
COx conversion to aromatics: a mini-review of nanoscale performance
Guo
Tian
,
Chenxi
Zhang
 * and
Fei
Wei
*
* and
Fei
Wei
*
Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: cxzhang@mail.tsinghua.edu.cn; wf-dce@mail.tsinghua.edu.cn
First published on 26th August 2022
Abstract
The conversion of COx into value-added green aromatics is considered as a promising route to achieve the world's decarbonization due to its considerable thermodynamic driving force and atomic economy where low H/C ratio aromatics are chosen as a product. It is enabled by bifunctional nano-catalysts composed of metal oxides with abundant oxygen vacancies and acid zeolites, thus realizing superior selectivity in hydrocarbons at the single pass of COx conversion. In this mini-review, we mainly provide some thought-provoking insights at the nanoscale of this complicated process including the proximity of active sites, reaction mechanism, asymmetric desorption behavior of intermediates and final products and overall thermodynamic analysis. The facile surface diffusion of intermediates owing to the proximity of active sites stimulates the reaction, which follows an autocatalytic process. This positive feedback attributed to the autocatalytic cycle accelerates the transformation of energy and materials in the thermodynamically optimal direction, making the reaction highly selective towards the final products. This complicated coupling process, like a nano-maze constituted by these micro-environment factors, is complicated in terms of the reaction pathway but highly selective to a fixed direction guided by overall thermodynamics. Deep understanding of such an autocatalytic cycle at the nanoscale paves the way for the rational design of next-generation high-performance catalysts.
 Guo Tian | Guo Tian obtained his Bachelor degree in Chemical Engineering and Technology from the School of Chemical Engineering, East China University of Science and Technology. He is currently a PhD candidate in the major of heterogeneous catalysis at the department of Chemical Engineering, Tsinghua University, under the supervision of Prof. Fei Wei. His research is focused on the rational design of next-generation high-performance catalysts for CO2 conversion to value-added chemicals. |
 Chenxi Zhang | Chenxi Zhang received his BE from Tongji University in 2013, and his PhD from Tsinghua University in 2017. He is now an assistant professor in Prof. Fei Wei's group. His research focuses on new catalytic materials and processes of CO2 hydrogenation to sustainable aviation fuel (CO2AFTM) and its commercialization. An acid–base heterojunction catalyst combined with a hydrogenation multiphase-flow reactor was developed that not only achieves ultra-high selectivity (80 wt%) for aviation fuel but also record-breaking in terms of 95% CO2 utilization. |
 Fei Wei | Fei Wei received his PhD degree from the China University of Petroleum in 1990 and has been a full professor in Tsinghua University since 1996. His research interests include the synthesis and properties of carbon nanotubes and other carbon materials, energy storage materials, zeolite catalysis engineering for methanol/syngas/CO2 conversion and mechanisms in gas–solid flow. |
Introduction
The conversion of COx to value-added chemicals—such as light olefins (C2–4), aromatics (C8–12) and oxygenates—has gained considerable attraction recently to achieve the decarbonisation goal across the globe.1–3 This route has played a significant role in replacing existing petroleum-derived products in the petroleum industry by employing coal-based syngas or waste CO2 as the feedstock. As a promising H2 storage technology, by introducing valuable H2, the waste carbon–based feedstock can be converted to upgraded products. Bao et al.1,4,5 first synthesised composite catalysts composed of partially reduced ZnCrOx and SAPO-34 for the conversion of syngas, with ASF-distributed break selectivity. Using a simple mixed physical method, the selectivity of lower olefins can reach 80% on a single-pass CO conversion of 17%, with a long lifetime of over 200 h. Since then, bifunctional catalysts composed of metal oxides and zeolites have been widely and deeply investigated for the conversion of COx.6,7Due to stoichiometric relationships and thermodynamics, the hydrogenation of carbon dioxide/carbon monoxide for the generation of high-carbon aromatic hydrocarbons leads to lower hydrogen consumption per unit of product while ensuring higher selectivity for products. Wang et al.8,9 used ZnO–ZrO2 and H-ZSM-5 catalysts for the highly efficient conversion of COx to aromatics. It is generally accepted that metal oxides mainly activate the C–O of COx and the dissociation of H2 in this process, while the C–C coupling is manipulated within the confined acidic pores of the zeolites.10 The primary COx molecule first undergoes hydrogenation to form C1 oxygenates, such as H2CO and CH3OH.11 Then, the C–C coupling reaction, such as aldol condensation or the Prins reaction, is opened at the acid zeolite sites. Due to the matching of the molecular dynamic diameter with the pore size, the final products can be selected by using different types of zeolites.12 Therefore, unveiling the nano-scale of this complicated catalysis system is beneficial for the rational design of catalysts, thus improving the catalytic activity.
In this challenging reaction system, the nano-scale microenvironment of composite catalysts has attracted much attention. Nano-sized metal-oxide-zeolite attachment is the key factor in catalyst activity; however, the reaction mechanism or, more precisely, the key intermediates of this process, remain ambiguous. Generally, the key intermediates are C1 oxygenates, such as adsorbed H2CO*, CH3O* and gas CH3OH; however, the desorption energy barrier determined by thermodynamic analysis is also key. From the perspective of thermodynamics, lower temperatures preferentially provide high selectivity for aromatics, while high temperatures favour the COx conversion.
The reaction process comprises an autocatalysis cycle, including C–C coupling and the choice of substrates, which ensure the desired transformation of energy and materials. Several studies have suggested that the ‘core engine’ of many (if not all) chemistry systems is formed by an autocatalytic set of species that promote each other through positive feedback loops.13,14 Autocatalytic sets were originally defined in terms of chemical species interacting in chemical/biochemical systems, where reactions between interacting species catalyse enough substrates for the next reaction and the whole set of chemical reactions is self-sustaining given sufficient input of energy and essential materials.15,16 As an analogue, the conversion of C1 molecular COx into single-ring aromatics may form an autocatalytic loop if each species produces the resources needed by the next species in the loop in such a configuration that the whole set of species is self-promoting and self-sustaining under the sufficient input of energy and essential materials (carbon). The autocatalytic loop enabled by a bifunctional metal oxide within a zeolite system shows superior conversion of COx and high selectivity for final products, which accelerates the transformation of energy and materials in the thermodynamically optimal direction.
For this mini review, we selected ZnCr2O4/ZnO–ZrO2/MnOx and ZSM-5 to represent the metal oxides and the zeolite, respectively. We mainly address the proximity of the active sites, the autocatalysis reaction mechanism, the desorption behaviour of intermediates versus final products and the overall thermodynamic analysis. The elemental steps, such as the adsorption, desorption and diffusion of intermediates, are controlled by the complex reaction network constituted by the nano-environment, which is responsible for the autocatalysis loop. Note that the formation of the autocatalysis loop by this nano-environment is the key factor for the highly efficient conversion of COx and its selectivity for aromatic products, where the transformation of materials (mainly devoted to the C–C coupling) and energy are in the optimal thermodynamic direction. This mini review depicts the interesting nano-scale behaviour of hot-spot bifunctional catalysts for the highly efficient conversion of COx under mild conditions, which may lead to more innovative research on the rational design of these complicated systems and promote their future development to meet the requirements of carbon reduction targets.
Proximity of the two active sites
The proximity of the two active sites is crucial for determining the catalyst activity.17 Researchers have mainly regulated the spatial arrangement of metal oxides and zeolites to influence the catalytic activity.18,19 Wei et al.17 systematically investigated the catalytic activity and product distribution over a physically mixed composite catalyst compared to separated/layered orientated catalysts using ZnCr2O4 and H-ZSM-5. Compared to layers 1 and 2, a significant increase in aromatic selectivity and CO conversion was observed over the physically mixed catalyst, which was attributed to the close proximity of the two active sites (Fig. 1a). Moreover, the same phenomenon was observed in the composite catalyst, consisting of MnOx and H-ZSM-5 during CO conversion to aromatics (Fig. 1b).20 Changing the proximity of the two active sites from microscale to nano-scale increased the aromatic selectivity by an order of magnitude. As one of the core energy barriers is created by the dissociation of H2 and the formation of surface CHxO species, the attachment of the metal oxides to H-ZSM-5 becomes the paramount factor when modulating the coupling effects in this bifunctional system. In addition, the close proximity of the active sites at the nano-scale enables the rapid diffusion of intermediates, thus connecting the different reactions at different active sites. Moreover, this long-chain growth reaction persists as an induction period for >3 h, which is considered typical autocatalysis behaviour. Overall, the proximity of active sites promotes the rapid diffusion of intermediates, the key step in this autocatalytic cycle, thus ensuring high activity and selectivity throughout the process.21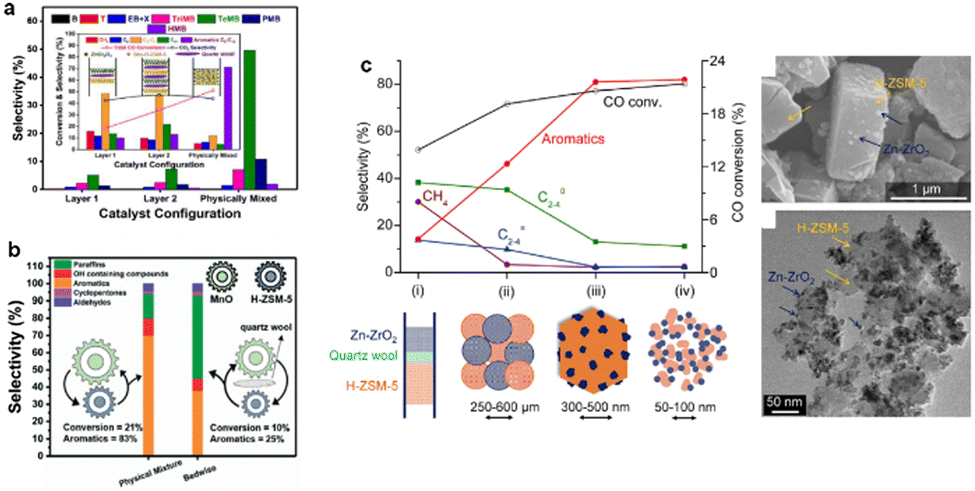 | ||
Fig. 1 (a) Product distribution and catalytic activity for different catalyst placement orientations over ZnCr2O4 and H-ZSM-5. (b) Effect of MnOx and H-ZSM-5 proximity. Reaction conditions: oxide/zeolite = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, temperature = 350 °C, pressure = 2 MPa, space velocity = 333 ml h−1 gcat−1 and syngas H2/CO = 2. (c) (i) Dual bed (0.33 g Zn–ZrO2 + 0.67 g H-ZSM-5); (ii) mixing of the two component's granules, 250–600 mm; (iii) Zn–ZrO2 nano-particles dispersed on micrometre-sized H-ZSM-5 crystallites and (iv) Zn–ZrO2 and H-ZSM-5 nano-composites. Reaction conditions: catalyst weight: 1.0 g; 673 K; H2/CO = 2:1; 3 MPa; 25 cm3 min−1. Copyright 2017, Elsevier. :1, temperature = 350 °C, pressure = 2 MPa, space velocity = 333 ml h−1 gcat−1 and syngas H2/CO = 2. (c) (i) Dual bed (0.33 g Zn–ZrO2 + 0.67 g H-ZSM-5); (ii) mixing of the two component's granules, 250–600 mm; (iii) Zn–ZrO2 nano-particles dispersed on micrometre-sized H-ZSM-5 crystallites and (iv) Zn–ZrO2 and H-ZSM-5 nano-composites. Reaction conditions: catalyst weight: 1.0 g; 673 K; H2/CO = 2:1; 3 MPa; 25 cm3 min−1. Copyright 2017, Elsevier. | ||
Controlling catalyst sizes, from the micro to the nano-scale, is an alternative method for controlling the proximity of active sites. Wang and co-workers9 prepared a series of Zn–ZrO2/H-ZSM-5 catalysts with differently sized Zn–ZrO2, ranging from 50 to 100 nm and 250 to 600 μm, respectively and found that the smaller particle sizes benefitted the generation of the final aromatic products (Fig. 1c). At nano-scale, the metal oxides attach to the surface of the H-ZSM-5 to form a local heterojunction, as shown in Fig. 1c. This indicates that the strong interaction between the metal oxides and the zeolites is key for the suppression of side products, such as CH4 and C2–4, which increases expected product yields. The Zn–ZrO2 nano-particle bifunctional catalyst containing the smallest particle size (4.8 nm) was well dispersed on the zeolites and exhibited the highest catalytic performance. The closer proximity of the two components enables the faster transfer of CH3OH/DME/H2CO intermediates formed on a typical methanol synthesis catalyst, such as ZnCr2O4/Zn–ZrO2, to H-ZSM-5 before the occurrence of unfavourable side reactions, thus favouring the selective formation of aromatics.
The main concepts of the mechanisms of catalytic performance from micro to nano-scale are as follows. Initially, the simple physical mixture composed of metal oxides and zeolites undergoes a relatively long reaction period of 3–5 h, indicating that structural evolution occurs in the attachment or migration of the two active species. Moreover, due to the metal oxide alkali sites and the zeolite acid sites, contact between the two species may form a heterojunction structure, which profoundly influences the diffusion of key intermediates. This nano-scale attachment, which forms the local novel structure, has a dramatic effect on the reaction conversion and selectivity, which is similar to an autocatalytic cycling mechanism activated by surface diffusion. Therefore, from the perspective of materials (C–C coupling), understanding the autocatalytic cycling of this reaction system is beneficial for innovative research in the C1 chemistry field.
Autocatalytic loop mechanism during COx conversion
To understand the autocatalytic loop mechanism of this complicated reaction system, some high-precision characterisation technologies, including in situ infrared transmission spectroscopy, SVUV-PIMS and GC-MS, have been used to identify the key intermediates of this reaction process.22,23 ZnCr2O4 and ZnO–ZrO2, typical methanol synthesis catalysts have been thoroughly and widely investigated for the conversion of COx to methanol.24 H2 is dissociated on metal sites and undergoes a reaction pathway with CO to form CO* → HCO* → H2CO* → H3CO* → H3COH.25,26 The reaction mechanism for solo metal oxides is clear, but when it is coupled with H-ZSM-5, the key intermediates are difficult to identify. Due to the catalytic nature of the metal oxide, H2CO or H3CO species are deemed to be the key active intermediates in this reaction system. Several studies demonstrated that the close proximity between metal oxide and H-ZSM-5 is beneficial for the diffusion of key intermediates such as H2CO/H3CO.5,27 At a relatively low temperature of 300–350 °C, these intermediates quickly diffuse into the straight pores of H-ZSM-5. Accordingly, Wei et al.28 determined that H2CO may be a crucial intermediate of the bifunctional catalyst using in situ Drifts technology. However, understanding the C–C coupling of the reaction intermediates is another challenge. The COx conversion to aromatics first undergoes the conversion of COx to oxygenates such as H2CO/CH3OH, then these intermediates go through some typical C–C coupling processes to form the final products. Therefore, the existing knowledge about the formation of the first C–C bond for C1 molecules like H2CO and CH3OH may guide the study concerning the formation of the first C–C bond in COx conversion over the bifunctional catalyst.Using multiple in situ spectroscopic and microscopic characterisation methods and theoretical calculations, Wei et al.28 found that the formation of aromatics followed an aldol–aromatic mechanism. As shown in Fig. 2(a), C1 oxygenates such as CH3OH, HCOOH and H2CO are first formed on the surface of ZnCr2O4 spinels. Because of the close proximity of ZnCr2O4 and H-ZSM-5, the C1 oxygenates quickly diffuse into ZSM-5, where they are transformed into a complex mixture of aldol condensates (C2+ species).29 These aldol species are then rapidly transformed into cyclic oxygenates (e.g. phenols, ketones and cyclic aldehydes) via an inter-/intra-molecular aldol cyclisation reaction inside the spatial constraint channels of H-ZSM-5. Furthermore, through hydrogenation/dehydrogenation and dehydration, these cyclic oxygenates are converted to single-ring aromatics (e.g. benzene, toluene and paraxylene) via an aldol–phenol-aromatic cycle inside the pores of H-ZSM-5. Therefore, an aldol–aromatic reaction pathway using a ZnCr2O4 and H-ZSM-5 bifunctional catalyst was demonstrated.
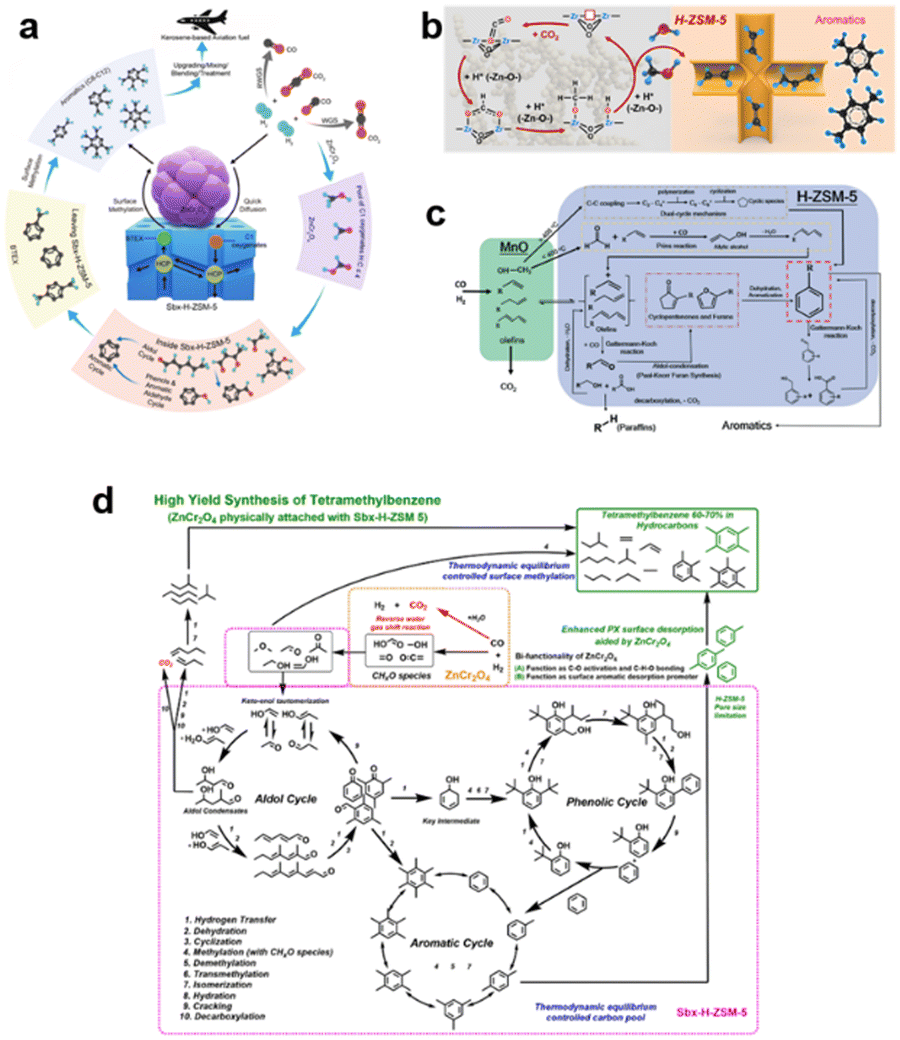 | ||
| Fig. 2 (a) Proposed reaction mechanism for the highly selective conversion of CO2 or syngas into aviation fuel precursors over the nano-sized bifunctional catalyst ZnCr2O4/Sbx-H-ZSM-5. (b) Possible mechanism for the direct hydrogenation of CO2 into aromatics over the bifunctional ZnO–ZrO2/H-ZSM-5 catalyst. (c) Proposed reaction mechanism over the MnOx/H-ZSM-5 catalyst. (d) Proposed autocatalytic mechanism of CO conversion to aromatics over ZnCr2O4/Sbx-H-ZSM-5. | ||
Wang et al.6 also systematically investigated the reaction mechanism of CO2 to C1 oxygenates for another methanol synthesis ZnO–ZrO2 catalyst. Initially, CO2 is preferentially adsorbed in the oxygen vacancies of the ZnO–ZrO2 to form 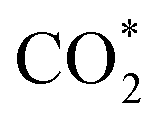 . Then, H2 dissociates on the surface of the ZnO species and H* is adsorbed on top of an O near the Zr. Subsequently, the adsorbed
. Then, H2 dissociates on the surface of the ZnO species and H* is adsorbed on top of an O near the Zr. Subsequently, the adsorbed 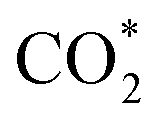 is stepwise hydrogenated to form formate (HCOO*). The HCOO* then reacts with H* to form adsorbed CH3O* species, which are further transferred into gaseous CH3OH and diffused into the H-ZSM-5 pores proceeding the C–C coupling reaction (Fig. 2b). Due to the matched straight channel (5.3 × 5.6 Å) of ZSM-5 with a kinetic diameter of benzene, toluene, and para-xylene (5.85 Å), the aromatic selectivity can be >80% in a single-pass COx conversion.30 It is generally accepted that the COx molecules are adsorbed first on the oxygen vacancies of metal oxides, and H2 is dissociated to form H* on the top metal sites. Then COx undergoes the hydrogenation reaction to form C1 oxygenates at the oxygen vacancies, final aromatics in the pores of H-ZSM-5.31
is stepwise hydrogenated to form formate (HCOO*). The HCOO* then reacts with H* to form adsorbed CH3O* species, which are further transferred into gaseous CH3OH and diffused into the H-ZSM-5 pores proceeding the C–C coupling reaction (Fig. 2b). Due to the matched straight channel (5.3 × 5.6 Å) of ZSM-5 with a kinetic diameter of benzene, toluene, and para-xylene (5.85 Å), the aromatic selectivity can be >80% in a single-pass COx conversion.30 It is generally accepted that the COx molecules are adsorbed first on the oxygen vacancies of metal oxides, and H2 is dissociated to form H* on the top metal sites. Then COx undergoes the hydrogenation reaction to form C1 oxygenates at the oxygen vacancies, final aromatics in the pores of H-ZSM-5.31
When the reaction temperature is over 400 °C, the C–C coupling reaction occurs on the surface of Mn-based oxides to form C2+ species, such as light olefins, which are transformed into cyclic species via the dual-cycle mechanism (Fig. 2c). Below 400 °C, the C–O bond is activated and hydrogenated to form CH* species when reacted with H2 from syngas. These identified hydrocarbons can be converted into aromatics on H-ZSM-5 at <300 °C through multiple reaction routes, including aldol and Prins reactions.
To more precisely describe the mechanism of the COx-to-aromatic reaction, Wei et al.17 proposed an autocatalytic cycle, including aldol, phenolic and aromatic cycles, as shown in Fig. 2d. The proposed aldol–aromatic process is an asymmetric autocatalytic cycle formed mainly by the aldol reaction.
If such an autocatalytic cycle is established, a positive feedback cycle driven by chemical potential towards aromatics is created. Discovered in the 1870s, the aldol condensation reaction is an important C–C bond formation reaction that is widely applied for the synthesis of important chemical intermediates.32,33 The reaction proceeds via a ketone enol or enolate, which attacks an aldehyde or another ketone to form a β-hydroxy aldehyde or β-hydroxy ketone (an aldol) over an acidic or basic catalytic site. This is followed by dehydration to produce an α,β-unsaturated ketone (aldol condensate). Each step of the aldol reaction is an equilibrium process, where the main products are determined by thermodynamics. This autocatalysis mechanism comprises three different cycles, with positive feedback driving the superior conversion of COx while retaining high selectivity towards the final aromatic product at relatively low temperatures.
This mechanism has been well established during the induction period, where most species are intermediates of phenols and multi-methylbenzenes. The emergence of autocatalysis in material cycles is due to the transformation of these species, which constitutes three autocatalytic loops, including aldol condensation and phenolic and aromatic loops (Fig. 3). The surface diffusion of intermediates, such as H2CO and H3CO/DME, enabled by the local heterojunction structure, opens the aldol condensation loop. Moreover, intramolecular aldol cyclisation forms single-ring oxygenates, such as phenols, which then rapidly form the phenolic loop. Under the action of the asymmetric autocatalytic behaviour of this loop, through the action of the aromatic loop, the remaining species are transferred into the aldol condensation loop. This category of autocatalytic mechanisms activated by surface diffusion accelerates the transformation of COx to aromatics both materially and energetically, which ensures that the process is selective in the optimal thermodynamic direction.
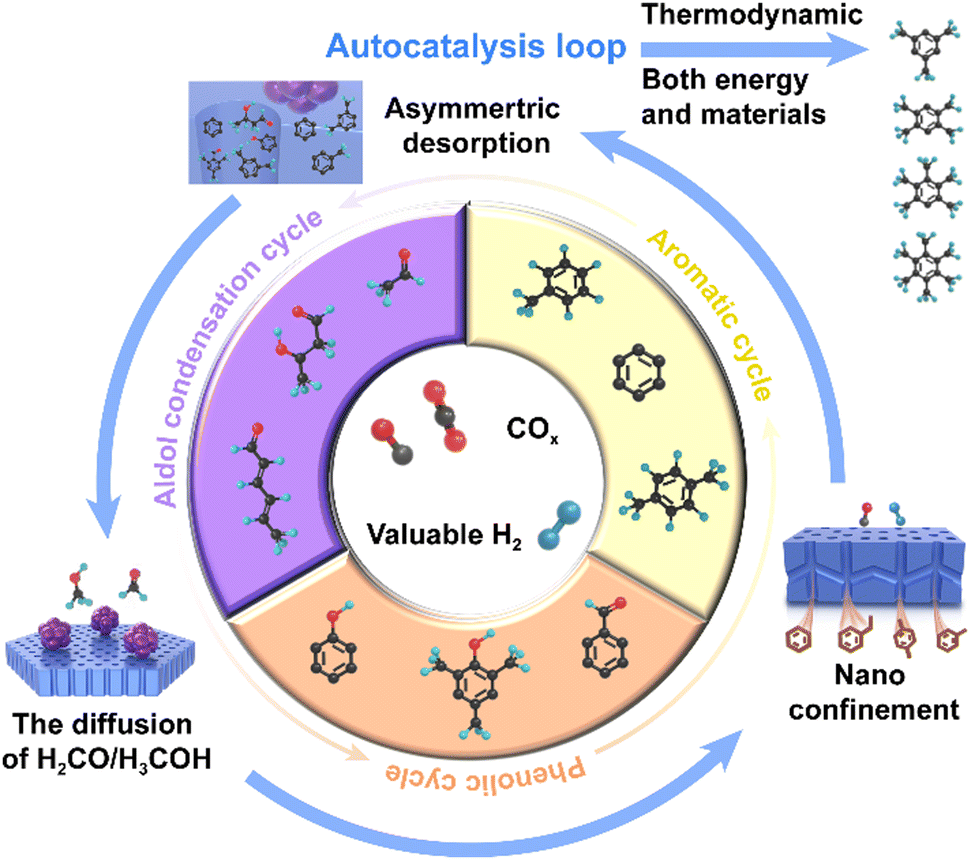 | ||
| Fig. 3 Schematic of an autocatalysis loop. | ||
Desorption behaviour between intermediates and products
From the perspective of elemental balance, the target aromatic product is located at the intersection of the C and H elements. Therefore, the O element should be discussed first. This complicated process includes oxygenate intermediates, such as formaldehyde, formate, methanol and phenols, while the final products lack oxygen.34,35 Based on this, the synthesis of aromatic processes should be highly selective towards diffusion or desorption behaviour between intermediates and products. It is hypothesised that the coupling effects of ZnCr2O4 and H-ZSM-5 can stimulate the in situ separation of oxygenated and nonoxygenated hydrocarbons.To describe the desorption behaviour of molecules confined to the straight pores of H-ZSM-5, Wei et al.28 developed thermogravimetric analysis technology combined with integrated differential phase contrast (iDPC)–STEM to quantitatively measure the activation energy of the molecules desorbed from H-ZSM-5. The TEM images reveal that the components of the composite catalysts (i.e. ZnCr2O4 and H-ZSM-5) are physically attached to each other, as shown in Fig. 4a. Due to this coupling effect of the two catalysts, C1 oxygenates, such as CHxO species, formed over the ZnCr2O4 catalyst, are rapidly transferred to H-ZSM-5 through surface diffusion and transform into carbon pool species. By profiling the carbon pool intensity in the pores of spent H-ZSM-5, Wei et al.28 identified the decay of carbon pool species by increasing the distance from the attached ZnCr2O4 particles by 20–25 nm, which indicates that the metal oxide can promote the desorption of aromatic hydrocarbons from H-ZSM-5. The enhancement of desorption causes carbon pool decay and prevents the over saturation of aromatic species in the carbon pool.36 In addition, through the inhibition of the formation of multi-ring/polycyclic aromatic compounds, the lifespans of the composite catalysts are extended, thus preventing pore blockages and de-alkylation side reactions.
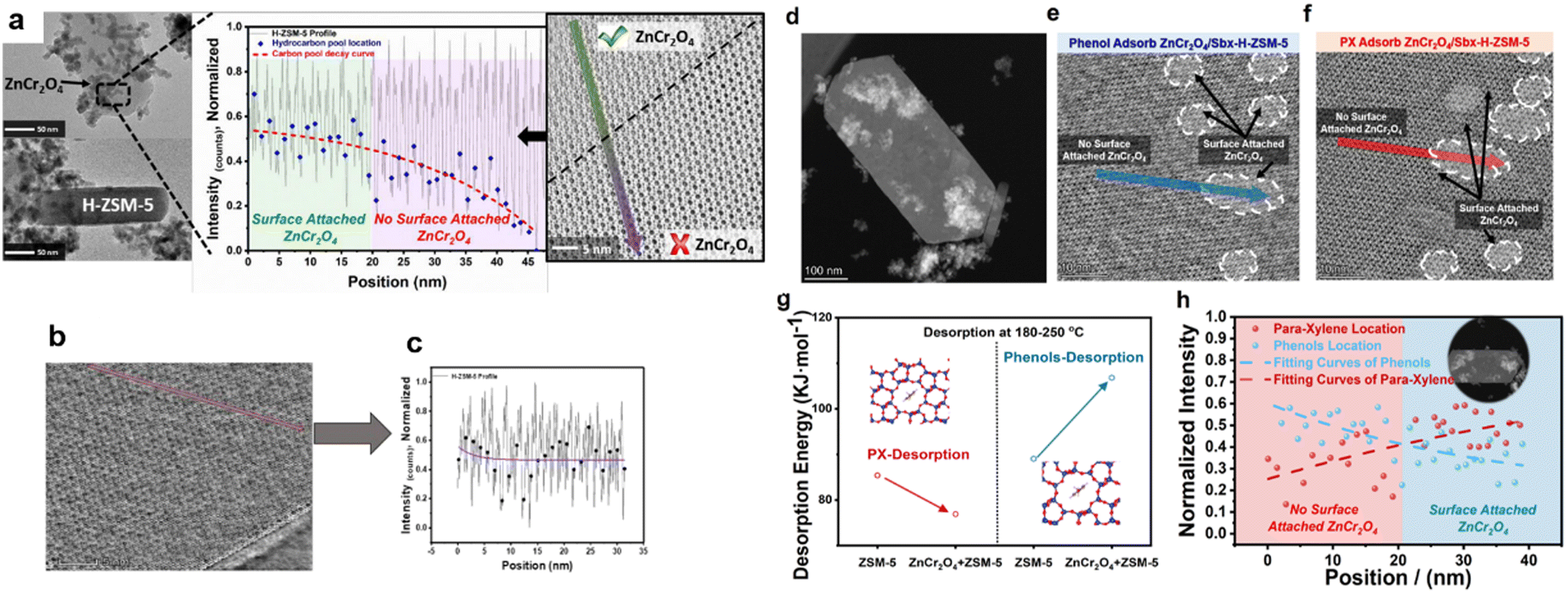 | ||
| Fig. 4 (a) TEM and integrated differential phase contrast (iDPC)–STEM images and intensity profile of the spent composite catalyst. (b and c) iDPC image of spent H-ZSM-5 showing filled pores and a carbon intensity profile within a single crystal of H-ZSM-5 and MnOx. (d) STEM image of the ZnCr2O4 and H-ZSM-5 physically mixed composite catalyst. (e) iDPC-STEM image of phenol adsorption on ZnCr2O4/H-ZSM-5. (f) iDPC-STEM image of paraxylene adsorption on ZnCr2O4/H-ZSM-5. (g) Calculated activation energies of phenol and paraxylene desorption from H-ZSM-5 with and without ZnCr2O4. (h) Intensity profiles of phenol and paraxylene in H-ZSM-5 with and without ZnCr2O4. | ||
We also investigated Mn-based oxide spent composite catalysts. The location of the H-ZSM-5 hydrocarbon pool is shown in Fig. 4b. Analysis of the carbon pool intensity profile shows that the hydrocarbon pool species are increased by surface-attached MnOx. From the perspective of desorption behaviour in aromatic products, the synergetic effects of the metal oxide and H-ZSM-5 promote the rapid desorption of the final products, which makes the reaction process highly selective for aromatics.
To confirm the separation effects of oxygenated and nonoxygenated hydrocarbons, Wei et al. selected phenol and paraxylene (PX) as the oxygenated and nonoxygenated hydrocarbon species, respectively, for adsorbtion on the catalyst. The STEM images (Fig. 4(d–f)) show the morphology of H-ZSM-5 with and without the attachment of ZnCr2O4. The desorption activation energy, determined using the thermogravimetric technique and Kissinger equation, shows that the attachment of ZnCr2O4 with H-ZSM-5 initially significantly decreases the desorption energy of PX from 85.4 to 76.9 kJ mol−1 at ∼200 °C, while the desorption energy of phenol increases from 89.05 to 106.83 kJ mol−1 (Fig. 4g). These results indicate an apparent trapping effect between the oxygenated and nonoxygenated hydrocarbons. The high desorption activation energies of the oxygenated hydrocarbons and the trapping effect of ZnCr2O4 retain the oxygenated species inside the H-ZSM-5 pores. The trapped oxygenated species increases the aromatic selectivity and ultimately enhances the activity and stability of the bifunctional catalyst. Furthermore, the iDPC-STEM result reveals the desorption behaviour of PX or phenol using the bifunctional catalyst. After the adsorption of PX and phenol, the intensity profile of the [010] surface of H-ZSM-5 shows an inverse trend when heated to 180 °C (Fig. 4h). The concentration of PX decreases when the distance from the attached ZnCr2O4 particle increases, which is the opposite to the trend observed in the phenol case. This phenomenon observed by Wei et al.28 indicates that the ZnCr2O4 oxide aids in the desorption of PX (nonoxygenated hydrocarbons) but does not play a role in the case of phenol (oxygenated hydrocarbons). Therefore, oxygenated hydrocarbons are retained inside the hydrocarbon pool and provide additional active sites for the formation of aromatics rather than becoming a part of the final product distribution. Thus, the configuration of a bifunctional catalyst (ZnCr2O4/H-ZSM-5) forms an in situ separation layer that selectively acts as a permeable membrane by only allowing the passage of desired products.
This unique distribution of metal oxide on the surface of the H-ZSM-5 leads to superior catalytic activity due to desorption between the intermediates and the final products. Such reaction complexity and high selectivity for a single product is similar to typical bioprocesses: highly complex biological reactions in terms of detailed reaction pathways or diffusion processes but very selective for the target product.37 Therefore, emphasising the key reaction steps of this process supports the rational design of metal oxide and zeolite combinations to improve the reactivity during COx conversion.
Thermodynamic control
Generally, the direction of a reaction is guided by its thermodynamics and adjusting the reaction temperature changes the product distribution and Gibbs free energy. The thermodynamic calculation of the conversion of COx to C1 oxygenates (e.g. CH3OH) can be used to analyse the conversion of COx to aromatics over bifunctional catalysts. Nowadays, the large-scale commercial production of methanol is mainly from syngas (a mixture of CO and H2), which is generated by fossil resources (mainly coal and natural gas) via processes including the gasification of coal and the steam reforming of natural gas.38 Small amounts of CO2 (about 2–8%) are typically added to the CO/H2 stream to bring the H/C ratio to the desired stoichiometry and to accelerate the reaction rate.Methanol formation from CO:
| CO + 2H2 = CH3OH ΔH298K = −90.6 kJ mol−1 |
| CO2 + 3H2 = CH3OH + H2O ΔH298K = −49.5 kJ mol−1 |
| CO2 + H2 = CO + H2O ΔH298K = 41.2 kJ mol−1 |
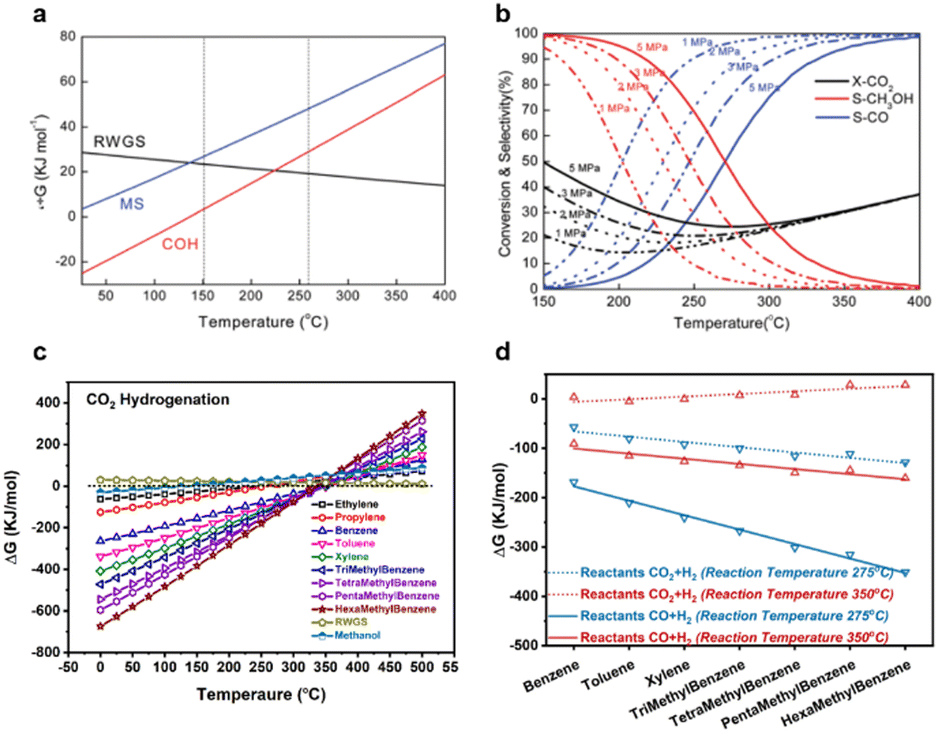 | ||
| Fig. 5 (a) Gibbs free energy of methanol synthesis from CO2, the RWGS and syngas hydrogenation. Copyright 2020, Royal Society of Chemistry. (b) Equilibrium conversion-selectivity values for the CO2-hydrogenation reaction at various pressures. Copyright 2020, Royal Society of Chemistry. (c) Gibbs free energy of products over 0–500 °C during CO2 hydrogenation. (d) Gibbs free energy of aromatic products at 275 °C and 350 °C during CO2 and CO hydrogenation reactions. | ||
Thermodynamic analysis shows that the optimised temperature of methanol synthesis is about 250 °C. As a key intermediate of the conversion of COx to aromatics, this implies that low temperatures may favour the final single-ring product. To investigate the influence of reaction temperature on product selectivity, Wei et al.28 calculated Gibbs free energy changes during the formation of C6–C12 aromatics in COx hydrogenation reactions. Fig. 5c shows that thermodynamics significantly determines the selectivity of products, as the distribution of products is changed by Gibbs free energy changes with respect to variations in the reaction temperature. As shown in Fig. 5d, the change in Gibbs free energy from benzene to hexamethylbenzene is reduced remarkably with the decreasing reaction temperature, from 350 °C to 275 °C, thus favouring the formation of aromatics and demonstrating that CH3OH is a key intermediate. The formation of aromatics at relatively low temperatures means that this process is not controlled by the typical dual-cycle reaction mechanism or carbene, which requires a reaction temperature >400 °C.44–47 This means that in contrast to a typical MTA mechanism, intermediate species, such as methoxy, formate and formaldehyde, are responsible for generating early HCP species, or strictly, the aldol-aromatic mechanism proposed by Wei et al.28,48 Thus, the proximity of the highly active nano-sized metal oxide-zeolite catalyst is key for the high selectivity of aromatics.
In summary, thermodynamic equilibrium controls the overall distribution of the final product, including regulating and guiding the direction of this complicated process. CO2 or biomass-derived syngas significantly enhances the capability to synthesise value-added aromatic products at high yields via control of the thermodynamic equilibrium.
Discussion
In general, some kinetic effects including the reaction temperature and pressure are very crucial to influence the reaction process, final product selectivity and total COx conversion. As a typical tandem reaction on a bifunctional catalyst composed of metal oxide and zeolite, it has been reported that high pressure can favour the CO conversion but can consequently enhance side reactions such as excessive hydrogenation to decrease aromatic selectivity.10,34 Xie et al.49 systematically investigated the reaction pressure on the CO conversion over the recrystallized catalyst comprising of Cr2O3 and H-ZSM-5. The CO conversion increased remarkably from 22.4% to 49.4% over the recrystallized catalyst, whereas a very slight increase in CO conversion with a considerable selectivity in CH4 and CH3OH was observed over the catalysts. Moreover, it is generally accepted that a relatively low temperature favours the total aromatic selectivity but sacrificing the single pass COx conversion.9,17 By lowering the reaction temperature from 350 °C to 275 °C, Wei et al.28 achieved the highest aromatic selectivity of >80% in total hydrocarbons for COx conversion to aromatics while the CO/CO2 conversion decreased from 34% to 16%, and 35% to 14%, respectively. Understanding the relationship of kinetic control on COx conversion paves the way for the rational design of next-generation catalyst to break the tradeoff between the activity and final product selectivity.Decreasing the proximity of metal oxide and zeolites from micro into nano size is an efficient method to improve the selectivity of aromatics. Deep investigation into this phenomenon indicates that the physical mixture composed of metal oxides and zeolites forms a heterojunction structure, which ensures the quick diffusion of key intermediates from the surface of metal oxides into the acidic pores of zeolites to open the C–C coupling reaction.27,50 Based on above acknowledgement, the core strategy to improve the selectivity of aromatics may be attributed to increase the attachment of metal oxides on the surface of zeolites to form more heterojunction structures. Using some typical surfactants such as polyvinylpyrrolidone (PVP, K-30) to initiate the self-assembly between metal oxides and zeolites to form the heterojunction structure may be a feasible method. Since surfactants, a unique class of surface-active molecules, possess remarkable ability to control the crystal growth and direct it in shape and size-controllable manner, the fine-tuning of the desired heterojunction structure may be achieved by controlling the surfactant architecture as well as its self-assembly behaviour. Moreover, encapsulating the metal oxides into the zeolites may be another method to achieve the selectivity of aromatics. By combining the ordered pore structure of zeolites and crystalline nature of metal oxides, it requires a closer diffusion distance between the two active sites. Therefore, the essential concept of designing next-generation high-performance catalyst for COx conversion is to strengthen the connection between the metal oxides and zeolites, further lowering the diffusion barrier of key intermediates.
The mechanism of CO and CO2 hydrogenation to aromatics is similar due to the diffusion of key intermediates H2CO from the surface of metal oxide to the zeolites via aldol reactions to open C–C coupling. However, the two processes have some differences. To begin with, owing to the water gas shift (WGS) reaction, the main by-product of CO hydrogenation is CO2 at a selectivity of 40–50% over the Cr-based catalyst. Moreover, as a result of the reverse water gas shift (RWGS) reaction, the main by-product of CO2 hydrogenation is CO at a selectivity of 70–80%.28 It indicates that the process of CO2 hydrogenation is accompanied by the production of considerable amounts of water. Several studies demonstrated that the competitive adsorption of water and reactant molecules over the active sites exhibits a certain influence on the conversion of reactants and the selectivity of products. Therefore, selective and rapid removal of water product from the CO2 hydrogenation system has been a highly desirable pathway toward boosting the catalytic performance that are restricted by water kinetically. Furthermore, there are some differences between the reactant molecules CO and CO2. In general, in comparison with CO, CO2 is an inherently more inert molecule due to the stable carbon![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) oxygen bond. As a result, the H2/COx ratio in the reaction system is different wherein CO hydrogenation requires less H2 (CO/H2 = 1:1–1:2) while CO2 is accompanied with more H2 (CO2/H2 = 1:3–1:4). Unveiling the differences between CO hydrogenation and CO2 hydrogenation provides a prototype to make these two processes more competitive for the existing industrial routes, further alleviating the environmental burden.
oxygen bond. As a result, the H2/COx ratio in the reaction system is different wherein CO hydrogenation requires less H2 (CO/H2 = 1:1–1:2) while CO2 is accompanied with more H2 (CO2/H2 = 1:3–1:4). Unveiling the differences between CO hydrogenation and CO2 hydrogenation provides a prototype to make these two processes more competitive for the existing industrial routes, further alleviating the environmental burden.
Challenges and perspectives
This paper has mainly reviewed bifunctional catalysts composed of metal oxides and zeolites, with a focus on ZnCr2O4 and H-ZSM-5 for COx conversion to value-added aromatics. This route provides an alternative method for producing aromatics via a carbon-neutral route for the conversion of COx, which can reduce carbon emissions. However, a ‘blackbox’, where the catalysis occurs, is required to make this a competitive process for existing industrial processes; therefore, there are vast opportunities accompanying this challenge.First, the proximity of the two active sites at the nano-scale is considered a key factor in this coupling effect. To regulate the distance of active sites above metal oxides and zeolites, many researchers have carried out a series of catalyst performance experiments, including physical mixture and layered attachment. However, there is no accurate descriptor for the proximity of two geometrically separated catalytic sites or, more strictly, the chemical nature of the proximity of two active sites. There may be interaction or chemical species changes between two functions upon close contact at the nano-scale, particularly for catalysts containing surface-migrating species, which can significantly modify the properties of the active sites and the reaction pathway. In addition, the formation of metal oxide nano-attachments with ZSM-5 benefits the diffusion of C1 oxygenate intermediates, such as H2CO/H3CO. The diffusion of C1 oxygenates from the surface of the metal oxide into the ZSM-5 pores opens the C–C coupling by aldol or Prins reactions. When the reaction species undergoes ring closure, the metal oxides can facilitate the desorption of the final products. This surface diffusion of species is responsible for the formation of an autocatalytic loop, further accelerating the conversion of COx into value-added aromatics under the direction of thermodynamics in terms of energy and materials. Understanding the formation process and limitations of this autocatalytic loop would provide a prototype for the enhanced conversion of C1 molecules, which would make it more competitive than the existing aromatic production route.
Second, taking the conversion of COx to aromatics as an example, the autocatalytic loop should be further investigated due to its complicated reaction conditions, such as the high reaction temperature and high pressure. Understanding the inhibition present in this autocatalytic loop will play a significant role in accelerating the entire process. To begin with, the local formation of the heterojunction structure under the interaction between metal oxides and zeolites is a crucial factor for limiting the autocatalytic loop, indicating that the surface diffusion of H2CO/CH3OH/DME is a bottleneck of the autocatalytic cycle. Moreover, the formation of C1 species also makes sense. Several studies indicate that by changing the ratio of metal oxides and zeolites, huge differences in product distribution are observed. Therefore, determining how to remove bottlenecks from the autocatalytic loop is key to accelerating the conversion and selectivity of this process. Developing DFT studies together with highly sensitive in situ/operando spectroscopic techniques may be a feasible method for discovering deeper insights, which will provide reliable results for both high temperature and pressure conditions. Revealing the mechanism of the first C–C bond and the reaction pathway of this complicated process will pave the way for improving the determining step of the reaction, which will significantly improve one-pass CO conversion without degrading selectivity and stability under high space velocity, which would make this process far more competitive than the existing industrial process.
Third, due to the physical mixture properties, the conversional design of such bifunctional catalysts often leads to uncontrolled and non-ideal spatial distributions of the metal oxides inside/on the zeolites, which limits their catalytic performance. The rapid diffusion of intermediates is enabled by the physical attachment of two species at the nano-scale, where this chemical structure is formed by the contribution of a reduction atmosphere, such as H2/CO/CO2. Note that co-precipitating the metal oxide and ZSM-5 precursors does not make sense because the heterojunction formed by the mutual migration of two catalysts at nano-scale improves the catalytic efficiency of the process by orders of magnitude. Therefore, in situ construction of the attachment structure that combines metal oxides and zeolites may be the next research issue, aimed at enhanced catalytic activity for a desired sequence.
Fourth, from the perspective of thermodynamic equilibrium, achieving the high selectivity of aromatics under relatively low temperatures <275 °C is feasible. However, in general, the kinetic conversion of reactants over bifunctional catalysts is low due to the weak activation ability of COx and H2 under mild reaction conditions. Introducing highly active metals, such as Fe, Co and Mn, to existing metal oxides to enhance C–O activation and H2 dissociation may be a promising strategy. Moreover, by combining machine learning and theoretical calculations, high-throughput screening may provide a novel prototype for the rational design of the desired catalysts. Therefore, developing catalysts with high catalytic activity at low temperatures on the basis of existing catalysts is the core of future academic research and industrialisation.
To summarise, the reaction process reviewed here can be linked to a nano-maze that includes nano-confinement, the proximity of active sites, reaction mechanisms and the asymmetric desorption of intermediates and products, which make this procedure more complicated but highly selective for the resulting products. This is similar to typical biomass or evolution processes, where the reaction is highly complex in terms of the reaction pathway but very selective for the target direction. Under overall thermodynamic control, the C1 molecules, including mainly CO and CO2, can find an efficient reaction pathway to form the final products over the bifunctional catalysts composed of ZnCr2O4 and H-ZSM-5. Therefore, it will not be surprising to see new processes developed for the selective synthesis of other high-value-added chemicals, especially aromatic oxygenates, in combination with the search for the thermodynamic and rational design of multiple functional catalysts.
Author contributions
Conceptualization and supervision: C. Z and F. W. Investigation, resources, and visualization: G. T and C. Z. Writing – original draft: G. T. Writing – review and editing: G. T, C. Z and F. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21908125 and 22005170), the National Key Research and Development Program of China (2018YFB0604801), the Key Research and Development Program of Inner Mongolia and Ordos, and CNPC Innovation Funds.References
- F. Jiao, et al., Selective conversion of syngas to light olefins, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- L. Zhong, et al., Cobalt carbide nanoprisms for direct production of lower olefins from syngas, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- B. Zhao, et al., Direct transformation of syngas to aromatics over Na-Zn-Fe5C2 and hierarchical HZSM-5 tandem catalysts, Chem, 2017, 3, 323–333 CAS.
- X. Pan, F. Jiao, D. Miao and X. Bao, Oxide–Zeolite-Based Composite Catalyst Concept That Enables Syngas Chemistry beyond Fischer–Tropsch Synthesis, Chem. Rev., 2021, 121, 6588–6609 CrossRef CAS PubMed.
- J. Yang, X. Pan, F. Jiao, J. Li and X. Bao, Direct conversion of syngas to aromatics, Chem. Commun., 2017, 53, 11146–11149, 10.1039/c7cc04768a.
- C. Zhou, et al., Highly Active ZnO–ZrO2 Aerogels Integrated with H-ZSM-5 for Aromatics Synthesis from Carbon Dioxide, ACS Catal., 2019, 10, 302–310, DOI:10.1021/acscatal.9b04309.
- J. Zuo, et al., Selective methylation of toluene using CO2 and H2 to para-xylene, Sci. Adv., 2020, 6(34) DOI:10.1126/sciadv.aba5433.
- W. Zhou, et al., New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- K. Cheng, et al., Bifunctional catalysts for one-step conversion of syngas into aromatics with excellent selectivity and stability, Chem, 2017, 3, 334–347 CAS.
- Z. Li, et al., Highly selective conversion of carbon dioxide to aromatics over tandem catalysts, Joule, 2019, 3, 570–583 CrossRef CAS.
- T. Li, T. Shoinkhorova, J. Gascon and J. Ruiz-Martinez, Aromatics production via methanol-mediated transformation routes, ACS Catal., 2021, 11, 7780–7819 CrossRef CAS.
- N. Li, et al., High-quality gasoline directly from syngas by dual metal oxide–zeolite (OX-ZEO) catalysis, Angew. Chem., 2019, 131, 7478–7482 CrossRef.
- P. A. Julien, et al., In situ monitoring of mechanochemical synthesis of calcium urea phosphate fertilizer cocrystal reveals highly effective water-based autocatalysis, Chem. Sci., 2020, 11, 2350–2355 RSC.
- L. Shi, et al., Combined prokaryotic–eukaryotic delivery and expression of therapeutic factors through a primed autocatalytic positive-feedback loop, J. Controlled Release, 2016, 222, 130–140 CrossRef CAS PubMed.
- M. Dorigo, V. Maniezzo and A. Colorni, The ant system: An autocatalytic optimizing process, ( 1991) Search PubMed.
- J. J. Tyson, Classification of instabilities in chemical reaction systems, J. Chem. Phys., 1975, 62, 1010–1015 CrossRef CAS.
- M. T. Arslan, et al., Selective Conversion of Syngas into Tetramethylbenzene via an Aldol-Aromatic Mechanism, ACS Catal., 2020, 10, 2477–2488, DOI:10.1021/acscatal.9b03417.
- M. T. Arslan, et al., Single-Step Conversion of H2-Deficient Syngas into High Yield of Tetramethylbenzene, ACS Catal., 2019, 9, 2203–2212, DOI:10.1021/acscatal.8b04548.
- X. Yang, X. Su, D. Chen, T. Zhang and Y. Huang, Direct conversion of syngas to aromatics: a review of recent studies, Chin. J. Catal., 2020, 41, 561–573 CrossRef CAS.
- S. Z. A. Gilani, et al., Two-way desorption coupling to enhance the conversion of syngas into aromatics by MnO/H-ZSM-5, Catal. Sci. Technol., 2020, 10, 3366–3375 RSC.
- M. P. Veldhuis, M. P. Berg, M. Loreau and H. Olff, Ecological autocatalysis: a central principle in ecosystem organization?, Ecol. Monogr., 2018, 88, 304–319 CrossRef.
- T. Wang, et al., ZnZrOx integrated with chain-like nanocrystal HZSM-5 as efficient catalysts for aromatics synthesis from CO2 hydrogenation, Appl. Catal., B, 2021, 286, 119929 CrossRef CAS.
- Y. Wang, et al., Direct and Oriented Conversion of CO2 into Value-Added Aromatics, Chemistry, 2019, 25, 5149–5153, DOI:10.1002/chem.201806165.
- J. Su, et al., Syngas to light olefins conversion with high olefin/paraffin ratio using ZnCrOx/AlPO-18 bifunctional catalysts, Nat. Commun., 2019, 10, 1297, DOI:10.1038/s41467-019-09336-1.
- Z. Zhang, et al., The active sites of Cu–ZnO catalysts for water gas shift and CO hydrogenation reactions, Nat. Commun., 2021, 12, 4331, DOI:10.1038/s41467-021-24621-8.
- S. Ma, S.-D. Huang and Z.-P. Liu, Dynamic coordination of cations and catalytic selectivity on zinc–chromium oxide alloys during syngas conversion, Nat. Catal., 2019, 2, 671–677, DOI:10.1038/s41929-019-0293-8.
- Y. Ji, et al., Oxygenate-based routes regulate syngas conversion over oxide–zeolite bifunctional catalysts, Nat. Catal., 2022, 1–11 Search PubMed.
- M. T. Arslan, et al., Highly Selective Conversion of CO2 or CO into Precursors for Kerosene-Based Aviation Fuel via an Aldol–Aromatic Mechanism, ACS Catal., 2022, 12, 2023–2033 CrossRef CAS.
- Y. Wang, et al., Rationally Designing Bifunctional Catalysts as an Efficient Strategy To Boost CO2 Hydrogenation Producing Value-Added Aromatics, ACS Catal., 2018, 9, 895–901, DOI:10.1021/acscatal.8b01344.
- H. Xiong, et al., In situ imaging of the sorption-induced subcell topological flexibility of a rigid zeolite framework, Science, 2022, 376, 491–496 CrossRef CAS PubMed.
- J. Zhang, et al., Hydrogenation of CO2 into aromatics over a ZnCrOx-zeolite composite catalyst, Chem. Commun., 2019, 55, 973–976, 10.1039/c8cc09019j.
- T. D. Machajewski and C. H. Wong, The catalytic asymmetric aldol reaction, Angew. Chem., Int. Ed., 2000, 39, 1352–1375 CrossRef CAS.
- J. I. Matsuo and M. Murakami, The Mukaiyama aldol reaction: 40 years of continuous development, Angew. Chem., Int. Ed., 2013, 52, 9109–9118 CrossRef CAS PubMed.
- Y. Ni, et al., Selective conversion of CO2 and H2 into aromatics, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- Y. Xu, et al., Selective production of aromatics from CO2, Catal. Sci. Technol., 2019, 9, 593–610 RSC.
- X. Cui, et al., Selective production of aromatics directly from carbon dioxide hydrogenation, ACS Catal., 2019, 9, 3866–3876 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Domino electroreduction of CO2 to methanol on a molecular catalyst, Nature, 2019, 575, 639–642, DOI:10.1038/s41586-019-1760-8.
- J. Zhong, et al., State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- T. Lin, et al., Direct Production of Higher Oxygenates by Syngas Conversion over a Multifunctional Catalyst, Angew. Chem., Int. Ed., 2019, 58, 4627–4631, DOI:10.1002/anie.201814611.
- S. Kasipandi and J. W. Bae, Recent Advances in Direct Synthesis of Value-Added Aromatic Chemicals from Syngas by Cascade Reactions over Bifunctional Catalysts, Adv. Mater., 2019, 31, e1803390, DOI:10.1002/adma.201803390.
- J. Hu, et al., Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol, Nat. Catal., 2021, 4, 242–250, DOI:10.1038/s41929-021-00584-3.
- Q. Sun, N. Wang and J. Yu, Advances in Catalytic Applications of Zeolite-Supported Metal Catalysts, Adv. Mater., 2021, 33, e2104442, DOI:10.1002/adma.202104442.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, Optimizing Binding Energies of Key Intermediates for CO2 Hydrogenation to Methanol over Oxide-Supported Copper, J. Am. Chem. Soc., 2016, 138, 12440–12450, DOI:10.1021/jacs.6b05791.
- J. Li, et al., A route to form initial hydrocarbon pool species in methanol conversion to olefins over zeolites, J. Catal., 2014, 317, 277–283, DOI:10.1016/j.jcat.2014.05.015.
- M. D. Porosoff, B. Yan and J. G. Chen, Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities, Energy Environ. Sci., 2016, 9, 62–73, 10.1039/c5ee02657a.
- A. M. Starik, N. S. Titova and S. A. Torokhov, Kinetics of oxidation and combustion of complex hydrocarbon fuels: Aviation kerosene, Combust., Explos. Shock Waves, 2013, 49, 392–408, DOI:10.1134/s0010508213040023.
- L. Yang, et al., Role of Acetaldehyde in the Roadmap from Initial Carbon–Carbon Bonds to Hydrocarbons during Methanol Conversion, ACS Catal., 2019, 9, 6491–6501, DOI:10.1021/acscatal.9b00641.
- Z. Chen, et al., Coupling of Methanol and Carbon Monoxide over H-ZSM-5 to Form Aromatics, Angew. Chem., Int. Ed., 2018, 57, 12549–12553, DOI:10.1002/anie.201807814.
- C. Liu, et al., Constructing directional component distribution in a bifunctional catalyst to boost the tandem reaction of syngas conversion, Chem. Catal., 2021, 1, 896–907 CrossRef.
- D. Zhao, et al., In situ formation of ZnOx species for efficient propane dehydrogenation, Nature, 2021, 599, 234–238, DOI:10.1038/s41586-021-03923-3.
| This journal is © The Royal Society of Chemistry 2022 |