
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single molecule localization-based analysis of clathrin-coated pit and caveolar dynamics†
Rui
Ma‡
a,
Martin
Štefl‡
 a and
Gerd Ulrich
Nienhaus
*abcd
a and
Gerd Ulrich
Nienhaus
*abcd
aInstitute of Applied Physics (APH), Karlsruhe Institute of Technology (KIT), Wolfgang Gaede-Strasse 1, 76131 Karlsruhe, Germany. E-mail: uli@uiuc.edu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), PO Box 3640, 76021 Karlsruhe, Germany
cInstitute of Biological and Chemical Systems (IBCS), Karlsruhe Institute of Technology (KIT), PO Box 3640, 76021 Karlsruhe, Germany
dDepartment of Physics, University of Illinois at Urbana–Champaign, Urbana, IL 61801, USA
First published on 10th March 2022
Abstract
Clathrin-coated pits and caveolae are nanoscale invaginations of the plasma membrane of cells, forming through the assembly of membrane coat and accessory proteins in a tightly regulated process. We have analyzed the development of these membrane coat structures with high spatial and temporal resolution and sensitivity using super-resolution single-molecule localization microscopy (SMLM) on live cells. To this end, we developed a sophisticated clustering and data analysis workflow that automatically extracts the relevant information from SMLM image sequences taken on live cells. We quantified lifetime distributions of clathrin-coated and caveolar structures, and analyzed their growth dynamics. Moreover, we observed hotspots in the plasma membrane where coat structures appear repeatedly. The stunningly similar temporal development of clathrin-coated and caveolar structures suggests that key accessory proteins, some of which are shared between the two types of membrane coat structures, orchestrate the temporal evolution of these complex architectures.
New conceptsClathrin-coated pits and caveolae are nanoscale invaginations of the plasma membrane of cells that are essential for key cellular processes. These intricate structures, formed by assembly of membrane coat proteins, are highly dynamic, undergoing continuous growth and decay. As conventional live-cell fluorescence microscopy does not provide adequate spatial resolution and optimal sensitivity, we have employed super-resolution single-molecule localization microscopy (SMLM) to study membrane coat dynamics. A sophisticated clustering and data analysis workflow was developed to extract quantitative information from SMLM image sequences for further analysis. These data allowed us to determine lifetime distributions (for caveolae for the first time) and to analyze the growth dynamics based on molecule numbers. We also identified hotspots, membrane locations where coat structures appear repeatedly. The stunningly similar dynamics of clathrin-coated and caveolar structures may indicate that accessory proteins common to both types of membrane coat structures play a crucial role in their temporal development. |
Introduction
Clathrin-coated pits (CCPs) and caveolae are plasma membrane invaginations with diameters of 100–200 nm1–3 and 50–80 nm,3–5 respectively. CCPs are complex structures that nucleate, grow and finally pinch off the membrane to form intracellular vesicles, thereby internalizing receptors and their cargo. This process, clathrin-mediated endocytosis (CME), is the main route of receptor-mediated internalization in mammalian cells.6,7 Caveolae also perform endocytosis; however, alternative functional roles have been identified in recent years, including mechanoprotection and stress signaling.5 Formation of these highly complex membrane-associated structures requires the coordinated assembly of a multitude of biomolecules.For CCP assembly, adaptor protein 2 (AP2) molecules associate with the plasma membrane and anchor clathrin triskelia consisting of clathrin light and heavy chains. The initially small and unstable complexes are stabilized by incorporating further AP2 and clathrin molecules as well as a wide range of endocytic accessory proteins.8,9 For clathrin-coated structures (CCSs) to develop into long-lived, mature CCPs that are competent to internalize cargo,10,11 timely recruitment of proteins is essential, or else, the growth process aborts prematurely within the first ∼25 s.12,13 A sequence of events, including initiation of CCSs, growth and maturation and, finally, fission and release of endocytic vesicles from the plasma membrane, has been revealed,9,12 and checkpoints have been proposed that have to be passed to proceed to the next step.12,14
Caveola formation starts with incorporation of aggregated caveolin proteins (mainly isoforms Cav1 or Cav3, depending on tissue type5) into the plasma membrane. In parallel, cavin proteins assemble into trimers and further into larger, interconnected oligomers.4 Cavin oligomers and Eps15 homology domain (EHD) proteins are recruited to caveolin-enriched regions for formation of caveolae, and other proteins including ROR1, pacsins and actin filaments associate with preformed caveolae to further stabilize them.5 Compared with CCSs, much less is known about the dynamics of caveolar structures (CAVSs). They have been reported to exist for seconds to minutes, with the majority having lifetimes <30 s.15,16
Even though the core and scaffold proteins of CCPs and caveolae are different, they share a range of associated proteins, including EHD2, dynamin 2, pacsin 25,11,17,18 and proteins associated with actin filaments and stress fibers, such as Arp2/3, N-WASP, and filamin A.18,19
Structure and development of membrane coat proteins were traditionally studied on fixed samples using electron microscopy and optical fluorescence microscopy.5,6,17 Over the last decade, our knowledge about CCP development has grown enormously thanks to classical (diffraction-limited) microscopy on live cells expressing CCS proteins fused to fluorescent proteins, which were visualized using total internal reflection fluorescence (TIRF) microscopy with single-particle tracking (SPT) analysis.8,9,15,20
Here we have addressed CCS and CAVS dynamics with super-resolution single-molecule localization microscopy (SMLM) with TIRF excitation, which offers key advantages for this research.21–23 To visualize membrane coat proteins, we employed fusion proteins with mEosFPthermo,24,25 a bright, monomeric green-to-red photoconverting fluorescent protein (FPs) of the EosFP clade of photoactivatable proteins.26 Upon expression in live cells, these fusion markers are sparsely and stochastically photoactivated with weak 405 nm light, and photons emitted by individual, red-converted mEosFP molecules upon 561 nm excitation are collected until photobleaching occurs. In each camera frame of a contiguous sequence of thousands of images taken over several minutes, single-molecule photon bursts are analyzed to localize the emitters. The typical localization precision of several ten nanometers allows for direct visualization of the nanometric membrane coat structures. The intrinsic single-molecule sensitivity of SMLM ensures a high dynamic range, enabling the study of small assemblies containing only a few fluorophores that escape detection by conventional TIRF microscopy. Moreover, in SMLM, fluorophore photobleaching can be controlled via the 405 nm photoactivation intensity, since only red-converted mEosFP species can be excited by 561 nm light and subsequently photobleached. Thus, the observation time can be adjusted to the dynamics of the process under study. SMLM yields quantitative information on protein complexes, including stoichiometry, location and localization precision, photon numbers and times of photon emission from each fluorescence emitter within the complex. SMLM imaging data are distinctly different from those of conventional camera-based imaging. Whereas the latter captures intensities of all pixels of each image, SMLM analysis initially generates a long list of coordinates of all localized molecules. In a second step, these point-cloud data are rendered to obtain images that scale linearly with the density of fluorophores. Analysis of extended structures containing multiple fluorophores such as membrane coat structures requires sophisticated clustering algorithms that swiftly process large SMLM data sets with inhomogeneous fluorophore density and background and with minimal user bias.27 To this end, we have developed robust and sophisticated clustering software, Local Outlier Factor – Density Based Spatial Clustering of Applications with Noise (LOF-DBSCAN) and integrated it in a workflow that links localizations in neighboring frames into clusters but separates spatially overlapped clusters forming at different times.
With this cluster analysis workflow, we have visualized and quantitatively analyzed CCS and CAVS development by SMLM. Hotspots,28,29 regions where these membrane coat structures form repeatedly, were found for both systems and characterized in detail. The remarkable similarity in the dynamics of CCS and CAVS formation suggests common mechanisms underlying their development.
Results
Image acquisition and cluster analysis
Sample preparation, microscopy methods and imaging procedures are described in Supplementary Methods (ESI†). To visualize the dynamics of CCSs and CAVSs in murine skeletal myoblast (C2C12) cells, we employed transient overexpression of the clathrin light chain a (CLCa) fused to mEosFPthermo (CLCa-mEosFPthermo) as well as (murine) mCav3-mEosFPthermo and (zebrafish) zfCav3-mEosFPthermo constructs, respectively. Details of the plasmids are compiled in Table S1 (ESI†). Overexpression of such fusion constructs always bears the risk that cellular processes might be affected, including those that are being studied. Previously, overexpression of a similar CLCa-FP fusion was carefully assessed and shown to not perturb CME.14 Furthermore, there were no abnormal cellular responses reported for overexpression of a caveolin–GFP fusion for live-cell imaging.30 Nevertheless, to ensure that our fusion constructs do not produce overexpression artifacts, we thoroughly inspected cells expressing our clathrin and caveolin fusion proteins at very different levels (Fig. S1, ESI†). Cells with low expression levels always showed the correct expression patterns and morphologies. Therefore, we only selected those cells for quantitative analysis. Conventional TIRF images of the basal plasma membrane, taken with TIRF excitation of the green form of mEosFPthermo, exhibit unresolved punctate features (Fig. 1a and d), as observed earlier.3,8,31 By contrast, SMLM images of the photoconverted red form of mEosFPthermo show well-resolved patches of 80–100 nm diameter (Fig. 1b and e, Table S2, ESI†) thanks to the roughly tenfold higher spatial resolution. Oftentimes, these structures (Fig. 1c and f) display donut-like shapes, as expected for stable and mature CCPs and caveolae. | ||
| Fig. 1 Fluorescence images of C2C12 cells expressing mEosFPthermo fused to membrane coat proteins. (a–c) CCS images. (a) Conventional TIRF microscopy image (30 ms exposure, averaged over 100 frames) with 473 nm excitation of the green FP species. Subsequently, a 5 min sequence of SMLM images (561 nm excitation of red-converted FP, 30 ms exposure) was taken. (b) Exemplary SMLM image from that sequence (close-up of the white square in panel a, 9 s total exposure). (c) Top: SMLM images of donut-shaped, mature CCPs, showing localizations (9 s interval of the 5 min sequence). To mimic an optical image, each localization was blurred by a Gaussian with a standard deviation equal to its localization precision. Bottom: Images resulting from LOF-DBSCAN segmentation. (d–f) CAVS images, captured as described for panels a–c. (d) Conventional TIRF image; (e) SMLM close-up (white square in d). (f) Top: SMLM images of donut-shaped, mature caveolae. Bottom: Results from LOF-DBSCAN segmentation. Scale bars (a and d), 10 μm; (b and e) 1 μm; (c and f) 100 nm. | ||
We studied the temporal evolution of these membrane coat structures by collecting SMLM image sequences (movies) of the cells. From all images, single-molecule locations (single-frame emission events) were extracted with a-livePALM software.32 They form irregular clusters embedded in background events (Fig. 2a). As none of the existing clustering algorithms could analyze the images in a robust fashion, which is of utmost importance to minimize user bias, we developed LOF-DBSCAN, a software that combines two popular clustering algorithms, LOF33 and DBSCAN34 in a sequential workflow comprising four steps, which are briefly sketched in the following. Details of the implementation are described in Supplementary Text (ESI†). (1) Identification and removal of localization events at the cluster boundaries (Fig. 2b). The LOF algorithm calculates the local density of events in the neighborhood of each localization and identifies events at the cluster edge (outliers) as those that have a lower density than its nearest neighbors within the cluster. Only events in regions with constant density, belonging to either clusters or background, are retained. (2) Density-based separation of cluster and background events (Fig. 2c and d). In LOF, the local reachability density (LRD), defined as the inverse of the average distance of a particular event from its neighbors, varies with the total number of localizations in an image. Therefore, the threshold for automatic removal of background must be adjusted accordingly. Analysis of synthetic images with randomly distributed localization events (see Supplementary Text, ESI†) revealed that the peaks of the LRD histograms (LRDmax) change with event numbers as a simple power law (Fig. 2c and Fig. S2a, b, ESI†). Thus, the best-fit power law curve to the simulated data was taken as the threshold separating cluster and background events in real images. (3) Assignment of non-background events to individual clusters (Fig. 2e), using DBSCAN, a reliable clustering algorithm for background-filtered images. (4) Expansion of each cluster (Fig. 2f). Events at cluster boundaries that were removed in step 1 are included again in their respective clusters by local cluster expansion using DBSCAN, as previously described.35
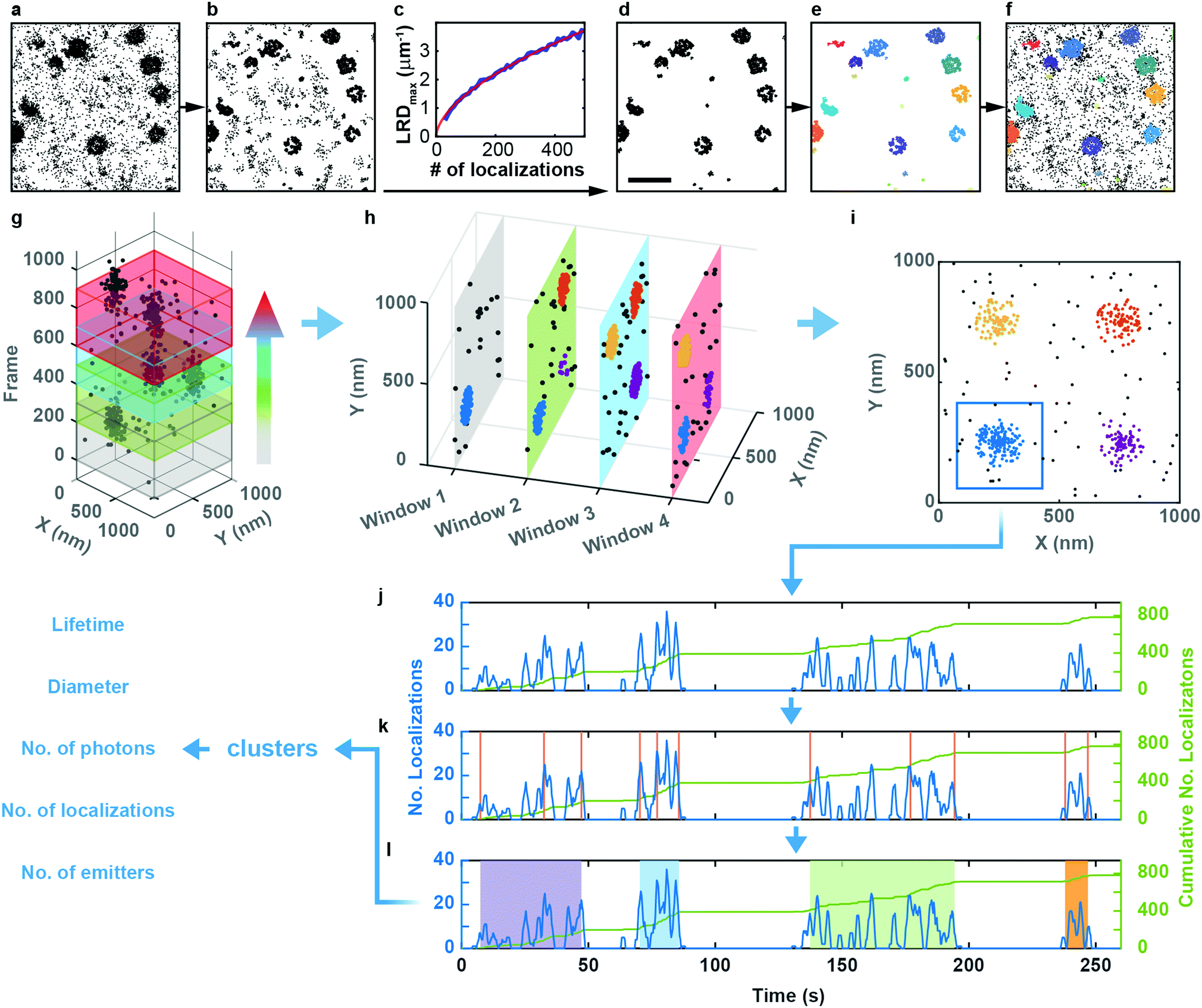 | ||
| Fig. 2 LOF-DBSCAN analysis pipeline and workflow of cluster merging and splitting in a SMLM time series. (a–f) LOF-DBSCAN analysis pipeline. (a) SMLM close-up image (2.18 × 2.18 μm2) of CCSs. (b) For each cluster, localization events at the cluster boundaries are identified and removed by LOF. (c) LRD values from three synthetic data sets with varying number of localizations; red line: fit with a power law used to set a LRD threshold for (d) background removal. (e) DBSCAN cluster identification. (f) Final LOF-DBSCAN clustering result after local expansion of clusters. Scale bar, 500 nm. (g) Schematic of the moving-window binning method to generate TOIs. Four partially overlapping windows are shown in different colors for illustration. (h) LOF-DBSCAN clustering results within each TOI. (i) Spatially overlapped clusters from all TOIs are merged into a single cluster. (j–l) Cluster merging and splitting workflow: (j) The time trace of the number of localizations in each frame (blue curve) is recast into a cumulative trace (green line). (k) A change point algorithm identifies time points in the trace where an abrupt change of slope occurs (vertical orange lines). (l) By setting a threshold for the slope change, individual clusters are identified (shaded regions). | ||
We carefully validated LOF-DBSCAN and compared it with DBSCAN using synthetic data sets (Fig. S2–S5, Table S3, ESI†), as described in Supplementary Methods (ESI†). We further tested LOF-DBSCAN with SMLM images taken on fixed NCI-H1703 cells, immunolabeled with antibodies against CLCa (Fig. S6a, ESI†). Representative examples of mature CCPs are shown in Fig. S6b–e (ESI†) as well as intensity profiles along the x- and y-directions (Fig. S6f–i, ESI†). LOF-DBSCAN also faithfully identified CCPs and caveolae in image sequences of live C2C12 cells (Fig. 1c and f).
Clustering algorithms are designed to sequentially process individual images and do not cope well with sequences of images with short exposure times (for good time resolution) and thus small signal-to-background ratio. Simply merging a large number of successive frames is not a good remedy for this problem because both signal and background accumulate in the merged images. Furthermore, the time resolution is lowered, and two or more transient structures appearing one after another in the same location may become subsumed in one cluster. Therefore, we adopted a ‘moving window binning’ approach,36 which splits the entire image sequence into (temporally partially overlapping) time-of-interest (TOI) intervals, in which the images are merged. Two parameters control this process, the number of frames in each TOI and the number-of-frames shift from one TOI to the next (Fig. 2g). These were set to 300 (corresponding to a 9 s interval) and 50 (1.5 s), respectively, as judged from their effects on the cluster lifetime statistics (Fig. S7a, Supplementary Text, ESI†). We inspected the clustering performance with a few TOIs first, optimized the LOF-DBSCAN parameters and kept those for cluster analysis of all other TOIs in the sequence (Fig. 2h).
Observation of an evolving membrane coat structure often extends over multiple TOIs, requiring clusters in successive TOIs to be linked. To this end, we first merge spatially overlapped clusters from all TOIs (Fig. 2i). As an example, Fig. 2j displays the number of localizations versus time for the blue patch in Fig. 2i. The cumulative number of localizations in a merged cluster region (Fig. 2j, green line) increases markedly whenever the structure grows. We identify the occurrence of such changes with a change point detection algorithm (Fig. 2k, orange vertical lines).37 Then, we calculate the slope for each interval between successive change points, and consecutive intervals with slopes above a pre-defined threshold are merged into individual clusters (Fig. 2l, Fig. S7b and c, Supplementary Text, ESI†). As a result, each single-molecule localization event of the entire time series is assigned to a particular cluster representing a membrane coat structure. The data set containing all clusters together with the associated localizations forms the basis for further quantitative analysis (Fig. 2l, Supplementary Text, ESI†).
The programs LOF-DBSCAN for cluster segmentation and ClusterAnalyzer for cluster separation are freely available via Github (https://github.com/NienhausLabKIT/MaRui).
Lifetime of membrane coat structures
To investigate the temporal evolution of CCSs and CAVSs on the plasma membrane of C2C12 cells, we acquired SMLM image sequences with TIRF excitation for 5 min by taking 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 camera frames (dwell time 30 ms) with optimized laser intensities (Fig. S8, Supplementary Text, ESI†). The individual localization events were identified and assigned to clusters as described above. Cluster occurrences were compiled in histograms according to their lifetime t, i.e., the time from appearance to disappearance of a cluster in the time series, using 1.5 s lifetime intervals. For easy comparison, we normalize all lifetime distributions to area 1 under the curves, which turns them into probability density functions (PDFs), quantifying the probability of finding a cluster with a lifetime in the respective interval (Fig. 3a).
000 camera frames (dwell time 30 ms) with optimized laser intensities (Fig. S8, Supplementary Text, ESI†). The individual localization events were identified and assigned to clusters as described above. Cluster occurrences were compiled in histograms according to their lifetime t, i.e., the time from appearance to disappearance of a cluster in the time series, using 1.5 s lifetime intervals. For easy comparison, we normalize all lifetime distributions to area 1 under the curves, which turns them into probability density functions (PDFs), quantifying the probability of finding a cluster with a lifetime in the respective interval (Fig. 3a).
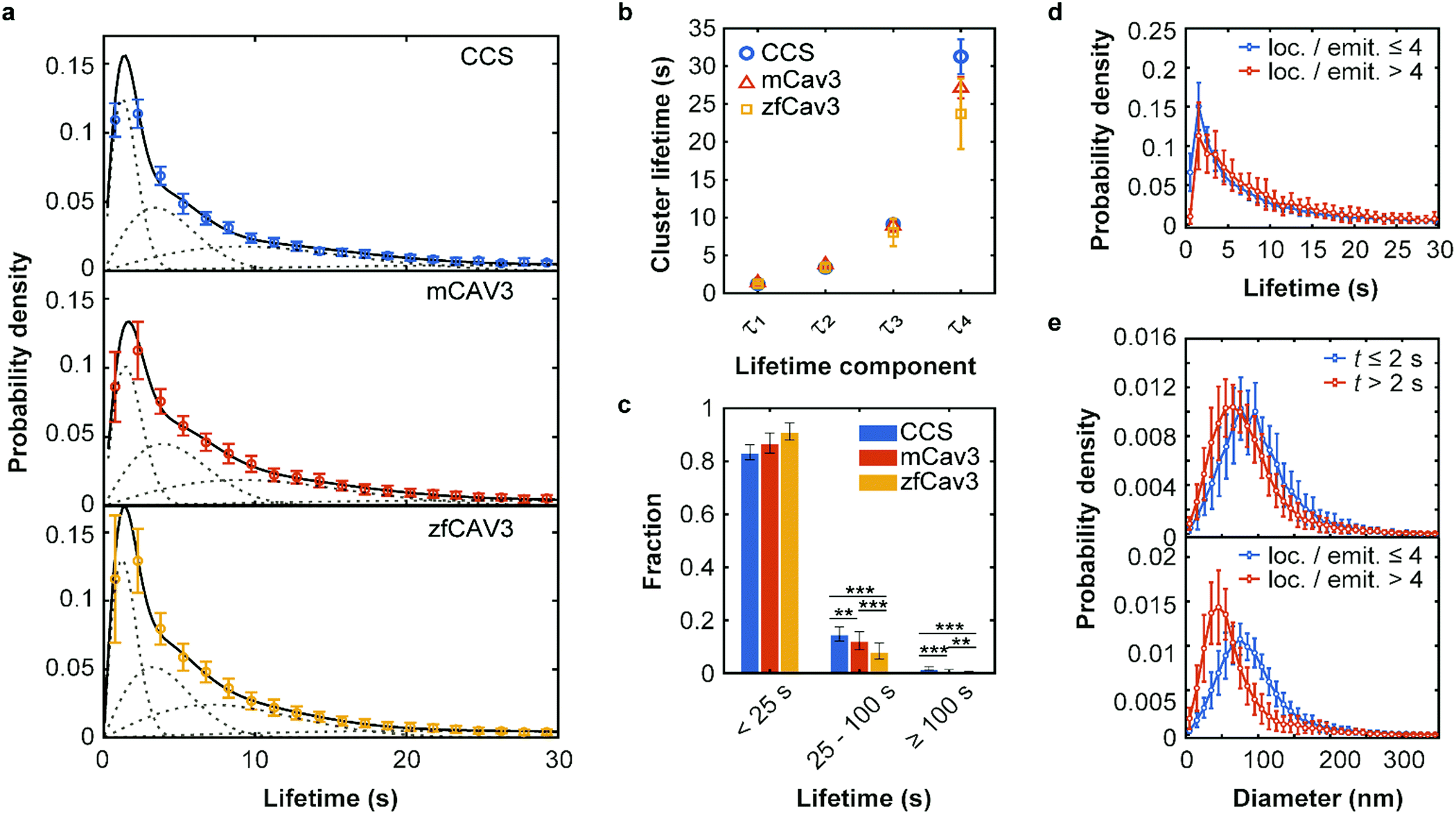 | ||
| Fig. 3 Lifetime distributions and sizes of membrane coat structures. (a) Data points: Probability of occurrence of (top) CCSs and CAVSs composed of (middle) mCav3 or (bottom) zfCav3 clusters within lifetime intervals (Δt = 1.5 s), plotted against the lifetime. For each Δt interval, the number of clusters was calculated as mean (data points) and standard deviation (SD, error bars) values, and the entire histogram was scaled to unit area to obtain PDFs. Solid lines: Fits with sums of four Rayleigh distributions; dashed lines: Individual Rayleigh subpopulations. (b) Peak lifetimes (mean ± SD) of the four subpopulations determined from three measurements. (c) Fractional populations with lifetimes t < 25 s, 25 s ≤ t < 100 s and t ≥ 100 s, calculated from the experimental distributions in (a). Error bars indicate SD values over all measured cells. Stars indicate P values of a two-sample t-test, **: P < 0.01, ***: P < 0.001 (see Supplementary Methods and Table S4 for details, ESI†). (d and e) PDFs of cluster lifetimes calculated with selected parameters for CAVSs composed of zfCav3. (d) The populations with the shortest lifetimes are markedly greater for emitters with four or less localizations than with more than four localizations. Cluster diameters are greater for (e top), t ≤ 2 s and (e bottom), four or less localizations of localizations per single emitter. | ||
The PDFs are broad and appear to consist of multiple subdistributions representing a small set of discrete processes. Their decomposition into Rayleigh and exponential subdistributions was introduced earlier for the analysis of CCS lifetimes with conventional TIRF microscopy, where the individual subdistributions were assigned to early abortive, late abortive and mature structures.12,13 Both types of distributions are special cases of the Weibull distribution, which is widely applied in reliability theory to estimate failures in a wide variety of systems. Specifically, the failure rate increases linearly in time for the Rayleigh distribution, describing aging or maturation scenarios, whereas it is constant for the exponential distribution. For various combinations of these model distributions, we evaluated their effectiveness in describing the experimental data by using the Bayesian information criterion38 (Fig. S9, ESI†). A sum of four Rayleigh distributions turned out to be optimal for all three data sets,
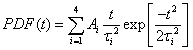 | (1) |
The best-fit cluster lifetime distributions are included in the lifetime histograms of CCS and CAVS clusters in Fig. 3a. The resulting four peak lifetimes, τi, are shown in Fig. 3b and compiled in Table 1 together with the associated fractional amplitudes, Ai. Remarkably, the shapes of the lifetime histograms and, consequently, the fit parameters are similar for the CCS and CAVS data. The largest variation was observed for the parameter τ4 of the longest-lived population, presumably representing mature membrane coat structures. For validation of our SMLM-based lifetime data, we also performed SPT measurements using eGFP-tagged CLCa and Cav3 constructs. SPT revealed essentially identical distributions (Fig. S10a–c, ESI†), albeit with lower time resolution (t ≥ 7.5 s). Following the approach by Senju et al.,15 we split the SMLM and SPT lifetime distributions into three populations, with t < 25 s, 25 s ≤ t < 100 s and t ≥ 100 s (Fig. 3c and Fig. S10d, ESI†). The overall greater fraction of long-lived CCSs suggests that they are less prone to being abortive than CAVSs.
| A 1 | τ 1 (s) | A 2 | τ 2 (s) | A 3 | τ 3 (s) | A 4 | τ 4 (s) | |
|---|---|---|---|---|---|---|---|---|
| Errors represent SDs from three independent measurements on different days. Top (rows 1–3): Fit with four Rayleigh distributions (eqn (1)). Bottom: Clusters were separated into hotspot and non-hotspot clusters and short-lived clusters (visitors, t < 2 s) were removed from the lifetime distributions. The resulting distributions were fitted with sums of three Rayleigh distributions. | ||||||||
| CCS | 0.25 ± 0.01 | 1.2 ± 0.1 | 0.26 ± 0.01 | 3.4 ± 0.1 | 0.27 ± 0.01 | 9.1 ± 0.5 | 0.23 ± 0.01 | 31.2 ± 1.9 |
| mCav3 CAVSs | 0.23 ± 0.04 | 1.4 ± 0.3 | 0.29 ± 0.03 | 3.8 ± 0.3 | 0.28 ± 0.03 | 8.9 ± 0.8 | 0.19 ± 0.03 | 27.1 ± 1.4 |
| zfCav3 CAVSs | 0.27 ± 0.02 | 1.3 ± 0.1 | 0.29 ± 0.07 | 3.4 ± 0.6 | 0.27 ± 0.05 | 8.0 ± 1.8 | 0.17 ± 0.03 | 23.7 ± 4.6 |
| CCSnon-hotspot | 0.55 ± 0.03 | 3.5 ± 0.1 | 0.29 ± 0.01 | 13.7 ± 1.0 | 0.15 ± 0.03 | 56.5 ± 6.9 | ||
| CCShotspot | 0.35 ± 0.03 | 4.4 ± 0.1 | 0.37 ± 0.02 | 11.7 ± 0.5 | 0.28 ± 0.02 | 32.8 ± 0.8 | ||
| mCav3 CAVSsnon-hotspot | 0.53 ± 0.03 | 3.8 ± 0.1 | 0.32 ± 0.01 | 12.0 ± 0.5 | 0.14 ± 0.02 | 39.6 ± 3.3 | ||
| mCav3 CAVSshotspot | 0.42 ± 0.02 | 4.3 ± 0.1 | 0.37 ± 0.01 | 10.7 ± 0.3 | 0.21 ± 0.02 | 30.1 ± 1.9 | ||
| zfCav3 CAVSsnon-hotspot | 0.65 ± 0.02 | 3.7 ± 0.1 | 0.32 ± 0.02 | 13.2 ± 0.6 | 0.04 ± 0.02 | 122 ± 53 | ||
| zfCav3 CAVSshotspot | 0.51 ± 0.02 | 4.5 ± 0.1 | 0.36 ± 0.01 | 12.7 ± 0.3 | 0.12 ± 0.01 | 36.9 ± 1.5 | ||
Next, we enquired into the nature of the shortest-lived components in the lifetime distributions (Fig. 3a). For this purpose, we separated our set of clusters according to their number of localizations per single emitter (≤4 and >4) and lifetimes (≤2 s and >2 s). The resulting lifetime histograms reveal that the shortest-lifetime components are selectively suppressed if the smallest clusters are excluded (Fig. 3d and Fig. S11a, ESI†). Moreover, in all cases, the histograms of cluster diameters shifted to greater values for the shortest-lived clusters and for the ones with a small number of localizations (Fig. 3e and Fig. S11b, c, ESI†). Taken together, these results suggest that short-lived clusters are largely ‘visitors’,14 coat proteins that are only transiently present but not firmly associated with compact structures in the plasma membrane. They may include small aggregates (e.g., CCSs with only two AP2 molecules and one triskelion8), or intracellular vesicles containing these proteins. Their locations in regions of weak excitation light (more distant from the surface) and movement inside the cells may result in fewer localizations per single emitter.
Dynamics of membrane coat structures
Our SMLM data analysis assigns localizations of individual fluorophores to clusters. The apparent number of detected emitters is obtained by combining localizations from the same positions in contiguous images. For technical reasons, this number has to be rescaled to represent the real number of proteins in the coat structure. Briefly, two major issues need to be accounted for (see Supplementary Methods for further details, ESI†). (1) Only a fraction of 0.61 ± 0.02 of all expressed mEosFPthermo fluorophores in a cluster photoconvert to the red form,39 causing undercounting of proteins. (2) Red-converted molecules may not permanently photobleach after an emissive period but rekindle after an extended dark period. This fluorophore intermittency (blinking) is typical of FPs40 and results in overcounting. A quantitative analysis yielded an average of 2.0+1.2−0.7 emission periods per red-converted mEosFPthermo fluorophore (Supplementary Text, Fig. S12, ESI†). As was noticed before,36 the two effects largely compensate one another and require only a small overall adjustment of the apparent numbers. The number of detected clusters, plotted in a two-dimensional histogram against their lifetime and the associated true number of mCav3 proteins (Fig. 4a) shows a high correlation of the two observables; linear regression yields an average mCav3 incorporation rate of 2.54 ± 0.01 s−1. A similar rate, 2.03 ± 0.01 s−1, was found for CCSs (Fig. 4b).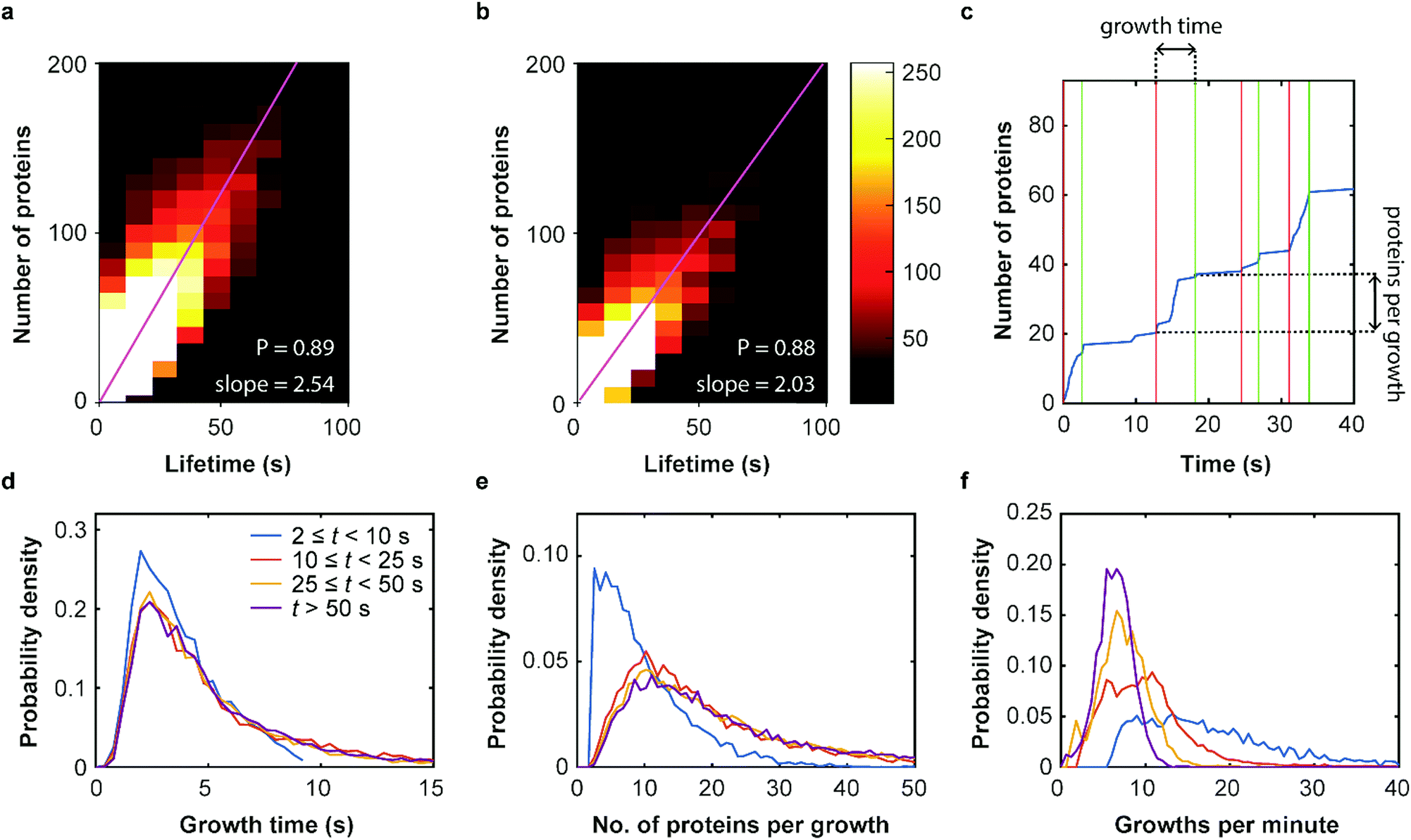 | ||
| Fig. 4 Dynamics of membrane coat structures. (a and b) Two-dimensional histogram showing the occurrence of (a) mCav3 CAVS and (b) CCS clusters, plotted against the (corrected) number of proteins and the cluster lifetime. Pearson's correlation coefficients of P = 0.89 (CAVS) and P = 0.88 reflect strong correlations; linear regressions yield 2.54 ± 0.01 s−1 (CAVS) and 2.03 ± 0.01 s−1 (CCS) (error: 95% confidence limits) as average rates of protein incorporation. (c) Exemplary cumulative growth curve, displaying the number of proteins in a cluster over time. Vertical red and green lines indicate start and end points of growth phases, respectively. (d–f) Analysis of growth phases of mCav3 CAVSs for four lifetime intervals comprising 2,313 (t ≥ 50 s), 4,569 (25 s ≤ t < 50 s), 10,691 (10 s ≤ t < 25 s) and 23,711 (2 s ≤ t < 10 s) clusters. PDFs of (d) the duration of growth phases (inset: color coding of intervals), (e) number of registered protein molecules per growth phase and (f) frequency of growth phases. | ||
We further investigated the dynamics of mCav3 incorporation into individual CAVSs. A representative example of the number of fluorescence markers detected in a cluster over time is shown in Fig. 4c. Apparently, the coat structure grows in discrete steps separated by phases of quiescence. We subdivided the clusters into four subsets with lifetime windows 2 s ≤ t < 10 s, 10 s ≤ t < 25 s, 25 s ≤ t < 50 s and t ≥ 50 s, and analyzed them separately (Fig. 4d–f, Supplementary Text, ESI†). For clusters with t ≥ 10 s, the PDFs are essentially identical, showing that growth phases lasted for 5.4 ± 0.5 s (mean ± SEM, 10 s ≤ t < 25 s), 5.6 ± 0.5 s (25 s ≤ t < 50 s) and 5.7 ± 0.3 s (t ≥ 50 s) (Fig. 4d); mean numbers of proteins per growth phase were 19.0 ± 1.1 (mean ± SEM, 10 s ≤ t < 25 s), 23.5 ± 1.2 (25 s ≤ t < 50 s) and 27.2 ± 1.0 (t ≥ 50 s) (Fig. 4e). For short-lived, abortive CAVSs (t < 10 s), there were shorter growth phases (3.9 ± 0.4 s) and fewer molecules (10.0 ± 0.8) were incorporated during a single growth phase. We further asked how often growth phases occur within 1 min and found a systematic shift of the distributions to higher frequencies for shorter lifetimes (Fig. 4f). Thus, short-lived CAVSs grow in smaller steps occurring quickly in succession, whereas long-lived CAVSs assemble by adding more proteins in a single step, after which there are longer periods of quiescence. We also analyzed CCS and zfCav3 CAVS development. The resulting PDFs for the lifetime window (shown for t > 50 s in Fig. S13, ESI†) are all very similar.
Hotspot analysis
It is well known that there are hotspots of CCS formation on the plasma membrane, distinct locations with a higher rate of CCS formation than expected for random initiation.28,29 We have observed that such hotspots of enhanced activity are also present for CAVSs, as shown exemplarily in Fig. 5a. For their characterization, we examined our cluster data set based on 5 min SMLM image sequences to distinguish clusters appearing only once (no hotspot) or multiple times (hotspot) in a particular location. Specifically, we assigned a location to a hotspot if there was spatial overlap between two or more time-separated clusters. To this end, we calculated cluster contours by connecting all border localizations, from which overlap between clusters can be assessed. Both non-hotspot and hotspot clusters appeared at an average rate of 0.21 ± 0.04 μm−2 min−1 for CCSs and an up to twofold greater rate for CAVSs (Fig. S14, ESI†). Our analysis shows that there is a majority (70–80%) of individual clusters (Fig. 5b). Hotspots with two and three clusters appeared with fractions of ca. 20% and 4–6%, respectively; those with more clusters were rarely observed. However, one has to be aware that some clusters may appear in sequence at the same spot simply by chance. A simulation that mimics the experimental conditions (Fig. S15, ESI†) yields a probability of ∼2% for accidental overlap of 100 nm clusters, which is far less than the observed fractions of clusters in hotspots (Fig. 5b). Thus, only a minority of registered hotspots are due to accidental overlap. We separated our cluster ensembles into two subsets, those associated with hotspots and those that are not, and calculated lifetime PDFs (Fig. S16, ESI†). After removing visitors, we fitted the PDFs with sums of three Rayleigh distributions, corresponding to early abortive, late abortive and mature clusters.12 The best-fit parameters of 3-Rayleigh fits (Fig. 5c) show that the fractions of early abortive events are strongly enhanced in comparison with the 4-Rayleigh fit of the full lifetime distribution (Table 1), since this subdistribution encompasses clusters from the two shortest-lifetime subdistributions of the 4-Rayleigh fit. Furthermore, the lifetimes of late abortive and mature clusters are greater in the 3-Rayleigh fit. Even without any fitting, comparison of hotspot and non-hotspot lifetime distributions (Fig. 5c, Table 1, Fig. S16 (ESI†)) yields an interesting result for all systems studied here: The fraction of the short-lived clusters is significantly reduced for clusters associated with a hotspot. Apparently, those clusters have a higher probability to survive the early stage of formation and enter the late abortive and mature phases. Thus, in the hotspot environment, growth of membrane coat structures up to maturation is more effective.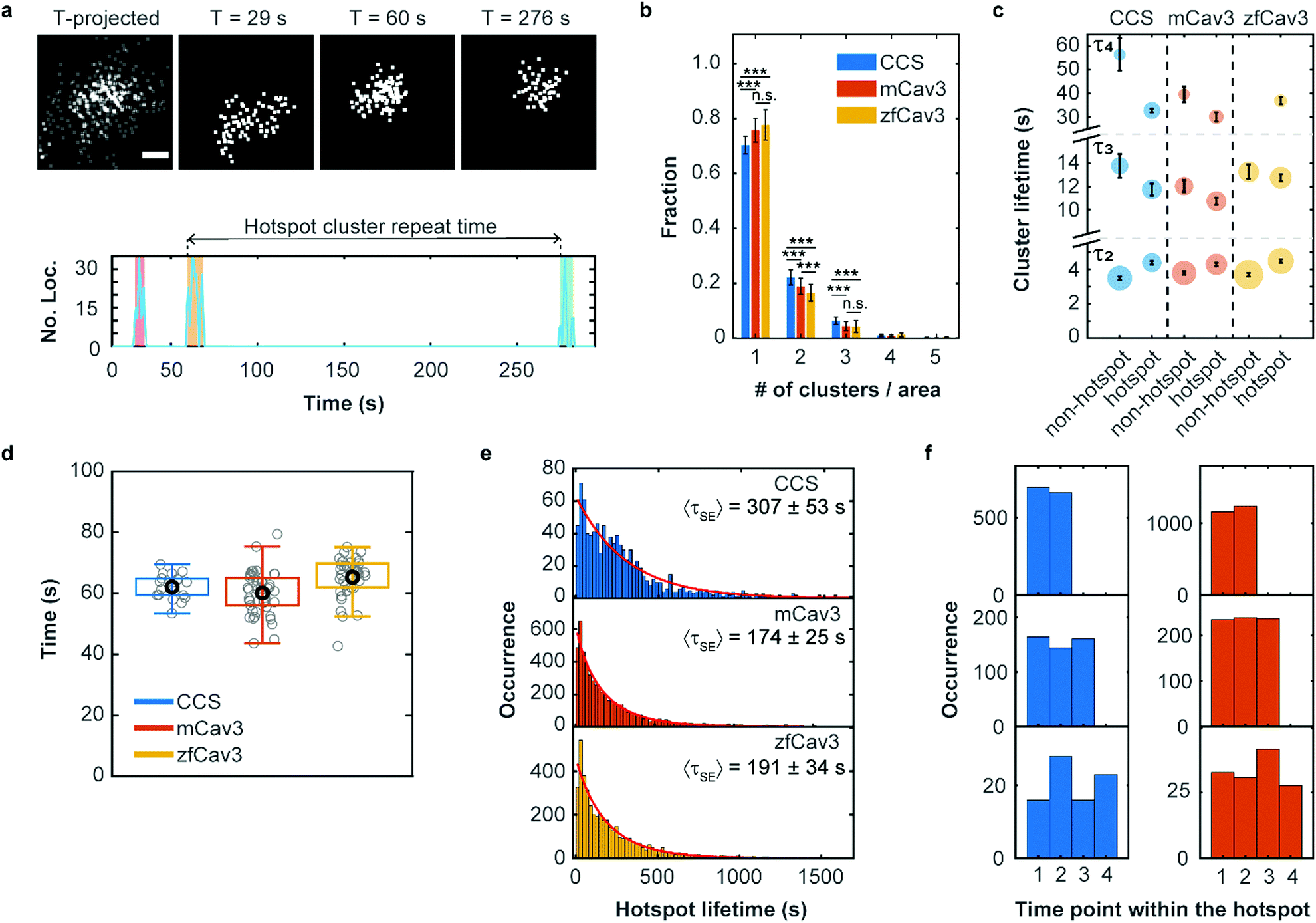 | ||
| Fig. 5 Hotspots of CCS and CAVS formation. (a) Top: Scatter plots of fluorophore localizations in an exemplary caveolar hotspot. The time-projected image shows all localizations occurring within 5 min. Scale bar, 100 nm. Images integrated over 9 s intervals (start times are given above the images). Bottom: The number of localizations at this site versus time shows discrete clusters (color shaded). The definition of cluster repeat time is also depicted. (b) Fractions of single or multiple clusters appearing at certain locations during a 5 min measurement. Stars represent P values of a two-sample t-test, ***: P < 0.001, n.s.: not significant (see Supplementary Methods and Table S4 for details, ESI†). (c) Scatter plot of lifetime parameters for hotspot and non-hotspot clusters, determined by fitting a sum of three Rayleigh distributions to the histograms and presented as mean (data points) and SD (error bars) over three independent measurements. Note that, for better visualization, the lifetime axis is broken. The areas of the circles encode the fractional weights of the subpopulations. (d) Boxplot of the hotspot cluster repeat times. Gray circles show mean values for individual cells, the black circle marks the mean over all cells and the box encloses the two central quartiles. Whiskers extend to the extreme data points of the distributions; outliers (points more than 1.5 times the interquartile range above or below the edges of the box) have been excluded. (e) Histograms of occurrences of hotspot lifetimes in 20 s intervals, based on data from nine (CCS), six (mCav3 CAVSs) and six (zfCav3 CAVSs) cells measured for 30 min, and fits with stretched exponentials (red lines). (f) Histograms of the appearance of (left) CCS and (right) mCav3 clusters with lifetimes >20 s in the time sequence of hotspot time traces, displayed for hotspots with two, three and four clusters (top to bottom). Clusters with t ≤ 2 s were excluded from the analysis. | ||
To quantify the dynamics of clusters in hotspots, we introduced a parameter denoted ‘hotspot cluster repeat (HCR) time’, which is the time between the start of a cluster and the beginning of the following cluster (see Fig. 5a). This parameter is similar for CCSs and CAVSs, with cluster repeat times averaged over individual cells, tHCR, ranging from 60–70 s (Fig. 5d). We then compiled histograms of hotspot cluster repeat times for the individual cells (Fig. S17, ESI†). Fits with a stretched exponential (SE), f(tHCR) = exp[−tHCR/τSE]β, yielded stretching exponents close to one within the error, suggesting that the observed dynamics is Markovian, meaning that the initiation of clusters in hotspots does not depend on the prior history. The parameters of the distributions are given in Table S5 (ESI†) as averages over the ensemble of cells. Hotspot cluster repeat times are clearly smaller than the 5 min measurement duration, so we can exclude artifacts arising from the limited measurement time. Moreover, we found only a weak dependence on the chosen degree of allowed cluster overlap within hotspots (Fig. S18, ESI†).
Next, we aimed to determine the hotspot lifetime, i.e., the period from the start of the first to the end of the last cluster in a hotspot. To this end, we collected image sequences for 30 min rather than 5 min, considering that tHCR ≈ 60 s. In the data analysis, we frequently noticed that some clusters that were assigned to the same hotspot had much longer time separations than expected from the previously determined cluster repeat distributions. This finding strongly suggests that unrelated clusters appear by chance at the particular location, which is more likely for longer measurement durations. For example, a hotspot might terminate and a new cluster (and even a new hotspot) may appear at the same location, artificially extending the hotspot lifetime. Evidence of such a scenario is clearly visible in the mCav3 CAVS hotspot lifetime histogram in Fig. S19a (ESI†). After a sharp drop at short lifetimes, there is a roughly constant occurrence of longer lifetimes, which is at odds with the random appearance of hotspots. To solve this problem, we introduced a maximum time gap, above which a newly appearing cluster is no longer assigned to the same hotspot as the previous one (but possibly to a new one). Varying the maximum gap between 2 and 10 × tHCR (Fig. S19b–f (ESI†), tHCR values taken from Table S5, ESI†) greatly changed the histogram, and best agreement with a stretched exponential distribution was found for 3 × tHCR. Notably, secondary peaks in the histogram, indicative of multiple independent processes, were minimal for this choice. The resulting hotspot lifetime distributions and fits are shown in Fig. 5e; fit parameters are compiled in Table S6 (ESI†). The average hotspot lifetime of CCSs (307 ± 53 s) is almost the double of those found for mCav3 CAVSs and zfCav3 CAVSs (Fig. 5e).
Finally, we asked if cluster lifetimes vary in a systematic way along the sequence of clusters in hotspots. In all hotspots containing two, three and four clusters, the longest-lived clusters in a hotspot appeared randomly in sequence for both CCSs and CAVSs, as seen in the histograms of the occurrence of the longest-lived cluster (for t > 20 s) at a particular point in the hotspot cluster sequence (Fig. 5f); analogous results were obtained for other lifetime thresholds (Fig. S20, ESI†). Thus, hotspots are membrane locations where clusters repeatedly yet independently form; lifetimes of newly created clusters in a hotspot do not depend on the prior hotspot history.
Discussion and conclusions
TIRF-based SMLM has distinct advantages over classical TIRF imaging for live-cell investigation of membrane coat structures. Its much higher spatial resolution greatly facilitates the assignment of the fluorescence emission from labeled proteins to a particular structure developing in time. Single-molecule detection sensitivity affords the widest-possible dynamic range, so that small and weakly fluorescent structures are visible. Notably, the higher dynamic range can also be traded in to achieve a better temporal resolution. In this work, we have presented a new clustering algorithm, LOF-DBSCAN, for automatic extraction of clusters associated with membrane coat structures from SMLM images. It is embedded in a workflow that processes image sequences, and combines and splits clusters appearing successively at the same location. This software allowed us to extract quantitative information about the dynamics of CCSs and CAVSs in the cell membrane.We validated our image analysis pipeline using CCSs as a well-established system. Comparison with previous reports from classical TIRF imaging12,13 yielded overall agreement but also differences. A sum of four Rayleigh distributions best described our data, whereas Loerke et al.12 used three subdistributions, assigned to early abortive and late abortive CCS and mature CCPs. Our additional, very short-lifetime component (τ ≈ 1 s) reflects the enhanced time resolution of SMLM. In previous work,14 this component was noticed (and filtered against), and attributed to transiently appearing small entities (visitors) that are not associated with proper CCSs, e.g., small assemblies or cytoplasmic vesicular structures. The lifetimes of the other three components (Fig. 3a–c, Table 1), however, were similar to those reported earlier from conventional TIRF imaging analyses.12,13 The intrinsic advantages of our method over classical imaging-based approaches should result in a higher precision; yet, it is satisfying to find good agreement of our lifetime distribution with earlier work. The parameters of CCS lifetime distributions vary considerably between earlier reports using classical microscopy,12,13 presumably due to different samples (cell lines, media) or imaging conditions (laser intensity, time resolution). Furthermore, decomposition of broad and featureless histograms into multiple subdistributions, possibly with different shapes (exponential or Rayleigh), is a mathematically ill-defined operation, and trade-off between parameters is unavoidable.
Extending our SMLM-based cluster analysis to CAVSs, we found stunningly similar lifetime distributions as for CCS (Fig. 3, Table 1). The almost identical lifetimes of abortive structures suggest that CCSs and CAVSs appear at the plasma membrane in a similar random initiation and maturation process. The great majority of attempts to assemble long-lived CCPs and caveolae fails; only a small fraction of initiated coat structures goes forward to the final, biologically active pit assembly. A wide body of evidence supports the existence of checkpoints in the formation of CCPs,14,41,42 at which progression of the assemblies is assessed and fate decisions are taken.12,29 These findings are likely also relevant for caveolae. It appears that the main structural proteins, clathrin and AP2 for CCSs, and caveolin and cavin for CAVSs, play the role of ‘brick and mortar’; they are the necessary building blocks of the protein assembly on the plasma membrane. Additional accessory proteins, however, some of which are common to both CCSs and CAVSs, may play decisive roles in orchestrating the assembly of these sophisticated architectures and lead to similar dynamics. Gene knockdown of altogether 67 accessory proteins has shown significant effects on CCP formation and CME.9 One of the key players is dynamin-2, a large GTPase that plays crucial roles in CCS initiation and maturation as well as the final membrane fission process.12,43 This protein is also involved in caveola formation and may be one of the common regulators of CCP and caveola formation. SMLM imaging of accessory proteins appears challenging, considering their smaller copy numbers compared to the coat proteins but could provide further insights.
SMLM has the unique advantage that individual molecules are registered. In our analysis, we detected 100–200 clathrin and Cav3 molecules in long-lived structures (Fig. 4a and b), whereas abortive structures contained <50 clathrin and Cav3 molecules. These figures agree well with the 150 Cav1 proteins found in mature caveolae of mammalian cells,44 and a recent cryo-EM study reporting between 28–37 triskelia per CCS, corresponding to 84–111 CLC molecules.2 Incorporation of proteins into membrane coat structures occurred in multiple, temporally separated growth steps, with average rates of 2 and 2.5 s−1 for CCS and CAVSs, respectively (Fig. 4c–f).
For CCS formation, it has long been known that there are locations in the plasma membrane with higher activity than expected for random occurrence. The presence of such hotspots has been associated with specialized cortical actin regions that enhance and retain essential resources for CCS formation, such as AP-2 and the phospholipid PIP2.28,29 Hotspots spatially organize endocytic activity at the cell surface and thus can play a role in cell polarization. Intriguingly, CAVS clusters are also formed repeatedly in hotspots. In the quantitative analysis of hotspot dynamics, one needs to be aware that an unrelated cluster may appear randomly at the same location where a cluster was registered earlier; so this location thus looks like a hotspot but it is not. This problem is of great concern for diffraction-limited microscopy but less so for super-resolution SMLM, due to its much sharper definition of ‘the same location’ for our nanoscale coat structures with 80–100 nm diameter (Table S2, ESI†). We found 20–30% of the clusters associated with hotspots (Fig. 5b). Cluster repeat time histograms are exponentially distributed, with an average time gap of ∼60 s (Fig. 5d). CAVS lifetimes of a few minutes have been reported based on SPT analysis.15,16 However, imaging with insufficient time resolution runs the risk of concatenating clusters, leading to artificially extended lifetimes. We further observed that early abortive events are markedly suppressed for hotspot-associated structures (Fig. S16 (ESI†), Table 1), so it is more likely for a cluster to reach the late abortive stage and long-lived pit formation in comparison to non-hotspot structures. This may reflect the enhancement of molecular resources ascribed to hotspots.28,29 Hotspots were found to persist on average for ∼5 min for CCSs and ∼3 min for CAVSs (Fig. 5e), and there was no systematic development of cluster lifetimes in a hotspot-associated cluster sequence, suggesting that the clusters are temporally independent from one another.
To conclude, we have quantified the dynamics of membrane coat structures in live cells using a super-resolution SMLM-based image analysis. The surprisingly similar dynamics of CCS and CAVS formation suggests that similar and possibly general principles underlie the development of such complex biomolecular architectures. Future work in this direction will address the great variety of accessory proteins, which may play a decisive role in membrane coat dynamics.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Dr Olivier Kassel for providing the C2C12 cells for this study, Dr Lucas Flatten for helpful discussions and Dr Volker Middel for the construction of zebrafish plasmids. This work was supported by the Helmholtz Association, Program Materials Systems Engineering (MSE), and the Chinese Scholarship Council (CSC).References
- M. Mettlen, P.-H. Chen, S. Srinivasan, G. Danuser and S. L. Schmid, Annu. Rev. Biochem., 2018, 87, 871–896 CrossRef CAS PubMed.
- K. L. Morris, J. R. Jones, M. Halebian, S. Wu, M. Baker, J. P. Armache, A. Avila Ibarra, R. B. Sessions, A. D. Cameron, Y. Cheng and C. J. Smith, Nat. Struct. Mol. Biol., 2019, 26, 890–898 CrossRef CAS PubMed.
- D. Li, L. Shao, B. C. Chen, X. Zhang, M. Zhang, B. Moses, D. E. Milkie, J. R. Beach, J. A. Hammer, M. Pasham, T. Kirchhausen, M. A. Baird, M. W. Davidson, P. Xu and E. Betzig, Science, 2015, 349, aab3500 CrossRef PubMed.
- M. Stoeber, P. Schellenberger, C. A. Siebert, C. Leyrat, A. Helenius and K. Grünewald, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8069–E8078 CrossRef CAS PubMed.
- R. G. Parton, Annu. Rev. Cell Dev. Biol., 2018, 34, 111–136 CrossRef CAS PubMed.
- M. S. Robinson, Traffic, 2015, 16, 1210–1238 CrossRef CAS PubMed.
- M. Lampe, S. Vassilopoulos and C. Merrifield, J. Struct. Biol., 2016, 196, 48–56 CrossRef CAS PubMed.
- E. Cocucci, F. Aguet, S. Boulant and T. Kirchhausen, Cell, 2012, 150, 495–507 CrossRef CAS PubMed.
- M. Bhave, R. E. Mino, X. Wang, J. Lee, H. M. Grossman, A. M. Lakoduk, G. Danuser, S. L. Schmid and M. Mettlen, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 31591–31602 CrossRef CAS PubMed.
- Y. Chen, J. Yong, A. Martínez-Sánchez, Y. Yang, Y. Wu, P. De Camilli, R. Fernández-Busnadiego and M. Wu, J. Cell Biol., 2019, 218, 3200–3211 CrossRef CAS PubMed.
- M. J. Taylor, D. Perrais and C. J. Merrifield, PLoS Biol., 2011, 9, e1000604 CrossRef CAS PubMed.
- D. Loerke, M. Mettlen, D. Yarar, K. Jaqaman, H. Jaqaman, G. Danuser and S. L. Schmid, PLoS Biol., 2009, 7, 628–639 CrossRef PubMed.
- A. V. Weigel, M. M. Tamkun and D. Krapf, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4591–E4600 CrossRef CAS PubMed.
- F. Aguet, C. N. Antonescu, M. Mettlen, S. L. Schmid and G. Danuser, Dev. Cell, 2013, 26, 279–291 CrossRef CAS PubMed.
- Y. Senju, E. Rosenbaum, C. Shah, S. Hamada-Nakahara, Y. Itoh, K. Yamamoto, K. Hanawa-Suetsugu, O. Daumke and S. Suetsugu, J. Cell Sci., 2015, 128, 2766–2780 CAS.
- J. Mohan, B. Morén, E. Larsson, M. R. Holst and R. Lundmark, J. Cell Sci., 2015, 128, 979–991 CAS.
- L. M. Traub, PLoS Biol., 2011, 9, 3–7 CrossRef PubMed.
- A. Echarri and M. A. Del Pozo, J. Cell Sci., 2015, 128, 2747–2758 CAS.
- G. M. Popowicz, M. Schleicher, A. A. Noegel and T. A. Holak, Trends Biochem. Sci., 2006, 31, 411–419 CrossRef CAS PubMed.
- X. Wang, Z. Chen, M. Mettlen, J. Noh, S. L. Schmid and G. Danuser, eLife, 2020, 9, 1–27 CAS.
- Y. M. Sigal, R. Zhou and X. Zhuang, Science, 2018, 361, 880–887 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, J. Mol. Biol., 2016, 428, 308–322 CrossRef CAS PubMed.
- M. Sauer and M. Heilemann, Chem. Rev., 2017, 117, 7478–7509 CrossRef CAS PubMed.
- J. Wiedenmann, S. Ivanchenko, F. Oswald, F. Schmitt, C. Röcker, A. Salih, K.-D. Spindler and G. U. Nienhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15905–15910 CrossRef CAS PubMed.
- J. Fuchs, S. Böhme, F. Oswald, P. N. Hedde, M. Krause, J. Wiedenmann and G. U. Nienhaus, Nat. Methods, 2010, 7, 627–630 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, RSC Chem. Biol., 2021, 2, 796–814 RSC.
- I. M. Khater, I. R. Nabi and G. Hamarneh, Patterns, 2020, 1, 100038 CrossRef PubMed.
- I. Gaidarov, F. Santini, R. A. Warren and J. H. Keen, Nat. Cell Biol., 1999, 1, 1–7 CrossRef CAS PubMed.
- D. Nunez, C. Antonescu, M. Mettlen, A. Liu, S. L. Schmid, D. Loerke and G. Danuser, Traffic, 2011, 12, 1868–1878 CrossRef CAS PubMed.
- K. A. McMahon, H. Zajicek, W. P. Li, M. J. Peyton, J. D. Minna, V. J. Hernandez, K. Luby-Phelps and R. G. W. Anderson, EMBO J., 2009, 28, 1001–1015 CrossRef CAS PubMed.
- B. Sinha, D. Köster, R. Ruez, P. Gonnord, M. Bastiani, D. Abankwa, R. V. Stan, G. Butler-Browne, B. Vedie, L. Johannes, N. Morone, R. G. Parton, G. Raposo, P. Sens, C. Lamaze and P. Nassoy, Cell, 2011, 144, 402–413 CrossRef CAS PubMed.
- Y. Li, Y. Ishitsuka, P. N. Hedde and G. U. Nienhaus, ACS Nano, 2013, 7, 5207–5214 CrossRef CAS PubMed.
- M. M. Breunig, H.-P. Kriegel, R. T. Ng and J. Sander, ACM SIGMOD Rec., 2000, 29, 93–104 CrossRef.
- M. Ester, H.-P. Kriegel, J. Sander and X. Xu, In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, 1996, pp. 226–231.
- J. Hou, H. Gao and X. Li, IEEE Trans. Image Process., 2016, 25, 3182–3193 Search PubMed.
- Y. Ishitsuka, N. Savage, Y. Li, A. Bergs, N. Grün, D. Kohler, R. Donnelly, G. U. Nienhaus, R. Fischer and N. Takeshita, Sci. Adv., 2015, 1, e1500947 CrossRef PubMed.
- R. Killick, P. Fearnhead and I. A. Eckley, J. Am. Stat. Assoc., 2012, 107, 1590–1598 CrossRef CAS.
- G. Schwarz, Ann. Stat., 1978, 6, 461–464 Search PubMed.
- N. Durisic, L. Laparra-Cuervo, Á. Sandoval-Álvarez, J. S. Borbely and M. Lakadamyali, Nat. Methods, 2014, 11, 156–162 CrossRef CAS PubMed.
- A. Schenk, S. Ivanchenko, C. Röcker, J. Wiedenmann and G. U. Nienhaus, Biophys. J., 2004, 86, 384 CrossRef CAS PubMed.
- M. A. Puthenveedu and M. von Zastrow, Cell, 2006, 127, 113–124 CrossRef CAS PubMed.
- A. G. Henry, J. N. Hislop, J. Grove, K. Thorn, M. Marsh and M. von Zastrow, Dev. Cell, 2012, 23, 519–532 CrossRef CAS PubMed.
- M. Bhave, M. Mettlen, X. Wang and S. L. Schmid, Mol. Biol. Cell, 2020, 31, 2035–2047 CrossRef CAS PubMed.
- L. Pelkmans and M. Zerial, Nature, 2005, 436, 128–133 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2nh00008c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |