R. Chen, B. Xia, W. Zhou, W. Guan, G. Zhang, C. Qin, J. Hu, L. Xiao and S. Jia, Opt. Express, 2021, 29, 1851–1869 CrossRef CAS.
- X. Tang, M. Brandl, B. May, I. Levchuk, Y. Hou, M. Richter, H. Chen, S. Chen, S. Kahmann and A. Osvet, J. Mater. Chem. A, 2016, 4, 15896–15903 RSC.
- Y. Yamada, T. Nakamura, M. Endo, A. Wakamiya and Y. Kanemitsu, J. Am. Chem. Soc., 2014, 136, 11610–11613 CrossRef CAS.
- M. B. Johnston and L. M. Herz, Acc. Chem. Res., 2016, 49, 146–154 CrossRef CAS.
- T. Schmidt, K. Lischka and W. Zulehner, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 8989 CrossRef.
- E. Mosconi, D. Meggiolaro, H. J. Snaith, S. D. Stranks and F. De Angelis, Energy Environ. Sci., 2016, 9, 3180–3187 RSC.
- X. Lü, W. Yang, Q. Jia and H. Xu, Chem. Sci., 2017, 8, 6764–6776 RSC.
- P. Postorino and L. Malavasi, J. Phys. Chem. Lett., 2017, 8, 2613–2622 CrossRef CAS PubMed.
- M. Anaya, J. F. Galisteo-López, M. E. Calvo, J. P. Espinós and H. Míguez, J. Phys. Chem. Lett., 2018, 9, 3891–3896 CrossRef CAS PubMed.
- M. Holland, A. Ruth, K. Mielczarek, V. V. Dhas, J. J. Berry and M. D. Irwin, ACS Appl. Mater. Interfaces, 2022, 14, 9352–9362 CrossRef PubMed.
- Q. Li, L. Zhang, Z. Chen and Z. Quan, J. Mater. Chem. A, 2019, 7, 16089–16108 RSC.
- Y. Jiang, S.-C. Yang, Q. Jeangros, S. Pisoni, T. Moser, S. Buecheler, A. N. Tiwari and F. Fu, Joule, 2020, 4, 1087–1103 CrossRef CAS.
- L. Zhang, K. Wang, Y. Lin and B. Zou, J. Phys. Chem. Lett., 2020, 11, 4693–4701 CrossRef CAS PubMed.
- T. W. Jones, A. Osherov, M. Alsari, M. Sponseller, B. C. Duck, Y.-K. Jung, C. Settens, F. Niroui, R. Brenes and C. V. Stan, Energy Environ. Sci., 2019, 12, 596–606 RSC.
- H.-S. Kim and N.-G. Park, NPG Asia Mater., 2020, 12, 78 CrossRef CAS.
- M. Gerhard, B. Louis, R. Camacho, A. Merdasa, J. Li, A. Kiligaridis, A. Dobrovolsky, J. Hofkens and I. G. Scheblykin, Nat. Commun., 2019, 10, 1698 CrossRef PubMed.
- V. S. Chirvony, S. González-Carrero, I. Suarez, R. E. Galian, M. Sessolo, H. J. Bolink, J. P. Martinez-Pastor and J. Perez-Prieto, J. Phys. Chem. C, 2017, 121, 13381–13390 CrossRef CAS.
- T. Guo, R. Bose, X. Zhou, Y. N. Gartstein, H. Yang, S. Kwon, M. J. Kim, M. Lutfullin, L. Sinatra and I. Gereige, J. Phys. Chem. Lett., 2019, 10, 6780–6787 CrossRef CAS PubMed.
- V. S. Chirvony, K. S. Sekerbayev, H. P. Adl, I. Suárez, Y. T. Taurbayev, A. F. Gualdrón-Reyes, I. Mora-Seró and J. P. Martínez-Pastor, J. Lumin., 2020, 221, 117092 CrossRef CAS.
- J. M. Ball and A. Petrozza, Nat. Energy, 2016, 1, 16149 CrossRef CAS.
- W.-J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- Z. Wang, Z. Huang, G. Liu, B. Cai, S. Zhang and Y. Wang, Adv. Opt. Mater., 2021, 9, 2100346 CrossRef CAS.
- A. Merdasa, Y. Tian, R. Camacho, A. Dobrovolsky, E. Debroye, E. L. Unger, J. Hofkens, V. Sundström and I. G. Scheblykin, ACS Nano, 2017, 11, 5391–5404 CrossRef CAS.
- M. Righetto, S. S. Lim, D. Giovanni, J. W. M. Lim, Q. Zhang, S. Ramesh, Y. K. E. Tay and T. C. Sum, Nat. Commun., 2020, 11, 2712 CrossRef CAS PubMed.
- Q. Sun, J. Gong, X. Yan, Y. Wu, R. Cui, W. Tian, S. Jin and Y. Wang, Nano Lett., 2022, 22, 2995–3002 CrossRef CAS PubMed.
- D. Meggiolaro, E. Mosconi and F. De Angelis, ACS Energy Lett., 2017, 2, 2794–2798 CrossRef CAS.
- H. Tsai, R. Asadpour, J.-C. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan and S. Tretiak, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- R. Brenes, C. Eames, V. Bulović, M. S. Islam and S. D. Stranks, Adv. Mater., 2018, 30, 1706208 CrossRef PubMed.
- X. Feng, H. Su, Y. Wu, H. Wu, J. Xie, X. Liu, J. Fan, J. Dai and Z. He, J. Mater. Chem. A, 2017, 5, 12048–12053 RSC.
- R. Brenes, D. Guo, A. Osherov, N. K. Noel, C. Eames, E. M. Hutter, S. K. Pathak, F. Niroui, R. H. Friend and M. S. Islam, Joule, 2017, 1, 155–167 CrossRef CAS.
- Y. Zhou, X. Hu, D. Xie and Y. Tian, J. Phys. Chem. C, 2020, 124, 3731–3737 CrossRef CAS.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00541g |
| This journal is © The Royal Society of Chemistry 2022 |