
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the mechanism of the peroxiredoxin decamer interaction with its reductase sulfiredoxin from the single molecule to the solution scale†
Audrey
Beaussart‡
 *a,
Florent
Canonico‡
b,
Hortense
Mazon
b,
Jorge
Hidalgo
b,
Sarah
Cianférani
cd,
Hélène
Le Cordier§
b,
Alexandre
Kriznik
be and
Sophie
Rahuel-Clermont
*be
*a,
Florent
Canonico‡
b,
Hortense
Mazon
b,
Jorge
Hidalgo
b,
Sarah
Cianférani
cd,
Hélène
Le Cordier§
b,
Alexandre
Kriznik
be and
Sophie
Rahuel-Clermont
*be
aUniversité de Lorraine, CNRS, LIEC, F-54000 Nancy, France. E-mail: audrey.beaussart@univ-lorraine.fr
bUniversité de Lorraine, CNRS, IMoPA, F-54000 Nancy, France
cLaboratoire de Spectrométrie de Masse BioOrganique, Université de Strasbourg, CNRS, IPHC UMR 7178, 67000 Strasbourg, France
dInfrastructure Nationale de Protéomique ProFI – FR2048 CNRS CEA, 67087 Strasbourg, France
eUniversité de Lorraine, CNRS, INSERM, UMS2008 IBSLor, Biophysics and Structural Biology core facility, F-54000 Nancy, France. E-mail: sophie.rahuel@univ-lorraine.fr
First published on 9th February 2022
Abstract
Peroxiredoxins from the Prx1 subfamily (Prx) are highly regulated multifunctional proteins involved in oxidative stress response, redox signaling and cell protection. Prx is a homodimer that associates into a decamer. The monomer C-terminus plays intricate roles in Prx catalytic functions, decamer stability and interaction with its redox partner, the small reductase sulfiredoxin (Srx), that regulates the switching between Prx cellular functions. As only static structures of covalent Prx–Srx complexes have been reported, whether Srx binding dissociates the decameric assembly and how Prx subunit flexibility impacts complex formation are unknown. Here, we assessed the non-covalent interaction mechanism and dynamics in the solution of Saccharomyces cerevisiae Srx with the ten subunits of Prx Tsa1 at the decamer level via a combination of multiscale biophysical approaches including native mass spectrometry. We show that the ten subunits of the decamer can be saturated by ten Srx molecules and that the Tsa1 decamer in complex with Srx does not dissociate in solution. Furthermore, the binding events of atomic force microscopy (AFM) tip-grafted Srx molecules to Tsa1 individual subunits were relevant to the interactions between free molecules in solution. Combined with protein engineering and rapid kinetics, the observation of peculiar AFM force–distance signatures revealed that Tsa1 C-terminus flexibility controls Tsa1/Srx two-step binding and dynamics and determines the force-induced dissociation of Srx from each subunit of the decameric complex in a sequential or concerted mode. This combined approach from the solution to the single-molecule level offers promising prospects for understanding oligomeric protein interactions with their partners.
New conceptsThe ubiquitous antioxidant enzyme peroxiredoxin (Prx) is a homodimer that associates into a decamer. The Prx monomer C-terminus conformation plays intricate roles in Prx decamer functions, stability and interactions with its redox partner, the small reductase sulfiredoxin (Srx), which controls the switching between its cellular functions as an antioxidant and a chaperone. Understanding the mechanism of Srx interaction with multiple Prx subunits requires (i) monitoring non-covalent dynamic complexes at the level of the decamer, (ii) assessing how Prx subunit flexibility impacts complex formation and dissociation and (iii) deciphering how the interaction affects the decameric assembly. The single-molecule atomic force microscopy approach using a tip sized to the decamer dimensions, decorated with multiple Srx molecules, allowed resolving up to five native interactions with Prx subunits within a decamer. Combining protein engineering and rapid kinetics allowed demonstrating a two-step mechanism of Srx binding to Prx and the importance of Prx C-terminus flexibility. AFM imaging and native mass spectrometry combined with solution scale techniques resolved the impact of Srx interactions on the dissociation of the decamer. By obtaining highly pertinent correlations between affinity, kinetics, and single molecule data, this multiscale strategy bridges the mechanistic understanding of protein supramolecular assembly from the structure to the single molecule and the solution behavior. |
Protein supramolecular assembly provides a means for regulation through multivalent interactions with partners, but designing experimental approaches for identifying these links is challenging. Peroxiredoxins are ubiquitous peroxide-reducing Cys enzymes, whose thiol redox reactivity, oligomeric structure and functions are intimately intertwined,1 that act as major players in oxidative stress defense and cell redox signaling networks.2–4 Peroxiredoxins from the Prx1 subfamily (hereafter referred to as Prxs) exist as homooligomers. The Prx active unit is a symmetrical head-to-tail dimer stabilized by interactions between two β-strands that define the B-interface (Fig. 1A). They act as essential peroxide-reducing enzymes operating with a catalytic cysteine (Cys CP), which reduces peroxide substrates with exquisite efficiency, leading to a sulfenic acid intermediate. Condensation with a resolving cysteine (Cys CR) located in the C-terminal tail of the symmetric monomer produces a disulfide, which is reduced by thioredoxin or other cellular reductant systems (Fig. S1, ESI†). Importantly, Prx dimers associate into toroid structures as decamers, defining a dimer–dimer interface named A (Fig. 1A). The stability of interface A is favored by the conformation of the C-terminal region in a fully folded FF state and disfavored in the locally unfolded LU conformation adopted in the disulfide state (Fig. 1 and Fig. S1, ESI†).5
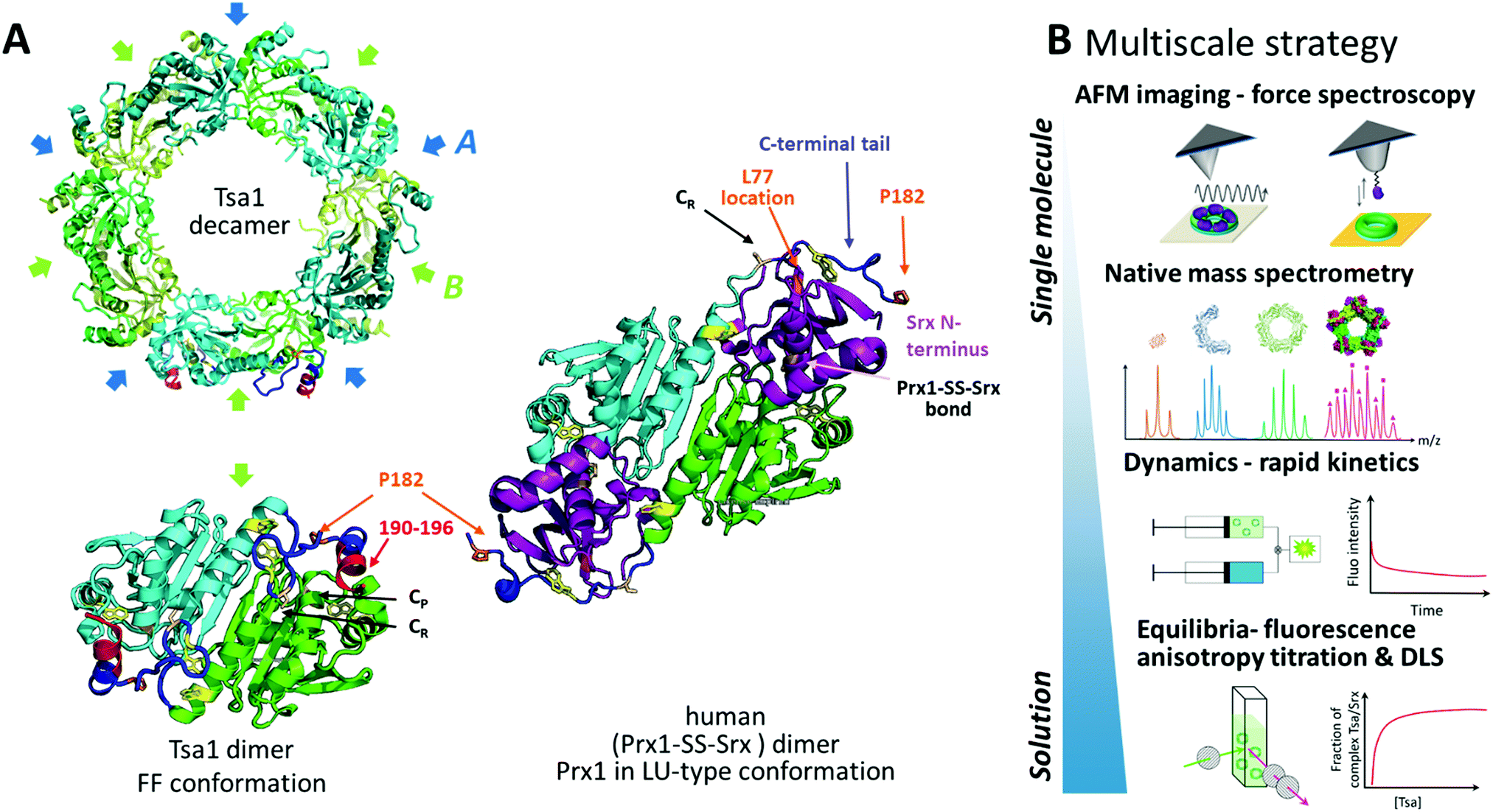 | ||
| Fig. 1 Assessing Prx/Srx interactions at multiscale levels. (A) Top, the cartoon scheme of the reduced Tsa1SH X-ray structure from the top view (pdb code: 3sbc); the A and B interfaces are indicated. Bottom, the cartoon scheme of an isolated dimer of the Tsa1 decamer shown above, with subunits in green and cyan. The dimer is rotated 90° compared to the top view. The catalytic and resolving Cys CP and CR are indicated in light pink, the C-terminal tails in dark blue, Trp residues in yellow and the location of the mutated residues in orange/red. Right, the cartoon scheme of the X-ray structure of the human dimeric Prx1–SS–Srx complex (pdb code: 2rii), stabilized by a disulfide bond between Prx CP and Srx catalytic Cys. The human Prx1 colors are similar to that of the TSaSH, i.e. subunits are green and cyan, the C-terminal tail is dark blue and P182 residues are in orange. Human Srx is in purple, and the human Prx1–SS–Srx disulfide bond and Prx1 Cys CR are in light pink. The human Srx residue corresponding to S. cerevisiae L77 on the backside interface is shown in orange. The location of Tsa1 Trp residues is shown in yellow. The Prx residues beyond P182 and the Srx residues preceding A40 (labelled the Srx N-terminus) are not defined in this structure. (B) Multiscale strategy developed in this study, based on the approaches providing equilibrium and kinetic information in solution to approaches allowing analyses at the level of single subunits within decamers (native mass spectrometry) or at the level of single decamers (AFM). | ||
Eukaryotic Prxs are sensitive to post-translational modification of the catalytic Cys CP to a sulfinic acid (also known as hyperoxidation), which occurs by reaction of a second peroxide molecule with the sulfenic acid intermediate and is critically regulated by competition with the FF/LU conformational transition along the Prx catalytic cycle (Fig. S1, ESI†).6–8 Prx CP sulfinylation inactivates the peroxidase activity, allowing local buildup of H2O2 levels,9 control of the redox state of reducing partners10 and regulation of noncovalent interactions with signaling partners.11 It also activates Prx chaperone function12,13 by stabilizing the FF conformation and Prx decamer state.14 Sulfinylated Prx is reduced by the ATP-dependent sulfinic acid reductase sulfiredoxin (Srx). By regulating the Prx sulfinylation level, Srx is associated with aging12 and diseases like cancer.15–17
The crystal structure of the human Prx1 dimer in complex with Srx revealed a double interface: the active site interface enables Prx sulfinylated CP to come in proximity to Srx catalytic Cys; the backside interface consists of the Prx C-terminal tail unfolded around Srx, in a conformation akin to the LU state (Fig. 1A, Fig. S1 and S2, ESI†).18 A similar structure was reported recently at the level of the decamer in the crystal state.19 The Prx/Srx complex formation thus implies unfolding of the Prx C-terminal region that could also destabilize the decamer A interface, thus raising the questions whether Srx binding dissociates the decameric assembly and how the Prx C-terminal subunit flexibility impacts complex formation. However, presently, the interaction mechanism and dynamics in the solution of the Prx decamer with its partner proteins like Srx are unknown. Their exploration requires monitoring non-covalent dynamic complexes at the level of the decamer using techniques that allow measurements at the oligomer scale, such as single-molecule and native mass spectrometry techniques.
Atomic force microscopy (AFM) has been instrumental in imaging proteins’ oligomeric state and supramolecular assembly at high resolution.20–24 In addition to electronic and X-ray crystallography, the technique allows the visualization of protein ultrastructures deposited on a substrate in liquid and in three dimensions in real time with minimal sample preparation. AFM also allows quantification of the forces involved in molecular interactions by decorating the tip with specific biomolecules and recording force–distance curves while the tip is moved up and down towards the substrate in the so-called single-molecule force spectroscopy (SMFS) mode at the level of individual subunits of a multimeric complex.25–28
We address here the mechanism of Srx interactions with the Prx decamer in solution and at multiple scale levels by combining AFM in imaging and force spectroscopy modes, native mass spectrometry, and bulk measurements via fluorescence anisotropy titration, dynamic light scattering and binding dynamics using rapid kinetics (Fig. 1B). We obtained the first images in liquid of decameric Tsa1, the major Prx in the yeast Saccharomyces cerevisiae, deposited on a substrate bound with ten Srx molecules. We show that binding of Srx does not induce dissociation of the decamer. Furthermore, SMFS using a tip grafted with multiple Srx molecules allowed us to observe binding events with the Prx decamer that are relevant to the interactions between free molecules in solution. Combined with protein engineering and rapid kinetics, the observation of peculiar force–distance signatures revealed that the Prx C-terminal tail plays a critical role in the rupture dynamics of the decameric Prx/Srx complex. Altogether our results highlight the wider potential of this multiscale methodology to obtain highly pertinent correlations from the structure to the single molecule and the solution behavior and answer mechanistic questions on the multivalent interactions of oligomeric proteins.
Results and discussion
Assessing the oligomeric architecture of Tsa1 redox forms in solution
We first assessed the behavior of Tsa1 redox forms, i.e. the reduced Tsa1SH, disulfide oxidized Tsa1SS and sulfinylated hyperoxidized Tsa1SO2 (Fig. S1, ESI†) by high resolution AFM imaging in buffer at physiological pH. Tsa1 solution was deposited on atomically flat freshly cleaved mica and the surface was then imaged under liquid conditions to avoid any artefacts that could be induced by a drying step, using peak force tapping AFM imaging (Fig. 2A). As shown in Fig. 2B, the hyperoxidized species Tsa1SO2 forms a layer of donut-shaped decameric rings homogeneously oriented flat, almost entirely covering the mica surface, similar to previous observations.29,30 Imaging these structures at higher resolution allows the 5 dimers constituting the decamers to be precisely distinguished (Fig. 2B, C and 3C). Imaging the other Tsa1 redoxomers – the reduced Tsa1SH and the oxidized CP–CR disulfide Tsa1SS forms – produced similar images (Fig. 2D and E), suggesting that, even at a concentration as low as 100 nM, purified Tsa1 primarily exists in solution as a decamer, including in the disulfide state. However, crescent-shaped structures were also observed amongst decamers (red arrows in Fig. 2C–E) with a frequency of 2.8, 2.7 and 12.5% in Tsa1SO2, Tsa1SH and Tsa1SS images, respectively. This suggests that the dissociation of the decamer into an octamer or a hexamer can occur in solution or as a result of tip scanning and that the decamer structure is destabilized in the disulfide form, compared to the other redox forms, which is consistent with previous observations on Prxs from various origins.31–34 We then analyzed the Tsa1 redox forms by native mass spectrometry which provides a resolution allowing the observation of intermediate oligomers for human Prx3.35Fig. 2F shows that, under low acceleration voltage (Vc = 80 V) conditions (with minimum perturbation of non-covalent interactions), the reduced Tsa1 and hyperoxidized Tsa1 are decamers, while Tsa1SS partially dissociates into dimers and a small fraction of tetramers and hexamers, a tendency confirmed under slightly more energetic conditions (Vc = 120 V; Fig. S3, ESI†). This supports that the crescent-shaped images correspond to oligomers with intermediate stoichiometry (2 to 6 subunits), and that the AFM images provide a snapshot of the oligomeric states of Tsa1 in solution discarding the hypothesis that the dissociation is induced by AFM tip scanning. However, we cannot exclude that – as suggested by their oriented deposition – the interactions of the decamers with mica contribute to stabilization of the decamers, compensating the dissociation that might occur at such low concentrations (0.1 μM).33 | ||
Fig. 2 Evaluation of the oligomeric state of the Tsa1 redox forms in solution. (A) The hyperoxidized Tsa1SO2 decamer or the Tsa1–SS–Srx decameric complex is deposited on a flat mica surface to image their molecular assembly at high resolution using peak force tapping AFM imaging under liquid conditions. (B) AFM image showing that Tsa1SO2 adsorbed on the mica surface forms an almost complete monolayer of donut-shaped decamers. High resolution imaging of Tsa1SO2 clearly reveals the 5 dimers composing the decamer. (C–E) Tsa1SO2 (C), Tsa1SH (D) and Tsa1SS (E) structures are mostly donut-shaped assemblies, containing a significant fraction of incomplete rings composed of octamers or hexamers, as highlighted by the red arrows. (F) Electrospray ionization mass spectra under native conditions of Tsa1 redox forms showing the dissociation of the decamer (experimental/theoretical mass for each form) (●; 215![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 031 ± 47 Da/214905 Da for Tsa1SO2, 214592 ± 9 Da/214585 Da for Tsa1SH and 214791 ± 142 Da/214575 Da for Tsa1SS) into hexamers ( 031 ± 47 Da/214905 Da for Tsa1SO2, 214592 ± 9 Da/214585 Da for Tsa1SH and 214791 ± 142 Da/214575 Da for Tsa1SS) into hexamers ( ; 128749 ± 18 Da/128745 Da for Tsa1SS), tetramers ( ; 128749 ± 18 Da/128745 Da for Tsa1SS), tetramers ( ; 85840 ± 23 Da/85830 Da for Tsa1SS) and dimers ( ; 85840 ± 23 Da/85830 Da for Tsa1SS) and dimers ( ; 42915 ± 2 Da/42915 Da for Tsa1SS) in the gas phase at Vc = 80 V. ; 42915 ± 2 Da/42915 Da for Tsa1SS) in the gas phase at Vc = 80 V. | ||
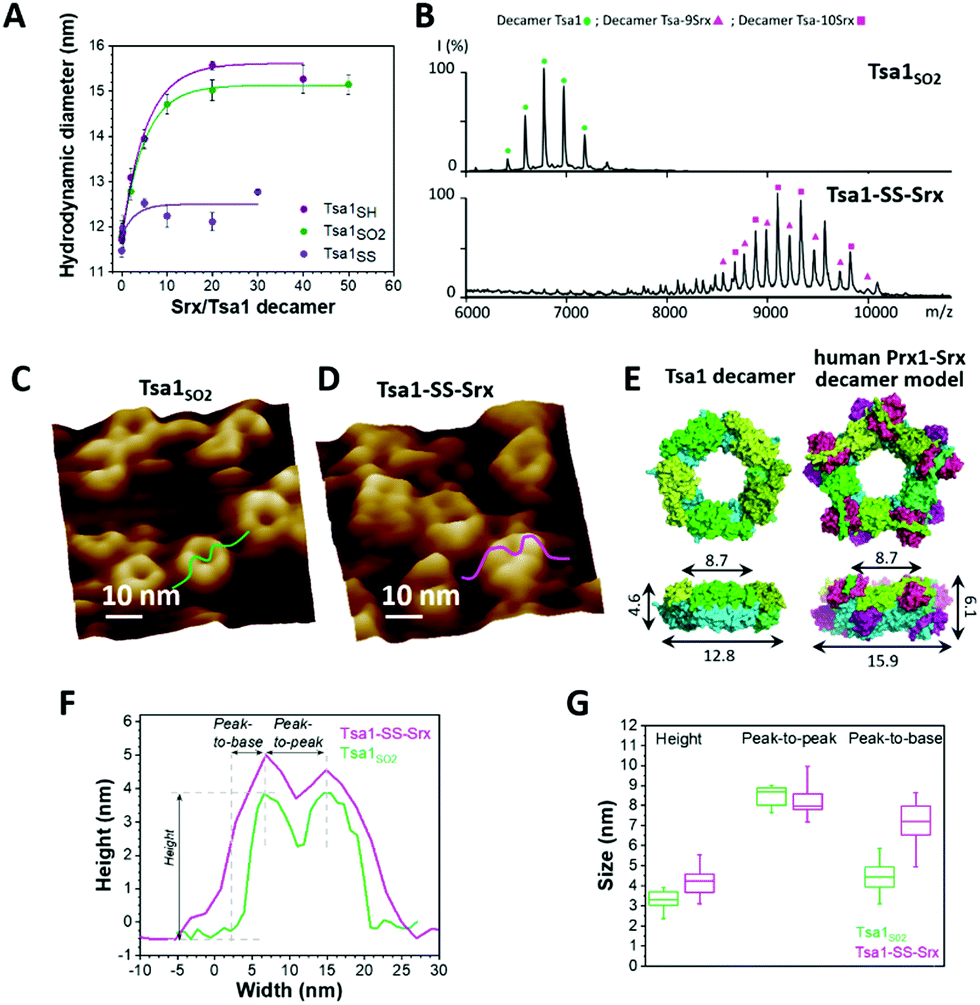 | ||
Fig. 3 Srx binding increases the width and height of the Tsa1 ring structure. (A) Hydrodynamic diameters of Tsa1SO2, Tsa1SH and Tsa1SS decamers measured by DLS as a function of the Srx:Tsa1 stoichiometry. The SrxC48S C84A C106V mutant was used with Tsa1SS to avoid disulfide bond exchange. Data are representative of n = 2 independent experiments using different protein samples. (B) Electrospray ionization mass spectra under native conditions of the Tsa1SO2 decamer ( ; 236587 ± 37 Da/236218 Da) and decameric Tsa1–SS–Srx complexes at Tsa1:Srx stoichiometries of 9 ( ; 236587 ± 37 Da/236218 Da) and decameric Tsa1–SS–Srx complexes at Tsa1:Srx stoichiometries of 9 ( ; 359819 ± 9 Da/358920 Da) and 10 ( ; 359819 ± 9 Da/358920 Da) and 10 ( ; 373393 ± 11 Da/372468 Da) in the gas phase at acceleration voltage Vc = 80 V (experimental/theoretical mass for each form). (C and D) High resolution AFM images of Tsa1SO2 (C) and the Tsa1–SS–Srx complex (D) with cross-sections used to determine decamer dimensions. (E) Left, top and side views of Tsa1SH (pdb code: 3sbc); right, top and side views of the human Prx1–SS–Srx decameric model in the surface mode, constructed by superimposition of the X-ray structure of five Prx1–SS–Srx dimers (pdb code: 2rii) (Fig. 1A) on the Tsa1SH structure. The height, width and peak-to-peak dimensions (defined in panel F) are indicated on the side views. Superimposition and measurements were performed using PyMol 2.2.2.62 (F) Typical cross-sections of the Tsa1SO2 decamer (green) and the Tsa1–SS–Srx complex (pink) extracted from AFM images in C and D. (G) Decamer dimensions shown by box charts. The bottom and top of the boxes represent the 25th and 75th percentiles; the whiskers, the 10th and 90th percentiles; and the horizontal bands, the median. Data were obtained from measurements on at least 25 donuts for Tsa1SO2 and 15 complexes for Tsa1–SS–Srx. Differences between Tsa1SO2 and Tsa1–SS–Srx were compared using two-sample t-tests. The peak-to-base difference was considered statistically significant (p <0.05). ; 373393 ± 11 Da/372468 Da) in the gas phase at acceleration voltage Vc = 80 V (experimental/theoretical mass for each form). (C and D) High resolution AFM images of Tsa1SO2 (C) and the Tsa1–SS–Srx complex (D) with cross-sections used to determine decamer dimensions. (E) Left, top and side views of Tsa1SH (pdb code: 3sbc); right, top and side views of the human Prx1–SS–Srx decameric model in the surface mode, constructed by superimposition of the X-ray structure of five Prx1–SS–Srx dimers (pdb code: 2rii) (Fig. 1A) on the Tsa1SH structure. The height, width and peak-to-peak dimensions (defined in panel F) are indicated on the side views. Superimposition and measurements were performed using PyMol 2.2.2.62 (F) Typical cross-sections of the Tsa1SO2 decamer (green) and the Tsa1–SS–Srx complex (pink) extracted from AFM images in C and D. (G) Decamer dimensions shown by box charts. The bottom and top of the boxes represent the 25th and 75th percentiles; the whiskers, the 10th and 90th percentiles; and the horizontal bands, the median. Data were obtained from measurements on at least 25 donuts for Tsa1SO2 and 15 complexes for Tsa1–SS–Srx. Differences between Tsa1SO2 and Tsa1–SS–Srx were compared using two-sample t-tests. The peak-to-base difference was considered statistically significant (p <0.05). | ||
The heights of the Tsa1SO2 decamers extracted from the AFM images (3.3 ± 0.5 nm, Fig. 3C, F, and G, green sections) were in agreement with those obtained from the dimensions of the Tsa1 decamer X-ray structure (4.6 nm, Fig. 3E, left), confirming the observation of a single layer of decamers. The subtle lower height value obtained from AFM measurements might correspond to a slight collapse of the structures when deposited on mica or a little indentation of the AFM tip while scanning the decamers. The vertical profiles of the rings were characterized by two summits, or peaks (Fig. 3F), allowing measurement of horizontal peak-to-peak dimensions of 8.5 ± 0.5 nm, perfectly consistent with the value of 8.7 nm deduced from the crystal structure (Fig. 3E, F, and G). No such direct comparison could be done on the total width of the domains, as the value from the AFM images can be distorted by the tip convolution. Indeed, the total width extracted from the AFM images (equal to the sum of the peak-to-peak distance plus twice the width of the peak-to-base, i.e. 17.3 nm for Tsa1SO2; Fig. 3F) is larger than the value of 12.8 nm from the X-ray structure (Fig. 3E, left). The overall dimensions of the reduced and disulfide forms of Tsa1 deposited on the surface were in the same range as Tsa1SO2 (Fig. S4, ESI†).
Altogether, the original combination of high-resolution AFM imaging and native mass spectrometry allows the sensing of heterogeneities between subunits within the toroidal structures, which prompted us to follow the same approach to probe the decamers in interaction with its partner Srx.
The Tsa1 decamer in complex with Srx does not dissociate in solution
The Prx/Srx complex formation involves unfolding of the Prx C-terminal region that also participates in stabilizing the decamer A interface (Fig. 1A). Whether the binding of Srx to Prx destabilizes or induces the dissociation of the decamer is presently unknown in solution. We thus studied Tsa1 structures in interaction with Srx using a similar experimental strategy as mentioned above. As a preliminary to the observation of the Tsa1/Srx complex, we first addressed the question whether the Tsa1 decamer could be non-covalently saturated by 10 Srx molecules using dynamic light scattering (DLS) (Fig. 3A). Titration of Tsa1SO2 by Srx resulted in an increase of the hydrodynamic diameter of Tsa1 from 11.8 nm to a plateau of ∼15.2 nm obtained for ratios above 10 Srx to one Tsa1 decamer, suggesting that Tsa1 can be saturated by 10 Srx molecules in solution. A similar result was obtained with Tsa1SH. The analogous experiment using Tsa1SS in which the C-terminal tail is locked by a disulfide bond between Cys CP and CR with SrxC48S C84A C106V (used here to avoid any disulfide bond exchange) resulted in a variation of the hydrodynamic diameter between 12 and 13 nm, indicating that Tsa1SS did not form specific non-covalent complexes, as expected.To support that the Tsa1/Srx complex exists as a decamer, we performed native mass spectrometry on the Tsa1 decamer in complex with Srx molecules (Fig. 3B), stabilized by a disulfide bond between the catalytic Cys residues of each Srx and Tsa1 Cys CP, which mimics the catalytic thiosulfinate intermediate of the Srx catalytic mechanism.36,37 This strategy was previously proved to be successful to resolve the X-ray structure of the human Prx1/Srx complex (Fig. 1).18,19 The specificity and completeness of the reaction between Tsa1 CP and Srx were ensured by using mutants that only retain the catalytic Cys residues, CP in Tsa1 and C84 in Srx. Reaction of Tsa1C171A CP pre-activated by 2,2′-dipyridyl disulfide (2PDS) with SrxC48A C106V yielded the Tsa1C171A–SS–SrxC48A C106V complex, referred to as Tsa1–SS–Srx, as investigated by non-reducing SDS-PAGE (Fig. S5 and Table S1, ESI†). Native mass spectrometry analysis showed that the Tsa1–SS–Srx solution is a mixture of decameric Tsa1 complexes with 9 and 10 Srx bound (Fig. 3B), in accordance with the DLS titration (Fig. 3A).
Next, to observe the morphology of the Tsa1 decamer in interaction with Srx, we imaged by AFM a Tsa1–SS–Srx solution deposited on the mica surface (Fig. 3D). Statistical analysis of the AFM-imaged decamer dimensions reveals that binding of Srx induces no significant changes in the peak-to-peak distances (8.2 ± 0.7 nm), but increases the height and peak-to-base dimensions of the rings by 0.9 nm (from 3.3 to 4.2 nm) and 2.7 nm (from 4.4 to 7.1 nm), respectively (Fig. 3F and G). Although the tip convolution might increase the peak-to-base length in absolute value, the relative difference measured before and after addition of Srx remains significant (Fig. 3F and G). These values were then compared with the dimensions deduced from a model of the decameric complex constructed by superposition of five duplicates of the human disulfide (Prx1–SS–Srx)2 complex18 and the Tsa1SH decamer X-ray structures.38 This model suggested an increase in height of 1.5 nm and peak-to-base dimension of 1.6 nm upon Srx binding (Fig. 3E, right), in good agreement with the tendency obtained by AFM (Fig. 3G). This supports that the mechanism of interaction between Srx and Tsa1 can be studied in solution at the level of the decamer.
C-terminus flexibility affects Tsa1/Srx two-step binding
The importance of the Prx C-terminal tail in the backside interface with Srx shown previously18 prompted us to investigate the role of this region in the dynamics of the interaction of the Tsa1 decamer with Srx. We prepared the P182L and Δ190–196 Tsa1 mutants (Table S1, ESI†), which exist as decamers (with hydrodynamic radii of 12.6 ± 0.5 and 13.7 ± 0.5 nm, respectively, similar to those of wild-type Tsa1). These mutants adopt primarily the LU conformation, as shown by their near-UV circular dichroism spectra similar to those of Tsa1SS which is locked in the LU conformation,7 suggesting increased C-terminus flexibility (Fig. S6, ESI†). For this reason these mutants are not susceptible to hyperoxidation and were used in their reduced form.7,39–41 The binding properties with Srx were thus compared in solution with reduced wild-type Tsa1SH. Srx bound to both P182L and Δ190–196 Tsa1SH with increased affinity, as shown by KD values of 5.9 ± 1.2 and 5.0 ± 1.7 μM, respectively, compared to 10.8 ± 3.0 μM measured for the wild-type by fluorescence anisotropy titration (Fig. 4A). To correlate these properties with the dynamics of interactions in solution, we measured the dissociation kinetics of the Tsa1/Srx complex by monitoring Tsa1 Trp intrinsic fluorescence upon rapid dilution of the Tsa1/Srx mixture in solution using stopped flow apparatus. Srx has no Trp residues while Tsa1 contains 3 Trp residues, one located close to the active site (W83), another in helix α4 preceding the C-terminal tail (W161), and the last in the C-terminal tail (W173), which are likely to be sensitive to the changes in the molecular environment induced by Srx binding (Fig. 1A and Fig. S2, ESI†). Indeed, using this probe to monitor dilution-induced dissociation, we observed that wild-type Tsa1SH dissociates from Srx by following biphasic kinetics. The data set was analyzed globally according to a two-step model, consisting of a first order step reflecting a conformational event occurring within the complex, modelled by an equilibrium between two conformer Tsa1·Srx and Tsa1·Srx* forms, and a second order step reflecting dissociation (Fig. S7A and B, ESI†). Using the same scheme to analyze the mirror association experiment performed under similar conditions by rapid mixing of Tsa1 in Srx solutions gave satisfactory fits and overall consistent rate constant values (Fig. S7C and Table S2, ESI†), confirming that the Trp fluorescence probe is suitable for the study of Tsa1/Srx interactions. These results suggested that wild-type Tsa1SH dissociates from Srx with a rate constant k−1 of 18.8 ± 2.8 s−1, which was reduced to 11.8 ± 3.0 s−1 and 7.5 ± 2.2 s−1 for the P182L and Δ190–196 Tsa1 mutants respectively (Fig. 4B and Table S2, ESI†). The same trend was observed from the association experiment, supporting that a slower bimolecular dissociation kinetics likely afforded by the more flexible C-terminal Tsa1 region contributes to the higher affinity of Srx for these mutants.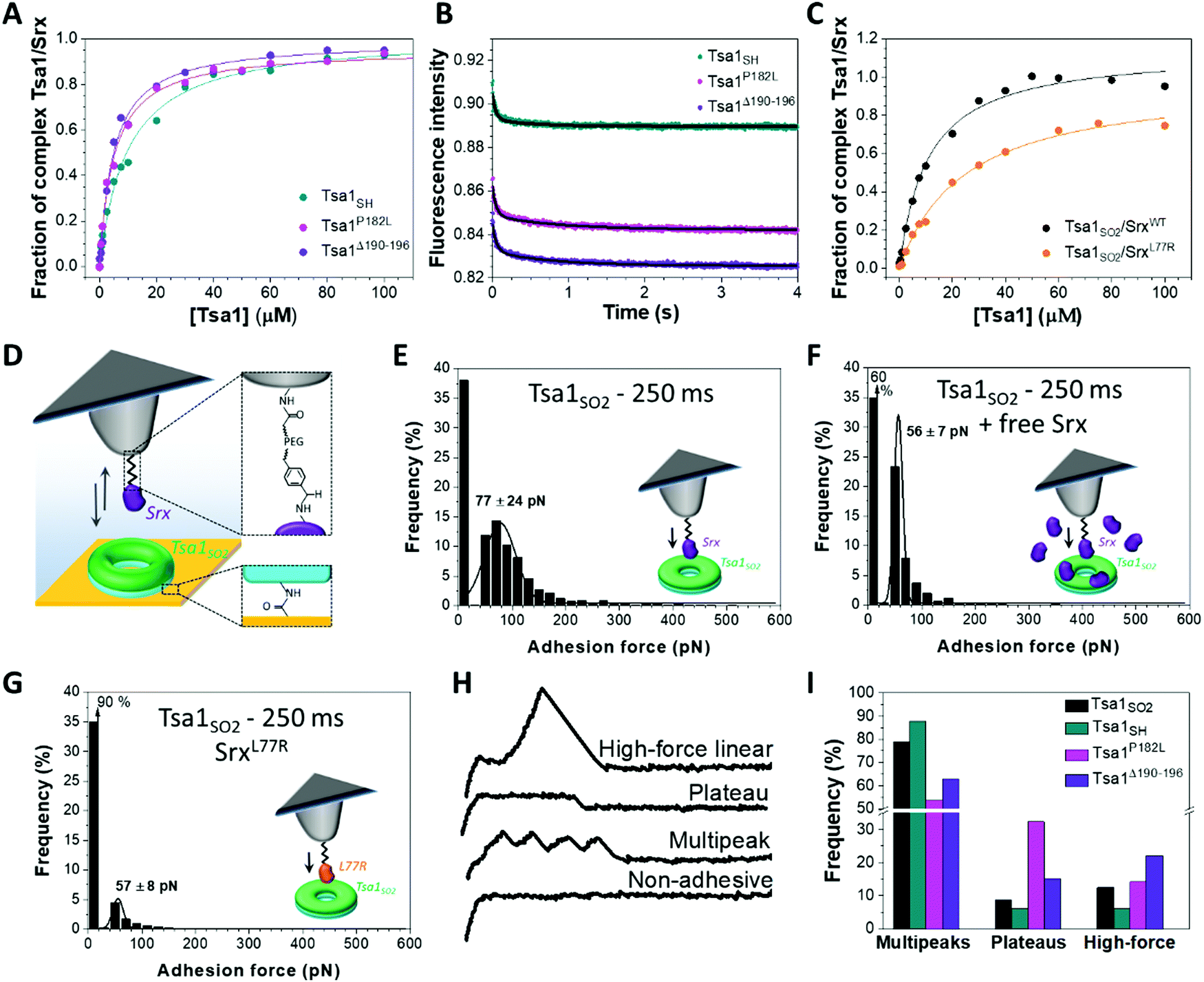 | ||
| Fig. 4 Impact of the Tsa1 C-terminal region flexibility on the Srx interaction mechanism. Fluorescence anisotropy titration of (A) Alexa Fluor labelled Srx by wild-type, P182L and Δ190–196 mutants Tsa1SH and (C) wild-type and L77R Srx N-terminally labelled with Alexa Fluor by Tsa1SO2. Data that are representative of n = 2 or 3 independent experiments using different protein batches are fitted using a single site model equation (solid lines). (B) Representative dilution-induced dissociation kinetics of the non-covalent Tsa1SH/Srx complexes in solution for wild-type, P182L and Δ190–196 Tsa1 monitored by intrinsic Trp fluorescence using stopped flow apparatus. The Tsa1 (subunits) and Srx concentrations are 2.5 and 20 μM, respectively. The curves are fitted as described in the ESI.† Data are representative of n = 3 independent experiments using different protein samples. (D) For AFM single-molecule force spectroscopy, Srx is covalently bound to the tip using an acetal–PEG linker. The tip is approached and retracted from Tsa1 covalently attached to the gold surface. (E) Adhesion force histograms of Srx binding to Tsa1SO2 with a 250 ms contact time. The specificity of the Srx/Tsa1SO2 interaction confirmed by significant reduction of the adhesion frequency in the presence of free competing Srx (F) or using the L77R Srx mutant (G). Each histogram is plotted from 3072 force curves obtained from 3 independent experiments with different functionalized tips. Black lines correspond to Gaussian fits and the values are mean ± standard deviation. Representative force–distance signatures (H) with their frequency (I) for wild-type Tsa1SH, Tsa1SO2, P182L and Δ190–196 mutants, obtained from 3072 force curves from 3 independent measurements for each form of Tsa1 and calculated relative to the total of the assigned curves, excluding the non-adhesive and hybrid profiles. | ||
To understand how these binding features translate at the level of the decamer, we addressed Srx interactions on Tsa1 single decamers by AFM in the force spectroscopy mode. Purified Tsa1 was bound to the gold surface modified with COOH-terminated thiols using NHS-EDC chemistry whereas Srx was covalently attached on AFM tips with an ∼6 nm long PEG–benzaldehyde linker (Fig. 4D). The grafting procedure to the tip was carried out under conditions that favor deprotonation of N-terminal amine (pH 8.5), thus leading to an oriented binding of the protein to the tip.18,42 Adhesion was then assessed using the Srx substrate, the hyperoxidized Tsa1SO2. Fig. 4E displays the adhesion force histograms extracted from 3072 force–distance curves each, obtained using 3 independent tips on different 1 × 1 μm2 areas of the Tsa1SO2 surfaces with a retraction speed of 1000 nm s−1. Only the last adhesion peak, corresponding to the force at rupture, was considered for the force histogram (Fig. S8A, ESI†). When Srx and Tsa1SO2 were set to interact for 250 ms, a substantial proportion of the force curves (63%, Fig. 4E) showed adhesive events between the two enzymes, with a mean adhesion force (which is displayed at the maximum of the Gaussian fit and corresponds to the force that is needed to break the interaction when Srx binds to Tsa1) of 77 ± 24 pN (Fig. 4E). Reducing the contact time between the two enzymes from 250 to 0 ms decreases the adhesion frequency from 63% (Fig. 4E) to 14% (Fig. S8B, ESI†) without changing the mean adhesion forces, at around 55 ± 8 pN.
At 250 ms contact time, a major reduction of the adhesion frequency was observed upon addition of free Srx in the AFM liquid set-up although the general shape of the histogram was not drastically modified (Fig. 4F). This suggested that a competition occurs between free and grafted Srx for binding to a subunit in a decamer and that the measured adhesion events in Fig. 4E correspond to the interactions between Srx on the tip and the Tsa1SO2 proteins bound to the surface. To substantiate this result, we used the SrxL77R mutant designed to impact the Srx backside interface with Tsa1 (Fig. 1A). Mutation of the corresponding residue in human Srx (Y92R) (Fig. S2, ESI†) was shown to severely alter the interaction with hyperoxidized human Prx1.18 Indeed, the dissociation constant of S. cerevisiae SrxL77R for Tsa1SO2 measured by fluorescence anisotropy titration increased from 12.6 ± 2.5 to 27.1 ± 1.3 μM (Fig. 4C). The AFM adhesion frequency for Tsa1SO2 at 250 ms contact time was reduced from 63% (Fig. 4E) to 10% for the L77R variant (Fig. 4G), whereas the maximum adhesion force was not affected. Furthermore, the adhesion frequencies of Srx-coated AFM tips to surfaces decorated with P182L and Δ190–196 Tsa1SH mutants were significantly higher as compared to those with wild-type Tsa1SH (Fig. S9, ESI†). This suggests that the alteration of the native Tsa1/Srx interaction on the backside interface is connected to the binding propensity, which translated into the adhesion probability as measured by AFM in the force spectroscopy mode. A very low adhesion frequency of 14% was observed under similar conditions for Tsa1SS (Fig. S10A, ESI†), in accordance with the DLS titration (Fig. 3A). Given the good correlation between the anisotropy titration and AFM results, we propose that the adhesion events measured by force spectroscopy in Fig. 4E reflect Srx/Tsa1SO2 interactions relevant to those occurring between the free proteins in solution.
To gain insight into the interaction mechanisms, we further analyzed the multiple force–distance curves recorded between Srx and Tsa1SO2. Distinct remarkable signatures were identified and classified into 5 force curve families: the non-adhesive curves, the multipeaks, the force plateaus, the high linear forces (see the representative force curves in Fig. 4H), and a last category of forces including undefined or hybrids of the profiles described above (Fig. S8C, ESI†). The proportion of the 3 well-defined profiles (i.e. the multipeaks, the force plateaus and the high linear forces) varies for the Tsa1 redox and mutant forms (Fig. 4I). The origin of these events is discussed below.
Peculiar signatures reveal two modes of force-induced Srx dissociation from the Tsa1 decamer
A first category of signatures consists of force profiles containing well-defined multipeaks, most frequently 1 to 4 peaks, and rare events with 5 peaks, of an almost constant adhesion force (<100 pN; Fig. 5A) and spaced by 15.7 ± 6.7 nm (Fig. S8D, ESI†). The chemistry that is used here to covalently attach Srx to the tip43 and the nominal value of the tip apex radius of 10 nm up to 40 nm (as given by the provider Bruker), although statistically resulting in the grafting of one single molecule at the apex of the tip,43,44 can lead to attachment of up to 13 Srx molecules on the tip (considering a half sphere geometry).43 Thus, we suggest that these events reflect the interaction of several Srx molecules decorating the tip with the upper ring of the decameric structure, in accordance with AFM imaging which showed that decamers lie flat on the mica surface (Fig. 2B). In the approach phase, the AFM tip might indent within the ring-shaped decamer structure whose dimensions are in the range of the tip diameter, also enabling other Srx molecules grafted on the side of the tip to interact with accessible Tsa1 subunits (Fig. 5B, case 1), yet we cannot exclude the interaction with neighboring decamers on the surface (Fig. 5B, case 2). The observation of reproducible multipeak patterns upon hundreds of force measurements on the adhesion maps (1024 force–distance curves recorded with each functionalized tip) suggests that the native folding of the tip-grafted Srx molecule is preserved and that the protein either does not unfold or unfolds and refolds on repeated association/dissociation events. Additionally, the force–distance signatures could not be adequately fitted using classical protein unfolding models (e.g. Worm-like-chain model45), supporting that the force-induced Tsa1/Srx binding and dissociation do not result in the unfolding of both enzymes upon pulling. Thus, owing to the N-terminal oriented Srx grafting on the tip, the observed force-induced dissociation could proceed via traction of the Tsa1 C-terminal tail by Srx bound at the backside interface (Fig. 5C) (>16 residues in length for Tsa1, ∼6 nm, Fig. S2, ESI†), which adds to the extension of the Srx N-terminal segment preceding those involved in the backside interface (∼10 residues, 4 nm, Fig. S2, ESI†) and the 6 nm linker, consistent with the ∼16 nm spacing between rupture events.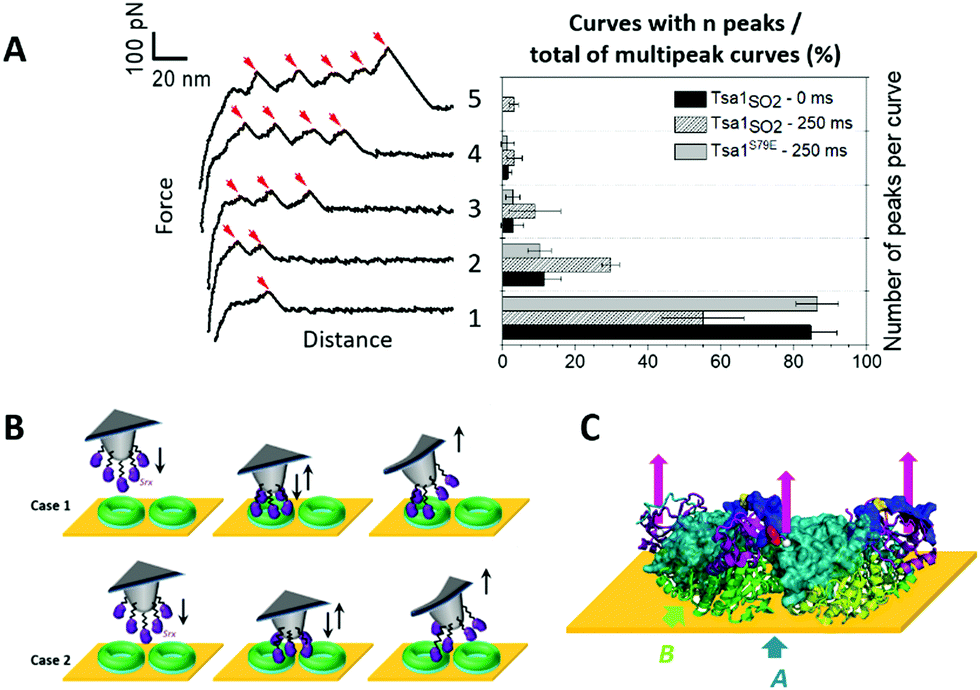 | ||
| Fig. 5 Srx interaction with the Tsa1 decamer reveals multipeak force–distance signatures. (A) Representative force–distance curves (left) obtained when recording force spectroscopy measurements between Srx and Tsa1SO2, revealing well-defined multipeaks ranging from 1 to 5 peaks. The corresponding frequencies (right) of the multipeak categories over the total number of multipeaks for Tsa1SO2 at 0 and 250 ms contact times, and for the dimeric Tsa1S79E mutant at a 250 ms contact time, obtained from 3072 force measurements for each contact time, from 3 independent experiments with different functionalized tips. The error bars correspond to the standard deviations between tips. (B) Proposed schematics of the interaction mechanism generating the multipeak signatures, where five Srx molecules bound to the AFM tip interact with the upper ring of one (case 1) or two (case 2) Tsa1 ring-shaped structures lying on the gold surface. (C) Illustration in cartoon mode of a composite decamer, in which the upper ring corresponds to human Prx1–SS–Srx subunits from the model shown in Fig. 3E, and the lower ring corresponds to the Tsa1SH subunits covalently bound to the surface. The human Prx1 subunit is shown in the surface mode. The colour code is the same as in Fig. 1A. Only the 3 dimers on the front are shown for clarity. The location of the last residue of the Srx N-terminus defined in the crystal structure is shown as a white ball and the arrows represent the traction of the AFM tip imposed by Srx grafted to the tip on the Prx C-terminal tail. | ||
The increasing rupture distances would then reflect successive dissociation of Srx molecules from the Prx C-terminus subunits (Fig. 5C). Indeed, the relatively high standard deviation (15.7 ± 6.7 nm) (Fig. S8D, ESI†) of the interpeak distance is consistent with Srx grafted at different heights on the tip and with the intrinsic rugosity of gold surfaces used for force spectroscopy measurements that may then slightly tilt the orientation of the decamers. Accordingly, increasing the contact time from 0 to 250 ms enhances the frequency of signatures with more than 1 peak (Fig. 5A), suggesting an increased probability of formation of more than one interaction with the accessible Tsa1 subunits of a single decamer before being broken upon tip retraction. To further support this interpretation, we studied the interaction between Srx and a dimeric Tsa1S79E mutant whose A interface is destabilized by introduction of two facing Glu residues,18,38 confirmed by its DLS hydrodynamic diameter of 6.3 nm (vs. 11.8 nm for the Tsa1SO2 decamer) and by native mass spectrometry (Fig. S10C, ESI†). This mutant showed a lower adhesion of 31% compared to Tsa1SO2 (Fig. S10B, ESI†) allowing analysis of the multiple force–distance curve profiles, which revealed mostly one- and two-peak signatures (Fig. 5A). This behavior is consistent with one or two Srx molecules grafted on the tip interacting with a dimer. Very rarely three- and four-peak events were observed, possibly explained by the interaction with neighboring dimers.
A second category of force–distance profiles was observed, consisting of well-defined plateaus occurring at a near constant force of ∼50 pN along distances ranging from 20 to 100 nm with a majority of curves extending at 40 nm (Fig. 6A and B). Strikingly, whereas the frequency of plateaus was low and almost similar for Tsa1SO2 and TsaSH (6.2 and 8.7%, respectively), they reached up to 15 and 32% for the Δ190–196 and P182L Tsa1 mutants, respectively, whose C-terminus flexibility is enhanced (Fig. 4I). If these peculiar signatures would reflect some nonspecific interactions of no biological origin (such as artifactual hydrophobic or electrostatic interactions), one would expect that they would be encountered with the same frequency for all Tsa1 forms. However, because the plateau signatures were correlated with the binding properties to Srx in solution, we speculate that the enhanced adhesion between Srx and the upper half-decamer (formed by the 5 Tsa1 monomers at the interface with the buffer solution; Fig. 6C, green subunits) could promote the concerted detachment from the 5 monomers covalently grafted on the gold surface (Fig. 6C, cyan subunits). However, the affinity of Srx with individual Tsa1 subunits is moderate (KD in the 10 μM range), and the β-strand interaction forming the B interface is likely tight. Thus, the synergistic traction of several Srx molecules on the C-terminal tails of Tsa1 subunits of one decamer, favored by mutations providing stronger interactions, would make the disruption of the B interface possible (and consequently the A interface) by the rupture of the β-strand H-bonds under constant force following an unzipping mechanism, an interpretation supported by the following arguments. Such plateaus are evocative of the signatures measured for the mechanical unzipping in other systems such as β-sheet interactions in pathological and functional amyloids.46,47 Although amyloid bonds are responsible for the very strong attachment of e.g. yeast to the epithelial layer, pulling on amyloid proteins with an AFM tip perpendicularly to the fiber axis was reported to allow mechanical unzipping of β-sheets at a low constant force of around 35 pN,47 similar to the average strength of the plateaus observed here around 50 pN (Fig. 6A). Because the force required to rupture β-sheets should be strongly dependent on the pulling direction,48 depending on the positioning of the AFM tip relative to the Tsa1 decamer, the upper half decamer could be lifted up at a constant low force as – schematically – would be a yogurt lid when peeled from the container. Accordingly, the measured average length of the plateaus is consistent with an unzipping mechanism occurring along the perimeter of a decamer of 6.4 nm radius (perimeter = 2π × 6.4 nm = 40.2 nm; Fig. 6C). In conclusion, the observation of such force-induced plateaus further highlights the critical role of the C-terminal tail in the interaction of Srx with the Tsa1 decamer.
 | ||
| Fig. 6 Srx interaction with the Tsa1 decamer shows constant force plateau force–distance signatures. (A) Representative force–distance curves of the linear plateaus. (B) Histogram of the frequency of the plateau length, measured from 128 force curves for wild-type Tsa1SO2. (C) Schematics of the suggested mechanism, where the synergistic traction of multiple Srx molecules bound to the upper half-decamer of the donut would induce unzipping from the bottom half-decamer covalently bound to the gold surface. | ||
Disulfide bond formation between Srx and Tsa1SO2 revealed by AFM in the force spectroscopy mode
Interestingly, another distinct, infrequent and peculiar force–distance profile was observed in force–distance experiments, corresponding to high linear force (HF) peaks ranging from 100 pN to >1 nN (Fig. 4I and 7A, B). With Tsa1SO2, the force distribution showed a maximum at 450 pN extending up to 1100 pN (Fig. 7B). Increasing the contact time between the tip and the surface from 250 ms to 1 s expanded the distribution to higher forces, with a significant number of occurrences above 1 nN up to 1.3 nN (Fig. 7B). Tsa1 and Srx are Cys-containing redox sensitive proteins prone to nonspecific oxidation by reactive oxygen species. We thus hypothesized that the Srx Cys residues involved in catalysis (C84 and C48)49 could oxidize to sulfenic acid or disulfide due to the reactive oxygen species present in the solution, and react with Tsa1SO2 residual Cys (CR) to form an intermolecular disulfide, a process favored by a longer contact delay to allow conformational orientation suitable for the Cys residues to react. Accordingly, the P182L and Δ190–196 Tsa1 mutants whose C-terminal region is likely more flexible show increased high force signature frequency (Fig. 4I).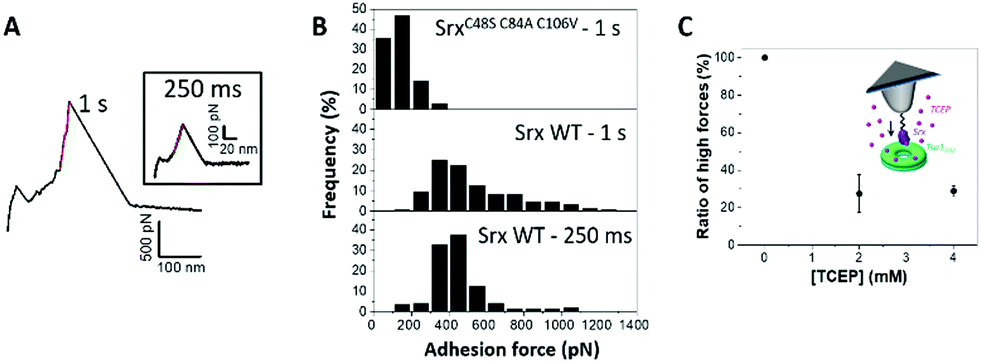 | ||
| Fig. 7 Srx interaction with the Tsa1 decamer reveals a high linear force (HF) signature consistent with disulfide bond formation. (A) Representative high linear forces obtained when Srx interacts with Tsa1SO2 with the contact time set to 1 s and 250 ms (a, inset). (B) Histograms of the adhesion force frequency of high linear forces for 250 ms (wild-type Srx) and 1 s contact times (SrxC48S C84A C106V and wild-type Srx). Histograms have been built from 168 HF curves for 250 ms and 160 HF curves for a 1s contact time. (C) Impact of the addition of the disulfide bond reducer TCEP in the AFM liquid cell on the frequency of appearance of HF signatures. Each point in C corresponds to the fraction of HF peaks over 2048 force curves obtained from 2 independent experiments with different functionalized tips. | ||
We showed that, for 1 s surface delay experiments, increasing the concentration of TCEP (tris(2-carboxyethyl)phosphine) – a reducing agent breaking disulfide bonds – from 0 to 4 mM decreases the number of HF peaks by 70% (Fig. 7C), thus supporting the HF peaks corresponding to disulfide bond rupture. Based on the literature,50,51 the formation and breaking of covalent bonds between both proteins would be reflected in the highest values of forces (above 1 nN). Although the origin of the linear forces ranging from 400 to 1000 pN cannot be precisely determined, we believe that they are associated with the interactions between the Cys residues of both Srx and Tsa1, as demonstrated by performing a similar experiment using the SrxC48S C84A C106V mutant in which all Cys residues are substituted. The global adhesion frequency was similar to that of wild-type Srx but it did not show any HF peaks above 400 pN, even at 1 s contact time (Fig. 7B). However, these controls do not allow us to discard the fact that the 400–1000 pN forces could also be due to the Srx–cysteine binding to the gold surface as this would result in the same range of forces.52
This observation provides significant opportunity to expand the use of single-molecule force-based techniques in thiol redox enzymology. AFM-based approaches have been used so far mostly to study the rupture of disulfide bonds by disulfide exchange reactions catalyzed by thioredoxin or related enzymes.53–55 Our results thus open the possibility to study the formation of covalent bonds involved in the enzyme-catalyzed reaction by AFM-based techniques.
Conclusions
Understanding how decameric Prxs interact with their partner proteins is essential to understand the regulation of their multiple functions. Indeed, in vivo full hyperoxidation of Prx decamers has been shown,56,57 which implies that all subunits of the decamer are substrates of Srx. Given the high concentration of cytosolic Prx from the Prx1 family (∼20 μM for Tsa158) and the relatively lower concentration of Srx (∼75-fold less than that of Tsa158), interactions of multiple Srx molecules on a single decamer could provide a biological advantage to regenerate a decamer to the fully reduced form vs. heterogeneous complexes of reduced and hyperoxidized subunits, which would exhibit e.g. lower peroxidase activity. This could be favored by Srx binding cooperatively to the decamer. Although this idea is speculative, the present study gives insight into the formation of these complexes at the single decamer level, which is necessary to further address such questions.Tsa1 decamer interactions with Srx have been resolved in solution at the decameric level by AFM imaging of a Tsa1(Srx)10 complex stabilized covalently, using native mass spectrometry and DLS, showing that the interaction of Srx does not induce dissociation of the decamer. Force spectroscopy assessment of the interaction between the surface-bound Tsa1 decamer and multiple tip-grafted Srx molecules appeared to be relevant to the interaction between free molecules in solution and allowed us to resolve the interaction of up to five Srx with Tsa1 subunits. Combining with the investigation of the dynamics of Tsa1/Srx interactions using rapid kinetics revealed a two-step binding mechanism and showed the critical role of the Prx C-terminal tail in the binding mechanism, in accordance with the sequential or concerted dissociation of tip-grafted Srx from the decamer shown by peculiar AFM force–distance signatures.
Overall, these highly pertinent correlations between affinity, kinetics, and single molecule data strongly support this multiscale methodology as a proficient method to further understand how protein supramolecular assembly regulates multivalent interactions with partners, such as the binding mechanisms of the interaction of Srx and newly discovered substrates.59 Finally, the formation of disulfide bonds could also be observed from specific rupture signatures, which should open avenues for the study of the mechanisms allowing Prxs to act as redox relays,60 and development of AFM as a new platform in thiol redox enzymology.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. A. B., H. M., S. C. and S. R. C. designed the research; A. B., F. C., H. M., J. H., H. L. C., and A. K. performed the research; A. B., F. C., H. M. and S. R. C. analyzed the data. A. B. and S. R. C. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge J. Charbonnel for efficient technical help. This work was supported by the ANR grant ANR-17-CE11-0034-02 to SRC, Ligue contre le Cancer to SRC, the ANR grant ANR-20-CE34-0005-01 to AB, the Université de Lorraine, Université de Strasbourg and CNRS. The authors acknowledge support of the group by the “Impact Biomolecules” project of the “Lorraine Université d’Excellence” (Investissements d’avenir-ANR). This work was granted access to the Biophysics and structural biology core facility of UMS2008 IBSLor CNRS-UL-INSERM and the French Proteomic Infrastructure ProFI (ANR-10-INBS-08-03 grant). This work was partly carried out in the Pôle de compétences Physico-Chimie de l’Environnement, LIEC laboratory UMR 7360 CNRS – Université de Lorraine.Notes and references
- A. Perkins, K. J. Nelson, D. Parsonage, L. B. Poole and P. A. Karplus, Trends Biochem. Sci., 2015, 40, 435–445 CrossRef CAS PubMed.
- S. Fourquet, M. E. Huang, B. D’Autreaux and M. B. Toledano, Antioxid. Redox Signaling, 2008, 10, 1565–1576 CrossRef CAS PubMed.
- S. G. Rhee, H. A. Woo and D. Kang, Antioxid. Redox Signaling, 2017, 28, 537–557 CrossRef PubMed.
- D. E. Fomenko, A. Koc, N. Agisheva, M. Jacobsen, A. Kaya, M. Malinouski, J. C. Rutherford, K.-L. Siu, D.-Y. Jin, D. R. Winge and V. N. Gladyshev, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2729–2734 CrossRef CAS PubMed.
- Z. A. Wood, L. B. Poole, R. R. Hantgan and P. A. Karplus, Biochemistry, 2002, 41, 5493–5504 CrossRef CAS PubMed.
- Z. A. Wood, L. B. Poole and P. A. Karplus, Science, 2003, 300, 650–653 CrossRef CAS PubMed.
- A. Kriznik, M. Libiad, H. Le Cordier, S. Boukhenouna, M. B. Toledano and S. Rahuel-Clermont, ACS Catal., 2020, 10, 3326–3339 CrossRef CAS PubMed.
- A. Perkins, K. J. Nelson, J. R. Williams, D. Parsonage, L. B. Poole and P. A. Karplus, Biochemistry, 2013, 52, 8708–8721 CrossRef CAS PubMed.
- I. S. Kil, S. K. Lee, K. W. Ryu, H. A. Woo, M.-C. Hu, S. H. Bae and S. G. Rhee, Mol. Cell, 2012, 46, 584–594 CrossRef CAS PubMed.
- A. M. Day, J. D. Brown, S. R. Taylor, J. D. Rand, B. A. Morgan and E. A. Veal, Mol. Cell, 2012, 45, 398–408 CrossRef CAS PubMed.
- B. Turner-Ivey, Y. Manevich, J. Schulte, E. Kistner-Griffin, A. Jezierska-Drutel, Y. Liu and C. A. Neumann, Oncogene, 2013, 32, 5302–5314 CrossRef CAS PubMed.
- S. Hanzén, K. Vielfort, J. Yang, F. Roger, V. Andersson, S. Zamarbide-Forés, R. Andersson, L. Malm, G. Palais, B. Biteau, B. Liu, M. B. Toledano, M. Molin and T. Nyström, Cell, 2016, 166, 140–151 CrossRef PubMed.
- H. H. Jang, K. O. Lee, Y. H. Chi, B. G. Jung, S. K. Park, J. H. Park, J. R. Lee, S. S. Lee, J. C. Moon, J. W. Yun, Y. O. Choi, W. Y. Kim, J. S. Kang, G. W. Cheong, D. J. Yun, S. G. Rhee, M. J. Cho and S. Y. Lee, Cell, 2004, 117, 625–635 CrossRef CAS PubMed.
- F. Teixeira, E. Tse, H. Castro, K. A. T. Makepeace, B. A. Meinen, C. H. Borchers, L. B. Poole, J. C. Bardwell, A. M. Tomás, D. R. Southworth and U. Jakob, Nat. Commun., 2019, 10, 659 CrossRef CAS PubMed.
- H. Jiang, L. Wu, J. Chen, M. Mishra, H. A. Chawsheen, H. Zhu and Q. Wei, Mol. Cancer Res., 2015, 13, 1554–1566 CrossRef CAS PubMed.
- Q. Wei, H. Jiang, Z. Xiao, A. Baker, M. R. Young, T. D. Veenstra and N. H. Colburn, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7004–7009 CrossRef CAS PubMed.
- H. Kim, G.-R. Lee, J. Kim, J. Y. Baek, Y.-J. Jo, S.-E. Hong, S. H. Kim, J. Lee, H. I. Lee, S.-K. Park, H. M. Kim, H. J. Lee, T.-S. Chang, S. G. Rhee, J.-S. Lee and W. Jeong, Free Radicals Biol. Med., 2016, 91, 264–274 CrossRef CAS PubMed.
- T. J. Jönsson, L. C. Johnson and W. T. Lowther, Nature, 2008, 451, 98–101 CrossRef PubMed.
- T. E. Forshaw, J. A. Reisz, K. J. Nelson, R. Gumpena, J. R. Lawson, T. J. Jönsson, H. Wu, J. E. Clodfelter, L. C. Johnson, C. M. Furdui and W. T. Lowther, Antioxidants, 2021, 10, 946 CrossRef CAS PubMed.
- Y. F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber and D. J. Müller, Nat. Nanotechnol., 2017, 12, 295–307 CrossRef PubMed.
- I. Munguira, I. Casuso, H. Takahashi, F. Rico, A. Miyagi, M. Chami and S. Scheuring, ACS Nano, 2016, 10, 2584–2590 CrossRef CAS PubMed.
- P. E. Milhiet, V. Vié, M.-C. Giocondi and C. L. Grimellec, Single Mol., 2001, 2, 109–112 CrossRef CAS.
- D. A. Heesterbeek, B. W. Bardoel, E. S. Parsons, I. Bennett, M. Ruyken, D. J. Doorduijn, R. D. Gorham Jr., E. T. Berends, A. L. Pyne, B. W. Hoogenboom and S. H. Rooijakkers, EMBO J., 2019, 38, e99852 CrossRef PubMed.
- J. Strasser, R. N. de Jong, F. J. Beurskens, G. Wang, A. J. R. Heck, J. Schuurman, P. W. H. I. Parren, P. Hinterdorfer and J. Preiner, Nano Lett., 2019, 19, 4787–4796 CrossRef CAS PubMed.
- B. Knoops, S. Becker, M. A. Poncin, J. Glibert, S. Derclaye, A. Clippe and D. Alsteens, Cell Chem. Biol., 2018, 25, 550–559 CrossRef CAS PubMed.
- H. Clausen-Schaumann, M. Seitz, R. Krautbauer and H. E. Gaub, Curr. Opin. Chem. Biol., 2000, 4, 524–530 CrossRef CAS PubMed.
- M. L. Hughes and L. Dougan, Rep. Prog. Phys., 2016, 79, 076601 CrossRef PubMed.
- M. Gaczynska and P. A. Osmulski, Curr. Opin. Colloid Interface Sci., 2008, 13, 351–367 CrossRef CAS PubMed.
- T. Haruyama, T. Uchihashi, Y. Yamada, N. Kodera, T. Ando and H. Konno, J. Mol. Biol., 2018, 430, 602–610 CrossRef CAS PubMed.
- R. Charoenwattanasatien, H. Tanaka, K. Zinzius, A. K. Hochmal, R. Mutoh, D. Yamamoto, M. Hippler and G. Kurisu, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2018, 74, 86–91 CrossRef CAS PubMed.
- M. A. B. Morais, P. O. Giuseppe, T. A. C. B. Souza, T. G. P. Alegria, M. A. Oliveira, L. E. S. Netto and M. T. Murakami, J. Biol. Chem., 2015, 290, 8582–8590 CrossRef CAS PubMed.
- C. A. Tairum, M. C. Santos, C. A. Breyer, R. R. Geyer, C. J. Nieves, S. Portillo-Ledesma, G. Ferrer-Sueta, J. C. Toledo, M. H. Toyama, O. Augusto, L. E. S. Netto and M. A. de Oliveira, Sci. Rep., 2016, 6, 33133 CrossRef CAS PubMed.
- S. Barranco-Medina, S. Kakorin, J. J. Lázaro and K.-J. Dietz, Biochemistry, 2008, 47, 7196–7204 CrossRef CAS PubMed.
- K. J. Nelson, A. Perkins, A. E. D. Van Swearingen, S. Hartman, A. E. Brereton, D. Parsonage, F. R. Salsbury, P. A. Karplus and L. B. Poole, Antioxid. Redox Signaling, 2018, 28, 521–536 CrossRef CAS PubMed.
- N. A. Yewdall, T. M. Allison, F. G. Pearce, C. V. Robinson and J. A. Gerrard, Chem. Sci., 2018, 9, 6099–6106 RSC.
- X. Roussel, G. Béchade, A. Kriznik, A. Van Dorsselaer, S. Sanglier-Cianferani, G. Branlant and S. Rahuel-Clermont, J. Biol. Chem., 2008, 283, 22371–22382 CrossRef CAS PubMed.
- T. J. Jönsson, A. W. Tsang, W. T. Lowther and C. M. Furdui, J. Biol. Chem., 2008, 283, 22890–22894 CrossRef PubMed.
- C. A. Tairum Jr., M. A. de Oliveira, B. B. Horta, F. J. Zara and L. E. S. Netto, J. Mol. Biol., 2012, 424, 28–41 CrossRef PubMed.
- K. H. Koo, S. Lee, S. Y. Jeong, E. T. Kim, H. J. Kim, K. Kim, K. Song and H. Z. Chae, Arch. Biochem. Biophys., 2002, 397, 312–318 CrossRef CAS PubMed.
- M. Jara, A. P. Vivancos and E. Hidalgo, Genes Cells, 2008, 13, 171–179 CrossRef CAS PubMed.
- L. M. Randall, J. Dalla Rizza, D. Parsonage, J. Santos, R. A. Mehl, W. T. Lowther, L. B. Poole and A. Denicola, Free Radicals Biol. Med., 2019, 141, 492–501 CrossRef CAS PubMed.
- A. Bersweiler, B. D’Autréaux, H. Mazon, A. Kriznik, G. Belli, A. Delaunay-Moisan, M. B. Toledano and S. Rahuel-Clermont, Nat. Chem. Biol., 2017, 13, 909–915 CrossRef CAS PubMed.
- L. Wildling, B. Unterauer, R. Zhu, A. Rupprecht, T. Haselgrübler, C. Rankl, A. Ebner, D. Vater, P. Pollheimer, E. E. Pohl, P. Hinterdorfer and H. J. Gruber, Bioconjugate Chem., 2011, 22, 1239–1248 CrossRef CAS PubMed.
- A. Ebner, L. Wildling, A. S. M. Kamruzzahan, C. Rankl, J. Wruss, C. D. Hahn, M. Hölzl, R. Zhu, F. Kienberger, D. Blaas, P. Hinterdorfer and H. J. Gruber, Bioconjugate Chem., 2007, 18, 1176–1184 CrossRef CAS PubMed.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109–1112 CrossRef CAS PubMed.
- M. S. Z. Kellermayer, L. Grama, Á. Karsai, A. Nagy, A. Kahn, Z. L. Datki and B. Penke, J. Biol. Chem., 2005, 280, 8464–8470 CrossRef CAS PubMed.
- D. Alsteens, C. B. Ramsook, P. N. Lipke and Y. F. Dufrêne, ACS Nano, 2012, 6, 7703–7711 CrossRef CAS PubMed.
- F. Gräter, J. Shen, H. Jiang, M. Gautel and H. Grubmüller, Biophys. J., 2005, 88, 790–804 CrossRef PubMed.
- X. Roussel, A. Kriznik, C. Richard, S. Rahuel-Clermont and G. Branlant, J. Biol. Chem., 2009, 284, 33048–33055 CrossRef CAS PubMed.
- M. Grandbois, Science, 1999, 283, 1727–1730 CrossRef CAS PubMed.
- S. Garcia-Manyes and A. E. M. Beedle, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- I. Popa, R. Berkovich, J. Alegre-Cebollada, C. L. Badilla, J. A. Rivas-Pardo, Y. Taniguchi, M. Kawakami and J. M. Fernandez, J. Am. Chem. Soc., 2013, 135, 12762–12771 CrossRef CAS PubMed.
- J. Alegre-Cebollada, R. Perez-Jimenez, P. Kosuri and J. M. Fernandez, J. Biol. Chem., 2010, 285, 18961–18966 CrossRef CAS PubMed.
- T. B. Kahn, J. M. Fernández and R. Perez-Jimenez, J. Biol. Chem., 2015, 290, 14518–14527 CrossRef CAS PubMed.
- J. Liang and J. M. Fernández, ACS Nano, 2009, 3, 1628–1645 CrossRef CAS PubMed.
- H. A. Woo, S. W. Kang, H. K. Kim, K.-S. Yang, H. Z. Chae and S. G. Rhee, J. Biol. Chem., 2003, 278, 47361–47364 CrossRef CAS PubMed.
- B. Biteau, J. Labarre and M. B. Toledano, Nature, 2003, 425, 980–984 CrossRef CAS PubMed.
- S. Ghaemmaghami, W.-K. Huh, K. Bower, R. W. Howson, A. Belle, N. Dephoure, E. K. O’Shea and J. S. Weissman, Nature, 2003, 425, 737–741 CrossRef CAS PubMed.
- S. Akter, L. Fu, Y. Jung, M. L. Conte, J. R. Lawson, W. T. Lowther, R. Sun, K. Liu, J. Yang and K. S. Carroll, Nat. Chem. Biol., 2018, 14, 995–1004 CrossRef CAS PubMed.
- S. Stöcker, M. Maurer, T. Ruppert and T. P. Dick, Nat. Chem. Biol., 2018, 14, 148–155 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2nh00037g |
| ‡ These authors contributed equally to this paper. |
| § Present address H. L. C.: LIEC, UMR 7360 CNRS-Université de Lorraine, Faculté des Sciences et Technologies, Bd des Aiguillettes, 54500 Vandœuvre-lès-Nancy, France. |
| This journal is © The Royal Society of Chemistry 2022 |