
Supramolecular nanocapsules as two-fold stabilizers of outer-cavity sub-nanometric Ru NPs and inner-cavity ultra-small Ru clusters†
Ernest Ubasart‡
 a,
Irene Mustieles Marin‡b,
Juan Manuel Asensiob,
Gabriel Menciab,
Ángela M. López-Vinascob,
Cristina García-Simóna,
Iker del Rosalb,
Romuald Poteau*b,
Bruno Chaudret*b and
Xavi Ribas*a
a,
Irene Mustieles Marin‡b,
Juan Manuel Asensiob,
Gabriel Menciab,
Ángela M. López-Vinascob,
Cristina García-Simóna,
Iker del Rosalb,
Romuald Poteau*b,
Bruno Chaudret*b and
Xavi Ribas*a
aInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, Campus Montilivi, E-17003 Girona, Catalonia, Spain. E-mail: xavi.ribas@udg.edu
bLaboratoire de Physique et Chimie des Nano-objets (LPCNO), INSA-CNRS, Université de Toulouse, 135 Ave. de Rangueil, 31077 Toulouse, France
First published on 1st April 2022
Abstract
The synthesis of metallic nanoparticles (MNP) with high surface area and controlled shape is of paramount importance to increase their catalytic performance. The detailed growing process of NP is mostly unknown and understanding the specific steps would pave the way for a rational synthesis of the desired MNP. Here we take advantage of the stabilization properties exerted by the tetragonal prismatic supramolecular nanocapsule 8·(BArF)8 to develop a synthetic methodology for sub-nanometric RuNP (0.6–0.7 nm). The catalytic properties of these sub-nanometric nanoparticles were tested on the hydrogenation of styrene, obtaining excellent selectivity for the hydrogenation of the alkene moiety. In addition, the encapsulation of [Ru5] clusters inside the nanocapsule is strikingly observed in most of the experimental conditions, as ascertained by HR-MS. Moreover, a thorough DFT study enlightens the nature of the [Ru5] clusters as tb-Ru5H2(η6-PhH)2(η6-pyz)3 (2) trapped by two arene moieties of the clip, or as tb-Ru5H2(η1-pyz)6(η6-pyz)3 (3) trapped between the two Zn–porphyrin units of the nanocapsule. Both options fulfill the Wade-Mingos counting rules, i.e. 72 CVEs for the closotb. The trapped [Ru5] metallic clusters are proposed to be the first-grown seeds of subsequent formation of the subnanometric RuNP. Moreover, the double role of the nanocapsule in stabilising ∼0.7 nm NPs and also in hosting ultra-small Ru clusters, is unprecedented and may pave the way towards the synthesis of ultra-small metallic clusters for catalytic purposes.
New conceptsDesigning new methodologies to control the size and shape of metallic nanoparticles is highly desirable. The stabilization of sub-nanometric NP or even metallic clusters will allow the access to ultra-small metallic species very active in catalysis. In order to do that, it is mandatory to gain control on the nucleation of the metallic atoms towards the formation of metallic clusters, which sequentially aggregate in the form of NPs, and there is an evident lack of detailed understanding of the NPs growth process. We introduce here a new supramolecular-controlled methodology for the direct observation of both sub-nanometric Ru NPs and [Ru5] metallic clusters. Our study combines experimental evidences with a thorough computational study for comprehending the formation of the small metallic clusters en-route to sub-nanometric aggregates, thanks to the stabilization effect in the supramolecular cavity, which is constituted by multiple arene rings and porphyrin units. The direct observation of the metallic cluster seeds for the formation of NP sheds light into this fundamental process, where the double role of the nanocapsule in stabilising 0.6–0.7 nm NPs in the outer-cavity and also host ultra-small Ru5 clusters inside the cavity is unprecedented. Moreover, the modularity of the supramolecular hosts and the use of other metal precursors renders a versatile strategy towards the synthesis of ultra-small metallic clusters for catalytic purposes. |
Introduction
The control of size and shape of metallic nanoparticles (MNPs) is mandatory to comprehend the implications of these parameters in their catalytic performance. In general terms, the quest for sub-nanometric MNPs in a reproducible form is appealing, owing to the fact that enhanced catalytic activity is expected for MNPs with larger surface/volume ratio, i.e. of the smaller size possible.1,2 In order to stabilize MNPs, organic molecules acting as ligands attached to the surface of the nanoparticles can be employed leading to a large variety of possibilities.3 In addition, trapping NPs (including Au, Pt, and NiCl2, PdCl2 and PtCl2)4–6 in the channels of MOFs has been demonstrated to be a successful strategy7,8 to enhance the selectivity of selected reactions.2,9 However, the stabilization of ultra-small MNPs is far more challenging. In this regard, some examples of organic or supramolecular cages capable of stabilizing MNPs have been reported, taking advantage of their putative affinity to the hollow cavity of the host. Examples of Pd NPs mainly, but also of Au NPs,10,11 Ag NPs,12 ferrihydrite NPs13 and Ru NPs14 have been reported. For instance, in 2014, Zhang and co-workers designed a 3D, shape-persistent organic molecular cage to act as a template for the growth of Au NPs with a small size (1.9 ± 0.4 nm),15 standing as one of the first examples of Au NPs synthesized in a confined space. However, most of the studies rely on the stabilization of Pd NPs, since their catalytic activity is of paramount interest. In 2018, Xu and co-workers reported a porous organic cage, RCC3, able to encapsulate active Pd NPs (Fig. 1a).16 The sub-nanometric NPs (0.72 nm) showed high stability and durability and demonstrated to be catalytically active in the hydrogenation of nitroarenes and reduction of organic dyes. On the other hand, Li and co-workers developed a porous organic nanocapsule that was used as an efficient heterogeneous catalyst for the carbonylation of aryl halides.17 Later, in 2019, Jiang's group developed Pd NPs (1.9 ± 0.4 nm) stabilized by an organic nanocapsule.18 The authors demonstrated the ability of a water soluble nanocapsule of sensing, controlling nanoparticle growth and developing a catalytic active platform for the hydroxylation of 4-nitrophenylboronic acid to 4-nitrophenol. Also in 2019, Beaumont and co-workers designed a porous organic cage that could incorporate very small Pd NPs (1.6 nm),19 being an effective system for the CO oxidation (Fig. 1b). More recently, Bharadwaj reported in 2020 a series of three positional isomers of organic cages (o-OC, m-OC and p-OC) capable to stabilize Pd NPs with a 1–2 nm of diameter (Fig. 1c).20 These NPs showed excellent catalysis of Tsuji–Trost allylation at room temperature. | ||
| Fig. 1 Pd-NPs encapsulated in porous organic cages for (A) catalytic hydrogenation of nitroarenes, (B) for CO oxidation and (C) for Tsuji–Trost allylation; (D) Ru NPs encapsulated in a porous coordination cage for dehydrogenation; (E) supramolecular nanocapsule 8·(BArF)8 for stabilization of outer-cavity sub-nanometric Ru NPs and inner-cavity ultra-small Ru clusters (this work). | ||
A sole example of the stabilization of Ru NPs in cages was reported by Zhou in 2018, by taking advantage of the accumulation of Ru3+ ions in an anionic porous coordination cage (PCC-2), in which Ru NPs (∼2 nm) were formed upon mild reduction with NaBH4 (Fig. 1d).14 The resulting Ru NPs@PCC-2 composite exhibited high catalytic activity in methanolysis of ammonia borane, which is relevant in chemical hydrogen storage.
Many of the above mentioned reports have argued or postulated that the NPs form within the cages, but this has been questioned since the 1.3–1.9 nm relative size range is far too big for the small cavity size (diameter < 1 nm), and an inter-pore arrangement among different cages is a more realistic scenario in many cases.19 The role of the cages is thus unclear and doubts are cast whether they promote external stabilization of NPs or incipient encapsulation of metallic cluster seeds.
On the other hand, we developed supramolecular tetragonal prismatic nanocapsules as hosts for large guests, as for example higher fullerenes and dumbbell-shaped azafullerene (C59N)2.21,22 In the present work, we targeted the formation of small Ru NPs stabilized by a tetragonal prismatic nanocapsule, via mild decomposition of the precursor Ru(cyclooctadiene)(cyclooctatriene). Remarkably, we have succeeded in obtaining sub-nanometric Ru NPs stabilized by nanocapsule 8·(BArF)8 throughout the outer-cavity surface. The small size is in agreement with the outcome of styrene hydrogenation catalysis. Surprisingly, in addition to acting as outer-surface NP-stabilizer, this nanocapsule can trap ultra-small ruthenium clusters ([Ru5], [Ru10] and [Ru15]) in the inner-cavity, as unequivocally indicated by HRMS studies. Moreover, the clusters’ nature has been revealed by a thorough DFT computational study, focusing on the evaluation of the relative thermodynamic stability of [Ru5] models. Within the framework of Wade–Mingos rules, possible [Ru5] clusters coated with hydrides and different ligands have been evaluated, including ligands from the solution or from possible adsorption or growth sites of the nanocapsule. The remarkable occurrence of the [Ru5] cluster in the mass spectrum is explained both in terms of electronic effects and of cavities’ size of the nanocapsule. The two-fold stabilization of sub-nanometric NPs and clusters is reported for the first time (Fig. 1e), and might pave the way towards the selective preparation and stabilization of metallic clusters for catalysis.
Results and discussion
The formation of Ru NPs was studied employing different nanocapsules displaying different cavity sizes. The supramolecular structures 6·(BArF)8 and 8·(BArF)8 (see Fig. 2) were synthesized by self-assembly of a Zn-porphyrin with a macrocyclic Pd-based complex that led to different cavity size from 16.8 to 18.4 Å. In Fig. 2 there is a schematic representation of the synthons of the nanocapsules. The synthesis of the nanoparticles was studied by decomposing Ru(COD)(COT) under 1 bar of H2 at room temperature, which have demonstrated to be successful conditions to synthesize Ru NPs.23 THF was chosen as solvent since both the nanocapsules and Ru(COD)(COT) complex are soluble. Different parameters such as the addition of an auxiliary ligand (pyrazine), reaction time and number of equivalents of the Ru precursor were studied. In addition, the Ru NP synthesis was also performed in the presence of the different nanocapsule fragments in order to study the influence of the different moieties in the NPs stabilization. The different conditions used are summarized in Table 1. The resulting Ru NPs were analysed by TEM and HR-ESI-MS (see details in the ESI†).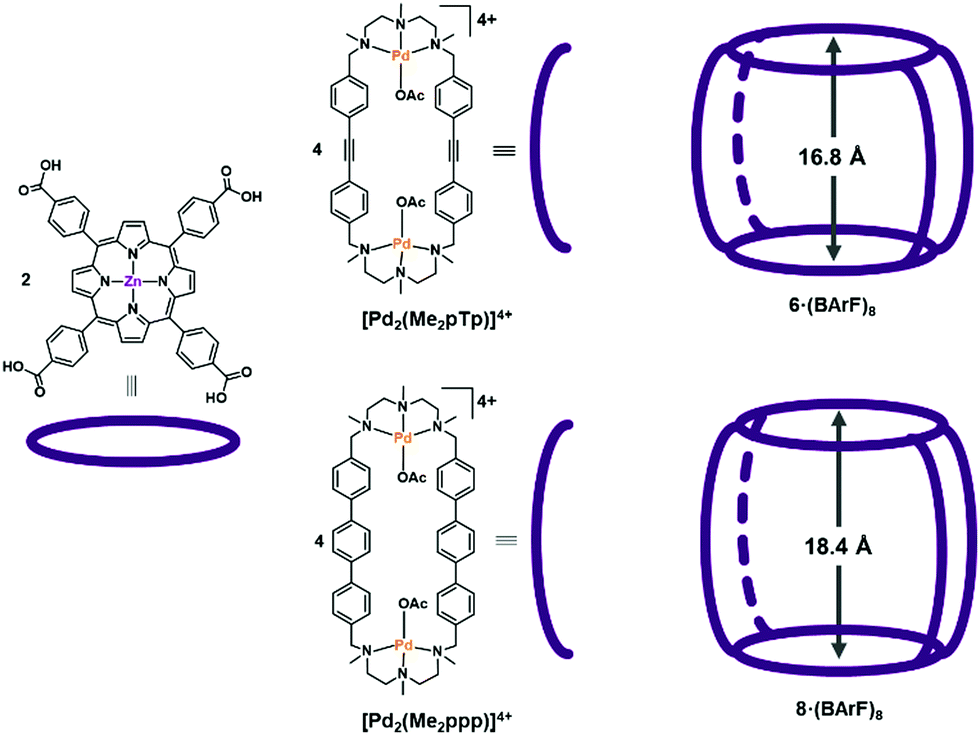 | ||
| Fig. 2 Tetragonal prismatic nanocapsules 6·(BArF)8 and 8·(BArF)8 used in this work, bearing the [Pd2(Me2pTp)]4+ and [Pd2(Me2ppp)]4+ clips, respectively. | ||
| Exp. | N·(BArF)8N = 6 or 8 | Ru equiv. | Pyz equiv. | Time (min) | Size Ru NP (nm) |
|---|---|---|---|---|---|
| E1 | 6 | 130 | 0 | 10 | 1.28 ± 0.17 |
| E2 | 6 | 130 | 0.2 | 10 | 0.68 ± 0.22 |
| E3 | 8 | 130 | 0 | 10 | 1.33 ± 0.24 |
| E4 | 8 | 130 | 0.2 | 10 | 0.65 ± 0.15 |
| E5 | 8 | 40 | 0.2 | 10 | 0.67 ± 0.19 |
| E6 | 8 | 20 | 0.2 | 10 | 0.74 ± 0.19 |
| E7 | 8 | 200 | 0.2 | 2 | 0.74 ± 0.18 |
| E8 | 8 | 130 | 0.2 | 2 | 0.72 ± 0.20 |
| E9 | 8 | 80 | 0.2 | 2 | 0.69 ± 0.18 |
| E10 | 8 | 60 | 0.2 | 2 | 0.71 ± 0.20 |
| E11 | 8 | 40 | 0.2 | 2 | 0.70 ± 0.21 |
| E12 | 8 | 130 | 1 | 2 | 0.60 ± 0.12 |
| E13 | [Pd2(Me2ppp)] clip | 32 | 0.2 | 10 | 0.96 ± 0.24 |
| E14 | ZnTCPP | 65 | 0.2 | 10 | >100 |
The synthesis of the Ru NPs was first studied in presence of the nanocapsules 6·(BArF)8 and 8·(BArF)8 in order to determine the influence of the cavity size (see Table 1, experiments E1 and E3, respectively). For these experiments 130 eq. of Ru precursor respect to the nanocapsule load and, after introduction of H2, a color change occurred almost immediately. The solution containing both nanocapsules changed from a pink solution to a dull dark solution. TEM images of the final solutions are present in Fig. 3a and c. In both cases amorphous NPs of a mean size of 1.3 nm were obtained (see Fig. S1, S3 and S16, ESI†), indicating that the cavity size had no influence, and furthermore, that the NPs were not encapsulated since the size obtained was bigger to that of the nanocapsules.
 | ||
| Fig. 3 TEM images of (a) E1, NPs synthesized with the 6·(BArF)8 nanocapsule, 130 eq. Ru(COD)(COT) and 0 eq. pyrazine; (b) E2 NPs synthesized with 6·(BArF)8 nanocapsule, 130 eq. Ru(COD)(COT) and 0.2 eq. pyrazine; (c) E3, NPs synthesized with 8·(BArF)8 nanocapsule, 130 eq. Ru(COD)(COT) and 0 eq. pyrazine; (d) E4, NPs synthesized with 8·(BArF)8 with 130 eq. Ru(COD)(COT) and 0.2 eq. pyrazine. | ||
To favour the formation of the small NPs an ancillary ligand was added to the reaction mixture. The addition of pyrazine to the solution was intended to help in the stabilization of the Ru nuclei during the decomposition. A small amount, only 0.2 eq. with respect to Ru, was employed to avoid the ligand saturation at the formed NPs surface. Thus, experiments E1 and E3 were repeated in presence of pyrazine (E2 and E4). TEM images are present in Fig. 3b and d. The NPs formed possess a mean size of 0.65 nm in the case of E4. Thus, the presence of pyrazine has a relevant role in the stabilization outside the nanocapsule, decreasing the nanoparticle size by half (Table 1). At this point, in order to determine the influence of the Ru concentration in the medium, we lowered the equivalents of Ru(COD)(COT) to 40 and 20 (experiments E5 and E6, respectively), instead of 130 equiv. TEM images showed the formation of NPs with a mean size of 0.67 and 0.74 nm, respectively (see Fig. S14, ESI†).
By keeping this amount of pyrazine (0.2 eq. respect to Ru), the time of reaction was decreased from 10 to 2 min and a larger range of Ru equivalents employed was screened: 200, 130, 80, 60 and 40 equiv., corresponding to experiments E7 to E11. The diminution of the reaction time hadn’t an impact in the size distribution of the NPs, which ranged between 0.65 and 0.74 nm, indicating that these sub-nanometric NPs formed at the first stage of the reaction. For instance, Ru NP size obtained for E4 (10 min, 130 eq. Ru) is 0.65 nm, very close to 0.72 nm for E8 (2 min, 130 eq. Ru). Likewise, E5 (10 min, 40 eq. Ru) afforded a mean size of 0.67 nm, also close to 0.74 for E11 (2 min, 40 eq. Ru). Further increasing the pyrazine concentration (1 eq. respect to Ru, E12 in Table 1) led within 2 minutes to the formation of uniform Ru NPs of even smaller size (0.6 ± 0.1 nm).
To obtain more information about the role of the nanocapsule moieties we performed blank experiments using the [Pd2(Me2ppp)]4+ clip or the ZnTCPP building blocks instead of the whole nanocapsule (see Fig. 2). By using the Pd-clip (experiment E13), sub-nanometric nanoparticles were obtained, while with the Zn-porphyrin (experiment E14) only aggregates were observed, confirming that the clip moiety has a prominent role if the synthesis and stabilization of this sub-nanometric Ru NPs (Fig. 4).
 | ||
| Fig. 4 TEM images of (a) experiment E13, with [Pd2(Me2ppp)]4+ clip, and (b) experiment E14, with Zn-TCPP. | ||
In order to get a more precise value of the size of the NPs formed, HR-TEM analysis was conducted for E4 (Fig. 5), confirming the subnanometric size (diameter of 0.65 nm average) for the nanoparticles, which showed a non-defined shape. The catalytic properties of these sub-nanometric nanoparticles were tested on the hydrogenation of styrene. Different volumes from the solutions E8 to E12 were taken to set a catalytic loading of 0.25 mol% and the reactions were performed under 3 bar of H2 at 50 °C (see Table S1, ESI†). As shown in Fig. 6, hydrogenation of the double bond occurs with high conversion and excellent selectivity for RuNPs in the range of 0.7 to 1.3 nm (experiments E3, E8–E10 and E13, and in moderate conversion and good selectivity in E11 (Fig. 6)). Catalytic reactions were also performed using the smallest RNPs obtained, i.e. 0.6 nm for E12, observing a conversion drop to 16.5% while keeping a 100% selectivity for ethylbenzene. The latter is rationalized by the hampered accessibility to the active surface of the nanoparticle due to the large amount of pyrazine molecules covering the nanoparticle. No hydrogenation of the aromatic ring was observed, which is a clear indication that nanoparticles are small enough, since larger size NPs favour surface interaction with the aromatic ring and full hydrogenation.23,24
 | ||
| Fig. 5 STEM and HR-TEM images for experiment E4. | ||
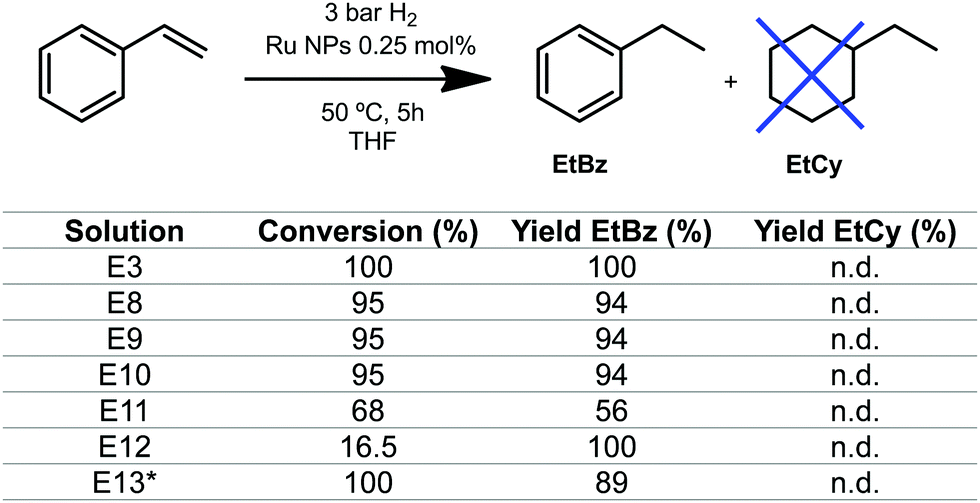 | ||
| Fig. 6 Hydrogenation of styrene catalysed by Ru NPs from experiments E8–E12. Conditions: 1 mmol of substrate, 0.25 mol% Ru, 5 h, 50 °C and 5.5 mL of THF. EtBz = ethylbenzene. EtCy = ethylcyclohexane *total volume 3 mL. See more details in the ESI.† | ||
At this point, we analysed the integrity of the nanocapsules at the end of the reaction by HRMS. The peak of the empty nanocapsule was detected at the end of the experiment in most of the cases, which indicates the stability of the cage throughout the reaction conditions. However, other peaks with higher molecular weight representing a 10% of the capsule content (comparing peak intensity) were found in samples E5, E6, E8, E9, E10, E11 and were assigned to (Ru5H2) ⊂ 8·(BArF)8 (Fig. 7 and Fig. S17, ESI†). Nanocapsule recovery was high in most of the samples (40% for E5, 95% for E6, >98% for E8, 76% for E9, 82% for E10 and 40% for E11). Furthermore, peaks corresponding to (Ru10H2) ⊂ 8·(BArF)8 and (Ru15H2) ⊂ 8·(BArF)8 were also detected in experiment E4 (see Fig. S18, ESI†).
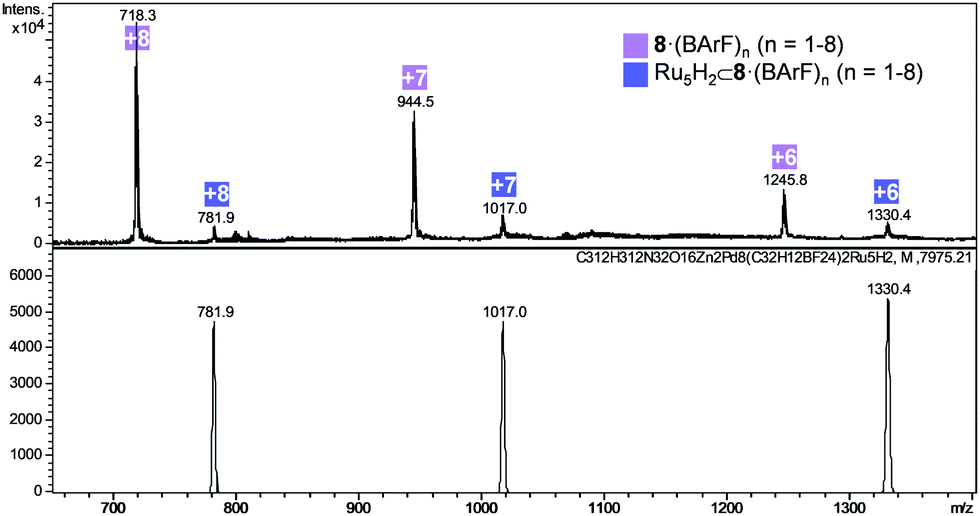 | ||
| Fig. 7 HRMS analysis of experiment E5 (addition of 40 eq. Ru(COD)(COT) and 0.2 eq. pyrazine in the presence of 2 mg of 8·(BArF)8). Calculated (bottom) and found (top) m/z peaks. | ||
This intriguing result prompted us to investigate theoretically the formation of ultrasmall Ru clusters. Computational studies were performed to gain insight into the putative structure of the ultra-small Ru clusters and the possibility to stabilize these nanoclusters with hydride anions and different ligands. The present observation of stable clusters made of 5, 10 and 15 Ruthenium atoms is reminiscent of magic numbers. Clusters of 3, 4, 5, 6, 9, 10 ruthenium atoms have been found in the past,25–31 most of them following the Wade–Mingos rules.32–35 According to the Wade–Mingos polyhedral skeletal electron pair (PSEP) theory, the total cluster valence electron (CVE) number of TM closo, nido and arachno deltahedral clusters is expected to be 14 m + 2, 14 m + 4 and 14 m + 6, respectively. It is both with the electron-counting rules in mind, and by using the ab initio thermodynamic methodology, that DFT calculations have been achieved to shed light on the present experimental results. They first aim at evaluating the relative thermodynamic stability of [Ru5] models coated with hydrides and ligands available either from the solution (pyrazine) or from possible adsorption or growth sites of a nanocapsule.
The most stable geometry of bare [Ru5] clusters was found to be a square pyramid (sp).36,37 The same result is found in the present study, with the trigonal bipyramid (tb) that lies at 26.4 kcal mol−1 above sp, in good agreement with the results reported in the literature.36 Given its occurrence in MS experiments, Ru5H has also been studied. All structures, magnetic moments and relative energies are reported in Fig. S20 and S21 (ESI†). The most stable one is an sp with a terminal H. According to the Wade-Mingos rules, [Ru5] clusters in solution are expected to have 74 CVEs for the nidosp and 72 CVEs for the closotb. The 24 and 22 additional CVEs to the 40 CVEs of the [Ru5] metal core can be brought by some hydrides, and mainly by the available ligands: either thanks to a π donation from the aromatic rings of the nanocapsule pillars or from the pyrazine solvent molecules, or either through the σ-donating ability of pyrazine. In the latter case, Ru atoms lying at an apex can be coordinated in a trigonal-pyramidal fashion to three η1-donating pyrazine ligands, that bring 6 CVEs. Some possible [Ru5] models that fulfill the ideal CVE count are reported in Fig. 8: sp-Ru5H4(η6-PhH)2(η6-pyz)3, 1, tb-Ru5H2(η6-PhH)2(η6-pyz)3, 2, tb-Ru5H2(η1-pyz)6(η6-pyz)3, 3, and tb-Ru5H4(η6-PhH)2(η6-pyz)2(η4-pyz), 4, where benzene molecules in compounds 1, 3 and 4 model phenylene groups of the Pd clip. Some energy clues can be obtained from these clusters, completed with all those considered in this work and reported in the ESI:† (i) the 74 CVEs sp cluster 1 is slightly more stable than the 72 CVEs tb cluster 4 by less than 2 kcal mol−1. Given the energies of the bare clusters, it means that the tb metal core is more strongly stabilized by the surface species than the sp core; (ii) the optimization of the 72 CVEs sp-Ru5H2(η6-PhH)2(η6-pyz)3 directly converges toward its tb counterpart, 2. Besides, it is shown in the (ESI†) that sp-Ru5H2(η6-PhH)2(η6-pyz)3, noted TS2−2′ in Fig. S22 (ESI†), is a transition state that allows an easy structural isomerization between 2 and a tb cluster, 2′, where the two η6-benzene ligands lie perpendicular to the vertical axis and not to the equatorial plane as in 2; (iii) the gas-phase substitution reaction 2 + 2 pyz → tb-Ru5H2(η6-pyz)5 + 2 PhH is endothermic by 11.1 kcal mol−1 per ligand as expected, given that benzene is a stronger π ligand than pyrazine. This value could be higher with ppp, that exhibits an extended conjugation; (iv) each benzene could be replaced in 2 by three σ-donating pyrazine ligands, given that the reaction 2 + 6 pyz → 3 + 2 PhH is exothermic by −26.8 cal mol−1 per Ru site. Of course, there must be no steric conflict that prevents the metal core from being stabilized by this bulky environment; (v) the π adsorption energy of pyrazine on Ru5H2, evaluated from the reaction Ru5H2 + 5 pyz → tb-Ru5H2(η6-pyz)5, is found to be −37.0 kcal mol−1 per ligand, indicating a fairly strong adsorption.
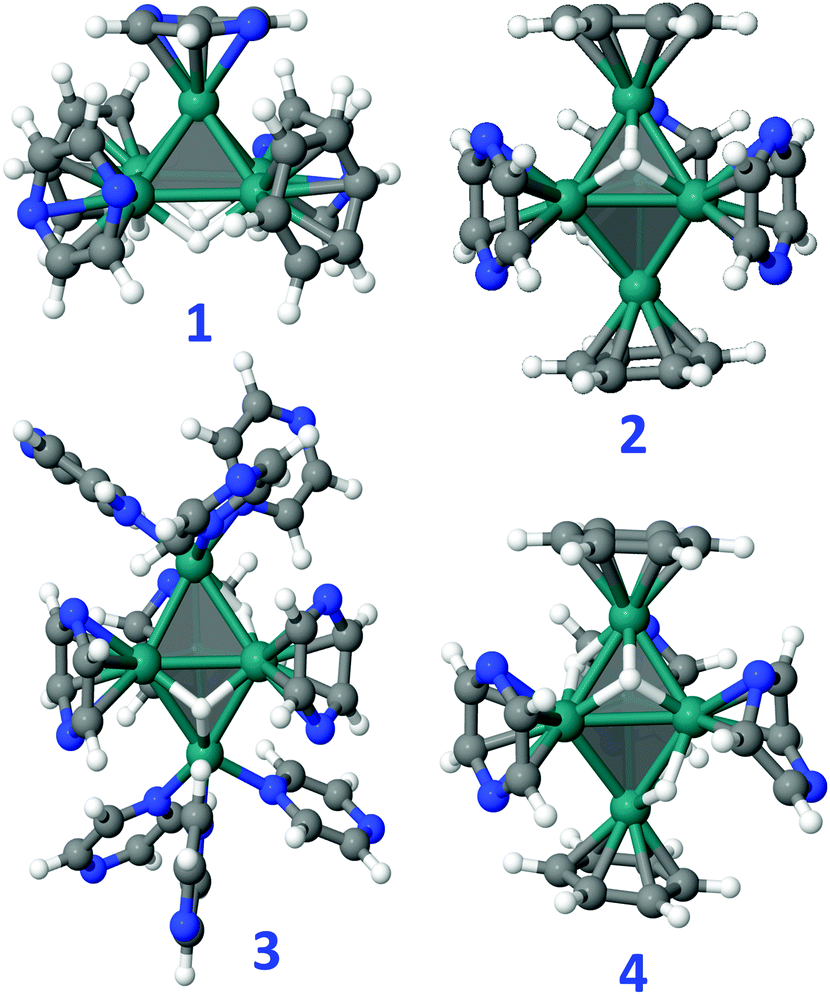 | ||
| Fig. 8 [Ru5] clusters stabilized by hydrides, benzene (PhH) and pyrazine (pyz) ligands. (1) sp-Ru5H4(η6-PhH)2(η6-pyz)3, (2) tb-Ru5H2(η6-PhH)2(η6-pyz)3, (3) tb-Ru5H2(η1-pyz)6(η6-pyz)3, and (4) tb-Ru5H4(η6-PhH)2(η6-pyz)2(η4-pyz). They all fulfill the Wade-Mingos counting rules, i.e. 74 CVEs for the nidosp and 72 CVEs for the closotb. While 4 has the same composition as 1, with one η4-pyrazine it is a 72 CVE cluster, thus having the optimal electron counting number of a closo structure. | ||
In order to identify the available growth/stabilizing zones of the nanocapsule 8·(BArF)8, it has been fully optimized at the DFT level of theory (see computational details in the ESI†). With a 1.2 nm height × 1.7 nm wide inner cavity (I site in Fig. 9), the resulting geometry is more compact than that found with Molecular Dynamics (MD) simulations using force fields.38 These MD simulations actually suggest an important flexibility involved by the palladium coordination geometry, that gives the possibility that the height of the inner cavity increases up to the ca. 18 Å value reported in Fig. 2. Other sites can also contribute to the stabilization of Ruthenium clusters or NPs (see Fig. 9). They can lie outside a ZnTCPP building block, with a ca. 1.2 nm width available space (Zn site). The pillars, which can provide through the phenylene groups six π electrons stabilizers to metal cores, are not equally separated. Small clusters can be trapped in either ∼0.6 or ∼0.8 nm coordination sites (P1 and P2 sites). Finally, pillars can also rotate around their main axis of inertia, thus favouring the binding of larger nanoclusters outside the cage (P3 site). With a ∼1.6–2.0 Å radius, [Ru5] clusters fit the P1 site, whereas ∼1 nm RuNPs could fit the larger coordination sites Zn, I and P3. We will see in the following that small clusters coated with η1-pyrazine ligands, such as in 3, can also be remarkably stabilized in site I.
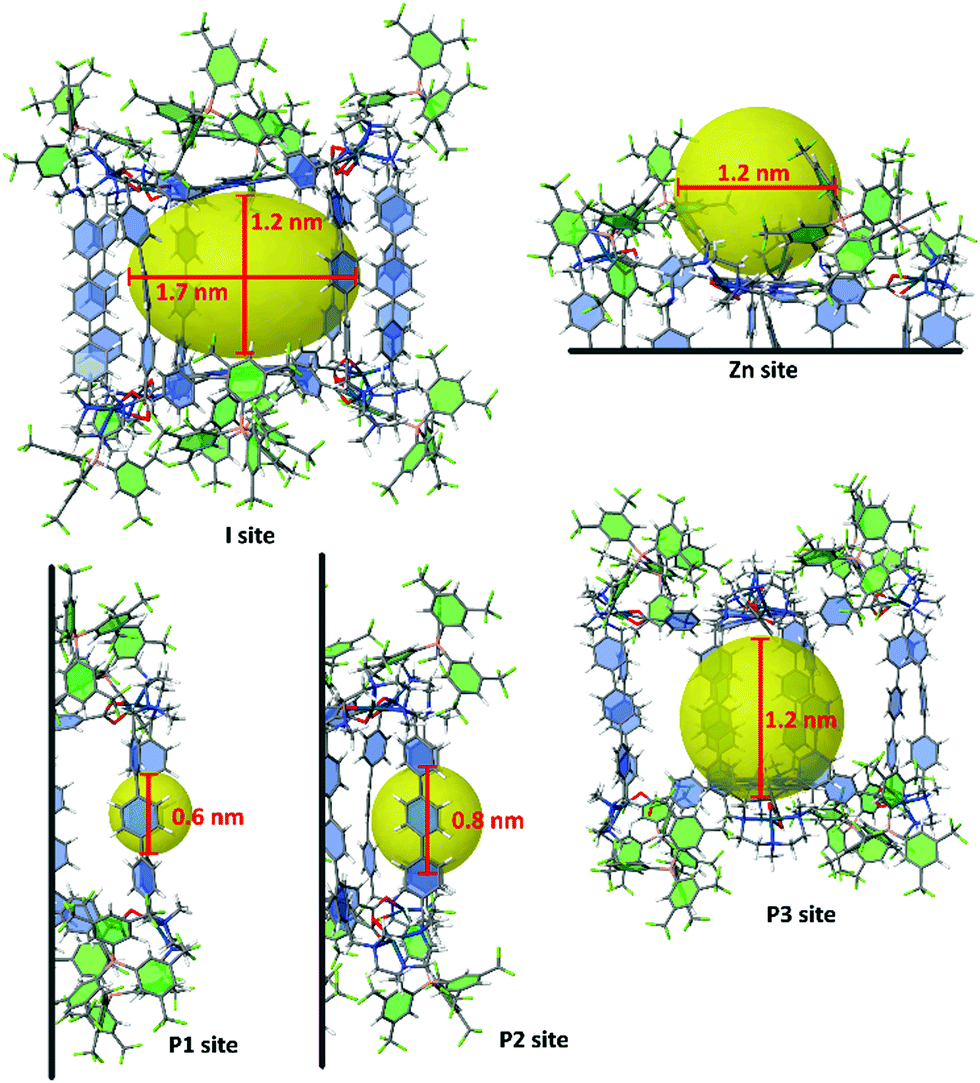 | ||
| Fig. 9 Possible Run clusters or RuNPs coordination sites in the 8·(BArF)8 nanocapsule. Given the overall plasticity of the cage, cavity sizes, shown as yellow spheres (sites Zn, P1–P3) or ellipsoids (site I), are only indicative. They are based on the optimization of a host-free cage. For the sake of clarity, aromatic rings of BArFs and of the ppp part of Pd-clips are highlighted in green and blue, respectively. | ||
We then focused on identifying the number of hydrides that could lie on the Ru5Hn(PhH)2(pyz)3 clusters. Their energies as well as the Ru–H vibrational properties and the μH2 chemical potential (see computational details in the ESI†) have been used to establish the stability diagram plotted in Fig. 10. Its purpose is to assess if the optimal number of hydrides that could lie on a [Ru5] core stabilized by the P1 site is consistent with the optimal 72 or 74 CVE numbers, with the reasonable assumption that in experimental conditions the actual number of valence electrons is likely to be reached first of all thanks to the π-coordination of five aromatic rings. 2 is remarkably stable over a wide pressure and temperature range, in agreement with the 72 CVEs of the closo metal core. All stable structures exhibit a tb metal core, with the exception of the most stable 74 CVE sp-Ru5H6(η6-PhH)2(η6-pyz)2(η4-pyz) cluster found in the very low temperature range. Interestingly, these remarkably stable structures are consistent with the second order energy differences plotted in Fig. S23 (ESI†), with the exception of the tb-Ru5H(η6-PhH)2(η6-pyz)3. This odd hydride-number cluster appears stable in a narrow domain, whereas such spin-doublet cluster is expected to be very reactive upon hydrogenation.
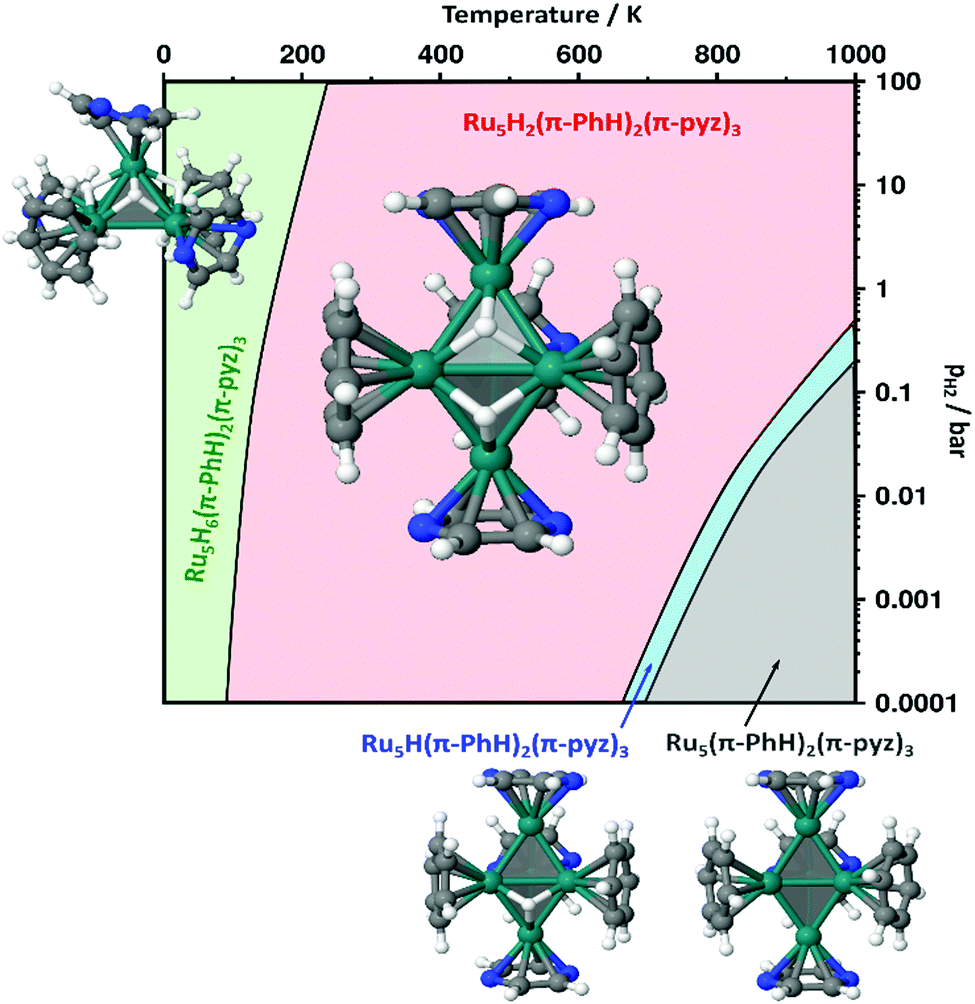 | ||
| Fig. 10 Stability diagram established for sp- or tb-Ru5Hn(PhH)2(pyz)3 (see text and computational details in the ESI†). The temperature range is set up to 1000 K, just to check the consistency of the model, i.e. that there is a gradual increase in the number of surface hydrides as the pressure increases and the temperature decreases. | ||
The adsorption energies of Ru5 and Ru57 clusters on the coordination sites I, Zn, P1 and P3 were also evaluated and schematized in Fig. 9. The adsorption of Ru5H2(η6-pyz)3 in the P1 site of a Pd clip has been first evaluated (Fig. 11a). It follows the Wade–Mingos rules, thanks to the coordination with two phenylene groups. The resulting cluster does correspond to cluster 2′, slightly less stable than 2, but which structure is better coordinated with both phenylene groups. Overall, Ru5H2 is stabilized by the two phenylene groups and the three pyrazine ligands by −209 kcal mol−1, i.e. −41.8 kcal mol−1 per ligand (to be compared to −202.9 kcal mol−1 in 2′). The resulting 0.7 nm wide geometry involves a very small spacing of the ppp pillars. Compound 3 could also be formed inside the cage (site I) and further stabilized thanks to the σ-donation of nitrogen atoms of pyrazine at the apex of the cluster toward zinc atoms of the zinc-porphyrin building blocks (Fig. 11b). The stabilization of the Ru5H2 moiety in site I is −38.1 kcal mol−1 per pyrazine ligand, with a moderate −18.6 kcal mol−1 energy per pyrazine-Zn coordination. With a final 1.6 nm inter-porphyrin width and given the adaptability of the whole cage, it appears feasible. Such coordination could also occur outside the nanocapsule, i.e. in the Zn site of Fig. 9.
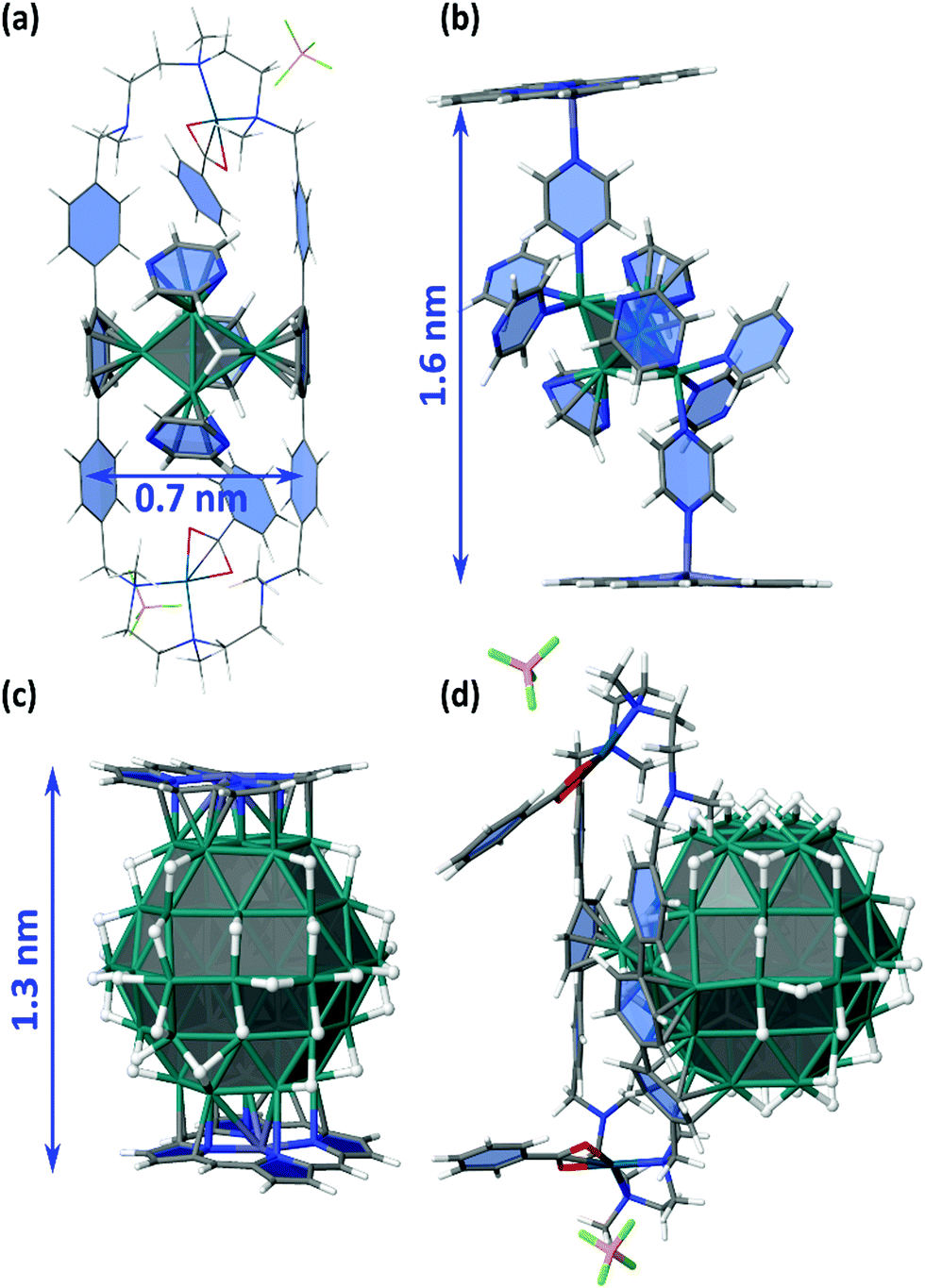 | ||
| Fig. 11 Study of the possible trapping of ligand-coated Ru5H2 clusters and Ru57H44 NPs in the nanocapsule (see also Fig. 9). (a) adsorption of Ru5H2(η6-pyz)3 in the P1 site of a Pd clip (with BF4 instead of BArF); (b) coordination of compound 3 between two Zn-porphyrin building blocks (model for site I); (c) coordination of a Ru57H44 NP between two Zn-porphyrin building blocks (model for site I); (d) stabilization of a Ru57H44 NP in the P3 site of a Pd-ppp clip. | ||
It is not obvious that the 0.8 nm spacing between the farthest ppp allows the diffusion of several precursors and seeds within the cage. Nevertheless, the possible growth of RuNPs inside the nanocapsule (Fig. 11c) cannot be discarded. Moreover, hydrides are very mobile and could move in order to favour the simultaneous stabilization of an NP by the Zn–porphyrin parts of the nanocage, as shown in Fig. 11c. Such encapsulation would be stabilized by −97.6 kcal mol−1 per interaction, with respect to the cage and to a free Ru57H44 with hydrides randomly scattered on all facets of this NP. NPs could also be stabilized outside the cage, in the Zn site, probably with additional pyrazine on the surface. The possible growth of RuNPs outside the cage, but stabilized in the P3 site, has also been considered (Fig. 11d). Phenylene groups can slightly rotate around the vertical ppp axis in order to maximize their coordination on the RuNP surface. On the contrary to the previous case, it does involve a concerted shift of several hydrides to favour this grafting. In this example, two facets of the Ru57H44 NP are firmly grafted to the ppp clip by a (η6-phenylene)(η6,μ3-phenylene)(η6,μ4-phenylene) motif. The resulting strong adsorption energy, −162.3 kcal mol−1, i.e. ca. −54 kcal mol−1 per phenylene, might be lower with the 8·(BArF)8 nanocapsule, as the pillars could be slightly less adaptive, and as the number of surface hydrides could be slightly higher. Even in this case, this external grafting is very likely to be significantly stable.
All the above DFT study shows the ability of the nanocapsule to stabilize both small [Ru]5 clusters at different trapping sites, but most likely, as (a) Ru5H2 stabilized at P1 site by the two phenylene groups and the three pyrazine ligands (i.e. compound 2, Fig. 8 and Fig. 11a or Fig. 11b) as compound 3 formed inside the cage (site I) and further stabilized thanks to the σ-donation of nitrogen atoms of pyrazine at the apex of the cluster toward zinc atoms of the zinc-porphyrin building blocks (Fig. 11b), with similar stabilization energies for both options. The remarkable occurrence of the [Ru5] cluster in the mass spectrum is thus explained both in terms of electronic effects and of cavities’ size of the nanocapsule. Moreover, the outer or inner-cavity stabilization of a ca. 1 nm RuNP model have also been considered. Given the combined experimental observations and computational insights, we propose that the trapped [Ru5] metallic clusters are the first-grown seeds of subsequent formation of the subnanometric RuNP, which are also stabilized by outer-nanocapsule interactions.
In conclusion, sub-nanometric Ru NPs were successfully synthesized and stabilized using the Pd-based tetragonal prismatic nanocapsule 8·(BArF)8 along with pyrazine as stabilizing agent. The data registered indicate that nanocapsule remains intact and the Ru NPs are stabilized through the outer-cavity of the nanocapsule, being the macrocyclic-based molecular clip moieties the ones mostly responsible for the stabilization of the ultra-small Ru NPs. Simultaneously, the same nanocapsule is able to trap in its inner-cavity ruthenium clusters [Ru5] as ascertained by HRMS, which is fully diagnostic for the mass identity of the encapsulated guests.21,39–41 Computational data point towards two main isoenergetic adsorption modes of the [Ru5] cluster: (a) adsorption of Ru5H2(η6-pyz)3 in the P1 site of a Pd clip (ca.tb-Ru5H2(η6-PhH)2(η6-pyz)3 (2)) or (b) coordination of tb-Ru5H2(η1-pyz)6(η6-pyz)3 (3) between two Zn–porphyrin building blocks (model for site I), both options fulfilling the Wade-Mingos counting rules, i.e. 72 CVEs for the closotb.
The direct observation of the metallic cluster seeds for the formation of NP sheds light into this fundamental process. Moreover, the double role of the nanocapsules in stabilising ∼0.7 nm NPs and also host ultra-small Ru clusters, is unprecedented and may pave the way towards the synthesis of ultra-small metallic clusters for catalytic purposes.
Methods
The synthesis of the different nanocapsules was performed following the reported procedure (see Fig. 1).22 The synthesis of the Ru NPs was performed in a Fisher–Porter bottle by decomposition of Ru(cyclooctadiene)(cyclooctatriene) in presence of the molecular cage and small amount of pyrazine.23,24 For this, in the glove box, 2 mg of nanocapsule were dissolved in 2 mL of THF, then the corresponding Ru equivalents (130, 80, 60, 40 or 20) were added from a 0.06 M solution of the complex, followed (in some cases) by 0.2 equivalents of pyrazine with respect to Ru from a 0.024 M solution. The final volume is 3.0–3.5 mL. Out of the glove box, the solution was then charged with 1 bar of H2 and stirred at room temperature for 2 min. At this time, the pressure was released and the Fisher–Porter opened to air quenching the decomposition. The NPs were characterized by Transmission Electron Microscopy (TEM) and High-Resolution Transmission Electron Microscopy (HRTEM). The solutions were also analysed by High Resolution Electrospray Ionization Mass Spectrometry (HR-ESI-MS).The catalytic tests were performed also in a Fisher-Porter bottle and were prepared at the air. To the solution of Ru NPs 135 μL (1 mmol) of styrene and 227 μL of dodecane (1 mmol, internal standard) were added. The volume of the Ru NPs solution utilised was different depending on the Ru equivalents employed in the initial solutions, and are specified in Table S1 (ESI†). In all the cases the Ru present in the solution constituted a 0.25 mol% with respect to the substrate.
All DFT calculations were performed with the VASP software,42,43 with the exchange–correlation potential approximated by the generalized gradient approach proposed by Perdew, Burke, and Ernzerhof (PBE),44,45 and using the projector-augmented wave (PAW) scheme to treat core electrons.46,47 Other computational details are given in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from MINECO-Spain (PID2019-104498GB-I00 and EQC2018-004422-P) and Generalitat de Catalunya (2017SGR264) to X. R. E. U. thanks UdG for a PhD grant. CNRS, INSAT and UT3-UPS are also acknowledged for financial support. I. d. R. and R. P. thank the HPC CALcul en MIdi-Pyrénées (CALMIP, OLYMPE machine, grant P0611) for a very generous allocation of computer time on this project. IMM, JMA and BC thank ERC Advanced Grant (MONACAT 2015-694159) for financial support. X. R. also thanks ICREA-Acadèmia awards. We thank STR-UdG for technical support.References
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- E. S. Gutterød, A. Lazzarini, T. Fjermestad, G. Kaur, M. Manzoli, S. Bordiga, S. Svelle, K. P. Lillerud, E. Skúlason, S. Øien-Ødegaard, A. Nova and U. Olsbye, J. Am. Chem. Soc., 2020, 142, 999–1009 CrossRef PubMed.
- Y. Liu, Y. Shen, W. Zhang, J. Weng, M. Zhao, T. Zhu, Y. R. Chi, Y. Yang, H. Zhang and F. Huo, Chem. Commun., 2019, 55, 11770–11773 RSC.
- M. I. Gonzalez, A. B. Turkiewicz, L. E. Darago, J. Oktawiec, K. Bustillo, F. Grandjean, G. J. Long and J. R. Long, Nature, 2020, 577, 64–68 CrossRef CAS PubMed.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- X. Yang and Q. Xu, Trends Chem., 2020, 2, 214–226 CrossRef CAS.
- L. M. Olaechea, L. Montero de Espinosa, E. Oveisi, S. Balog, P. Sutton, S. Schrettl and C. Weder, J. Am. Chem. Soc., 2020, 142, 342–348 CrossRef CAS PubMed.
- Q.-T. Fu, X. Yan, X.-Y. Zhang, Y. He, W.-D. Zhang, Y. Liu, Y. Li and Z.-G. Gu, Dalton Trans., 2020, 49, 12145–12149 RSC.
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- G.-J. Chen, W.-L. Xin, J.-S. Wang, J.-Y. Cheng and Y.-B. Dong, Chem. Commun., 2019, 55, 3586–3589 RSC.
- M. Nihei, H. Ida, T. Nibe, A. M. P. Moeljadi, Q. T. Trinh, H. Hirao, M. Ishizaki, M. Kurihara, T. Shiga and H. Oshio, J. Am. Chem. Soc., 2018, 140, 17753–17759 CrossRef CAS PubMed.
- Y. Fang, J. Li, T. Togo, F. Jin, Z. Xiao, L. Liu, H. Drake, X. Lian and H.-C. Zhou, Chem, 2018, 4, 555–563 CAS.
- R. McCaffrey, H. Long, Y. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef CAS PubMed.
- X. Yang, J.-K. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS.
- Y. Zhang, Y. Xiong, J. Ge, R. Lin, C. Chen, Q. Peng, D. Wang and Y. Li, Chem. Commun., 2018, 54, 2796–2799 RSC.
- N. Sun, C. Wang, H. Wang, L. Yang, P. Jin, W. Zhang and J. Jiang, Angew. Chem., Int. Ed., 2019, 58, 18011–18016 CrossRef CAS PubMed.
- S. Jiang, H. J. Cox, E. I. Papaioannou, C. Tang, H. Liu, B. J. Murdoch, E. K. Gibson, I. S. Metcalfe, J. S. O. Evans and S. K. Beaumont, Nanoscale, 2019, 11, 14929–14936 RSC.
- V. Sharma, D. De, R. Saha, P. K. Chattaraj and P. K. Bharadwaj, ACS Appl. Mater. Interfaces, 2020, 12, 8539–8546 CrossRef CAS PubMed.
- E. Ubasart, O. Borodin, C. Fuertes-Espinosa, Y. Xu, C. García-Simón, L. Gómez, J. Juanhuix, F. Gándara, I. Imaz, D. Maspoch, M. von Delius and X. Ribas, Nat. Chem., 2021, 13, 420–427 CrossRef CAS PubMed.
- E. Ubasart, C. García-Simón, M. Pujals, K. Asad, N. Chronakis, T. Parella and X. Ribas, Org. Chem. Front., 2021, 8, 4101–4105 RSC.
- L. M. Martínez-Prieto and B. Chaudret, Acc. Chem. Res., 2018, 51, 376–384 CrossRef PubMed.
- L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Cámpora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647–4650 RSC.
- M. I. Bruce, J. Organomet. Chem., 1990, 394, 365–384 CrossRef CAS.
- P. J. Bailey, M. J. Duer, B. F. G. Johnson, J. Lewis, G. Conole, M. McPartlin, H. R. Powell and C. E. Anson, J. Organomet. Chem., 1990, 383, 441–461 CrossRef CAS.
- J. W. Benson, T. Ishida, K. Lee, S. R. Wilson and J. R. Shapley, Organometallics, 1997, 16, 4929–4932 CrossRef CAS.
- J. A. Cabeza, I. del Río, P. García-Álvarez and D. Miguel, Inorg. Chem., 2006, 45, 6020–6027 CrossRef CAS PubMed.
- I. del Rosal, F. Jolibois, L. Maron, K. Philippot, B. Chaudret and R. Poteau, Dalton Trans., 2009, 2142–2156, 10.1039/B817055J.
- G. Meister, G. Rheinwald, H. Stoeckli-Evans and G. Süss-Fink, J. Chem. Soc., Dalton Trans., 1994, 3215–3223, 10.1039/DT9940003215.
- R. Gautier, F. Chérioux, G. Süss-Fink and J.-Y. Saillard, Inorg. Chem., 2003, 42, 8278–8282 CrossRef CAS PubMed.
- D. M. P. Mingos, J. Chem. Soc., Chem. Commun., 1985, 1352–1354, 10.1039/C39850001352.
- M. A. Fox and K. Wade, Pure Appl. Chem., 2003, 75, 1315–1323 CrossRef CAS.
- J.-Y. Saillard and J.-F. Halet, in The Chemical Bond I: 100 Years Old and Getting Stronger, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2016, pp. 157–179. DOI:10.1007/430_2015_210.
- D. M. P. Mingos and D. J. Wales, Introduction to Cluster Chemistry, Prentice Hall, 1990 Search PubMed.
- W. Zhang, H. Zhao and L. Wang, J. Phys. Chem. B, 2004, 108, 2140–2147 CrossRef CAS.
- F. Aguilera-Granja, L. C. Balbás and A. Vega, J. Phys. Chem. A, 2009, 113, 13483–13491 CrossRef CAS PubMed.
- C. García-Simón, C. Colomban, Y. A. Çetin, A. Gimeno, M. Pujals, E. Ubasart, C. Fuertes-Espinosa, K. Asad, N. Chronakis, M. Costas, J. Jiménez-Barbero, F. Feixas and X. Ribas, J. Am. Chem. Soc., 2020, 142, 16051–16063 CrossRef PubMed.
- C. Fuertes-Espinosa, C. García-Simón, M. Pujals, M. Garcia-Borràs, L. Gómez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem, 2020, 6, 169–186 CAS.
- C. Fuertes-Espinosa, A. Gómez-Torres, R. Morales-Martínez, A. Rodríguez-Fortea, C. García-Simón, F. Gándara, I. Imaz, J. Juanhuix, D. Maspoch, J. M. Poblet, L. Echegoyen and X. Ribas, Angew. Chem., Int. Ed., 2018, 57, 11294–11299 CrossRef CAS PubMed.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2127407. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1nh00677k |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |